
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 01 March 2023
Sec. Genitourinary Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1129140
 Katie Joanna Miller†Isla Henry†
Katie Joanna Miller†Isla Henry† Zoe Maylin*
Zoe Maylin* Christopher SmithEinthavy Arunachalam
Christopher SmithEinthavy Arunachalam Hardev Pandha
Hardev Pandha Mohammad Asim*
Mohammad Asim*Persistent androgen receptor (AR) signalling is the main driver of prostate cancer (PCa). Truncated isoforms of the AR called androgen receptor variants (AR-Vs) lacking the ligand binding domain often emerge during treatment resistance against AR pathway inhibitors such as Enzalutamide. This review discusses how AR-Vs drive a more aggressive form of PCa through the regulation of some of their target genes involved in oncogenic pathways, enabling disease progression. There is a pressing need for the development of a new generation of AR inhibitors which can repress the activity of both the full-length AR and AR-Vs, for which the knowledge of differentially expressed target genes will allow evaluation of inhibition efficacy. This review provides a detailed account of the most common variant, AR-V7, the AR-V7 regulated genes which have been experimentally validated, endeavours to understand their relevance in aggressive AR-V driven PCa and discusses the utility of the downstream protein products as potential drug targets for PCa treatment.
Prostate cancer (PCa) is the second most common cancer in men worldwide, accounting for over 1.4 million new cases every year, leaving one in every six being affected by the disease within their lifetime (1) (2). The prostate is a small walnut-sized gland within the male reproductive system, located underneath the bladder and surrounding the urethra. The growth of prostate epithelial cells is regulated by the actions of androgens which manifest their biological effects via the androgen receptor, a member of the nuclear hormone receptor superfamily (3). The AR is a ligand-activated transcription factor (TF) and drives genes associated with proliferation and cell survival along with other target genes such as KLK3 and TMPRSS2 (2) (Figure 1). Under physiological conditions, AR signalling is responsible for the development and maintenance of the male reproductive system, as well as cell proliferation and apoptosis of prostate epithelial cells (4, 5). However, the uncontrolled overstimulation of AR signalling is considered a major driver of PCa through the regulation of an oncogenic gene signature which supports uncontrolled cell growth. This can further progress to a pre-cancerous condition known as prostate intraepithelial neoplasia (PIN) and subsequent tumour formation (2, 6).
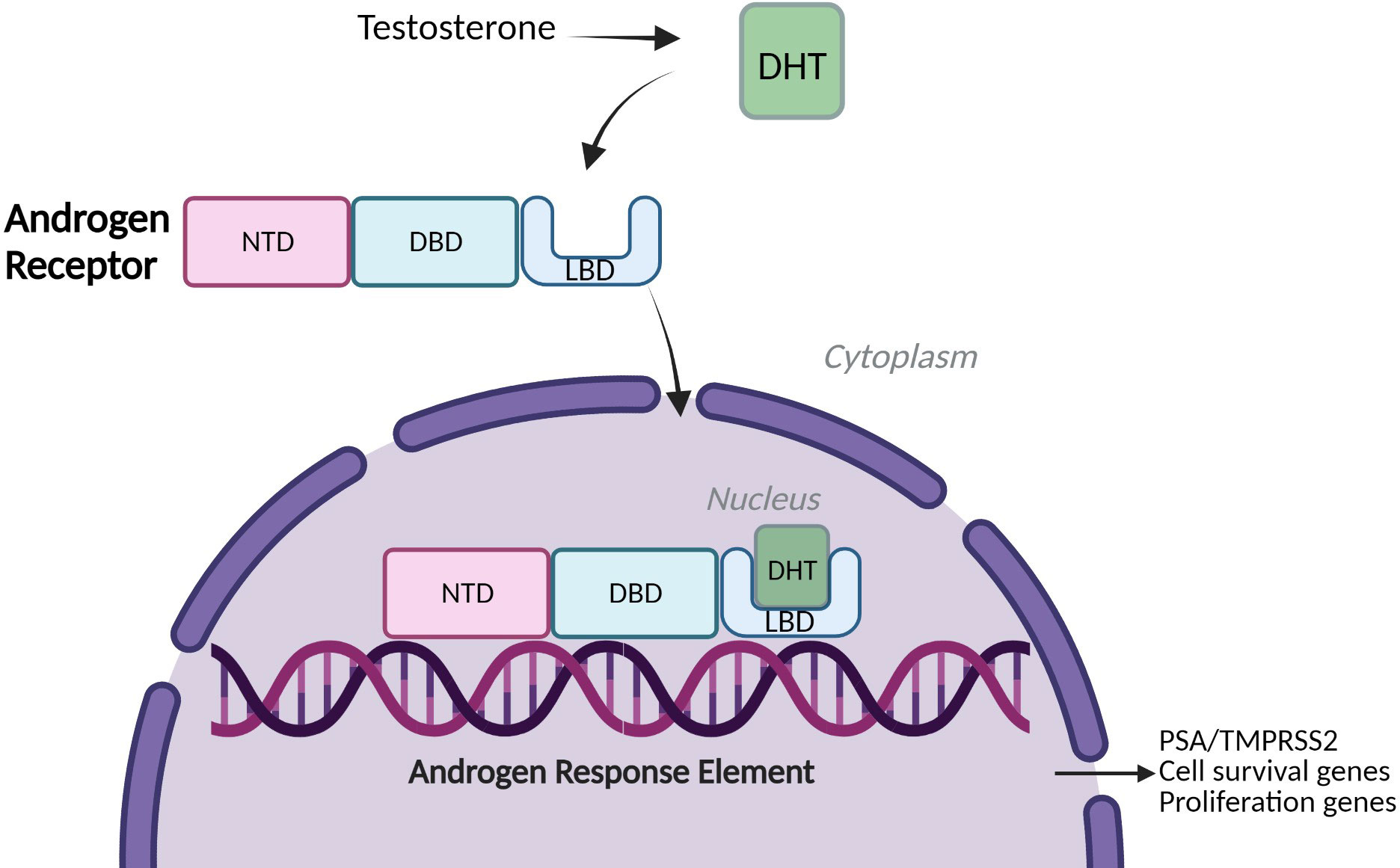
Figure 1 Androgen Receptor signalling. A simplified schematic of Androgen Receptor (AR) Signalling: Full-Length Androgen Receptor, a transcription factor binds the testosterone derivative, Dihydrotestosterone (DHT) in the cytoplasm. The AR translocates to the nucleus, binds to Androgen Response Elements (AREs) on DNA and drives expression of cell survival and proliferation genes and target genes such as PSA (KLK3) and TMPRSS2 (2). NTD= amino-terminal domain, DBD= DNA-binding domain, LBD= Ligand-binding domain.
The androgen receptor itself is regulated by a number of transcription factors and related proteins. Transcription factors CREB, Myc, Sp1, Foxo3a, LEF1, NF-ĸB, Twist1, E2F, SREBP-1 have shown to positively regulate AR expression while c-Jun, p53 and Purα demonstrated negative regulation of AR expression. Other transcription cofactors, cytokines, signal transducers and RNA-binding proteins such as β-Catenin, Rb1, Smad2 and 3 and EBP1 also play a role in direct regulation of the AR. Further to this, there are known compounds and agents that alter AR expression by acting on transcription, translation and post-translational activity for example polyphenols acting on hormones and transcription factors (7). It is also well known that the AR and androgens autoregulate AR expression whereby mRNA levels of the AR are unchanged or opposite to AR protein expression levels (8–10).
Given the mitogenic effects of androgens on prostate epithelial cells, the drugs used in the treatment of advanced PCa aim to suppress AR signalling. Androgen deprivation therapy (ADT) either surgically or chemically castrates patients to reduce the levels of free androgens in the blood (2), and thus decrease AR signalling. While ADT is very effective, many patients develop an ADT-resistant, lethal form of the disease known as castration-resistant prostate cancer (CRPC). ADT is often combined with direct AR inhibitors (ARIs) for the treatment of the castration-resistant form of the disease, whereby AR signalling is maintained despite the low levels of androgens. Some ADT drugs such as Abiraterone acetate inhibit adrenal gland enzymes, involved in androgen biosynthesis, and subsequently reduce AR signalling by androgen deficit (2). The majority of AR inhibitors (ARIs), such as Enzalutamide and Bicalutamide, competitively antagonise the binding of androgens to the ligand-binding domain (LBD) of the AR (2, 11). ARIs therefore, reduce the activity of AR signalling often through the inhibition of its nuclear translocation and chromatin binding of the AR leading to the inhibition of AR target gene expression, subsequently causing the suppression of tumour growth (2, 11). ARIs have shown to reduce tumour size and prolong the survival of patients with Castration-resistant Prostate Cancer (CRPC) who had failed chemotherapy (12). Enzalutamide reduces the disease burden as well as prolongs progression-free survival with metastatic hormone-sensitive prostate cancer (mHSPC) (11, 13). However, in metastatic disease, resistance to ARIs usually occurs within 18-36 (14) months due to a transition to CRPC, which has a median survival of 14 months (15). Among others, resistance can occur through several AR centric mechanisms: AR amplification, increased AR promiscuity, increased activation of androgen-independent activation of AR, and AR variants (AR-Vs) (2).
There are ca. 20 different known AR-Vs which are truncated forms of the full-length AR (AR-FL) (Figure 2) (13, 16). Clinically relevant AR-Vs are almost always expressed with AR-FL and may coordinate gene expression via heterodimerisation (17, 18). In the context of AR-FL, the LBD represses the AF1 function residing within the NTD when androgens are not bound to the LBD restricting its activation when the androgen trigger is absent (19). However, the lack of a functional LBD results in the NTD of AR-Vs no longer being repressed thus rendering AR-Vs constitutively active. As most clinically employed ARIs target the LBD, AR-Vs represent a treatment escape mechanism and thus their emergence in tumours can predict tumour response to ARI drugs such as Enzalutamide (2). Since AR is known to interact with a wide array of factors that form its transcriptional network, losing the LBD may modulate the AR interactome and could affect its transcriptional output resulting in a change in the AR cistrome, with evidence discussed in this review that there is some differential gene regulation between AR-FL and AR-Vs (20, 21).
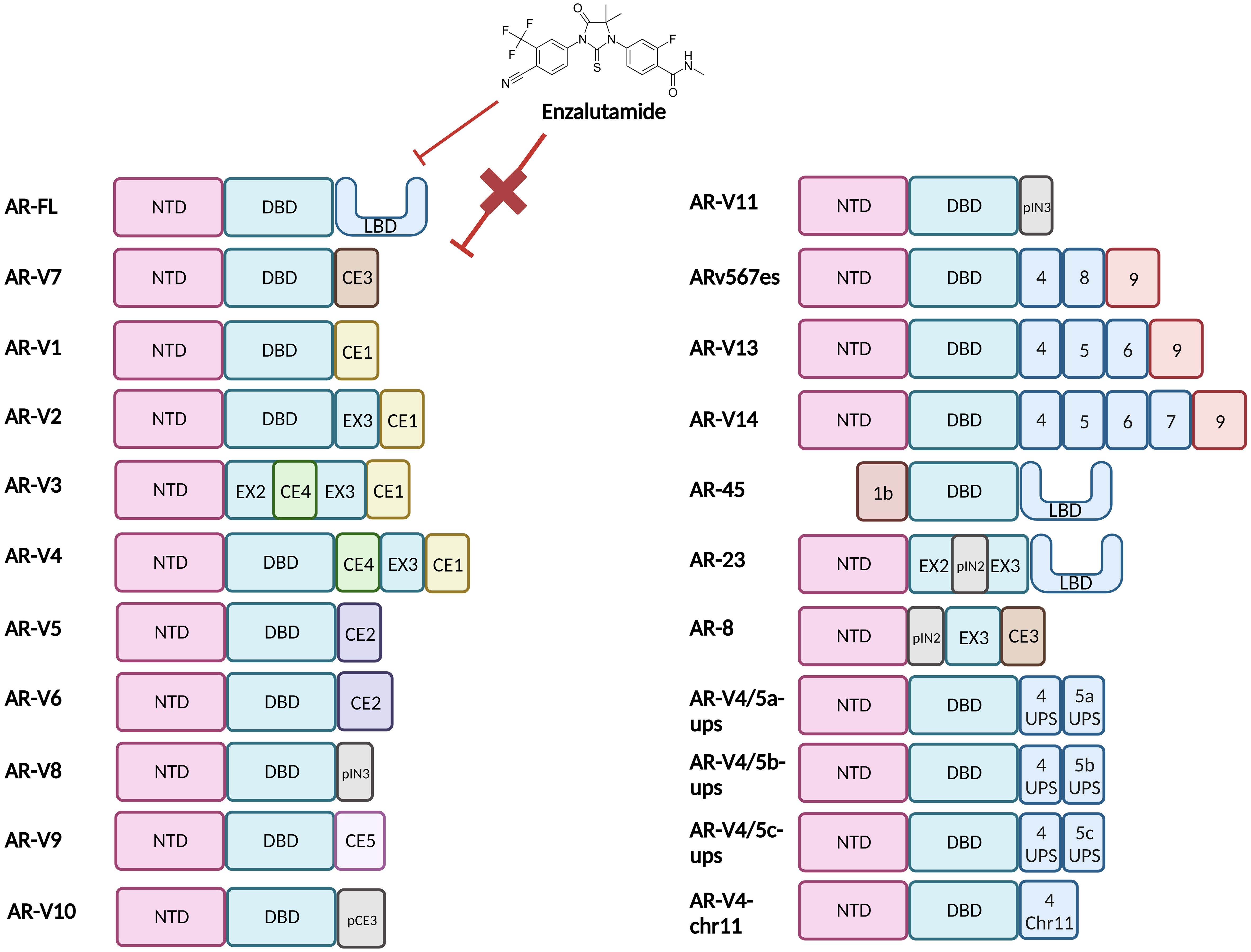
Figure 2 Structural representation of the Androgen Receptor and Androgen Receptor Variants. Full length Androgen Receptor (AR-FL) protein consists of three functional domains: an amino-terminal domain (NTD), DNA-binding domain (DBD) and Ligand-binding domain (LBD). Between the DBD and the LBD is a small hinge region (not shown). The AR gene resides on the X chromosome with 8 exons coding for the protein. Exon 1 codes for the N-terminal domain (NTD), exons 2 & 3 code for the DNA-binding domain (DBD) and exons 4-8 code for the hinge region (HR) and the ligand-binding domain (LBD). The NTD is the primary driver of AR-dependent transcription, the charged DBD allows interaction of AR with the DNA and the hydrophobic LBD facilitates androgen binding. Truncated AR variants (AR-Vs) lack the LBD (e.g. AR-V1 to AR-V14, inclusive of ARv567es) and instead, contain cryptic exons or partial LBD-coding exons or are non-functional and/or carry out alternate functions. In all cases these alterations diminish the ability of the AR to bind to ligands and subsequently be inhibited by LBD-targeting drugs such as Enzalutamide. CE, Cryptic Exon; EX, Exon; IN, Intron; p, Partial.
The selection pressure of AR-targeting treatments induces genomic rearrangements and/or alternative splicing that form the AR-Vs (22–24). The most researched variant is AR-V7, which is correlated with ARI resistance (25) and reduced survival (26). Sharp and colleagues demonstrated that whilst <1% of primary PCa (hormone naive) had AR-V7, over 75% of ADT-treated tumours show positivity for AR-V7 (25). Similar findings were reported in the SU2C dataset of metastatic PCa (n=125) where 90% tumours showed AR-V7 expression (27). Other variants include AR-V567es, which have a transcriptomic overlap with AR-FL and AR-V7 and may produce an intermediate phenotype whereby target gene regulation and thus aggressiveness of disease is intermediary of AR-FL and AR-V7-driven diseases (28).
There are a few studies that discuss how AR-Vs and specifically AR-V7, are regulated. Hu et al. concluded that AR-V protein expression is negatively regulated by AR-FL signalling (29); Jones et al. determined that Aurora A kinase regulates AR-V7 expression in a feedback loop mechanism (30) while Li et al. showed protein phosphatase-1 and AKT kinase drive AR-V7 expression (31). The alternative splicing that synthesises these AR-Vs thus also contributes to AR-V7 expression where splicing factors have been shown to increase in expression after hormone therapy (2, 32). Interestingly, AR-Vs have been shown to attenuate AR-FL transactivation (33).
AR-V7 is produced in PCa cells as a result of aberrant splicing. It retains exons 1, 2, and 3 which code for the NTD, DBD, and part of the hinge region, as well as the cryptic exon 3 (CE3) that codes for an additional stretch of sixteen further amino acids (Figure 2). The splicing of pre-mRNA requires the recruitment of RNA splicing factors to 3’ and 5’ splice sites, which control intron removal and their abrupt functioning can lead to alternative splicing of the mRNA generating AR-Vs (34). Identification of which RNA splicing factors are differentially recruited to the pre-mRNA of AR to induce the formation of different AR-V transcripts may be an avenue for therapeutic targeting outside the realm of posttranslational AR modifications. Liu et al. identified an alteration in splicing factors U2AF65 and ASF/SF2 binding the 3’ site splice site of AR-V7 pre-mRNA under Enzalutamide-induced castrate conditions (23). Miller et al. suggested that SRPK2 may be a key factor in increased availability of ASF/SF2 for this binding, but this requires experimental validation (35).
Constitutive activation of AR-V7 results from the predominant localisation of AR-V7 to the nucleus, and the loss of LBD mediated repression of NTD activation in the absence of hormone binding (25, 36). Other differences between AR-FL and AR-V7 may lie in their interactions with DNA and TFs. In most cases, AR-FL will homodimerise upon ligand activation and bind to DNA. Whilst it is widely agreed that AR-V7 will primarily heterodimerise with AR-FL, the protein can also homodimerise and have more transient DNA interactions (17, 37–39) (Figure 3). Özgün et al. discovered that the genomic locations of heterodimers and AR-FL homodimers did not change but their chromatin binding dynamics were independent of each other (17), while AR-V7 homodimer DNA-binding locations have not been fully explored, although it has been determined that AR-Vs bind to constitutively open chromatin sites that are known to overlap AR-FL binding sites (40). Although AR-FL and AR-V7 bind to similar response elements within the DNA, AR-V7 is not simply a constitutively active version of AR but has a unique cistrome, which can be used to experimentally quantify AR-V7 activity. This distinct transcriptomic signature may be explained by different cofactor interactions. AR-V7 exhibits binding to Zinc finger protein x-linked (ZFX) at AR-V7 solo sites (20), increased binding to NCoR corepressors which could alter H3K27ac regulation (37), and a reduced affinity for forkhead box protein A1 (FOXA1) (41), which may potentially alter the AR-V interactome.
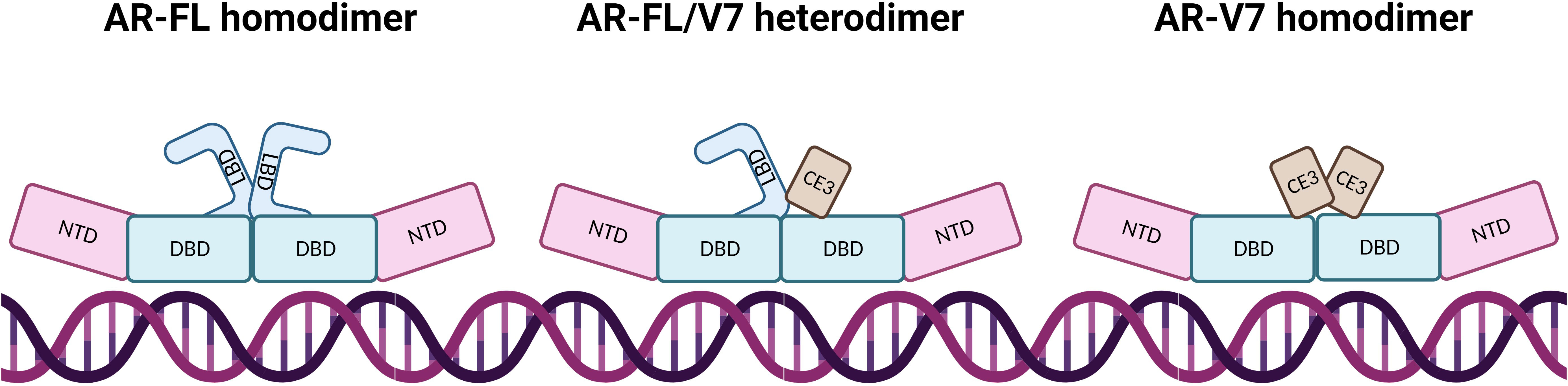
Figure 3 AR dimerisation and interaction with DNA. In AR-FL homodimers, the proteins intermolecularly associate by N/C interaction which is lost after DNA binding, allowing co-activator binding due to NTD and LBD separation. AR-FL/V7 heterodimers interact via N/C and D-Box interaction whereby the N/C interaction is also lost upon DNA binding. The AR-FL protein is immobilised on the DNA for a long time whereas AR-V7 protein demonstrates fast DNA-binding dynamics at high-affinity binding sites and dissociates from the dimer shortly after binding. AR-V7 homodimers have a short immobilisation time on the DNA, where dimers have close proximity via the DBD.
Since AR-Vs confer resistance to AR inhibition therapies, recent research has been focussing on how to inhibit their activity in CRPC. Targeting of AR-Vs has proven challenging, with new ways of inhibition being required for control in disease progression (2). Novel emerging therapies that target both AR-FL and AR-Vs do not bind and inhibit the AR-LBD. The new treatments belong to four main classes: NTD-targeting, DBD-targeting, AR degraders and AR dimerisation inhibitors. NTD-targeting drugs (e.g. Anitens and Sintokamides) inhibit the AR transcriptional activation while DBD-targeting drugs (e.g. VPC compounds) inhibit AR-DNA interactions. AR degraders (e.g Niclosamide) degrade AR protein by proteasome or non-proteasome-dependent mechanisms to reduce AR protein abundance and AR dimerisation inhibitors (e.g.VPC-17005) impede AR dimerisation and thus interrupts AR interactions with DNA (2).
Successful inhibition of AR-V7 can be experimentally verified by examining the gene expression signature of AR-V7 target genes and therefore, there is a need to identify AR-V7 exclusive target genes. This review will provide a comprehensive account of some of the key AR-V7 target genes and their relevance in ARI-resistant PCa.
Table 1 summarises AR-V7 target genes and whether they are upregulated or downregulated with AR-V7 expression.
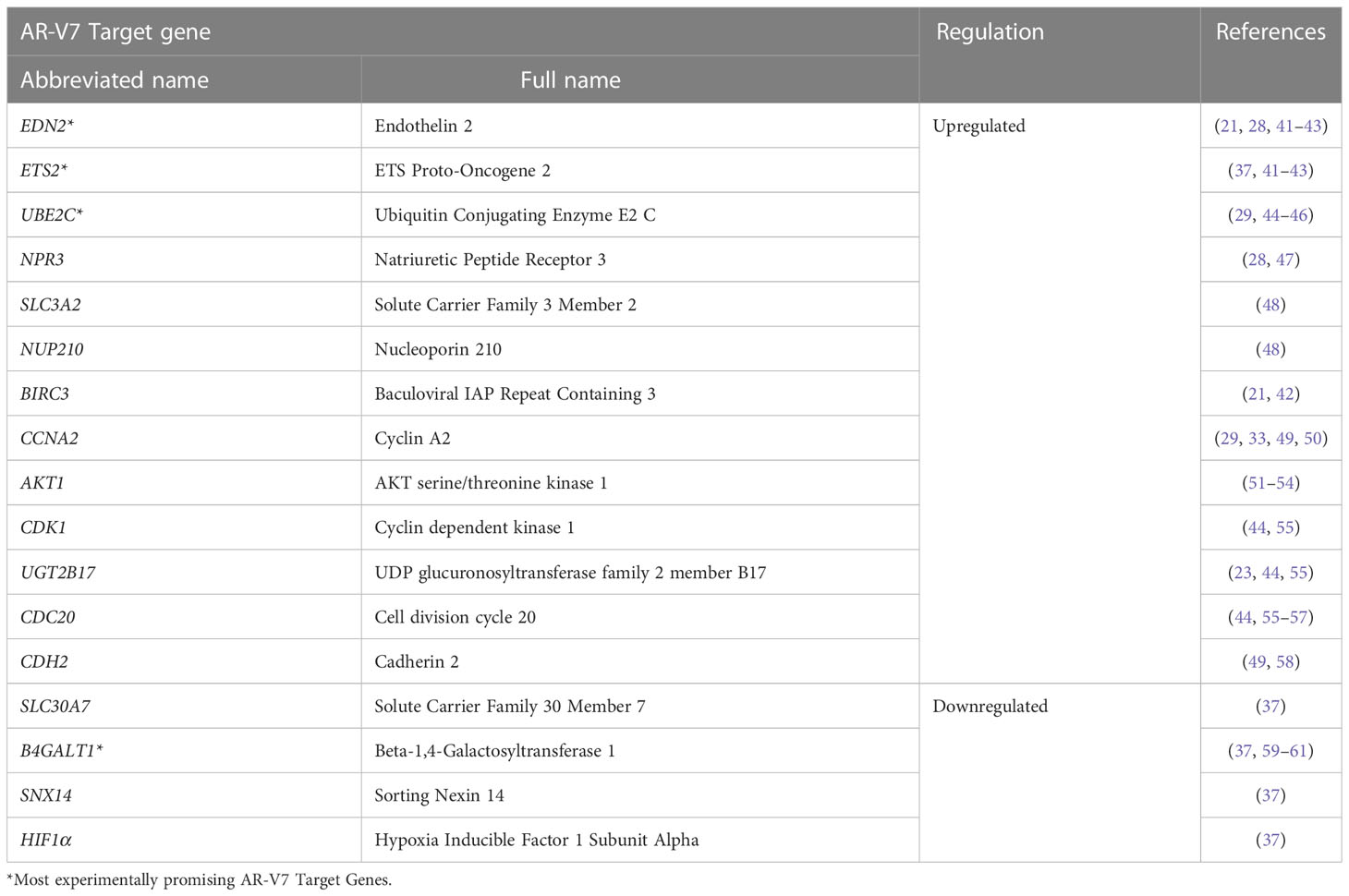
Table 1 Summary of AR-V7 target genes defined in the literature.
Endothelin-2 (EDN2) was first identified as an AR-V7 target gene in 2014 by Krause et al. The creation of AR-V7 expressing cell lines by transfection with lentivirus containing doxycycline (Dox)-inducible expression of AR-V7 enabled quantification of AR-V7-specific target gene expression by qPCR and therefore identified transcriptional up-regulation of EDN2 by AR-V7 (41).
EDN2 encodes the protein endothelin-2 (ET-2) which belongs to a group of secretory vasoconstrictive peptides that are part of the endothelin protein family and is co-regulated by the TF FOXA1 [FOXA1 has been described to co-regulate the expression of many genes with the AR (62)]. The preproprotein (preproendothelin-2) undergoes processing, resulting in a mature form that acts as a ligand for the endothelin receptors. Upon ligand-receptor binding, intracellular signalling cascades leads to vasoconstrictive activity (63). There is a paucity of information in the literature regarding the function of EDN2 in PCa. However, Grimshaw et al., reported that under hypoxic conditions in breast cancer cell lines, an increase in tumour survival was observed with increases in ET-2 expression levels. The presence of ET-2 has also been shown to increase the migratory and invasive abilities of breast cancer cells (64, 65).
ET-2 receptor antagonists are often used to treat pulmonary arterial hypertension, with approved treatments known as besentan, ambrisentan and macitentan, while other experimental drugs are in the clinical trials (66, 67). The drug tezosentan had been used in clinical trials as an endothelin receptor blocker in the treatment of congestive heart failure. Therefore, there is a potential to repurpose these drugs as a cancer treatment, specifically for AR-V7-expressing CRPC.
EDN2 expression remains unaltered by synthetic androgen R1881 (41) and is therefore not AR-FL regulated, however there are discrepancies in the literature as to whether EDN2 is regulated by a wider range of variants than just AR-V7. Several studies have shown that EDN2 expression can be upregulated by both AR-V7 and AR-V567es (21, 28, 41–43) therefore further investigation is required to identify whether EDN2 is specific to certain variants or widely regulated by AR-Vs.
Alongside EDN2, Krause et al. also identified ETS proto-oncogene 2 (ETS2) as another AR-V7 target gene (41). ETS2 is a TF that specifically binds the DNA GGAA/T core motif of the Ets-binding site to activate transcription (68). ETS2 regulates genes that are involved in angiogenesis, cell cycle control and cell proliferation, such as plasminogen activator urokinase and c-met (69). Interestingly, ETS2 is known to upregulate the expression of genes involved in prostate carcinogenesis, such as those controlling anchorage-independent growth (68).
There are no known clinically available ETS2 inhibitors, however an experimental molecule has been mentioned in the literature for the treatment of PCa. Carbone et al. designed a triplex-forming oligonucleotide targeting a homopurine:homopyrimidine sequence in the ETS2 promoter to inhibit transcription (70). This experimental therapeutic holds promise for the treatment of AR-V7-expressing CRPC.
ETS2 is not an AR-FL target gene as its expression is not induced by R1881 (41), however, whether AR-V7 is not the sole regulator of ETS2 expression needs further investigation. Studies have demonstrated that upon the induction of AR-V7 expression in cell line models of PCa, ETS2 expression was upregulated. However, this may not be specific to AR-V7, since when LNCaP were made to express a pan AR-V model, ETS2 was also upregulated (41–43), indicating ETS2 upregulation may be a general mechanism by AR-Vs.
Ubiquitin-conjugating enzyme 2C (UBE2C) was first mentioned as an AR-V gene in metastatic PCa tissue (44) and then later showed a significant correlation between UBE2C and AR-V7 expression levels. In vivo LNCaP-95CR xenografts treated with Abiraterone showed a significant correlation between UBE2C and AR-V7 transcript levels. Immunohistochemical staining also validated this significant correlation (29), indicating UBE2C is a potential AR-V7 target gene.
As a component of the anaphase-promoting complex/cyclosome (APC/C) enzyme, UBE2C is essential for ubiquitination (71, 72). UBE2C has been described as a hub gene of PCa i.e., a gene with high connectivity in the genetic interaction network and is involved in many oncogenic pathways involved in PCa progression such as NOTCH signalling and WNT-β-Catenin signalling (71), while also being positively correlated with the progression of other cancers types such as breast and ovarian, whereby its activity promotes a more aggressive tumour phenotype (73, 74). Under normal conditions, the UBE2C protein is necessary for the eradication of mitotic cyclins for the progression through the cell cycle (45, 75). However, in PCa, UBE2C drives cell cycle progression by inactivating the M-phase checkpoint. Alternatively, it can also increase the level of active APC/C (76).
As UBE2C is commonly upregulated in multiple cancers, inhibition of this gene has been explored, however inhibitors specifically for this protein are not widely used in the clinic. Bortezomib however is an FDA approved treatment for myeloma and has been shown to also prevent cell cycle progression in Colorectal Carcinoma (77). Therefore, there is potential for this drug to be repurposed as a CRPC treatment. Furthermore, a 2011 study discovered that an mTOR inhibitor, CC1-779 was able to inhibit UBE2C mRNA and protein expression in CRPC cell line models (78), therefore there should be further investigation to fully explore the therapeutic potential of this drug as a treatment for CRPC.
UBE2C has often been shown as an AR-V7-regulated target gene (45, 46). Interestingly, studies have shown that in hormone-sensitive PCa (HSPC), there is a significant correlation between the expression of UBE2C and the expression of AR-FL. However, as the PCa progresses this correlation shifts to AR-V7 instead of AR-FL, further pointing towards an indispensable role of UBE2C in PCa growth and progression (29, 76). In addition to AR-V7 and AR-FL, AR-V567es can also induce the expression of UBE2C and the growth of PCa cells; although this occurs in the presence of mediator complex subunit 1 (MED1) (75, 76).
AR-V7 and AR-V567es expressing LNCaP and VCaP cells were generated by Nagandla et al. via lentivirus-mediated transduction. The gene expression mediated by AR-V7 or AR-V567es, relative to variant gene expression in 22Rν1 cells (expressing AR-FL and AR-Vs) was compared to identify differentially expressed genes between the AR-Vs and AR-FL. These data identified Natriuretic Peptide Receptor 3 (NPR3) as an AR-V7- upregulated target gene (28).
NPR3 encodes for natriuretic peptide clearance receptor (NPCR). NPCR represents approximately 94% of natriuretic peptide receptors (NPRs) (79), and removes natriuretic peptides from circulation, thus reducing binding to the other two NPR which have guanylate cyclase activity (80). NPCR has the greatest affinity for the Atrial Natriuretic peptide (ANP) (81), which has been linked to controlling PCa progression (82), therefore an increase in NPCR expression may enhance PCa progression. One of the anticancer effects of long-lasting ANP is the inhibition of phosphorylation and activation of MEK1/2 and ERK1/2 via cyclic GMP activation (83, 84), which is required for the activation of AR signalling. This mechanism is also immunomodulatory in the tumour microenvironment of PCa (85). Indeed, the MEK1/2 inhibitor, Trametinib is under Phase II trials for CRPC (NCT02881242) (86). However, as NPCR lacks guanylate cyclase activity, it would competitively inhibit the inhibitory effects of MEK1/2 on ANP thereby reducing the ANP levels in the circulation. However, when ANP binds to NPCR, the oncogenic Vascular Endothelial Growth Factor (VEGF) is inhibited (87). Thus, AR-V7-upregulated NPCR plays an oncogenic role in PCa but can be inhibited by the binding of ANP, which has the potential to be exploited therapeutically to reduce PCa progression.
Inhibition of natriuretic peptide receptors has been investigated in literature, and a few antagonists of the receptors have been identified. Isatin has demonstrated inhibition of atrial natriuretic peptide receptor binding in mammalian cell models (88), while peptides ANP4-23 and AP-811 have shown to be selective atrial NPCR antagonists (89). Along with other experimental molecules, these therapies could have the potential to be used as CRPC therapeutics in a clinical setting.
Further contradiction occurs as to when NPR3 expression increases in PCa, and if NPR3 is specifically AR-V7 regulated. AR-V7 regulation of NPR3 expression, as described by Nagandla et al, would implicate NPR3 expression is upregulated in CRPC and is corroborated by Terada et al. (47). However, this contrasts with findings of increased NPCR during PIN to PCa transitions (90), and in a CWR22 xenograft model, which is derived from primary PCa and in which AR-V7 expression is rare (91). Furthermore, NPCR was identified in PC-3, a PCa cell line derived from bone metastases (91) which also does not express AR-V7 (36). Therefore, whilst the role of NPR3 in PCa needs further validation, it is unlikely to be specifically regulated by AR-V7.
Solute Carrier Family 3 Member 2 (SLC3A2) was identified as an AR-V7 target gene by Sugiura et al. following AR-V7-specific ChIP-sequencing investigating LNCaP-95 (PCa cell line expressing both AR-FL and AR-V7) vs LNCaP (PCa cell line expressing AR-FL only), where expression levels were more than two-fold higher in LNCaP-95 (48). Furthermore, the expression of SLC3A2 was significantly downregulated following AR-V7 knockdown by RNAi assumed to target the AR-V7-specific domain, CE3, indicating it is regulated by CE3-containing AR (AR-V7, AR-V8) (48).
SLC3A2 encodes for 4F2 heavy chain, aka CD98hc, a subunit of several heterometric amino acid transporters (92, 93). 4F2hc also has a role in glucose availability (94), as well as nucleotide availability to facilitate cell cycle progression (94). The actions on the cell cycle are at least partially due to 4F2hc regulation of other genes including S-Phase Kinase Associated Protein 2 (SKP2) (95). Other activities include involvement in integrin signalling (93). Oncogenicity of SLC3A2 is due to its involvement in amino acid transport adhesion, and proliferation. Indeed, increased SLC3A2 expression has been observed in several cancers (96–99), including PCa, lung and liver cancers (95). Whilst SLC3A2 is ubiquitously expressed, its increased expression is linked to poor PCa prognosis as evident from shorter progression-free survival, and association with a higher Gleason score (95, 100).
BCH (2-amino-2-norbornane-carboxylic acid), a non-metabolisable amino acid, has shown to inhibit the disulphide bond-linked heterodimer that consists of CD98hc plus a catalytic light chain (101). This was demonstrated in a panel of breast cancer cell lines, which consequently showed reduced viability and therefore it is conceivable that this inhibition could also be seen in PCa cells and may be used to target AR-V7-expressing CRPC tumours.
Although Sugiura et al. identified the SLC3A2 gene as specifically regulated by AR-V7 in LNCaP-95, an increase in SLC3A2 expression by androgen R1881 treatment has been reported which would suggest regulation by AR-FL as well (48, 102). Additionally, SLC3A2 is also regulated by Activating Transcription Factor 4 (ATF4) (102), another TF for which protein levels are increased upon androgen induction (103). Therefore, SLC3A2 may be directly regulated by AR-V7, but also indirectly regulated by AR through direct regulation by ATF4. Regulation by multiple TFs indicates the essentiality of SLC3A2 given its vital role in supporting tumour proliferation.
Nucleoporin complex 210 (NUP210) was also identified as an AR-V7 gene by Sugiura et al., as it was expressed more than 2-fold higher in AR-V7-expressing LNCaP-95 than LNCaP. Specific knockdown of AR-V7 in LNCaP-95 caused a significant decrease in NUP210 expression. Furthermore, in the TCGA-PRAD dataset, NUP210 expression positively correlated with AR-V7 expression (48). In line, there was a significant increase in NUP210 expression in the Grasso dataset (GSE35988) between benign prostate and primary PCa, where there is little AR-V7 expression (48). However, the increase in NUP210 expression between benign prostate and CRPC was more significant. Whilst AR-FL does directly regulate NUP210, AR-V7 appears to have a stronger impact on NUP210 expression.
Functionally, the encoded protein of NUP210, also named NUP210, anchors the nuclear pore complex (NPC) - which regulates nucleocytoplasmic transport, although it also has non-structural functions (104, 105). For example NUP210 has been linked to the modulation of gene expression during differentiation (106, 107) and to T cell homeostasis (108). In multiple PCa cell lines, NUP210 has been identified as the regulator of mTOR transport into the nucleus (109), an interactor of AR (110) and plays a role in PI3K-Akt signalling pathways which are often hyperactivated in PCa (111). Nuclear mTOR modulates gene transcription in CRPC to favour proliferation and migration in the absence of AR-FL and is correlated with PCa aggressiveness and disease progression (109).
Direct targeting of NUP210 protein has not been demonstrated, however targeting of the transcriptional and epigenetic regulator, Bromodomian-containing protein 4 (BRD4) with an aminocyclopropenone (ACP) has been explored, ACP-1n has demonstrated inhibition of NUP210 expression in colorectal cancer cells. This is due to NUP210 being a BRD4-driven nuclear complex component (112). Reducing the expression of NUP210 caused a decrease in cancer cell growth and nucleus size, and therefore the application of this inhibitory mechanism in the context of AR-V7-driven CRPC tumours could result in a similar outcome and therefore represent a novel treatment strategy.
Baculoviral IAP Repeat Containing 3 (BIRC3), which encodes for Inhibitor of Apoptosis 2 (IAP-2) was first experimentally validated by Basil et al. (21, 42). To identify genes which were differentially bound by AR-FL and AR-V7 on chromatin, Basil et al. utilised LNCaP and VCaP cell lines stably expressing Dox-inducible AR-V7. ChIP-exo was performed using an antibody specific for both AR-FL and AR-V7 isoforms in conditions either recruiting AR-FL and AR-V7 to chromatin or only AR-FL to chromatin. Along with the ChIP-exo data, ChIP-qPCR confirmed that AR-V7 bound with greater affinity to BIRC3, and whilst BIRC3 expression was marginally induced by AR-FL, it was significantly increased with AR-V7 recruiting treatment, indicating that BIRC3 is positively regulated to a greater extent by AR-V7 than AR-FL (21).
The encoded BIRC3 protein, IAP-2 has several roles throughout cell death, immunity, inflammation, and proliferation. The role of IAP-2 in evading cell death is not primarily through direct inhibition of caspases (113, 114), but through negative regulation of the necrosome, a protein complex that can carry out both caspase-dependent and independent cell death pathways (113, 115). IAP-2 is associated with the NF-κB pathway, crucial for inflammation and immune responses, in which IAP-2 is required for TNF activation of the NF-κB pathway, but is also a negative regulator of non-canonical NF-κB signalling (114). However, the role of BIRC3 in cancer is paradoxical, being reported as both a tumour suppressor and pro-oncogenic, even within the same cancer type (116). There is paucity in literature on the roles of BIRC3 or IAP-2 in PCa, however it has been identified that BIRC3 is upregulated with a knockdown of Elongation Factor For RNA Polymerase II 2 (ELL2) (117), which in itself is downregulated by AR-FL and AR-V7 (21, 118), and drives proliferation in LNCaP cells (117). Therefore, increased AR signalling reduces ELL2 activity and thus upregulates BIRC3.
The endogenous antagonist of IAPs, second mitochondria-derived activator of caspase (SMAC) has been employed for drug design since the early 2000s and are now in their fourth generation. The fourth-generation optimised non-alanine agonist, ASTX600, showed growth repression in breast cancer mouse models and are now in phase I/II clinical trials (119, 120). Similarly, there at least 8 small molecule IAP antagonists in clinical trials for the treatment of solid cancers including ASTX600, namely: GDC-0152, LCL-161, Debio-1143, Birinapant, GDC-0917 and APG-1387 (119). This pathway has shown it can be inhibited and exploited in terms of cancer treatment, thus giving it great potential to be used to treat AR-V7-expressing CRPC tumours.
Although the data presented by Basil et al. that BIRC3 is AR-V7 upregulated rather than AR-FL is compelling, BIRC3 can be regulated by other TFs in PCa. Wang et al. explored IAP-2 protein levels in several PCa cell lines including LNCaP, LNCaP-95 and VCaP (117), but although the latter two contain AR-V7, it was found that LNCaP cells not expressing AR-V7 had the greatest amount of IAP-2. This increase may result from ELL2 downregulation which is commonly observed in PCa whereby ELL2 expression inversely correlates with Gleason scores (121). Furthermore, BIRC3 has also been identified as a glucocorticoid receptor (GR) regulated gene (122, 123), although this was in the context of bronchial and pulmonary epithelial cells. The GR is also correlated with CRPC and PCa progression (124), whereby androgens can bind the GR to activate proliferation genes due to high homology between the AR and GR (125). Therefore, BIRC3 is not solely regulated by AR-V7 and crosstalk between the GR pathway should be considered.
Cyclin A2 (CCNA2) was experimentally shown to be an AR-V7 target gene in a variety of PCa cell lines including 22Rν1 and C4-2B-ENZ (49, 50). Hu et al. first identified CCNA2 as AR-V7 regulated by transiently inducing AR-V7 expression in LNCaP cells independent of AR-FL (29). Furthermore, AR-V7 depletion resulted in inhibition of CCNA2 expression but not when AR-FL was depleted in 22Rv1 cell line (33).
The protein encoded by CCNA2, cyclin A2, is a key regulator of both the G1/S and the G2/M checkpoints of the cell cycle through interaction with cyclin-dependent kinase (CDK) 2 and 1 respectively (126), and has further roles in cytokinesis and cell mobility (127). CCNA2 has been identified as a hub gene for metastasis in PCa (56), and also has significant correlation to reduced overall survival, decreased disease-free survival, and increased biochemical recurrence of prostate cancer (128, 129).
Generally, exclusive cyclin A2 antagonists are much less common, rather inhibitors of its interaction complexes are widely used. Cyclin A2 forms complexes with the cell cycle proteins, cyclin-dependent kinases (CDK1, 2 and 3) and the transcription factor, E2F-1, along with other proteins. Nitrogen-containing bisphosphonates (N-BPs) however, seem to solely target and inhibit the expression of cyclin A2 at the transcriptional and consequently translational level in epithelial cell models (130). RXL peptides have been shown to bind a hydrophobic site on the surface of cyclin A and thus inhibit kinase activity of CDK2, causing cells to undergo apoptosis (131). There are many general cyclin A/CDK inhibitors available commercially such as Indirubin and JNJ-7706621, often targeting the ATP site of CDK (132). Exclusive cyclin A2 or cyclin complex-targeting molecules are important in inhibiting the cell cycle and thus cancer cell proliferation, therefore these inhibitors might be useful for the treatment of AR-V7-expressing CRPC.
It is questionable whether CCNA2 is solely regulated by AR-V7. Cao et al. demonstrated that CCNA2 expression did not decrease with AR-FL knockdown or Enzalutamide (33), but an investigational NTD inhibitor EPI-7170 decreased CCNA2 expression levels. This decrease, however, may not only be due to AR-V7 inhibition but the culmination of multiple NTD-containing AR-Vs in conjunction with AR-V7. In fact, induced expression of AR-V567es in LNCaP also increased CCNA2 expression (29, 133), although AR-V7 appeared to cause higher CCNA2 expression (133). Furthermore, CCNA2 may be regulated by the TF FOXA1 (134), which is also increased in mCRPC (62). Therefore, whilst CCNA2 expression is not induced by AR-FL, it may be induced by multiple AR-Vs other than AR-V7 as well as FOXA1.
The AKT1 gene encodes a serine-threonine protein kinase and was one of the first AR-V7 genes to be identified. It was the focal AR-V7 upregulated gene in the first paper on AR3 aka AR-V7 by Guo et al. (51). This conclusion was drawn by the culmination of several findings. Firstly, AKT1 expression decreased following the knockdown of AR-V7 but not following AR-FL knockdown. Furthermore, investigation into the protein levels of the AKT1 kinase showed higher AKT1 protein levels in xenograft tumours of LNCaP transduced with AR-V7 than in the control LNCaP. Finally, ChIP-sequencing for AR-V7 and AR-FL identified that whilst AR-V7 could bind at two sites of the AKT1 gene, AR-FL did not bind to AKT1 (51). AKT1 has since been used as an AR-V7 target gene in multiple cell lines for example, LNCaP treated with Enzalutamide, LNCaP-95, and 22Rν1 to assess AR-V7 expression (52–54).
AKT1 specifically encodes for AKT1, commonly referred to as protein kinase B, a regulatory kinase implicated in several signalling pathways including PI3K/AKT/mTOR, AR, and MAPK (111). As such, AKT1 is involved in multiple pathways including cell cycle progression and cell survival, and is hyperphosphorylated, which activates AKT1 in over 50% of human cancers (135). Thus, AKT1 inhibition is a treatment option undergoing clinical trials for several cancers, including PCa (111, 136, 137). In PCa, AKT1 is part of several inhibitory feedback loops with the AR (111). AKT1 can also directly regulate AR by phosphorylation of serine 213 in the NTD and serine 791 in the LBD with contrasting outcomes on nuclear translocation and transcriptional activation of AR (138). Despite the generally antagonistic crosstalk between PI3K/AKT pathway, interestingly, AKT1 promotes AR-V7 transcriptional activity (139). Perhaps, therefore, AKT1 can differentially regulate AR-FL and AR-Vs, although this requires experimental validation and is beyond the scope of this review.
AKT inhibitors have been widely discussed in literature as potential anti-cancer therapies, with several clinical trials already taking place for the treatment of mCRPC. Ipatasertib treatment demonstrated a 3-month median survival benefit in a phase II trial and showed consistent results in combination with abiraterone in a phase II trial. Capivasertib and MK2206 are also undergoing clinical trials for PCa (140), while Deguelin, GSK690693, Celebcoxib and Genistein treatment have been explored in pre-clinical studies (141). These AKT inhibitors show encouraging results for the treatment of CRPC and have the potential to become standard therapy in combination with current treatments in the clinic.Whilst AKT1 is often used experimentally as an AR-V7 target gene, it may also be regulated by other AR-Vs. AKT1 expression was increased with AR-V expression compared to AR-FL (44), which could be due to multiple AR-V expression. Furthermore, although AKT1 may not be AR-FL-regulated at the expression level, it is worth considering that the PI3K/AKT pathway can be upregulated by ADT due to inhibitory feedback loops within AR signalling (111), which may indirectly increase AKT1 expression, independent of AR-V7.
Cyclin-Dependent Kinase 1 (CDK1) was first shown as an AR-V7 target gene by Jones et al., although had also been previously identified as a hub gene for AR-Vs (30, 44, 55). In 22Rv1 cells grown in ADT conditions, Jones et al. silenced AR-V7 and observed a significant decrease in CDK1 expression when compared to the control, indicating AR-V7 positively regulates CDK1 expression.
CDK1 encoded protein, CDK1 is known as a master regulator of the cell cycle (142–146), alongside having other roles, such as in DNA replication (147), and regulation of several TFs (148–150) including the AR and FOXO1 (151, 152). As a result, increased CDK1 expression has been linked to the progression of several cancers, including PCa and breast cancer (56, 152–156). One of the mechanisms by which CDK1 promotes PCa is by directly phosphorylating the AR on serine residues 81 and 515, which increases AR transactivation, nuclear localisation, and stability (151, 157–159), thus increased CDK1 expression will increase oncogenic AR signalling.
As mentioned with inhibition of cyclin A2, CDK inhibitors are widely used to treat cancer. There is however, a well-known inhibitor, RO3306, that is specific to CDK1 and has shown inhibition of the kinase in many cancer models including PCa (160–162). There are no known clinical trials with this drug in mCRPC, but there is scope for this to be used as a prospective treatment for AR-V7-expressing tumours.
Whilst Jones et al. (30) found a significant decrease in CDK1 expression during AR-V7 knockdown, utilisation of siRNA targeted to AR exon one, which is common to all transcriptionally competent AR isoforms including AR-FL, caused a significantly greater decrease in CDK1 expression. This indicates that although CDK1 expression is regulated by AR-V7, it is not specific to AR-V7. Given that Zhang et al. found a significant decrease in CDK1 expression in cells expressing AR-FL compared to AR-Vs, it appears that AR-Vs are the primary drivers of CDK1 expression rather than AR-FL (44). Thus, whilst CDK1 is useful to differentiate AR-FL from AR-Vs, CDK1 should not be treated as AR-V7 specific.
UDP Glucuronosyltransferase Family 2 Member B17 (UGT2B17) was first described as an AR-V target gene in a study by Liu et al. where metastatic PCa patient samples were collected and split into groups of low and high expression of AR-Vs and then gene profiles of each group were compared (23, 163). This showed that UGT2B17 was differentially upregulated by AR-Vs (55). Total RNA from human primary and metastatic PCa tissue was quantitatively analysed to find that UGT2B17 transcript expression was higher in patients with high AR-V expression compared to low expression. Moreover, AR-V-expressing LuCaP 86.2 cells expressed higher UGT2B17 than LuCaP 35 cells which only expressed AR-FL, thus validating that UGT2B17 is positively regulated by AR-Vs (44). The correlation between UGT2B17 and AR-V7 levels was made when VCaP cells were transfected with siRNA targeting AR-V7 and expressed reduced UGT2B17, indicating the dependency of UGT2B17 expression on AR-V7 (23).
UGT2B17 encodes for an enzyme, UGT2B17, which has an important role in the intermediate step of testosterone metabolism known as glucuronidation. The function of UGT2B17 is to catalyse the transferal of glucuronic acid from UDP-glucuronic acid to steroid hormones, such as testosterone. This catalysation increases testosterone renal excretion, aiding in the maintenance of androgen homeostasis in the prostate (164, 165). In CRPC progression, UGT2B17 may be actively involved in AR signalling via increasing c-Src activation, a tyrosine kinase protein normally activated in a ligand-independent manner but also by EGF and cytokine signalling. Therefore, an increase of UGT2B17 protein can increase ligand-independent AR signalling (mainly determined by constitutively active AR-Vs) and androgen insensitivity in castrate conditions by inducing increased testosterone excretion (164, 165) and thus progressing the disease.
Imatinib is known as a selective UGT2B17 inhibitor (166) and has been in phase II clinical trials for hormone refractory PCa in combination with docetaxel (167). There were however issues with toxicity (168) and the trial was terminated. Imatinib has also been purposed as a platelet-derived growth factor receptor inhibitor, however in clinical trials, the treatment proved ineffective for PCa patients (169). Recently however, salicylic acid has been discovered as an uncompetitive inhibitor of UGT2B17 (170). This avenue could be explored for the treatment of AR-V7-expressing CRPC.
Under conditions of androgen deprivation, AR-V7 is necessary for the expression of UGT2B17 in VCaP PCa cells, but UGT2B17 is suppressed by DHT-activated AR-FL (23). However, most literature considers UGT2B17 to be a more general AR-V regulated gene. Therefore, further investigation is required to find if this gene is dominantly or solely regulated by AR-V7 relative to other AR-Vs (23, 55).
Gu et al. identified that the Cell Division Cycle 20 (CDC20) gene was specifically regulated by AR-V7 in CRPC LNCaP-95 cells (56). Following treatment of LBD-targeting Enzalutamide or NTD-targeting EPI-001, CDC20 expression only decreased with EPI-001 treatment but not with Enzalutamide treatment. The inhibition of AR-FL by Enzalutamide would allow AR-V7 to remain activated in LNCaP-95 cells and drive CDC20 expression, indicating that persistent expression of CDC20 following Enzalutamide treatment but not EPI-001 treatment is a result of AR-V7-driven expression of CDC20 (56). Further studies evaluating the expression level of CDC20 following AR-V inhibition/knockdown validated CDC20 to be a target gene of AR-V7 (44, 55, 57).
CDC20 is involved in cell division and associates to the anaphase-promoting complex/cyclosome (APC/C), activating the APC/CDC20 complex (171). The APC/C is an enzyme consisting of multiple subunits and is responsible for mediating the ubiquitin-mediated degradation of securin, the anaphase inhibitor. The onset of anaphase allows the activation of separase, a cysteine protease, which then permits the separation of sister-chromatids during mitosis (172, 173). In CRPC, the abnormal upregulation of CDC20 leads to the failure of mitotic arrest and progression through the cell cycle and division of cancer cells with aneuploidy (171, 174), thus increasing oncogenic proliferation and driving disease progression.
Inhibition of CDC20 has been demonstrated in multiple cancer cell models including PCa by the compound Apcin (175–177). Mechanistically, it acts as an APC-CDC20 E3 ligase inhibitor, thus interfering with CDC20 binding to its substrates, causing a blockade of mitotic exit and consequently apoptosis. Apcin-based compounds have been developed further to improve antitumour activity, with encouraging results (178). To date, these compounds have not been used in clinical trials, however, they show great potential for the treatment of AR-V7 expressing CRPC tumours.
Whilst some studies have mentioned CDC20 to be generally upregulated by AR-Vs, none have specified which AR-Vs other than AR-V7 regulate this gene. Thus, further investigation is necessary in order to determine whether CDC20 is solely AR-V7 regulated (45, 179, 180).
Cottard et al. first identified Cadherin-2 (CDH2) as an AR-V target gene (58, 181) by overexpressing AR-FL, ARQ640X [an AR-V model (182)] or AR-V7 in LNCaP cells. Cottard et al. discovered that AR-V7 and ARQ640X increased CDH2 expression while AR-FL induction in these AR mutant models repressed this expression, highlighting differential regulation of AR mutants to AR-FL (58).
The protein product of CDH2, N-cadherin is a calcium-dependent cell adhesion protein which allows neighbouring cells to adhere to one another through dimerization of their N-cadherin chains (181). In CRPC, AR-V7 upregulates N-cadherin, however requires DBC1 (deleted in breast cancer 1) as a coactivator to carry out its enhanced transcriptional activities (183). N-cadherin is a mesenchymal cell marker, and often seen increased in expression in many cancers including CRPC during epithelial-mesenchymal transition (EMT) (184). This transition in turn assists with the progression and metastasis of PCa as the cells are able to migrate and invade into other tissues or organs more easily (185, 186).
N-cadherin antagonists have been widely researched over the past three decades and comprise of monoclonal antibody therapy, short synthetic peptides containing the cadherin CAR sequence, tryptophan-containing peptides (12-mers) and synthetic cyclic peptides containing the cadherin CAR sequence HAV (e.g ADH-1) (187, 188). ADH-1 was able to activate apoptosis, inhibit cell migration and demonstrated tumour growth inhibition in pre-clinical animal models and anti-tumour activity in phase I clinical trials for the treatment of solid cancers. Further derivatives of these peptides have been synthesised and tested. ADH-1 has been tested in preclinical AR negative PCa models (189) but further testing in AR-V7-expressing tumours is worth exploring.
CDH2 is not expressed in HSPC (183) and while Cottard et al. demonstrated that its upregulation is driven by AR-Vs, research has shown and reported CDH2 to be an AR-V7-specific gene (49, 58, 183).
Cato et al. identified four AR-V7 repressed genes through several steps (37). First, they used differential expression analysis to identify genes that had increased expression when AR-V7 was knocked down in LNCaP-95. The genes they identified also required an AR-V7 binding site within 50kb of their transcription start site. The expression of these genes was analysed in patient samples and demonstrated significantly different expression levels between CRPC with high AR-V7 expression levels vs low AR-V7 expression levels. These genes were then compared with positively selected genes from a Genome-wide CRISPR knockout screen in LNCaP-95 cells. Four of these genes were found to have a negative effect on CRPC cell proliferation (37). The first of these was Solute Carrier Family 30 Member 7 (SLC30A7), which encodes for zinc transporter member 7 (ZNT7) (37).
ZNT7 transports zinc from the cytoplasm into the Golgi apparatus for storage and incorporation as a cofactor into alkaline phosphatases (190, 191), and was found in vivo to be important for dietary zinc absorption and body fat accumulation (192). Whilst a null-mutation of SLC30A7 in the Transgenic Adenocarcinoma of Mouse Prostate (TRAMP) mouse model of PCa accelerated prostate tumour formation (193), the role of SLC30A7 in CRPC has not been extensively studied. Zinc deficiency however, has shown to induce DNA damage in normal human prostate epithelial cells (194), and thus downregulation of ZNT7 by AR-V7 could indicate an altered DNA damage response facilitating increased genomic instability.
ZNT7 agonists or stimulators have not been developed and it is unlikely that upregulating this protein is possible for the treatment of AR-V7 expressing CRPC tumours. Current literature does however show that hyperglycaemic states can increase ZNT7 protein expression (195). The mechanism behind this could be explored for potential drug treatments, however caution should be given as hyperglycaemia can have many other health implications for patients and this mechanism therefore may not be clinically relevant.
There are conflicting findings in the literature around SLC30A7 activity and expression in PCa. Firstly, Cato et al. found SLC30A7 expression was lower in PCa patients that developed metastasis vs patients that did not develop metastasis (37). This was supported by Tepaamorndech et al. who showed a null-mutation of SLC30A7 caused acceleration of prostate tumour formation (193). However, a reduction in SLC30A7 expression would imply that alkaline phosphatases would decrease in activity, however the enzyme alkaline phosphatase is a serum-biomarker of bone metastasis in PCa (196), an event in which AR-V7 is highly expressed (55). Moreover, alkaline phosphatase activity was found to be increased in the metastatic epithelial ARCaPM PCa cell line in comparison to the paired and non-metastatic ARCaPE mesenchymal PCa cell line (197). However, alkaline phosphatase levels remain high in many cancers and the high levels in serum may be produced elsewhere, while the metastatic mesenchymal PCa cell line is not fully representative of the majority of mCRPC which are derived from epithelial cells. More experimental validation is required to identify the role of SLC30A7 in PCa, and to confirm SLC30A7 as an AR-V7 regulated gene.
The second of the AR-V7 target genes Cato et al. identified was β-1,4-galactosyltransferase 1 (B4GALT1) (37). B4GALT1 is an important enzyme in the post-translational modification of proteins such as PD-L1 through the formation of complex glycans (37). Indeed, galactose is involved in the synthesis of the core O-linked glycans and in some N-linked glycans, and dysregulation of glycosylation occurs in many cancers including prostate cancer (198, 199). B4GALT1 has increased expression in several cancers including glioblastoma, breast cancer, and leukaemia (200–203), but shows decreased expression in colorectal, endometrial, and metastatic prostate cancers (59, 60, 204). A conflicting report by Radhakrishnan however, states upregulation of B4GALT1 can be seen in PCa by TNFα stimulation to increase motility and invasiveness of the disease (205) and multiple reports show upregulation of B4GALT1 increases drug resistance in cancers (206–209).
There are no known B4GALT1 agonists, however one study found upregulation of B4GALT1 at the mRNA level by lectin stimulation (specifically Concanavalin A) (210). This was however, demonstrated in immune cells and therefore investigation into whether this would translate in PCa cells should be explored. There is potential, however if this mechanism can be proved in PCa, for Concanavalin A to upregulate B4GALT1 and be used as therapy for AR-V7-expressing CRPC.
Wang et al. found that B4GALT1 was downregulated by both Aldo-keto Reductase Family 1 member C3 (AKR1C3, an androgenic enzyme) and AR-V7, indicating a complex formed by these two proteins, transcriptionally repress B4GALT1 (59–61) and allows tumour progression. This complex may explain why Al-Obaide et al. (207) did not identify any AR binding sites in the B4GALT1 promoter. AKR1C3 can also bind and colocalise with AR-FL, however this phenomenon is often seen with ADT when AKR1C3 expression is higher to promote CRPC development (211). This downregulation of B4GALT1 could reduce NOTCH signalling due to the reduced galactose activity (212) and thus inhibit the tumour suppressor activity of NOTCH signalling such as reducing the innate and adaptive immune response or reducing the p53 checkpoint protein (213). Although B4GALT1 is an AR-V7 repressed gene, the extent to which it may be regulated by AR-FL while lesser, is not fully understood. Furthermore, the repressive effect of other AR-Vs on B4GALT1 has not yet been investigated. Despite this, B4GALT1 is the most promising AR-V7-repressed target gene.
Sorting Nexin 14 (SNX14) is the third gene that was found downregulated by AR-V7 by Cato et al. (37) and encodes the protein, SNX14, which is primarily involved in intracellular trafficking. SNX14 binds to phosphoinositides on endosomal membranes for trafficking to the endoplasmic reticulum, and also plays a role in lipid metabolism in the endoplasmic reticulum (214, 215), in the nervous system and in bone metabolism (216, 217). SNX14 deficiency has been linked with an upregulation of proinflammatory cytokines such as TNF and IL-1β (218). TNFα as previously stated induces PCa migration to induce metastasis while IL-1 has also been positively correlated with bone metastasis, and thus this is a possible mechanism by which SNX14 downregulation drives the progression of PCa (219, 220).
SNX14 agonists or upregulators have not been widely explored. However, one study showed that protein kinase A-mediated phosphorylation of SNX14 inhibited its activity (221) and therefore PKA inhibitors could increase SNX14 in cells. Furthermore, another study demonstrated that the release of miR1324 inhibited expression of SNX14, therefore a degrader of miR1324 could upregulate SNX14 expression. However, considering the role that SNX14 plays biologically, manipulation of these processes may be a risk to patient health.
In PCa, the genomic region chromosome 6q14-q22, which contains SNX14, is commonly deleted (222), and therefore suggested by Dong et al. to contain at least one tumour suppressor gene. The genes within this region were identified via deletion mapping and did include SNX14. SNX14 was removed from the analysis as Dong et al. stated that SNX14 expression was not detected in normal prostate and concluded that SNX14 did not have a role in the normal function or the structure of the prostate (222). However, Cato et al. and the NCBI gene database do show that there is expression of SNX14 within normal prostate (37, 223), thus investigations into SNX14 and its role in PCa and AR-V7 driven disease could be reopened for insight into SNX14 as a target gene.
Hypoxia Inducible Factor 1 Subunit Alpha (HIF1α) is the last of the four AR-V7 downregulated genes identified by Cato et al. HIF1α is the alpha subunit of the HIF-1 TF and plays a large role in regulating the cellular homeostatic response in the event of hypoxia, through the transcriptional initiation of genes involved in angiogenesis, apoptosis, and increasing oxygen transportation (37). Given the role of HIF1α, it is generally considered an oncogene and has indeed been identified with promote cancer progression in several cancers such as pancreatic cancer, colon cancer and renal cell carcinoma (224–226). In PCa, the overall literature also points to HIF1α’s role as an oncogene, and is seen overexpressed in CRPC (37, 227–230). Furthermore, it is known that AR-FL and AR-V7 can heterodimerise with HIF1α to drive transcription (231) and therefore it is expected that AR and HIF1α transcript levels would correlate, which is in contrast to the findings of Cato et al.
Hypoxia-mimetic agents are well known HIF1α activators used to treat ischemic stroke (232). Iron chelators such as Deferoxamine mesylate (DFO), HIF-prolyl hydroxylases (PHD) inhibitors such as FG-4497 and the antioxidant, N-acetylcysteine treatment have all shown to increase HIF1α expression via various mechanisms. There is potential in using these agents to treat AR-V7 expressing CRPC, however due to the conflicting reports in literature often stating HIF1α upregulation in many cancers, this may not be the ideal strategy for treatment.
Tran et al. performed immunohistochemical staining of HIF1α on CRPC tumour biopsies, revealing that there were high levels of HIF1α expression present in CRPC (229). Zhong et al. confirmed an overexpression of HIF1α in prostate adenocarcinomas compared to normal prostate tissues (230). The overexpression of HIF1α under normoxic conditions has been shown to promote the progression of CRPC (233), and Carnell et al. have suggested that a hypoxic microenvironment within the prostate is a primary contributor to secondary genetic alterations, which result in enhanced malignancy and a more aggressive PCa phenotype (234, 235). Therefore, due to the collection of literature contrary to the downregulation of HIF1α in PCa metastasis and its potential role as a tumour suppressor, we do not consider HIF1α a promising AR-V7 target gene. HIF1α is often seen activated in larger hypoxic tumours, and thus we hypothesise that in tumours with decreased tumour burden often seen immediately after Enzalutamide treatment, there may not be an upregulation of HIF1α due to the absence of hypoxic environments and thus why we see HIF1α downregulation as reported by Cato. Furthermore, it was discovered recently that FOXA1 can bind and repress HIF1α, where this complex regulates macrophage invasion and PCa cell invasion and thus driving disease progression. Therefore, increased FOXA1 co-expression with the AR may contribute to this HIF1α repression (236).
Based on the available research, it appears that the majority of the AR-FL regulated genes are also co-regulated by AR-Vs, presumably due to: the co-existence of both forms in a majority of CRPC tumours; their similar structure; and the fact that they can heterodimerise to drive transcription (21, 28, 37, 237). Thus, defining sole AR-V7 targets to study its transcriptional activity is challenging. Additionally, other TFs such as the glucocorticoid receptor (28) or other AR interactors may also alter AR signalling and gene expression (35, 62, 238, 239). Furthermore, research discussed in this review used AR-V models experimentally before validating AR-V7-specific regulation. The truncated AR models used lack the LBD, but do not contain the cryptic exons that most variants additionally contain, therefore these models could be broader models encompassing most of the AR-Vs, and therefore it is possible that genes validated as AR-V7-regulated may also be regulated by other AR-Vs since there is not yet the evidence to prove otherwise.
To verify these genes as AR-V7 target genes, analysis can be carried out using publicly available datasets, however this is beyond the scope of this review, although this review does use all available literature that discusses AR-V7 target genes in the studies carried out. The ideal dataset would be a large RNA-seq study with patient samples, some of which have CRPC and available AR-V7 expression data. However, there is a deficit of such datasets; on cBioPortal there are 8 datasets for “prostate cancer” that have RNA-seq data, and the only one with CRPC samples is the SU2C dataset (27, 240). Although AR-V7 data is also available in the TCGA Cell 2015 dataset, only benign prostate and primary PCa tissue are represented, with AR-V7 only present at very low levels in 42 of the 333 samples (241). On the GEO, there are 29 returns for “prostate cancer” with “AR-V7” and “Expression profiling with RNA-seq”, and whilst the majority of these assess novel inhibitors, 2 contain a small number CRPC patient-derived samples and 10 directly assess the impact of AR-V7 inhibition in PCa cell lines with siRNA, antibody, or shRNA (Table 2). However, the small number of clinical samples and the cell lines will not be as representative of most patients as the large clinical SU2C dataset.
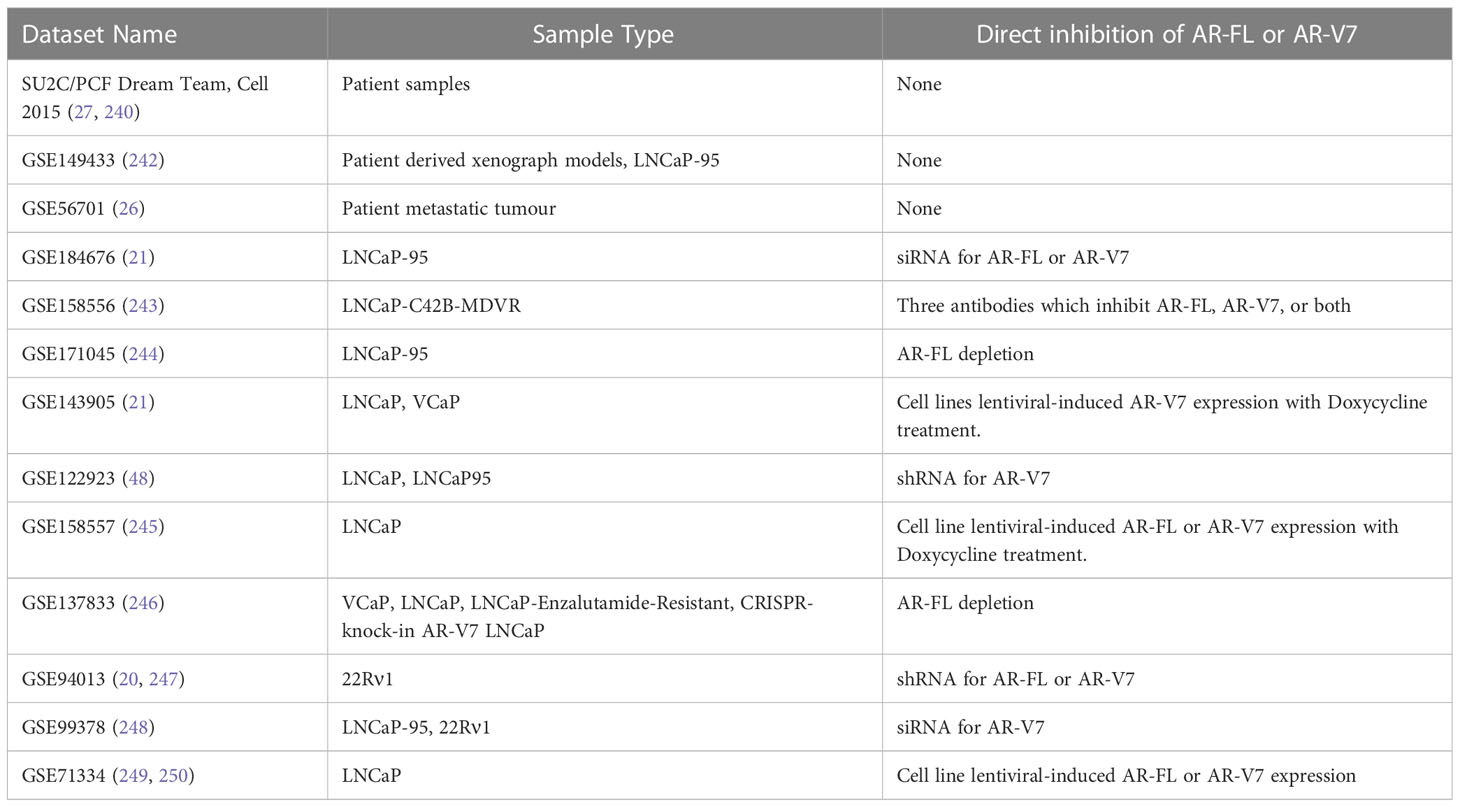
Table 2 Datasets that can be utilised to determine AR-V7 target genes.
Practically, target gene expression is analysed to determine whether drugs have inhibitory activity, and in this case to determine whether drugs can inhibit AR-Vs lacking a complete LBD. Therefore, it is recommended that target genes that are used experimentally should have a distinction between regulation by AR-FL and AR-Vs and as little regulation by other TFs. From our literature search, we deduce the most promising AR-V7 target genes for experimental use are UBE2C, EDN2, ETS2 and B4GALT1, as these have been thoroughly validated from multiple sources and show the least regulation from other TFs.
A few other genes such as OLAH, SDC1, SRD5A1, ORM1, HSP27, C-MYC and HES1 (21, 29, 42) have been discussed in literature as also being AR-V7 target genes, however there is not yet enough evidence to be certain that these are true AR-V7 target genes. Furthermore, Lu et al. reviewed several studies (251) that discuss AR-V-regulated genes, including research from Guo et al. and Li et al. Guo listed genes “preferentially regulated” by AR3 (AR-V7) after data was collected during overexpression of AR-V7 in LNCaP cells such as WNK1 and ADCY6 (51) while Li compiled a list of AR-V upregulated genes after siRNA targeting of exon 1 and 7 of the AR including genes such as NPTX and APOD (133). It is questionable however, whether these are also AR-FL or general AR-V regulated and therefore further validation is needed to prove whether these are AR-V7 specific.
Moreover, there are a few other interactors of AR-V7 that should be investigated further. FOXA1 as previously mentioned, is a transcription factor that is positively correlated with CRPC, and is found to co-regulate genes with the AR (62). Krause et al. discovered that there was differential co-regulation of genes with FOXA1 when AR-FL was present vs when AR-V7 was present. They showed that previously mentioned genes EDN2 and ETS2 that are initially negatively regulated by AR-FL are then positively regulated by AR-V7 (41). Therefore, there is scope to further explore additional genes that follow this pattern. Moreover, the HOXB13 gene encodes a homeodomain-containing TF that is important for normal prostate development and function, and it is primarily expressed in the nuclei of luminal epithelial cells within the prostatic epithelium (252, 253). Elevated expression levels of HOXB13 have been shown in CRPC tumours when compared to hormone-sensitive tumours (253), and it has been demonstrated that HOXB13 interacts with the DBD of the AR, regulating the transcriptional activity of genes containing AREs (254). Furthermore, recent evidence has emerged demonstrating that HOXB13 mediates AR-V7 genomic-binding activity, and that silencing HOXB13 decreased CRPC growth both in-vitro and in-vivo (248, 255). These results highlight an interesting avenue to explore in terms of the discovery of new AR-V7 target genes that are coregulated by HOXB13.
The AR-V7 target genes mentioned in this review are linked with a more aggressive phenotype of PCa (Figure 4), aligning with clinical evidence concerning prognosis and diagnosis; whereby if a patient’s cancer expresses these truncated AR-Vs, the patient is more likely to have a more aggressive form of PCa, compared to patients with tumours only expressing AR-FL (26, 256). These target genes are involved in and drive many biological pathways that determine this more aggressive phenotype, for example the overexpression of AKT1 drives cell cycle progression and cell survival of these cancer cells thus increasing cell proliferation (135). Moreover, CCNA2’s role in metastasis means that its upregulation will increase cell migration of tumour cells and thus increase the likelihood of secondary tumour formation (127). As AR-V7 decreases the activity of the protein encoded by SLC30A7 that transports zinc (190), this could cause more DNA damage and impair DNA repair and thus allows new mutations in cancer, causing disease progression (194) (257),. To summarise, AR-V7 is responsible for the significant upregulation of several cancer hallmark pathways. Sugiura et al. claimed the epithelial mesenchymal transition (EMT) pathway, which plays a role in migration and metastasis, to have been the most significantly upregulated by AR-V7 in CRPC (48, 258). However, there is a lack of information in the literature to make claims about the most prominent pathway that is regulated by AR-V7.
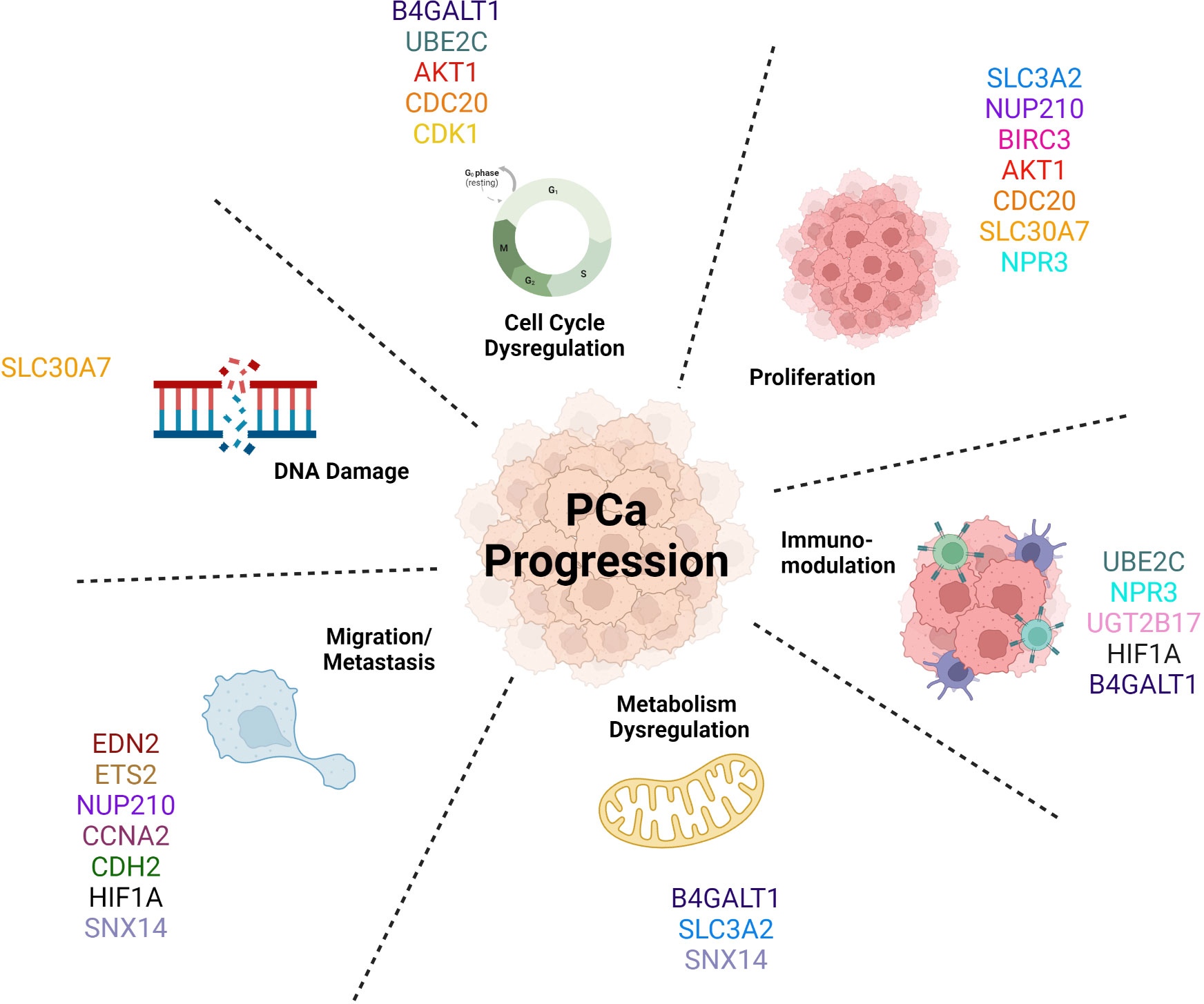
Figure 4 AR-V7 target genes promote Prostate Cancer progression by driving a more aggressive phenotype by activating multiple biological pathways.
The fact that AR-Vs promote a more aggressive phenotype highlights the need for new therapeutics to inhibit them since the antiandrogen drugs currently in the clinic only target AR-FL. Many experimental drugs are currently being tested to determine their efficacy, such as those named in the review by Maylin et al. (2) and thus exploring the expression of the mentioned target genes in PCa models under these novel drug treatments can be a helpful experimental tool. Furthermore, there is also scope for targeting the protein products of the AR-V7 target genes, with a plethora of options available, summarised in Table 3, which have the potential to inhibit growth of AR-V7-expressing CRPC cells.
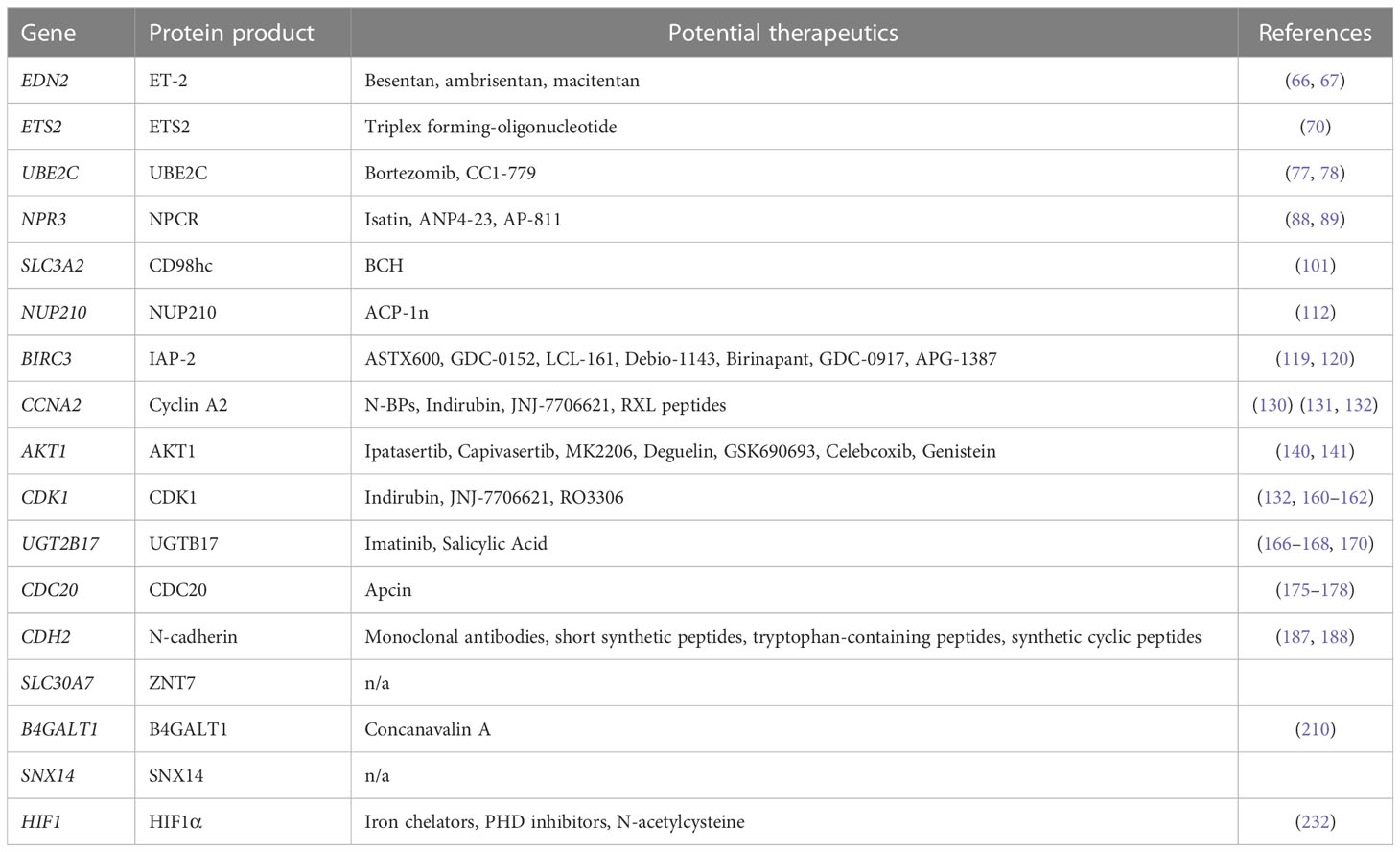
Table 3 Druggable strategies targeting protein products of AR-V7 target genes.
KM and IH wrote the majority of the manuscript with planning, direction and editing from ZM. ZM and CS wrote the discussion. ZM and IH prepared the figures. EA, CS, HP and MA further edited the manuscript and provided guidance throughout. All authors contributed to the article and approved the submitted version.
This work was supported by a doctoral college of the University of Surrey through their PhD studentship award (ZM), the Medical Research Council (UK), the Prostate Cancer Foundation (USA) and the Prostate Project (MA), by studentship/postdoctoral placement supported by HOX therapeutics ltd. funding (EA, IH) and by Univar studentship funding (CS). The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Figures were created using BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Prostate cancer incidence statistics. Cancer Res UK (2015). Available from: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/prostate-cancer/incidence
2. Maylin ZR, Nicolescu RC, Pandha H, Asim M. Breaking androgen receptor addiction of prostate cancer by targeting different functional domains in the treatment of advanced disease. Transl Oncol (2021) 14(8):101115. doi: 10.1016/j.tranon.2021.101115
3. Prostate Cancer: Symptoms, Diagnosis & Treatment - Urology Care Foundation [Internet]. [cited 2022 Jun 30]. Available from: https://www.urologyhealth.org/urologic-conditions/prostate-cancer
4. Cooke PS, Young P, Cunha GR. Androgen receptor expression in developing male reproductive organs. Endocrinology (1991) 128(6):2867–73. doi: 10.1210/endo-128-6-2867
5. Isaacs JT. Antagonistic effect of androgen on prostatic cell death. Prostate (1984) 5(5):545–57. doi: 10.1002/pros.2990050510
6. Davey RA, Grossmann M. Androgen receptor structure, function and biology: From bench to bedside. Clin Biochem Rev (2016) 37(1):3–15.
7. Shiota M, Yokomizo A, Naito S. Increased androgen receptor transcription: a cause of castration-resistant prostate cancer and a possible therapeutic target. J Mol Endocrinol (2011) 47(1):R25–41. doi: 10.1530/JME-11-0018
8. Mora GR, Mahesh VB. Autoregulation of the androgen receptor at the translational level: testosterone induces accumulation of androgen receptor mRNA in the rat ventral prostate polyribosomes. Steroids (1999) 64(9):587–91. doi: 10.1016/S0039-128X(99)00037-9
9. Takeda H, Nakamoto T, Kokontis J, Chodak GW, Chang C. Autoregulation of androgen receptor expression in rodent prostate: immunohistochemical and in situ hybridization analysis. Biochem Biophys Res Commun (1991) 177(1):488–96. doi: 10.1016/0006-291X(91)92010-H
10. Yeap BB, Krueger RG, Leedman PJ. Differential posttranscriptional regulation of androgen receptor gene expression by androgen in prostate and breast cancer cells*. Endocrinology (1999) 140(7):3282–91. doi: 10.1210/endo.140.7.6769
11. Prostate Cancer - Types of Treatment [Internet]. Cancer.Net. (2012). [cited 2022 Jun 30]. Available from: https://www.cancer.net/cancer-types/prostate-cancer/types-treatment
12. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. New Engl J Med (2012) 367(13):1187–97. doi: 10.1056/NEJMoa1207506
13. Treatments expanding for metastatic prostate cancer (2019). Available at: https://www.cancer.gov/news-events/cancer-currents-blog/2019/enzalutamide-apalutamide-metastatic-prostate-cancer (Accessed 2022 Jun 30). NCI.
14. Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res (2006) 12(6):1665–71. doi: 10.1158/1078-0432.CCR-06-0067
15. Mehtälä J, Zong J, Vassilev Z, Brobert G, Gabarró MS, Stattin P, et al. Overall survival and second primary malignancies in men with metastatic prostate cancer. PloS One (2020) 15(2):e0227552. doi: 10.1371/journal.pone.0227552
16. Lu C, Brown LC, Antonarakis ES, Armstrong AJ, Luo J. Androgen receptor variant-driven prostate cancer II: advances in laboratory investigations. Prostate Cancer Prostatic Dis (2020) 23(3):381–97. doi: 10.1038/s41391-020-0217-3
17. Özgün F, Kaya Z, Morova T, Geverts B, Abraham TE, Houtsmuller AB, et al. DNA Binding alters ARv7 dimer interactions. J Cell Sci (2021) 134(14):jcs258332. doi: 10.1242/jcs.258332
18. Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. (2010) 107(39):16759–65. doi: 10.1073/pnas.1012443107
19. Callewaert L, Van Tilborgh N, Claessens F. Interplay between two hormone-independent activation domains in the androgen receptor. Cancer Res (2006) 66(1):543–53. doi: 10.1158/0008-5472.CAN-05-2389
20. Cai L, Tsai YH, Wang P, Wang J, Li D, Fan H, et al. ZFX mediates non-canonical oncogenic functions of the androgen receptor splice variant 7 (AR-V7) in castrate-resistant prostate cancer. Mol Cell (2018) 72(2):341–354.e6. doi: 10.1016/j.molcel.2018.08.029
21. Basil P, Robertson MJ, Bingman WE, Dash AK, Krause WC, Shafi AA, et al. Cistrome and transcriptome analysis identifies unique androgen receptor (AR) and AR-V7 splice variant chromatin binding and transcriptional activities. Sci Rep (2022) 12(1):5351. doi: 10.1038/s41598-022-09371-x
22. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res (2008) 68(13):5469–77. doi: 10.1158/0008-5472.CAN-08-0594
23. Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene (2014) 33(24):3140–50. doi: 10.1038/onc.2013.284
24. Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res (2011) 71(6):2108–17. doi: 10.1158/0008-5472.CAN-10-1998
25. Sharp A, Coleman I, Yuan W, Sprenger C, Dolling D, Rodrigues DN, et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest (2019) 129(1):192–208. doi: 10.1172/JCI122819
26. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med (2014) 371(11):1028–38. doi: 10.1056/NEJMoa1315815
27. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell (2015) 161(5):1215–28. doi: 10.1016/j.cell.2015.05.001
28. Nagandla H, Robertson MJ, Putluri V, Putluri N, Coarfa C, Weigel NL. Isoform-specific activities of androgen receptor and its splice variants in prostate cancer cells. Endocrinology (2021) 162(3):bqaa227. doi: 10.1210/endocr/bqaa227
29. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res (2012) 72(14):3457–62. doi: 10.1158/0008-5472.CAN-11-3892
30. Jones D, Noble M, Wedge SR, Robson CN, Gaughan L. Aurora a regulates expression of AR-V7 in models of castrate resistant prostate cancer. Sci Rep (2017) 7:40957. doi: 10.1038/srep40957
31. Li Y, Xie N, Gleave ME, Rennie PS, Dong X. AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget (2015) 6(32):33743–54. doi: 10.18632/oncotarget.5608
32. Zhu Y, Luo J. Regulation of androgen receptor variants in prostate cancer. Asian J Urol (2020) 7(3):251–7. doi: 10.1016/j.ajur.2020.01.001
33. Cao B, Qi Y, Zhang G, Xu D, Zhan Y, Alvarez X, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget (2014) 5(6):1646–56. doi: 10.18632/oncotarget.1802
34. Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci (2006) 119(Pt 13):2635–41. doi: 10.1242/jcs.03053
35. Miller KJ, Asim M. Unravelling the role of kinases that underpin androgen signalling in prostate cancer. Cells (2022) 11(6):952. doi: 10.3390/cells11060952
36. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone refractory prostate cancer. Cancer Res (2009) 69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764
37. Cato L, de Tribolet-Hardy J, Lee I, Rottenberg JT, Coleman I, Melchers D, et al. ARv7 represses tumor-suppressor genes in castration-resistant prostate cancer. Cancer Cell (2019) 35(3):401–413.e6. doi: 10.1016/j.ccell.2019.01.008
38. Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res (2015) 75(17):3663–71. doi: 10.1158/0008-5472.CAN-15-0381
39. Roggero CM, Jin L, Cao S, Sonavane R, Kopplin NG, Ta H, et al. A detailed characterization of stepwise activation of the androgen receptor variant 7 in prostate cancer cells. Oncogene (2021) 40(6):1106–17. doi: 10.1038/s41388-020-01585-5
40. He Y, Lu J, Ye Z, Hao S, Wang L, Kohli M, et al. Androgen receptor splice variants bind to constitutively open chromatin and promote abiraterone-resistant growth of prostate cancer. Nucleic Acids Res (2018) 46(4):1895–911. doi: 10.1093/nar/gkx1306
41. Krause WC, Shafi AA, Nakka M, Weigel NL. Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol (2014) 54:49–59. doi: 10.1016/j.biocel.2014.06.013
42. Luo J, Attard G, Balk SP, Bevan C, Burnstein K, Cato L, et al. Role of androgen receptor variants in prostate cancer: Report from the 2017 mission androgen receptor variants meeting. Eur Urol (2018) 73(5):715–23. doi: 10.1016/j.eururo.2017.11.038
43. Monaghan AE, McEwan IJ. A sting in the tail: the n-terminal domain of the androgen receptor as a drug target. Asian J Androl (2016) 18(5):687–94. doi: 10.4103/1008-682X.181081
44. Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PloS One (2011) 6(11):e27970. doi: 10.1371/journal.pone.0027970
45. Shimizu Y, Tamada S, Kato M, Hirayama Y, Takeyama Y, Iguchi T, et al. Androgen receptor splice variant 7 drives the growth of castration resistant prostate cancer without being involved in the efficacy of taxane chemotherapy. J Clin Med (2018) 7(11):444. doi: 10.3390/jcm7110444
46. Banuelos CA, Tavakoli I, Tien AH, Caley DP, Mawji NR, Li Z, et al. Sintokamide a is a novel antagonist of androgen receptor that uniquely binds activation function-1 in its amino-terminal domain. J Biol Chem (2016) 291(42):22231–43. doi: 10.1074/jbc.M116.734475
47. Terada N, Shimizu Y, Kamba T, Inoue T, Maeno A, Kobayashi T, et al. Identification of EP4 as a potential target for the treatment of castration-resistant prostate cancer using a novel xenograft model. Cancer Res (2010) 70(4):1606–15. doi: 10.1158/0008-5472.CAN-09-2984
48. Sugiura M, Sato H, Okabe A, Fukuyo M, Mano Y, Shinohara K i, et al. Identification of AR-V7 downstream genes commonly targeted by AR/AR-V7 and specifically targeted by AR-V7 in castration resistant prostate cancer. Trans Oncol (2021) 14(1):100915. doi: 10.1016/j.tranon.2020.100915
49. Moon SJ, Jeong BC, Kim HJ, Lim JE, Kim HJ, Kwon GY, et al. Bruceantin targets HSP90 to overcome resistance to hormone therapy in castration-resistant prostate cancer. Theranostics (2021) 11(2):958–73. doi: 10.7150/thno.51478
50. Hirayama Y, Tam T, Jian K, Andersen RJ, Sadar MD. Combination therapy with androgen receptor n-terminal domain antagonist EPI-7170 and enzalutamide yields synergistic activity in AR-V7-positive prostate cancer. Mol Oncol (2020) 14(10):2455–70. doi: 10.1002/1878-0261.12770
51. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is upregulated during prostate cancer progression and promotes androgen-depletion-resistant growth. Cancer Res (2009) 69(6):2305–13. doi: 10.1158/0008-5472.CAN-08-3795
52. Jillson LK, Rider LC, Rodrigues LU, Romero L, Karimpour-Fard A, Nieto C, et al. MAP3K7 loss drives enhanced androgen signaling and independently confers risk of recurrence in prostate cancer with joint loss of CHD1. Mol Cancer Res (2021) 19(7):1123–36. doi: 10.1158/1541-7786.MCR-20-0913
53. Han Y, Huang W, Liu J, Liu D, Cui Y, Huang R, et al. Triptolide inhibits the AR signaling pathway to suppress the proliferation of enzalutamide resistant prostate cancer cells. Theranostics (2017) 7(7):1914–27. doi: 10.7150/thno.17852
54. Kawamura N, Nimura K, Saga K, Ishibashi A, Kitamura K, Nagano H, et al. SF3B2-mediated RNA splicing drives human prostate cancer progression. Cancer Res (2019) 79(20):5204–17. doi: 10.1158/0008-5472.CAN-18-3965
55. Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PloS One (2011) 6(4):e19059. doi: 10.1371/journal.pone.0019059
56. Gu P, Yang D, Zhu J, Zhang M, He X. Bioinformatics analysis identified hub genes in prostate cancer tumorigenesis and metastasis. Math Biosci Eng (2021) 18(4):3180–96. doi: 10.3934/mbe.2021158
57. Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S, et al. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin Cancer Res (2016) 22(17):4466–77. doi: 10.1158/1078-0432.CCR-15-2901
58. Cottard F, Asmane I, Erdmann E, Bergerat JP, Kurtz JE, Céraline J. Constitutively active androgen receptor variants upregulate expression of mesenchymal markers in prostate cancer cells. PloS One (2013) 8(5):e63466. doi: 10.1371/journal.pone.0063466
59. Wang ZH, Zhang YZ, Wang YS, Ma XX. Identification of novel cell glycolysis related gene signature predicting survival in patients with endometrial cancer. Cancer Cell Int (2019) 19:296. doi: 10.1186/s12935-019-1001-0
60. Narayanan R, Yepuru M, Wu Z, Barrett C, Kim J, Penning TM, et al. Aldo-keto reductase (AKR) 1C3 as an androgen receptor coactivator. JCO (2013) 31(6_suppl):100–0. doi: 10.1200/jco.2013.31.6_suppl.100
61. Wang B, Wu S, Fang Y, Sun G, He D, Hsieh JT, et al. The AKR1C3/AR-V7 complex maintains CRPC tumour growth by repressing B4GALT1 expression. J Cell Mol Med (2020) 24(20):12032–43. doi: 10.1111/jcmm.15831
62. Yang YA, Yu J. Current perspectives on FOXA1 regulation of androgen receptor signaling and prostate cancer. Genes Dis (2015) 2(2):144–51. doi: 10.1016/j.gendis.2015.01.003
63. GeneCards. EDN2 Gene- Endothelin 2 [Internet]. The Human Gene Database. (2022). Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=EDN2
64. Ling L, Maguire JJ, Davenport AP. Endothelin-2, the forgotten isoform: emerging role in the cardiovascular system, ovarian development, immunology and cancer. Br J Pharmacol (2013) 168(2):283–95. doi: 10.1111/j.1476-5381.2011.01786.x
65. Grimshaw MJ, Hagemann T, Ayhan A, Gillett CE, Binder C, Balkwill FR. A role for endothelin-2 and its receptors in breast tumor cell invasion. Cancer Res (2004) 64(7):2461–8. doi: 10.1158/0008-5472.CAN-03-1069
66. Enevoldsen FC, Sahana J, Wehland M, Grimm D, Infanger M, Krüger M. Endothelin receptor antagonists: Status quo and future perspectives for targeted therapy. J Clin Med (2020) 9(3):824. doi: 10.3390/jcm9030824
67. Rich S, McLaughlin VV. Endothelin receptor blockers in cardiovascular disease. Circulation (2003) 108(18):2184–90. doi: 10.1161/01.CIR.0000094397.19932.78
68. UniProt. P15036 · ETS2_HUMAN (2022). Available at: https://www.uniprot.org/uniprotkb/P15036/entry (Accessed 2022 Aug 1).
69. Sementchenko VI, Schweinfest CW, Papas TS, Watson DK. ETS2 function is required to maintain the transformed state of human prostate cancer cells. Oncogene (1998) 17(22):2883–8. doi: 10.1038/sj.onc.1202220
70. Carbone GM, McGuffie EM, Collier A, Catapano CV. Selective inhibition of transcription of the Ets2 gene in prostate cancer cells by a triplex-forming oligonucleotide. Nucleic Acids Res (2003) 31(3):833–43. doi: 10.1093/nar/gkg198
71. Wang Y, Wang J, Tang Q, Ren G. Identification of UBE2C as hub gene in driving prostate cancer by integrated bioinformatics analysis. PloS One (2021) 16(2):e0247827. doi: 10.1371/journal.pone.0247827
72. Liu G, Sprenger C, Wu PJ, Sun S, Uo T, Haugk K, et al. MED1 mediates androgen receptor splice variant induced gene expression in the absence of ligand. Oncotarget (2015) 6(1):288–304. doi: 10.18632/oncotarget.2672
73. Liu Y, Zhao R, Chi S, Zhang W, Xiao C, Zhou X, et al. UBE2C is upregulated by estrogen and promotes epithelial–mesenchymal transition via p53 in endometrial cancer. Mol Cancer Res (2020) 18(2):204–15. doi: 10.1158/1541-7786.MCR-19-0561
74. Li J, Zhi X, Shen X, Chen C, Yuan L, Dong X, et al. Depletion of UBE2C reduces ovarian cancer malignancy and reverses cisplatin resistance via downregulating CDK1. Biochem Biophys Res Commun (2020) 523(2):434–40. doi: 10.1016/j.bbrc.2019.12.058
75. GeneCards. UBE2C Gene-Ubiquitin Conjugating Enzyme E2 C [Internet]. The Human Gene Database. (2022) Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=UBE2C.
76. Lee CH, Ku JY, Ha JM, Bae SS, Lee JZ, Kim CS, et al. Transcript levels of androgen receptor variant 7 and ubiquitin-conjugating enzyme 2C in hormone sensitive prostate cancer and castration-resistant prostate cancer. Prostate (2017) 77(1):60–71. doi: 10.1002/pros.23248
77. Bavi P, Uddin S, Ahmed M, Jehan Z, Bu R, Abubaker J, et al. Bortezomib stabilizes mitotic cyclins and prevents cell cycle progression via inhibition of UBE2C in colorectal carcinoma. Am J Pathol (2011) 178(5):2109–20. doi: 10.1016/j.ajpath.2011.01.034
78. Wang H, Zhang C, Rorick A, Wu D, Chiu M, Thomas-Ahner J, et al. CCI-779 inhibits cell-cycle G2–m progression and invasion of castration-resistant prostate cancer via attenuation of UBE2C transcription and mRNA stability. Cancer Res (2011) 71(14):4866–76. doi: 10.1158/0008-5472.CAN-10-4576
79. Leitman DC, Andresen JW, Kuno T, Kamisaki Y, Chang JK, Murad F. Identification of multiple binding sites for atrial natriuretic factor by affinity cross-linking in cultured endothelial cells. J Biol Chem (1986) 261(25):11650–5. doi: 10.1016/S0021-9258(18)67292-8
80. Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, et al. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. PNAS (1999) 96(13):7403–8. doi: 10.1073/pnas.96.13.7403
81. Bennett BD, Bennett GL, Vitangcol RV, Jewett JR, Burnier J, Henzel W, et al. Extracellular domain-IgG fusion proteins for three human natriuretic peptide receptors. hormone pharmacology and application to solid phase screening of synthetic peptide antisera. J Biol Chem (1991) 266(34):23060–7.
82. Mezzasoma L, Peirce MJ, Minelli A, Bellezza I. Natriuretic peptides: The case of prostate cancer. Molecules (2017) 22(10):E1680. doi: 10.3390/molecules22101680
83. Sun Y, Eichelbaum EJ, Wang H, Vesely DL. Atrial natriuretic peptide and long acting natriuretic peptide inhibit MEK 1/2 activation in human prostate cancer cells. Anticancer Res (2007) 27(6B):3813–8.
84. Vesely BA, Alli AA, Song SJ, Gower Jr.WR, Sanchez-Ramos J, Vesely DL. Four peptide hormones’ specific decrease (up to 97%) of human prostate carcinoma cells. Eur J Clin Invest (2005) 35(11):700–10. doi: 10.1111/j.1365-2362.2005.01569.x
85. Mezzasoma L, Talesa VN, Costanzi E, Bellezza I. Natriuretic peptides regulate prostate cells inflammatory behavior: Potential novel anticancer agents for prostate cancer. Biomolecules (2021) 11(6):794. doi: 10.3390/biom11060794
86. Jonsson Comprehensive Cancer Center. A single-arm, open-label, two-stage phase II study of the MEK 1/2 inhibitor trametinib in men with progressive metastatic castrate resistant prostate cancer (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT02881242 (Accessed 2022 Jul 13). clinicaltrials.govReport No.: NCT02881242.
87. Pedram A, Razandi M, Levin ER. Natriuretic peptides suppress vascular endothelial cell growth factor signaling to angiogenesis*. Endocrinology (2001) 142(4):1578–86. doi: 10.1210/endo.142.4.8099
88. Glover V, Medvedev A, Sandler M. Isatin is a potent endogenous antagonist of guanylate cyclase-coupled atrial natriuretic peptide receptors. Life Sci (1995) 57(22):2073–9. doi: 10.1016/0024-3205(95)02189-p
89. Veale CA, Alford VC, Aharony D, Banville DL, Bialecki RA, Brown FJ, et al. The discovery of non-basic atrial natriuretic peptide clearance receptor antagonists. part 1. Bioorganic Medicinal Chem Letters (2000) 10(17):1949–52. doi: 10.1016/S0960-894X(00)00387-5
90. Ashida S, Nakagawa H, Katagiri T, Furihata M, Iiizumi M, Anazawa Y, et al. Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: Genome-wide gene-expression profiles of prostate cancers and PINs. Cancer Res (2004) 64(17):5963–72. doi: 10.1158/0008-5472.CAN-04-0020
91. Pressly ED, Pierce RA, Connal LA, Hawker CJ, Liu Y. Nanoparticle PET/CT imaging of natriuretic peptide clearance receptor in prostate cancer. Bioconjug Chem (2013) 24(2):196–204. doi: 10.1021/bc300473x
92. Chillarón J, Roca R, Valencia A, Zorzano A, Palacín M. Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am J Physiol Renal Physiol (2001) 281(6):F995–1018. doi: 10.1152/ajprenal.2001.281.6.F995
93. Fenczik CA, Zent R, Dellos M, Calderwood DA, Satriano J, Kelly C, et al. Distinct domains of CD98hc regulate integrins and amino acid transport. J Biol Chem (2001) 276(12):8746–52. doi: 10.1074/jbc.M011239200
94. Cano-Crespo S, Chillarón J, Junza A, Fernández-Miranda G, García J, Polte C, et al. CD98hc (SLC3A2) sustains amino acid and nucleotide availability for cell cycle progression. Sci Rep (2019) 9(1):14065. doi: 10.1038/s41598-019-50547-9
95. Maimaiti M, Sakamoto S, Sugiura M, Kanesaka M, Fujimoto A, Matsusaka K, et al. The heavy chain of 4F2 antigen promote prostate cancer progression via SKP-2. Sci Rep (2021) 11(1):11478. doi: 10.1038/s41598-021-90748-9
96. Satoh T, Kaira K, Takahashi K, Takahashi N, Kanai Y, Asao T, et al. Prognostic significance of the expression of CD98 (4F2hc) in gastric cancer. Anticancer Res (2017) 37(2):631–6. doi: 10.21873/anticanres.11357
97. Kaira K, Kawashima O, Endoh H, Imaizumi K, Goto Y, Kamiyoshihara M, et al. Expression of amino acid transporter (LAT1 and 4F2hc) in pulmonary pleomorphic carcinoma. Hum Pathol (2019) 84:142–9. doi: 10.1016/j.humpath.2018.09.020
98. Kaira K, Ohde Y, Endo M, Nakagawa K, Okumura T, Takahashi T, et al. Expression of 4F2hc (CD98) in pulmonary neuroendocrine tumors. Oncol Rep (2011) 26(4):931–7. doi: 10.3892/or.2011.1384
99. Wu B, Wang Y, Yang XM, Xu BQ, Feng F, Wang B, et al. Basigin-mediated redistribution of CD98 promotes cell spreading and tumorigenicity in hepatocellular carcinoma. J Exp Clin Cancer Res (2015) 34(1):110. doi: 10.1186/s13046-015-0226-6
100. Fort J, Nicolàs-Aragó A, Palacín M. The ectodomains of rBAT and 4F2hc are fake or orphan α-glucosidases. Molecules (2021) 26(20):6231. doi: 10.3390/molecules26206231
101. Shennan DB, Thomson J. Inhibition of system l (LAT1/CD98hc) reduces the growth of cultured human breast cancer cells. Oncol Rep (2008) 20(4):885–9.
102. Wang Q, Tiffen J, Bailey CG, Lehman ML, Ritchie W, Fazli L, et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: effects on cell cycle, cell growth, and tumor development. J Natl Cancer Inst (2013) 105(19):1463–73. doi: 10.1093/jnci/djt241
103. Sheng X, Arnoldussen YJ, Storm M, Tesikova M, Nenseth HZ, Zhao S, et al. Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO Mol Med (2015) 7(6):788–801. doi: 10.15252/emmm.201404509
104. Rabut G, Doye V, Ellenberg J. Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat Cell Biol (2004) 6(11):1114–21. doi: 10.1038/ncb1184
105. Bindra D, Mishra RK. In pursuit of distinctiveness: Transmembrane nucleoporins and their disease associations. Front Oncol (2021) 11:784319. doi: 10.3389/fonc.2021.784319
106. Raices M, Bukata L, Sakuma S, Borlido J, Hernandez LS, Hart DO, et al. Nuclear pores regulate muscle development and maintenance by assembling a localized Mef2C complex. Dev Cell (2017) 41(5):540–554.e7. doi: 10.1016/j.devcel.2017.05.007
107. Sharma M, Johnson M, Brocardo M, Jamieson C, Henderson BR. Wnt signaling proteins associate with the nuclear pore complex: implications for cancer. Adv Exp Med Biol (2014) 773:353–72. doi: 10.1007/978-1-4899-8032-8_16
108. Borlido J, Sakuma S, Raices M, Carrette F, Tinoco R, Bradley LM, et al. Nuclear pore complex-mediated modulation of TCR signaling is required for naïve CD4+ T cell homeostasis. Nat Immunol (2018) 19(6):594–605. doi: 10.1038/s41590-018-0103-5
109. Dufour CR, Scholtes C, Yan M, Chen Y, Han L, Li T, et al. The mTOR chromatin-bound interactome in prostate cancer. Cell Rep (2022) 38(12):110534. doi: 10.1016/j.celrep.2022.110534
110. Audet-Walsh É, Dufour CR, Yee T, Zouanat FZ, Yan M, Kalloghlian G, et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev (2017) 31(12):1228–42. doi: 10.1101/gad.299958.117
111. Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci (2020) 21(12):4507. doi: 10.3390/ijms21124507
112. Kondo H, Mishiro K, Iwashima Y, Qiu Y, Kobayashi A, Lim K, et al. Discovery of a novel aminocyclopropenone compound that inhibits BRD4-driven nucleoporin NUP210 expression and attenuates colorectal cancer growth. Cells (2022) 11(3):317. doi: 10.3390/cells11030317
113. Silke J, Meier P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect Biol (2013) 5(2):a008730. doi: 10.1101/cshperspect.a008730
114. Silke J, Vucic D. Chapter two - IAP family of cell death and signaling regulators, in: Methods in enzymology (2014). Academic Press. Available at: https://www.sciencedirect.com/science/article/pii/B9780128014301000020 (Accessed 2022 Jul 15). Regulated Cell Death Part B.
115. Geserick P, Hupe M, Moulin M, Wong WWL, Feoktistova M, Kellert B, et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol (2009) 187(7):1037–54. doi: 10.1083/jcb.200904158
116. Frazzi R. BIRC3 and BIRC5: multi-faceted inhibitors in cancer. Cell Biosci (2021) 11(1):8. doi: 10.1186/s13578-020-00521-0
117. Wang Z, Zhong M, Song Q, Pascal LE, Yang Z, Wu Z, et al. Anti-apoptotic factor Birc3 is up-regulated by ELL2 knockdown and stimulates proliferation in LNCaP cells. Am J Clin Exp Urol (2019) 7(4):223–31.
118. Wang G, Jones SJM, Marra MA, Sadar MD. Identification of genes targeted by the androgen and PKA signaling pathways in prostate cancer cells. Oncogene (2006) 25(55):7311–23. doi: 10.1038/sj.onc.1209715
119. Cong H, Xu L, Wu Y, Qu Z, Bian T, Zhang W, et al. Inhibitor of apoptosis protein (IAP) antagonists in anticancer agent discovery: Current status and perspectives. J Med Chem (2019) 62(12):5750–72. doi: 10.1021/acs.jmedchem.8b01668
120. Ward GA, Lewis EJ, Ahn JS, Johnson CN, Lyons JF, Martins V, et al. ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 and XIAP, potently induces TNFα-dependent apoptosis in cancer cell lines and inhibits tumor growth. Mol Cancer Ther (2018) 17(7):1381–91. doi: 10.1158/1535-7163.MCT-17-0848
121. Qiu X, Pascal LE, Song Q, Zang Y, Ai J, O’Malley KJ, et al. Physical and functional interactions between ELL2 and RB in the suppression of prostate cancer cell proliferation, migration, and invasion. Neoplasia (2017) 19(3):207–15. doi: 10.1016/j.neo.2017.01.001
122. Kan M, Diwadkar AR, Shuai H, Joo J, Wang AL, Ong MS, et al. Multiomics analysis identifies BIRC3 as a novel glucocorticoid response-associated gene. J Allergy Clin Immunol (2022) 149(6):1981–91. doi: 10.1016/j.jaci.2021.11.025
123. Mostafa MM, Rider CF, Shah S, Traves SL, Gordon PMK, Miller-Larsson A, et al. Glucocorticoid-driven transcriptomes in human airway epithelial cells: commonalities, differences and functional insight from cell lines and primary cells. BMC Med Genomics (2019) 12(1):29. doi: 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A2126
124. Puhr M, Hoefer J, Eigentler A, Ploner C, Handle F, Schaefer G, et al. The glucocorticoid receptor is a key player for prostate cancer cell survival and a target for improved antiandrogen therapy. Clin Cancer Res (2018) 24(4):927–38. doi: 10.1158/1078-0432.CCR-17-0989
125. Hoshi S, Meguro S, Imai H, Matsuoka Y, Yoshida Y, Onagi A, et al. Upregulation of glucocorticoid receptor-mediated glucose transporter 4 in enzalutamide-resistant prostate cancer. Cancer Sci (2021) 112(5):1899–910. doi: 10.1111/cas.14865
126. Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin a is required at two points in the human cell cycle. EMBO J (1992) 11(3):961–71. doi: 10.1002/j.1460-2075.1992.tb05135.x
127. Arsic N, Bendris N, Peter M, Begon-Pescia C, Rebouissou C, Gadéa G, et al. A novel function for cyclin A2: control of cell invasion via RhoA signaling. J Cell Biol (2012) 196(1):147–62. doi: 10.1083/jcb.201102085
128. Yang R, Du Y, Wang L, Chen Z, Liu X. Weighted gene co-expression network analysis identifies CCNA2 as a treatment target of prostate cancer through inhibiting cell cycle. J Cancer (2020) 11(5):1203–11. doi: 10.7150/jca.38173
129. Shen H, Guo YL, Li GH, Zhao W, Zhang L. Gene expression analysis reveals key genes and signalings associated with the prognosis of prostate cancer. Comput Math Methods Med (2021) 2021:9946015. doi: 10.1155/2021/9946015
130. Lee RS, Sohn S, Shin KH, Kang MK, Park NH, Kim RH. Bisphosphonate inhibits the expression of cyclin A2 at the transcriptional level in normal human oral keratinocytes. Int J Mol Med (2017) 40(3):623–30. doi: 10.3892/ijmm.2017.3066
131. Mendoza N, Fong S, Marsters J, Koeppen H, Schwall R, Wickramasinghe D. Selective cyclin-dependent kinase 2/Cyclin a antagonists that differ from ATP site inhibitors block tumor growth. Cancer Res (2003) 63(5):1020–4.
132. Santa Cruz Animal Health. Cyclin a inhibitors (2023). Available at: https://www.scbt.com/browse/cyclin-a-Inhibitors/_/N-jkc83c (Accessed 2023 Jan 26).
133. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KAT, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res (2013) 73(2):483–9. doi: 10.1158/0008-5472.CAN-12-3630
134. Zhang C, Wang L, Wu D, Chen H, Chen Z, Thomas-Ahner JM, et al. Definition of a FoxA1 cistrome that is crucial for G1 to s-phase cell-cycle transit in castration-resistant prostate cancer. Cancer Res (2011) 71(21):6738–48. doi: 10.1158/0008-5472.CAN-11-1882
135. McKenna M, Balasuriya N, Zhong S, Li SSC, O’Donoghue P. Phospho-form specific substrates of protein kinase b (AKT1). Front Bioeng Biotechnol (2021) 8:619252. doi: 10.3389/fbioe.2020.619252
136. Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, et al. AKT inhibitors: New weapons in the fight against breast cancer? Front Pharmacol (2021) 12:662232. doi: 10.3389/fphar.2021.662232
137. Revathidevi S, Munirajan AK. Akt in cancer: Mediator and more. Semin Cancer Biol (2019) 59:80–91. doi: 10.1016/j.semcancer.2019.06.002
138. Wen S, Niu Y, Huang H. Posttranslational regulation of androgen dependent and independent androgen receptor activities in prostate cancer. Asian J Urol (2020) 7(3):203–18. doi: 10.1016/j.ajur.2019.11.001
139. Mediwala SN, Sun H, Szafran AT, Hartig SM, Sonpavde G, Hayes TG, et al. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate (2013) 73(3):267–77. doi: 10.1002/pros.22566
140. Gasmi A, Roubaud G, Dariane C, Barret E, Beauval JB, Brureau L, et al. Overview of the development and use of akt inhibitors in prostate cancer. J Clin Med (2021) 11(1):160. doi: 10.3390/jcm11010160
141. Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: Inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets (2009) 9(2):237–49. doi: 10.2174/156800909787580999
142. Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div (2010) 5:11. doi: 10.1186/1747-1028-5-11
143. Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol (1997) 13:261–91. doi: 10.1146/annurev.cellbio.13.1.261
144. Malumbres M. Revisiting the ‘Cdk-centric’ view of the mammalian cell cycle. Cell Cycle (2005) 4(2):206–10. doi: 10.4161/cc.4.2.1406
145. Kalous J, Jansová D, Šušor A. Role of cyclin-dependent kinase 1 in translational regulation in the m-phase. Cells (2020) 9(7):1568. doi: 10.3390/cells9071568
146. Santamaría D, Barrière C, Cerqueira A, Hunt S, Tardy C, Newton K, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature (2007) 448(7155):811–5. doi: 10.1038/nature06046
147. Liao H, Ji F, Geng X, Xing M, Li W, Chen Z, et al. CDK1 promotes nascent DNA synthesis and induces resistance of cancer cells to DNA-damaging therapeutic agents. Oncotarget (2017) 8(53):90662–73. doi: 10.18632/oncotarget.21730
148. Bertoli C, Skotheim JM, de Bruin RAM. Control of cell cycle transcription during G1 and s phases. Nat Rev Mol Cell Biol (2013) 14(8):518–28. doi: 10.1038/nrm3629
149. Haase SB, Wittenberg C. Topology and control of the cell-Cycle-Regulated transcriptional circuitry. Genetics (2014) 196(1):65–90. doi: 10.1534/genetics.113.152595
150. Landry BD, Mapa CE, Arsenault HE, Poti KE, Benanti JA. Regulation of a transcription factor network by Cdk1 coordinates late cell cycle gene expression. EMBO J (2014) 33(9):1044–60. doi: 10.1002/embj.201386877
151. Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci U S A. (2006) 103(43):15969–74. doi: 10.1073/pnas.0604193103
152. Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene (2008) 27(34):4733–44. doi: 10.1038/onc.2008.104
153. Bury M, Le Calvé B, Lessard F, Dal Maso T, Saliba J, Michiels C, et al. NFE2L3 controls colon cancer cell growth through regulation of DUX4, a CDK1 inhibitor. Cell Rep (2019) 29(6):1469–1481.e9. doi: 10.1016/j.celrep.2019.09.087
154. Wu CX, Wang XQ, Chok SH, Man K, Tsang SHY, Chan ACY, et al. Blocking CDK1/PDK1/β-catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics (2018) 8(14):3737–50. doi: 10.7150/thno.25487
155. Kang J, Sergio CM, Sutherland RL, Musgrove EA. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer (2014) 14(1):32. doi: 10.1186/1471-2407-14-32
156. Yasukawa M, Ando Y, Yamashita T, Matsuda Y, Shoji S, Morioka MS, et al. CDK1 dependent phosphorylation of hTERT contributes to cancer progression. Nat Commun (2020) 11(1):1557. doi: 10.1038/s41467-020-15289-7
157. Gao X, Liang J, Wang L, Zhang Z, Yuan P, Wang J, et al. Phosphorylation of the androgen receptor at Ser81 is co-sustained by CDK1 and CDK9 and leads to AR-mediated transactivation in prostate cancer. Mol Oncol (2021) 15(7):1901–20. doi: 10.1002/1878-0261.12968
158. Liu X, Gao Y, Ye H, Gerrin S, Ma F, Wu Y, et al. Positive feedback loop mediated by protein phosphatase 1α mobilization of p-TEFb and basal CDK1 drives androgen receptor in prostate cancer. Nucleic Acids Res (2017) 45(7):3738–51. doi: 10.1093/nar/gkw1291
159. Willder JM, Heng SJ, McCall P, Adams CE, Tannahill C, Fyffe G, et al. Androgen receptor phosphorylation at serine 515 by Cdk1 predicts biochemical relapse in prostate cancer patients. Br J Cancer (2013) 108(1):139–48. doi: 10.1038/bjc.2012.480
160. Xia Q, Cai Y, Peng R, Wu G, Shi Y, Jiang W. The CDK1 inhibitor RO3306 improves the response of BRCA-proficient breast cancer cells to PARP inhibition. Int J Oncol (2014) 44(3):735–44. doi: 10.3892/ijo.2013.2240
161. Zhang B, Zhang M, Li Q, Yang Y, Shang Z, Luo J. TPX2 mediates prostate cancer epithelial-mesenchymal transition through CDK1 regulated phosphorylation of ERK/GSK3β/SNAIL pathway. Biochem Biophys Res Commun (2021) 546:1–6. doi: 10.1016/j.bbrc.2021.01.106
162. Kojima K, Shimanuki M, Shikami M, Andreeff M, Nakakuma H. Cyclin-dependent kinase 1 inhibitor RO-3306 enhances p53-mediated bax activation and mitochondrial apoptosis in AML. Cancer Sci (2009) 100(6):1128–36. doi: 10.1111/j.1349-7006.2009.01150.x
163. GeneCards. UGT2B17 Gene- UDP Glucuronosyltransferase Family 2 Member B17 [Internet]. The Human Gene Database. (2022). Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=UGT2B17
164. Zhu AZX, Cox LS, Ahluwalia JS, Renner CC, Hatsukami DK, Benowitz NL, et al. Genetic and phenotypic variation in UGT2B17, a testosterone-metabolizing enzyme, is associated with BMI in males. Pharmacogenetics Genomics (2015) 25(5):263–9. doi: 10.1097/FPC.0000000000000135
165. Li H, Xie N, Chen R, Verreault M, Fazli L, Gleave ME, et al. UGT2B17 expedites progression of castration-resistant prostate cancers by promoting ligand-independent AR signaling. Cancer Res (2016) 76(22):6701–11. doi: 10.1158/0008-5472.CAN-16-1518
166. Zhang H, Basit A, Busch D, Yabut K, Bhatt DK, Drozdzik M, et al. Quantitative characterization of UDP-glucuronosyltransferase 2B17 in human liver and intestine and its role in testosterone first-pass metabolism. Biochem Pharmacol (2018) 156:32–42. doi: 10.1016/j.bcp.2018.08.003
167. ClinicalTrials.gov. A phase II study of imatinib and docetaxel in metastatic hormone refractory prostate cancer (2017). Available at: https://clinicaltrials.gov/ct2/show/study/NCT00251225 (Accessed 2023 Jan 26). clinicaltrials.govReport No.: study/NCT00251225.
168. Detchokul S, Frauman AG. Recent developments in prostate cancer biomarker research: therapeutic implications. Br J Clin Pharmacol (2011) 71(2):157–74. doi: 10.1111/j.1365-2125.2010.03766.x
169. Rosenberg A, Mathew P. Imatinib and prostate cancer: lessons learned from targeting the platelet-derived growth factor receptor. Expert Opin Investigational Drugs (2013) 22(6):787–94. doi: 10.1517/13543784.2013.787409
170. Salhab H, Naughton DP, Barker J. Potential assessment of UGT2B17 inhibition by salicylic acid in human supersomes in vitro. Molecules (2021) 26(15):4410. doi: 10.3390/molecules26154410
171. Wu F, Sun Y, Chen J, Li H, Yao K, Liu Y, et al. The oncogenic role of APC/C activator protein Cdc20 by an integrated pan-cancer analysis in human tumors. Front Oncol (2021) 11:721797. doi: 10.3389/fonc.2021.721797
172. GeneCards. CDC-20 Gene- Cell Division Cycle 20 [Internet]. The Human Gene Database. (2022). Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CDC20
173. Yu H. Cdc20: A WD40 activator for a cell cycle degradation machine. Mol Cell (2007) 27(1):3–16. doi: 10.1016/j.molcel.2007.06.009
174. Zhang Q, Huang H, Liu A, Li J, Liu C, Sun B, et al. Cell division cycle 20 (CDC20) drives prostate cancer progression via stabilization of β-catenin in cancer stem-like cells. EBioMedicine (2019) 42:397–407. doi: 10.1016/j.ebiom.2019.03.032
175. Ding Y, Zhang C, He L, Song X, Zheng C, Pan Y, et al. Apcin inhibits the growth and invasion of glioblastoma cells and improves glioma sensitivity to temozolomide. Bioengineered (2021) 12(2):10791–8. doi: 10.1080/21655979.2021.2003927
176. Gao Y, Zhang B, Wang Y, Shang G. Cdc20 inhibitor apcin inhibits the growth and invasion of osteosarcoma cells. Oncol Rep (2018) 40(2):841–8. doi: 10.3892/or.2018.6467
177. Wu F, Dai X, Gan W, Wan L, Li M, Mitsiades N, et al. Prostate cancer-associated mutation in SPOP impairs its ability to target Cdc20 for poly-ubiquitination and degradation. Cancer Letters (2017) 385:207–14. doi: 10.1016/j.canlet.2016.10.021
178. Bhuniya R, Yuan X, Bai L, Howie KL, Wang R, Li W, et al. Design, synthesis, and biological evaluation of apcin-based CDC20 inhibitors. ACS Med Chem Lett (2022) 13(2):188–95. doi: 10.1021/acsmedchemlett.1c00544
179. Seki M, Kajiwara D, Mizutani H, Minamiguchi K. Analysis of novel enzalutamide-resistant cells: upregulation of testis-specific y-encoded protein gene promotes the expression of androgen receptor splicing variant 7. Trans Cancer Res (2020) 9(10):6232–45. doi: 10.21037/tcr-20-1463
180. Yu B, Liu Y, Luo H, Fu J, Li Y, Shao C. Androgen receptor splicing variant 7 (ARV7) inhibits docetaxel sensitivity by inactivating the spindle assembly checkpoint. J Biol Chem (2021) 296:100276. doi: 10.1016/j.jbc.2021.100276
181. GeneCards. CDH2 Gene- Cadherin 2 [Internet]. The Human Gene Database. (2022). Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CDH2
182. Streicher W, Zengerling F, Laschak M, Weidemann W, Höpfner M, Schrader AJ, et al. AR-Q640X, a model to study the effects of constitutively active c-terminally truncated AR variants in prostate cancer cells. World J Urol (2012) 30(3):333–9. doi: 10.1007/s00345-012-0842-0
183. Moon SJ, Jeong BC, Kim HJ, Lim JE, Kwon GY, Kim JH. DBC1 promotes castration-resistant prostate cancer by positively regulating DNA binding and stability of AR-V7. Oncogene (2018) 37(10):1326–39. doi: 10.1038/s41388-017-0047-5
184. Kim YS, Yi BR, Kim NH, Choi KC. Role of the epithelial–mesenchymal transition and its effects on embryonic stem cells. Exp Mol Med (2014) 46(8):e108–8. doi: 10.1038/emm.2014.44
185. Clarke NW, Hart CA, Brown MD. Molecular mechanisms of metastasis in prostate cancer. Asian J Androl (2009) 11(1):57–67. doi: 10.1038/aja.2008.29
186. Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol (2011) 8(8):428–39. doi: 10.1038/nrurol.2011.85
187. Blaschuk OW. N-cadherin antagonists as oncology therapeutics. Philos Trans R Soc Lond B Biol Sci (2015) 370(1661):20140039. doi: 10.1098/rstb.2014.0039
188. Blaschuk OW. Potential therapeutic applications of n-cadherin antagonists and agonists. Front Cell Dev Biol (2022) 10:866200. doi: 10.3389/fcell.2022.866200
189. Li H, Price DK, Figg WD. ADH1, an n-cadherin inhibitor, evaluated in preclinical models of angiogenesis and androgen-independent prostate cancer. Anticancer Drugs (2007) 18(5):563–8. doi: 10.1097/CAD.0b013e328020043e
190. Kirschke CP, Huang L. ZnT7, a novel mammalian zinc transporter, accumulates zinc in the golgi apparatus. J Biol Chem (2003) 278(6):4096–102. doi: 10.1074/jbc.M207644200
191. Suzuki T, Ishihara K, Migaki H, Matsuura W, Kohda A, Okumura K, et al. Zinc transporters, ZnT5 and ZnT7, are required for the activation of alkaline phosphatases, zinc-requiring enzymes that are glycosylphosphatidylinositol-anchored to the cytoplasmic membrane. J Biol Chem (2005) 280(1):637–43. doi: 10.1074/jbc.M411247200
192. Huang L, Yu YY, Kirschke CP, Gertz ER, Lloyd KKC. Znt7 (Slc30a7)-deficient mice display reduced body zinc status and body fat accumulation. J Biol Chem (2007) 282(51):37053–63. doi: 10.1074/jbc.M706631200
193. Tepaamorndech S, Huang L, Kirschke CP. A null-mutation in the Znt7 gene accelerates prostate tumor formation in a transgenic adenocarcinoma mouse prostate model. Cancer Lett (2011) 308(1):33–42. doi: 10.1016/j.canlet.2011.04.011
194. Yan M, Song Y, Wong CP, Hardin K, Ho E. Zinc deficiency alters DNA damage response genes in normal human prostate epithelial cells. J Nutr (2008) 138(4):667–73. doi: 10.1093/jn/138.4.667
195. Tuncay E, Bitirim C, Olgar Y, Durak A, Rutter G, Turan B. Zn 2+ -transporters ZIP7 and ZnT7 play important role in progression of cardiac dysfunction via affecting sarco(endo)plasmic reticulum-mitochondria coupling in hyperglycemic cardiomyocytes. Mitochondrion (2018) 44:41–52. doi: 10.1016/j.mito.2017.12.011
196. Li D, Lv H, Hao X, Hu B, Song Y. Prognostic value of serum alkaline phosphatase in the survival of prostate cancer: evidence from a meta-analysis. Cancer Manag Res (2018) 10:3125–39. doi: 10.2147/CMAR.S174237
197. Rao SR, Snaith AE, Marino D, Cheng X, Lwin ST, Orriss IR, et al. Tumour-derived alkaline phosphatase regulates tumour growth, epithelial plasticity and disease-free survival in metastatic prostate cancer. Br J Cancer (2017) 116(2):227–36. doi: 10.1038/bjc.2016.402
198. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer (2015) 15(9):540–55. doi: 10.1038/nrc3982
199. Samaržija I. Post-translational modifications that drive prostate cancer progression. Biomolecules (2021) 11(2):247. doi: 10.3390/biom11020247
200. Wang P, Li X, Xie Y. B4GalT1 regulates apoptosis and autophagy of glioblastoma in vitro and In vivo. Technol Cancer Res Treat (2020) 19. 1533033820980104. doi: 10.1177/1533033820980104
201. Wu J, Xiao L, Zhou H, Liu H, Ge Y, Yang J, et al. ZFX modulates the growth of human leukemic cells via B4GALT1. Acta Biochim Biophys Sin (Shanghai) (2018) 50(5):522. doi: 10.1093/abbs/gmy012
202. Choi HJ, Chung TW, Kim CH, Jeong HS, Joo M, Youn B, et al. Estrogen induced β-1,4-galactosyltransferase 1 expression regulates proliferation of human breast cancer MCF-7 cells. Biochem Biophys Res Commun (2012) 426(4):620–5. doi: 10.1016/j.bbrc.2012.08.140
203. Liu YX, Wang L, Liu WJ, Zhang HT, Xue JH, Zhang ZW, et al. MiR-124-3p/B4GALT1 axis plays an important role in SOCS3-regulated growth and chemo-sensitivity of CML. J Hematol Oncol (2016) 9(1):69. doi: 10.1186/s13045-016-0300-3
204. Poeta ML, Massi E, Parrella P, Pellegrini P, De Robertis M, Copetti M, et al. Aberrant promoter methylation of beta-1,4 galactosyltransferase 1 as potential cancer-specific biomarker of colorectal tumors. Genes Chromosomes Cancer (2012) 51(12):1133–43. doi: 10.1002/gcc.21998
205. Radhakrishnan P, Chachadi V, Lin MF, Singh R, Kannagi R, Cheng PW. TNFα enhances the motility and invasiveness of prostatic cancer cells by stimulating the expression of selective glycosyl- and sulfotransferase genes involved in the synthesis of selectin ligands. Biochem Biophys Res Commun (2011) 409(3):436–41. doi: 10.1016/j.bbrc.2011.05.019
206. Zhou H, Zhang Z, Liu C, Jin C, Zhang J, Miao X, et al. B4GALT1 gene knockdown inhibits the hedgehog pathway and reverses multidrug resistance in the human leukemia K562/adriamycin-resistant cell line. IUBMB Life (2012) 64(11):889–900. doi: 10.1002/iub.1080
207. Al-Obaide MAI, Alobydi H, Abdelsalam AG, Zhang R, Srivenugopal KS. Multifaceted roles of 5’-regulatory region of the cancer associated gene B4GALT1 and its comparison with the gene family. Int J Oncol (2015) 47(4):1393–404. doi: 10.3892/ijo.2015.3136
208. Zhou H, Ma H, Wei W, Ji D, Song X, Sun J, et al. B4GALT family mediates the multidrug resistance of human leukemia cells by regulating the hedgehog pathway and the expression of p-glycoprotein and multidrug resistance-associated protein 1. Cell Death Dis (2013) 4(6):e654–4. doi: 10.1038/cddis.2013.186
209. Chen Y, Su L, Huang C, Wu S, Qiu X, Zhao X, et al. Galactosyltransferase B4GALT1 confers chemoresistance in pancreatic ductal adenocarcinomas by upregulating n-linked glycosylation of CDK11p110. Cancer Letters (2021) 500:228–43. doi: 10.1016/j.canlet.2020.12.006
210. Nilius V, Killer MC, Timmesfeld N, Schmitt M, Moll R, Lorch A, et al. High β-1,4-Galactosyltransferase-I expression in peripheral T-lymphocytes is associated with a low risk of relapse in germ-cell cancer patients receiving high-dose chemotherapy with autologous stem cell reinfusion. Oncoimmunology (2018) 7(5):e1423169. doi: 10.1080/2162402X.2017.1423169
211. Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, et al. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res (2013) 19(20):5613–25. doi: 10.1158/1078-0432.CCR-13-1151
212. Hou X, Tashima Y, Stanley P. Galactose differentially modulates lunatic and manic fringe effects on Delta1-induced NOTCH signaling. J Biol Chem (2012) 287(1):474–83. doi: 10.1074/jbc.M111.317578
213. Aster JC, Pear WS, Blacklow SC. The varied roles of notch in cancer. Annu Rev Pathol (2017) 12:245–75. doi: 10.1146/annurev-pathol-052016-100127
214. GeneCards. SNX14 Gene- Sorting Nexin 14 [Internet]. The Human Gene Database. (2022). Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=SNX14
215. Bryant D, Liu Y, Datta S, Hariri H, Seda M, Anderson G, et al. SNX14 mutations affect endoplasmic reticulum-associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20. Hum Mol Genet (2018) 27(11):1927–40. doi: 10.1093/hmg/ddy101
216. Vieira N, Rito T, Correia-Neves M, Sousa N. Sorting out sorting nexins functions in the nervous system in health and disease. Mol Neurobiol (2021) 58(8):4070–106. doi: 10.1007/s12035-021-02388-9
217. Zhu Y, Yang K, Cheng Y, Liu Y, Gu R, Liu X, et al. Apoptotic vesicles regulate bone metabolism via the miR1324/SNX14/SMAD1/5 signaling axis. Small (2023), 2205813. doi: 10.1002/smll.202205813
218. Zhang H, Hong Y, Yang W, Wang R, Yao T, Wang J, et al. SNX14 deficiency-induced defective axonal mitochondrial transport in purkinje cells underlies cerebellar ataxia and can be reversed by valproate. Natl Sci Rev (2021) 8(7):nwab024. doi: 10.1093/nsr/nwab024
219. Maolake A, Izumi K, Natsagdorj A, Iwamoto H, Kadomoto S, Makino T, et al. Tumor necrosis factor-α induces prostate cancer cell migration in lymphatic metastasis through CCR7 upregulation. Cancer Sci (2018) 109(5):1524–31. doi: 10.1111/cas.13586
220. Tong Y, Cao Y, Jin T, Huang Z, He Q, Mao M. Role of interleukin-1 family in bone metastasis of prostate cancer. Front Oncol (2022) 12:951167. doi: 10.3389/fonc.2022.951167
221. Ha CM, Park D, Kim Y, Na M, Panda S, Won S, et al. SNX14 is a bifunctional negative regulator for neuronal 5-HT6 receptor signaling. J Cell Sci (2015) 128(9):1848–62. doi: 10.1242/jcs.169581
222. Dong XY, Rodriguez C, Guo P, Sun X, Talbot JT, Zhou W, et al. SnoRNA U50 is a candidate tumor-suppressor gene at 6q14.3 with a mutation associated with clinically significant prostate cancer. Hum Mol Genet (2008) 17(7):1031–42. doi: 10.1093/hmg/ddm375
223. SNX14 sorting nexin 14 [Homo sapiens (human)] - gene - NCBI (2022). Available at: https://www.ncbi.nlm.nih.gov/gene/57231.
224. Jin X, Dai L, Ma Y, Wang J, Liu Z. Implications of HIF-1α in the tumorigenesis and progression of pancreatic cancer. Cancer Cell Int (2020) 20. doi: 10.1186/s12935-020-01370-0
225. Shyu KG, Hsu FL, Wang MJ, Wang BW, Lin S. Hypoxia-inducible factor 1alpha regulates lung adenocarcinoma cell invasion. Exp Cell Res (2007) 313(6):1181–91. doi: 10.1016/j.yexcr.2007.01.013
226. Gudas LJ, Fu L, Minton DR, Mongan NP, Nanus DM. The role of HIF1α in renal cell carcinoma tumorigenesis. J Mol Med (Berl) (201) 92(8):825–36. doi: 10.1007/s00109-014-1180-z
227. Ranasinghe WKB, Xiao L, Kovac S, Chang M, Michiels C, Bolton D, et al. The role of hypoxia-inducible factor 1α in determining the properties of castrate-resistant prostate cancers. PloS One (2013) 8(1):e54251. doi: 10.1371/journal.pone.0054251
228. Vergis R, Corbishley CM, Norman AR, Bartlett J, Jhavar S, Borre M, et al. Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol (2008) 9(4):342–51. doi: 10.1016/S1470-2045(08)70076-7
229. Tran MGB, Bibby BAS, Yang L, Lo F, Warren AY, Shukla D, et al. Independence of HIF1a and androgen signaling pathways in prostate cancer. BMC Cancer (2020) 20(1):469. doi: 10.1186/s12885-020-06890-6
230. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res (1999) 59(22):5830–5.
231. Fruehauf JP, Farrokhian N, Sarkissian S, Kim JH. Blockade of ARV7:HIF1α heterodimers after topotecan reverses enzalutamide resistance in 22Rv1 cells. JCO (2016) 34(15_suppl):e16594–4. doi: 10.1200/JCO.2016.34.15_suppl.e16594
232. Davis CK, Jain SA, Bae ON, Majid A, Rajanikant GK. Hypoxia mimetic agents for ischemic stroke. Front Cell Dev Biol (2019) 6:175. doi: 10.3389/fcell.2018.00175
233. Ranasinghe WKB, Baldwin GS, Shulkes A, Bolton D, Patel O. Normoxic regulation of HIF-1α in prostate cancer. Nat Rev Urol (2014) 11(7):419. doi: 10.1038/nrurol.2013.110-c2
234. Carnell DM, Smith RE, Daley FM, Saunders MI, Bentzen SM, Hoskin PJ. An immunohistochemical assessment of hypoxia in prostate carcinoma using pimonidazole: implications for radioresistance. Int J Radiat Oncol Biol Phys (2006) 65(1):91–9. doi: 10.1016/j.ijrobp.2005.11.044
235. GeneCards. HIF1A gene- hypoxia inducible factor 1 subunit alpha. Hum Gene Database (2022). Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=HIF1A
236. Wang X, Brea L, Lu X, Gritsina G, Park SH, Xie W, et al. FOXA1 inhibits hypoxia programs through transcriptional repression of HIF1A. Oncogene (2022) 41(37):4259–70. doi: 10.1038/s41388-022-02423-6
237. Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem (2012) 287(23):19736–49. doi: 10.1074/jbc.M112.352930
238. Yao M, Rosario ER, Soper JC, Pike CJ. Androgens regulate tau phosphorylation through phosphatidylinositol 3-Kinase-Protein kinase b-glycogen synthase kinase 3β signaling. Neuroscience (2022). S0306-4522(22)00335-9. doi: 10.1016/j.neuroscience.2022.06.034
239. Stella S, Vitale SR, Massimino M, Motta G, Longhitano C, Lanzafame K, et al. Molecular analysis of luminal androgen receptor reveals activated pathways and potential therapeutic targets in breast cancer. Cancer Genomics Proteomics (2022) 19(4):464–76. doi: 10.21873/cgp.20333
240. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. (2019) 116(23):11428–36. doi: 10.1073/pnas.1902651116
241. The Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell (2015) 163(4):1011–25. doi: 10.1016/j.cell.2015.10.025
242. Zhu Y, Dalrymple SL, Coleman I, Zheng SL, Xu J, Hooper JE, et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene (2020) 39(45):6935–49. doi: 10.1038/s41388-020-01479-6
243. Liu C, Armstrong CM, Ning S, Yang JC, Lou W, Lombard AP, et al. ARVib suppresses growth of advanced prostate cancer via inhibition of androgen receptor signaling. Oncogene (2021) 40(35):5379–92. doi: 10.1038/s41388-021-01914-2
244. Liang J, Wang L, Poluben L, Nouri M, Arai S, Xie L, et al. Androgen receptor splice variant 7 functions independently of the full length receptor in prostate cancer cells. Cancer Lett (2021) 519:172–84. doi: 10.1016/j.canlet.2021.07.013
245. Erdmann É, Ould Madi Berthélémy P, Cottard F, Angel CZ, Schreyer E, Ye T, et al. Androgen receptor-mediated transcriptional repression targets cell plasticity in prostate cancer. Mol Oncol (2022) 16(13):2518–36. doi: 10.1002/1878-0261.13164
246. Kregel S, Wang C, Han X, Xiao L, Fernandez-Salas E, Bawa P, et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia (2020) 22(2):111–9. doi: 10.1016/j.neo.2019.12.003
247. Xu C, Tsai YH, Galbo PM, Gong W, Storey AJ, Xu Y, et al. Cistrome analysis of YY1 uncovers a regulatory axis of YY1:BRD2/4-PFKP during tumorigenesis of advanced prostate cancer. Nucleic Acids Res (2021) 49(9):4971–88. doi: 10.1093/nar/gkab252
248. Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao P, Zhang Q, et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc Natl Acad Sci U S A. (2018) 115(26):6810–5. doi: 10.1073/pnas.1718811115
249. Schreyer E, Barthélémy P, Cottard F, Ould Madi-Berthélémy P, Schaff-Wendling F, Kurtz JE, et al. [Androgen receptor variants in prostate cancer]. Med Sci (Paris) (2017) 33(8–9):758–64. doi: 10.1051/medsci/20173308021
250. Cottard F, Madi-Berthélémy PO, Erdmann E, Schaff-Wendling F, Keime C, Ye T, et al. Dual effects of constitutively active androgen receptor and full-length androgen receptor for n-cadherin regulation in prostate cancer. Oncotarget (2017) 8(42):72008–20. doi: 10.18632/oncotarget.18270
251. Lu J, van der Steen T, Tindall DJ. Are androgen receptor variants a substitute for the full-length receptor? Nat Rev Urol (2015) 12(3):137–44. doi: 10.1038/nrurol.2015.13
252. Javed S, Langley SEM. Importance of HOX genes in normal prostate gland formation, prostate cancer development and its early detection. BJU Int (2014) 113(4):535–40. doi: 10.1111/bju.12269
253. Kim YR, Oh KJ, Park RY, Xuan NT, Kang TW, Kwon DD, et al. HOXB13 promotes androgen independent growth of LNCaP prostate cancer cells by the activation of E2F signaling. Mol Cancer (2010) 9:124. doi: 10.1186/1476-4598-9-124
254. Norris JD, Chang CY, Wittmann BM, Kunder RS, Cui H, Fan D, et al. The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol Cell (2009) 36(3):405–16. doi: 10.1016/j.molcel.2009.10.020
255. Navarro HI, Goldstein AS. HoxB13 mediates AR-V7 activity in prostate cancer. Proc Natl Acad Sci (2018) 115(26):6528–9. doi: 10.1073/pnas.1808196115
256. Wang Z, Shen H, Ma N, Li Q, Mao Y, Wang C, et al. The prognostic value of androgen receptor splice variant 7 in castration-resistant prostate cancer treated with novel hormonal therapy or chemotherapy: A systematic review and meta-analysis. Front Oncol (2020) 10:572590. doi: 10.3389/fonc.2020.572590
257. Dhawan DK, Chadha VD. Zinc: A promising agent in dietary chemoprevention of cancer. Indian J Med Res (2010) 132(6):676–82.
Keywords: AR-V7, prostate, castration, gene, cancer
Citation: Miller KJ, Henry I, Maylin Z, Smith C, Arunachalam E, Pandha H and Asim M (2023) A compendium of Androgen Receptor Variant 7 target genes and their role in Castration Resistant Prostate Cancer. Front. Oncol. 13:1129140. doi: 10.3389/fonc.2023.1129140
Received: 21 December 2022; Accepted: 13 February 2023;
Published: 01 March 2023.
Edited by:
Sakthivel Muniyan, University of Nebraska Medical Center, United StatesReviewed by:
Shuai Gao, New York Medical College, United StatesCopyright © 2023 Miller, Henry, Maylin, Smith, Arunachalam, Pandha and Asim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zoe Maylin, ei5tYXlsaW5Ac3VycmV5LmFjLnVr; Mohammad Asim, bS5hc2ltQHN1cnJleS5hYy51aw==
†These authors contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.