 Yu-Chen Yang
Yu-Chen Yang Can-Jun Zhao1†
Can-Jun Zhao1† Li-Tian Ma
Li-Tian Ma- 1Department of Traditional Chinese Medicine, Tangdu Hospital, Air Force Medical University, Xi’an, China
- 2School of Psychology, Weifang Medical University, Weifang, China
- 3Department of Gastroenterology, Tangdu Hospital, Air Force Medical University, Xi’an, China
Lung cancer is one of the most common malignant tumours worldwide, with the highest mortality rate. Approximately 1.6 million deaths owing to lung cancer are reported annually; of which, 85% of deaths occur owing to non-small-cell lung cancer (NSCLC). At present, the conventional treatment methods for NSCLC include radiotherapy, chemotherapy, targeted therapy and surgery. However, drug resistance and tumour invasion or metastasis often lead to treatment failure. The ubiquitin–proteasome pathway (UPP) plays an important role in the occurrence and development of tumours. Upregulation or inhibition of proteins or enzymes involved in UPP can promote or inhibit the occurrence and development of tumours, respectively. As regulators of UPP, ubiquitin-specific proteases (USPs) primarily inhibit the degradation of target proteins by proteasomes through deubiquitination and hence play a carcinogenic or anticancer role. This review focuses on the role of USPs in the occurrence and development of NSCLC and the potential of corresponding targeted drugs, PROTACs and small-molecule inhibitors in the treatment of NSCLC.
1 Introduction
Lung cancer is one of the most common primary malignant tumours. Non-small-cell lung cancer (NSCLC) refers to any type of epithelial lung cancer except for small cell lung cancer. It accounts for approximately 85% of the total lung cancer cases. NSCLC is primarily divided into two types: lung squamous cell carcinoma (LUSC) and lung adenocarcinoma (LUAD) (1). Recent studies have demonstrated that uncontrolled intracellular protein homeostasis, especially abnormality of the ubiquitin–proteasome pathway (UPP), plays an important role in the progression of NSCLC. Deubiquitinating enzymes (DUBs) play an important role in UPP, which is responsible for removing the ubiquitin chain of protein substrate in cells. Abnormal activity or expression of DUBs can cause functional changes in key carcinogenic/tumour suppressor proteins, leading to malignant transformation of cells (2, 3). Ubiquitin-specific proteases (USPs), the largest subset of the DUBs family, are expressed in different cancer cells and mediate deubiquitination (4). Various USPs are abnormally expressed in NSCLC cells, with most USPs having evident carcinogenic effects and only a few USPs having beneficial effects (5–9). Targeting USPs with carcinogenic effects may benefit patients with NSCLC. Furthermore, E3 ligases mediate the binding of target proteins to the proteasome, and enhancing or inhibiting their activity can affect the degradation of target proteins. Therefore, E3 ligases may participate in regulating the activity of tumour cells (10). In this review, we summarised the role and mechanisms of action of USPs in NSCLC, the effects of enhancing/reducing the activity of target proteins through UPP and the potential of small-molecule inhibitors and PROTACs technology in the treatment of NSCLC.
2 Ubiquitination and deubiquitination
Eukaryotic cells recognize and degrade proteins through UPP. Substrate proteins bound to ubiquitin chains are directed to the 26S proteasome for degradation. UPP is a major protein breakdown mechanism in mammalian cells. The 26S proteasome consists of a 19S regulatory complex, which is responsible for recognizing ubiquitinated proteins, and a 20S protein breakdown core, which is primarily responsible for catalysing protein degradation (11). Ubiquitination can be classified as monoubiquitination, in which a Ub molecule is directly added to the lysine residue of a protein, and polyubiquitination, in which a Ub chain is formed from a single lysine residue on a substrate. The lysine residue and the N-terminal methionine residue (M1) of Ub can serve as ubiquitination sites and bind to Ub molecules to form different types of Ub chains. Seven lysine residues, namely, K6, K11, K27, K29, K33, K48 and K63, are known to be amenable to ubiquitination. K48- and K11-linked polyubiquitin chains are mainly related to proteasomal degradation, whereas K63-linked polyubiquitin chains are mainly involved in signal transduction in cells (12, 13). E1 enzymes initiate the modification of ubiquitination by adenylating Ub using ATP to form a high-energy thioester bond between the C-terminal carboxyl group of Ub and the thiol group of the cysteine residue of E1. The activated Ub is subsequently transferred to the cysteine residue of an E2 enzyme to form a similar thioester bond. Finally, an E3 ligase recruits the E2 enzyme to facilitate the specific transfer of Ub to substrate proteins, resulting in degradation of target proteins by the proteasome (14). Ubiquitination is involved in several biological processes, including but not limited to: enzyme activation, regulatory interaction between proteins, signal transduction and regulation of transcription (15).
E3 ligases are the key enzymes in ubiquitination, which determine the reaction rate and substrate properties (16). E3 ligases found in mammalian cells are categorised into three types according to their structural characteristics and mechanisms of action: really interesting new genes (RINGs), homologous to E6AP C-terminus (HECT) and RING-between-RING (RBR) ligases (17). RING E3 ligases catalyse the direct transfer of Ub from E2 enzymes to the substrate, whereas HECT and RBR E3 ligases contain a catalytic form of cysteine that receives Ub from E2 enzymes and transfers it to the target protein. Studies have demonstrated that E3 ligases are closely related to tumour development and may be involved in tumour invasion, cell proliferation, apoptosis, DNA damage and repair, tumour metabolism and immunity (10). The effects of E3 ligases vary based on the target protein they bind to. For example, Pirh2, the product of the human RCHY1 gene, is an E3 ligase containing a ring structure that stabilises c-MYC and promotes the growth, invasive ability and migratory ability of NSCLC (H1299) cells (18). Homologous to the E6-associated protein carboxyl terminus domain-containing 3 (HECTD3) is a member of the HECT E3 ligase family. Its overexpression primarily regulates K63 polyubiquitination and promotes MALT1 stabilisation, which promotes the proliferation of angiotensin II receptor-positive breast cancer cells (19, 20). Therefore, E3 ligases may play an oncogenic role. Knockdown of the E3 ligase MKRN3 can increase the proliferation of NSCLC cells, whereas upregulation of recombinant MKRN3 can directly inhibit the growth and proliferation of NSCLC cells both in vitro and in vivo. Therefore, E3 ligases may mediate the action of MKRN3 and have certain tumour-suppressing effects (21). E3 ligases mediate the degradation of substrate proteins and exert both protective and detrimental effects. However, whether the degradation of substrate proteins mediated by E3 ligases promotes or inhibits tumours remains unclear.
Ubiquitination and deubiquitination are two mechanisms for modifying protein homeostasis in cells. These mechanisms have controllable and reciprocal characteristics and cooperate to dynamically regulate various cellular processes (22). DUBs catalyse deubiquitination and disrupt the bond between Ub molecules and substrate proteins, thus acting as proofreaders for protein degradation and preventing abnormal hydrolysis of active proteins. The human genome encodes approximately 100 DUBs, which can be roughly divided into two categories based on their catalytic domain: cysteine proteases and metalloproteinases. The seven subfamilies of DUBs are as follows: USPs, ubiquitin carboxyl-terminal hydrolases (UCHs), ovarian tumour domain proteases (OTUs), Machado–Joseph disease protein domain-containing proteases (MJDs/Josephins), motif− interacting with ubiquitin−containing novel DUB family (MINDYs) and zinc finger-containing ubiquitin peptidase1 (ZUP1) belong to cysteine proteases, whereas JAB1/MPN/Mov34 metalloenzymes (JAMMs) belong to metalloproteinases (23–25). The deubiquitination mechanism of cysteine proteases depends on the conserved catalytic triad of amino acids, His-Cys-Asn/Asp. Histidine (His) residues reduce the pKa of cysteine (Cys) residues and promote the nucleophilic attack of Cys residues on isopeptide bonds, whereas Asn/Asp residues usually play a role in the localisation and polarisation of His residues (26). The deubiquitination mechanism of metalloproteinases depends on the coordination of His, aspartic acid and serine residues with zinc ions (27).
USPs are the largest subset of the DUBs family, and approximately 70 USPs have been identified in humans. The functions of USPs are similar to those of DUBs. USPs can recognise various substrates with Lys48-, Lys63- and Met1-linked Ub chains, which are involved in several processes related to tumour progression, including epithelial–mesenchymal transition (EMT), tumour metastasis, alterations in the tumour microenvironment, DNA damage repair and protein dysfunction (4, 28, 29). USPs possess a highly conserved USP domain consisting of three subdomains that resemble the palm, thumb and finger of the right hand, respectively. The catalytic site of USPs is located between the palm and thumb regions, and the catalytic centre of USPs includes a C-terminal His Box and an N-terminal Cys Box with catalytic His and Cys residues, respectively, at the interface between the thumb and palm subdomains. The finger region is responsible for interaction with Ub (30). In vivo studies have reported that weak interactions between the Ub-binding domain and monoubiquitin chains are passively regulated by various mechanisms such as aggregation of Ub, modification of Ub and concatenation of domains (31). Given that the substrate proteins of USPs contain numerous cell homeostasis regulators, oncoproteins and tumour suppressor proteins, some USPs may serve as targets for the development of anti-tumour drugs.
The ubiquitin-proteasome pathway has been shown in Figure 1.
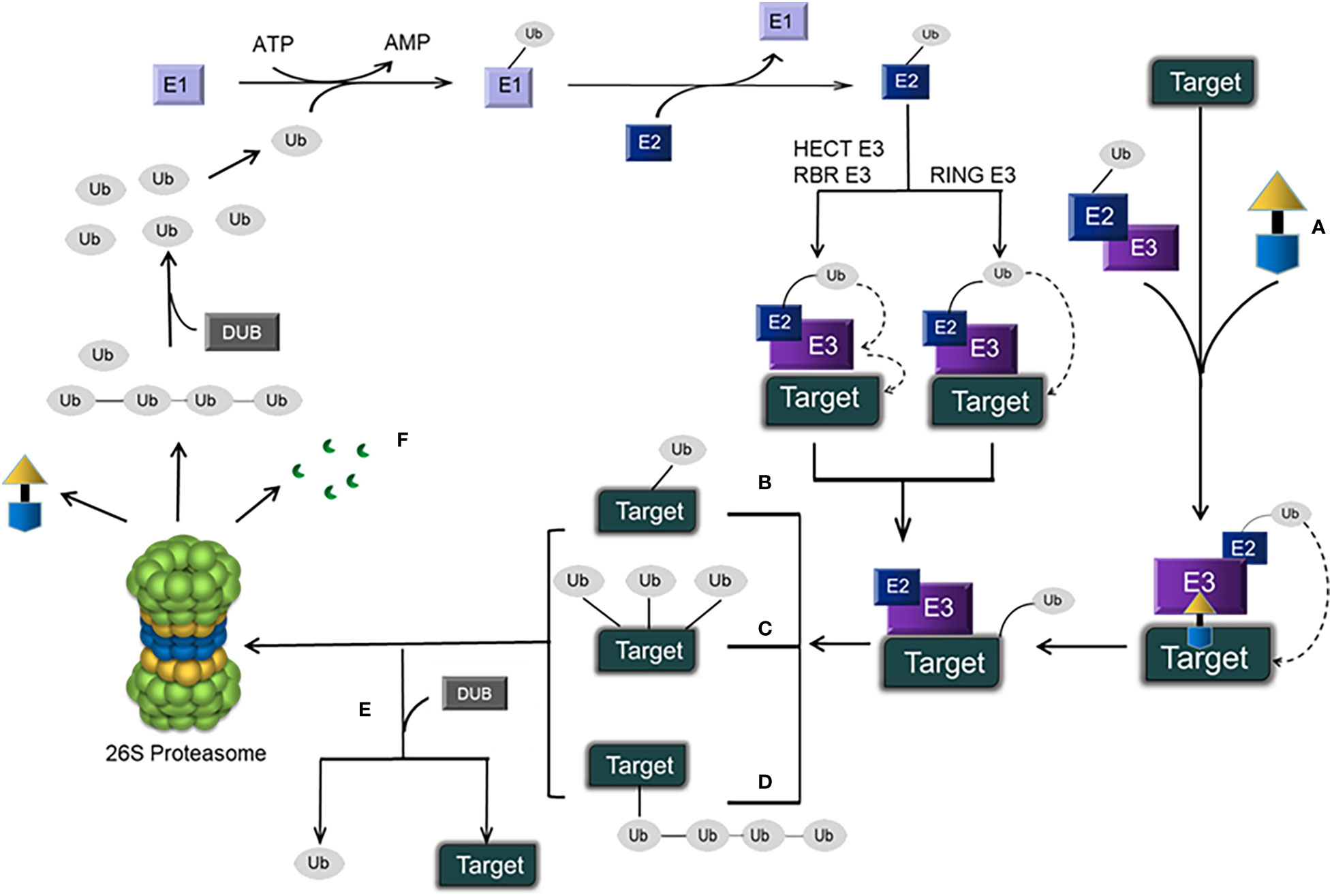
Figure 1 Schematic representation of the ubiquitin–proteasome pathway and PROTACs. (A) A PROTAC, which has an E3 ligase ligand at one end and a protein ligand at the other end, is connected by a linker in the middle to form a ternary complex: target protein–PROTAC–E3. Eventually, the target protein is degraded by the 26S proteasome, and the PROTAC is retained. (B) Monoubiquitination. (C) Multiubiquitination. (D) Polyubiquitination. (E) DUB-mediated deubiquitination (which disconnects target proteins from Ub). (F) Protein degradation products.
3 PROTACs
Proteolysis-targeting chimaeras (PROTACs) are emerging small molecules that use UPP to degrade target proteins and have promoted unprecedented innovation in drug research and development. Multiple PROTACs are undergoing evaluation in phase I and II clinical trials (32–35). Advances in PROTAC technology may lead to the development of novel strategies for targeted therapy of cancer.
PROTAC is a chemical molecule containing different ligands at two ends. One end binds to E3 ligases, whereas the other end binds to intracellular proteins. The two ends are subsequently connected by a linker to form a ternary complex: target protein-PROTAC-E3 ligase. Such chemical molecules can be polyubiquitinated by recruiting targeted proteins to the vicinity of E3 ligases, resulting in the degradation of target proteins by the 26S proteasome. Owing to their recyclability, PROTACs are good candidates for cancer therapy (36, 37). For example, PROTAC ARV-110, which uses UPP to degrade the androgen receptor (AR) protein, has demonstrated some efficacy in the treatment of prostate cancer, especially metastatic castration-resistant prostate cancer (mCRPC). The mechanism of action of ARV-110 differs from that of traditional inhibitors. Because ARV-110 acts by degrading AR, instead of inhibiting AR function, it may help to overcome drug resistance (38).
PROTACs have several advantages over small-molecule inhibitors. First, PROTACs act by degrading target proteins instead of inhibiting their activity. Second, PROTACs may be used for selective targeting of target subtypes. They can inhibit the scaffold function of target proteins and improve the efficiency of the warhead. Third, because the protective effects of PROTACs can be achieved at lower doses, their use decreases the risk of dose-dependent toxicity and promotes long-term adherence (39). However, PROTACs have some disadvantages. Compared with the common small-molecule Ub ligase inhibitors, PROTACs have larger molecular weight and poor tissue and cell permeability in the human body. Moreover, their clinical safety as drugs warrants further investigation (40, 41).
The content of PROTACs has been shown in Figure 1.
4 Abnormally expressed USPs in tumour cells
To date, many studies have focused on evaluating the functions and substrates of proteins and the role of USPs in specific diseases. Mutations in USPs can disrupt cellular metabolism in vivo, resulting in an increased predisposition to cancer. Therefore, USPs can be used as targets for developing anticancer drugs (28). The common abnormal USPs associated with NSCLC and other tumours are summarised below.
4.1 USP4
The human USP4 gene was identified as a proto-oncogene related to Tre2/Tre17(USP6). In USP4, its catalytic domain is composed of a Cys box and His box, whereas its non-catalytic structure is composed of a USP domain (DUSP) and a Ub-like domain (UBL) (42). Studies have demonstrated that USP4 is closely associated with various malignant tumours, such as colorectal, breast, liver and lung cancers (43–46).
In breast cancer, USP4 directly interacts with and deubiquitinates TβRI and promotes the proliferation of tumor cells (44). In liver cancer, the low expression of microRNA-148a can increase the expression of the downstream molecule USP4, thereby promoting the proliferation and migration of liver cancer cells (45). In human osteosarcoma cells, USP4 can directly bind to and deubiquitinate ARF-BP1, which stabilises ARF-BP1 and reduces p53 levels, thereby promoting cancer cell proliferation (47).
USP4 may act as a tumour suppressor protein in some tumours. In head and neck squamous cell carcinoma, USP4 inhibits carcinogenesis by deubiquitinating receptor-interacting protein 1 (RIP1) and promoting apoptosis induced by tumour necrosis factor-α (TNF-α) (48). Downregulation of endogenous USP4 can promote TNF-α-induced migration of LUAD (A549) cells, indicating that USP4 is a negative regulator of cell migration. Mechanistically, USP4 can directly bind to the TRAF domain to inhibit TRAF2/TRAF6 activity through deubiquitination in an activity-dependent manner and negatively regulate IL-1β and TNF-α-induced NF-κB activation, thereby inhibiting lung cancer metastasis (46).
Therefore, USP4 plays different roles in different types of cancer cells, and whether it promotes or suppresses cancer remains controversial.
4.2 USP7
To date, USP7 is the most extensively investigated DUB and is the first DUB that can be specifically targeted and inhibited by drugs with promising therapeutic effects (49). It contains 1102 amino acid residues, has a molecular weight of approximately 135 kDa, and contains seven domains: an N-terminal TRAF-like (Tumor necrosis factor Receptor-Associated Factor) domain, a catalytic domain and five C-terminal ubiquitin-like domains (50, 51). MDM2 is one of the substrates of USP7. USP7 has a high affinity for MDM2, which often leads to p53 degradation and increases the risk of cancer. Therefore, USP7 can be considered an oncogene (52).
USP7 can deubiquitinate CCDC6, a tumour suppressor protein, in cells with DNA damage to protect it from degradation by the tumour suppressor FBXW7, thus improving the prognosis of NSCLC (53, 54).
Both USP7 and Ki-67 are highly expressed in NSCLC. In particular, Ki-67 expression is high in the G1, S, G2 and mitotic phases of the cell cycle, which usually indicates a poor prognosis. USP7 acts by binding to Ki-67 and stabilizing its expression to play a pro-carcinogenic role (55).
Fat mass and obesity associated protein (FTO), a demethylase that is significantly upregulated in NSCLC, can promote the expression of USP7 by activating its m6A demethylase activity, reducing the m6A level of USP7 and improving the stability of its mRNA (56).
In addition, USP7 is associated with phosphatase and tensin homologue (PTEN). In chronic myeloid leukaemia (CML), BCR-ABL can enhance USP7-induced deubiquitination of PTEN, which is conducive to nuclear rejection (57). NPM1 is frequently mutated and can interact with USP7 in acute myeloid leukaemia (AML). It prevents USP7-mediated deubiquitination in the nucleus and promotes the translocation of PTEN to the cytoplasm, leading to polyubiquitination and degradation of PTEN in the cytoplasm (58). Patients with acute promyelocytic leukaemia (PML) harbour a PML-RARα translocation in which USP7 deubiquitinates PTEN primarily in the cytoplasm, promoting nuclear rejection (59).
4.3 USP10
The target proteins of USP10 include p53 (60), PTEN (61), P14ARF (62). Similar to that of USP7, the most prominent physiological function of USP10 is to specifically deubiquitinate p53 and neutralise the effects of MDM2, eventually stabilising p53 in normal cells (63). However, USP10 can stabilise oncogenes such as FLT3 (64), RAF1 (65) and Musashi2 (66). USP10 overexpression in breast cancer and neuroblastoma is found to be associated with a poor prognosis (67, 68).
USP10 expression is downregulated in some cancers, including gastric cancer and NSCLC. Downregulation of USP10 results in a poor prognosis and shortened survival in gastric cancer. In NSCLC, USP10 deficiency is not significantly associated with clinical outcomes and prognosis, indicating that USP10 may not be directly involved in tumour progression (69, 70). However, USP10 can interact with PTEN to reduce K63-linked ubiquitination of PTEN mediated by the E3 ligase TRIM25, restore PTEN activity and reduce PIP3 production, thereby inhibiting the signal transduction of mammalian target of rapamycin (mTOR) in NSCLC cells (71). Although USP10 may act as a tumour suppressor, its role in different tumours remains controversial.
4.4 USP22
USP22 is highly expressed in various tumours such as lung (72), colorectal (73) and gastric cancers (74), and is involved in DNA transcription, malignant transformation of cells and cell cycle progression (75).
USP22 can be inhibited by shRNA, activates the p53 pathway in tumours and downregulates MDMX protein, thereby inducing apoptosis in NSCLC cells (9). In LUAD, knocking down USP22 may induce ubiquitin C (UBC) expression, which promotes cell cycle and ubiquitin-mediated protein degradation, and inhibits the occurrence of lysosomal autophagy, thereby promoting the development and progression of LUAD (76).
In addition, USP22 can promote hypoxia-induced generation of liver cancer stem cells through the HIF1α/USP22 positive feedback loop after p53 inactivation, and lipoprotein complexes targeting USP22 can inhibit the growth of liver cancer cells and enhance sorafenib sensitivity (77).
Therefore, USP22 may be a potential drug target for cancer therapy.
4.5 Other USPs
USP3 can interact with KLF5 and stabilize KLF5 through deubiquitination. USP3 knockdown inhibited the proliferation of breast cancer cells, while ectopic expression of KLF5 partially rescued this inhibitory effect (78). As one of the factors responsible for NSCLC cell proliferation, USP3 may lead to Ub-mediated degradation by targeting RBM4, which is a tumour suppressor gene and a key molecule for RNA splicing (79).
USP5 can enhance the stability of cyclin D1 (CCND1) protein, significantly prolong the half-life of CCND1 and reduce the degradation of CCND1. Additionally, it can promote the proliferative, migratory and colony-forming abilities of NSCLC cells. The USP5 inhibitor G9 can lead to cell cycle arrest in NSCLC cells and can significantly downregulate phosphorylated retinoblastoma (RB) protein, thereby inhibiting the growth of NSCLC xenografts (80). USP5 can activate the Wnt/β-catenin signalling pathway, and its expression is higher in NSCLC tissues than in normal tissues. It promotes nuclear accumulation and signalling of β-catenin by deubiquitinating it, and its increased expression is associated with large tumour size, poor differentiation, advanced tumour stage and poor patient survival (81).
Overexpression of USP14 promotes tumour cell proliferation and is associated with the poor prognosis of NSCLC. The mRNA expression of USP14, accumulation of β-catenin protein and activation of the Wnt pathway are upregulated in NSCLC, resulting in shorter survival (82). Therefore, USP14 acts as a tumour promoter and may serve as a promising therapeutic target for NSCLC.
USP17 is highly expressed in NSCLC, and its expression in squamous cell carcinoma is significantly higher than that in adenocarcinoma. The high expression of USP17 in squamous cell carcinoma may be attributed to differences in transcriptional regulation or protein turnover (83).
Overexpression of USP19 increases the migratory and invasive abilities of breast cancer (MCF7) cells. These effects of USP19 are closely related to its catalytic activity and transmembrane domain. Knockdown of USP19 reduces tumour aggressiveness, suggesting that USP19 plays a key role in the migration of breast cancer cells (84).
Overexpression of USP21 is associated with the progression of pancreatic ductal adenocarcinoma (PDAC). Overexpression of USP21 can accelerate PDAC growth in mice and promote the progression of pancreatic intraepithelial neoplasia (PanIN) to PDAC in immortalised human pancreatic ductal cell models. However, the loss of USP21 impairs PDAC cell growth. Mechanistically, USP21 promotes cancer cell proliferation by deubiquitinating and stabilising the TCF/LEF transcription factor TCF7 (85).
USP28 is upregulated in NSCLC and is associated with a poor prognosis of NSCLC (86). The lower expression of miR-3940-5p in NSCLC tissues than in paired adjacent normal tissues usually indicates tumor growth and proliferation (87). miR-3940-5p can target CCND1 and USP28 to inhibit the growth of NSCLC cells, and overexpression of CCND1 and USP28 can attenuate miR-3940-5P-induced proliferation and apoptosis of NSCLC cells (88). Additionally, USP28 plays an important role in maintaining the protein expression of c-MYC, c-JUN and Δp63 in laryngeal squamous cell carcinoma (LSCC). Inhibition of USP28 can reduce the expression of these proteins in mice with LSCC and induce tumour cell death (89).
USP29 can promote the migration of gastric cancer cells by preventing Snail degradation, thereby maintaining high protein expression of Snail in cancer cells. Snail is a major inducer of EMT and metastasis, and its high expression is associated with poor survival and prognosis (90). In addition, USP29 overexpression induced by chemotherapy and oxidative stress treatment can promote chemotherapy resistance by hijacking the USP29/Snail1 axis, which is associated with enhanced cancer stem cell properties, poor prognosis and drug resistance in lung cancer (91).
USP43, an H2BK120 deubiquitinase, exerts strong inhibitory effects on the growth and metastasis of breast cancer in vivo. It is phosphorylated by AKT in the cytoplasm and subsequently binds to the 14-3-3β/ε heterodimer to be retained in the cytoplasm, resulting in a marked reduction in USP43 levels in the nucleus. Low levels of USP43 in the nucleus are associated with EGFR accumulation, AKT hyperactivation, higher histological grades and a poor prognosis (92).
A list of USPs upregulated/downregulated in NSCLC is provided in Table 1.
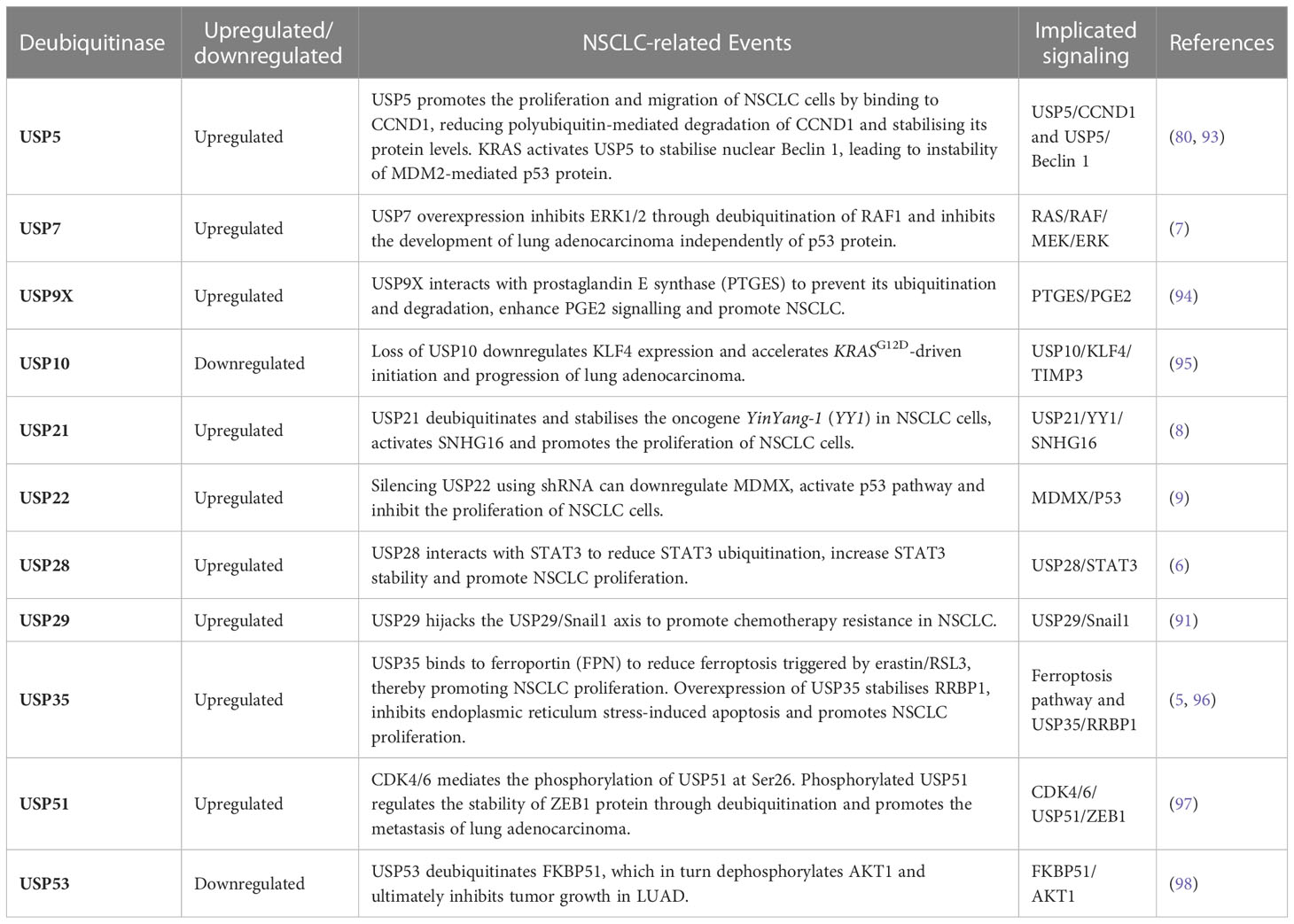
Table 1 Ubiquitin-specific proteases upregulated/downregulated in NSCLC.
5 NSCLC-related targets and treatment
Signalling pathways associated with the development of NSCLC should be identified and examined to understand the pathogenesis of NSCLC. The PI3K/AKT/mTOR (99), Ras/Raf/Mek/Erk (100), Wnt/β-catenin (101), NF-κB pathways (102), are closely related to the occurrence and development of NSCLC. Inhibiting key proteins involved in these pathways may represent an effective therapeutic strategy for NSCLC. Similarly, tumour suppressor genes are often aberrantly expressed in NSCLC. For example, downregulation of p53 and PTEN promotes cancer cell proliferation and indicates a poor prognosis (103). Specific inhibitors can be used to target and degrade oncoproteins through UPP and activate tumour suppressor proteins, which can effectively block downstream signal transduction and prevent the proliferation of cancer cells. In recent years, PROTACs have demonstrated anti-tumour effects in cellular or preclinical models of NSCLC. The USPs and PROTAC targets related to NSCLC-associated signalling pathways are summarised below.
5.1 Targeting key proteins in tumour-related signalling pathways and application of PROTACs
5.1.1 EGFR
Overexpression of Epidermal growth factor receptor (EGFR) is associated with the poor prognosis of several cancers, including oesophageal, breast, head and neck squamous cell, and lung cancers (104–107). In Asian and non-Asian populations, EGFR mutations are observed in approximately 40% and 20% of patients with NSCLC, respectively (108). Therefore, EGFR is one of the key therapeutic targets for NSCLC. To date, several generations of epithelial growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) have been used for the treatment of NSCLC. EGFR-TKIs can inhibit the proliferation and metastasis of tumour cells by mimicking ATP configuration and occupying ATP-binding sites, thereby blocking downstream signal transduction (109, 110). However, owing to mutations and signal bypass pathways, resistance to EGFR-TKIs has become a common problem in clinical settings (111). In lung adenocarcinoma, USP22 can induce EGFR-TKI resistance by regulating the endocytosis and transport of EGFR through deubiquitination modification, stabilising the intracellular EGFR protein levels and promoting the continuous activation of EGFR-dependent signalling pathways (112). Similarly, tripartite motif 25 (TRIM25), an E3 ligase, can promote the proliferation of cancer cells by promoting the K63-linked ubiquitination of EGFR, increasing the stability of EGFR protein and promoting the continuous activation of downstream signalling pathways (113).
In recent years, PROTACs have demonstrated good EGFR-targeting ability. Yu et al. developed MS 9427, an EGFR-specific PROTAC degrader that effectively induces efficient degradation of mutant EGFR in a concentration- and time-dependent manner through UPP and the autophagy-lysosome pathway and exerts a strong inhibitory effect on NSCLC cell proliferation (114). Cheng et al. reported that the E3 ligases VHL and CRBN can recruit the EGFR degraders MS 39 and MS 154, respectively, to effectively induce the degradation of mutant EGFR in NSCLC in an E3 ligase-dependent manner (115). In addition, Zhao et al. reported that compound P3, which contains a purine structure that recruits VHL, can significantly induce degradation of mutant EGFRdel 19, promote cell apoptosis, arrest the cell cycle and inhibit colony formation (116). The PROTACs SIAIS 125 and SIAIS 126 can selectively degrade EGFRL858R+T790M resistance proteins in H1975 cells and exhibit potent and selective anti-tumour effects against EGFR-TKI-resistant lung cancer. Selective degradation of EGFREx19del mutant protein in NSCLC (PC9) cells can induce apoptosis (117). Zhang et al. reported that the orally available PROTAC HJM-561 can overcome EGFR triple mutation-mediated resistance in NSCLC by specifically degrading mutant EGFR proteins. Additionally, HJM-561 can exhibit potent antitumour effects against EGFRDel19/T790M/C797S-driven Ba/F3 cell-derived xenograft (CDX) and patient-derived xenograft (PDX) models of NSCLC resistant to osimertinib (118). Overall, PROTACs targeting EGFR are excellent therapeutic candidates for overcoming EGFR-TKI-induced resistance.
5.1.2 PI3K/AKT/mTOR
The PI3K/AKT/mTOR pathway plays a key role in tumour growth, invasion, metastasis, and angiogenesis (119). Activation of PI3K by upstream signalling molecules, such as EGFR or other receptor tyrosine kinases, can catalyse PIP3 production, thereby triggering the activation of the AKT/mTOR signalling cascade (120). AKT plays an important role in tumor growth and development, such as ovarian, breast and gastric cancers (121–123). K63-linked polyubiquitination of AKT at lysine 8 and 14, which is mediated by the E3 ligase TRAF6 or SKP2, can localise AKT to the cell membrane, thereby resulting in AKT activation and enhanced intracellular signalling (124).
USPs and E3 ligases are involved in the PI3K/AKT/mTOR pathway. As a tumour suppressor, USP53 can deubiquitinate FK506-binding protein 51 (FKBP51) to dephosphorylate AKT1 and inactivate downstream signalling pathways, thereby inhibiting the progression of LUAD (98).
Calcium- and integrin-binding protein 1 (CIB1) is an oncogene that regulates cell adhesion, migration and differentiation (125). The E3 ligase CHIP can activate K48-linked polyubiquitination, which targets CIB1 to inhibit AKT/mTOR signalling and EMT in LUAD (126). Although significant progress has been achieved in the development of inhibitors targeting specific proteins in the PI3K/AKT/mTOR pathway to treat NSCLC, acquired drug resistance remains inevitable. However, targeted degradation of AKT may help to overcome drug resistance in NSCLC, and may exert long-term pharmacological effects when compared with their inhibition (127, 128). PROTACs can control tumour growth and invasion by inducing the ubiquitination and degradation of AKT. In the first PROTAC targeting AKT, INY-03-041, GDC-0068 was used as the target head compound and lenalidomide was used as an E3 ligase ligand. It can effectively induce degradation of the three isoforms of AKT proteins (AKT1/2/3) (128). Similarly, MS 143, a potent AKT degrader, can effectively inhibit tumour growth in xenograft mouse models by hijacking UPP to induce rapid and stable AKT degradation in a concentration- and time-dependent manner (129). However, to date, PROTACs targeting AKT in NSCLC have not been developed and warrant further exploration.
5.1.3 RAS/RAF/MEK/ERK
The RAS/RAF/MEK/ERK signalling pathway, one of the MAPK cascades in humans, plays a key role in regulating various processes, including cell proliferation, differentiation and apoptosis (130, 131). After EGFR is activated by TGF-α, RAS, one of the downstream signals of EGFR, is activated initially through SOS and Grb2 and subsequently through a cascade of RAF and MEK1/2, eventually resulting in activation of ERK1/2 (132).
Studies have shown that USP7 may be involved in the regulation of RAF1 in lung adenocarcinoma cells in this signaling pathway. Overexpression of USP7 can regulate ERK1/2 signaling pathway, increase P53 expression, and inhibit G2/M transformation and proliferation of LUAD cells by deubiquitinating the M1, K6, K11, K27, K33, and K48 polyubiquitin chains of Raf-1 to reduce the level of RAF1 phosphorylation (7). In chronic myeloid leukemia cells, Leucine zipper-like transcriptional regulator 1 (LZTR1) participates in the formation of the junction of Cullin 3 (CUL3) ubiquitin ligase complex, mediates CUL3 and thus causes RAS ubiquitination, And regulate downstream MEK, ERK and other signals, and finally achieve the purpose of affecting BCR-ABL TKI (133).
KRAS is the predominant mutant subtype of the RAS family, accounting for approximately 85% of the total RAS mutations. KRASG12C mutation accounts for approximately half of KRAS mutations in patients with LUAD (134, 135). KRAS is difficult to target owing to its lack of a deep binding pocket, its high affinity for GTP and the presence of abundant intracellular levels of GTP (136, 137). PROTACs can be used to overcome KRAS12C-induced resistance and target other KRAS mutants. For example, a VHL-based PROTAC has been designed using MRTX849 as a covalent KRASG12C warhead. The lead molecule LC-2 in this PROTAC can induce KRAS degradation and impair downstream MAPK signalling, resulting in reduced pERK levels in KRASG12C mutant-positive human lung cancer cell lines (138). Lu et al. developed a reversible covalent PROTAC that can efficiently target the degradation of endogenous KRASG12C in KRASG12C-mutated H358 and H23 lung cancer cells (139).
Furthermore, PROTACs have demonstrated great potential in targeting MEK protein for tumour therapy. The PROTAC MS432 has been synthesised by linking the major portion of PD0325901, a non-ATP competitive MEK inhibitor, to VHL or CRBN E3 ligase. It can effectively degrade MEK1/2 protein and effectively inhibit the proliferation of BRAF-mutated colorectal cancer (HT-29) cells and melanoma (SK-MEL-28) cells (140).
5.1.4 Wnt/β-catenin
Wnt signalling was first found to play a role in tumour and embryonic development. Recent studies have revealed that it also plays a role in tissue regeneration in adult bones, skin and intestines (141, 142). Wnt/β-catenin is one of the branches of the Wnt signalling pathway. The modification and degradation of β-catenin protein is the key function of this signalling pathway, which can be regulated through ubiquitination and deubiquitination. The interaction between β-catenin and E-cadherin may be associated with the prognosis of NSCLC (143–145).
FBXW2, an E3 ligase, binds to β-catenin via EGF-AKT1-mediated phosphorylation at Ser552 and promotes its ubiquitination and degradation. Functionally, FBXW2 overexpression inhibits the migratory and invasive abilities of lung cancer cells by blocking β-catenin driven trans-activation of MMPs, whereas FXBW2 knockdown promotes the migratory and invasive abilities of lung cancer cells and tumour metastasis in vitro and in vivo (146), indicating that FBXW2 acts as a tumour suppressor.
USP44 may serve as a tumour suppressor protein in colorectal cancer (CRC). USP44 is significantly downregulated in CRC and enhances the apoptosis of CRC cells. Its overexpression increases the expression of Axin1 protein, a scaffolding protein that promotes negative regulation of the Wnt signalling pathway; reduces the expression of β-catenin, c-myc and cyclin D1 and inhibits Wnt/β-catenin signalling-mediated activation of apoptosis in CRC cells (147).
As an activator of the Wnt signalling pathway, USP7 may serve as a specific therapeutic target for APC-mutated CRC. USP7 stabilizes β-catenin through deubiquitination and activates the Wnt signalling pathway, whereas the β-catenin inhibitory domain of APC protects β-catenin from deubiquitination by USP7 (148). P5091, a specific inhibitor of UPS7, inhibits the growth of CRC cells by promoting ubiquitination and degradation of β-catenin. Therefore, inhibition of USP7 can inhibit the activation of Wnt signalling by regulating the ubiquitination of β-catenin (149).
Chen and Hu et al. reported that the PROTAC xStAx-VHLL, which is designed to target the Wnt pathway, can trigger β-catenin degradation and effectively inhibit the survival of patient-derived CRC organoids through Wnt signalling blockade. Therefore, xStAx-VHLL can be used in the treatment of CRC (150).
5.1.5 NF-κB
Nuclear factor-κb (NF-κB) is a family of transcription factors that play an important role in regulating immune response and inflammation. NF-κB regulates the expression of a variety of genes and plays a key role in the occurrence and development of tumors, such as proliferation, migration and apoptosis. Inhibition of NF-κB signaling can inhibit cancer progression (151). Ubiquitination/deubiquitination plays a key role in the activation of the NF-κB signalling pathway (152).
USP20 plays an important role in TNFα-induced NF-κB signalling by stabilising p62. Knockdown of USP20 and p62 in HeLa cells decreases cell viability and number while increasing the expression of cleaved caspase-8, caspase-3 and PARP (153).
USP4 can inhibit NF-κB activation through deubiquitination of TRAF2 and TRAF6, thereby promoting cell migration and invasion in lung cancer (46). Overexpression of wild-type USP4 in Hela cells can inhibit TAK1 polyubiquitination and NF-κB activation, whereas its knockdown can enhance polyubiquitination of TAK1 and phosphorylated IκBα and negatively regulate NF-κB activation induced by IL-1β, LPS and TGFβ (154).
USP18 interacts with the TAK1-TAB1 and IKKα/β-Nemo complexes to cleave the K63-linked polyubiquitin chain attached to TAK1 and inhibit NEMO ubiquitination, respectively, in a protein-dependent manner, thereby negatively regulating NF-κB (155).
5.1.6 PTEN
PTEN is a tumour inhibitor that can antagonise PI3K function by dephosphorylating PIP3 to PIP2, thereby inactivating downstream oncogenic signalling and exerting tumour-suppressing effects (156, 157). PTEN inhibits cell proliferation and survival by inducing cell cycle arrest through the PI3K/AKT pathway or by downregulating cyclin D1 and decreasing its accumulation in the nucleus (158). However, owing to gene mutations, deletions or promoter methylation, PTEN expression is significantly reduced in NSCLC, which promotes tumour growth and invasion (159). Post-translational modifications (PTMS) of PTEN, including ubiquitination of PTEN, may represent an effective strategy for regulating PTEN function and serve as therapeutic targets for cancer (160).
Casein kinase 1-alpha (CK1α) competitively antagonises PTEN ubiquitination mediated by the E3 ligase NEDD4-1 by binding to the C-terminus of PTEN. Additionally, it positively regulates autophagy and inhibits the growth of NSCLC cells (161). Similarly, the deubiquitylase OTUD3 can interact with PTEN and deubiquitinate it, thus stabilising PTEN and inhibiting the development of breast cancer (162). However, in lung cancer, OTUD3 promotes tumour proliferation and metastasis and inhibits apoptosis through deubiquitination of GRP78 protein and is closely related to drug resistance (163). Therefore, most deubiquitinating enzymes play different roles in different tumours, which should be comprehensively investigated for the precise treatment of tumours.
5.1.7 MDM2/p53
p53 is a typical intracellular tumour suppressor that is activated in response to DNA damage or oncogene activation and can act as a negative regulator to induce cell cycle arrest, apoptosis and senescence in cancer cells, thereby inhibiting growth and proliferation. MDM2 suppresses p53 primarily by inhibiting its transcriptional activity and inducing its nuclear export and Ub-mediated degradation, thereby decreasing the p53 protein level to maintain normal cell function (164). Ubiquitination of p53 is a key regulatory event in the p53 pathway and can be reversed by DUBs (165). Therefore, reactivation of p53 in cancer cells by blocking the MDM2/p53 pathway is an effective approach for targeted therapy of cancer.
Several studies have demonstrated that USPs can regulate MDM2/p53. Abraxas brother 1 (ABRO1), a component of the BRCC36-containing isopeptidase complex (BRISC), stabilises p53 by promoting its interaction with USP7 to reduce p53 ubiquitination (166). USP15 knockdown can delay Ub-mediated degradation of p53 by accelerating MDM2 degradation in melanoma (A375) cells (167). USP2a regulates the p53 pathway by stabilising MDM2 and MdmX to promote the growth of testicular embryonal carcinoma (NTERA-2) cells and breast cancer (MCF7) cells (168).
Compound 11a, a PROTAC that induces MDM2 degradation, can induce proteasome-dependent degradation of MDM2 in NSCLC (A549) cells and effectively inhibit tumour growth in xenograft mouse models of LUAD (169).
5.1.8 ALK
ALK rearrangement genes are detected in 3%–7% of NSCLC cases, with EML4-ALK being the most important fusion genes (170). ALK TKIs are the first-line treatment agents in patients with lung cancer who harbour ALK rearrangements. To date, six small-molecule inhibitors of ALK have been used to treat ALK-positive NSCLC: crizotinib, ceritinib, alectinib, brigatinib, ensartinib and lorlatinib (171). Although these inhibitors have demonstrated promising efficacy in clinical settings, serious challenges such as drug resistance and brain metastasis may emerge with their long-term use (172, 173).
PROTACs have unique advantages in targeting ALK. Gray and Jin et al. reported two sets of PROTACs: TL13-12 and TL13-112 and MS4077 and MS4078, which can mediate ubiquitination and degradation of NPM-ALK and EML4-ALK in vitro (174, 175). Xu et al. developed PROTACs compounds (36 and 37) with a novel skeleton based on the second-generation small-molecule inhibitor alectinib. These PROTACs induced strong anti-proliferative activity in H3122 (NSCLC cells expressing the EML4-ALK fusion protein) and Karpas 299 (anaplastic large cell lymphoma cells expressing the NPM-ALK fusion protein) cells without causing toxicity in A549 and HFL-1 cells (cell without ALK expression). These results indicated that the PROTACs had good selectivity (176).
Therefore, in addition to traditional chemical inhibitors, PROTACs are promising therapeutic agents for NSCLC and can delay the emergence of resistance mutations, thereby improving patient outcomes.
5.1.9 The roles of PROTACs in other targets associated with NSCLC
Research into the use of PROTACs for targeting NSCLC-associated genes is gradually emerging.
SHP2 is frequently activated in lung cancer (177). The SHP2-targeting PROTAC SHP2-D26 can induce rapid and efficient degradation of SHP2 and is 10- to 100-fold more effective than allosteric SHP inhibitors in inhibiting ERK activity in cancer cells (178).
Nuclear focal adhesion kinase (FAK) is overexpressed in various tumours, including lung cancer (179). The Fak-targeted PROTAC GSK215 can significantly inhibit the migration of A549 cells, and induce rapid and long-term degradation of FAK, with lasting effects on FAK levels (180).
The PROTAC MD13, which targets macrophage migration inhibitory factor (MIF), can effectively inhibit the proliferation of A549 cells, resulting in cell cycle arrest at the G2/M phase (181).
Bromodomain-containing protein 4 (BRD4) is abnormally upregulated in lung cancer (182). The BRD4-targeting PROTAC CREATE can reshape the tumour microenvironment to directly reduce TAMs and effectively treat lung cancer (183).
In conclusion, PROTACs represent a novel therapeutic modality for NSCLC and other cancers.
5.2 Application of USP-related small-molecule inhibitors in cancer
Various partial and specific inhibitors of USPs have been developed for the treatment of cancer. Some USP inhibitors used in the treatment of NSCLC are shown in Table 2.

Table 2 Specific protease inhibitors used in the treatment of NSCLC.
LCA hydroxyamide (LCAHA) is a USP2a inhibitor that induces G0/G1-phase arrest by inhibiting the deubiquitinase USP2a and destabilising cyclin D1 and is independent of p53 status (189). FT671, a noncovalent inhibitor of USP7, can increase the degradation of the Ub ligase MDM2 and the expression of p53 to exert tumour-suppressing effects in vivo (190). In addition, USP7 inhibitor P22077 can effectively induce apoptosis of cancer cells in neuroblastoma cells with intact USP7/HDM2/p53 axis (191). WP1130 is an inhibitor of DUBs that inhibits activities such as USP5, UCH-L1, USP9X, USP14, and UCH37 (192). Combined treatment with WP1130 and cisplatin can synergistically inhibit the viability of LUAD (A549 and HCC827) cells (186). EOAI3402143(G9) is a non-specific USP inhibitor that inhibits USP5, USP9X and USP24 in a dose-dependent manner. Compared with WP1130, G9 has greater solubility and inhibitory activity against USP5 and USP9X (80, 193, 194), and the therapeutic role of G9 has been investigated in human pancreatic cancer (195). GSK2643943A, an inhibitor of USP20, has been investigated in oral squamous cell carcinoma (OSCC) (196).
5.3 Targeting of E3 ligases
Proteasome inhibitors, such as bortezomib, have demonstrated good efficacy in the treatment of multiple myeloma, which indicates the feasibility of research and development of drugs targeting UPP. However, because these inhibitors inhibit all 26S proteasome-dependent protein degradation pathways without discrimination, they can cause damage to normal cells and have significant side effects (197). Among the potential drug targets of ubiquitin-protease system, E3 ligases have outstanding advantages: the specific structure of some E3 ligases determines that they have better selectivity for screening small molecule inhibitors or developing specific targeted antibodies.
The E3 ligase CRBN is the target of many drugs, and several multi-target PROTACs based on CRBN have been developed (198). For example, ARV-825, a CRBN-based anti-BET PROTAC, exploits the association between CRBN and OTX015 to promote BRD4 degradation and inhibit cancer cell progression (199). Lenalidomide exerts significant activity by inhibiting CRBN, which forms a new substrate bound by the cullin-RING ligase 4 (CRL4) complex, leading to ubiquitination and proteasome-dependent degradation, resulting in antimyeloma activity (200).
The E3 ligase Skp2 is overexpressed in many human cancers and can regulate tumorigenesis (201). The Skp2 inhibitor SMIP004 can improve the efficacy of radiotherapy; upregulate programmed cell death protein 4 (PDCD4) expression levels, the target of Skp2; and inhibit the proliferation of breast cancer cells (202).
Compound A1874 synthesised using mouse double minute 2 (MDM2) as an E3 ligase receptor can degrade bromodomain-containing protein 4 (BRD4), upregulate the tumour suppressor p53, activate caspase-related apoptosis of colon cancer cells and effectively inhibit the viability of colon cancer cells (203, 204).
AMG-232, a small-molecule inhibitor of piperidinones that targets the E3 ligase, induces p53 activity, thereby arresting the cell cycle and inhibiting tumour cell proliferation in melanoma (205, 206).
The PROTAC ARV-471 is a protein degrader that binds to E3 ligases and oestrogen receptor (ER) and has potential applications in the treatment of breast cancer (207).
In conclusion, targeting E3 ligases represents an effective strategy for the treatment of NSCLC.
6 Concluding remarks
In this review, we summarised the role of USPs and E3 ligases in UPP and NSCLC-associated signalling pathways and discussed the research progress of corresponding targeted drugs, PROTACs and small-molecule inhibitors. Significant advancements have been made in the field of USPs; however, the role of USPs and E3 ligase-mediated PROTACs in the signal transduction of target protein degradation warrants further investigation. USPs and E3 ligases are aberrantly expressed in many tumours and can be used to regulate the activity of cancer cells. To date, studies have majorly focused on the following aspects: identifying oncogenic proteins or enzymes and understanding their pathogenic mechanisms and targeting and inducing ubiquitination and degradation of related substrates for the treatment of cancer. Given that promoting the degradation of USPs and enhancing the activity of E3 ligases are beneficial approaches for cancer therapy, the high expression of USPs in cells can be inhibited to shift the intracellular balance to E3 ligases that promote the degradation of target proteins. However, this viewpoint is complicated because target proteins in different cancers play different roles, and the corresponding specific USPs or E3 ligases also act as tumour promoters and/or suppressors. In addition, most related studies are based on carcinogenic USPs, whereas studies on tumour-suppressing USPs are limited. Moreover, the mechanisms underlying USP-induced drug resistance in tumours remain unclear. USPs are involved in not only cancer but also other diseases, such as neurodegenerative diseases, immune diseases, alcohol-associated liver disease and chronic kidney disease (208–211). When considering the function of USPs as an oncogene or tumor inhibitor, this knowledge needs to be combined with a variety of unknown substrate proteins and tissue and/or cell-specific aspects that will improve our understanding of the pathogenesis of NSCLC and may contribute to the development of future therapies.
Author contributions
JZ and L-TM participated in supervision, conception, and editing. Y-CY, C-JZ and Z-FJ participated in writing the manuscript, writing-review. All authors contributed to the article and approved the submitted version.
Funding
Key clinical research projects of the Second Affiliated Hospital of Air Force Military Medical University (2021LCYJ016 to JZ) and Social Talent Fund Funding Program Project (2021SHRC044 to C-JZ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature (2018) 553(7689):446–54. doi: 10.1038/nature25183
2. Fan Q, Wang Q, Cai R, Yuan H, Xu M. The ubiquitin system: Orchestrating cellular signals in non-Small-Cell lung cancer. Cell Mol Biol Lett (2020) 25:1. doi: 10.1186/s11658-019-0193-6
3. Jin JO, Puranik N, Bui QT, Yadav D, Lee PC. The ubiquitin system: An emerging therapeutic target for lung cancer. Int J Mol Sci (2021) 22(17):9629. doi: 10.3390/ijms22179629
4. Young MJ, Hsu KC, Lin TE, Chang WC, Hung JJ. The role of ubiquitin-specific peptidases in cancer progression. J Biomed Sci (2019) 26(1):42. doi: 10.1186/s12929-019-0522-0
5. Tang Z, Jiang W, Mao M, Zhao J, Chen J, Cheng N. Deubiquitinase Usp35 modulates ferroptosis in lung cancer Via targeting ferroportin. Clin Trans Med (2021) 11(4):e390. doi: 10.1002/ctm2.390
6. Li P, Huang Z, Wang J, Chen W, Huang J. Ubiquitin-specific peptidase 28 enhances Stat3 signaling and promotes cell growth in non-Small-Cell lung cancer. OncoTargets Ther (2019) 12:1603–11. doi: 10.2147/ott.S194917
7. Park HB, Hwang S, Baek KH. Usp7 regulates the Erk1/2 signaling pathway through deubiquitinating raf-1 in lung adenocarcinoma. Cell Death Dis (2022) 13(8):698. doi: 10.1038/s41419-022-05136-6
8. Xu P, Xiao H, Yang Q, Hu R, Jiang L, Bi R, et al. The Usp21/Yy1/Snhg16 axis contributes to tumor proliferation, migration, and invasion of non-Small-Cell lung cancer. Exp Mol Med (2020) 52(1):41–55. doi: 10.1038/s12276-019-0356-6
9. Ding F, Bao C, Tian Y, Xiao H, Wang M, Xie X, et al. Usp22 promotes nsclc tumorigenesis Via mdmx up-regulation and subsequent P53 inhibition. Int J Mol Sci (2014) 16(1):307–20. doi: 10.3390/ijms16010307
10. Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer (2018) 18(2):69–88. doi: 10.1038/nrc.2017.105
11. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature (2003) 426(6968):895–9. doi: 10.1038/nature02263
12. Ye P, Chi X, Cha JH, Luo S, Yang G, Yan X, et al. Potential of E3 ubiquitin ligases in cancer immunity: Opportunities and challenges. Cells (2021) 10(12):3309. doi: 10.3390/cells10123309
13. Liu J, Cheng Y, Zheng M, Yuan B, Wang Z, Li X, et al. Targeting the Ubiquitination/Deubiquitination process to regulate immune checkpoint pathways. Signal Transduct Targeted Ther (2021) 6(1):28. doi: 10.1038/s41392-020-00418-x
14. Zhang W, Sidhu SS. Development of inhibitors in the ubiquitination cascade. FEBS Lett (2014) 588(2):356–67. doi: 10.1016/j.febslet.2013.11.003
15. Morrow JK, Lin HK, Sun SC, Zhang S. Targeting ubiquitination for cancer therapies. Future Med Chem (2015) 7(17):2333–50. doi: 10.4155/fmc.15.148
16. Zheng N, Shabek N. Ubiquitin ligases: Structure, function, and regulation. Annu Rev Biochem (2017) 86:129–57. doi: 10.1146/annurev-biochem-060815-014922
17. Toma-Fukai S, Shimizu T. Structural diversity of ubiquitin E3 ligase. Molecules (2021) 26(21):6682. doi: 10.3390/molecules26216682
18. Daks A, Petukhov A, Fedorova O, Shuvalov O, Merkulov V, Vasileva E, et al. E3 ubiquitin ligase Pirh2 enhances tumorigenic properties of human non-small cell lung carcinoma cells. Genes Cancer (2016) 7(11-12):383–93. doi: 10.18632/genesandcancer.123
19. Ekambaram P, Lee JL, Hubel NE, Hu D, Yerneni S, Campbell PG, et al. The Carma3-Bcl10-Malt1 signalosome drives nfκb activation and promotes aggressiveness in angiotensin ii receptor-positive breast cancer. Cancer Res (2018) 78(5):1225–40. doi: 10.1158/0008-5472.Can-17-1089
20. Li Y, Chen X, Wang Z, Zhao D, Chen H, Chen W, et al. The Hectd3 E3 ubiquitin ligase suppresses cisplatin-induced apoptosis Via stabilizing Malt1. Neoplasia (2013) 15(1):39–48. doi: 10.1593/neo.121362
21. Li K, Zheng X, Tang H, Zang YS, Zeng C, Liu X, et al. E3 ligase Mkrn3 is a tumor suppressor regulating Pabpc1 ubiquitination in non-small cell lung cancer. J Exp Med (2021) 218(8):e20210151. doi: 10.1084/jem.20210151
22. Sun T, Liu Z, Yang Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol Cancer (2020) 19(1):146. doi: 10.1186/s12943-020-01262-x
23. Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat Rev Drug Discov (2018) 17(1):57–78. doi: 10.1038/nrd.2017.152
24. Clague MJ, Urbe S, Komander D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol (2019) 20(6):338–52. doi: 10.1038/s41580-019-0099-1
25. Mennerich D, Kubaichuk K, Kietzmann T. Dubs, hypoxia, and cancer. Trends Cancer (2019) 5(10):632–53. doi: 10.1016/j.trecan.2019.08.005
26. Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbe S. Deubiquitylases from genes to organism. Physiol Rev (2013) 93(3):1289–315. doi: 10.1152/physrev.00002.2013
27. Ambroggio XI, Rees DC, Deshaies RJ. Jamm: A metalloprotease-like zinc site in the proteasome and signalosome. PloS Biol (2004) 2(1):E2. doi: 10.1371/journal.pbio.0020002
28. Cruz L, Soares P, Correia M. Ubiquitin-specific proteases: Players in cancer cellular processes. Pharm (Basel Switzerland) (2021) 14(9):848. doi: 10.3390/ph14090848
29. Li Y, Reverter D. Molecular mechanisms of dubs regulation in signaling and disease. Int J Mol Sci (2021) 22(3):986. doi: 10.3390/ijms22030986
30. Komander D, Clague MJ, Urbé S. Breaking the chains: Structure and function of the deubiquitinases. Nat Rev Mol Cell Biol (2009) 10(8):550–63. doi: 10.1038/nrm2731
31. Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem J (2006) 399(3):361–72. doi: 10.1042/bj20061138
32. Han X, Wei W, Sun Y. Protac degraders with ligands recruiting Mdm2 E3 ubiquitin ligase: An updated perspective. Acta Mater Med (2022) 1(2):244–59. doi: 10.15212/amm-2022-0010
33. Hu Z, Crews CM. Recent developments in protac-mediated protein degradation: From bench to clinic. Chembiochem (2022) 23(2):e202100270. doi: 10.1002/cbic.202100270
34. Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: Expanding the toolbox. Nat Rev Drug Discov (2019) 18(12):949–63. doi: 10.1038/s41573-019-0047-y
35. Nieto-Jiménez C, Morafraile EC, Alonso-Moreno C, Ocaña A. Clinical considerations for the design of protacs in cancer. Mol Cancer (2022) 21(1):67. doi: 10.1186/s12943-022-01535-7
36. Li X, Song Y. Proteolysis-targeting chimera (Protac) for targeted protein degradation and cancer therapy. J Hematol Oncol (2020) 13(1):50. doi: 10.1186/s13045-020-00885-3
37. Zeng S, Huang W, Zheng X, Liyan C, Zhang Z, Wang J, et al. Proteolysis targeting chimera (Protac) in drug discovery paradigm: Recent progress and future challenges. Eur J Med Chem (2021) 210:112981. doi: 10.1016/j.ejmech.2020.112981
38. Jia X, Han X. Targeting androgen receptor degradation with protacs from bench to bedside. Biomed pharmacother (2023) 158:114112. doi: 10.1016/j.biopha.2022.114112
39. Churcher I. Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones? J med Chem (2018) 61(2):444–52. doi: 10.1021/acs.jmedchem.7b01272
40. Atilaw Y, Poongavanam V, Svensson Nilsson C, Nguyen D, Giese A, Meibom D, et al. Solution conformations shed light on protac cell permeability. ACS Med Chem Lett (2021) 12(1):107–14. doi: 10.1021/acsmedchemlett.0c00556
41. Békés M, Langley DR, Crews CM. Protac targeted protein degraders: The past is prologue. Nat Rev Drug Discov (2022) 21(3):181–200. doi: 10.1038/s41573-021-00371-6
42. Faesen AC, Luna-Vargas MP, Sixma TK. The role of ubl domains in ubiquitin-specific proteases. Biochem Soc Trans (2012) 40(3):539–45. doi: 10.1042/BST20120004
43. Xing C, Lu XX, Guo PD, Shen T, Zhang S, He XS, et al. Ubiquitin-specific protease 4-mediated deubiquitination and stabilization of prl-3 is required for potentiating colorectal oncogenesis. Cancer Res (2016) 76(1):83–95. doi: 10.1158/0008-5472.Can-14-3595
44. Zhang L, Zhou F, Drabsch Y, Gao R, Snaar-Jagalska BE, Mickanin C, et al. Usp4 is regulated by akt phosphorylation and directly deubiquitylates tgf-B type I receptor. Nat Cell Biol (2012) 14(7):717–26. doi: 10.1038/ncb2522
45. Heo MJ, Kim YM, Koo JH, Yang YM, An J, Lee SK, et al. Microrna-148a dysregulation discriminates poor prognosis of hepatocellular carcinoma in association with Usp4 overexpression. Oncotarget (2014) 5(9):2792–806. doi: 10.18632/oncotarget.1920
46. Xiao N, Li H, Luo J, Wang R, Chen H, Chen J, et al. Ubiquitin-specific protease 4 (Usp4) targets Traf2 and Traf6 for deubiquitination and inhibits tnfalpha-induced cancer cell migration. Biochem J (2012) 441(3):979–86. doi: 10.1042/BJ20111358
47. Zhang X, Berger FG, Yang J, Lu X. Usp4 inhibits P53 through deubiquitinating and stabilizing arf-Bp1. EMBO J (2011) 30(11):2177–89. doi: 10.1038/emboj.2011.125
48. Hou X, Wang L, Zhang L, Pan X, Zhao W. Ubiquitin-specific protease 4 promotes tnf-A-Induced apoptosis by deubiquitination of Rip1 in head and neck squamous cell carcinoma. FEBS Lett (2013) 587(4):311–6. doi: 10.1016/j.febslet.2012.12.016
49. Kim RQ, van Dijk WJ, Sixma TK. Structure of Usp7 catalytic domain and three ubl-domains reveals a connector A-helix with regulatory role. J Struct Biol (2016) 195(1):11–8. doi: 10.1016/j.jsb.2016.05.005
50. Pozhidaeva A, Bezsonova I. Usp7: Structure, substrate specificity, and inhibition. DNA Repair (2019) 76:30–9. doi: 10.1016/j.dnarep.2019.02.005
51. Kim RQ, Sixma TK. Regulation of Usp7: A high incidence of E3 complexes. J Mol Biol (2017) 429(22):3395–408. doi: 10.1016/j.jmb.2017.05.028
52. Al-Eidan A, Wang Y, Skipp P, Ewing RM. The Usp7 protein interaction network and its roles in tumorigenesis. Genes Dis (2022) 9(1):41–50. doi: 10.1016/j.gendis.2020.10.004
53. Laxmi A, Gupta P, Gupta J. Ccdc6, a gene product in fusion with different protoncogenes, as a potential chemotherapeutic target. Cancer Biomarkers: Section A Dis Markers (2019) 24(4):383–93. doi: 10.3233/cbm-181601
54. Morra F, Luise C, Merolla F, Poser I, Visconti R, Ilardi G, et al. Fbxw7 and Usp7 regulate Ccdc6 turnover during the cell cycle and affect cancer drugs susceptibility in nsclc. Oncotarget (2015) 6(14):12697–709. doi: 10.18632/oncotarget.3708
55. Zhang C, Lu J, Zhang QW, Zhao W, Guo JH, Liu SL, et al. Usp7 promotes cell proliferation through the stabilization of ki-67 protein in non-small cell lung cancer cells. Int J Biochem Cell Biol (2016) 79:209–21. doi: 10.1016/j.biocel.2016.08.025
56. Li J, Han Y, Zhang H, Qian Z, Jia W, Gao Y, et al. The M6a demethylase fto promotes the growth of lung cancer cells by regulating the M6a level of Usp7 mrna. Biochem Biophys Res Commun (2019) 512(3):479–85. doi: 10.1016/j.bbrc.2019.03.093
57. Morotti A, Panuzzo C, Crivellaro S, Pergolizzi B, Familiari U, Berger AH, et al. Bcr-abl disrupts pten nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of hausp. Leukemia (2014) 28(6):1326–33. doi: 10.1038/leu.2013.370
58. Noguera NI, Song MS, Divona M, Catalano G, Calvo KL, Garcia F, et al. Nucleophosmin/B26 regulates pten through interaction with hausp in acute myeloid leukemia. Leukemia (2013) 27(5):1037–43. doi: 10.1038/leu.2012.314
59. Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, et al. The deubiquitinylation and localization of pten are regulated by a hausp-pml network. Nature (2008) 455(7214):813–7. doi: 10.1038/nature07290
60. Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of P53 by regulating the deubiquitination activity of Usp10 and Usp13. Cell (2011) 147(1):223–34. doi: 10.1016/j.cell.2011.08.037
61. Sun J, Li T, Zhao Y, Huang L, Sun H, Wu H, et al. Usp10 inhibits lung cancer cell growth and invasion by upregulating pten. Mol Cell Biochem (2018) 441(1-2):1–7. doi: 10.1007/s11010-017-3170-2
62. Ko A, Han SY, Choi CH, Cho H, Lee MS, Kim SY, et al. Oncogene-induced senescence mediated by c-myc requires Usp10 dependent deubiquitination and stabilization of P14arf. Cell Death Differ (2018) 25(6):1050–62. doi: 10.1038/s41418-018-0072-0
63. Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. Usp10 regulates P53 localization and stability by deubiquitinating P53. Cell (2010) 140(3):384–96. doi: 10.1016/j.cell.2009.12.032
64. Weisberg EL, Schauer NJ, Yang J, Lamberto I, Doherty L, Bhatt S, et al. Inhibition of Usp10 induces degradation of oncogenic Flt3. Nat Chem Biol (2017) 13(12):1207–15. doi: 10.1038/nchembio.2486
65. Chen Q, Hang Y, Zhang T, Tan L, Li S, Jin Y. Usp10 promotes proliferation and migration and inhibits apoptosis of endometrial stromal cells in endometriosis through activating the raf-1/Mek/Erk pathway. Am J Physiol Cell Physiol (2018) 315(6):C863–c72. doi: 10.1152/ajpcell.00272.2018
66. Ouyang SW, Liu TT, Liu XS, Zhu FX, Zhu FM, Liu XN, et al. Usp10 regulates musashi-2 stability Via deubiquitination and promotes tumour proliferation in colon cancer. FEBS Lett (2019) 593(4):406–13. doi: 10.1002/1873-3468.13323
67. Shi J, Zhang Q, Yin X, Ye J, Gao S, Chen C, et al. Stabilization of Igf2bp1 by Usp10 promotes breast cancer metastasis Via Cpt1a in an M6a-dependent manner. Int J Biol Sci (2023) 19(2):449–64. doi: 10.7150/ijbs.76798
68. Tong CW, Wang JL, Jiang MS, Hsu CH, Chang WT, Huang AM. Novel genes that mediate nuclear respiratory factor 1-regualted neurite outgrowth in neuroblastoma imr-32 cells. Gene (2013) 515(1):62–70. doi: 10.1016/j.gene.2012.11.026
69. Zeng Z, Wu HX, Zhan N, Huang YB, Wang ZS, Yang GF, et al. Prognostic significance of Usp10 as a tumor-associated marker in gastric carcinoma. Tumour Biol (2014) 35(4):3845–53. doi: 10.1007/s13277-013-1509-1
70. Zeng Z, Li D, Yu T, Huang Y, Yan H, Gu L, et al. Association and clinical implication of the Usp10 and Msh2 proteins in non-small cell lung cancer. Oncol Lett (2019) 17(1):1128–38. doi: 10.3892/ol.2018.9702
71. He Y, Jiang S, Mao C, Zheng H, Cao B, Zhang Z, et al. The deubiquitinase Usp10 restores pten activity and inhibits non-small cell lung cancer cell proliferation. J Biol Chem (2021) 297(3):101088. doi: 10.1016/j.jbc.2021.101088
72. Hu J, Liu YL, Piao SL, Yang DD, Yang YM, Cai L. Expression patterns of Usp22 and potential targets bmi-1, pten, p-akt in non-Small-Cell lung cancer. Lung Cancer (Amsterdam Netherlands) (2012) 77(3):593–9. doi: 10.1016/j.lungcan.2012.05.112
73. Liu YL, Yang YM, Xu H, Dong XS. Aberrant expression of Usp22 is associated with liver metastasis and poor prognosis of colorectal cancer. J Surg Oncol (2011) 103(3):283–9. doi: 10.1002/jso.21802
74. Yang DD, Cui BB, Sun LY, Zheng HQ, Huang Q, Tong JX, et al. The Co-expression of Usp22 and bmi-1 may promote cancer progression and predict therapy failure in gastric carcinoma. Cell Biochem Biophys (2011) 61(3):703–10. doi: 10.1007/s12013-011-9229-x
75. Guo J, Zhao J, Fu W, Xu Q, Huang D. Immune evasion and drug resistance mediated by Usp22 in cancer: Novel targets and mechanisms. Front Immunol (2022) 13:918314. doi: 10.3389/fimmu.2022.918314
76. Han B, Sun Y, Yang D, Zhang H, Mo S, Chen X, et al. Usp22 promotes development of lung adenocarcinoma through ubiquitination and immunosuppression. Aging (2020) 12(8):6990–7005. doi: 10.18632/aging.103056
77. Ling S, Shan Q, Zhan Q, Ye Q, Liu P, Xu S, et al. Usp22 promotes hypoxia-induced hepatocellular carcinoma stemness by a Hif1α/Usp22 positive feedback loop upon Tp53 inactivation. Gut (2020) 69(7):1322–34. doi: 10.1136/gutjnl-2019-319616
78. Wu Y, Qin J, Li F, Yang C, Li Z, Zhou Z, et al. Usp3 promotes breast cancer cell proliferation by deubiquitinating Klf5. J Biol Chem (2019) 294(47):17837–47. doi: 10.1074/jbc.RA119.009102
79. Liao XH, Wang Y, Zhong B, Zhu SY. Usp3 promotes proliferation of non-small cell lung cancer through regulating Rbm4. Eur Rev Med Pharmacol Sci (2020) 24(6):3143–51. doi: 10.26355/eurrev_202003_20681
80. Zhang Z, Cui Z, Xie Z, Li C, Xu C, Guo X, et al. Deubiquitinase Usp5 promotes non-small cell lung cancer cell proliferation by stabilizing cyclin D1. Trans Lung Cancer Res (2021) 10(10):3995–4011. doi: 10.21037/tlcr-21-767
81. Ma X, Qi W, Pan H, Yang F, Deng J. Overexpression of Usp5 contributes to tumorigenesis in non-small cell lung cancer Via the stabilization of B-catenin protein. Am J Cancer Res (2018) 8(11):2284–95.
82. Wu N, Liu C, Bai C, Han YP, Cho WC, Li Q. Over-expression of deubiquitinating enzyme Usp14 in lung adenocarcinoma promotes proliferation through the accumulation of B-catenin. Int J Mol Sci (2013) 14(6):10749–60. doi: 10.3390/ijms140610749
83. McFarlane C, McFarlane S, Paul I, Arthur K, Scheaff M, Kerr K, et al. The deubiquitinating enzyme Usp17 is associated with non-small cell lung cancer (Nsclc) recurrence and metastasis. Oncotarget (2013) 4(10):1836–43. doi: 10.18632/oncotarget.1282
84. Rossi FA, Enriqué Steinberg JH, Calvo Roitberg EH, Joshi MU, Pandey A, Abba MC, et al. Usp19 modulates cancer cell migration and invasion and acts as a novel prognostic marker in patients with early breast cancer. Oncogenesis (2021) 10(3):28. doi: 10.1038/s41389-021-00318-x
85. Hou P, Ma X, Zhang Q, Wu CJ, Liao W, Li J, et al. Usp21 deubiquitinase promotes pancreas cancer cell stemness via wnt pathway activation. Genes Dev (2019) 33(19-20):1361–6. doi: 10.1101/gad.326314.119
86. Zhang L, Xu B, Qiang Y, Huang H, Wang C, Li D, et al. Overexpression of deubiquitinating enzyme Usp28 promoted non-small cell lung cancer growth. J Cell Mol Med (2015) 19(4):799–805. doi: 10.1111/jcmm.12426
87. Sun Y, Su B, Zhang P, Xie H, Zheng H, Xu Y, et al. Expression of mir-150 and mir-3940-5p is reduced in non-small cell lung carcinoma and correlates with clinicopathological features. Oncol Rep (2013) 29(2):704–12. doi: 10.3892/or.2012.2152
88. Ren K, Li Y, Lu H, Li Z, Han X. Mir-3940-5p functions as a tumor suppressor in non-small cell lung cancer cells by targeting cyclin D1 and ubiquitin specific peptidase-28. Trans Oncol (2017) 10(1):80–9. doi: 10.1016/j.tranon.2016.11.004
89. Ruiz EJ, Pinto-Fernandez A, Turnbull AP, Lan L, Charlton TM, Scott HC, et al. Usp28 deletion and small-molecule inhibition destabilizes c-myc and elicits regression of squamous cell lung carcinoma. eLife (2021) 10:e71596. doi: 10.7554/eLife.71596
90. Qian W, Li Q, Wu X, Li W, Li Q, Zhang J, et al. Deubiquitinase Usp29 promotes gastric cancer cell migration by cooperating with phosphatase Scp1 to stabilize snail protein. Oncogene (2020) 39(44):6802–15. doi: 10.1038/s41388-020-01471-0
91. Wu Y, Zhang Y, Wang D, Zhang Y, Zhang J, Zhang Y, et al. Usp29 enhances chemotherapy-induced stemness in non-small cell lung cancer Via stabilizing Snail1 in response to oxidative stress. Cell Death Dis (2020) 11(9):796. doi: 10.1038/s41419-020-03008-5
92. He L, Liu X, Yang J, Li W, Liu S, Liu X, et al. Imbalance of the reciprocally inhibitory loop between the ubiquitin-specific protease Usp43 and Egfr/Pi3k/Akt drives breast carcinogenesis. Cell Res (2018) 28(9):934–51. doi: 10.1038/s41422-018-0079-6
93. Li J, Wang Y, Luo Y, Liu Y, Yi Y, Li J, et al. Usp5-beclin 1 axis overrides P53-dependent senescence and drives kras-induced tumorigenicity. Nat Commun (2022) 13(1):7799. doi: 10.1038/s41467-022-35557-y
94. Wang T, Jing B, Sun B, Liao Y, Song H, Xu D, et al. Stabilization of ptges by deubiquitinase Usp9x promotes metastatic features of lung cancer Via Pge(2) signaling. Am J Cancer Res (2019) 9(6):1145–60.
95. Wang X, Xia S, Li H, Wang X, Li C, Chao Y, et al. The deubiquitinase Usp10 regulates Klf4 stability and suppresses lung tumorigenesis. Cell Death Differ (2020) 27(6):1747–64. doi: 10.1038/s41418-019-0458-7
96. Wang W, Wang M, Xiao Y, Wang Y, Ma L, Guo L, et al. Usp35 mitigates endoplasmic reticulum stress-induced apoptosis by stabilizing Rrbp1 in non-small cell lung cancer. Mol Oncol (2022) 16(7):1572–90. doi: 10.1002/1878-0261.13112
97. Li J, Xiao X, Wang H, Wang W, Ou Y, Wang Z, et al. Cdk4/6-Usp51 axis regulates lung adenocarcinoma metastasis through Zeb1. Cancer Gene Ther (2022) 29(8-9):1181–92. doi: 10.1038/s41417-021-00420-7
98. Zhao X, Wu X, Wang H, Yu H, Wang J. Usp53 promotes apoptosis and inhibits glycolysis in lung adenocarcinoma through Fkbp51-Akt1 signaling. Mol Carcinog (2020) 59(8):1000–11. doi: 10.1002/mc.23230
99. Tan AC. Targeting the Pi3k/Akt/Mtor pathway in non-small cell lung cancer (Nsclc). Thorac Cancer (2020) 11(3):511–8. doi: 10.1111/1759-7714.13328
100. Avery TY, Köhler N, Zeiser R, Brummer T, Ruess DA. Onco-immunomodulatory properties of pharmacological interference with ras-Raf-Mek-Erk pathway hyperactivation. Front Oncol (2022) 12:931774. doi: 10.3389/fonc.2022.931774
101. Wang R, Liu J, Li K, Yang G, Chen S, Wu J, et al. An Setd1a/Wnt/B-catenin feedback loop promotes nsclc development. J Exp Clin Cancer res: CR (2021) 40(1):318. doi: 10.1186/s13046-021-02119-x
102. Liu W, Wang H, Bai F, Ding L, Huang Y, Lu C, et al. Il-6 promotes metastasis of non-Small-Cell lung cancer by up-regulating Tim-4 Via nf-Kb. Cell Prolif (2020) 53(3):e12776. doi: 10.1111/cpr.12776
103. Andjelkovic T, Bankovic J, Stojsic J, Milinkovic V, Podolski-Renic A, Ruzdijic S, et al. Coalterations of P53 and pten tumor suppressor genes in non-small cell lung carcinoma patients. Trans Res: J Lab Clin Med (2011) 157(1):19–28. doi: 10.1016/j.trsl.2010.09.004
104. Hanawa M, Suzuki S, Dobashi Y, Yamane T, Kono K, Enomoto N, et al. Egfr protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int J Cancer (2006) 118(5):1173–80. doi: 10.1002/ijc.21454
105. Lee HJ, Seo AN, Kim EJ, Jang MH, Kim YJ, Kim JH, et al. Prognostic and predictive values of egfr overexpression and egfr copy number alteration in Her2-positive breast cancer. Br J Cancer (2015) 112(1):103–11. doi: 10.1038/bjc.2014.556
106. Maiti GP, Mondal P, Mukherjee N, Ghosh A, Ghosh S, Dey S, et al. Overexpression of egfr in head and neck squamous cell carcinoma is associated with inactivation of Sh3gl2 and Cdc25a genes. PloS One (2013) 8(5):e63440. doi: 10.1371/journal.pone.0063440
107. Araújo A, Ribeiro R, Azevedo I, Coelho A, Soares M, Sousa B, et al. Genetic polymorphisms of the epidermal growth factor and related receptor in non-small cell lung cancer–a review of the literature. Oncologist (2007) 12(2):201–10. doi: 10.1634/theoncologist.12-2-201
108. Gelatti ACZ, Drilon A, Santini FC. Optimizing the sequencing of tyrosine kinase inhibitors (Tkis) in epidermal growth factor receptor (Egfr) mutation-positive non-small cell lung cancer (Nsclc). Lung Cancer (Amsterdam Netherlands) (2019) 137:113–22. doi: 10.1016/j.lungcan.2019.09.017
109. Beyett TS, To C, Heppner DE, Rana JK, Schmoker AM, Jang J, et al. Molecular basis for cooperative binding and synergy of atp-site and allosteric egfr inhibitors. Nat Commun (2022) 13(1):2530. doi: 10.1038/s41467-022-30258-y
110. To C, Beyett TS, Jang J, Feng WW, Bahcall M, Haikala HM, et al. An allosteric inhibitor against the therapy-resistant mutant forms of egfr in non-small cell lung cancer. Nat Cancer (2022) 3(4):402–17. doi: 10.1038/s43018-022-00351-8
111. Ciardiello F, Tortora G. Egfr antagonists in cancer treatment. New Engl J Med (2008) 358(11):1160–74. doi: 10.1056/NEJMra0707704
112. Zhang H, Han B, Lu H, Zhao Y, Chen X, Meng Q, et al. Usp22 promotes resistance to egfr-tkis by preventing ubiquitination-mediated egfr degradation in egfr-mutant lung adenocarcinoma. Cancer Lett (2018) 433:186–98. doi: 10.1016/j.canlet.2018.07.002
113. Zhou D, Yu J, Hu Z, Hua F. Trim25 enhances egfr stability and signaling activity to promote lung cancer progression. Acta Pharm Sin (2019) 54(06):1026–35. doi: 10.16438/j.0513-4870.2019-0289
114. Yu X, Cheng M, Lu K, Shen Y, Zhong Y, Liu J, et al. Exploring degradation of mutant and wild-type epidermal growth factor receptors induced by proteolysis-targeting chimeras. J Med Chem (2022) 65(12):8416–43. doi: 10.1021/acs.jmedchem.2c00345
115. Cheng M, Yu X, Lu K, Xie L, Wang L, Meng F, et al. Discovery of potent and selective epidermal growth factor receptor (Egfr) bifunctional small-molecule degraders. J Med Chem (2020) 63(3):1216–32. doi: 10.1021/acs.jmedchem.9b01566
116. Zhao HY, Yang XY, Lei H, Xi XX, Lu SM, Zhang JJ, et al. Discovery of potent small molecule protacs targeting mutant egfr. Eur J Med Chem (2020) 208:112781. doi: 10.1016/j.ejmech.2020.112781
117. Qu X, Liu H, Song X, Sun N, Zhong H, Qiu X, et al. Effective degradation of Egfr(L858r+T790m) mutant proteins by crbn-based protacs through both proteosome and Autophagy/Lysosome degradation systems. Eur J Med Chem (2021) 218:113328. doi: 10.1016/j.ejmech.2021.113328
118. Du Y, Chen Y, Wang Y, Chen J, Lu X, Zhang L, et al. Hjm-561, a potent, selective, and orally bioavailable egfr protac that overcomes osimertinib-resistant egfr triple mutations. Mol Cancer Ther (2022) 21(7):1060–6. doi: 10.1158/1535-7163.Mct-21-0835
119. Xia P, Xu XY. Pi3k/Akt/Mtor signaling pathway in cancer stem cells: From basic research to clinical application. Am J Cancer Res (2015) 5(5):1602–9.
120. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the Pi3k/Akt pathway for cancer drug discovery. Nat Rev Drug Discov (2005) 4(12):988–1004. doi: 10.1038/nrd1902
121. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the Pi3k/Akt/Mtor signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol (2019) 59:147–60. doi: 10.1016/j.semcancer.2019.05.012
122. Miricescu D, Totan A, Stanescu-Spinu II, Badoiu SC, Stefani C, Greabu M. Pi3k/Akt/Mtor signaling pathway in breast cancer: From molecular landscape to clinical aspects. Int J Mol Sci (2020) 22(1):173. doi: 10.3390/ijms22010173
123. Fattahi S, Amjadi-Moheb F, Tabaripour R, Ashrafi GH, Akhavan-Niaki H. Pi3k/Akt/Mtor signaling in gastric cancer: Epigenetics and beyond. Life Sci (2020) 262:118513. doi: 10.1016/j.lfs.2020.118513
124. Tsai PJ, Lai YH, Manne RK, Tsai YS, Sarbassov D, Lin HK. Akt: A key transducer in cancer. J Biomed Sci (2022) 29(1):76. doi: 10.1186/s12929-022-00860-9
125. Leisner TM, Freeman TC, Black JL, Parise LV. Cib1: A small protein with big ambitions. FASEB J (2016) 30(8):2640–50. doi: 10.1096/fj.201500073R
126. Liu Y, Zhou Y, Zhang P, Li X, Duan C, Zhang C. Chip-mediated Cib1 ubiquitination regulated epithelial-mesenchymal transition and tumor metastasis in lung adenocarcinoma. Cell Death Differ (2021) 28(3):1026–40. doi: 10.1038/s41418-020-00635-5
127. Xu F, Zhang X, Chen Z, He S, Guo J, Yu L, et al. Discovery of isoform-selective Akt3 degraders overcoming osimertinib-induced resistance in non-small cell lung cancer cells. J Med Chem (2022) 65(20):14032–48. doi: 10.1021/acs.jmedchem.2c01246
128. You I, Erickson EC, Donovan KA, Eleuteri NA, Fischer ES, Gray NS, et al. Discovery of an akt degrader with prolonged inhibition of downstream signaling. Cell Chem Biol (2020) 27(1):66–73e7. doi: 10.1016/j.chembiol.2019.11.014
129. Yu X, Xu J, Shen Y, Cahuzac KM, Park KS, Dale B, et al. Discovery of potent, selective, and in vivo efficacious akt kinase protein degraders Via structure-activity relationship studies. J Med Chem (2022) 65(4):3644–66. doi: 10.1021/acs.jmedchem.1c02165
130. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. Erk/Mapk signalling pathway and tumorigenesis. Exp Ther Med (2020) 19(3):1997–2007. doi: 10.3892/etm.2020.8454
131. Rocca A, Braga L, Volpe MC, Maiocchi S, Generali D. The predictive and prognostic role of ras-Raf-Mek-Erk pathway alterations in breast cancer: Revision of the literature and comparison with the analysis of cancer genomic datasets. Cancers (2022) 14(21):5306. doi: 10.3390/cancers14215306
132. Roberts PJ, Der CJ. Targeting the raf-Mek-Erk mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene (2007) 26(22):3291–310. doi: 10.1038/sj.onc.1210422
133. Bigenzahn JW, Collu GM, Kartnig F, Pieraks M, Vladimer GI, Heinz LX, et al. Lztr1 is a regulator of ras ubiquitination and signaling. Sci (New York NY) (2018) 362(6419):1171–7. doi: 10.1126/science.aap8210
134. Reck M, Carbone DP, Garassino M, Barlesi F. Targeting kras in non-Small-Cell lung cancer: Recent progress and new approaches. Ann Oncol (2021) 32(9):1101–10. doi: 10.1016/j.annonc.2021.06.001
135. Kadota K, Sima CS, Arcila ME, Hedvat C, Kris MG, Jones DR, et al. Kras mutation is a significant prognostic factor in early-stage lung adenocarcinoma. Am J Surg Pathol (2016) 40(12):1579–90. doi: 10.1097/pas.0000000000000744
136. Papke B, Der CJ. Drugging ras: Know the enemy. Sci (New York NY) (2017) 355(6330):1158–63. doi: 10.1126/science.aam7622
137. Simanshu DK, Nissley DV, McCormick F. Ras proteins and their regulators in human disease. Cell (2017) 170(1):17–33. doi: 10.1016/j.cell.2017.06.009
138. Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted degradation of oncogenic Kras(G12c) by vhl-recruiting protacs. ACS Cent Sci (2020) 6(8):1367–75. doi: 10.1021/acscentsci.0c00411
139. Yang F, Wen Y, Wang C, Zhou Y, Zhou Y, Zhang ZM, et al. Efficient targeted oncogenic Kras(G12c) degradation Via first reversible-covalent protac. Eur J Med Chem (2022) 230:114088. doi: 10.1016/j.ejmech.2021.114088
140. Wei J, Hu J, Wang L, Xie L, Jin MS, Chen X, et al. Discovery of a first-in-Class mitogen-activated protein kinase kinase 1/2 degrader. J Med Chem (2019) 62(23):10897–911. doi: 10.1021/acs.jmedchem.9b01528
141. Steinhart Z, Angers S. Wnt signaling in development and tissue homeostasis. Dev (Cambridge England) (2018) 145(11):dev146589. doi: 10.1242/dev.146589
142. Nusse R, Clevers H. Wnt/B-catenin signaling, disease, and emerging therapeutic modalities. Cell (2017) 169(6):985–99. doi: 10.1016/j.cell.2017.05.016
143. Zhao H, Ming T, Tang S, Ren S, Yang H, Liu M, et al. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol Cancer (2022) 21(1):144. doi: 10.1186/s12943-022-01616-7
144. Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Institute (2014) 106(1):djt356. doi: 10.1093/jnci/djt356
145. Clevers H, Nusse R. Wnt/B-catenin signaling and disease. Cell (2012) 149(6):1192–205. doi: 10.1016/j.cell.2012.05.012
146. Yang F, Xu J, Li H, Tan M, Xiong X, Sun Y. Fbxw2 suppresses migration and invasion of lung cancer cells Via promoting beta-catenin ubiquitylation and degradation. Nat Commun (2019) 10(1):1382. doi: 10.1038/s41467-019-09289-5
147. Huang T, Zhang Q, Ren W, Yan B, Yi L, Tang T, et al. Usp44 suppresses proliferation and enhances apoptosis in colorectal cancer cells by inactivating the Wnt/Beta-catenin pathway Via Axin1 deubiquitination. Cell Biol Int (2020) 44(8):1651–9. doi: 10.1002/cbin.11358
148. Novellasdemunt L, Foglizzo V, Cuadrado L, Antas P, Kucharska A, Encheva V, et al. Usp7 is a tumor-specific wnt activator for apc-mutated colorectal cancer by mediating B-catenin deubiquitination. Cell Rep (2017) 21(3):612–27. doi: 10.1016/j.celrep.2017.09.072
149. An T, Gong Y, Li X, Kong L, Ma P, Gong L, et al. Usp7 inhibitor P5091 inhibits wnt signaling and colorectal tumor growth. Biochem Pharmacol (2017) 131:29–39. doi: 10.1016/j.bcp.2017.02.011
150. Liao H, Li X, Zhao L, Wang Y, Wang X, Wu Y, et al. A protac peptide induces durable beta-catenin degradation and suppresses wnt-dependent intestinal cancer. Cell Discov (2020) 6:35. doi: 10.1038/s41421-020-0171-1
151. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. Nf-kb in development and progression of human cancer. Virchows Archiv (2005) 446(5):475–82. doi: 10.1007/s00428-005-1264-9
152. Mooney EC, Sahingur SE. The ubiquitin system and A20: Implications in health and disease. J Dental Res (2021) 100(1):10–20. doi: 10.1177/0022034520949486
153. Ha J, Kim M, Seo D, Park JS, Lee J, Lee J, et al. The deubiquitinating enzyme Usp20 regulates the tnfα-induced nf-Kb signaling pathway through stabilization of P62. Int J Mol Sci (2020) 21(9):3116. doi: 10.3390/ijms21093116
154. Fan YH, Yu Y, Mao RF, Tan XJ, Xu GF, Zhang H, et al. Usp4 targets Tak1 to downregulate tnfα-induced nf-Kb activation. Cell Death Differ (2011) 18(10):1547–60. doi: 10.1038/cdd.2011.11
155. Yang Z, Xian H, Hu J, Tian S, Qin Y, Wang RF, et al. Usp18 negatively regulates nf-kappab signaling by targeting Tak1 and nemo for deubiquitination through distinct mechanisms. Sci Rep (2015) 5:12738. doi: 10.1038/srep12738
156. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discovery (2009) 8(8):627–44. doi: 10.1038/nrd2926
157. Zito CR, Jilaveanu LB, Anagnostou V, Rimm D, Bepler G, Maira SM, et al. Multi-level targeting of the phosphatidylinositol-3-Kinase pathway in non-small cell lung cancer cells. PloS One (2012) 7(2):e31331. doi: 10.1371/journal.pone.0031331
158. Lee YR, Chen M, Pandolfi PP. The functions and regulation of the pten tumour suppressor: New modes and prospects. Nat Rev Mol Cell Biol (2018) 19(9):547–62. doi: 10.1038/s41580-018-0015-0
159. Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, et al. Pten expression in non-Small-Cell lung cancer: Evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol (2005) 36(7):768–76. doi: 10.1016/j.humpath.2005.05.006
160. Chen L, Liu S, Tao Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct Targeted Ther (2020) 5(1):90. doi: 10.1038/s41392-020-0196-9
161. Cai J, Li R, Xu X, Zhang L, Lian R, Fang L, et al. Ck1α suppresses lung tumour growth by stabilizing pten and inducing autophagy. Nat Cell Biol (2018) 20(4):465–78. doi: 10.1038/s41556-018-0065-8
162. Yuan L, Lv Y, Li H, Gao H, Song S, Zhang Y, et al. Deubiquitylase Otud3 regulates pten stability and suppresses tumorigenesis. Nat Cell Biol (2015) 17(9):1169–81. doi: 10.1038/ncb3218
163. Du T, Li H, Fan Y, Yuan L, Guo X, Zhu Q, et al. The deubiquitylase Otud3 stabilizes Grp78 and promotes lung tumorigenesis. Nat Commun (2019) 10(1):2914. doi: 10.1038/s41467-019-10824-7
164. Hassin O, Oren M. Drugging P53 in cancer: One protein, many targets. Nat Rev Drug Discovery (2022) 22(2):127–44. doi: 10.1038/s41573-022-00571-8
165. Chao CC. Mechanisms of P53 degradation. Clin Chim Acta (2015) 438:139–47. doi: 10.1016/j.cca.2014.08.015
166. Zhang J, Cao M, Dong J, Li C, Xu W, Zhan Y, et al. Abro1 suppresses tumourigenesis and regulates the DNA damage response by stabilizing P53. Nat Commun (2014) 5:5059. doi: 10.1038/ncomms6059
167. Zou Q, Jin J, Hu H, Li HS, Romano S, Xiao Y, et al. Usp15 stabilizes Mdm2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat Immunol (2014) 15(6):562–70. doi: 10.1038/ni.2885
168. Allende-Vega N, Sparks A, Lane DP, Saville MK. Mdmx is a substrate for the deubiquitinating enzyme Usp2a. Oncogene (2010) 29(3):432–41. doi: 10.1038/onc.2009.330
169. He S, Ma J, Fang Y, Liu Y, Wu S, Dong G, et al. Homo-protac mediated suicide of Mdm2 to treat non-small cell lung cancer. Acta Pharm Sin B (2021) 11(6):1617–28. doi: 10.1016/j.apsb.2020.11.022
170. Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: A paradigm for precision cancer medicine. Clin Cancer Res (2015) 21(10):2227–35. doi: 10.1158/1078-0432.Ccr-14-2791
171. Zhang SS, Nagasaka M, Zhu VW, Ou SI. Going beneath the tip of the iceberg. identifying and understanding Eml4-alk variants and Tp53 mutations to optimize treatment of alk fusion positive (Alk+) nsclc. Lung Cancer (Amsterdam Netherlands) (2021) 158:126–36. doi: 10.1016/j.lungcan.2021.06.012
172. Qiao M, Zhao C, Liu Q, Wang Y, Shi J, Ng TL, et al. Impact of alk variants on brain metastasis and treatment response in advanced nsclc patients with oncogenic alk fusion. Trans Lung Cancer Res (2020) 9(4):1452–63. doi: 10.21037/tlcr-19-346
173. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-Small-Cell lung cancer who harbor Eml4-alk. J Clin Oncol (2009) 27(26):4247–53. doi: 10.1200/jco.2009.22.6993
174. Powell CE, Gao Y, Tan L, Donovan KA, Nowak RP, Loehr A, et al. Chemically induced degradation of anaplastic lymphoma kinase (Alk). J med Chem (2018) 61(9):4249–55. doi: 10.1021/acs.jmedchem.7b01655
175. Zhang C, Han XR, Yang X, Jiang B, Liu J, Xiong Y, et al. Proteolysis targeting chimeras (Protacs) of anaplastic lymphoma kinase (Alk). Eur J Med Chem (2018) 151:304–14. doi: 10.1016/j.ejmech.2018.03.071
176. Xie S, Sun Y, Liu Y, Li X, Li X, Zhong W, et al. Development of alectinib-based protacs as novel potent degraders of anaplastic lymphoma kinase (Alk). J Med Chem (2021) 64(13):9120–40. doi: 10.1021/acs.jmedchem.1c00270
177. Zhang J, Zhang F, Niu R. Functions of Shp2 in cancer. J Cell Mol Med (2015) 19(9):2075–83. doi: 10.1111/jcmm.12618
178. Wang M, Lu J, Wang M, Yang CY, Wang S. Discovery of Shp2-D26 as a first, potent, and effective protac degrader of Shp2 protein. J Med Chem (2020) 63(14):7510–28. doi: 10.1021/acs.jmedchem.0c00471
179. Sulzmaier FJ, Jean C, Schlaepfer DD. Fak in cancer: Mechanistic findings and clinical applications. Nat Rev Cancer (2014) 14(9):598–610. doi: 10.1038/nrc3792
180. Law RP, Nunes J, Chung CW, Bantscheff M, Buda K, Dai H, et al. Discovery and characterisation of highly cooperative fak-degrading protacs. Angewandte Chemie (International ed English) (2021) 60(43):23327–34. doi: 10.1002/anie.202109237
181. Xiao Z, Song S, Chen D, van Merkerk R, van der Wouden PE, Cool RH, et al. Proteolysis targeting chimera (Protac) for macrophage migration inhibitory factor (Mif) has anti-proliferative activity in lung cancer cells. Angewandte Chemie (International ed English) (2021) 60(32):17514–21. doi: 10.1002/anie.202101864
182. Duan Y, Guan Y, Qin W, Zhai X, Yu B, Liu H. Targeting Brd4 for cancer therapy: Inhibitors and degraders. MedChemComm (2018) 9(11):1779–802. doi: 10.1039/c8md00198g
183. Zhang HT, Peng R, Chen S, Shen A, Zhao L, Tang W, et al. Versatile nano-Protac-Induced epigenetic reader degradation for efficient lung cancer therapy. Advanced Sci (Weinheim Baden-Wurttemberg Germany) (2022) 9(29):e2202039. doi: 10.1002/advs.202202039
184. Castella M, Jacquemont C, Thompson EL, Yeo JE, Cheung RS, Huang JW, et al. Fanci regulates recruitment of the fa core complex at sites of DNA damage independently of Fancd2. PloS Genet (2015) 11(10):e1005563. doi: 10.1371/journal.pgen.1005563
185. Dexheimer TS, Rosenthal AS, Luci DK, Liang Q, Villamil MA, Chen J, et al. Synthesis and structure-activity relationship studies of n-Benzyl-2-Phenylpyrimidin-4-Amine derivatives as potent Usp1/Uaf1 deubiquitinase inhibitors with anticancer activity against nonsmall cell lung cancer. J med Chem (2014) 57(19):8099–110. doi: 10.1021/jm5010495
186. Wang X, Bao Y, Dong Z, Chen Q, Guo H, Ziang C, et al. Wp1130 attenuates cisplatin resistance by decreasing P53 expression in non-small cell lung carcinomas. Oncotarget (2017) 8(30):49033–43. doi: 10.18632/oncotarget.16931
187. Moghadami A-A, Aboutalebi Vand Beilankouhi E, Kalantary-Charvadeh A, Hamzavi M, Mosayyebi B, Sedghi H, et al. Inhibition of Usp14 induces er stress-mediated autophagy without apoptosis in lung cancer cell line A549. Cell Stress chaperones (2020) 25(6):909–17. doi: 10.1007/s12192-020-01125-w
188. Han KH, Kwak M, Lee TH, Park MS, Jeong IH, Kim MJ, et al. Usp14 inhibition regulates tumorigenesis by inducing autophagy in lung cancer in vitro. Int J Mol Sci (2019) 20(21):5300. doi: 10.3390/ijms20215300
189. Magiera K, Tomala M, Kubica K, De Cesare V, Trost M, Zieba BJ, et al. Lithocholic acid hydroxyamide destabilizes cyclin D1 and induces G(0)/G(1) arrest by inhibiting deubiquitinase Usp2a. Cell Chem Biol (2017) 24(4):458–70.e18. doi: 10.1016/j.chembiol.2017.03.002
190. Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL, et al. Molecular basis of Usp7 inhibition by selective small-molecule inhibitors. Nature (2017) 550(7677):481–6. doi: 10.1038/nature24451
191. Fan YH, Cheng J, Vasudevan SA, Dou J, Zhang H, Patel RH, et al. Usp7 inhibitor P22077 inhibits neuroblastoma growth Via inducing P53-mediated apoptosis. Cell Death Dis (2013) 4(10):e867. doi: 10.1038/cddis.2013.400
192. Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule Wp1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res (2010) 70(22):9265–76. doi: 10.1158/0008-5472.Can-10-1530
193. Akiyama H, Umezawa Y, Ishida S, Okada K, Nogami A, Miura O. Inhibition of Usp9x induces apoptosis in Flt3-Itd-Positive aml cells cooperatively by inhibiting the mutant kinase through aggresomal translocation and inducing oxidative stress. Cancer Lett (2019) 453:84–94. doi: 10.1016/j.canlet.2019.03.046
194. He H, Yi L, Zhang B, Yan B, Xiao M, Ren J, et al. Usp24-gsdmb complex promotes bladder cancer proliferation Via activation of the Stat3 pathway. Int J Biol Sci (2021) 17(10):2417–29. doi: 10.7150/ijbs.54442
195. Pal A, Dziubinski M, Di Magliano MP, Simeone DM, Owens S, Thomas D, et al. Usp9x promotes survival in human pancreatic cancer and its inhibition suppresses pancreatic ductal adenocarcinoma in vivo tumor growth. Neoplasia (2018) 20(2):152–64. doi: 10.1016/j.neo.2017.11.007
196. Lu R, Wu G, Chen M, Ji D, Liu Y, Zhou GG, et al. Usp18 and Usp20 restrict ohsv-1 replication in resistant human oral squamous carcinoma cell line Scc9 and affect the viability of Scc9 cells. Mol Ther Oncol (2021) 23:477–87. doi: 10.1016/j.omto.2021.11.004
197. Fricker LD. Proteasome inhibitor drugs. Annu Rev Pharmacol Toxicol (2020) 60:457–76. doi: 10.1146/annurev-pharmtox-010919-023603
198. Ishida T, Ciulli A. E3 ligase ligands for protacs: How they were found and how to discover new ones. SLAS Discov (2021) 26(4):484–502. doi: 10.1177/2472555220965528
199. Li Z, Lim SL, Tao Y, Li X, Xie Y, Yang C, et al. Protac bromodomain inhibitor arv-825 displays anti-tumor activity in neuroblastoma by repressing expression of mycn or c-myc. Front Oncol (2020) 10:574525. doi: 10.3389/fonc.2020.574525
200. Ito T, Handa H. Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs. Int J Hematol (2016) 104(3):293–9. doi: 10.1007/s12185-016-2073-4
201. Cai Z, Moten A, Peng D, Hsu CC, Pan BS, Manne R, et al. The Skp2 pathway: A critical target for cancer therapy. Semin Cancer Biol (2020) 67(Pt 2):16–33. doi: 10.1016/j.semcancer.2020.01.013
202. Li C, Du L, Ren Y, Liu X, Jiao Q, Cui D, et al. Skp2 promotes breast cancer tumorigenesis and radiation tolerance through Pdcd4 ubiquitination. J Exp Clin Cancer res: CR (2019) 38(1):76. doi: 10.1186/s13046-019-1069-3
203. Hines J, Lartigue S, Dong H, Qian Y, Crews CM. Mdm2-recruiting protac offers superior, synergistic antiproliferative activity Via simultaneous degradation of Brd4 and stabilization of P53. Cancer Res (2019) 79(1):251–62. doi: 10.1158/0008-5472.Can-18-2918
204. Qin AC, Jin H, Song Y, Gao Y, Chen YF, Zhou LN, et al. The therapeutic effect of the Brd4-degrading protac A1874 in human colon cancer cells. Cell Death Dis (2020) 11(9):805. doi: 10.1038/s41419-020-03015-6
205. Rew Y, Sun D. Discovery of a small molecule Mdm2 inhibitor (Amg 232) for treating cancer. J Med Chem (2014) 57(15):6332–41. doi: 10.1021/jm500627s
206. Moschos SJ, Sandhu S, Lewis KD, Sullivan RJ, Puzanov I, Johnson DB, et al. Targeting wild-type Tp53 using amg 232 in combination with mapk inhibition in metastatic melanoma; a phase 1 study. Investigational New Drugs (2022) 40(5):1051–65. doi: 10.1007/s10637-022-01253-3
207. Lin X, Xiang H, Luo G. Targeting estrogen receptor A for degradation with protacs: A promising approach to overcome endocrine resistance. Eur J Med Chem (2020) 206:112689. doi: 10.1016/j.ejmech.2020.112689
208. Yan Y, Wang X, Chaput D, Shin MK, Koh Y, Gan L, et al. X-Linked ubiquitin-specific peptidase 11 increases tauopathy vulnerability in women. Cell (2022) 185(21):3913–30.e19. doi: 10.1016/j.cell.2022.09.002
209. Zhang Q, Tang Z, An R, Ye L, Zhong B. Usp29 maintains the stability of cgas and promotes cellular antiviral responses and autoimmunity. Cell Res (2020) 30(10):914–27. doi: 10.1038/s41422-020-0341-6
210. Yan R, Chu J, Zhou Y, Shan W, Hu Y, Lin M, et al. Ubiquitin-specific protease 22 ameliorates chronic alcohol-associated liver disease by regulating Brd4. Pharmacol Res (2021) 168:105594. doi: 10.1016/j.phrs.2021.105594
Keywords: NSCLC, USPs, PROTACs, E3 ligase, signalling pathway
Citation: Yang Y-C, Zhao C-J, Jin Z-F, Zheng J and Ma L-T (2023) Targeted therapy based on ubiquitin-specific proteases, signalling pathways and E3 ligases in non-small-cell lung cancer. Front. Oncol. 13:1120828. doi: 10.3389/fonc.2023.1120828
Received: 10 December 2022; Accepted: 01 February 2023;
Published: 09 March 2023.
Edited by:
Xinying Xue, Beijing Shijitan Hospital, Capital Medical University, ChinaReviewed by:
Xiong Chen, Nanjing General Hospital of Nanjing Military Command, ChinaPengda Liu, University of North Carolina at Chapel Hill, United States
Copyright © 2023 Yang, Zhao, Jin, Zheng and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Tian Ma, bWFsaXRpYW4xMjM0QDE2My5jb20=; Jin Zheng, empkZGxuQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship