 Simone Feurstein
Simone Feurstein
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 06 April 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1128533
This article is part of the Research Topic Advances in Predisposition to Bone Marrow Failure and Hematopoietic Neoplasms View all 11 articles
Inherited bone marrow failure (BMF) syndromes are genetically diverse — more than 100 genes have been associated with those syndromes and the list is rapidly expanding. Risk assessment and genetic counseling of patients with recently discovered BMF syndromes is inherently difficult as disease mechanisms, penetrance, genotype-phenotype associations, phenotypic heterogeneity, risk of hematologic malignancies and clonal markers of disease progression are unknown or unclear. This review aims to shed light on recently described BMF syndromes with sparse concise data and with an emphasis on those associated with germline variants in ADH5/ALDH2, DNAJC21, ERCC6L2 and MECOM. This will provide important data that may help to individualize and improve care for these patients.
Bone marrow failure (BMF) syndromes are defined by decreased production of one or more hematopoietic lineages, which leads to diminished or absent hematopoietic precursors in the bone marrow and subsequent cytopenia in the peripheral blood. BMF can be distinguished into an acquired form and an inherited form. The acquired form, which is likely caused by an autoimmune reaction (1), may be successfully treated with immunosuppressant regimens. Inherited BMF syndromes include a broad spectrum of heterogenous diseases such as Fanconi anemia, telomere biology disorders, Shwachman-Diamond syndrome, Diamond-Blackfan anemia, congenital cytopenia, immunodeficiency and others (2). In excess of 100 genes have been associated with inherited BMF to date (3–8). The first inherited BMF syndrome, Fanconi anemia, was described in 1927 by the Swiss pediatrician Guido Fanconi, who reported a family with three boys with physical birth defects and a condition resembling pernicious anemia (9). The first causative gene, FANCC, was successfully cloned in 1992 (10). A number of genes have emerged as new bona fide genes associated with the development of BMF in the past ten years: In 2014, variants in ERCC6L2 were shown to cause autosomal recessive BMF and predisposition to myeloid malignancies (11). MECOM as causative gene for inherited BMF has been described in 2015 (12), but its association with a predisposition to hematologic malignancies was only reported three years later (13, 14). Homozygous/compound heterozygous variants in DNAJC21 were linked to a Shwachman-Diamond-like BMF with additional telomeropathy-like features in 2016 (15). In 2020, a digenic ADH5/ALD2H2 deficiency causing severe BMF, early-onset myelodysplastic syndrome (MDS), short stature and intellectual disability was connected to the inability to detoxify formaldehyde (16).
Particularly for the recently described syndromes, data on disease mechanism, penetrance, overall risk of developing hematologic malignancies, and molecular or cytogenetic factors indicating a risk of worsening cytopenia, development of bone marrow dysplasia or leukemogenesis is sparse. This is an important and incomplete pillar for counseling patients and providing them with the most complete and up-to-date information specific to their underlying condition (17). Penetrance, risk of hematologic malignancy and phenotypic heterogeneity may influence the decision towards early (preventive) hematopoietic stem cell transplantation (HSCT) versus ‘watch and wait’ and a more specific follow-up program tailored to the early detection of clonal evolution and disease progression. Amino acid hotspots, genotype-phenotype correlations and disease mechanisms based on reported variants are crucial to determine the strength and validity of the underlying genetic diagnosis and the expected/predicted phenotype and course of disease. This review is therefore based on the recently described syndromes with germline variants in ERCC6L2, MECOM, DNAJC21, and ADH5/ALDH2 that lack concise reviews at this point in time.
In 2014, whole-exome sequencing (WES) of three children and young adults (ages nine to nineteen years old) with BMF and neurological abnormalities (microcephaly, developmental delay) and a history of consanguinity revealed homozygous ERCC6L2 variants in two index patients (11). ERCC6L2 belongs to the Snf2-like ERCC6 family, which also includes ERCC6 and ERCC6L. Functional studies revealed that the molecular mechanism of ERCC6L2 deficiency is an impaired nucleotide excision repair mechanism and an increased amount of reactive oxygen species via a defect in the mitochondrial function of ERCC6L2 (11). The short ERCC6L2 isoform contains an N-terminal TUDOR and a C-terminal DEAD/DEAH ATP-helicase domain. Zhang et al. (18) later reported an alternative ERCC6L2 transcript translating a new protein, Hebo (helicase mutated in BMF), which differs from the ERCC6L2 protein by an 850-amino acid sequence and an additional HEBO domain. Hebo is ubiquitously expressed and is recruited to sites of DNA damage (18). A subsequent study by Tummala et al. postulated the underlying mechanism as primary transcription deficiency rather than a DNA repair defect based on patients being defective in the repair of transcription-associated DNA damage leading to genomic instability (19). Liu et al. described that ERCC6L2 clusters with core subunit non-homologous end joining (NHEJ) genes. ERCC6L2-deficient cells were depleted upon treatment with γ-irradiation, zeocin and etoposide inducing double-strand breaks, lending itself to a similar, but less severe phenotype than that observed in cells lacking the NHEJ ligase LIG4. They could also demonstrate that Ercc6l2−/− mice were viable and ERCC6L2 deficiency resulted in an approximately 50% reduction in orientation-specific class switch recombination of antibody genes (20). A CRISPR-Cas9 screen against genotoxic agents also identified ERCC6L2 as a canonical NHEJ pathway factor (21). SFPQ, a member of the SFPQ-NONO complex that has recently been attributed a putative function in NHEJ, has been described as novel interaction partner of ERCC6L2 (22). Somatic ERCC6L2 variants have been described in a variety of hematologic and solid malignancies, most commonly in patients with uterine corpus endometrial carcinoma. Upon treatment with radiotherapy, these patients showed a strikingly longer disease-free and overall survival than patients with wild-type ERCC6L2, indicating that ERCC6L2 loss may be clinically relevant (22). The most recent study described an impaired clonogenic capacity and erythroid differentiation in ERCC6L2-silenced HSPCs and a probable impact on mesenchymal stromal cells and their differentiation potential (23).
Consanguinity has been described in at least 8 of the 24 families (33%) reported to date (Table 1). The disease is caused by loss-of-function (LOF) variants and all but two variants, D272N and S658N (NM_020207.7) (7, 19), are truncating variants affecting both isoforms or just the long isoform with its HEBO domain (Tables 1, S1 and Figure 1). Two variants, R644* and I475fs, have been found in more than one family and are present in the heterozygous state in gnomAD (https://gnomad.broadinstitute.org) with significant allele frequencies in the European (Non-Finnish) and Finnish subpopulation, respectively (Table 1 and Figure 1). One copy number variant (CNV), a homozygous intragenic deletion of exon 11, has been reported as the causative allele in a patient with BMF (Figure 1) (24). The association with neurological abnormalities such as microcephaly, congenital mirror movements and developmental delay of various degrees was discovered in three studies (18, 24, 25) and may be part of the phenotype or could be an independent effect of the underlying consanguinity in these cases. Of note, ataxia, microcephaly, and developmental delay have also been described in diseases associated with variants in other NHEJ factors such as ATM, MRE11, NBS1, NHEJ1, PRKDC, RAD50, and XRCC4 (26).

Table 1 Overview of emerging bone marrow failure syndromes including genetic and phenotypic features.
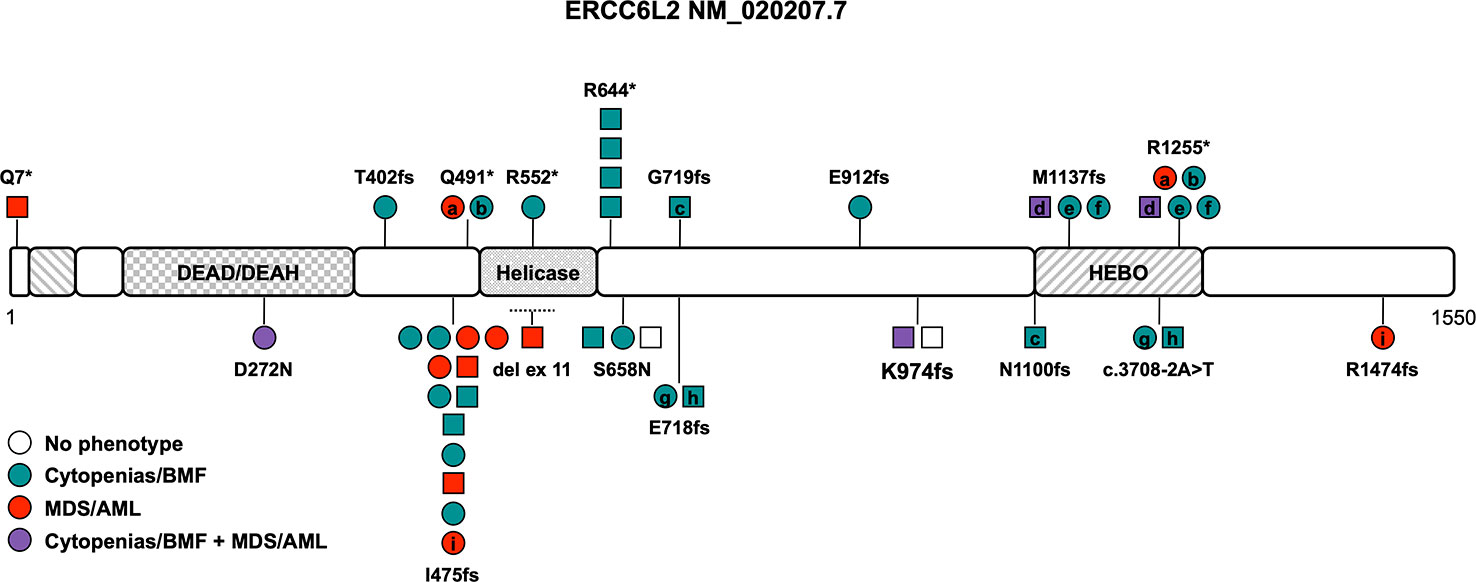
Figure 1 Schematic of the ERCC6L2 transcript (NM_020207.7) and its protein domains with the location of all reported germline variants. Circles represent females and squares represent males. Symbols on the same horizontal level are individuals from the same family. Letters within the symbols indicate individuals with compound heterozygous genotype, while symbols without letters indicate homozygosity for the variant. The color fill depicts different phenotypes, no color fill designates healthy individuals carrying homozygous/compound heterozygous causative variants. The dotted line shows the location of the sole described copy number variant. AML, acute myeloid leukemia; BMF, bone marrow failure; HEBO, helicase mutated in bone marrow failure; MDS, myelodysplastic syndrome.
There is no known genotype-phenotype association and the phenotype ranges from mild cytopenia to severe BMF in childhood and/or development of MDS/acute myeloid leukemia (AML) (Table 1). The overall penetrance is high with an estimate of 94% with two asymptomatic homozygotes still being very young (Table 1). Cytopenia and/or overt BMF develop early at an average age of 14 years and were reported in 24 out of 36 patients (66%, range 2 to 47 years (n=24), Table 1) (7, 11, 18, 19, 23, 25, 27, 28). The development of hematologic malignancies (MDS/AML) has been described in approximately 31% of ERCC6L2 germline-mutated patients at an average age of 35 years (range 2-59 years (n=12), Table 1) (7, 19, 24, 28–30). Importantly, only one in four showed signs of cytopenia or BMF beforehand, which is in line with reports that cytopenia can be subtle, intermittent and go unnoticed. Close to all patients with MDS fell into high-risk groups with a complex karyotype or isolated monosomy 7 and co-occurring (often multi-hit) TP53 alterations (Table 1) (7, 19, 24, 28–30). Several patients with acute erythroid leukemia have been reported, either progressing from MDS or as isolated disease, leading to the assumption that this AML subtype seems to be much more prevalent in ERCC6L2 germline-mutated patients (Table 1) (28). Acute erythroid leukemia, defined by excess of maturation-arrested primitive erythroblasts, is a rare subtype of AML, occurring in about 3% of all AML patients (31). It is characterized by a significantly higher frequency of TP53 variants (36%), especially bi-allelic/multi-hit TP53 alterations with relatively lower somatic mutational burden compared to other AML subtypes (31, 32). While acute erythroid leukemia by itself does not seem to carry an additional prognostic impact as independent risk factor (33), its frequent association with complex karyotypes and multi-hit TP53 alterations does confer to a dismal outcome in at least the subset of cases with these abnormalities (34). Cytogenetic abnormalities in the twelve ERCC6L2 patients with AML and MDS presented often as a complex or monosomal karyotype with loss of chromosomes 5, 7 or 17 or isolated monosomy 7. In addition, multi-hit TP53 alterations were reported in seven out of twelve patients with MDS/AML. An assessment of the allelic state of these TP53 alterations was not performed (Table 1) (7, 19, 24, 28–30). The prognosis of MDS/AML in patients with ERCC6L2 germline variants is poor, especially when progression to acute erythroid leukemia is noted, with no known survivors of this AML subtype so far (28). HSCT was performed in at least ten individuals (28%), either because of severe, transfusion-dependent BMF or after MDS/AML development (Table 1). Given the high frequency of monosomy 7, complex/monosomal karyotypes and TP53 variants, which may be associated with disease progression and development of MDS/AML, HSCT should be considered early, especially when these aberrations are discovered in the context of clonal evolution and bone marrow dysplasia.
Heterozygous variants in HOXA11 are known to cause radioulnar synostosis (RUS) (a congenital proximal fusion of the radius and ulna) with amegakaryocytic thrombocytopenia (RUSAT) (35). However, families with RUSAT without HOXA11 variants were reported (36), suggesting that additional candidate genes/loci exist. Consequently, Niihori et al. (12) performed WES on three individuals with RUSAT without an identified variant in HOXA11 and detected heterozygous missense variants in MECOM in all three patients. This was the first time RUSAT has been linked to variants in MECOM.
The MDS1-EVI1 complex locus (MECOM) gives rise to several transcripts through alternative splicing of the N-terminus that encode at least three different isoforms: full length EVI1-145 kDa, EVI1-Δ324, which lacks zinc fingers 6 and 7, and MDS1-EVI1. MDS1-EVI1 comprises an N-terminal so-called PRDF1-RIZ homology domain, two C2H2 zinc finger DNA binding domains, one at the N-terminus including seven zinc fingers, and the other at the C-terminus including three zinc fingers, a proline-rich repressor domain and a small aspartate/glutamate-rich acidic region located in the C-terminal region (Figure 2) (37). MECOM acts as crucial transcription factor in hematopoiesis, playing an important role in the formation and self-renewal of long-term hematopoietic stem and progenitor cells (HSPCs) (38) as well as myeloid differentiation through interaction with transcription factors including GATA1 (39), SPI1 (40), RUNX1 (41) and others (42). The inversion or translocation of chromosome 3 drives inv(3)/t(3;3) AML via structural rearrangement of an enhancer that upregulates transcription of EVI1. It is associated with poor overall survival in AML patients and HSCT is usually mandated whenever possible (43). Furthermore, overexpression of EVI1 has been reported in 6 to 11% of AML patients without 3q aberrations (44).
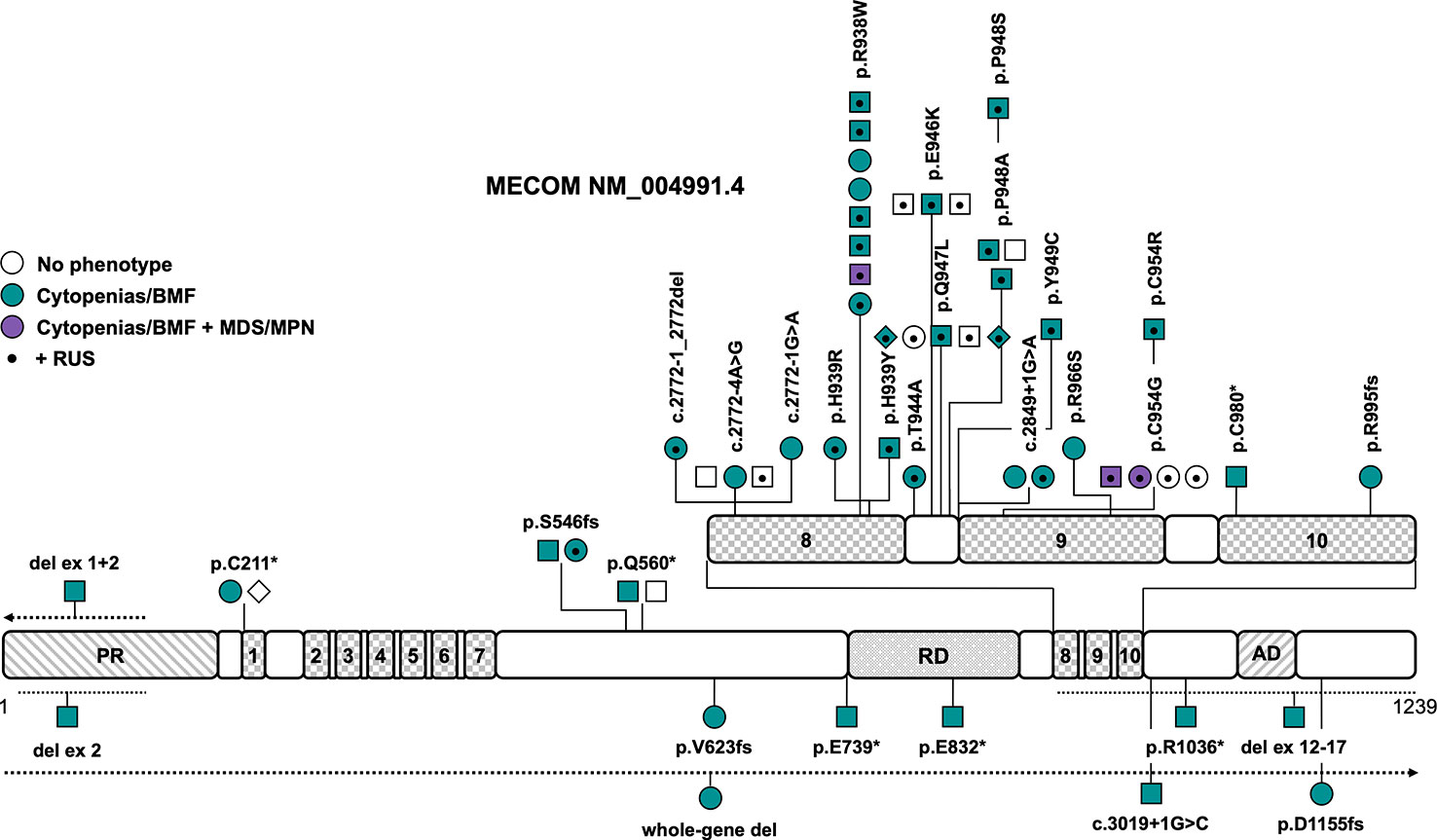
Figure 2 Schematic of the MECOM transcript (NM_004991.4) and its protein domains with the location of all reported germline variants. Checkered domains with the numbers 1 to 10 stand for zinc fingers 1 to 10. Circles represent females, squares represent males and diamonds represent unknown gender. Symbols on the same horizontal level are individuals from the same family. The color fill depicts different phenotypes, no color fill indicates healthy individuals carrying a causative variant. A dot in the middle of the symbol designates the presence of RUS in these individuals. The dotted lines show the location of the described copy number variants, the arrow denotes that the copy number variant extends in this direction. AD, acidic domain; BMF, bone marrow failure; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm; PR, PRDF1-RIZ domain; RD, repression domain; RUS, radioulnar synostosis.
Later reports broadened the phenotype caused by germline MECOM variants, including BMF without RUS (7, 13, 45–50), predisposition to myeloid malignancies (14, 51), abnormalities of other organ systems (7, 13, 14, 45–47, 49–55), and RUS without any other phenotypic features (56). Causative variants include LOF variants that are scattered across the entire gene and missense variants that are solely located in zinc fingers 8 and 9 (Figure 2 and Tables 1, S1). Confirmed de novo variants have been reported in 16% of patients (7, 48–51). The variants R938W and P948A (NM_004991.4) as well as variants affecting the splice sites between exons 7 and 8 have been described in more than one family (Figure 2 and Table 1). Interestingly, there seems to be a rather distinct genotype-phenotype association — all but one variant associated with the co-presentation of RUS and hematologic disease cluster in the region spanning zinc fingers 8 and 9, which includes mostly missense, but also canonical and non-canonical splice variants (Figure 2 and Table 1). Specific missense variants in zinc fingers 8 and 9 (namely R969C/H/L, I971T, and Q965E) have been described in 21 individuals from 6 families with RUS and finger malformations without hematological abnormalities (56). Little is known about this association of RUS with missense variants in zinc fingers 8 and 9. It was shown that the Evi1 expression pattern is temporally and spatially restricted in mouse embryos with a transient expression in the emerging limb buds (57). Junbo mice with an Evi1 variant affecting zinc finger 9 had extra digits on their forelimbs, suggesting that the C-terminal zinc finger domain may be relevant in digit development (58). Both the MECOM missense variants H939R and R969C have displayed attenuated suppression of TGFB1 (12, 56), which has been previously shown to play a role in digit formation during mouse development (59). LOF variants in MECOM seem to cause BMF but not RUS. CNVs have been described in four patients and were all confirmed or presumed de novo (Figure 2) (46, 48–50). One 751kb 3q26 microdeletion encompassing the entire MECOM gene and the pseudogene EGFEM1P was reported in one patient with BMF without other phenotypic features (48). Intragenic deletions of exons 1 + 2 (49), exon 2 (50) and exons 12-17 (46), affecting zinc finger and acidic domains, were reported in three patients with BMF at birth and a range of congenital skeletal and/or heart abnormalities. The intragenic deletion of exons 1 and 2 extended to other genes and also included the telomere biology gene TERC, so that specific phenotypic features cannot be attributed to either gene in this case (49). Overall penetrance of any related features (RUS/other skeletal abnormalities, deafness, cytopenia/BMF) is high at an estimated 96% (Table 1). Cytopenia/BMF was present in 80% of patients with an average age of onset at birth/in infancy (n=43) (7, 12–14, 45–55, 60). Although cytopenia can be severe and present at birth, even leading to intrauterine death, and may require early HSCT, three cases (5%) with spontaneous resolution or improvement of cytopenia/BMF have been reported (13, 45, 51). RUS was the most frequent non-hematopoietic feature in 54% of patients (7, 12–14, 51–55, 60), followed by brachy-, campto-, and clinodactyly and/or other finger abnormalities in 38% of patients (7, 12, 13, 45, 46, 51–53). Other frequent abnormalities included sensorineural hearing impairment/congenital deafness in 20% (12–14, 51), cardiac abnormalities such as atrial/ventricular septal defects, tetralogy of Fallot, aortic coarctation, pulmonary stenoses/atresias, pulmonary venous return anomaly, patent ductus arteriosus and myocardial atrophy in 18% (7, 13, 47, 49–51, 54) and prematurity, hydrops fetalis or polyhydramnios in 13% of patients (12, 47, 50, 53–55). Less frequent phenotypic features are micro- or macrocephaly, structural brain abnormalities or developmental delay/cognitive impairment in 11% (12, 13, 46, 51, 52), patellar hypoplasia (13, 14), metatarsus adductus, hallux valgus and other toe abnormalities (13, 14) in 9% and clubfoot (7, 51, 52) and renal abnormalities (7, 13, 55) in 7% of patients each (Table 1). Other features such as hip dysplasia (13, 52, 53), cleft palate (12, 13, 49), early-onset ischemic insults (13, 14, 51), facial dysmorphia (7, 49, 51), precocious puberty/gynecomastia in infancy (13), and paralysis of the larynx/laryngomalacia (45, 49) have been described in only two to three individuals and consequently the association with germline MECOM variants may not be entirely clear or proven in these cases. Three patients (5%) were reported to develop hematologic malignancies, specifically MDS with refractory cytopenia with multilineage dysplasia at 37 years (51), MDS with excess blasts-2 at 73 years with interstitial deletion of the long arm of chromosome 9 (14) and MDS/myeloproliferative disease- unclassifiable at 42 years with a translocation t(1;14)(q44;q32) (Table 1) (14). All patients reportedly had a history of thrombocytopenia or BMF with earlier onset (14, 51). HSCT has been performed in 27 out of 55 patients (50%) because of severe BMF.
DNAJC21 is ubiquitously expressed and encodes a protein with 531 amino acids, containing a highly conserved N-terminal DnaJ molecular chaperone homology domain, a central coiled coil region as well as two zinc fingers (Figure 3). The first studies in yeast showed that it functions together with the cytoplasmic zinc finger protein Znf622 to stimulate the ATPase activity of the Hsp70 chaperone protein Hspa8, thereby initiating the removal/recycling process of Arx1, a ribosome maturation factor (61, 62). In 2016, Tummala et al. screened a cohort of 28 unrelated individuals with BMF and syndromal features by WES and identified 3 individuals with homozygous variants in DNAJC21. By targeted re-sequencing of DNAJC21 in patients with similar phenotype, a fourth patient with a homozygous DNAJC21 variant was found (15). Functional studies on patient-derived lymphoblastoid cell lines implicated involvement of DNAJC21 in rRNA biogenesis and 60S ribosome maturation — thereby resembling the function of SBDS — leading to decreased interaction with HSPA8, ZNF622 and PA2G4 and increased cell death in patients with DNAJC21 deficiency (15). So far, 19 patients from 14 different families have been described in the literature (7, 15, 63–67), in 8 families (57%) a history of consanguinity was reported (Table 1). There is no confirmed case of a de novo variant in DNAJC21 reported to date.
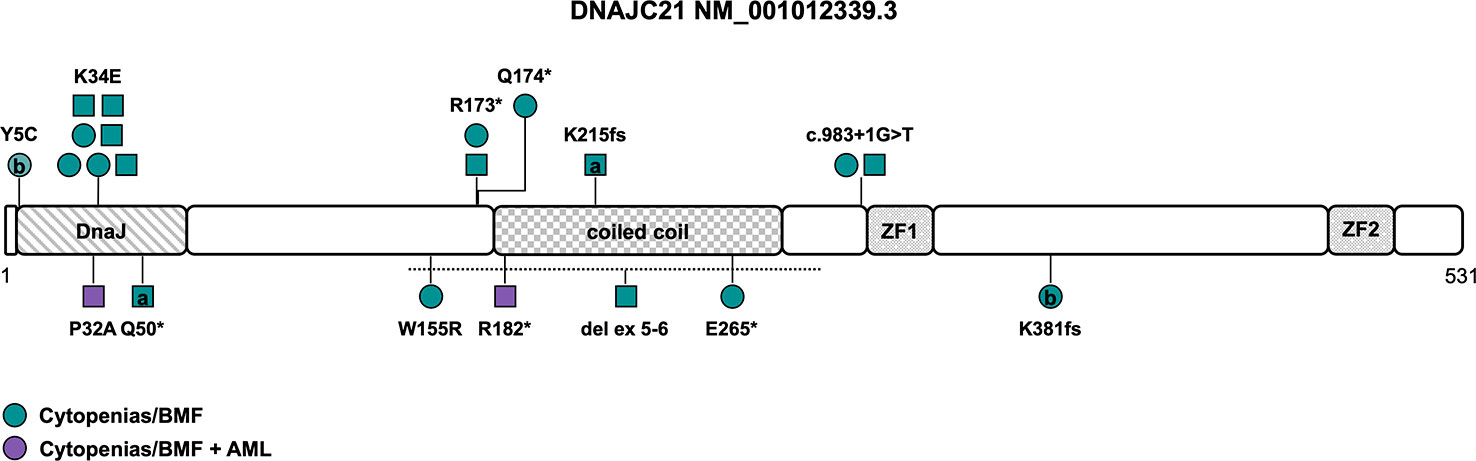
Figure 3 Schematic of the DNAJC21 transcript (NM_001012339.3) and its protein domains with the location of all reported germline variants. Circles represent females and squares represent males. Symbols on the same horizontal level are individuals from the same family. Letters within the symbols indicate individuals with compound heterozygous genotype, while symbols without letters indicate homozygosity for the variant. The color fill depicts different phenotypes. The dotted line shows the location of the sole described copy number variant. AML, acute myeloid leukemia; BMF, bone marrow failure; ZF, zinc finger.
Causative variants include missense variants, particularly within the N-terminal DnaJ-domain affecting the amino acids 5, 32 and 34 (Y5C, K34E, P32A, NM_001012339.3), with two of those located in the universally conserved HPD motif (H33-P34-D35), which is essential for stimulation of ATPase activity. The K34E variant is the most common variant described in seven individuals from four families (Figure 3 and Tables 1, S1) (64, 65), reversing the surface charge of a key amino acid adjacent to the HPD motif and also likely disrupting the interaction with HSPA8 (68). The P32A variant potentially alters the fold of the HPD motif, disrupting the interaction with HSPA8 and stimulation of its ATPase activity (68). Truncating variants encompassing nonsense, frameshift and canonical splice site variants (Table S1) are predicted to undergo nonsense mediated decay, leading to significant reduction of DNAJC21 protein expression (7, 15, 65–67). While most patients were homozygous for a causative DNAJC21 variant, two individuals were found to be compound heterozygous (7, 66). Besides single-nucleotide variants, one individual with a homozygous intragenic deletion of exons 5 and 6 was reported as well (Figure 3 and Table 1) (65).
While some features such as exocrine pancreatic dysfunction are consistent with a (classic) Shwachman-Diamond phenotype (as has been described in patients with bi-allelic inactivation of SBDS and less likely of EFL1 or heterozygous variants in SRP45 (69)), other features such as skin hypopigmentation, dental and retinal abnormalities seem to resemble characteristics of telomeropathies (64). There is no known genotype-phenotype association. The penetrance of a hematologic phenotype in the sense of single- or multiple lineage cytopenia/BMF seems to be complete (Table 1). The average age of onset for BMF is two years (range 0 to 15 years, n=19). Spontaneous, intermittent or prolonged improvement of cytopenia was reported in at least four patients (64–66), while four patients (21%) needed a HSCT (Table 1). AML developed in two patients (11%) at the age of twelve and fifteen years, respectively (15, 67). Somatic cytogenetic or molecular alterations were unknown or not reported. However, one case of acute megakaryoblastic leukemia was described (15). Other cytogenetic abnormalities, including a complex karyotype (7), a derivative chromosome 15 with translocation t(1;15) (65), a deletion of 17p13 (64) and a deletion of 20q (64) have been reported in patients with BMF without hematologic malignancy and consequently their significance in disease progression and development of MDS/AML is unclear. Growth delay and/or short stature as the most frequent non-hematopoietic feature has been described in all but one patient (95%). Other frequent abnormalities included developmental delay/intellectual disability and/or neurological abnormalities (68%), skeletal abnormalities (particularly hip dysplasia, thoracic deformities, genu valgum and metaphyseal dysplasia) and skin abnormalities (mainly hypopigmentation and palmoplantar cutis laxa) at 63% each, as well as microcephaly (42%), facial dysmorphia (37%), dental abnormalities (32%), and osteopenia/osteoporosis (32%). Pancreas lipomatosis was reported in five cases (26%), with four out of the five patients suffering from exocrine pancreatic dysfunction with preserved endocrine function. Intriguingly, while (high) myopia, astigmatism and other visual field defects were described in six patients (32%), rare features of retinal (rod-cone) dystrophy and other retinal abnormalities were also identified in six patients (32%), albeit one patient developed symptoms after HSCT (65) and another patient carried a homozygous variant in PCARE, known to cause an autosomal recessive form of retinitis pigmentosa (Table 1) (65). This suggests retinal abnormalities may be part of the developing phenotype. Due to the low number of reported patients, specific phenotypic features need to be defined and refined over time.
Sixteen Italian patients with Shwachman-Diamond syndrome and bi-allelic SBDS variants were screened for additional variants in DNAJC21, EFL1, and SRP45. One of the two germline-mutated SBDS patients with compound heterozygous SBDS variants and an additional heterozygous DNAJC21 variant was reported to suffer from a more severe hematologic phenotype, in particular severe neutropenia (70). Both DNAJC21 variants identified in this study (70), E276K and V342M, are reasonably rare in 0,32% and 0,0016% of the gnomAD population but ensemble in-silico predictions are contradicting or in favor of no significant impact on the protein structure.
Formaldehyde is a ubiquitous endogenous and environmental metabolite, which has been classified as a group I human carcinogen by the International Agency for Research on Cancer as it may cause nasopharyngeal cancer, lung cancer and is associated with development of AML (71). Specifically, it was shown that formaldehyde exposure induces chromosomal aneuploidy, in particular aberrations of chromosomes 5, 7, and 8, which are frequently seen in AML (72, 73). It was also reported to induce hematopoietic toxicity to both mature and stem/progenitor cells in the bone marrow of mice exposed to formaldehyde by affecting myeloid progenitor growth and survival through oxidative damage apoptosis and dysregulation of colony stimulating factor receptors (74).
ADH5, located in the cytoplasm, is the most widely expressed alcohol dehydrogenase and the main formaldehyde-detoxifying enzyme (75). It contains a catalytic and zinc-binding domain of the alcohol dehydrogenase (Figure 4). ALDH2, a mitochondrial enzyme oxidizing acetaldehyde to acetate, is important in ethanol metabolism, and deficiency of this enzyme is very common in humans, leading to facial flushing, nausea, headaches, cardiac palpitations, and overall discomfort in response to drinking alcohol (76). The ALDH2*2 allele, defined by the c.1510G>A (NM_000690.4) variant (rs671), encoding an E504K amino acid substitution, reduces enzyme activity to less than 50% in heterozygotes and less than 4% in homozygotes in a dominant-negative manner (76). It is common in the East Asian population with an allele frequency of about 25% in the gnomAD (https://gnomad.broadinstitute.org/) population. ALDH2, active mainly in detoxifying acetaldehyde, also takes part in formaldehyde detoxification (75).
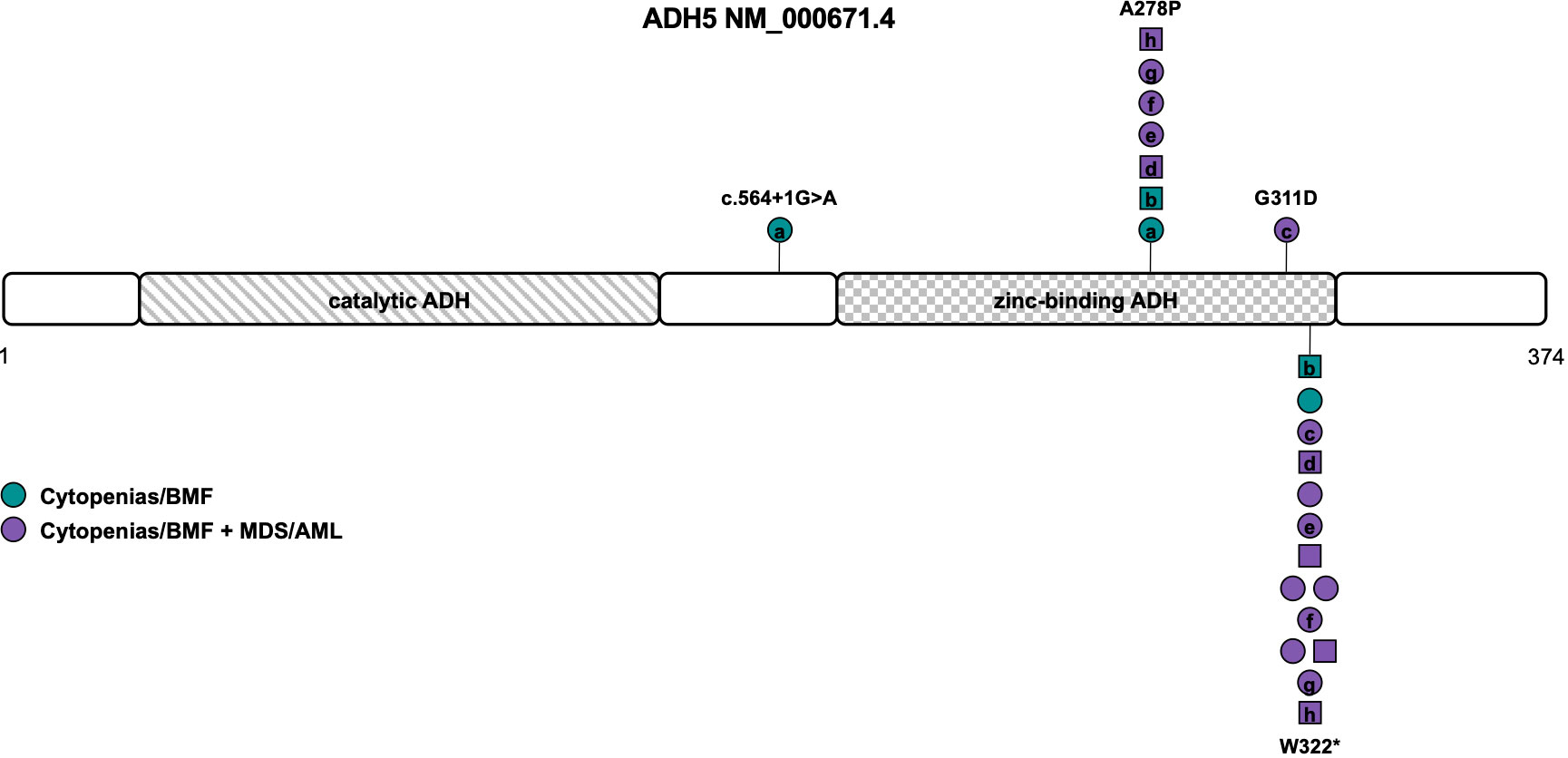
Figure 4 Schematic of the ADH5 transcript (NM_000671.4) and its protein domains with the location of all reported germline variants. Circles represent females and squares represent males. Symbols on the same horizontal level are individuals from the same family. Letters within the symbols indicate individuals with compound heterozygous genotype, while symbols without letters indicate homozygosity for the variant. The color fill depicts different phenotypes. ADH, alcohol dehydrogenase; AML, acute myeloid leukemia; BMF, bone marrow failure; MDS, myelodysplastic syndrome.
Dingler et al. described the first seven children/young adults with homozygous or compound heterozygous ADH5 variants and the heterozygous ALDH2*2 allele associated with BMF and predisposition to MDS/AML that is solely driven by formaldehyde accumulation (16). A subsequent study by Oka et al. reported seven individuals from five different families with BMF and development of MDS/AML at young age (77). Of those, four individuals were heterozygous and three homozygous for the ALDH2*2 allele. The three homozygotes were reported to harbor more severe phenotypes including neurological deterioration and early death (77). Because of its association with short stature and intellectual disability, it was subsequently also called AMeD syndrome (for Anemia, Mental retardation and Dwarfism) (10). One male patient with a history of anemia since the age of 8 years who developed MDS at 18 years old was diagnosed with ADH5/ALDH2 deficiency and sensorineural hearing loss based on concurrent compound heterozygous variants in ADGRV1 (78).
Of the 15 individuals from 13 families described so far, 8 were compound heterozygous and seven homozygous for variants in ADH5. Consanguinity or occurrence of de novo variants was not reported (Table 1). Causative variants include LOF variants c.564+1G>A (NM_000671.4), resulting in retention of intron 5 (L188fs), and the recurrent variant W322*, that has been described in all but one individual (Table S1). Missense variants were located within the zinc-binding domain of ADH5, namely the G311D variant and the recurrent A278P variant found in seven patients (Tables 1, S1 and Figure 4) (16, 77, 78). Penetrance is complete with all patients diagnosed with either BMF or early-onset MDS/AML and nine out of twelve patients (75%) undergoing HSCT (Table 1). Based on the published data, MDS/AML was diagnosed in 12 out of 15 patients (80%) at an average age of 7 years (range 0 to 18 years (n=9), Table 1, Figure 4). In eight out of eleven patients (73%) with reported cytogenetic information, a gain of the long arm of chromosome 1, frequently seen in Fanconi anemia (79, 80) was described. Other recurrent cytogenetic alterations included monosomy 7, trisomy 8 and 21p alterations (Table 1) (16, 77, 78). All patients with reported additional phenotypical data were short of stature and displayed mild to moderate intellectual disability. Microcephaly was described in 67% and abnormal skin pigmentation was found in 58% of patients (Table 1). Although the phenotype mimics Fanconi anemia, radial ray defects have not been detected so far and chromosomal breakage tests are negative. Other features such as retinal degeneration, facial dysmorphia, skeletal, and endocrine abnormalities have only been described in single individuals so that the clinical association remains unclear to date (16, 77, 78).
Using a CRISPR-Cas9 functional screen, ADH5 was (together with ESD and the FANC family genes) described as a top candidate gene dramatically increasing cellular formaldehyde sensitivity when disrupted (81). Concordantly, Adh5-/- Aldh2-/- double-deficient mice recapitulated some of the hematopoietic phenotypes seen in these patients such as reduced proliferation of HSPCs and loss of differentiation (16, 77). Another group reported that Adh5-/- deficient mice with wildtype Aldh2 are born and develop normally, while double-deficient mice showed significantly lower body weight, which mimics the short stature seen in humans (82).
Formaldehyde also triggers a cellular redox imbalance that can lead to reactive oxygen species accumulation and cytotoxicity, which may cause BMF development even in the presence of functional DNA repair mechanisms by overwhelming the DNA-repair capacity in HSPCs (83). Using patient-derived lymphoblasts, fibroblasts, induced pluripotent stem cells (iPSCs), and CRISPR/Cas9-engineered cell lines, Mu et al. were able to demonstrate that patient-derived iPSCs were sensitive to exogenous treatment with formaldehyde, which induced drastically defective cell expansion when stimulated into hematopoietic differentiation and increased levels of DNA damage. This phenotype was attenuated upon expression of ADH5 and less so by addition of an ALDH2 activator (84). Therapies aiming to lower endogenous formaldehyde could be a promising treatment strategy for ADH5/ALDH2 deficiency. C1, a new small molecule acting as agonist of ALDH2, was well tolerated and able to partially reverse the HSPC expansion/differentiation defect in iPSCs in vitro (84). The combination of a formaldehyde scavenger such as metformin and glutathione precursors (for instance N-acetyl-L-cysteine) (83) may also benefit patients with Fanconi anemia.
In whose transcriptional reprogramming during differentiation of HSPCs may lead to acute accumulation of endogenous DNA damage, most likely arising from formaldehyde, an obligate by-product during transcriptional regulation (85). Further studies are needed to determine if aldehydes are the major cause of pathology in Fanconi anemia patients, who have functional ALDH2 and ADH5 to mediate aldehyde metabolism.
There may be a confounding bias for all described syndromes by the short period of clinical observations since these syndromes have been discovered. The likelihood of developing hematologic malignancies and the penetrance of these diseases may be estimated as too low since many patients are still children or young adults and others already underwent HSCT to treat severe early-onset BMF.
Suspicion of an inherited BMF syndrome should arise when patients are diagnosed with BMF in infancy/early-childhood (7) and/or MDS at young age (below 40 years old) (29, 30). A positive family history and other organ manifestations also point towards an inherited rather than acquired BMF syndrome (2, 7, 17). Germline BMF panel-based next-generation sequencing (NGS) is a reasonable first-tier option (86). WES or whole-genome sequencing (WGS) should be considered when suspicion of an inherited BMF syndrome is high and initial panel-based results are negative. WES covers all coding genes, however, if genes have not been described as candidate genes at the time of the analysis, the genetic variant causing the phenotype may be missed. Regular re-analysis of WES, as was done in one patient with BMF and bi-allelic DNAJC21 variants (66) should be included (87). Even if this initially may only increase the number of variants of unknown significance (88), these could be upgraded over time when new information such as observation in multiple probands, segregation with disease, or functional impact of the variant becomes available. Intragenic and whole-gene CNVs were described in DNAJC21 (65), ERCC6L2 (24), and MECOM (46, 48–50), so that high-density microarrays or bioinformatic analysis of panel-based NGS/WES data have to be incorporated (89). Non-canonical, deeply intronic or exonic synonymous splice variants may require additional RNA sequencing to unravel the effects on splicing and prove pathogenicity of these unusual but not infrequent variants (89–91).
Crucially, unrecognized inherited BMF syndromes may lead to use of related donors carrying the same variants (29), as well as excessive death upon HSCT, which can be reduced using tailored conditioning regimens (92). Adapted non-myeloablative conditioning protocols have been used successfully as conditioning regimens in telomere biology disorders and Fanconi anemia (93, 94). A small case series of six patients with germline MECOM variants reported that reduced-intensity conditioning was an effective treatment and reduced toxicity-related morbidities (95). However, comprehensive HSCT data from patients with germline ERCC6L2, MECOM, DNAJC21, and ADH5/ALDH2 variants are lacking to date, including donor choice, conditioning regimens and non-relapse morbidity and mortality.
A clear genotype-phenotype correlation has so far only been established for MECOM variants, where the co-presentation of RUS and hematologic disease appears to be caused by variants spanning zinc fingers 8 and 9. Although there is indication that homozygosity for the ALDH2*2 allele may lead to a more severe (neurological) phenotype in patients with ADH5/ALDH2 deficiency (77), the number of individuals is too low to draw comprehensive conclusions at this time. The discovery of additional mechanisms of disease and amino acid hotspots may help predict the individual, variant-based risk of hematologic malignancies, severe BMF and other phenotypic features.
Some data suggest clonal genetic markers of disease progression such as (multi-hit) TP53 variants in patients with ERCC6L2 germline variants may be indicative of disease progression (24, 28–30) and given the poor prognosis of MDS/acute erythroid leukemia in these patients, early HSCT should be performed. Similar data have been reported for TP53 variants in other inherited BMF syndromes (96, 97), although a clear link to disease progression in patients with somatic TP53 variants in the absence of other high-risk molecular or cytogenetic markers is unclear (98). Early pre-emptive HSCT comes also with the potential of HSCT-related mortality and morbidity so that more valid early markers of disease progression are needed (99).
In summary, this review provides new insights into four distinct and recently described BMF syndromes and will thereby improve the clinical management for these patients. New data will over time further refine these syndromes and add more pieces to the yet unsolved puzzle of inherited BMF syndromes.
SF conceptualized and designed the study and wrote the manuscript.
SF receives funding from the Olympia Morata Program of the Medical Faculty Heidelberg.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1128533/full#supplementary-material
1. Giudice V, Selleri C. Aplastic anemia: Pathophysiology. Semin Hematol (2022) 59(1):13–20. doi: 10.1053/j.seminhematol.2021.12.002
2. Savage SA, Dufour C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin Hematol (2017) 54(2):105–14. doi: 10.1053/j.seminhematol.2017.04.004
3. Pecci A, Balduini CL. Inherited thrombocytopenias: an updated guide for clinicians. Blood Rev (2021) 48:100784. doi: 10.1016/j.blre.2020.100784
4. Sieff C. Diamond-blackfan anemia. Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW and Amemiya A, editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle (1993).
5. Revy P, Kannengiesser C, Bertuch AA. Genetics of human telomere biology disorders. Nat Rev Genet (2023) 24(2):86–108. doi: 10.1038/s41576-022-00527-z
6. Mehta PA, Ebens C. Fanconi anemia. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al, editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle (1993). Available at: http://www.ncbi.nlm.nih.gov/pubmed/20301575. GeneReviews®.
7. Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood (2018) 131(7):717–32. doi: 10.1182/blood-2017-09-806489
8. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
9. Lobitz S, Velleuer E. Guido Fanconi (1892-1979): a jack of all trades. Nat Rev Cancer (2006) 6(11):893–8. doi: 10.1038/nrc2009
10. Strathdee CA, Gavish H, Shannon WR, Buchwald M. Cloning of cDNAs for fanconi’s anaemia by functional complementation. Nature (1992) 356(6372):763-7. doi: 10.1038/356763a0
11. Tummala H, Kirwan M, Walne AJ, Hossain U, Jackson N, Pondarre C, et al. ERCC6L2 mutations link a distinct bone-Marrow-Failure syndrome to DNA repair and mitochondrial function. Am J Hum Genet (2014) 94(2):246–56. doi: 10.1016/j.ajhg.2014.01.007
12. Niihori T, Ouchi-Uchiyama M, Sasahara Y, Kaneko T, Hashii Y, Irie M, et al. Mutations in MECOM, encoding oncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. Am J Hum Genet (2015) 97(6):848–54. doi: 10.1016/j.ajhg.2015.10.010
13. Germeshausen M, Ancliff P, Estrada J, Metzler M, Ponstingl E, Rütschle H, et al. MECOM-associated syndrome: a heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Adv (2018) 2(6):586–96. doi: 10.1182/bloodadvances.2018016501
14. Ripperger T, Hofmann W, Koch JC, Shirneshan K, Haase D, Wulf G, et al. MDS1 and EVI1 complex locus (MECOM): a novel candidate gene for hereditary hematological malignancies. Haematologica (2018) 103(2):e55–8. doi: 10.3324/haematol.2017.178723
15. Tummala H, Walne AJ, Williams M, Bockett N, Collopy L, Cardoso S, et al. DNAJC21 mutations link a cancer-prone bone marrow failure syndrome to corruption in 60S ribosome subunit maturation. Am J Hum Genet (2016) 99(1):115–24. doi: 10.1016/j.ajhg.2016.05.002
16. Dingler FA, Wang M, Mu A, Millington CL, Oberbeck N, Watcham S, et al. Two aldehyde clearance systems are essential to prevent lethal formaldehyde accumulation in mice and humans. Mol Cell (2020) 80(6):996–1012.e9. doi: 10.1016/j.molcel.2020.10.012
17. Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol (2017) 177(4):526–42. doi: 10.1111/bjh.14535
18. Zhang S, Pondarre C, Pennarun G, Labussiere-Wallet H, Vera G, France B, et al. A nonsense mutation in the DNA repair factor hebo causes mild bone marrow failure and microcephaly. J Exp Med (2016) 213(6):1011–28. doi: 10.1084/jem.20151183
19. Tummala H, Dokal AD, Walne A, Ellison A, Cardoso S, Amirthasigamanipillai S, et al. Genome instability is a consequence of transcription deficiency in patients with bone marrow failure harboring biallelic ERCC6L2 variants. Proc Natl Acad Sci (2018) 115(30):7777–82. doi: 10.1073/pnas.1803275115
20. Liu X, Liu T, Shang Y, Dai P, Zhang W, Lee BJ, et al. ERCC6L2 promotes DNA orientation-specific recombination in mammalian cells. Cell Res (2020) 30(9):732–44. doi: 10.1038/s41422-020-0328-3
21. Olivieri M, Cho T, Álvarez-Quilón A, Li K, Schellenberg MJ, Zimmermann M, et al. A genetic map of the response to DNA damage in human cells. Cell (2020) 182(2):481–96. doi: 10.1016/j.cell.2020.05.040
22. Francica P, Mutlu M, Blomen VA, Oliveira C, Nowicka Z, Trenner A, et al. Functional radiogenetic profiling implicates ERCC6L2 in non-homologous end joining. Cell Rep (2020) 32(8):108068. doi: 10.1016/j.celrep.2020.108068
23. Armes H, Bewicke-Copley F, Rio-Machin A, di Bella D, Philippe C, Wozniak A, et al. Germline ERCC excision repair 6 like 2 (ERCC6L2) mutations lead to impaired erythropoiesis and reshaping of the bone marrow microenvironment. Br J Haematol (2022) 199(5):754–64. doi: 10.1111/bjh.18466
24. Thams S, Islam M, Lindefeldt M, Nordgren A, Granberg T, Tesi B, et al. Heterozygous variants in DCC. Neurol Genet (2020) 6(6):e526. doi: 10.1212/NXG.0000000000000526
25. Shabanova I, Cohen E, Cada M, Vincent A, Cohn RD, Dror Y. ERCC6L2-associated inherited bone marrow failure syndrome. Mol Genet Genomic Med (2018) 6(3):463–8. doi: 10.1002/mgg3.388
26. Slatter MA, Gennery AR. Update on DNA-double strand break repair defects in combined primary immunodeficiency. Curr Allergy Asthma Rep (2020) 20(10):57. doi: 10.1007/s11882-020-00955-z
27. Järviaho T, Halt K, Hirvikoski P, Moilanen J, Möttönen M, Niinimäki R. Bone marrow failure syndrome caused by homozygous frameshift mutation in the ERCC6L2 gene. Clin Genet (2018) 93(2):392–5. doi: 10.1111/cge.13125
28. Douglas SPM, Siipola P, Kovanen PE, Pyörälä M, Kakko S, Savolainen ER, et al. ERCC6L2 defines a novel entity within inherited acute myeloid leukemia. Blood (2019) 133(25):2724–8. doi: 10.1182/blood-2019-01-896233
29. Feurstein SK, Trottier AM, Estrada-Merly N, Pozsgai MJ, McNeely KE, Drazer MW, et al. Germline predisposition variants occur in myelodysplastic syndrome patients of all ages. Blood (2022) 140(24):2533–48. doi: 10.1182/blood.2022015790
30. Feurstein S, Churpek JE, Walsh T, Keel S, Hakkarainen M, Schroeder T, et al. Germline variants drive myelodysplastic syndrome in young adults. Leukemia (2021) 35(8):2439–44. doi: 10.1038/s41375-021-01137-0
31. Rose D, Haferlach T, Schnittger S, Perglerová K, Kern W, Haferlach C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia (2017) 31(1):11–7. doi: 10.1038/leu.2016.163
32. Montalban-Bravo G, Benton CB, Wang SA, Ravandi F, Kadia T, Cortes J, et al. More than 1 TP53 abnormality is a dominant characteristic of pure erythroid leukemia. Blood (2017) 129(18):2584–7. doi: 10.1182/blood-2016-11-749903
33. Santos FPS, Faderl S, Garcia-Manero G, Koller C, Beran M, O’Brien S, et al. Adult acute erythroleukemia: an analysis of 91 patients treated at a single institution. Leukemia (2009) 23(12):2275–80. doi: 10.1038/leu.2009.181
34. Boddu P, Benton CB, Wang W, Borthakur G, Khoury JD, Pemmaraju N. Erythroleukemia-historical perspectives and recent advances in diagnosis and management. Blood Rev (2018) 32(2):96–105. doi: 10.1016/j.blre.2017.09.002
35. Thompson AA, Nguyen LT. Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat Genet (2000) 26(4):397–8. doi: 10.1038/82511
36. Castillo-Caro P, Dhanraj S, Haut P, Robertson K, Dror Y, Sharathkumar AA. Proximal radio-ulnar synostosis with bone marrow failure syndrome in an infant without a HOXA11 mutation. J Pediatr Hematol Oncol (2010) 32(6):479–85. doi: 10.1097/MPH.0b013e3181e5129d
37. Maicas M, Vázquez I, Alis R, Marcotegui N, Urquiza L, Cortés-Lavaud X, et al. The MDS and EVI1 complex locus (MECOM) isoforms regulate their own transcription and have different roles in the transformation of hematopoietic stem and progenitor cells. Biochim Biophys Acta (BBA) - Gene Regul Mech (2017) 1860(6):721–9. doi: 10.1016/j.bbagrm.2017.03.007
38. Kataoka K, Sato T, Yoshimi A, Goyama S, Tsuruta T, Kobayashi H, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med (2011) 208(12):2403–16. doi: 10.1084/jem.20110447
39. Laricchia-Robbio L, Fazzina R, Li D, Rinaldi CR, Sinha KK, Chakraborty S, et al. Point mutations in two EVI1 zn fingers abolish EVI1-GATA1 interaction and allow erythroid differentiation of murine bone marrow cells. Mol Cell Biol (2006) 26(20):7658–66. doi: 10.1128/MCB.00363-06
40. Laricchia-Robbio L, Premanand K, Rinaldi CR, Nucifora G. EVI1 impairs myelopoiesis by deregulation of PU.1 function. Cancer Res (2009) 69(4):1633–42. doi: 10.1158/0008-5472.CAN-08-2562
41. Senyuk V, Sinha KK, Li D, Rinaldi CR, Yanamandra S, Nucifora G. Repression of RUNX1 activity by EVI1: A new role of EVI1 in leukemogenesis. Cancer Res (2007) 67(12):5658–66. doi: 10.1158/0008-5472.CAN-06-3962
42. Voit RA, Tao L, Yu F, Cato LD, Cohen B, Fleming TJ, et al. A genetic disorder reveals a hematopoietic stem cell regulatory network co-opted in leukemia. Nat Immunol (2023) 24(1):69–83. doi: 10.1038/s41590-022-01370-4
43. Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood (2022) 140(12):1345–77. doi: 10.1182/blood.2022016867
44. Hinai AA, Valk PJM. Review: Aberrant EVI1 expression in acute myeloid leukaemia. Br J Haematol (2016) 172(6):870–8. doi: 10.1111/bjh.13898
45. Weizmann D, Pincez T, Roussy M, Vaillancourt N, Champagne J, Laverdière C. New MECOM variant in a child with severe neonatal cytopenias spontaneously resolving. Pediatr Blood Cancer (2020) 67(5). doi: 10.1002/pbc.28215
46. van der Veken LT, Maiburg MC, Groenendaal F, van Gijn ME, Bloem AC, Erpelinck C, et al. Lethal neonatal bone marrow failure syndrome with multiple congenital abnormalities, including limb defects, due to a constitutional deletion of 3′ MECOM. Haematologica (2018) 103(4):e173–6. doi: 10.3324/haematol.2017.185033
47. Deliloğlu B, Tüfekçi Ö, Tüzün F, Aykut A, Ceylan Eİ, Durmaz A, et al. A novel mecom gene mutation associated with amegakaryocytic thrombocytopenia in a premature infant. Turk J Pediatr (2022) 64(4):736. doi: 10.24953/turkjped.2021.4855
48. Nielsen M, Vermont CL, Aten E, Ruivenkamp CAL, van Herrewegen F, Santen GWE, et al. Deletion of the 3q26 region including the EVI1 and MDS1 genes in a neonate with congenital thrombocytopenia and subsequent aplastic anaemia. J Med Genet (2012) 49(9):598–600. doi: 10.1136/jmedgenet-2012-100990
49. Bouman A, Knegt L, Gröschel S, Erpelinck C, Sanders M, Delwel R, et al. Congenital thrombocytopenia in a neonate with an interstitial microdeletion of 3q26.2q26.31. Am J Med Genet A (2016) 170A(2):504–9. doi: 10.1002/ajmg.a.37451
50. Kjeldsen E, Veigaard C, Aggerholm A, Hasle H. Congenital hypoplastic bone marrow failure associated with a de novo partial deletion of the MECOM gene at 3q26.2. Gene (2018) 656:86–94. doi: 10.1016/j.gene.2018.02.061
51. Walne A, Tummala H, Ellison A, Cardoso S, Sidhu J, Sciuccati G, et al. Expanding the phenotypic and genetic spectrum of radioulnar synostosis associated hematological disease. Haematologica (2018) 103(7):e284–7. doi: 10.3324/haematol.2017.183855
52. Niihori T, Tanoshima R, Sasahara Y, Sato A, Irie M, Saito-Nanjo Y, et al. Phenotypic heterogeneity in individuals with MECOM variants in 2 families. Blood Adv (2022) 6(18):5257–61. doi: 10.1182/bloodadvances.2020003812
53. Lord S v, Jimenez JE, Kroeger ZA, Patrick CS, Sanchez-Pena I, Ziga E, et al. A MECOM variant in an African American child with radioulnar synostosis and thrombocytopenia. Clin Dysmorphol (2018) 27(1):9–11. doi: 10.1097/MCD.0000000000000200
54. Coste T, Vincent-Delorme C, Stichelbout M, Devisme L, Gelot A, Deryabin I, et al. COL4A1/COL4A2 and inherited platelet disorder gene variants in fetuses showing intracranial hemorrhage. Prenat Diagn (2022) 42(5):601–10. doi: 10.1002/pd.6113
55. Al-Abboh H, Zahra A, Adekile A. A novel MECOM variant associated with congenital amegakaryocytic thrombocytopenia and radioulnar synostosis. Pediatr Blood Cancer (2022) 69(12):e29761. doi: 10.1002/pbc.29761
56. Shen F, Yang Y, Zheng Y, Li P, Luo Z, Fu Y, et al. MECOM-related disorder: Radioulnar synostosis without hematological aberration due to unique variants. Genet Med (2022) 24(5):1139–47. doi: 10.1016/j.gim.2022.01.021
57. Wieser R. The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene (2007) 396(2):346–57. doi: 10.1016/j.gene.2007.04.012
58. Parkinson N, Hardisty-Hughes RE, Tateossian H, Tsai HT, Brooker D, Morse S, et al. Mutation at the Evi1 locus in junbo mice causes susceptibility to otitis media. PLoS Genet (2006) 2(10):e149. doi: 10.1371/journal.pgen.0020149
59. Sader F, Roy S. Tgf-β superfamily and limb regeneration: Tgf-β to start and bmp to end. Dev Dyn (2022) 251(6):973–87. doi: 10.1002/dvdy.379
60. Loganathan A, Munirathnam D, Ravikumar T. A novel mutation in the MECOM gene causing radioulnar synostosis with amegakaryocytic thrombocytopenia (RUSAT-2) in an infant. Pediatr Blood Cancer (2018) 66(4):e27574. doi: 10.1002/pbc.27574
61. Meyer AE, Hoover LA, Craig EA. The cytosolic J-protein, Jjj1, and Rei1 function in the removal of the pre-60 s subunit factor Arx1. J Biol Chem (2010) 285(2):961–8. doi: 10.1074/jbc.M109.038349
62. Meyer AE, Hung NJ, Yang P, Johnson AW, Craig EA. The specialized cytosolic J-protein, Jjj1, functions in 60S ribosomal subunit biogenesis. Proc Natl Acad Sci U.S.A. (2007) 104(5):1558–63. doi: 10.1073/pnas.0610704104
63. Alsavaf MB, Verboon JM, Dogan ME, Azizoglu ZB, Okus FZ, Ozcan A, et al. A novel missense mutation outside the DNAJ domain of DNAJC21 is associated with shwachman–diamond syndrome. Br J Haematol (2022) 197(6):e88–93. doi: 10.1111/bjh.18112
64. D’Amours G, Lopes F, Gauthier J, Saillour V, Nassif C, Wynn R, et al. Refining the phenotype associated with biallelic DNAJC21 mutations. Clin Genet (2018) 94(2):252–8. doi: 10.1111/cge.13370
65. Dhanraj S, Matveev A, Li H, Lauhasurayotin S, Jardine L, Cada M, et al. Biallelic mutations in DNAJC21 cause shwachman-diamond syndrome. Blood (2017) 129(11):1557–62. doi: 10.1182/blood-2016-08-735431
66. Chirita-Emandi A, Petrescu CAM, Zimbru CG, Stoica F, Marian C, Ciubotaru A, et al. Case report: Novel biallelic variants in DNAJC21 causing an inherited bone marrow failure spectrum phenotype: An odyssey to diagnosis. Front Genet (2022) 13:870233/full. doi: 10.3389/fgene.2022.870233/full
67. Ji J, Shen L, Bootwalla M, Quindipan C, Tatarinova T, Maglinte DT, et al. A semiautomated whole-exome sequencing workflow leads to increased diagnostic yield and identification of novel candidate variants. Mol Case Stud (2019) 5(2):a003756. doi: 10.1101/mcs.a003756
68. Warren AJ. Molecular basis of the human ribosomopathy shwachman-diamond syndrome. Adv Biol Regul (2018) 67:109–27. doi: 10.1016/j.jbior.2017.09.002
69. Nelson A, Myers K. Shwachman-diamond syndrome Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW and Amemiya A, editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle (1993). Available at: http://www.ncbi.nlm.nih.gov/pubmed/20301722.
70. Morini J, Nacci L, Babini G, Cesaro S, Valli R, Ottolenghi A, et al. Whole exome sequencing discloses heterozygous variants in the DNAJC21 and EFL1 genes but not in SRP54 in 6 out of 16 patients with shwachman-diamond syndrome carrying biallelic SBDS mutations. Br J Haematol (2019) 185(3):627–30. doi: 10.1111/bjh.15594
71. Protano C, Buomprisco G, Cammalleri V, Pocino RN, Marotta D, Simonazzi S, et al. The carcinogenic effects of formaldehyde occupational exposure: A systematic review. Cancers (Basel) (2021) 14(1):165. doi: 10.3390/cancers14010165
72. Zhang L, Tang X, Rothman N, Vermeulen R, Ji Z, Shen M, et al. Occupational exposure to formaldehyde, hematotoxicity, and leukemia-specific chromosome changes in cultured myeloid progenitor cells. Cancer Epidemiol Biomarkers Prev (2010) 19(1):80–8. doi: 10.1158/1055-9965.EPI-09-0762
73. Lan Q, Smith MT, Tang X, Guo W, Vermeulen R, Ji Z, et al. Chromosome-wide aneuploidy study of cultured circulating myeloid progenitor cells from workers occupationally exposed to formaldehyde. Carcinogenesis (2015) 36(1):160–7. doi: 10.1093/carcin/bgu229
74. Wei C, Wen H, Yuan L, McHale CM, Li H, Wang K, et al. Formaldehyde induces toxicity in mouse bone marrow and hematopoietic stem/progenitor cells and enhances benzene-induced adverse effects. Arch Toxicol (2017) 91(2):921–33. doi: 10.1007/s00204-016-1760-5
75. Edenberg HJ, McClintick JN. Alcohol dehydrogenases, aldehyde dehydrogenases, and alcohol use disorders: A critical review. Alcohol Clin Exp Res (2018) 42(12):2281–97. doi: 10.1111/acer.13904
76. Matsumura Y, Stiles KM, Reid J, Frenk EZ, Cronin S, Pagovich OE, et al. Gene therapy correction of aldehyde dehydrogenase 2 deficiency. Mol Ther Methods Clin Dev (2019) 15:72–82. doi: 10.1016/j.omtm.2019.08.004
77. Oka Y, Hamada M, Nakazawa Y, Muramatsu H, Okuno Y, Higasa K, et al. Digenic mutations in ALDH2 and ADH5 impair formaldehyde clearance and cause a multisystem disorder, AMeD syndrome. Sci Adv (2020) 6(51). doi: 10.1126/sciadv.abd7197
78. Kinoshita S, Ando M, Ando J, Ishii M, Furukawa Y, Tomita O, et al. Trigenic ADH5/ALDH2/ADGRV1 mutations in myelodysplasia with usher syndrome. Heliyon (2021) 7(8):e07804. doi: 10.1016/j.heliyon.2021.e07804
79. Sebert M, Gachet S, Leblanc T, Rousseau A, Bluteau O, Kim R, et al. Clonal hematopoiesis driven by chromosome 1q/MDM4 trisomy defines a canonical route toward leukemia in fanconi anemia. Cell Stem Cell (2023) 30(2):153–70. doi: 10.1016/j.stem.2023.01.006
80. Merfort LW, Lisboa M de O, Cavalli LR, Bonfim CMS. Cytogenetics in fanconi anemia: The importance of follow-up and the search for new biomarkers of genomic instability. Int J Mol Sci (2022) 23(22):14119. doi: 10.3390/ijms232214119
81. Zhao Y, Wei L, Tagmount A, Loguinov A, Sobh A, Hubbard A, et al. Applying genome-wide CRISPR to identify known and novel genes and pathways that modulate formaldehyde toxicity. Chemosphere (2021) 269:128701. doi: 10.1016/j.chemosphere.2020.128701
82. Nakamura J, Holley DW, Kawamoto T, Bultman SJ. The failure of two major formaldehyde catabolism enzymes (ADH5 and ALDH2) leads to partial synthetic lethality in C57BL/6 mice. Genes Environ (2020) 42(1):21. doi: 10.1186/s41021-020-00160-4
83. Umansky C, Morellato AE, Rieckher M, Scheidegger MA, Martinefski MR, Fernández GA, et al. Endogenous formaldehyde scavenges cellular glutathione resulting in redox disruption and cytotoxicity. Nat Commun (2022) 13(1):745. doi: 10.1038/s41467-022-28242-7
84. Mu A, Hira A, Niwa A, Osawa M, Yoshida K, Mori M, et al. Analysis of disease model iPSCs derived from patients with a novel fanconi anemia–like IBMFS ADH5/ALDH2 deficiency. Blood (2021) 137(15):2021–32. doi: 10.1182/blood.2020009111
85. Shen X, Wang R, Kim MJ, Hu Q, Hsu CC, Yao J, et al. A surge of DNA damage links transcriptional reprogramming and hematopoietic deficit in fanconi anemia. Mol Cell (2020) 80(6):1013–24. doi: 10.1016/j.molcel.2020.11.040
86. Gálvez E, Vallespín E, Arias-Salgado EG, Sánchez-Valdepeñas C, Giménez Y, Navarro S, et al. Next-generation sequencing in bone marrow failure syndromes and isolated cytopenias: Experience of the Spanish network on bone marrow failure syndromes. Hemasphere (2021) 5(4):e539. doi: 10.1097/HS9.0000000000000539
87. Robertson AJ, Tan NB, Spurdle AB, Metke-Jimenez A, Sullivan C, Waddell N. Re-analysis of genomic data: An overview of the mechanisms and complexities of clinical adoption. Genet Med (2022) 24(4):798–810. 10.1016/j.gim.2021.12.011
88. Mørup SB, Nazaryan-Petersen L, Gabrielaite M, Reekie J, Marquart H v, Hartling HJ, et al. Added value of reanalysis of whole exome- and whole genome sequencing data from patients suspected of primary immune deficiency using an extended gene panel and structural variation calling. Front Immunol (2022) 13:906328/full. doi: 10.3389/fimmu.2022.906328/full
89. Feurstein S, Drazer M, Godley LA. Germline predisposition to hematopoietic malignancies. Hum Mol Genet (2021) 30(R2):R225–35. doi: 10.1093/hmg/ddab141/6294510
90. Blombery P, Fox L, Ryland GL, Thompson ER, Lickiss J, McBean M, et al. Utility of clinical comprehensive genomic characterization for diagnostic categorization in patients presenting with hypocellular bone marrow failure syndromes. Haematologica (2020) 106(1):64–73. doi: 10.3324/haematol.2019.237693
91. Wlodarski MW. The arrival of personalized genomics in bone marrow failure. Haematologica (2021) 106(1):11–3. doi: 10.3324/haematol.2020.265124
92. McReynolds LJ, Rafati M, Wang Y, Ballew BJ, Kim J, Williams V v, et al. Genetic testing in severe aplastic anemia is required for optimal hematopoietic cell transplant outcomes. Blood (2022) 140(8):909–21. doi: 10.1182/blood.2022016508
93. Fink O, Even-Or E, Avni B, Grisariu S, Zaidman I, Schejter YD, et al. Two decades of stem cell transplantation in patients with fanconi anemia: Analysis of factors affecting transplant outcomes. Clin Transplant (2023) 37(1). doi: 10.1111/ctr.14835
94. Tummala H, Walne A, Dokal I. The biology and management of dyskeratosis congenita and related disorders of telomeres. Expert Rev Hematol (2022) 15(8):685–96. doi: 10.1080/17474086.2022.2108784
95. Irie M, Niihori T, Nakano T, Suzuki T, Katayama S, Moriya K, et al. Reduced-intensity conditioning is effective for allogeneic hematopoietic stem cell transplantation in infants with MECOM-associated syndrome. Int J Hematol (2022). doi: 10.1007/s12185-022-03505-7
96. Toki T, Yoshida K, Wang R, Nakamura S, Maekawa T, Goi K, et al. De novo mutations activating germline TP53 in an inherited bone-Marrow-Failure syndrome. Am J Hum Genet (2018) 103(3):440–7. doi: 10.1016/j.ajhg.2018.07.020
97. Kennedy AL, Myers KC, Bowman J, Gibson CJ, Camarda ND, Furutani E, et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in shwachman-diamond syndrome. Nat Commun (2021) 12(1):1334. doi: 10.1038/s41467-021-21588-4
98. Choijilsuren HB, Park Y, Jung M. Mechanisms of somatic transformation in inherited bone marrow failure syndromes. Hematol Am Soc Hematol Educ Program (2021) 2021(1):390–8. doi: 10.1182/hematology.2021000271
Keywords: bone marrow failure, ADH5, ALDH2, DNAJC21, ERCC6L2, MECOM, early onset myeloid malignancies
Citation: Feurstein S (2023) Emerging bone marrow failure syndromes- new pieces to an unsolved puzzle. Front. Oncol. 13:1128533. doi: 10.3389/fonc.2023.1128533
Received: 20 December 2022; Accepted: 22 March 2023;
Published: 06 April 2023.
Edited by:
Sushree Sahoo, St. Jude Children’s Research Hospital, United StatesReviewed by:
Serine Avagyan, University of California, San Francisco, United StatesCopyright © 2023 Feurstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simone Feurstein, c2ltb25la3Jpc3RpbmEuZmV1cnN0ZWluQG1lZC51bmktaGVpZGVsYmVyZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.