 Yongfeng Xu1
Yongfeng Xu1 Qunli Xiong
Qunli Xiong Junhong Han
Junhong Han Qing Zhu
Qing Zhu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 27 April 2023
Sec. Cancer Genetics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1116061
This article is part of the Research Topic Current Understanding of Genomic and Chromosomal Instabilities in Solid Malignancies View all 9 articles
The p53 family is made up of three transcription factors: p53, p63, and p73. These proteins are well-known regulators of cell function and play a crucial role in controlling various processes related to cancer progression, including cell division, proliferation, genomic stability, cell cycle arrest, senescence, and apoptosis. In response to extra- or intracellular stress or oncogenic stimulation, all members of the p53 family are mutated in structure or altered in expression levels to affect the signaling network, coordinating many other pivotal cellular processes. P63 exists as two main isoforms (TAp63 and ΔNp63) that have been contrastingly discovered; the TA and ΔN isoforms exhibit distinguished properties by promoting or inhibiting cancer progression. As such, p63 isoforms comprise a fully mysterious and challenging regulatory pathway. Recent studies have revealed the intricate role of p63 in regulating the DNA damage response (DDR) and its impact on diverse cellular processes. In this review, we will highlight the significance of how p63 isoforms respond to DNA damage and cancer stem cells, as well as the dual role of TAp63 and ΔNp63 in cancer.
In most human malignancies, tumors develop through a series of genetic alterations, in contrast to the normal function of the p53 family gene (1–3); this procedure was identified as a tumor-driven progression to promote invasion, proliferation, cell survival, and drug resistance (4–6). Moreover, owing to the similar structure of p53 family members, p63 shares the function of p53 (e.g., activation of the apoptosis-related signal pathway in response to genome stress) (7–10), and the p53 homolog p63 is also capable of binding to the majority of p53-responsive promoters and initiating transcription (e.g., p21, Bax, MDM2, etc.) in development and homeostasis (11–16). There is increasing evidence that p53 and p63 can modulate resistance to cancer chemotherapy and DNA damage (17–19). Interestingly, some reports revealed that p63 is expressed in two main multiple isoforms, which often play very opposite functions in cancer progression.
TAp63 (a subtype of p63), widely known as a synergistic effector with p53, promotes cancer cell apoptosis after chemotherapy and is involved in cell cycle arrest, apoptosis, and DNA repair (20–23). In contrast, oddly, ΔNp63 (another main subtype of p63) serves more like an oncogene, presenting a phenotype to resist chemotherapy, inducing cell proliferation, and driving stem cell formation (24, 25). In addition, some reports also indicated that TAp63-/- mice have an increasing number of breast hyperplastic cells with highly disordered, polarity defects, resulting in fragile skin, blisters, wounds that never heal, and alopecia. Nevertheless, ΔNp63-/- transgenic mice showed a significantly accelerated keratinocyte differentiation through direct regulation of the Notch signaling pathway. This intricate phenotype relies on the defective proliferation and senility of dermal and epidermal precursors and indicates that both TAp63 and ΔNp63 may play roles in the development of skin stem cells. Why different subtypes of the same molecule play very different molecular and functional different molecular and functional. Hence, in this review, we focus on the latest developments in comprehending the regulatory network through which two p63 subtypes could potentially modulate molecular signaling pathways.
The p53 family members have a series of similar gene frameworks and are constituted by three main domains: an N-terminal transactivation domain (TAD), a central DNA-binding domain (DBD), and an oligomerization domain (OD) (26, 27). Those highly similar homeodomains among p53 family members allow binding transactivation of the same gene promoters. Similar to the structure of p53, p63 is capable of recognizing and binding to the TAD of p53 response elements two or more tandem repeats of RRRCWWGYYY and initiating transcription of various genes (elaborated below), hence replacing a portion of the functions with p53 (such as cell cycle arrest and activation of apoptosis) (28). Unlike p53, p63 has two different promoters: the first promoter drives the transcription of TAp63, while the second promoter triggers the transcriptional activation of ΔNp63 isotypes; therefore, the p63 protein can be divided into two isoforms, depending on the different domains. The TA forms include the TAD, whereas the ΔN isoforms do not (Figure 1) (27).
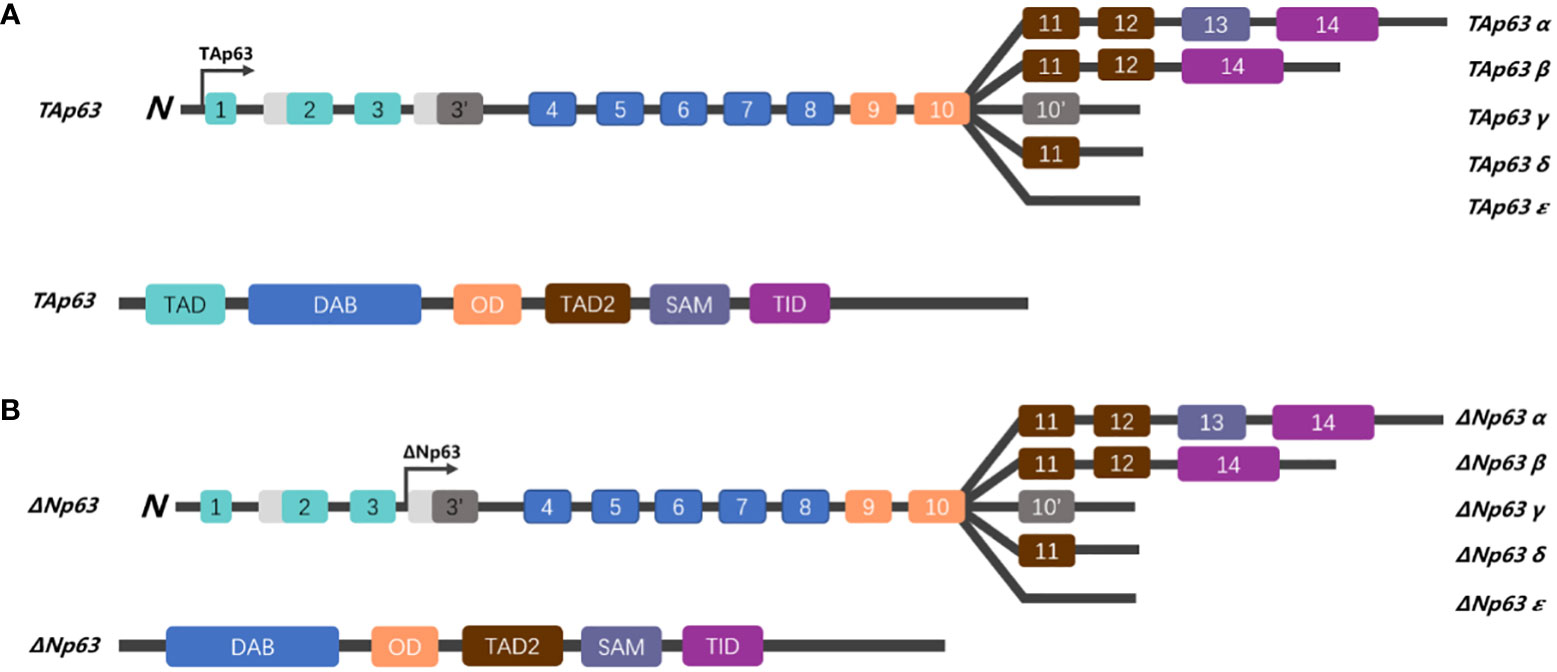
Figure 1 Schematic representation of the gene structure of p63. (A) The human p63 gene is located on chromosome 3q27 and spans over 250 kb, comprising 14 exons. Alternative splicing generates five isoforms (α, β, γ, δ, and ε), which differ in their C-terminus. The TAp63 isoforms, which contain a trans-activating domain (TAD), are encoded by exons 1, 2, and 3. (B) While ΔNp63 isoforms lack the TAD, TAp63 and ΔNp63 share a DNA binding domain (DBD), oligomerization domain (OD), sterile alpha domain (SAM), and a transactivation inhibitory domain (TID) in the C-terminal region.
It is generally understood that the two subtypes of p63 have different functions. Some researchers believe that the TAp63 subtype has a longer acidic N-terminal trans-transcriptional activation region similar to p53, which can undoubtedly transactivate p53-related downstream target genes, arrest the cell cycle, and induce apoptosis, resulting in p53-like biological effects and function as a tumor suppressor (29–31). In contrast, the biological characteristics of ΔNp63 are opposite to those of the TAp63 isomer. ΔNp63 lacks the TAD and, therefore, cannot induce transcription, loses the transactivating function of p53 downstream target genes, and has opposite results to p53-related cell cycle arrest and apoptosis (32–34). Furthermore, both TAp63 and ΔNp63 can compete for the DBD to directly activate or inactivate downstream cell proliferation or apoptosis (such as the p53–p21 signaling pathway, which can inhibit Cyclin E/Cdk2 to mediate cell senescence and restrain cell proliferation) (26, 35–37). In summary, similar to p53, TAp63 plays a role in cancer suppression; in contrast, ΔNp63 plays opposite roles in cell cycle regulation and apoptosis.
The tumor protein p53 is a main transcriptional regulator in multiple significant signaling and programmed cell death pathways, in response to diverse genome stresses, such as DNA mutation, reactive oxygen species (ROS) injury, oncogene activation, or others, affecting a series of cellular processes, including DNA repair, cell cycle arrest, senescence, apoptosis, and differentiation (38–40). However, once the p53 pathway has been hijacked, it will not fulfill the normal obligations of the “guardian of the genome” to induce aging, or apoptosis in response to genotoxic stress. What was worse, mutated p53 does not perform a normal protective function but promotes cell proliferation and anti-apoptosis, causes tumor formation, and even leads to chemotherapy resistance and radiotherapy resistance for cancer survival (6, 41–43). Specifically, according to data from The Cancer Genome Atlas (TCGA) platform, more than half of cancer patients experienced mutation of p53 (44). Most p53 mutations are located within the DBD (96–293 aa) at several hotspots, such as R175, R248, R273, and R282, leading further to “gain of function” (GOF), which plays a significant role in promoting cancer progression and chemoresistance (45–48).
In contrast to the comparatively infrequent mutations of the p63 gene, p53 is often downregulated or mutated in tumors (49). To be specific, more than 95% of p53 mutations lie in the DBD, a mutated hotspot located in the DBD (such as R175H), resulting in structural instability and polarity disorder (50). In fact, when the DBD is mutated, hydrophobic core fragments 251–257 are exposed, resulting in p53 aggregation. Even worse, aggregation of mutant p53 (mut-p53) not only interfered with the transcriptional activity of wild-type p53 (wt-p53) in the nucleus but also congregated to p63 and p73, leading to coprecipitation, misfolding, and loss of normal function of wt-p53, p63, and p73, contributing to tumorigenesis. Interestingly, wt-p53 did not interact detectably with either p63 or p73, but mut-p53 coimmunoprecipitated with wt-p53, p63, and p73, then mut-p53 significantly counteracted the wt-p53/p63-induced growth inhibition (51–55).
Intriguingly, these aggressive behaviors of mut-p53 render us reminiscent of prion-like properties. Similar to prion, the infectious nature of mut-p53 is characterized by (i) mutated p53 attacking the wt-p53 protein, (ii) misfolding and assembly into amyloid granules, (iii) nucleic acid free, and (iv) propagating to other cells like virus (56).
Because of this prion-like behavior, mut-p53 amyloid can possess a “seeding” capacity and transmit to other cells. Once mut-p53 is internalized in other cells, the amount of mut-p53 amyloid seeds can misfold and aggregate with wt-p53/p63/p73 (56), acting similarly to amyloid-associated diseases, such as Alzheimer’s and Parkinson’s disease (57, 58). Meanwhile, numerous studies have established that the aggregation of mut-p53 assembled TAp63/TAp73, and aggregated and inactivated them into perinuclear amyloid oligomers, and this aggregation behavior can be suppressed by treatment with nocodazole, a small chemical that disrupts microtubule assembly (51, 54, 59, 60). This prion-like behavior of oncogenic mut-p53 provides an explanation for its binding and inactivation of TAp63, which is involved in the regulation of progression and apoptosis, and increases the drug resistance and invasion ability of tumors.
The identification of the p21 gene as a target of induction by wt-p53 protein was the first one on record (61). Cyclin E and cyclin A/CDK genes are associated with the p53-dependent cell cycle arrest that occurs at the G1/S transition in response to various factors, such as oncogenes or chemotherapies. However, once wt-p53 is affected by some factors and mutates, mutant p53 is highly susceptible to misfolding, leading to its accumulation as large aggregates inside the cell, which results in the loss of its physiological function as a tumor-suppressor protein.
The amyloid structures of p53 can penetrate cells and trigger the formation of amyloid aggregates of endogenous wt-p53 and TAp63. Loss of the native function of genes can cause genomic instability, which is a critical contributor to cancer development. What is worse, the results confirmed that p53 amyloid can be internalized and has the ability to “seed” the formation of amyloid structures. Once inside the cell, even a small number of mut-p53 amyloid seeds can promote the formation of amyloid aggregates of wt-p53 and TAp63, in a manner akin to prions. This templating ability of p53 fibrillar seeds renders wt-p53 and TAp63 nonfunctional and keeps ΔNp63 relatively highly expressed (62). Hence, we hypothesized that mut-p53 amyloids might spread between cells in a prion-like manner, which could have detrimental effects on cellular integrity. This spread could lead to the pervasive loss of wt-p53 function in tissues, effectively converting the guardian of the genome, p53, into a prion-like protein (Figure 2).
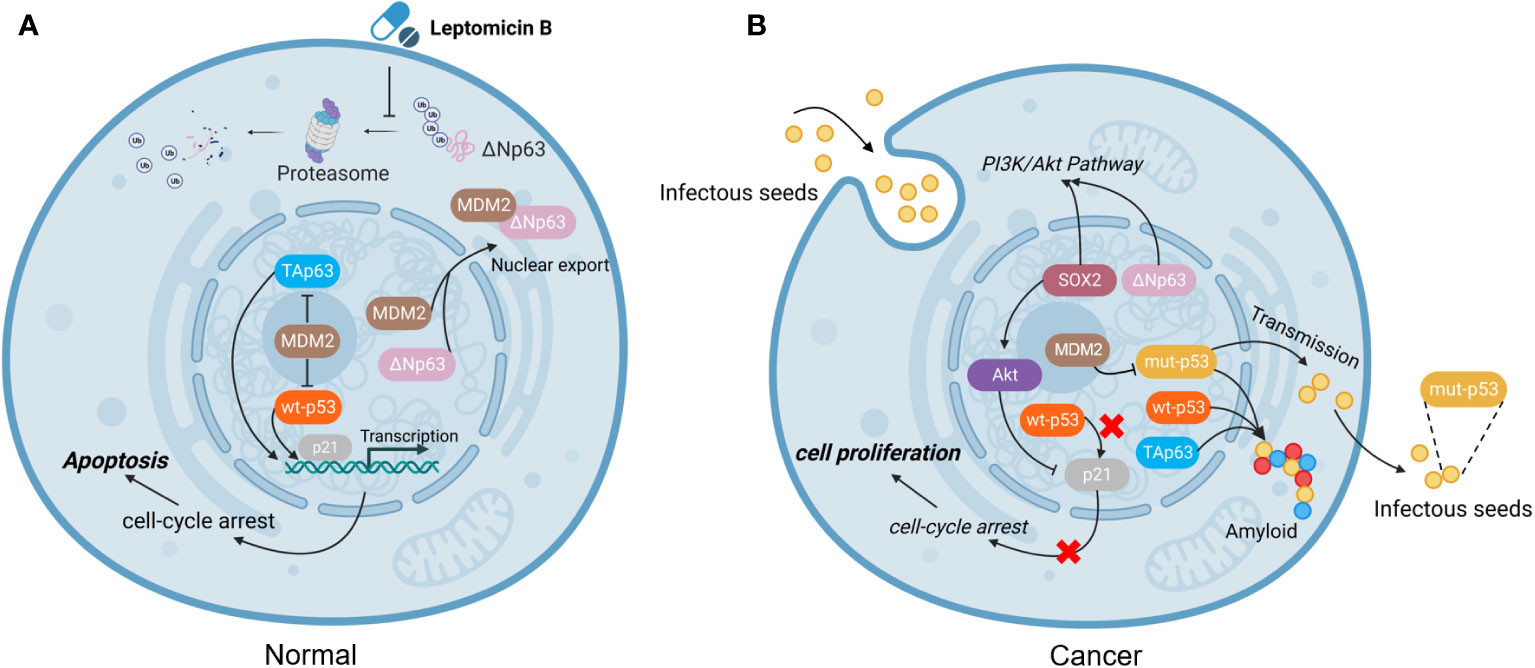
Figure 2 The function of mutated p53. (A) In normal cells, MDM2 plays an important role in regulating apoptosis. MDM2 binds to ΔNp63 and promotes its entry into the cytoplasm for degradation via proteasome, which can be blocked by the drug Leptomycin B (63). (B) In tumor cells, mut-p53 behaves in a prion-like behavior, not only possessing “seeding” ability and spreading to other cells, but also forming amyloid aggregates with TAp63 and wt-p53, leading to inactivation and promoting cell proliferation. However, MDM2 can block the aggregation of mut-p53, prevent mut-p53 from binding to TAp63, and alleviate mut-p53-related suppression to TAp63 (13, 64). (Created with Biorender.com).
As we mentioned above, mut-p53 aggregated with wt-p53 and TAp63, resulting in the loss of their surveillant function in cancer formation. In addition, mut-p53 bound more efficiently to TAp63 than to the corresponding ΔNp63 isoforms (52), TAp63 and ΔNp63 are a well-known pair of contradictions, and TAp63 acts as a transcription factor, both functionally and structurally similar to the tumor suppressor p53, to induce cell apoptosis and suppress tumorigenesis, and is involved in the regulation of cell cycle arrest for DNA damage and aging. On the contrary, ΔNp63 acts in the opposite manner with wt-p53 and TAp63, and overexpression of ΔNp63 induces the accelerated growth of transformed cells in vitro and in vivo. Immunoprecipitation experiment showed that ΔNp63 interacts effectively with wt-p53 but not mut-p53, then ΔNp63 continuously inhibits p53-mediated transactivation by competitive binding of transcription factors to the same promoter regions of the target sequence (65). Interestingly, in combination with the above-reported prion-like behavior of mut-p53, it binds with TAp63 but not ΔNp63, resulting in amyloid precipitation, which makes TAp63 unable to perform its normal tumor suppressor function and renders a relatively high expression level of ΔNp63 in mut-p53 cells.
The occurrence of senescence is a multi-procedural process; cells suffered from DNA damage under the genome pressure (such as ionizing radiation, ROS, and chemical agents). Unchecked DNA damage is an unexpected event for cells, resulting in mutation, chromosomal breakage, and cell cycle halting during S-phase; however, loss of the ability to monitor cell cycle checkpoints induced by DNA damage is a hallmark of cancer cells (66).
TAp63 as a widely known carcinogenesis and progression suppressor, nevertheless inactivate TAp63 by forming amyloid particles with mut-p53 or other signs of progression, including chemoresistance and antagonistic with senescence (67). Furthermore, some researchers believe that forced expression of TAp63 results in a synergistic effect to induce chemotherapeutic treatment-relative apoptosis in hepatoma cells. Additionally, Gressner’s group found that TAp63 has the ability to activate both death receptor- and mitochondria-mediated apoptosis pathways, and both signaling processes are renowned for reinforcing sensitivity to chemotherapy; on the contrary, blocking TAp63 function leads to enhanced chemoresistance (68).
Meanwhile, p63 has also been described to play a significant role in regulating the apoptotic response following DNA damage agents (69–72). Some reports indicated that ΔNp63 expression levels decreased after treatment for 24 h with ultraviolet radiation (73); in contrast, exposure to cisplatin for 24 h induced DNA damage, and although the total TAp63 protein level did not change, the phosphorylation level of TAp63 increased. This genomic injury activated phospho-TAp63Ser395, induced the SAPK/JNK signaling pathway, and triggered apoptosis in oocytes and granulosa cells. In addition, Gressner’s group also measured the expression levels of the CD95, TNF-R, and TRAIL-R cell death-related NF-κB pathways, and found that stimulation of TAp63 can trigger each of these death receptors and consequently sensitize tumor cells toward apoptosis (68).
TAp63 proteins are degraded via the ubiquitin–proteasome pathway under normal cellular circumstances (74, 75). While other reports also indicated that genotoxic agents, including ultraviolet (UV) irradiation, actinomycin D, bleomycin, and etoposide, led to elevated expression of TAp63 protein levels, the interaction with Cables1 is responsible for the stabilization of the TAp63 isoform structure, which enables apoptosis of cells in response to genotoxic agents (76). According to this, it appears that under the stress of DNA damage, cells promote high expression of Tap63 and stabilize the structure of Tap63, thereby promoting a series of reactions that induce cell apoptosis.
The primary p63 isoform, known as “ΔNp63,” has been shown to impede the transactivation of p53, TAp63, and TAp73 by the specific formation of inhibitory heterogenous complexes to competitively bind the promoters and affect their downstream target genes (26, 27, 36, 77). ΔNp63 is often found highly expressed in various cancers; ΔNp63 protein levels are significantly repressed after UV irradiation, and exotic expression of ΔNp63 alleviates the UV-induced apoptosis (65, 78). In addition, ΔNp63 also triggers a variety of survival signaling cascades, for example, the epithelial growth factor receptor (EGFR), transforming growth factor (TGFβ), and hepatocyte growth factor receptor (HGFR) pathways to drive tumor invasiveness and metastasis (79–82), and activate a set of DNA damage repair-related genes [such as CDK12 (83) and SMG1 (84) proteins], thus promoting cancer cell survival and proliferation. Meaningfully, the kinases CDK12 and SMG1 are recruited to chromatin upon ultraviolet (UV) irradiation only in the presence of ΔNp63 (85), suggesting ΔNp63 as a trigger in the DNA repair signaling pathway. Moreover, reduced ΔNp63 level promoted recruitment of the DNA damage responsive proteins (e.g., FANCI and Lsh) to chromatin, and promoted the expression of γH2A.X, indicating ongoing DNA damage and repair (85). Additionally, ΔNp63 is consequently phosphorylated after DNA damage by ATM, CDK2, and p70s6K. Exposure to DNA damage (such as cisplatin)-induced phosphorylation of ΔNp63 (S385, T397, and S466) leads to a rapid degradation of ΔNp63 protein levels in cancer cells, and results in the transfer of cisplatin-resistant cells into cisplatin-sensitive cells (75). ATM, CDK2, and p70s6K are key regulators in the cellular response to DNA injury, whose activation/homodimerization causes ATM to bind to and phosphorylate its sequencing protein targets and impact DNA repair, apoptosis, and cell cycle checkpoints (86, 87). Of particular importance, although ΔNp63 was degraded under the exposure of genotoxic substances, interaction with YAP1 to stabilize ΔNp63 protects cancer cells from UV-induced apoptosis (88). Liefer et al. also found that the number of apoptotic cells in ΔNp63 transgenic mice decreased by 40%–45% compared with non-transgenic mice under the exposure of UV (69). Hence, ΔNp63 inhibits receptor-mediated and chemotherapy-induced mitochondrial apoptosis pathways, and may assist in predicting cancer cells in response to various genotoxic stresses (89).
Previous interesting studies presented that cancer cells exposed to genotoxic stress agents (such as UV-irradiation and etoposide) accumulated expression of TAp63 (90). Furthermore, TAp63 is degraded via the lysosomal degradation pathway under normal cellular circumstances but stabilized under genotoxic stress (91). Another report showed that under the exposure to UV-B irradiation, increasing genotoxic pressure mediated the downregulation of ΔNp63 protein levels (69). Together, these two different P63 family proteins regulate different cell homeostasis under genotoxic stress (Figure 3).
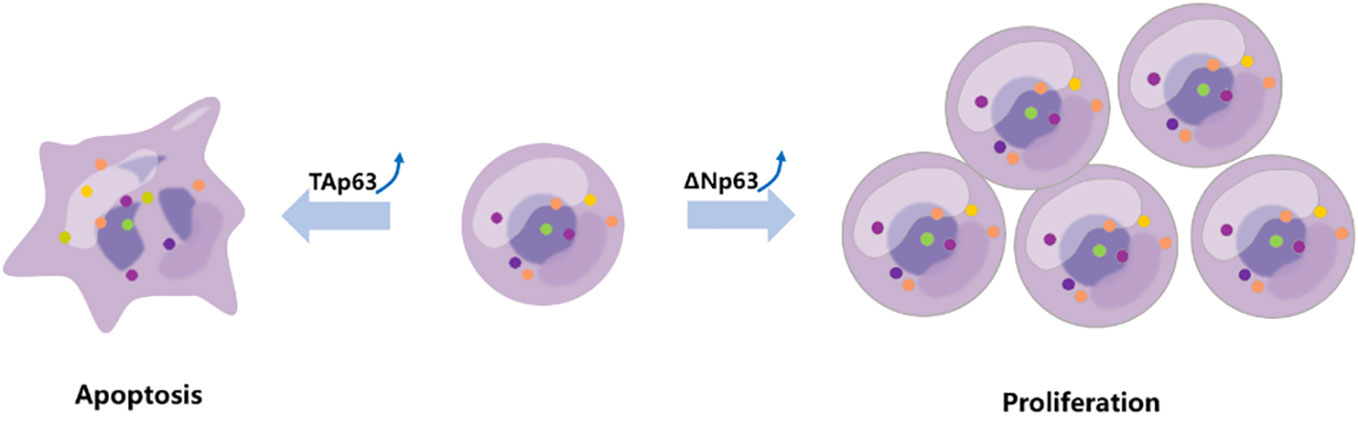
Figure 3 TAp63 and ΔNp63 exert different roles in response to chemotherapy. The TAp63 isoform is frequently associated with tumor suppression involved in cell cycle arrest, apoptosis, and DNA repair. In contrast, the ΔNp63 isoform serves as an oncogene, repressing proapoptotic genes, increasing chemotherapeutic resistance, and inducing cell proliferation.
Tumor recurrence after chemotherapy is still a troublesome problem for physicians. This is frequently brought out by the multidrug resistance (MDR) reaction to chemotherapy drugs (92). The effectiveness of malignancies in response to chemotherapeutic agents depends on many factors, most of which are currently unknown. Some scholars believe that the molecular mechanism of resistant response occurs through DNA damage repair and subsequent p53 overexpression, further induction of apoptosis, or cell cycle arrest (93).
P53, as a guidance of the genome, protects cells from radiation or other genotoxic stresses and ultimately causes cell apoptosis. However, the mutant form of p53 can confer resistance to chemotherapy-induced apoptosis, thereby reducing tumor cell susceptibility to cell death. As we mentioned above, the prion-like behavior of mut-p53 binds with TAp63 but not ΔNp63, resulting in amyloid precipitation, which makes TAp63 unable to perform its normal tumor suppressor function and induce the apoptosis of tumor cells under chemotherapy. Interestingly, Flores et al. showed that, compared with p53+/- mice alone, p53+/- and TAp63+/- transgenic mice spontaneously formed cancer at a dramatically higher incidence. Moreover, p53+/- and TAp63+/- double knockdown mice presented a shorter life span, formed multiple primary tumors (such as bladder, breast, and esophageal cancers), and a much higher tendency to promote metastasis of cancer, and were linked to organismal aging (94). Moreover, another independent research reported that like p53, the TAp63 isoform is an outstanding mediator to monitor tumorigenesis and aging in vivo; p53-/- and p63-/- compared with p53-/- and p73-/- mice presented a higher resistance to DNA damage-induced apoptosis, but transfection of TAp63 into p63-/- mutant mice caused a significant increase in doxorubicin-induced cellular senescence (70). In addition, Guo et al. also found that high expression of TAp63 is an excellent indicator of senescence, independent of p53, prohibits Ras-mediated cancer development, and promotes doxycycline-induced cellular senescence, whereas loss of TAp63 results in aggressive tumor phenotypes, accelerated proliferation, and relieved senescence in cancer cells (37). Furthermore, TAp63 is degraded via the lysosomal degradation pathway under normal cellular circumstances but stabilized under genotoxic stress (91).
Taken as a whole, the TAp63 isoform may limit tumor growth by controlling senescence through p53-independent mechanisms. Inhabitation of senescence is one of the hallmarks of cancer, therefore reactivating senescence in tumor cells, especially those resistant to genotoxicity-induced death, so TAp63 should be considered as a significant mediator in senescence that inhibits tumorigenesis and provides a new foundation on anticancer treatment.
Previous interesting studies presented that cancer cells exposed under genotoxic stress agents (such as UV irradiation and etoposide) accumulated expression of TAp63 but mediated downregulation of ΔNp63 proteins (69, 90). Thus, another subtype of p63, ΔNp63, should also be discussed in the next section.
The p53 family member p63 plays an important role in the cell cycle checkpoint. The ΔN isoform of p63 (ΔNp63), a dominant inactivated form of P63, promotes the proliferation of tumor cells by inhibiting the transcription of the cell cycle regulators (p21, cyclin B2, and cdc2) that influence downstream signaling pathways and resist apoptosis, and can be considered as a prognostic indicator (36, 95–97).
A number of reports have shown that the expression of ΔNp63 is associated with cancer proliferation and drug resistance (98–100). The ΔNp63 protein level has a negative correlation with the concentration of bortezomib, and silencing ΔNp63 significantly reduces the volume of cancer and enhances the survival of mice treated with bortezomib. In contrast, mice treated with bortezomib showed a higher cancer load and a shorter life span following forced ΔNp63 expression (101). Another report indicated that knockdown of ΔNp63 in BRAFi-resistant cells promoted resensitization of these resistant cells in response to vemurafenib and directly enhanced the activation of p53-dependent mitochondrial apoptotic pathways; on the contrary, overexpression of ΔNp63 promoted cell proliferation and enhanced cell resistance to genotoxicity-induced apoptosis (102, 103), and resistance to MAPK inhibitors (18). In squamous cell carcinoma of the head and neck (HNSCC), after the treatment of cisplatin, ΔNp63 is phosphorylated at S385G (p-ΔNp63S385G) by ATM and degraded following DNA damage (104); then, ΔNp63 also downregulates the expression of mir-181a, mir-519a, and mir-374a, leading to a series of mRNAs involved in apoptosis, rendering cancer cells more sensitive to DNA damage agents (Figure 4) (75, 107, 108). Moreover, ΔNp63 also activated phospho-EGFR (Y1086); promoted EGF-mediated activation of ERK, Akt, and JNK signaling; stimulated cancer proliferation, motility, and invasion; and enhanced resistance to cisplatin-induced apoptosis; on the other hand, when a missense mutation was introduced into the ΔNp63 DBD at position 202, it will downregulate the expression of EGFR (109, 110). In order to promote the accumulation of ΔNp63 in cancer, the deubiquitylate USP28 stabilizes ΔNp63 by counteracting its proteasome-mediated degradation, promoting cancer cell survival under the treatment of chemotherapy (99). Furthermore, some findings highlight that p63 plays an important role in controlling ROS. Overexpression of ΔNp63 cooperates with the BCL-2 family to prevent etoposide-induced ROS accumulation, leading to ferroptosis independent of p53 (111, 112). Moreover, a report also indicated that with the increased dose of H2O2-induced ROS, ΔNp63 increased gradually (113), and this procedure provides a way for tumor cells to inhibit oxidative stress-induced cell death and promote survival.
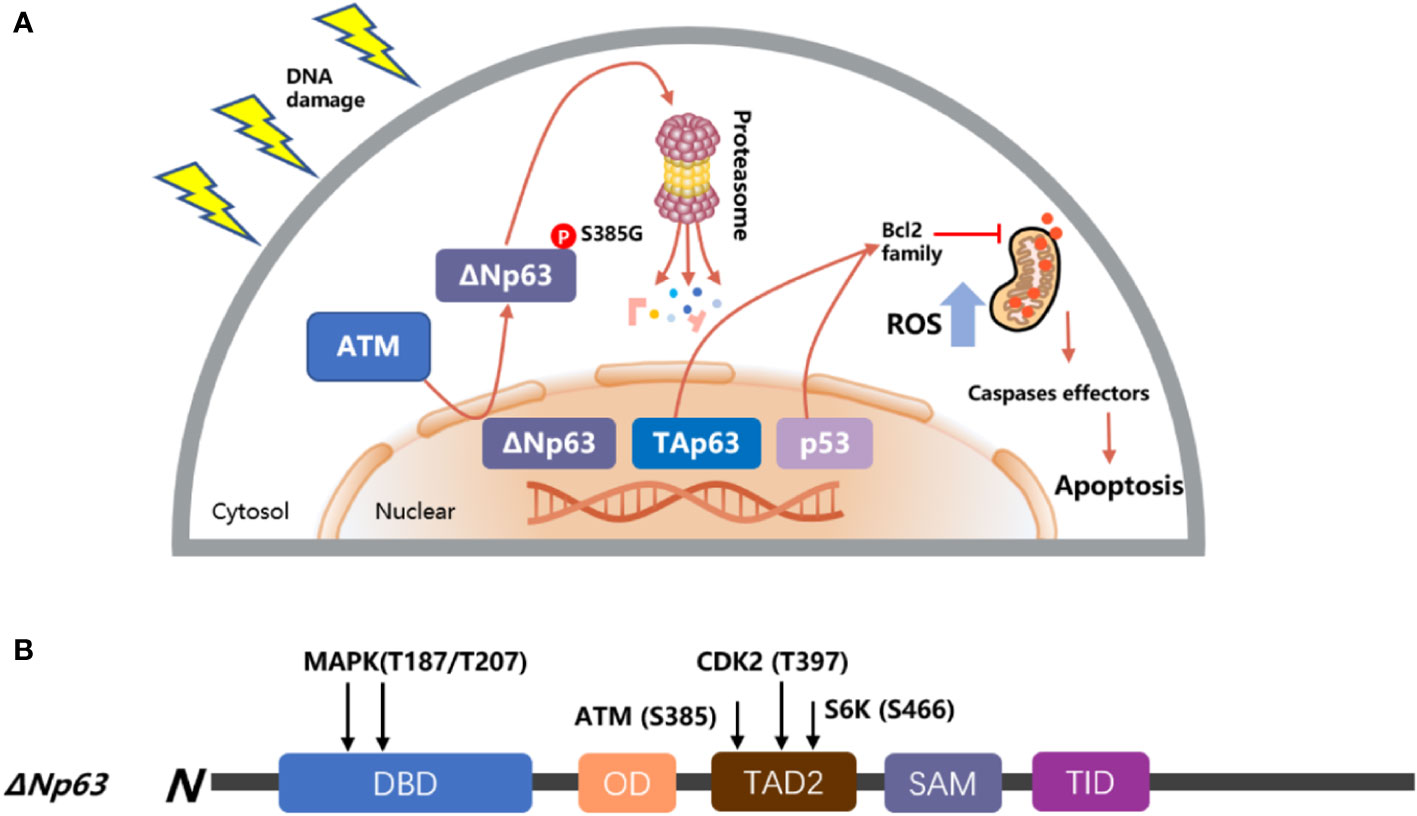
Figure 4 Model of p53 and p63 in response to DNA damage. (A) TAp63 and p53 induce apoptosis pathways to activate the mitochondrial cascades, and ΔNp63 is phosphorylated at S385G (p-ΔNp63S385G) by ATM and degraded following DNA damage. (B) The modular structure of ΔNp63 with putative phosphorylation sites. The arrows indicate the newly identified phosphorylation sites for MAPK (T187/T207), ATM (S385), CDK2 (T397), and p70s6K (S466) kinases (75, 105, 106).
Therefore, it can be concluded that the TA-type ΔN homolog of the P63 gene has the opposite biological function in chemotherapy response. The TA-type homolog has the potential activity of a tumor suppressor gene while the ΔN-type homolog has the function of an oncogene to promote the survival of cancer cells.
It has been hypothesized that only a small group of cancer cells, known as cancer stem cells (CSCs) or cancer-initiating cells (CICs), capable of infinite self-renewal capacity, rapid reproduction, and resistance to chemotherapy, are responsible for tumor initiation, progression, and metastasis (114–116). Furthermore, p63 has been implicated in body development, it is highly expressed in the proliferating basal cell layer, which contains a large number of epithelial progenitor cells responsible for normal replacement function of cells in tissues such as skin, prostate, eyelid, and jaw. Therefore, p63 may be involved in the maintenance of stemness and serve as a potential marker for identifying stem cells (117–121).
Since TAp63 shares the abilities of the “guardian of the genome” p53 to induce cell cycle arrest and apoptosis, TAp63 may thus act as a tumor suppressor. Meaningfully, the knockdown of TAp63 can significantly affect the function of the cells and even GOF to promote cell proliferation. Some reports have indicated that TAp63 participated in the regulation of stem cells via transcriptional regulation of LKB1, further affecting the Hippo pathway effector TAZ, which has previously been demonstrated to have crucial functions in the progression of stem cells and metastasis. Loss of regulation of LKB1 in TAp63-deficient mammary epithelial cells resulted in a loss of Scrib expression and activation of the Hippo pathway through TAZ and a subsequent loss of cell polarity and accumulation of cancer stem cells (122, 123). In addition, some animal studies have confirmed the role of TAp63 in tumor stemness suppressor. TAp63-/- mice showed significantly increased proliferation and self-renewal, resulting in overproliferation of stem cells; however, as normal stem cells are not immortalized in proliferation progress, this procedure will exhaust normal adult stem-cell function and result in depletion of normal stem cells, and eventually resulting in TAp63-/- mice having fragile skin, blisters, wounds that never heal, and alopecia (124–126). Furthermore, another report indicated that TAp63-/- mice have an increasing number of breast hyperplastic cells with highly disordered and polarity defects, and the TAp63-/- mice had 75% ± 1% Ki67-positive cells in the mammary gland, while WT mice only had 38% ± 2% Ki67-positive cells in the mammary gland. Moreover, 8% of TAp63-/- mice also spontaneously form mammary adenocarcinoma at 9–16 months of age, and TAp63-/- mice have also been shown to express high levels of Sox2 and BMP4, which are known markers for cancer stem cells (67, 123).
Very interestingly, unlike the downregulation of TAp63 to promote stem cell proportion, ΔNp63 acts like an oncotarget promoter to enhance cell proliferation, directly interacts with the Hippo effector YAP1, and is a mediator of YAP1 function to promote cancer cell spheroid formation, invasion, migration, and enhance cancer stem cell survival (127, 128). Moreover, ΔNp63 acts as an oncogene that positively participates in the Hedgehog signaling pathway by directly binding to Shh, Gli2, and Ptch1 gene regulatory regions and influencing stemness, contributing to enhancing CSCs’ self-renewal potential (129). In addition, ΔNp63 increases the expression of the Wnt receptor Frizzled 7, thereby combining with β-catenin to enhance Wnt signaling, which leads to promotion of normal mammary stem cell activity and tumor-initiating activity in the basal-like subtype of breast cancer (130, 131). Several additional in vitro and in vivo experiments also suggest that ΔNp63 drives the stem cell formation and differentiation in normal tissues. SETDB2 interacts with ΔNp63 and methylates and stabilizes the ΔNp63 protein, and SOX2 activates ΔNp63 by directly binding the enhancer site and rescued the cancer stem cell maintenance (132, 133). ΔNp63-/- transgenic mice showed a significantly accelerated keratinocyte differentiation through direct regulation of the Notch signaling pathway (134). Liu et al. identified that ΔNp63 was upregulated in 100 of 173 (58%) breast cancer patients and was associated with poorer survival in patients with ER-/HER2+ breast cancer (135).
Moreover, many researchers currently recognize that CSCs are characterized by high expression of CD29, CD44, CD82, or CD133, which are associated with tumor progression and stemness in various cancers (136–140). In prostate cancer, ΔNp63, as a key regulator of CSC-related genes, cooperates with CD82 and is involved in tumor metastatic adhesion (141, 142). Additionally, overexpression of ΔNp63 promotes the expression of CD44 through an indirect way in HNSCC (143). In another report, according to the expression level of CD29, Li et al. divided the breast cancer cell population into CD29 high- and low-expression groups (CD29high and CD29low) and found that TAp63 was highly expressed in CD29low cells; in contrast, ΔNp63 was highly expressed in CD29high cells (144). Meng’s group also identified that ΔNp63 directly activates Notch signaling pathway to induce cancer cells to acquire CSC-like properties, and the expression levels of ΔNp63 were positively correlated with CD133 to affect the self-renewal capacity of cancer cells (145). Hence, ΔNp63 could be considered a biomarker of certain epithelial stem cells and CSCs, and understanding the relationship between p63 isoforms and CSCs is helpful to understand the occurrence and development of tumor cells.
Considering the great influence of post-transcriptional modification on protein function and structure, p63-related interactome is a significant parameter of the p73 activity (Table 1). P63 can be posttranslationally regulated by RNA-binding proteins (RBPs) RBM24, RPM38, and HuR via mRNA stability and protein translation (163–165); to be specific, RBM24 has the ability to bind to multiple sites within the 3’ untranslated region of p63 and destabilize the transcript, resulting in decreased p63 expression levels. Ectopic expression of RBM24 shortens the half-life of both TAp63 and ΔNp63 mRNA levels. This is due to RBM24 binding to multiple regions in the 3′UTR of the p63 transcript, which is essential for TAp63 expression. The RNA-binding domain in RBM24 is composed of two RNA recognition submotifs, RNP1 and RNP2, and in the absence of either RNP, RBM24 cannot bind to p63; thus, the RNA-binding domain of RBM24 is essential for binding to the p63 transcript, which leads to the inhibition of p63 expression. Other types of kinases have also been found to be involved in the activation process of Tap63, such as Cables1, TLR3, PML, and PlK1. The activation process of ΔNp63 also involves the participation of many other kinases, including ATM (75), CDK2 (75), HIPK2 (166), p38 (167), p70s6K (75), and Raf1 (168).
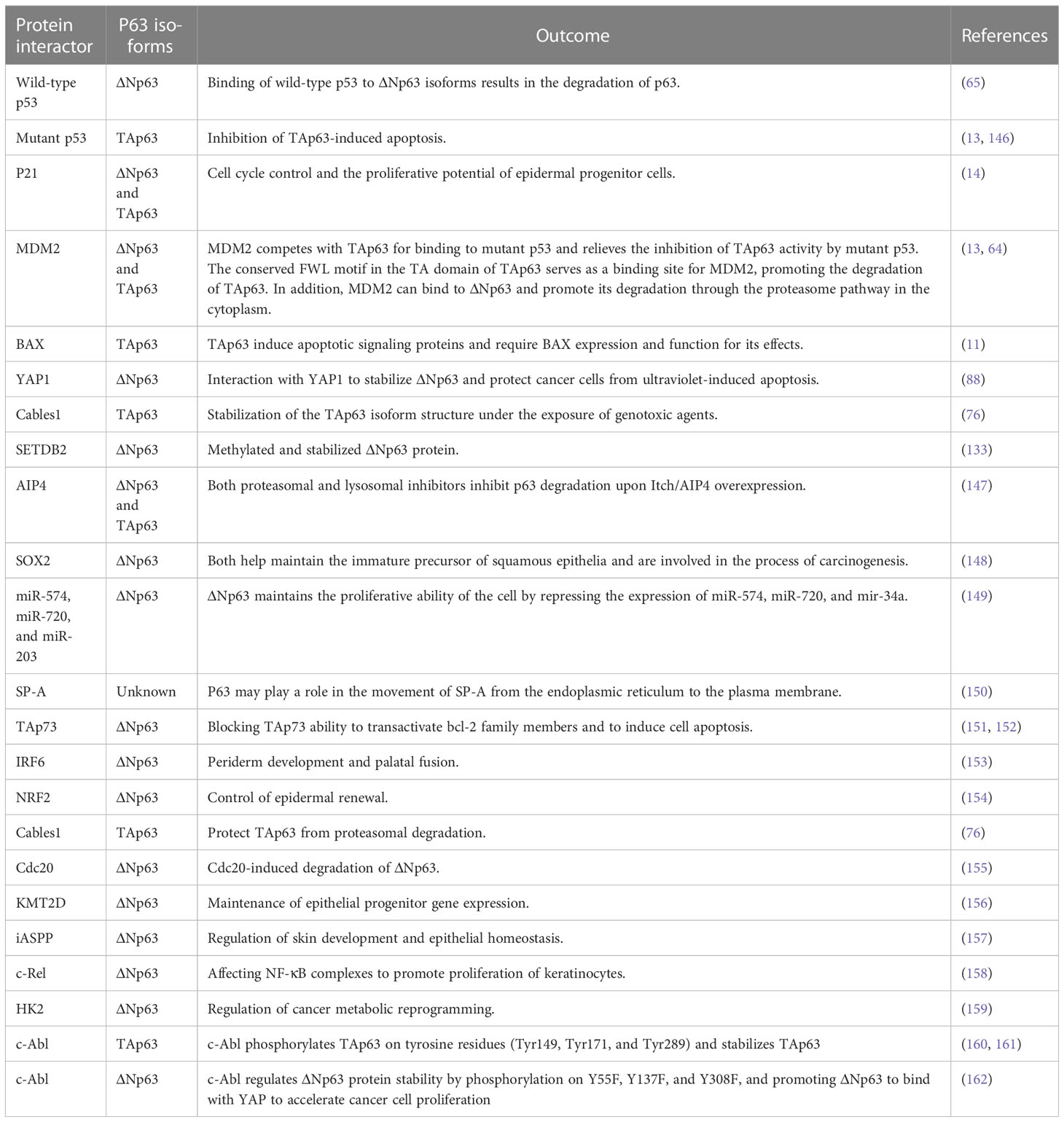
Table 1 Interacting partners of p63 isoforms and their effect on p63 function.
Significantly, after cisplatin treatment, in response to DNA damage, c-Abl kinase detects the signal and phosphorylates TAp63 on specific tyrosine residues (Tyr149, Tyr171, and Tyr289) and stabilizes TAp63, consistent with c-Abl nuclear accumulation toward apoptotic genes (160, 161). Moreover, repression of this process by imatinib, a BCR-ABL inhibitor used to clinically treat chronic myelogenous leukemia, results in the abolition of TAp63 activation and protection of mouse oocytes from cisplatin chemotherapy (160).
Interestingly, in cancer, c-Abl also regulates ΔNp63 protein stability by phosphorylation on Y55F, Y137F, and Y308F, and promoting ΔNp63 to bind with YAP to accelerate cancer cell proliferation (162). Taken as a whole, c-Abl phosphorylates and stabilizes TAp63 to perform its apoptotic function in normal cells under the exposure of chemotherapy, but, after the treatment of cisplatin in cancer cells, c-Abl phosphorylates and stabilizes ΔNp63 to promote cancer cell survival.
Furthermore, Hsp70 (heat-shock protein 70) and CHIP (C-terminus of Hsc-70 interacting protein) have been reported as critical switches for TAp63 and ΔNp63 ubiquitination and degradation (169, 170), and both Hsp70 and CHIP are involved in the process of ubiquitin ligase activity.
CHIP, as a cochaperone ubiquitin ligase, is a highly conserved ubiquitin E3 ligase containing a U-box domain responsible for chaperone partners. CHIP has an N-terminal tetratricopeptide repeat (TPR) domain involved in protein–protein interactions (PPIs) with Hsp70 and Hsp90. Moreover, CHIP has also been proven to conserve the ubiquitin E3 ligase of p53 (171), c-Myc (172), PRMT5 (173), and EGFR (174). Moreover, Wu et al. also proved that the stability of TAp63 and ΔNp63 is regulated by CHIP/Hsp70-mediated ubiquitin–proteasome degradation (170).
Hsp70 acts as a crucial switch to control the CHIP-mediated ubiquitination and degradation of both TAp63 and ΔNp63 isoforms. Hsp70 depletion by siRNA enhanced the interaction of CHIP with ΔNp63 but reduced the interaction with TAp63, thus promoting ΔNp63 degradation, increasing the expression of TAp63, and downregulating the expression of ΔNp63 in cancer cells. Furthermore, the author also used a small-molecule inhibitor of Hsp70, called Ver-155008, and a similar result has been observed in which an increase in ΔNp63 ubiquitination and an accompanying decrease in ΔNp63 protein levels suggest that Hsp70 is involved in CHIP-mediated p63 degradation. Thus, c-Abl, Hsp70, and CHIP seem to play a dual role in tumor and normal cells, and this vague condition in response to chemotherapy requires further studies to elucidate the potential mechanisms underlying these effects.
The p53 family proteins exert a crucial dual role in cancer development and chemotherapy. P63, as a significant regulatory factor similar to p53, is involved in tissue proliferation and differentiation, acts as a transcriptional regulator of tumorigenesis, and is highly expressed in the basal cells where a majority of human epithelial neoplasm develop. Here, we reviewed the influence of two major p63 subtypes (ΔNp63 and TAp63) in pathological conditions, such as cancer stem cells, DNA damage, and drug resistance. Consistently, the balance between TAp63 and ΔNp63 isoforms appears to be important in regulating cellular fates, the ability of tumor suppressors vs. oncogenes, sensitivity vs. drug resistance, and apoptosis vs. proliferation. p63 is widely expressed in cancer tissues and is essential for the survival of cancer cells under the exposure of DNA damage agents. However, there is currently no antitumor drug that targets p63 in DrugBank or other databases. We also reflect on whether p63 is a potential therapeutic target in light of the significant role that p63 plays in the progression of tumors. Taken together, TAp63 isoforms can potentially emulate wt-p53 functions in cancer cells by promoting apoptosis in response to DNA damage, while ΔNp63 isoforms imitate the ability of mut-p53 to initiate cell proliferation and resist DNA replication stress. In this regard, estimating the rankings of the specific p63 isoforms in various cancer patients is of high relevance as it may have a promising impact on patient prognosis and therapeutic outcomes.
Contributions: (I) Conception and design: YX; (II) Administrative support: QZ and JH; (III) Provision of study materials: XY; (IV) Collection and assembly of data: QX; (V) Data analysis and interpretation: YX; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors.
This research was funded by the Sichuan Science and Technology Program (2019YFS0042) and the 1.3.5 project for Disciplines of Excellence, West China Hospital (ZYJC21042), Sichuan University for QZ.
We thank our colleagues for the critical reading and constructive criticism of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol (2011) 12(4):259–65. doi: 10.1038/nrm3086
2. Van Nostrand JL, Bowen ME, Vogel H, Barna M, Attardi LD. The p53 family members have distinct roles during mammalian embryonic development. Cell Death Differ (2017) 24(4):575–9. doi: 10.1038/cdd.2016.128
3. Wang Q, Zou Y, Nowotschin S, Kim SY, Li QV, Soh CL, et al. The p53 family coordinates wnt and nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell (2017) 20(1):70–86. doi: 10.1016/j.stem.2016.10.002
4. Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ (2018) 25(1):154–60. doi: 10.1038/cdd.2017.180
5. Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol (2018) 15(1):13–30. doi: 10.1038/nrclinonc.2017.151
6. Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer (2020) 20(8):471–80. doi: 10.1038/s41568-020-0262-1
7. Arrowsmith CH. Structure and function in the p53 family. Cell Death Differ (1999) 6(12):1169–73. doi: 10.1038/sj.cdd.4400619
8. Zhang Y, Yan W, Chen X. P63 regulates tubular formation via epithelial-to-mesenchymal transition. Oncogene (2014) 33(12):1548–57. doi: 10.1038/onc.2013.101
9. Fisher ML, Balinth S, Mills AA. p63-related signaling at a glance. J Cell Sci (2020) 133(17). doi: 10.1242/jcs.228015
10. Blanchet A, Bourgmayer A, Kurtz JE, Mellitzer G, Gaiddon C. Isoforms of the p53 family and gastric cancer: a menage a trois for an unfinished affair. Cancers (Basel) (2021) 13(4). doi: 10.3390/cancers13040916
11. Jacobs WB, Govoni G, Ho D, Atwal JK, Barnabe-Heider F, Keyes WM, et al. p63 is an essential proapoptotic protein during neural development. Neuron (2005) 48(5):743–56. doi: 10.1016/j.neuron.2005.10.027
12. Ohtani N, Imamura Y, Yamakoshi K, Hirota F, Nakayama R, Kubo Y, et al. Visualizing the dynamics of p21(Waf1/Cip1) cyclin-dependent kinase inhibitor expression in living animals. Proc Natl Acad Sci U.S.A. (2007) 104(38):15034–9. doi: 10.1073/pnas.0706949104
13. Stindt MH, Muller PA, Ludwig RL, Kehrloesser S, Dotsch V, Vousden KH. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene (2015) 34(33):4300–10. doi: 10.1038/onc.2014.359
14. Suzuki D, Sahu R, Leu NA, Senoo M. The carboxy-terminus of p63 links cell cycle control and the proliferative potential of epidermal progenitor cells. Development (2015) 142(2):282–90. doi: 10.1242/dev.118307
15. Aberg E, Saccoccia F, Grabherr M, Ore WYJ, Jemth P, Hultqvist G. Evolution of the p53-MDM2 pathway. BMC Evol Biol (2017) 17(1):177. doi: 10.1186/s12862-017-1023-y
16. Taniguchi Y, Kawata M, Ho Chang S, Mori D, Okada K, Kobayashi H, et al. Regulation of chondrocyte survival in mouse articular cartilage by p63. Arthritis Rheumatol (2017) 69(3):598–609. doi: 10.1002/art.39976
17. Ratovitski EA. Phospho-DeltaNp63alpha-responsive microRNAs contribute to the regulation of necroptosis in squamous cell carcinoma upon cisplatin exposure. FEBS Lett (2015) 589(12):1352–8. doi: 10.1016/j.febslet.2015.04.020
18. Patel A, Garcia LF, Mannella V, Gammon L, Borg TM, Maffucci T, et al. Targeting p63 upregulation abrogates resistance to MAPK inhibitors in melanoma. Cancer Res (2020) 80(12):2676–88. doi: 10.1158/0008-5472.CAN-19-3230
19. Van Sciver N, Ohashi M, Nawandar DM, Pauly NP, Lee D, Makielski KR, et al. DeltaNp63alpha promotes Epstein-Barr virus latency in undifferentiated epithelial cells. PloS Pathog (2021) 17(11):e1010045. doi: 10.1371/journal.ppat.1010045
20. Li D, Li C, Wu M, Chen Q, Wang Q, Ren J, et al. PKCdelta stabilizes TAp63 to promote cell apoptosis. FEBS Lett (2015) 589(16):2094–9. doi: 10.1016/j.febslet.2015.06.014
21. Awais R, Spiller DG, White MR, Paraoan L. p63 is required beside p53 for PERP-mediated apoptosis in uveal melanoma. Br J Cancer (2016) 115(8):983–92. doi: 10.1038/bjc.2016.269
22. Wang H, Yuan Q, Niu M, Zhang W, Wen L, Fu H, et al. Transcriptional regulation of P63 on the apoptosis of male germ cells and three stages of spermatogenesis in mice. Cell Death Dis (2018) 9(2):76. doi: 10.1038/s41419-017-0046-z
23. Wang J, Thomas HR, Li Z, Yeo NCF, Scott HE, Dang N, et al. Puma, noxa, p53, and p63 differentially mediate stress pathway induced apoptosis. Cell Death Dis (2021) 12(7):659. doi: 10.1038/s41419-021-03902-6
24. He Y, Wu X, Tang W, Tian D, Luo C, Yin Z, et al. Impaired delta NP63 expression is associated with poor tumor development in transitional cell carcinoma of the bladder. J Korean Med Sci (2008) 23(5):825–32. doi: 10.3346/jkms.2008.23.5.825
25. Huang Y, Chuang AY, Romano RA, Liegeois NJ, Sinha S, Trink B, et al. Phospho-DeltaNp63alpha/NF-Y protein complex transcriptionally regulates DDIT3 expression in squamous cell carcinoma cells upon cisplatin exposure. Cell Cycle (2010) 9(2):328–38. doi: 10.4161/cc.9.2.10432
26. Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell (1998) 2(3):305–16. doi: 10.1016/s1097-2765(00)80275-0
27. Mangiulli M, Valletti A, Caratozzolo MF, Tullo A, Sbisa E, Pesole G, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res (2009) 37(18):6092–104. doi: 10.1093/nar/gkp674
28. Osada M, Park HL, Nagakawa Y, Yamashita K, Fomenkov A, Kim MS, et al. Differential recognition of response elements determines target gene specificity for p53 and p63. Mol Cell Biol (2005) 25(14):6077–89. doi: 10.1128/MCB.25.14.6077-6089.2005
29. Vakonaki E, Soulitzis N, Sifakis S, Papadogianni D, Koutroulakis D, Spandidos DA. Overexpression and ratio disruption of DeltaNp63 and TAp63 isoform equilibrium in endometrial adenocarcinoma: correlation with obesity, menopause, and grade I/II tumors. J Cancer Res Clin Oncol (2012) 138(8):1271–8. doi: 10.1007/s00432-012-1200-8
30. Yao JY, Chen JK. Roles of p63 in epidermal development and tumorigenesis. BioMed J (2012) 35(6):457–63. doi: 10.4103/2319-4170.104410
31. Nekulova M, Holcakova J, Nenutil R, Stratmann R, Bouchalova P, Muller P, et al. Characterization of specific p63 and p63-n-terminal isoform antibodies and their application for immunohistochemistry. Virchows Arch (2013) 463(3):415–25. doi: 10.1007/s00428-013-1459-4
32. Missero C, Antonini D. Crosstalk among p53 family members in cutaneous carcinoma. Exp Dermatol (2014) 23(3):143–6. doi: 10.1111/exd.12320
33. Lu JH, Liao WT, Lee CH, Chang KL, Ke HL, Yu HS. DeltaNp63 promotes abnormal epidermal proliferation in arsenical skin cancers. Toxicol In Vitro (2018) 53:57–66. doi: 10.1016/j.tiv.2018.07.011
34. Osterburg C, Dotsch V. Structural diversity of p63 and p73 isoforms. Cell Death Differ (2022) 29(5):921–37. doi: 10.1038/s41418-022-00975-4
35. Westfall MD, Mays DJ, Sniezek JC, Pietenpol JA. The delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by hay-wells syndrome-derived mutations. Mol Cell Biol (2003) 23(7):2264–76. doi: 10.1128/MCB.23.7.2264-2276.2003
36. Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ (2006) 13(6):962–72. doi: 10.1038/sj.cdd.4401914
37. Guo X, Keyes WM, Papazoglu C, Zuber J, Li W, Lowe SW, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol (2009) 11(12):1451–7. doi: 10.1038/ncb1988
38. Cao J, Liu X, Yang Y, Wei B, Li Q, Mao G, et al. Decylubiquinone suppresses breast cancer growth and metastasis by inhibiting angiogenesis via the ROS/p53/ BAI1 signaling pathway. Angiogenesis (2020) 23(3):325–38. doi: 10.1007/s10456-020-09707-z
39. Boutelle AM, Attardi LD. p53 and tumor suppression: it takes a network. Trends Cell Biol (2021) 31(4):298–310. doi: 10.1016/j.tcb.2020.12.011
40. Pawge G, Khatik GL. p53 regulated senescence mechanism and role of its modulators in age-related disorders. Biochem Pharmacol (2021) 190:114651. doi: 10.1016/j.bcp.2021.114651
41. Agupitan AD, Neeson P, Williams S, Howitt J, Haupt S, Haupt Y. P53: a guardian of immunity becomes its saboteur through mutation. Int J Mol Sci (2020) 21(10). doi: 10.3390/ijms21103452
42. Zhang C, Liu J, Xu D, Zhang T, Hu W, Feng Z. Gain-of-function mutant p53 in cancer progression and therapy. J Mol Cell Biol (2020) 12(9):674–87. doi: 10.1093/jmcb/mjaa040
43. Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, et al. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J Hematol Oncol (2021) 14(1):157. doi: 10.1186/s13045-021-01169-0
44. Tomczak K, Czerwinska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) (2015) 19(1A):A68–77. doi: 10.5114/wo.2014.47136
45. Clarke SL, Thompson LR, Dandekar E, Srinivasan A, Montgomery MR. Distinct TP53 mutation subtypes differentially influence cellular iron metabolism. Nutrients (2019) 11(9). doi: 10.3390/nu11092144
46. Wang H, Liao P, Zeng SX, Lu H. It takes a team: a gain-of-function story of p53-R249S. J Mol Cell Biol (2019) 11(4):277–83. doi: 10.1093/jmcb/mjy086
47. Hassin O, Nataraj NB, Shreberk-Shaked M, Aylon Y, Yaeger R, Fontemaggi G, et al. Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nat Commun (2022) 13(1):2800. doi: 10.1038/s41467-022-30481-7
48. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol (2010) 2(1):a001008. doi: 10.1101/cshperspect.a001008
49. Novelli F, Ganini C, Melino G, Nucci C, Han Y, Shi Y, et al. p63 in corneal and epidermal differentiation. Biochem Biophys Res Commun (2022) 610:15–22. doi: 10.1016/j.bbrc.2022.04.022
50. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer (2009) 9(12):862–73. doi: 10.1038/nrc2763
51. Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol (1999) 19(2):1438–49. doi: 10.1128/MCB.19.2.1438
52. Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol (2001) 21(5):1874–87. doi: 10.1128/MCB.21.5.1874-1887.2001
53. Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem (2002) 277(21):18817–26. doi: 10.1074/jbc.M201405200
54. Ano Bom AP, Rangel LP, Costa DC, de Oliveira GA, Sanches D, Braga CA, et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J Biol Chem (2012) 287(33):28152–62. doi: 10.1074/jbc.M112.340638
55. Wang G, Fersht AR. Propagation of aggregated p53: cross-reaction and coaggregation vs. seeding. Proc Natl Acad Sci U.S.A. (2015) 112(8):2443–8. doi: 10.1073/pnas.1500262112
56. Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol (2010) 11(4):301–7. doi: 10.1038/nrm2873
57. Irwin DJ, Lee VM, Trojanowski JQ. Parkinson's disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci (2013) 14(9):626–36. doi: 10.1038/nrn3549
58. Jeremic D, Jimenez-Diaz L, Navarro-Lopez JD. Past, present and future of therapeutic strategies against amyloid-beta peptides in alzheimer's disease: a systematic review. Ageing Res Rev (2021) 72:101496. doi: 10.1016/j.arr.2021.101496
59. Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol (2011) 7(5):285–95. doi: 10.1038/nchembio.546
60. Wilcken R, Wang G, Boeckler FM, Fersht AR. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc Natl Acad Sci U.S.A. (2012) 109(34):13584–9. doi: 10.1073/pnas.1211550109
61. Holland TA, Elder J, McCloud JM, Hall C, Deakin M, Fryer AA, et al. Subcellular localisation of cyclin D1 protein in colorectal tumours is associated with p21(WAF1/CIP1) expression and correlates with patient survival. Int J Cancer (2001) 95(5):302–6. doi: 10.1002/1097-0215(20010920)95:5<302::aid-ijc1052>3.0.co;2-#
62. Ghosh S, Salot S, Sengupta S, Navalkar A, Ghosh D, Jacob R, et al. p53 amyloid formation leading to its loss of function: implications in cancer pathogenesis. Cell Death Differ (2017) 24(10):1784–98. doi: 10.1038/cdd.2017.105
63. Galli F, Rossi M, D'Alessandra Y, De Simone M, Lopardo T, Haupt Y, et al. MDM2 and Fbw7 cooperate to induce p63 protein degradation following DNA damage and cell differentiation. J Cell Sci (2010) 123(Pt 14):2423–33. doi: 10.1242/jcs.061010
64. Ying H, Chang DL, Zheng H, McKeon F, Xiao ZX. DNA-Binding and transactivation activities are essential for TAp63 protein degradation. Mol Cell Biol (2005) 25(14):6154–64. doi: 10.1128/MCB.25.14.6154-6164.2005
65. Ratovitski EA, Patturajan M, Hibi K, Trink B, Yamaguchi K, Sidransky D. p53 associates with and targets delta Np63 into a protein degradation pathway. Proc Natl Acad Sci U.S.A. (2001) 98(4):1817–22. doi: 10.1073/pnas.98.4.1817
66. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
67. Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, et al. TAp63 suppresses metastasis through coordinate regulation of dicer and miRNAs. Nature (2010) 467(7318):986–90. doi: 10.1038/nature09459
68. Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H, et al. TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J (2005) 24(13):2458–71. doi: 10.1038/sj.emboj.7600708
69. Liefer KM, Koster MI, Wang XJ, Yang A, McKeon F, Roop DR. Down-regulation of p63 is required for epidermal UV-b-induced apoptosis. Cancer Res (2000) 60(15):4016–20.
70. Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature (2002) 416(6880):560–4. doi: 10.1038/416560a
71. Fomenkov A, Zangen R, Huang YP, Osada M, Guo Z, Fomenkov T, et al. RACK1 and stratifin target DeltaNp63alpha for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle (2004) 3(10):1285–95. doi: 10.4161/cc.3.10.1155
72. Rinaldi VD, Bloom JC, Schimenti JC. Oocyte elimination through DNA damage signaling from CHK1/CHK2 to p53 and p63. Genetics (2020) 215(2):373–8. doi: 10.1534/genetics.120.303182
73. Ferguson-Yates BE, Li H, Dong TK, Hsiao JL, Oh DH. Impaired repair of cyclobutane pyrimidine dimers in human keratinocytes deficient in p53 and p63. Carcinogenesis (2008) 29(1):70–5. doi: 10.1093/carcin/bgm244
74. Katoh I, Aisaki KI, Kurata SI, Ikawa S, Ikawa Y. p51A (TAp63gamma), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene (2000) 19(27):3126–30. doi: 10.1038/sj.onc.1203644
75. Huang Y, Sen T, Nagpal J, Upadhyay S, Trink B, Ratovitski E, et al. ATM Kinase is a master switch for the delta Np63 alpha phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle (2008) 7(18):2846–55. doi: 10.4161/cc.7.18.6627
76. Wang N, Guo L, Rueda BR, Tilly JL. Cables1 protects p63 from proteasomal degradation to ensure deletion of cells after genotoxic stress. EMBO Rep (2010) 11(8):633–9. doi: 10.1038/embor.2010.82
77. Wu G, Nomoto S, Hoque MO, Dracheva T, Osada M, Lee CC, et al. DeltaNp63alpha and TAp63alpha regulate transcription of genes with distinct biological functions in cancer and development. Cancer Res (2003) 63(10):2351–7.
78. Marchbank A, Su LJ, Walsh P, DeGregori J, Penheiter K, Grayson TB, et al. The CUSP DeltaNp63alpha isoform of human p63 is downregulated by solar-simulated ultraviolet radiation. J Dermatol Sci (2003) 32(1):71–4. doi: 10.1016/s0923-1811(03)00040-9
79. Muller PA, Trinidad AG, Timpson P, Morton JP, Zanivan S, van den Berghe PV, et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene (2013) 32(10):1252–65. doi: 10.1038/onc.2012.148
80. Balboni AL, Cherukuri P, Ung M, DeCastro AJ, Cheng C, DiRenzo J. p53 and DeltaNp63alpha coregulate the transcriptional and cellular response to TGFbeta and BMP signals. Mol Cancer Res (2015) 13(4):732–42. doi: 10.1158/1541-7786.MCR-14-0152-T
81. Zhao G, Li R, Cao Y, Song M, Jiang P, Wu Q, et al. DeltaNp63alpha-induced DUSP4/GSK3beta/SNAI1 pathway in epithelial cells drives endometrial fibrosis. Cell Death Dis (2020) 11(6):449. doi: 10.1038/s41419-020-2666-y
82. Niu M, He Y, Xu J, Ding L, He T, Yi Y, et al. Noncanonical TGF-beta signaling leads to FBXO3-mediated degradation of DeltaNp63alpha promoting breast cancer metastasis and poor clinical prognosis. PloS Biol (2021) 19(2):e3001113. doi: 10.1371/journal.pbio.3001113
83. Wang C, Wang H, Lieftink C, du Chatinier A, Gao D, Jin G, et al. CDK12 inhibition mediates DNA damage and is synergistic with sorafenib treatment in hepatocellular carcinoma. Gut (2020) 69(4):727–36. doi: 10.1136/gutjnl-2019-318506
84. Roberts TL, Ho U, Luff J, Lee CS, Apte SH, MacDonald KP, et al. Smg1 haploinsufficiency predisposes to tumor formation and inflammation. Proc Natl Acad Sci U.S.A. (2013) 110(4):E285–294. doi: 10.1073/pnas.1215696110
85. Bamberger C, Pankow S, Yates JR 3rd. SMG1 and CDK12 link DeltaNp63alpha phosphorylation to RNA surveillance in keratinocytes. J Proteome Res (2021) 20(12):5347–58. doi: 10.1021/acs.jproteome.1c00427
86. Bakkenist CJ, Kastan MB. DNA Damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature (2003) 421(6922):499–506. doi: 10.1038/nature01368
87. Huang RX, Zhou PK. DNA Damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther (2020) 5(1):60. doi: 10.1038/s41392-020-0150-x
88. Tomlinson V, Gudmundsdottir K, Luong P, Leung KY, Knebel A, Basu S. JNK phosphorylates yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis (2010) 1:e29. doi: 10.1038/cddis.2010.7
89. Mundt HM, Stremmel W, Melino G, Krammer PH, Schilling T, Muller M. Dominant negative (DeltaN) p63alpha induces drug resistance in hepatocellular carcinoma by interference with apoptosis signaling pathways. Biochem Biophys Res Commun (2010) 396(2):335–41. doi: 10.1016/j.bbrc.2010.04.093
90. Kerr JB, Hutt KJ, Michalak EM, Cook M, Vandenberg CJ, Liew SH, et al. DNA Damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of puma and noxa. Mol Cell (2012) 48(3):343–52. doi: 10.1016/j.molcel.2012.08.017
91. Okada Y, Osada M, Kurata S, Sato S, Aisaki K, Kageyama Y, et al. p53 gene family p51(p63)-encoded, secondary transactivator p51B(TAp63alpha) occurs without forming an immunoprecipitable complex with MDM2, but responds to genotoxic stress by accumulation. Exp Cell Res (2002) 276(2):194–200. doi: 10.1006/excr.2002.5535
92. Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye WC, et al. Autophagy and multidrug resistance in cancer. Chin J Cancer (2017) 36(1):52. doi: 10.1186/s40880-017-0219-2
93. Quintanal-Villalonga A, Chan JM, Yu HA, Pe'er D, Sawyers CL, Sen T, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol (2020) 17(6):360–71. doi: 10.1038/s41571-020-0340-z
94. Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell (2005) 7(4):363–73. doi: 10.1016/j.ccr.2005.02.019
95. Foschini MP, Gaiba A, Cocchi R, Pennesi MG, Gatto MR, Frezza GP, et al. Pattern of p63 expression in squamous cell carcinoma of the oral cavity. Virchows Arch (2004) 444(4):332–9. doi: 10.1007/s00428-003-0969-x
96. Testoni B, Mantovani R. Mechanisms of transcriptional repression of cell-cycle G2/M promoters by p63. Nucleic Acids Res (2006) 34(3):928–38. doi: 10.1093/nar/gkj477
97. Loljung L, Coates PJ, Nekulova M, Laurell G, Wahlgren M, Wilms T, et al. High expression of p63 is correlated to poor prognosis in squamous cell carcinoma of the tongue. J Oral Pathol Med (2014) 43(1):14–9. doi: 10.1111/jop.12074
98. Hilmarsdottir B, Briem E, Sigurdsson V, Franzdottir SR, Ringner M, Arason AJ, et al. MicroRNA-200c-141 and Np63 are required for breast epithelial differentiation and branching morphogenesis. Dev Biol (2015) 403(2):150–61. doi: 10.1016/j.ydbio.2015.05.007
99. Prieto-Garcia C, Hartmann O, Reissland M, Braun F, Fischer T, Walz S, et al. Maintaining protein stability of Np63 via USP28 is required by squamous cancer cells. EMBO Mol Med (2020) 12(4):e11101. doi: 10.15252/emmm.201911101
100. Prieto-Garcia C, Hartmann O, Reissland M, Fischer T, Maier CR, Rosenfeldt M, et al. Inhibition of USP28 overcomes cisplatin-resistance of squamous tumors by suppression of the fanconi anemia pathway. Cell Death Differ (2022) 29(3):568–84. doi: 10.1038/s41418-021-00875-z
101. Zhou P, Zhang C, Song X, Zhang D, Zhu M, Zheng H. DeltaNp63alpha promotes bortezomib resistance via the CYGB-ROS axis in head and neck squamous cell carcinoma. Cell Death Dis (2022) 13(4):327. doi: 10.1038/s41419-022-04790-0
102. Li X, Chen J, Yi Y, Li C, Zhang Y. DNA Damage down-regulates DeltaNp63alpha and induces apoptosis independent of wild type p53. Biochem Biophys Res Commun (2012) 423(2):338–43. doi: 10.1016/j.bbrc.2012.05.126
103. Li C, Chang DL, Yang Z, Qi J, Liu R, He H, et al. Pin1 modulates p63alpha protein stability in regulation of cell survival, proliferation and tumor formation. Cell Death Dis (2013) 4:e943. doi: 10.1038/cddis.2013.468
104. Ratovitski EA. Phospho-DeltaNp63alpha-dependent microRNAs modulate chemoresistance of squamous cell carcinoma cells to cisplatin: at the crossroads of cell life and death. FEBS Lett (2013) 587(16):2536–41. doi: 10.1016/j.febslet.2013.06.020
105. Finlan LE, Hupp TR. p63: the phantom of the tumor suppressor. Cell Cycle (2007) 6(9):1062–71. doi: 10.4161/cc.6.9.4162
106. Perez CA, Pietenpol JA. Transcriptional programs regulated by p63 in normal epithelium and tumors. Cell Cycle (2007) 6(3):246–54. doi: 10.4161/cc.6.3.3801
107. Zangen R, Ratovitski E, Sidransky D. DeltaNp63alpha levels correlate with clinical tumor response to cisplatin. Cell Cycle (2005) 4(10):1313–5. doi: 10.4161/cc.4.10.2066
108. Huang Y, Chuang A, Hao H, Talbot C, Sen T, Trink B, et al. Phospho-DeltaNp63alpha is a key regulator of the cisplatin-induced microRNAome in cancer cells. Cell Death Differ (2011) 18(7):1220–30. doi: 10.1038/cdd.2010.188
109. Danilov AV, Neupane D, Nagaraja AS, Feofanova EV, Humphries LA, DiRenzo J, et al. DeltaNp63alpha-mediated induction of epidermal growth factor receptor promotes pancreatic cancer cell growth and chemoresistance. PloS One (2011) 6(10):e26815. doi: 10.1371/journal.pone.0026815
110. Holcakova J, Nekulova M, Orzol P, Nenutil R, Podhorec J, Svoboda M, et al. DeltaNp63 activates EGFR signaling to induce loss of adhesion in triple-negative basal-like breast cancer cells. Breast Cancer Res Treat (2017) 163(3):475–84. doi: 10.1007/s10549-017-4216-6
111. Latina A, Viticchie G, Lena AM, Piro MC, Annicchiarico-Petruzzelli M, Melino G, et al. DeltaNp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene (2016) 35(12):1493–503. doi: 10.1038/onc.2015.222
112. Wang GX, Tu HC, Dong Y, Skanderup AJ, Wang Y, Takeda S, et al. DeltaNp63 inhibits oxidative stress-induced cell death, including ferroptosis, and cooperates with the BCL-2 family to promote clonogenic survival. Cell Rep (2017) 21(10):2926–39. doi: 10.1016/j.celrep.2017.11.030
113. Wang Y, Li J, Gao Y, Luo Y, Luo H, Wang L, et al. Hippo kinases regulate cell junctions to inhibit tumor metastasis in response to oxidative stress. Redox Biol (2019) 26:101233. doi: 10.1016/j.redox.2019.101233
114. Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol (2015) 31:28–35. doi: 10.1016/j.semcancer.2014.07.001
115. Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol (2016) 11:47–76. doi: 10.1146/annurev-pathol-012615-044438
116. Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauss A, et al. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol (2020) 11:1280. doi: 10.3389/fimmu.2020.01280
117. Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature (1999) 398(6729):714–8. doi: 10.1038/19539
118. Dellavalle RP, Egbert TB, Marchbank A, Su LJ, Lee LA, Walsh P. CUSP/p63 expression in rat and human tissues. J Dermatol Sci (2001) 27(2):82–7. doi: 10.1016/s0923-1811(01)00105-0
119. Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U.S.A. (2001) 98(6):3156–61. doi: 10.1073/pnas.061032098
120. Di Como CJ, Urist MJ, Babayan I, Drobnjak M, Hedvat CV, Teruya-Feldstein J, et al. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res (2002) 8(2):494–501.
121. Rizzo S, Attard G, Hudson DL. Prostate epithelial stem cells. Cell Prolif (2005) 38(6):363–74. doi: 10.1111/j.1365-2184.2005.00356.x
122. Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA, et al. A temporal requirement for hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev (2014) 28(5):432–7. doi: 10.1101/gad.233676.113
123. Su X, Napoli M, Abbas HA, Venkatanarayan A, Bui NHB, Coarfa C, et al. TAp63 suppresses mammary tumorigenesis through regulation of the hippo pathway. Oncogene (2017) 36(17):2377–93. doi: 10.1038/onc.2016.388
124. Beretta C, Chiarelli A, Testoni B, Mantovani R, Guerrini L. Regulation of the cyclin-dependent kinase inhibitor p57Kip2 expression by p63. Cell Cycle (2005) 4(11):1625–31. doi: 10.4161/cc.4.11.2135
125. Candi E, Dinsdale D, Rufini A, Salomoni P, Knight RA, Mueller M, et al. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle (2007) 6(3):274–85. doi: 10.4161/cc.6.3.3797
126. Paris M, Rouleau M, Puceat M, Aberdam D. Regulation of skin aging and heart development by TAp63. Cell Death Differ (2012) 19(2):186–93. doi: 10.1038/cdd.2011.181
127. Zhao R, Fallon TR, Saladi SV, Pardo-Saganta A, Villoria J, Mou H, et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells. Dev Cell (2014) 30(2):151–65. doi: 10.1016/j.devcel.2014.06.004
128. Fisher ML, Kerr C, Adhikary G, Grun D, Xu W, Keillor JW, et al. Transglutaminase interaction with alpha6/beta4-integrin stimulates YAP1-dependent DeltaNp63alpha stabilization and leads to enhanced cancer stem cell survival and tumor formation. Cancer Res (2016) 76(24):7265–76. doi: 10.1158/0008-5472.CAN-16-2032
129. Memmi EM, Sanarico AG, Giacobbe A, Peschiaroli A, Frezza V, Cicalese A, et al. p63 sustains self-renewal of mammary cancer stem cells through regulation of sonic hedgehog signaling. Proc Natl Acad Sci U.S.A. (2015) 112(11):3499–504. doi: 10.1073/pnas.1500762112
130. Drewelus I, Gopfert C, Hippel C, Dickmanns A, Damianitsch K, Pieler T, et al. p63 antagonizes wnt-induced transcription. Cell Cycle (2010) 9(3):580–7. doi: 10.4161/cc.9.3.10593
131. Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, et al. DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and wnt signalling. Nat Cell Biol (2014) 16(10):1004–1015, 1001-1013. doi: 10.1038/ncb3040
132. Bhattacharya S, Serror L, Nir E, Dhiraj D, Altshuler A, Khreish M, et al. SOX2 regulates P63 and Stem/Progenitor cell state in the corneal epithelium. Stem Cells (2019) 37(3):417–29. doi: 10.1002/stem.2959
133. Ying L, Fei X, Jialun L, Jianpeng X, Jie W, Zhaolin M, et al. SETDB2 promoted breast cancer stem cell maintenance by interaction with and stabilization of DeltaNp63alpha protein. Int J Biol Sci (2020) 16(12):2180–91. doi: 10.7150/ijbs.43611
134. Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development (2012) 139(4):772–82. doi: 10.1242/dev.071191
135. Liu Y, Nekulova M, Nenutil R, Horakova I, Appleyard MV, Murray K, et al. Np63/p40 correlates with the location and phenotype of basal/mesenchymal cancer stem-like cells in human ER(+) and HER2(+) breast cancers. J Pathol Clin Res (2020) 6(1):83–93. doi: 10.1002/cjp2.149
136. Geng S, Guo Y, Wang Q, Li L, Wang J. Cancer stem-like cells enriched with CD29 and CD44 markers exhibit molecular characteristics with epithelial-mesenchymal transition in squamous cell carcinoma. Arch Dermatol Res (2013) 305(1):35–47. doi: 10.1007/s00403-012-1260-2
137. Durko L, Wlodarski W, Stasikowska-Kanicka O, Wagrowska-Danilewicz M, Danilewicz M, Hogendorf P, et al. Expression and clinical significance of cancer stem cell markers CD24, CD44, and CD133 in pancreatic ductal adenocarcinoma and chronic pancreatitis. Dis Markers (2017) 2017:3276806. doi: 10.1155/2017/3276806
138. Liu X, Taftaf R, Kawaguchi M, Chang YF, Chen W, Entenberg D, et al. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discovery (2019) 9(1):96–113. doi: 10.1158/2159-8290.CD-18-0065
139. Li J, Xu J, Li L, Ianni A, Kumari P, Liu S, et al. MGAT3-mediated glycosylation of tetraspanin CD82 at asparagine 157 suppresses ovarian cancer metastasis by inhibiting the integrin signaling pathway. Theranostics (2020) 10(14):6467–82. doi: 10.7150/thno.43865
140. Saito-Reis CA, Balise VD, Pascetti EM, Jiminez M, Gillette JM. Tetraspanin CD82 regulates S1PR1-mediated hematopoietic stem and progenitor cell mobilization. Stem Cell Rep (2021) 16(10):2422–31. doi: 10.1016/j.stemcr.2021.08.009
141. Portillo-Lara R, Alvarez MM. Enrichment of the cancer stem phenotype in sphere cultures of prostate cancer cell lines occurs through activation of developmental pathways mediated by the transcriptional regulator DeltaNp63alpha. PloS One (2015) 10(6):e0130118. doi: 10.1371/journal.pone.0130118
142. Di Giacomo V, Tian TV, Mas A, Pecoraro M, Batlle-Morera L, Noya L, et al. DeltaNp63alpha promotes adhesion of metastatic prostate cancer cells to the bone through regulation of CD82. Oncogene (2017) 36(31):4381–92. doi: 10.1038/onc.2017.42
143. Boldrup L, Coates PJ, Gu X, Nylander K. DeltaNp63 isoforms regulate CD44 and keratins 4, 6, 14 and 19 in squamous cell carcinoma of head and neck. J Pathol (2007) 213(4):384–91. doi: 10.1002/path.2237
144. Li N, Singh S, Cherukuri P, Li H, Yuan Z, Ellisen LW, et al. Reciprocal intraepithelial interactions between TP63 and hedgehog signaling regulate quiescence and activation of progenitor elaboration by mammary stem cells. Stem Cells (2008) 26(5):1253–64. doi: 10.1634/stemcells.2007-0691
145. Xie C, Zhu J, Jiang Y, Chen J, Wang X, Geng S, et al. Sulforaphane inhibits the acquisition of tobacco smoke-induced lung cancer stem cell-like properties via the IL-6/DeltaNp63alpha/Notch axis. Theranostics (2019) 9(16):4827–40. doi: 10.7150/thno.33812
146. Kehrloesser S, Osterburg C, Tuppi M, Schafer B, Vousden KH, Dotsch V. Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ (2016) 23(12):1952–60. doi: 10.1038/cdd.2016.75
147. Rossi M, De Simone M, Pollice A, Santoro R, La Mantia G, Guerrini L, et al. Itch/AIP4 associates with and promotes p63 protein degradation. Cell Cycle (2006) 5(16):1816–22. doi: 10.4161/cc.5.16.2861
148. Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W, et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest (2014) 124(4):1636–45. doi: 10.1172/JCI71545
149. Candi E, Amelio I, Agostini M, Melino G. MicroRNAs and p63 in epithelial stemness. Cell Death Differ (2015) 22(1):12–21. doi: 10.1038/cdd.2014.113
150. Gupta N, Manevich Y, Kazi AS, Tao JQ, Fisher AB, Bates SR. Identification and characterization of p63 (CKAP4/ERGIC-63/CLIMP-63), a surfactant protein a binding protein, on type II pneumocytes. Am J Physiol Lung Cell Mol Physiol (2006) 291(3):L436–446. doi: 10.1152/ajplung.00415.2005
151. Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell (2006) 9(1):45–56. doi: 10.1016/j.ccr.2005.12.013
152. Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene (2007) 26(36):5169–83. doi: 10.1038/sj.onc.1210337
153. Thomason HA, Zhou H, Kouwenhoven EN, Dotto GP, Restivo G, Nguyen BC, et al. Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J Clin Invest (2010) 120(5):1561–9. doi: 10.1172/JCI40266
154. Kurinna S, Seltmann K, Bachmann AL, Schwendimann A, Thiagarajan L, Hennig P, et al. Interaction of the NRF2 and p63 transcription factors promotes keratinocyte proliferation in the epidermis. Nucleic Acids Res (2021) 49(7):3748–63. doi: 10.1093/nar/gkab167
155. Rokudai S, Li Y, Otaka Y, Fujieda M, Owens DM, Christiano AM, et al. STXBP4 regulates APC/C-mediated p63 turnover and drives squamous cell carcinogenesis. Proc Natl Acad Sci U.S.A. (2018) 115(21):E4806–14. doi: 10.1073/pnas.1718546115
156. Lin-Shiao E, Lan Y, Coradin M, Anderson A, Donahue G, Simpson CL, et al. KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev (2018) 32(2):181–93. doi: 10.1101/gad.306241.117
157. Chikh A, Matin RN, Senatore V, Hufbauer M, Lavery D, Raimondi C, et al. iASPP/p63 autoregulatory feedback loop is required for the homeostasis of stratified epithelia. EMBO J (2011) 30(20):4261–73. doi: 10.1038/emboj.2011.302
158. King KE, Ponnamperuma RM, Allen C, Lu H, Duggal P, Chen Z, et al. The p53 homologue DeltaNp63alpha interacts with the nuclear factor-kappaB pathway to modulate epithelial cell growth. Cancer Res (2008) 68(13):5122–31. doi: 10.1158/0008-5472.CAN-07-6123
159. Viticchie G, Agostini M, Lena AM, Mancini M, Zhou H, Zolla L, et al. p63 supports aerobic respiration through hexokinase II. Proc Natl Acad Sci U.S.A. (2015) 112(37):11577–82. doi: 10.1073/pnas.1508871112
160. Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med (2009) 15(10):1179–85. doi: 10.1038/nm.2033
161. Amelio I, Grespi F, Annicchiarico-Petruzzelli M, Melino G. p63 the guardian of human reproduction. Cell Cycle (2012) 11(24):4545–51. doi: 10.4161/cc.22819
162. Yuan M, Luong P, Hudson C, Gudmundsdottir K, Basu S. C-abl phosphorylation of DeltaNp63alpha is critical for cell viability. Cell Death Dis (2010) 1(1):e16. doi: 10.1038/cddis.2009.15
163. Zhang J, Jun Cho S, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc Natl Acad Sci U.S.A. (2010) 107(21):9614–9. doi: 10.1073/pnas.0912594107
164. Yan W, Zhang Y, Zhang J, Cho SJ, Chen X. HuR is necessary for mammary epithelial cell proliferation and polarity at least in part via DeltaNp63. PloS One (2012) 7(9):e45336. doi: 10.1371/journal.pone.0045336
165. Xu E, Zhang J, Zhang M, Jiang Y, Cho SJ, Chen X. RNA-Binding protein RBM24 regulates p63 expression via mRNA stability. Mol Cancer Res (2014) 12(3):359–69. doi: 10.1158/1541-7786.MCR-13-0526
166. Lazzari C, Prodosmo A, Siepi F, Rinaldo C, Galli F, Gentileschi M, et al. HIPK2 phosphorylates DeltaNp63alpha and promotes its degradation in response to DNA damage. Oncogene (2011) 30(48):4802–13. doi: 10.1038/onc.2011.182
167. Hildesheim J, Belova GI, Tyner SD, Zhou X, Vardanian L, Fornace AJ Jr. Gadd45a regulates matrix metalloproteinases by suppressing DeltaNp63alpha and beta-catenin via p38 MAP kinase and APC complex activation. Oncogene (2004) 23(10):1829–37. doi: 10.1038/sj.onc.1207301
168. Di Costanzo A, Festa L, Duverger O, Vivo M, Guerrini L, La Mantia G, et al. Homeodomain protein Dlx3 induces phosphorylation-dependent p63 degradation. Cell Cycle (2009) 8(8):1185–95. doi: 10.4161/cc.8.8.8202
169. Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature (2006) 440(7083):551–5. doi: 10.1038/nature04600
170. Wu HH, Wang B, Armstrong SR, Abuetabh Y, Leng S, Roa WHY, et al. Hsp70 acts as a fine-switch that controls E3 ligase CHIP-mediated TAp63 and DeltaNp63 ubiquitination and degradation. Nucleic Acids Res (2021) 49(5):2740–58. doi: 10.1093/nar/gkab081
171. Esser C, Scheffner M, Hohfeld J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J Biol Chem (2005) 280(29):27443–8. doi: 10.1074/jbc.M501574200
172. Paul I, Ahmed SF, Bhowmik A, Deb S, Ghosh MK. The ubiquitin ligase CHIP regulates c-myc stability and transcriptional activity. Oncogene (2013) 32(10):1284–95. doi: 10.1038/onc.2012.144
173. Zhang HT, Zeng LF, He QY, Tao WA, Zha ZG, Hu CD. The E3 ubiquitin ligase CHIP mediates ubiquitination and proteasomal degradation of PRMT5. Biochim Biophys Acta (2016) 1863(2):335–46. doi: 10.1016/j.bbamcr.2015.12.001
Keywords: p63, p53, DNA damage, cancer progression, protein-protein interactions (PPI), stem cell
Citation: Xu Y, Yang X, Xiong Q, Han J and Zhu Q (2023) The dual role of p63 in cancer. Front. Oncol. 13:1116061. doi: 10.3389/fonc.2023.1116061
Received: 05 December 2022; Accepted: 13 April 2023;
Published: 27 April 2023.
Edited by:
Elzbieta Pluciennik, Medical University of Lodz, PolandReviewed by:
Lindsey D. Mayo, Purdue University Indianapolis, United StatesCopyright © 2023 Xu, Yang, Xiong, Han and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Zhu, bmV3emh1cWluZzE5NzJAeWFob28uY29t; Junhong Han, aGp1bmhvbmdAc2N1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.