
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 24 April 2023
Sec. Cancer Molecular Targets and Therapeutics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1115405
 Laura Schubert1†
Laura Schubert1† Andrew Elliott2†
Andrew Elliott2† Anh T. Le1
Anh T. Le1 Adriana Estrada-Bernal1Robert C. Doebele1
Adriana Estrada-Bernal1Robert C. Doebele1 Emil Lou3
Emil Lou3 Hossein Borghaei4Michael J. Demeure5Razelle Kurzrock6
Hossein Borghaei4Michael J. Demeure5Razelle Kurzrock6 Joshua E. Reuss7Sai-Hong Ignatius Ou8David R. Braxton9Christian A. Thomas10Sourat Darabi5Wolfgang Michael Korn11Wafik S. El-Deiry12Stephen V. Liu7*
Joshua E. Reuss7Sai-Hong Ignatius Ou8David R. Braxton9Christian A. Thomas10Sourat Darabi5Wolfgang Michael Korn11Wafik S. El-Deiry12Stephen V. Liu7*Purpose: Gene fusions involving receptor tyrosine kinases (RTKs) define an important class of genomic alterations with many successful targeted therapies now approved for ALK, ROS1, RET and NTRK gene fusions. Fusions involving the ERBB family of RTKs have been sporadically reported, but their frequency has not yet been comprehensively analyzed and functional characterization is lacking on many types of ERBB fusions.
Materials and methods: We analyzed tumor samples submitted to Caris Life Sciences (n=64,354), as well as the TCGA (n=10,967), MSK IMPACT (n=10,945) and AACR GENIE (n=96,324) databases for evidence of EGFR, ERBB2 and ERBB4 gene fusions. We also expressed several novel fusions in cancer cell lines and analyzed their response to EGFR and HER2 tyrosine kinase inhibitors (TKIs).
Results: In total, we identified 1,251 ERBB family fusions, representing an incidence of approximately 0.7% across all cancer types. EGFR, ERBB2, and ERBB4 fusions were most frequently found in glioblastoma, breast cancer and ovarian cancer, respectively. We modeled two novel types of EGFR and ERBB2 fusions, one with a tethered kinase domain and the other with a tethered adapter protein. Specifically, we expressed EGFR-ERBB4, EGFR-SHC1, ERBB2-GRB7 and ERBB2-SHC1, in cancer cell lines and demonstrated that they are oncogenic, regulate downstream signaling and are sensitive to small molecule inhibition with EGFR and HER2 TKIs.
Conclusions: We found that ERBB fusions are recurrent mutations that occur across multiple cancer types. We also establish that adapter-tethered and kinase-tethered fusions are oncogenic and can be inhibited with EGFR or HER2 inhibitors. We further propose a nomenclature system to categorize these fusions into several functional classes.
Knowledge of the genetic landscape of cancers has grown increasingly critical to guide proper treatment choice. Chromosomal rearrangements involving receptor tyrosine kinases (RTKs) result in the aberrant activation of proliferative and pro-survival pathways. This is often accomplished through the contribution of a dimerization domain which allows constitutive activation of RTKs in the absence of ligand binding and/or enhanced expression from the new 5’ gene partner. Improved identification and understanding of these oncogenic fusions have resulted in the development of many successful targeted therapies over the last several years. ALK, ROS1, NTRK, FGFR and RET fusions now define an important subset of non-small cell lung cancers (NSCLC) and other malignancies with FDA-approved targeted inhibitors (1–9).
Activation of the ERBB/HER family of proteins has been repeatedly implicated in oncogenesis and can be induced by various molecular alterations, including EGFR point mutations or short insertions or deletions (indels) in NSCLC and ERBB2 amplification in breast cancer (10, 11). HER family RTKs (EGFR, HER2, HER3 and HER4) are normally activated by ligand binding, dimerization and eventual activation of downstream pathways including RAS/MAPK and PI3K/AKT. HER family receptors can form both homodimers and heterodimers. In particular, HER2 and HER3 often form heterodimers because HER2 lacks a ligand binding domain and HER3 does not have a functional ATP binding pocket (12).
ERBB family fusions involving the EGFR, ERBB2 and ERBB4 genes are emerging therapeutic targets in several cancer types. Recurrent EGFR-SEPT14 fusions have previously been identified in glioblastoma and are capable of activating proliferative signaling in the absence of EGFR ligands (13). Other EGFR fusions have also been rarely reported in lung adenocarcinoma as well as colorectal adenocarcinoma (14–16). Rearrangements of the ERBB2 gene have been found in breast as well as gastric cancers and ERBB4 fusions have been reported in lung adenocarcinomas and ovarian cancer cell lines (17–19). Fusions involving ERBB family ligands, such as NRG1, have also been identified in NSCLC as well as other cancer types (20, 21). Despite the identification of several gene fusions in this family, many of these have never been functionally validated as oncogenes in vitro.
In this study, we sought to comprehensively analyze the frequency and characteristics of ERBB fusions across cancer types. We queried a large cohort of patient samples profiled at Caris Life Sciences as well as several publicly available data sets (TCGA, MSK IMPACT and AACR GENIE) for EGFR, ERBB2 and ERBB4 fusions. We further investigated the biology of several novel EGFR and ERBB2 fusions in an in vitro cell line model.
Formalin-fixed paraffin-embedded (FFPE) patient samples (n=64,354) were submitted to a commercial CLIA-certified laboratory (Caris Life Sciences, Phoenix, AZ). The present study was conducted in accordance with guidelines of the Declaration of Helsinki, Belmont Report, and U.S. Common Rule. With compliance to policy 45 CFR 46.101(b) (4), this study was conducted using retrospective, de-identified clinical data. Therefore, the present study was considered IRB exempt, and patient consent was not required.
Gene fusion detection was performed on mRNA isolated from a FFPE tumor sample using the Illumina NovaSeq platform (Illumina, Inc., San Diego, CA) and Agilent SureSelect Human All Exon V7 bait panel (Agilent Technologies, Santa Clara, CA). FFPE specimens underwent pathology review to diagnose percent tumor content and tumor size; a minimum of 10% of tumor content in the area for microdissection was required to enable enrichment and extraction of tumor-specific RNA. Qiagen RNA FFPE tissue extraction kit was used for extraction, and the RNA quality and quantity was determined using the Agilent TapeStation. Biotinylated RNA baits were hybridized to the synthesized and purified cDNA targets and the bait-target complexes were amplified in a post capture PCR reaction. The resultant libraries were quantified, normalized and the pooled libraries are denatured, diluted and sequenced; the reference genome used was GRCh37/hg19 and analytical validation of this test demonstrated ≥97% Positive Percent Agreement (PPA), ≥99% Negative Percent Agreement (NPA) and ≥99% Overall Percent Agreement (OPA) with a validated comparator method.
Fusion topologies were assessed by mapping gene fusion sequences to the reference genome GRCh37/hg19 to identify chromosome number, sense/antisense strand, and genomic breakpoint coordinates for gene-pairs of each fusion event detected. For each tumor, detection of multiple isoforms of a unique gene-pair with the same 5’-3’ gene order, presumed to result from alternative splicing, and reciprocal fusion isoforms were scored as a single event in the topology distribution.
NGS was performed on genomic DNA isolated from FFPE tumor samples using the NextSeq platform (Illumina, Inc., San Diego, CA). Matched normal tissue was not sequenced. A custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). All variants were detected with >99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of >500X and an analytic sensitivity of 5%. Prior to molecular testing, tumor enrichment was achieved by harvesting targeted tissue using manual microdissection techniques. Genetic variants identified were interpreted by board-certified molecular geneticists and categorized as ‘pathogenic,’ ‘presumed pathogenic,’ ‘variant of unknown significance,’ ‘presumed benign,’ or ‘benign,’ according to the American College of Medical Genetics and Genomics (ACMG) standards. When assessing mutation frequencies of individual genes, ‘pathogenic,’ and ‘presumed pathogenic’ were counted as mutations while ‘benign’, ‘presumed benign’ variants and ‘variants of unknown significance’ were excluded.
The datasets presented in this article from Caris Life Sciences are not publicly available because the raw data is protected proprietary information – these datasets are available for qualified researchers upon reasonable request and with permission of Caris Life Sciences.
We queried the TCGA PanCancer Analyses, MSK Impact and AACR GENIE data bases through cBioPortal (accession dates 6/18/20, 8/8/20, 8/8/20 respectively) for EGFR, ERBB2 and ERBB4 fusions (22–25).
H3122 and Ba/F3 cell lines were acquired through Dr. John Minna (The University of Texas Southwestern Medical Center, Dallas, TX) and Dr. Dan Theodorescu (University of Colorado Comprehensive Cancer Center, Aurora, CO), respectively. Cells were grown in RPMI-1640 with 10% FBS. The HEK293T cell line was purchased from ATCC. Alectinib was purchased from Selleck Chemicals. AKT pS437 (4058), AKT (2920), ERK pT202/Y204 (9101), ERK (9107), EGFR pY845 (6963) and HER2 pY877 (2241) were purchased from Cell Signaling Technology. EGFR (610017) and HER2 (610161) antibodies were purchased from BD Biosciences and anti-GAPDH was from Millipore.
Lentiviral constructs for EGFR and ERBB2 fusions were manufactured by Vector Builders Inc (Chicago, IL) in their pLV lentiviral vector under CMV promoters. Transduction into H3122 was performed as previously described (26) with the following additions: after puromycin selection, H3122 cells were cultured in the presence of 100nM alectinib to inhibit ALK signaling. Cell viability assays and signaling analysis via immunoblotting were performed in the presence of 100nM alectinib.
Cell proliferation assays were performed using the CellTiter 96 Aqueous One Solution from Promega according to the manufacturer’s instructions after 72 hours of incubation with indicated inhibitors.
Cells were lysed in T-PER (Thermo Scientific) with Halt Protease and Phosphatase Inhibitor (Thermo Scientific). Proteins were separated with SDS-PAGE, transferred to nitrocellulose membranes and stained with indicated antibodies. Secondary IR-Dye anti-mouse or anti rabbit IgG (LI-COR) antibodies were then added. Imaging was performed with the Odyssey Imager and Odyssey Image Studio software (LI-COR).
We identified a total of 1,251 ERBB family fusions across the four data sets including a total of 182,590 samples. EGFR fusions (n=811) were the most common overall, followed by ERBB2 (n=287) and ERBB4 fusions (n=153). We estimated that the frequency of EGFR fusions across data sets was 0.4%, ERBB2 was 0.2% and ERBB4 was 0.1% (Figure 1A). We did not assess the frequency of ERBB3 fusions in this study, as the HER3 protein is unable to form functional homodimers (12). For the Caris samples, profiled by whole transcriptome sequencing, we further analyzed the topology of ERBB fusion transcripts to determine if the ERBB fusions resulted from deletion, inversion, duplication or translocation events. The majority of EGFR and ERBB2 fusions were the result of inversions, while ERBB4 fusions were most often translocations (Figure 1B). We also found that ~80% of EGFR fusions were both in-frame and retained a kinase domain. Approximately 50% of ERBB4 and 40% of ERBB2 fusions had these two features (Figure 1C).
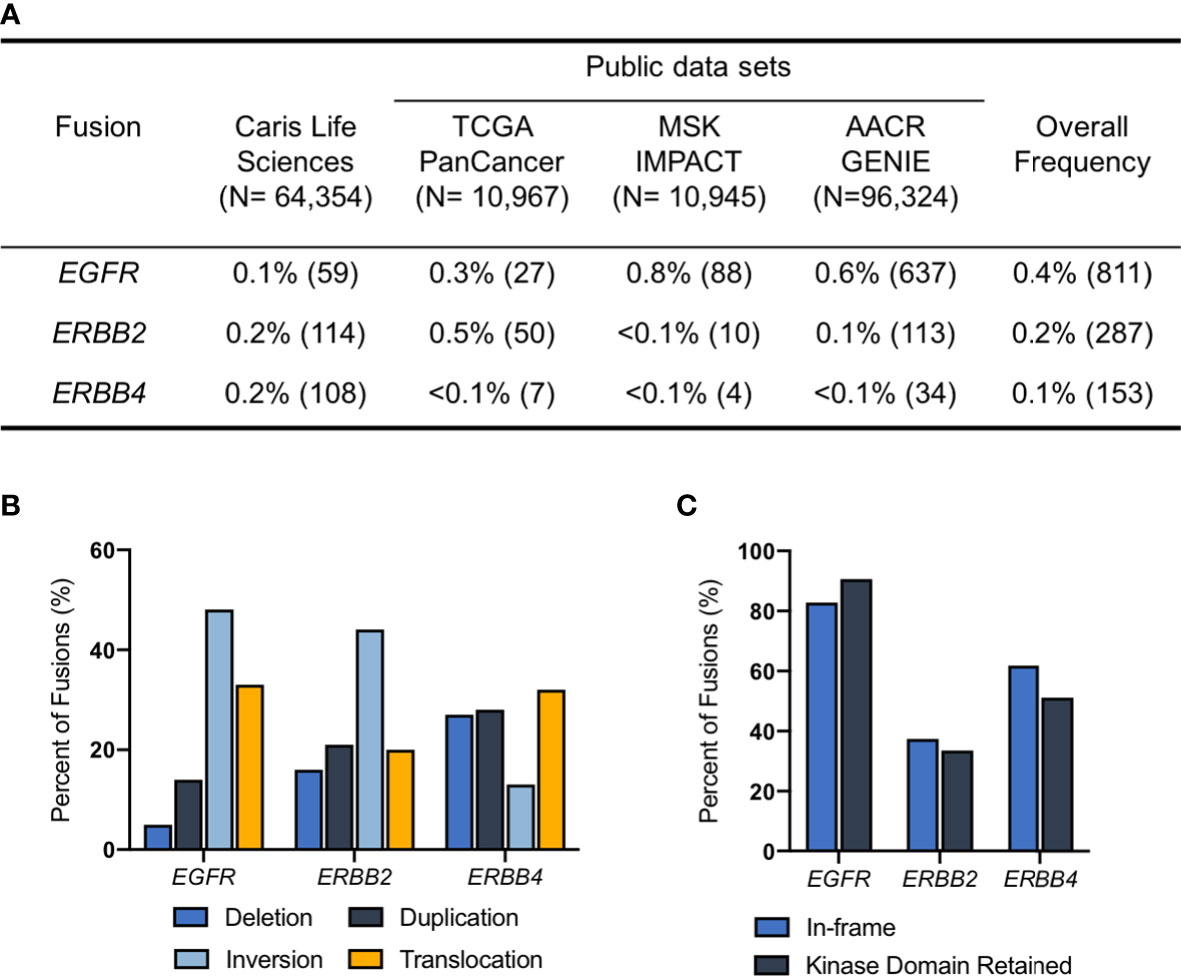
Figure 1 ERBB fusions are rare, recurrent alterations. (A) Frequency of EGFR, ERBB2 and ERBB4 fusions in Caris Life Sciences, TCGA PanCancer Atlas, MSK IMPACT and AACR GENIE datasets. (B) Fusion architecture in Caris Life Sciences datasets. (C) Percentage of ERBB fusions that are in-frame and/or have retained kinase domains in Caris Life Sciences datasets.
Next, we analyzed the identity of the ERBB gene fusion partners. EGFR-SEPT14 was the most common EGFR fusion, along with the reciprocal fusion SEPT14-EGFR that also results from the associated inversion event. However, based on the common breakpoint at EGFR exons 24-25, which retains the extracellular and kinase domains in the EGFR-SEPT14 fusion, the SEPT14-EGFR reciprocal fusion would harbor only a small C-terminal portion of the EGFR protein, and thus, potentially have different or no oncogenic consequences compared to EGFR-SEPT14 fusions. ERBB2-IKZF3 and ERBB2-PPP1R1B were the two most common ERBB2 fusions. IKZF2-ERBB4 was the most common ERBB4 fusion (Table 1).
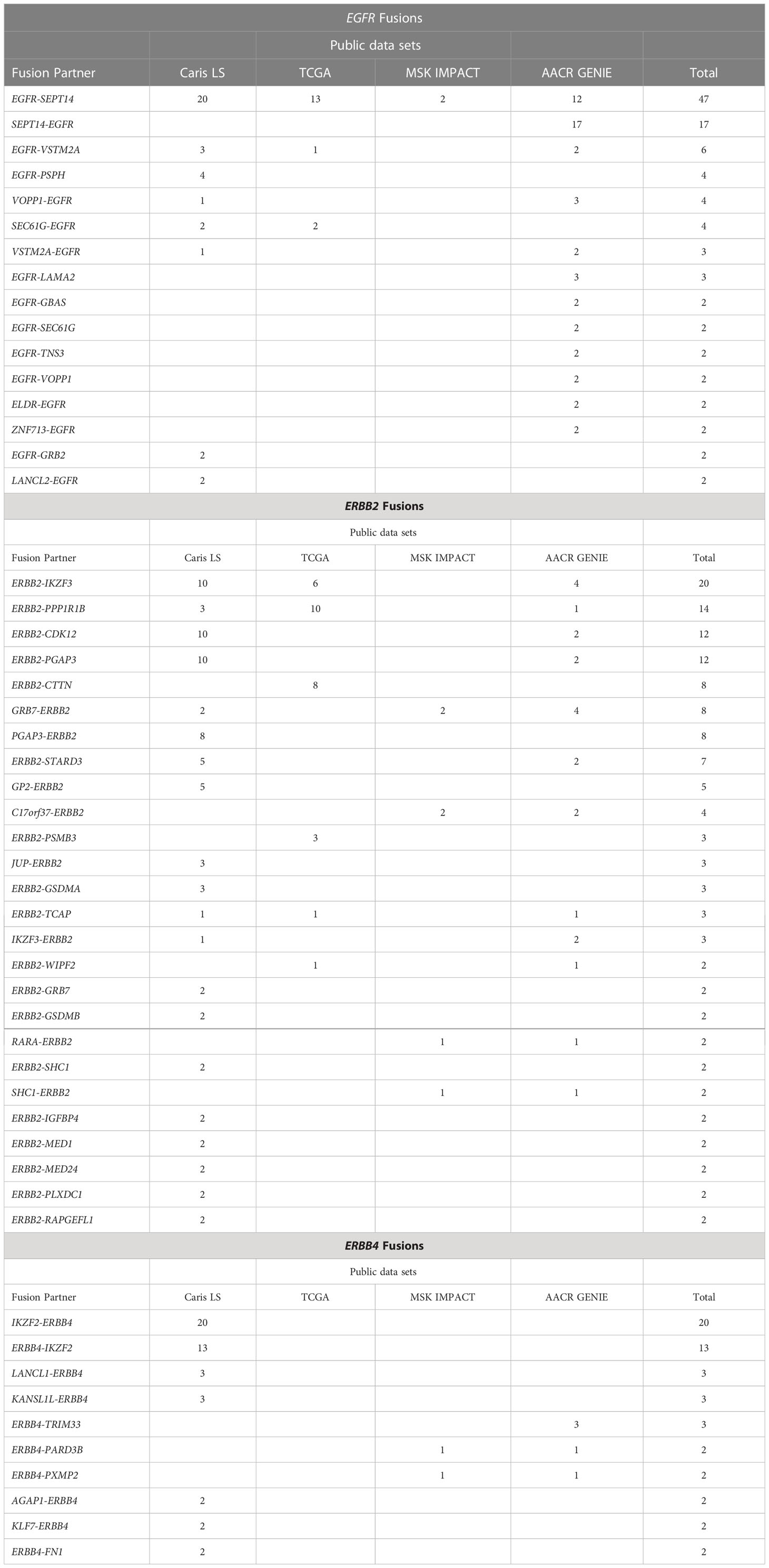
Table 1 Recurrent EGFR, ERBB2 and ERBB4 fusions. Fusions observed more than once are reported above.
We assessed the most common cancer types harboring ERBB fusions. The majority of EGFR fusions were found in glioblastomas or gliomas, followed by NSCLC. ERBB2 fusions were most frequently observed in breast cancer followed by gastroesophageal cancers. Depending on the dataset, ERBB4 fusions were most often found in ovarian cancers, breast cancers or NSCLC (Figure 2).
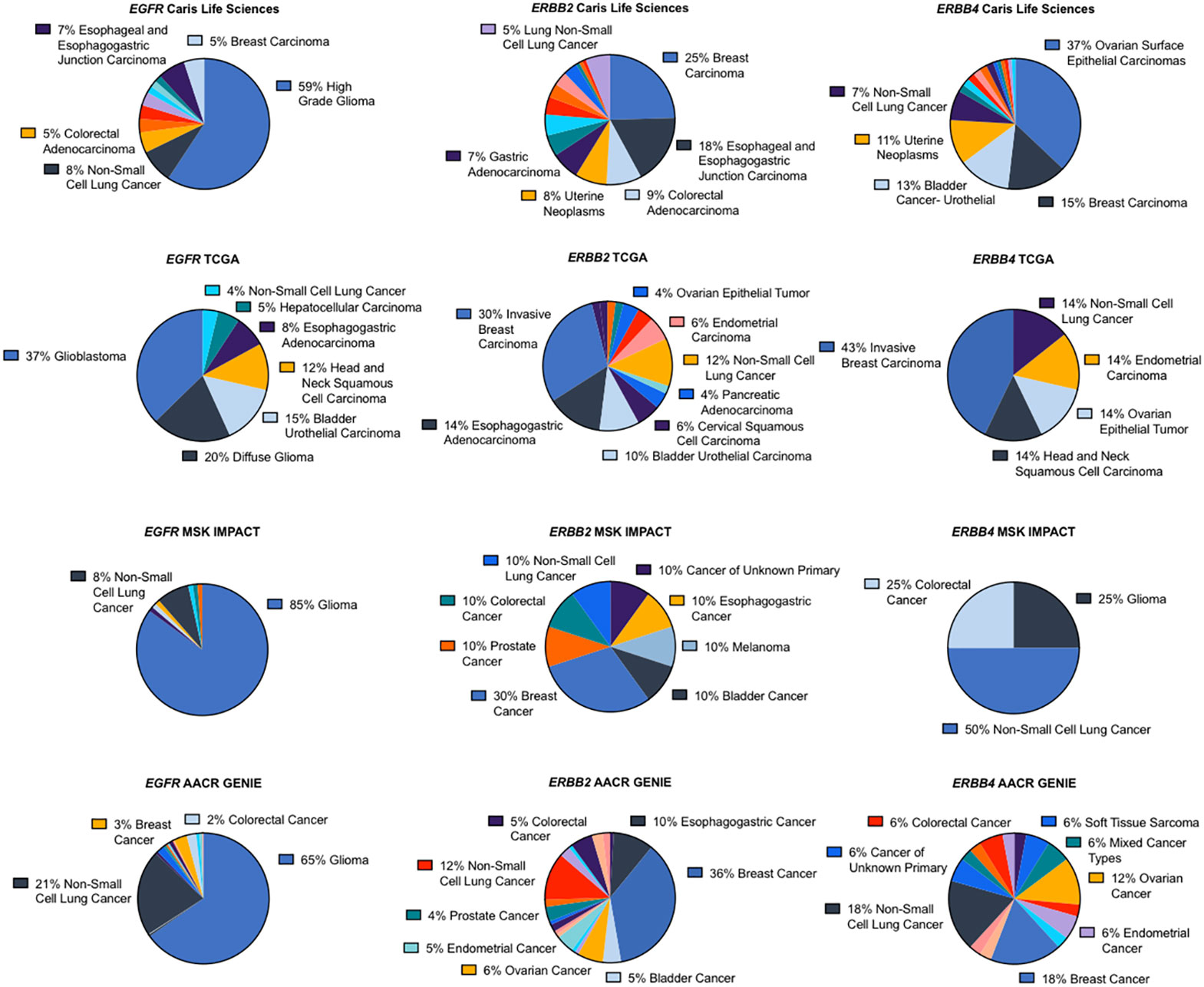
Figure 2 ERBB family fusions frequency by cancer type.
We categorized the fusions that we identified based on their genomic architecture and proposed mechanism of activation. The first class, classical 3’ fusions, have a 5’ gene fusion partner that introduces a new 5’ promoter that likely enhances expression and typically has protein features, such as coiled-coil domains, that may promote dimerization of the fusion kinase. Class II fusions have a 3’ gene fusion partner that potentially allows constitutive dimerization. Class III fusions are tandemly linked to another kinase domain that may promote activation, presumably through enhanced dimerization. Lastly, Class IV fusions are tethered to an adapter protein that we propose allows direct activation of downstream pathways (Figure 3A). We assigned each fusion we identified to a class and found that overall Class II fusions were the most common among ERBB fusions, followed by Class I. Interestingly, we noticed that ERBB2 fusions had an increased rate of Class III and class IV fusions, compared to EGFR or ERBB4 fusions (Figure 3B).
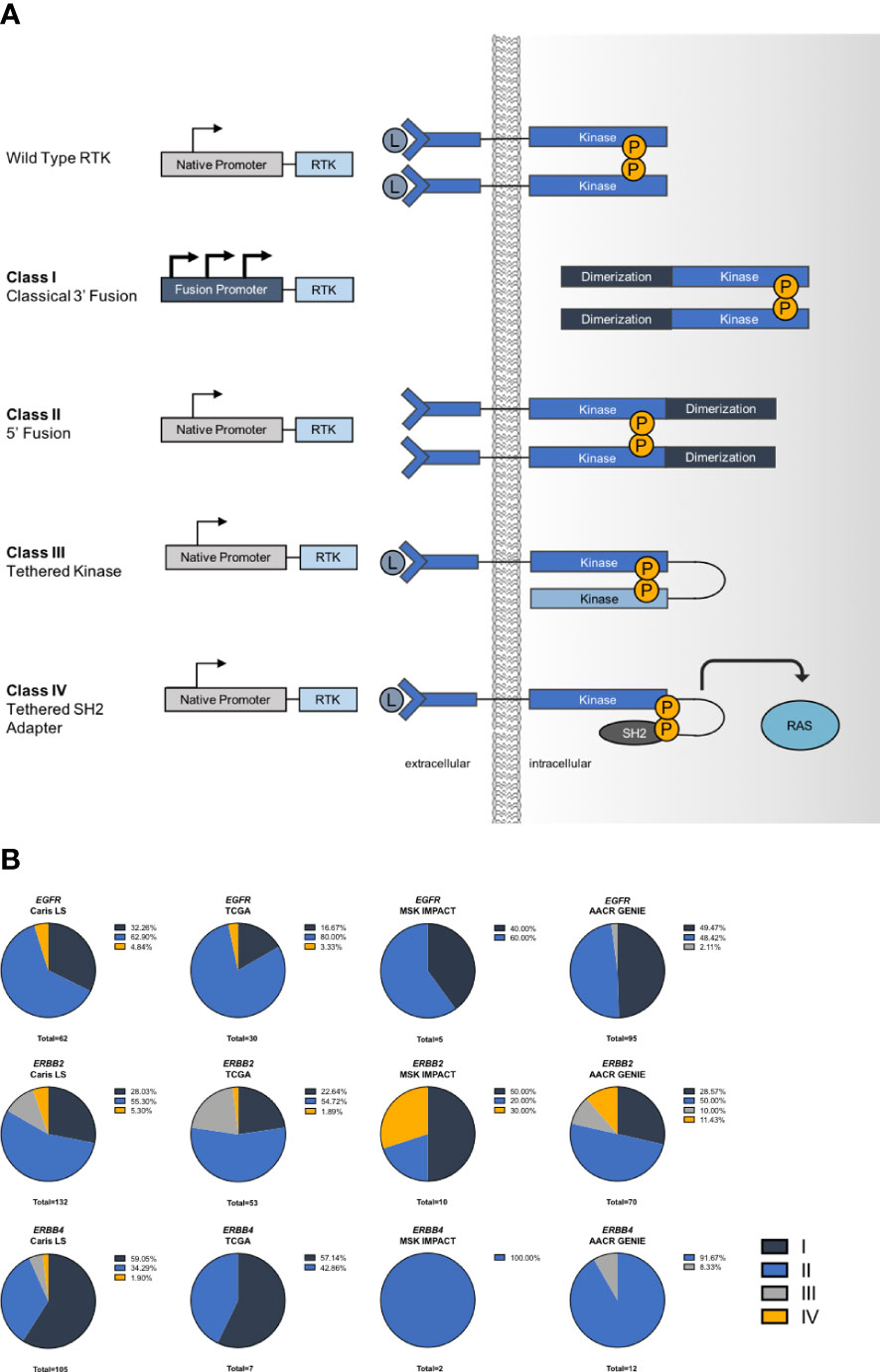
Figure 3 Mechanisms of activation in ERBB fusions. (A) Schematic describing classes of fusion oncogenes. (B) Frequency of each class of ERBB fusions.
We then analyzed which mutations co-occur with ERBB fusions. EGFR fusions frequently co-occurred with TERT mutations (>60%) and EGFR mutations (~30%). We also observed that ERBB2 and ERBB4 fusions co-occurred with TP53 mutations (~60-80%) at a rate that was higher than the mutation rate found in the data sets queried (36-42%) (Figure 4).
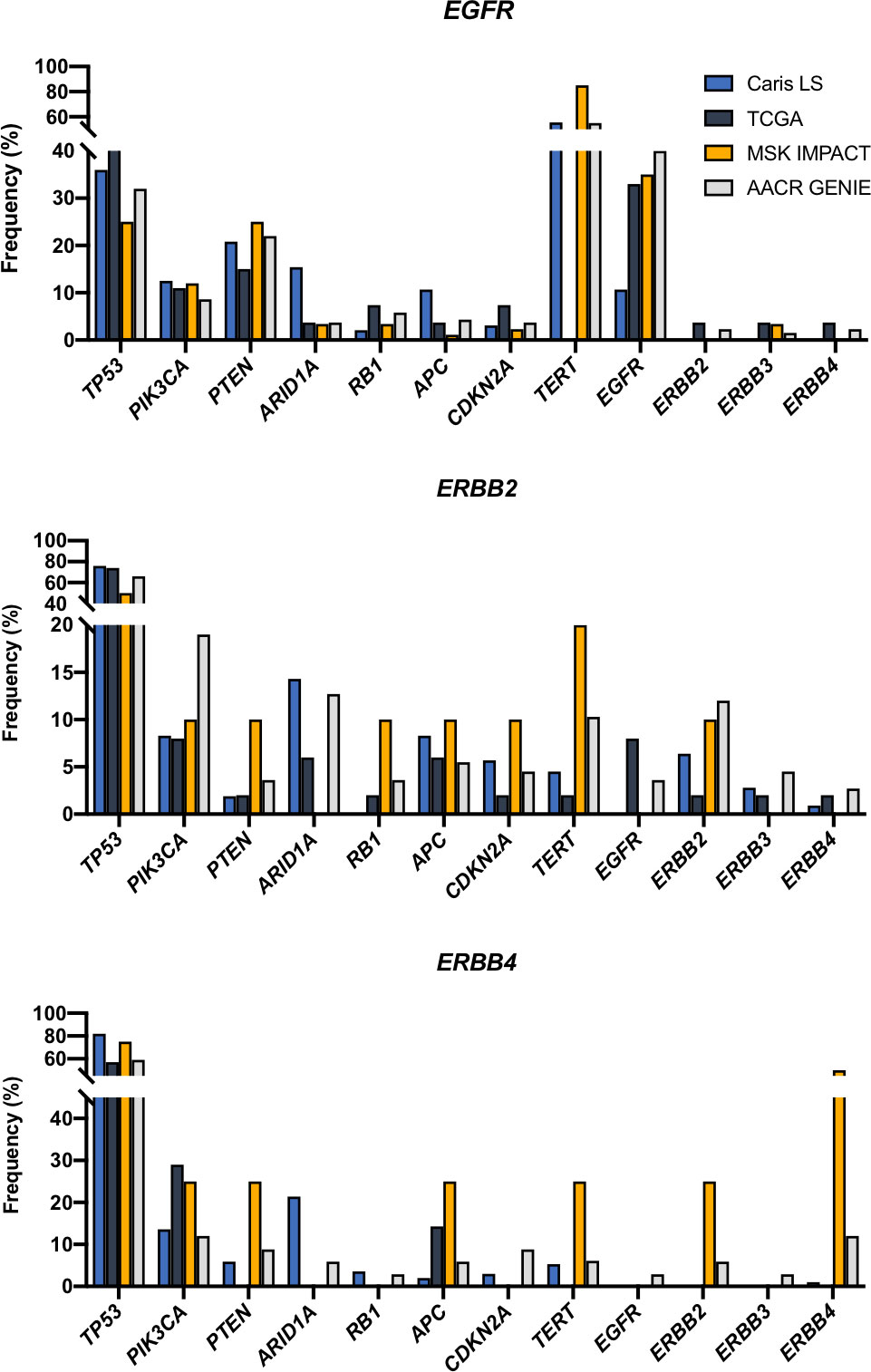
Figure 4 Co-occurring mutations in selected cancer-related genes.
Class I and Class II fusions have been extensively characterized and evidence supports oncogenic activity (13, 27, 28). Given the lack of functional characterization of Class III and IV fusions, we selected several of the EGFR and ERBB2 fusions from the novel Class III and IV groups to analyze in vitro, including EGFR-ERBB4 (Class III), EGFR-SHC1 (Class IV), ERBB2-SHC1 (Class IV) and ERBB2-GRB7 (Class IV) (Figure 5A). First, we attempted to express these fusions in the Ba/F3 cell line, which is a murine B-cell cell line that is dependent on the addition of exogenous IL3 for proliferation but can become IL3-independent following oncogenic transformation. Ultimately, we were unable to transform the Ba/F3 cells with introduction of ERBB fusions. We hypothesized that because Ba/F3 cells may be lacking expression of signaling proteins necessary for these ERBB family fusions to successfully transform cells and therefore attempted to introduce ERBB fusions into a lung adenocarcinoma cell line. Previously we have demonstrated that H3122 can serve as model cells for novel fusions such as NRG1 fusions (29). Parental H3122 cells without ERBB fusions are insensitive to EGFR or HER2 inhibition (29). Other groups have also used similar techniques to model novel oncogenes in lung adenocarcinoma cell lines (30). We transduced the novel ERBB fusions into the EML4-ALK fusion expressing H3122 cell line using lentivirus. We then performed short term selection with the ALK inhibitor alectinib to eliminate non-transduced cells and to select for cells containing ERBB fusions, which should inhibit proliferation of untransformed cells that remain dependent on ALK signaling, but not those cells expressing the ERBB fusions (Figure 5B). Once the cells were able to proliferate in 100nM alectinib we assessed their sensitivity to several EGFR/HER2 inhibitors. We found that both the ERBB2-GRB7+ and ERBB2-SHC1+ cells were sensitive to the HER2 inhibitors afatinib, lapatinib and tarloxotinib suggesting both that the cells were now reliant on the ERBB2 fusion for proliferation and that they are sensitive to HER2 inhibition. The transformed cells were less sensitive to the EGFR inhibitor gefitinib (Figure 5C).
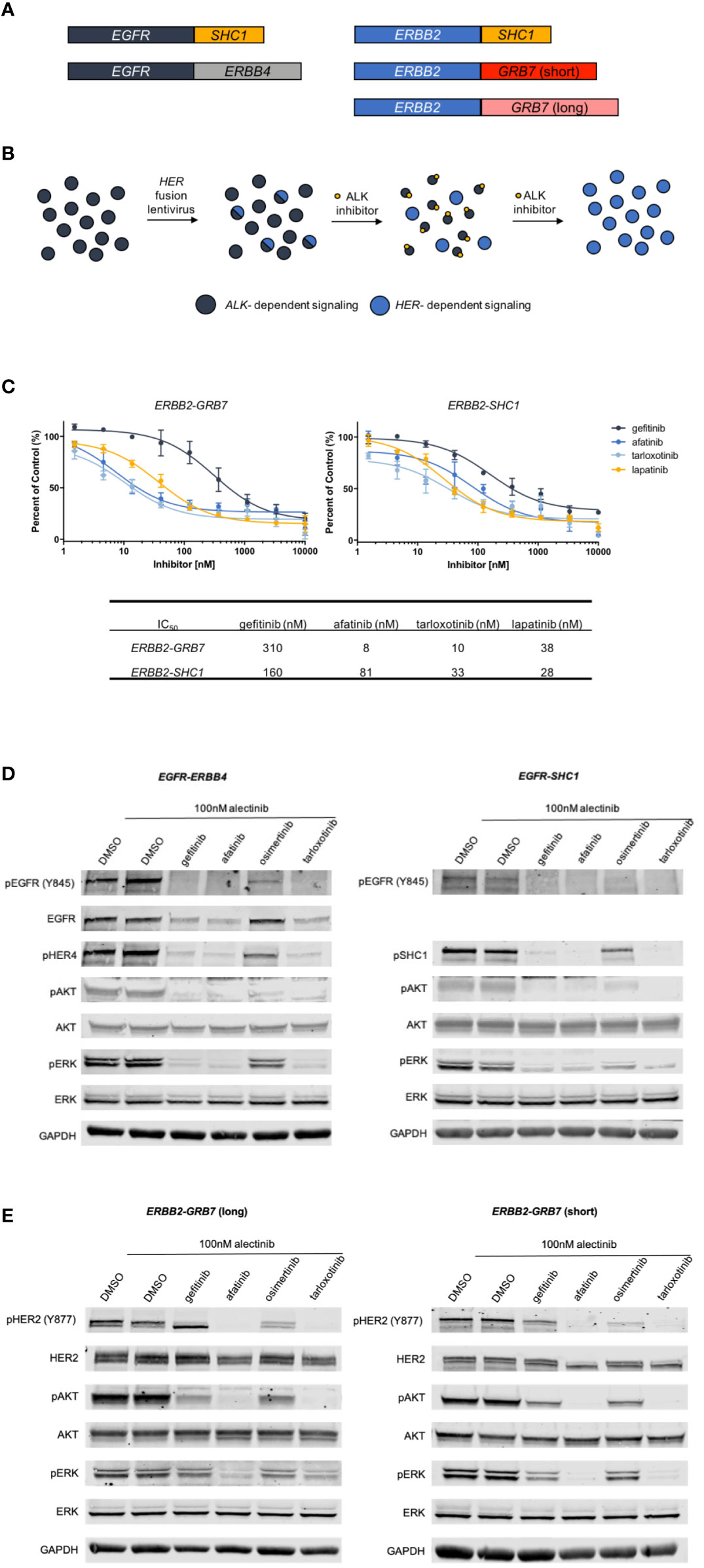
Figure 5 Derivation of EGFR and ERBB2 fusion expressing cell lines. (A). Schematic of ERBB fusions modeled in in vitro experiments. (B) Schematic describing the generation and selection of cells with EGFR or ERBB2 fusions. ALK+ H3122 cells were transduced with ERBB fusion lentivirus, then treated with increasing doses of the ALK inhibitor alectinib to select for cells expressing ERBB fusions. (C) MTS proliferation assays of ERBB2-GRB7 (short) and ERBB2-SHC1 expressing cells treated with increasing concentrations of gefitinib, afatinib, tarloxotinib or lapatinib for two hours in the presence of 100nM alectinib to suppress ALK signaling. Error bars = ±SEM, N=3. (D) Western blot analysis of cells expressing EGFR-ERBB4 and EGFR-SHC1 fusions treated with 100nM alectinib and gefitinib, afatinib, osimertinib or tarloxotinib. (E). Western blot analysis of ERBB2-GRB7 (short) or ERBB2-SHC1 expressing cells treated with 100nM alectinib and 1000 nM gefitinib, 100nM afatinib, 100nM osimertinib or 100nM tarloxotinib for two hours.
Next, we characterized the signaling changes in our ERBB+ cells in response to the EGFR inhibitors gefitinib, afatinib, osimertinib and tarloxotinib. Cells expressing either EGFR-ERBB4 or EGFR-SHC1 fusions showed a decrease in pEGFR following treatment, as well as decreased activation of the downstream signaling proteins pERK and pAKT. Additionally, we saw inhibition of the 3’ fusion partners pHER4 and pSHC1. As expected, the mutant-selective EGFR inhibitor osimertinib was less effective at inhibiting EGFR and downstream signaling, given the wild-type sequence of the EGFR kinase domain in these constructs (Figure 5D). We observed a similar pattern in our ERBB2 fusion cell lines where treatment with HER2 inhibitors (afatinib and tarloxotinib) resulted in decreased HER2 phosphorylation and decreased pERK and pAKT. Gefitinib and osimertinib, which have less activity against HER2, resulted in less inhibition of HER2 and downstream signaling (Figure 5E).
We found that ERBB family fusions involving EGFR, ERBB2 and ERBB4 are rare, but that these recurrent alterations collectively represent a notable patient population that may benefit from available targeted therapies or those under development. We estimate that ERBB fusions may be present in up to 0.7% of cancers, which represents about 12,646 patients based on the 2020 cancer incidence of 1,806,569 cases (31). These fusions were detected across four different data sets and a wide range of cancer types. We believe that the synthesis of data from several different datasets allowed us to capture more rare genetic events and alterations that may be present in different types of sequencing analyses as well as build a cohort of samples that incorporates more rare diseases and patient population characteristics. Several TKIs now have demonstrated activity across cancer types, so called “agnostic” indications, including the TRK inhibitor larotrectinib, the TRK/ROS1 inhibitor entrectinib, and the RET inhibitors selpercatinib and pralsetinib (5, 6, 32–34). Consequently, larotrectinib and entrectinib have been granted tumor-agnostic FDA approvals and selpercatinib and pralsetinib are approved for both NSCLC and papillary or medullary thyroid cancers with RET fusions or mutations, respectively. FGFR inhibitors have similarly been approved for use in both cholangiocarcinoma and urothelial cancers with FGFR alterations (7–9). This highlights the potential utility of comprehensive NGS assays for patients that can broadly detect ERBB fusions, as many of these alterations are in theory targetable with currently available drugs or those in development.
When we analyzed the mutations that co-occurred with ERBB family fusions we observed that ERBB2 and ERBB4 fusions had a higher-than-average rate of TP53 mutations while EGFR fusions had a lower-than-average rate of TP53 mutations. This suggests that TP53 mutations may be cooperative with ERBB2 and ERBB4 fusions and facilitate oncogenesis or provide a selective advantage. We also noticed many co-occurring TERT mutations in EGFR fusion samples, however, this is likely due to the high representation of glioblastomas, which frequently have TERT mutations (35). We also found that EGFR fusions co-occurred with EGFR point mutations in ~35% of samples.
We classified ERBB family fusions into several distinct classes based on genetic architecture and/or their proposed mechanism of activation. Most of the well characterized fusions, such as ALK, NTRK, ROS1 and RET fall into Class I. Here, we describe several additional structural types of gene fusions. EGFR fusions were most commonly detected in glioblastoma or glioma, most of which were EGFR-SEPT14 fusions. The majority of EGFR fusions we identified, including EGFR-SEPT14, were Class II (3’ fusions) which are likely activated by the addition of a dimerization domain. EGFR-SEPT14 fusions have previously been reported in glioblastoma, where they can activate STAT3 signaling and are sensitive to treatment with lapatinib or erlotinib (13). This study suggested that Class II EGFR fusions are likely functional, activating oncogenic events.
We identified a unique class of fusions that are activated through tethering to either a kinase domain or an adapter protein (Class III and Class IV). Recruitment of adapter proteins is a crucial step in activation of downstream signaling via RTKs. We found that Class III and IV fusions were more highly represented in ERBB2 fusions. We chose three adapter-linked fusions to model in vitro: EGFR-SHC1, ERBB2-SHC1, and ERBB2-GRB7. Both SHC1 and GRB7 are SH2 domain-containing adapter proteins responsible for activating MAPK signaling (36, 37). While these fusions have been previously reported, there have not been any functional studies of adapter protein-linked fusions (38). It is interesting to note that these fusions did not successfully transform Ba/F3 cells, suggesting that some ERBB fusions may require specific downstream proteins or expression profiles to transform cells. Because of this, we developed a novel approach for modeling oncogenic changes by using a cell line with a known driver oncogene (ALK) that is sensitive to an inhibitor (alectinib), which allowed us to select for cells containing EGFR or ERBB2 fusions. We have previously employed a similar approach with CRISPR/Cas9 generated fusions in ALK+ cells (29). We believe this approach will be widely useful and can be easily adapted for other cancer types or cell types and should be considered when model cell lines, such as Ba/F3, fail to support oncogenic transformation of putative novel oncogenes. These data suggest that there may only be certain cell types or conditions that are permissive for ERBB fusions to transform cells. Our data show that adapter-tethered EGFR and ERBB2 fusions are capable of sustaining cancer cell growth and proliferation and, importantly, are sensitive to inhibition with EGFR and/or HER2 inhibitors. We also developed an in vitro model of a Class IV fusion, EGFR-ERBB4. We found that this fusion behaved similarly to the Class III fusions and was capable of regulating downstream signaling through pERK and pAKT. It is also interesting to note that while the EGFR-ERBB4 chromosomal rearrangement contains EGFR exons 1-27, exons 26 and 27 are spliced out in the RNA transcript in order to maintain a proper reading frame. We have previously observed a similar mechanism in ROS1 fusions (39) (Supplementary Figure 2).
Based on our in vitro data, we expect ERBB fusions can be successfully targeted with TKIs. These fusions do present a potential therapeutic challenge because they retain a wild-type, rather than mutated, kinase domain that may be more challenging to target specifically as has been the case with EGFR and ERBB2 exon 20 mutations, which lack the significant therapeutic window observed for classical EGFR del 19 and L858R mutations. However, it has been previously shown that a patient with an EGFR kinase domain duplication, similar to the Class IV fusions, had a partial response to the EGFR inhibitor afatinib (40). Furthermore, there are two examples of patients with tumors harboring EGFR-RAD51 fusions being responsive to afatinib or erlotinib (15, 16). The hypoxia-activated EGFR/HER2 inhibitor tarloxotinib, may be another mechanism of more specifically targeting EGFR or ERBB2 fusions in only tumor cells (26). There is additional evidence that in patients harboring fusions, targeting other mutations without the fusion results in responses equivalent to patients with un-matched treatments; further suggesting that fusions are frequently key driver mutations (41). There is further evidence that gene fusions may also play an important role in tumor evolution and adaptation over time. Chromosomal instability is a hallmark of cancer that facilitates generation of new gene fusions, which are important genetic changes that enhance cellular fitness and facilitate oncogenesis (42). Furthermore, gene fusions may be a way to rapidly adapt to changes in the microenvironment; an area which warrants further study (43). Similarly gene fusions are already observed as resistance mechanisms to TKIs, further supporting their role in tumor evolution and response to changes in the microenvironment (29, 44).
Overall, we demonstrate that EGFR, ERBB2 and ERBB4 fusions are present across numerous cancer types and importantly are excellent candidates for targeted therapy development. We also propose a nomenclature system to help categorize fusion oncogenes. Finally, we show that the novel adapter-tethered and kinase-tethered fusions are oncogenic and sensitive to TKIs. These data show that ERBB family fusions are important therapeutic targets and warrant further study of their biology.
The datasets presented in this article from Caris Life Sciences are not publicly available because the raw data is protected proprietary information – these datasets are available for qualified researchers upon reasonable request and with permission of Caris Life Sciences.
LS, AE, RD and SV contributed to the conception and design of the study. LS and AE performed data mining and analysis. AL and AE-B performed experiments. EL, HB, MD, RK, JR, SIO, DB, CT, SD, WK and WE-D performed data acquisition, analysis and interpretation. The first draft manuscript was written by LS. All authors contributed to the article and approved the submitted version.
RD is currently employed by Rain Oncology and shareholder in Rain Oncology.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1115405/full#supplementary-material
Supplementary Figure 1 | (A) MTS proliferation assay for cells expressing a longer variant of the ERBB2-GRB7 fusion containing all exons for the GRB7 gene. Cells were treated with increasing concentrations of gefitinib, afatinib, tarloxotinib or lapatinib for two hours. Error bars = ±SEM, N=3. (B) Western blot analysis of ERBB2-GRB7 (long) expressing cells treated with 100nM alectinib and 1000 nM gefitinib, 100nM afatinib, 100nM osimertinib or 100nM tarloxotinib for two hours
Supplementary Figure 2 | EGFR exons 26 and 27 are skipped to maintain reading frame in fusion transcript of EGFR-ERBB4 fusion. Upper: Genomic sequencing of EGFR-ERBB4 fusion including exons 1-27 of EGFR and exons 18-28 of ERBB4. Lower: RNA sequencing of EGFR-ERBB4 fusion transcript and schematic illustrating skipping of exons 26 and 27.
1. Hong DS, Bauer TM, Lee JJ, Dowlati A, Brose MS, Farago AF, et al. Larotrectinib in adult patients with solid tumours: a multi-centre, open-label, phase I dose-escalation study. Ann Oncol (2019) 30(2):325–31. doi: 10.1093/annonc/mdy539
2. Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus crizotinib in untreated ALK-positive non-Small-Cell lung cancer. N Engl J Med (2017) 377(9):829–38. doi: 10.1056/NEJMoa1704795
3. Shaw AT, Ou SHI, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non–Small-Cell lung cancer. N Engl J Med (2014) 371(21):1963–71. doi: 10.1056/NEJMoa1406766
4. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med (2014) 371(23):2167–77. doi: 10.1056/NEJMoa1408440
5. Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, et al. Efficacy of selpercatinib in RET fusion-positive non-Small-Cell lung cancer. N Engl J Med (2020) 383(9):813–24. doi: 10.1056/NEJMoa2005653
6. Gainor JF, Lee DH, Curigliano G, Doebele RC, Kim D-W, Baik CS, et al. Clinical activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients (pts) with advanced RET-fusion+ non-small cell lung cancer (NSCLC). J Clin Oncol (2019) 37(15_suppl):9008. doi: 10.1200/JCO.2019.37.15_suppl.9008
7. Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol (2018) 36(3):276–82. doi: 10.1200/JCO.2017.75.5009
8. Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol (2020) 21(5):671–84. doi: 10.1016/S1470-2045(20)30109-1
9. Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med (2019) 381(4):338–48. doi: 10.1056/NEJMoa1817323
10. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science (2004) 304(5676):1497–500. doi: 10.1126/science.1099314
11. Venter DJ, Tuzi NL, Kumar S, Gullick WJ. Overexpression of the c-erbB-2 oncoprotein in human breast carcinomas: immunohistological assessment correlates with gene amplification. Lancet (1987) 2(8550):69–72. doi: 10.1016/s0140-6736(87)92736-x
12. Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res (2014) 79:34–74. doi: 10.1016/j.phrs.2013.11.002
13. Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet (2013) 45(10):1141–9. doi: 10.1038/ng.2734
14. Li Y, Zhang HB, Chen X, Yang X, Ye Y, Bekaii-Saab T, et al. A rare EGFR-SEPT14 fusion in a patient with colorectal adenocarcinoma responding to erlotinib. Oncol (2020) 25(3):203–7. doi: 10.1634/theoncologist.2019-0405
15. Zhu YC, Wang WX, Xu CW, Song ZB, Du KQ, Chen G, et al. EGFR-RAD51 fusion variant in lung adenocarcinoma and response to erlotinib: a case report. Lung Cancer (2018) 115:131–4. doi: 10.1016/j.lungcan.2017.12.001
16. Konduri K, Gallant JN, Chae YK, Giles FJ, Gitlitz BJ, Gowen K, et al. EGFR fusions as novel therapeutic targets in lung cancer. Cancer Discov (2016) 6(6):601–11. doi: 10.1158/2159-8290.CD-16-0075
17. Nakaoku T, Tsuta K, Ichikawa H, Shiraishi K, Sakamoto H, Enari M, et al. Druggable oncogene fusions in invasive mucinous lung adenocarcinoma. Clin Cancer Res (2014) 20(12):3087–93. doi: 10.1158/1078-0432.CCR-14-0107
18. Guenzi E, Pluvy J, Guyard A, Nguenang M, Rebah K, Benrahmoune Z, et al. A new KIF5B-ERBB4 gene fusion in a lung adenocarcinoma patient. ERJ Open Res (2021) 7(1):00582–2020. doi: 10.1183/23120541.00582-2020
19. Papp E, Hallberg D, Konecny GE, Bruhm DC, Adleff V, Noë M, et al. Integrated genomic, epigenomic, and expression analyses of ovarian cancer cell lines. Cell Rep (2018) 25(9):2617–33. doi: 10.1016/j.celrep.2018.10.096
20. Jonna S, Feldman RA, Swensen J, Gatalica Z, Korn WM, Borghaei H, et al. Detection of NRG1 gene fusions in solid tumors. Clin Cancer Res (2019) 25(16):4966–72. doi: 10.1158/1078-0432.CCR-19-0160
21. Fernandez-Cuesta L, Plenker D, Osada H, Sun R, Menon R, Leenders F, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov (2014) 4(4):415–22. doi: 10.1158/2159-8290.CD-13-0633
22. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
23. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6(269):pl1. doi: 10.1126/scisignal.2004088
24. AACR Project GENIE Consortium. AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov (2017) 7(8):818–31. doi: 10.1158/2159-8290.CD-17-0151
25. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med (2017) 23(6):703–13. doi: 10.1038/nm.4333
26. Estrada-Bernal A, Le AT, Doak AE, Tirunagaru VG, Silva S, Bull MR, et al. Tarloxotinib is a hypoxia-activated pan-HER kinase inhibitor active against a broad range of HER-family oncogenes. Clin Cancer Res (2021) 27(5):1463–75. doi: 10.1158/1078-0432.CCR-20-3555
27. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature (2007) 448(7153):561–6. doi: 10.1038/nature05945
28. Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med (2012) 18(3):378–81. doi: 10.1038/nm.2658
29. McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non-small cell lung cancer. Clin Cancer Res (2018) 24(14):3334–47. doi: 10.1158/1078-0432.CCR-17-2452
30. Floc'h N, Martin MJ, Riess JW, Orme JP, Staniszewska AD, Ménard L, et al. Antitumor activity of osimertinib, an irreversible mutant-selective EGFR tyrosine kinase inhibitor, in NSCLC harboring EGFR exon 20 insertions. Mol Cancer Ther (2018) 17(5):885–96. doi: 10.1158/1535-7163.MCT-17-0758
31. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
32. Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol (2020) 21(4):531–40. doi: 10.1016/S1470-2045(19)30856-3
33. Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol (2020) 21(2):271–82. doi: 10.1016/S1470-2045(19)30691-6
34. Drilon A, Siena S, Dziadziuszko R, Barlesi F, Krebs MG, Shaw AT, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1-2 trials. Lancet Oncol (2020) 21(2):261–70. doi: 10.1016/S1470-2045(19)30690-4
35. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
36. Ravichandran KS. Signaling via shc family adapter proteins. Oncogene (2001) 20(44):6322–30. doi: 10.1038/sj.onc.1204776
37. Daly RJ. The Grb7 family of signalling proteins. Cell Signalling (1998) 10(9):613–8. doi: 10.1016/S0898-6568(98)00022-9
38. Hechtman JF, Zehir A, Yaeger R, Wang L, Middha S, Zheng T, et al. Identification of targetable kinase alterations in patients with colorectal carcinoma that are preferentially associated with wild-type RAS/RAF. Mol Cancer Res (2016) 14(3):296–301. doi: 10.1158/1541-7786.MCR-15-0392-T
39. Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res (2012) 18(17):4570–9. doi: 10.1158/1078-0432.CCR-12-0550
40. Gallant JN, Sheehan JH, Shaver TM, Bailey M, Lipson D, Chandramohan R, et al. EGFR kinase domain duplication (EGFR-KDD) is a novel oncogenic driver in lung cancer that is clinically responsive to afatinib. Cancer Discov (2015) 5(11):1155–63. doi: 10.1158/2159-8290.CD-15-0654
41. Nikanjam M, Okamura R, Barkauskas DA, Kurzrock R. Targeting fusions for improved outcomes in oncology treatment. Cancer (2020) 126(6):1315–21. doi: 10.1002/cncr.32649
42. Glenfield C, Innan H. Gene duplication and gene fusion are important drivers of tumourigenesis during cancer evolution. Genes (2021) 12(9):1376. doi: 10.3390/genes12091376
43. Luo W. Nasopharyngeal carcinoma ecology theory: cancer as multidimensional spatiotemporal “unity of ecology and evolution” pathological ecosystem. Theranostics (2023) 13(5):1607–31. doi: 10.7150/thno.82690
Keywords: gene fusion, oncogene, EGFR, ERBB2, ERBB4 words: 3, 500 ERBB family fusions across cancer types
Citation: Schubert L, Elliott A, Le AT, Estrada-Bernal A, Doebele RC, Lou E, Borghaei H, Demeure MJ, Kurzrock R, Reuss JE, Ou S-HI, Braxton DR, Thomas CA, Darabi S, Korn WM, El-Deiry WS and Liu SV (2023) ERBB family fusions are recurrent and actionable oncogenic targets across cancer types. Front. Oncol. 13:1115405. doi: 10.3389/fonc.2023.1115405
Received: 03 December 2022; Accepted: 05 April 2023;
Published: 24 April 2023.
Edited by:
Daekyu Sun, University of Arizona, United StatesReviewed by:
Gajanan Bhat, Spectrum Pharmaceuticals, United StatesCopyright © 2023 Schubert, Elliott, Le, Estrada-Bernal, Doebele, Lou, Borghaei, Demeure, Kurzrock, Reuss, Ou, Braxton, Thomas, Darabi, Korn, El-Deiry and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen V. Liu, c3RlcGhlbi5saXVAZ3VuZXQuZ2VvcmdldG93bi5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.