 Mengxue Yu
Mengxue Yu Kaiyu Qian
Kaiyu Qian Gang Wang
Gang Wang Yu Xiao
Yu Xiao Yuan Zhu3*
Yuan Zhu3* Lingao Ju
Lingao Ju- 1Department of Biological Repositories, Zhongnan Hospital of Wuhan University, Wuhan, China
- 2Department of Urology, Zhongnan Hospital of Wuhan University, Wuhan, China
- 3Human Genetic Resources Preservation Center of Hubei Province, Wuhan, China
- 4Wuhan Research Center for Infectious Diseases and Cancer, Chinese Academy of Medical Sciences, Wuhan, China
- 5Medical Research Institute, Wuhan University, Wuhan, China
SET domain-containing 2 (SETD2) is a lysine methyltransferase that catalyzes histone H3 lysine36 trimethylation (H3K36me3) and has been revealed to play important roles in the regulation of transcriptional elongation, RNA splicing, and DNA damage repair. SETD2 mutations have been documented in several cancers, including clear cell renal cell carcinoma (ccRCC). SETD2 deficiency is associated with cancer occurrence and progression by regulating autophagy flux, general metabolic activity, and replication fork speed. Therefore, SETD2 is considered a potential epigenetic therapeutic target and is the subject of ongoing research on cancer-related diagnosis and treatment. This review presents an overview of the molecular functions of SETD2 in H3K36me3 regulation and its relationship with ccRCC, providing a theoretical basis for subsequent antitumor therapy based on SETD2 or H3K36me3 targets.
1 Introduction
Renal cell carcinoma (RCC) is one of the most prevalent malignancies with a case-fatality rate among urinary tract tumors (1, 2). There are several pathological types of renal cancer, such as clear cell RCC (ccRCC), papillary RCC (pRCC), and chromophobe RCC (chRCC). In the WHO classification, with a list of RCC defined molecularly, including TFE3-rearranged RCC, TFEB-rearranged RCC, ELOC (TCEB1)-mutated RCC, Fumarate hydratase (FH)-deficient RCC, Succinate dehydrogenase (SDH)-deficient RCC, ALK-rearranged RCC, SMARCB1-deficient RCC, and so on (3), a molecular perspective to define RCC is necessary. ccRCC is the major type with a high incidence rate and poor prognosis. Remarkably, several secondary mutations of tumor suppressor genes and chromatin regulators have been identified near von Hippel-Lindau (VHL), including PBRM1, BAP1, and SETD2 (4). Furthermore, metastatic ccRCC occurs in about 30% of patients, and there are few effective treatment options available (5). Despite advances in chemotherapeutic drugs, chemotherapy resistance remains a problem in ccRCC treatment; therefore, there is an urgent need to understand the regulatory mechanism underlying the recurrence and metastasis of ccRCC, identify possible therapeutic targets and develop new therapeutic options.
Epigenetic regulation, including histone modification, plays a crucial role in maintaining eukaryotic genome stability, gene expression regulation, and chromatin structure. Histone H3 lysine 36 trimethylation (H3K36me3) is involved in the regulation of transcriptional activation and RNA splicing, as well as DNA repair and recombination (6). In mammalian cells, SETD2 is the main H3K36me3 methyltransferase (7), and genomic profiling of ccRCC clinical samples revealed high-frequency SETD2 mutations. SETD2 has been reported to accelerate ccRCC progression (4, 8) and is a potential prognostic and predictive marker in both localized and metastatic RCC (9). This paper reviews the multiple roles and functions of SETD2 in the occurrence and progression of ccRCC.
2 Protein structure of SETD2
The human SETD2 gene is located in the p21.31 region of chromosome 3, where the copy number is frequently lost in many tumors. Thus, SETD2 is generally considered a tumor suppressor. The human SETD2 protein consists of several conserved functional domains, containing the AWS (associated with SET)-SET-PS (post-SET) domains, WW domains, SRI (Set2-Rpb1 Interacting domain), SETD2-hnRNP interaction (SHI) domains, and a large unstructured N-terminal domain (Figure 1).
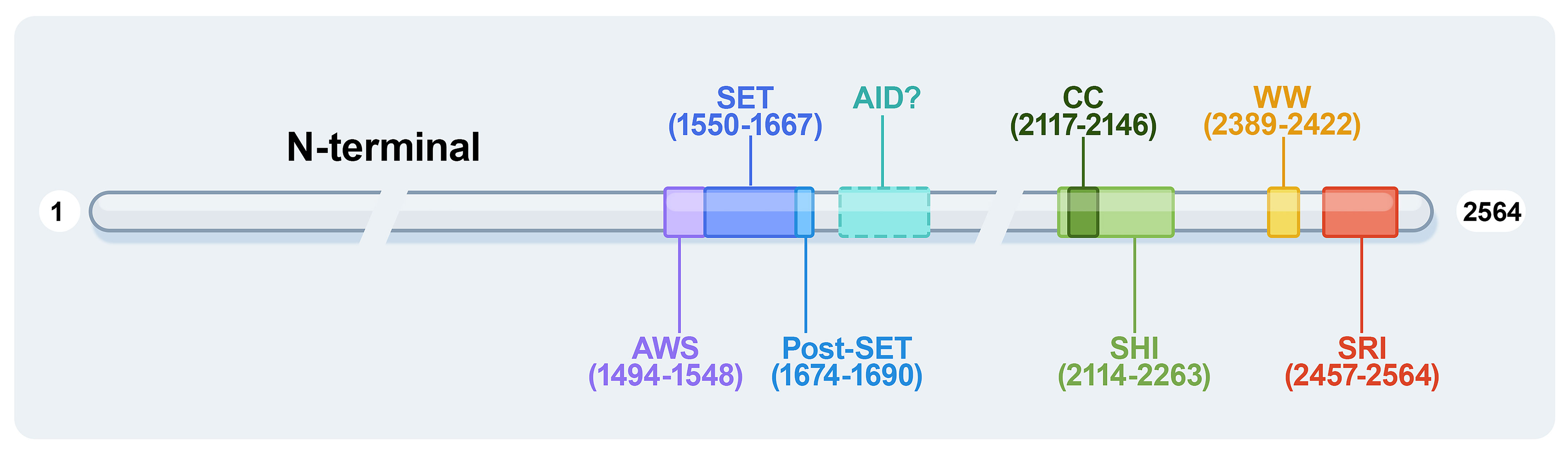
Figure 1 The protein domains of human SETD2. AWS, associated with-SET domain; AID, auto-inhibitory domain; WW, tryptophan-tryptophan domain; CC, coiled-coil domain; SHI, SETD2-hnRNP interaction domain; SRI, Set2-Rpb1 interacting domain.
2.1 The AWS-SET-PS domains
The AWS-SET-PS domains are essential as a catalytic methyltransferase domain for H3K36me3; the AWS and post-SET domains are flanked onto the SET domain at the N- and C-terminally, respectively. All methylation of H3K36me2 to H3K36me3 depends on the SET domain, with S-adenosylmethionine (SAM) as the cofactor, providing an additional methyl (10). It is reported that the H3K36M oncohistone mutation inhibits SETD2 methyltransferase activity; the structure of the SETD2-H3K36M-SAM complex suggests that SAM indirectly affects the SETD2-H3K36M interaction and maintains the SET domain in the proper fold state (11). The AWS-SET-PS domains of SETD2 recognize the α-N helix of histone H3 and bind to the nucleosome DNA by cryo-EM analyses (12).
2.2 The Set2-Rpb1 interacting domain
The SRI domain of 108 amino acids at the C-terminal end is the main region that interacts with RNA polymerase II (RNAPII), entering a transcription elongation phase. The SRI domain binds to RNAPII-C terminal domain (RNAPII-CTD) Ser5P and Ser2P (13) and promotes SETD2 activity to modify H3K36me3, particularly along the 3’ end of the coding sequences of long genes (Figure 2). This association is crucial for SETD2 activity and stability. In addition, the SRI domain of SETD2 is also required for microtubule lysine 40 trimethylation (α-TubK40me3) (14, 15) (Figure 2). Molenaar et al. recently reported that overexpression of the SRI domain significantly inhibited H3K36me3 and enlarged cell size (16).
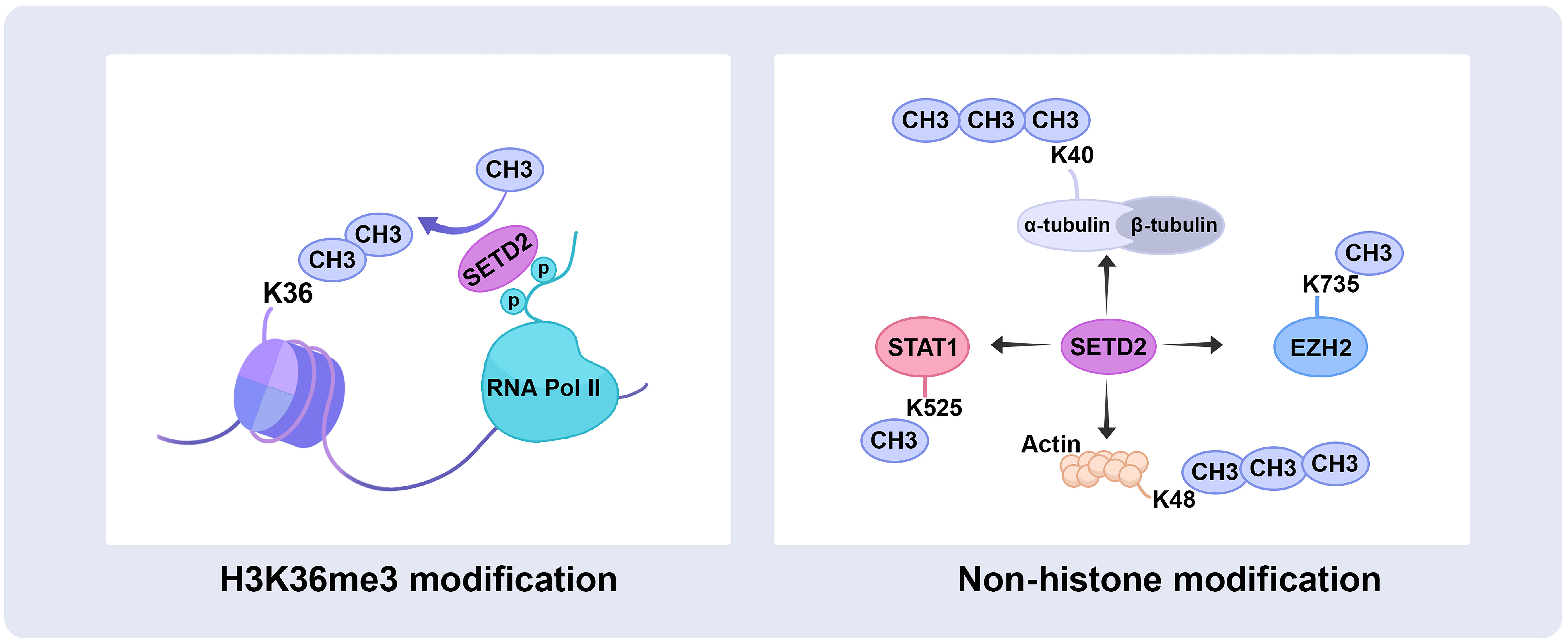
Figure 2 SETD2 catalyzes histone and non-histone substrate methylation. SETD2 has initially identified a methyltransferase that trimethylates H3 histones on lysine 36, also occurs on the non‐histone substrate, trimethylates α-tubulin at lysine 40 and actin at lysine 68, as well as methylates STAT1 at lysine 525 and EZH2 at lysine 735. RNA Pol II, RNA polymerase II; STAT1, signal transducer and activator of transcription 1; EZH2, enhancer of zeste homolog 2.
2.3 The WW domain
The WW domain comprises two conserved tryptophan (W) residues in the SETD2 C-terminus. The WW domain interacts with proline-rich or proline-containing motifs of other proteins mediating protein-protein interactions (17). For example, the WW domain interacts with the Huntingtin (HTT) proline-rich region (PRR) and the actin-binding adaptor HTT-interacting protein 1-related protein (HIP1R), leading to SETD2 tri-methylating actin at lysine 68 (ActK68me3) (Figure 2). The SETD2-HTT-HIP1R axis modifies actin, which increases actin polymerization and promotes cell migration (18).
2.4 The SHI domain
The structure of the coiled-coil (CC) domain has been predicted by in-silico calculations to be a conserved motif that participates in protein-protein interactions in yeast and promotes protein homodimerization. In human SETD2, the predicted structure of the CC domain is much shorter (19). The SHI domain contains the CC domain and adjacent unstructured sequences in a recently identified region. The histone mark H3K36me3 is known to regulate splicing (8). The SHI region interacts with heterogeneous nuclear ribonucleoprotein L (hnRNP L), RNA-recognition motif 2 (RRM2), as well as other splicing-related factors associated with RNA processing (20). Crystallographic analysis revealed that the Leu-Leu in the SHI domain is important for the interaction (21). Moreover, the double mutant that lacks both the SHI and SRI domains lost practically all catalyzing H3K36me3 activity, indicating that these domains are regulators of SETD2 activity. SETD2 activity toward H3K36me3 modification is similarly influenced by the SHI domain (20).
2.5 Auto-inhibitory domain
The AID domain in the middle region of Set2 (a yeast ortholog of human SETD2) suppresses SET domain activity, and the AID domain suppresses its catalytic activity when the SRI domain is lost. AID mutations usually lead to excessive activity of Set2 in vivo and promote the abnormal methylation of Set2 to histones (22). The AID and SRI domains synergistically control the catalytic SET domain, with AID mutations resulting in changes in Set2 protein stability and binding to RNAPII-CTD and variable H3K36me3 levels. In summary, Set2 AID exerts repressive effects requiring the presence of the SRI domain and Set2 SRI to interact with RNAPII and histones, ensuring that H3K36 methylation occurs explicitly on the active transcript chromatin. Therefore, under specific growth conditions, the Set2 autoinhibitory domain may serve as a target for other regulators (23). It would be intriguing to ascertain whether the Set2 AID interacts with any proteins and whether this interaction infuses Set2 activity via the AID-SRI axis (19). All the above studies are implemented in yeast, but there are no reports about the structure and function of human AID as yet.
2.6 The large unstructured N−terminal domain
Human SETD2 has an extended N-terminal region with unknown function (~1400 amino acids) and is unstructured. SETD2 is an unstable protein that depends on the degradation of the proteasome (24). It was recently reported that the N-terminal region regulates its half-life by the proteasome system, and removal of the N-terminal region leads to SETD2 stabilization (25), and a segment (aa 1104-1403) of the N-terminal region contributes to SETD2 degradation by the proteasome (24). SETD2 is an intrinsically aggregation-prone protein, and the N-terminal region contributes to SETD2 droplet formation in vivo, a property that is enhanced by its reduced degradation. The N-terminal region is conducive to the liquid-liquid phase separation of the protein, and the phase separation behavior of SETD2 intensifies with the removal of the N-terminal fragment (26). Thus, the N-terminal fragment of SETD2 regulates the amount of SETD2 protein required in the cell and may contribute to its role in regulating transcription and splicing.
3 SETD2 and clear cell renal cell carcinomas
3.1 SETD2 mutation and ccRCC
VHL inactivation occurs in 90% of all ccRCCs, and several mutations in tumor suppressor genes on chromosome 3p have been identified: PBRM1, BAP1, and SETD2 (4, 27). SETD2 mutations occur in about 15% of ccRCC (4). Mono-allelic and bi-allelic mutations in SETD2 are observed in many cancers, including ccRCC (28–30). Bi-allelic mutations in SETD2 cause loss of H3K36me3 in ccRCC (31). SETD2 gene inactivation mutations are a prevalent molecular feature, and SETD2 deficiency is associated with ccRCC recurrence and poor prognosis (Tables 1, 2). Moreover, SETD2 mutations are more frequently found in late-stage ccRCC tumors, which is related to a higher and earlier risk of relapse and poor survival outcomes (9, 50).
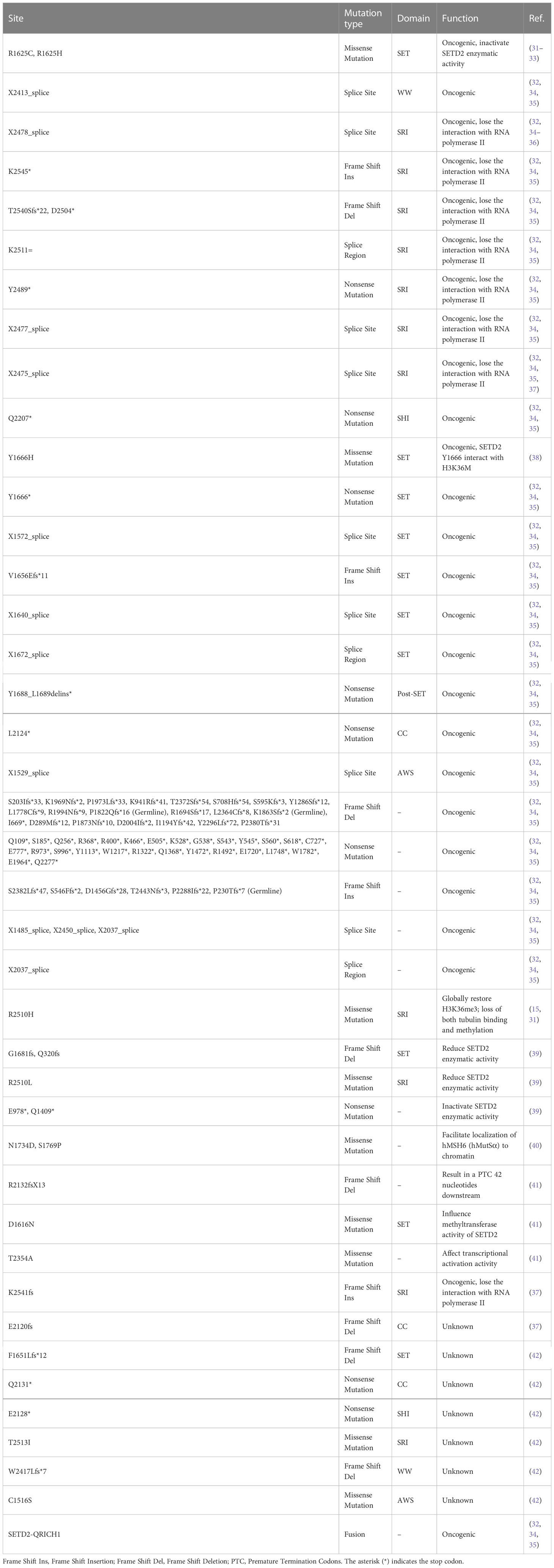
Table 1 List of SETD2 mutations reported in ccRCC.
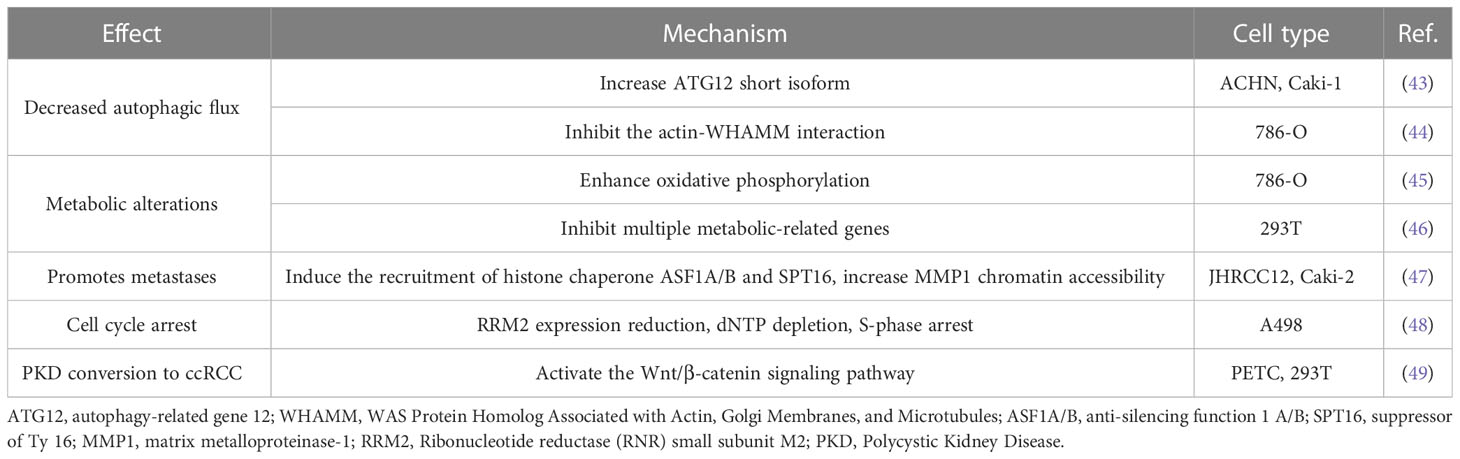
Table 2 Effects and mechanisms of SETD2 deficiency in ccRCC.
Referenced by cBioPortal database and reported research (31, 32, 34, 36–38, 42), SETD2 mutations were identified in ccRCC predominantly inactivating, containing nonsense mutations, missense mutations, frame shift, and fusion, which lead to loss of function, such as mutations R1625C or R1625G, resulting in a complete loss of SETD2 enzymatic activity (31, 33) (Table 1). The presence of intratumor heterogeneity was confirmed in metastatic renal-cell carcinoma tumors, which demonstrated independent and different SETD2 mutations in different sections of an individual tumor (51). Thus, SETD2 plays a critical role in the development and progression of ccRCC.
3.2 SETD2 serves as a tumor-suppressor gene in ccRCC
3.2.1 Cryptic transcription
Cryptic transcription initiates transcription from a downstream “promoter-like” region and produces short and meaningless transcripts in gene bodies. Previous studies have demonstrated that SETD2 suppresses cryptic transcription initiation from within several active gene bodies (52, 53). The histone chaperone FACT and its subunits SPT16 and SPT6 promote transcriptional elongation through nucleosome recombination, and deletion of SETD2 reduces recruitment to FACT and plays a critical role in repressing cryptic intragenic transcriptional initiation (52). In yeast, Set2-mediated prevention of cryptic intragenic transcription is independent on histone deacetylation (54). In mammalian cells, SETD2-mediated H3K36me3 recruits DNA-methyltransferase 3B (DNMT3B), resulting in a high density of DNA methylation, and thus represses transcription from alternate intragenic promoters or initiation of cryptic transcription (55), protecting RNAPII from inappropriate transcription re-initiation and enforced silence intragenic transcription (53, 56). In conclusion, SETD2 is crucial in maintaining active gene bodies dormant in mammalian cells (Figure 3).
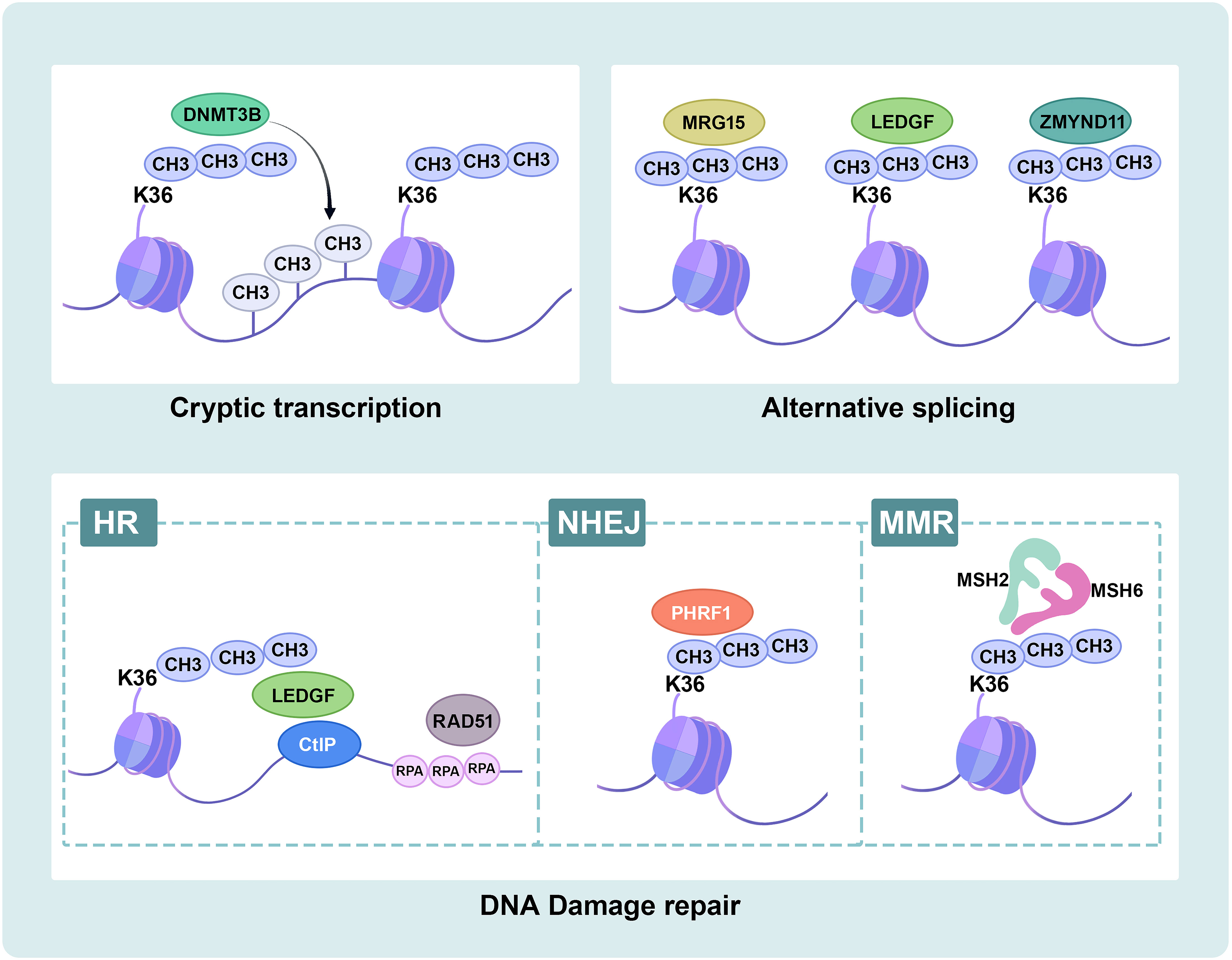
Figure 3 Schematic overview of SETD2 functions. SETD2-mediated H3K36me3 plays important biological roles in ccRCC. Cryptic transcription: SETD2-mediated H3K36me3 recruits DNMT3B to target intragenic DNA methylation. RNA splicing: SETD2-mediated H3K36me3 recruits splice factors MRG15, LEDGF and ZMYND11 to modulate alternative splicing events. DNA damage repair: SETD2-mediated H3K36me3 recruits LEDGF, and LEDGF binds CtIP at the break site to promote HR repair. SETD2-mediated H3K36me3 recruits PHRF1 to modulate NHEJ repair. SETD2-mediated H3K36me3 recruits hMutSα (hMSH2-hMSH6) to MMR repair. DNMT3B, DNA-methyltransferase 3B; MRG15, MORF4-related gene on chromosome 15; LEDGF, lens epithelium-derived growth factor; ZMYND11, Zinc finger MYND-domain containing 11; HR, Homologous recombination; CtIP, C-terminal binding protein interacting protein; RPA, replication protein A; NHEJ, non-homologous end-joining; PHRF1, plant homeodomain of Ring finger domains 1; MMR, mismatch repair; MSH2, MutSα homolog 2; MSH6, MutSα homolog 6.
3.2.2 RNA splicing
SETD2 is linked to the RNA splicing process. Compared to controls, SHI domain deletion mutation lost interaction with hnRNP L and did not affect splicing events (21). SETD2-deficient ccRCC is susceptible to mis-splicing. Gene set enrichment analysis (GSEA) shows that SETD2-deficient negatively enriched the gene related to the mRNA splicing pathway (57). A genome-wide transcript profile for SETD2-deficient primary ccRCC tumors demonstrated that altered splicing patterns or splicing defects, including intron retention and variation in exon utilization, are widely present in SETD2-deficient cancers. Notably, active genes also revealed increased chromatin accessibility (39). The increased chromatin accessibility of upstream abnormally spliced exons and decreased occupancy of nearby nucleosomes significantly contribute to the splicing defect in tumors with H3K36me3 deficiency (58).
Proteins containing the Pro-Trp-Trp-Pro (PWWP) domain play an important role in recognizing H3K36me3. MORF4-related gene on chromosome 15 (MRG15) can bind to H3K36me3 (59, 60) and recruit polypyrimidine tract-binding protein (PTB) to its target alternatively spliced exon sites (61). Lens epithelium-derived growth factor (LEDGF) binds to H3K36me3 (62), as well as to both chromatin and multiple regulators, to modulate alternative splicing events and influence transcription elongation (63, 64). Zinc finger MYND-domain containing 11 (ZMYND11) directly binds to H3K36me3 and H3K36me3-modified chromatin to regulate RNA splicing and Pol II elongation (65, 66). Furthermore, the deficiency of SETD2-mediated H3K36me3 reduces the recruitment of readers, resulting in splicing defects (Figure 3).
3.2.3 DNA damage and repair signaling
SETD2 is vital in the DNA damage response (DDR) by generating H3K36me3. Cell death occurs if DNA repair fails, and tumor development may arise from incorrect DNA repair. SETD2 facilitates DNA double-strand breaks (DSBs) repair by homologous recombination (HR), activating replication protein A (RPA) single-stranded DNA (ssDNA)-binding protein complex loading and the formation of RAD51 presynaptic filaments (35, 62, 67, 68). ATM is activated in DSB, then phosphorylates a variety of downstream effector proteins, such as p53. SETD2-deficient cancer cells failed to activate p53 and displayed lower cell survival in DNA damage (62, 67, 68). Ectopic expression of demethylase KDM4A decreased H3K36me3 levels and resulted in HR (62). Consistent with this, LEDGF recruits and binds C-terminal binding protein interacting protein (CtIP), promoting HR by CtIP-dependent DNA end resection (69). Accordingly, the loss of SETD2 obstructs HR repair (70, 71). Also, SETD2 promotes DSB repair via combination with plant homeodomain (PHD) of Ring finger domains 1 (PHRF1), modulating non-homologous end-joining (NHEJ) and stabilizing genomic integrity (72). SETD2 has also been proven to trigger DNA mismatch repair (MMR). Specifically, the mismatch recognition protein hMutSα (hMSH2-hMSH6), hMSH6 contains a PWWP domain that recruits and interacts with H3K36me3 like many other H3K36me3 effector proteins. hMSH6 foci are reduced in SETD2 knockdown cancer cells (40). The crystal structure modeling revealed that H3G34R/V mutations block the SETD2 catalytic activity and inhibit H3K36me3-MSH6 interaction from inducing tumorigenesis (73). SETD2-deficient cells exhibit microsatellite instability (MSI) with a high frequency of spontaneous mutations (40). Compared to introns and non-transcribed regions, H3K36me3 and MutS are more enriched in exons as well as active transcriptional regions and transcriptionally protect against actively transcribed genes (74). Recent studies suggest that targeting DDR is feasible to achieve immunotherapy in ccRCC (75, 76) (Figure 3).
3.2.4 Autophagy
Autophagy is involved in physiological and pathological processes and tightly regulated by a network of autophagy-related genes (ATG). Also, the actin cytoskeleton regulates autophagy dynamics (77). Autophagy is an intracellular degradation system procedure associated with cytoplasmic events, and key epigenetic events are recognized to be significant for this progression. De facto, histone post-translational modification plays a central role in regulating transcriptional programs and epigenetic networks during autophagy (78–83).
Autophagy is an important regulatory process in ccRCC (84–86). The deficiency of SETD2 in ccRCC cells reduces LC3-II expression, which is linked with abnormal cumulative ATG12 in free and complexes containing ATG12, except for the ATG5-ATG12 complex. Furthermore, SETD2-loss deregulates alternative splicing, which is related to increased ATG12 short isoform and reduced conventional ATG12 long isoform (43). Another research confirms that SETD2 knockdown causes a decreased expression of ATG14 long isoform in HeLa cells (87). Whether ATG14 long isoforms expression is down-regulated in ccRCC cells with a high-frequency mutation in SETD2 remains to be further investigated.
Autophagy also involves the actin cytoskeleton. As mentioned before, SETD2 trimethylates actin (ActK68me3), cells lacking SETD2 have decreased interaction of the actin nucleation-promoting factor WHAMM with its target actin, actin filaments are required for initiation of autophagy in ccRCC, and autophagy markers LC3-II and p62 are decreased (44).
Recent studies display that the components of the autophagic system play a central role in regulating the innate immune system (88, 89). In pancreatic ductal adenocarcinoma cells, autophagy deficiency results in increased MHC-I expression and increased infiltration of CD8+ T cells. Inhibition of autophagy or lysosomal production increases MHC-I expression, enhances the adaptive immune response, and inhibits the generation of tumors (90). Thus, tumor-autonomous autophagy can alter tumor growth by regulating immune responses. SETD2 promotes autophagy flux. Therefore, further understanding the pathways inhibited by SETD2 deficiency in ccRCC may help identify immunotherapy targets.
3.2.5 Cancer metabolism
ccRCC is considered a metabolic disease and involves several inactivated genes (91), such as VHL, controlled tumor energetics and biosynthesis, and the hypoxia pathway (92). The KEGG pathway-based study identified compounds that were present in varied abundance in tumor and normal kidney tissues. Remarkably, most of the upregulated pathways in tumor tissues were engaged in carbohydrate metabolism, whereas the deregulated pathways involved amino acid metabolism (93).
However, the influence of inactivated SETD2 on metabolic reprogramming is unclear. Compared to parental 786-O cells, SETD2-deficient cells promote PGC1α, increase oxidative phosphorylation, and elevate mitochondrial oxidative metabolism. Acetyl-CoA is a pivotal substance in biochemical metabolism, which enters the TCA cycle for oxidation and catabolism, and also as a source of fatty acid synthesis, given fatty acid metabolism is always associated with metastasis. Liu et al., hypothesized that enhanced TCA metabolite acetyl-CoA may shunt fatty acid synthesis, resulting in cancer metastasis (45). Compared to wild-type cells, SETD2 knockout cells inhibit multiple metabolic-related genes in the various metabolic pathways (46). Therefore, tumor metastasis accompanied by metabolic alterations and further metabolic pathways analysis of SETD2 inactivated in ccRCC will have the potential to discover new therapeutics for precision medicine.
3.2.6 Metastases
Previous studies identified an association between SETD2 mutations and the prognosis of patients with localized ccRCC. The mono-allelic mutant of SETD2 is insignificant in H3K36me3 modification. SETD2 loss-of-function mutations were revealed in 10%~20% of primary ccRCC tumors, increasing to 30%~60% of metastatic ccRCC tumors. A significant reduction in H3K36 methylation was also found in both ccRCC cell lines and patient samples, suggesting the potential involvement of SETD2 in driving ccRCC metastatic progression (8, 9). In the TCGA cohort, SETD2 mutations were correlated with poorer cancer-specific survival in ccRCC patients (50). Immunohistochemical staining displayed a gradually decreasing H3K36me3 modification with distant metastases from primary ccRCC tumors. During the progression of ccRCC, H3K36me3 is reduced in distant metastases, and regional H3K36me3 alterations influence alternative splicing in ccRCC (94–97). The H3K36me3 dysregulation axis is linked to an increased risk of death from RCC. Specifically, this connection is substantial, especially for patients with low-risk malignancies (98); however, the mechanism by which SETD2 causes cell metastasis has not been fully elucidated.
The activation of enhancer elements that promote metastatic carcinoma progression has been proven in several cancers, including ccRCC (99–101). Increased chromatin accessibility containing activating enhancers is regulated by aberrant histone chaperone recruitment and activity (102, 103). A recent study has shown that SETD2 deficiency mediated reduction of H3K36me3 induced the recruitment of histone chaperone ASF1A/B and SPT16, increased MMP1 chromatin accessibility, and activated enhancers to drive genes involved in metastasis, promoted ccRCC metastasis (47).
3.2.7 Cell proliferation and cell cycle regulation
SETD2 stabilization increases cell proliferation contrary to its canonical role as a tumor suppressor (25). According to Li et al., decreased SETD2 reduces cell proliferation and can be restored by CDK1 knockdown. Multiple SETD2-regulated cellular pathways suppress cancer development and uncover mechanisms underlying aberrant cell cycle regulation in SETD2-depleted cells (46). SETD2 is a tumor suppressor in renal primary tubular epithelial cells (PTECs). The proliferative capacity of SETD2-knockdown PTECs was higher than that of SETD2 wild-type PTECs, indicating that SETD2 inactivation enables PTECs to facilitate a malignant transformation toward ccRCC (67).
Generally, DNA damage could cause cell cycle arrest. The abundance of H3K36me3 ensures the recruitment of DNA damage repair key proteins during DNA replication to restore genome integrity in G1 and early S phase (40, 104). Replication fork speed is also decreased in ccRCC cells when SETD2 is depleted (35). Throughout the cell cycle, the SETD2 protein level is minimal in G1 and maximal in G2/M. Both H3K36me3 and WEE1 are critical in DNA replication and promote ribonucleotide reductase subunit (RRM2) expression, respectively. In SETD2-deficient cells, WEE1 inhibition reduces dNTP and RRM2 with higher sensitivity, resulting in S-phase arrest (48).
In recent studies, Helena et al. and Zhu et al. found SETD2 can also catalyze H3K37me1 and H3K14me3, H3K14me3 recruits the RPA complex to active Ataxia telangiectasia and Rad3 related (ATR) during replication stress, which plays a crucial role in the DNA replication stress response and negatively regulates replication initiation, the deletion of SETD2 reduces replication stress in the absence of H3K37me1 and H3K14me3 (105, 106). In conclusion, SETD2 controls the proper course of the S-phase, and catalyzes H3K37me1 and H3K14me3 to regulate the replication progress. However, the detailed correlation between SETD2 and cell cycle regulation is still incomplete and requires further exploration.
3.2.8 Non-histone substrates of SETD2
SETD2 is the main H3K36me3 methyltransferase in mammalian cells. Recent studies have suggested that SETD2 could also catalyze non-histone substrate methylation. During ccRCC mitosis, SETD2 trimethylates α-TubK40me3 and maintains genomic stability. Mono-allelic mutation of SETD2 results in α-TubK40me3 deficiency, leading to chromosome abnormalities and genomic instability exhibiting multipolar spindle formation, chromosome bridges, micronuclei, polyploidy, and multiple nuclei (14). SETD2, as a chromatocytoskeletal remodeler, trimethylates ActK68me3. The SETD2-HTT-HIP1R axis modifies actin, which increases actin polymerization and promotes ccRCC migration (18). In addition, SETD2 methylates STAT1 on lysine 525 promotes IFNα-dependent antiviral immunity (107), and methylates EZH2 on lysine 735 inhibits prostate cancer metastasis (33). Since SETD2 and EZH2 commonly occur abnormally in urological cancers, the SETD2-EZH2 axis may also be promising targets for pharmacological intervention in ccRCC. In order to search the specificity substrate sequence of SETD2, the amino acid specificity profile of the SETD2 substrate sequence was determined by the peptide SPOT arrays and find the super-matching methylation site on K666 of FBN-1 (108). Further cytological work is still needed to demonstrate that FBN1 is a methylated substrate of SETD2.
A recent study reported that SETD2 could indirectly methylate non-histone substrates, loss of SETD2 increases protein translation-related gene expression and decreases eEF1A1 K165me3 and K318me1 in ccRCC, but SETD2 is associated with eEF1A1 methylation indirectly, SET domain of SETD2 regulated the expression of EEF1AKMT2 and EEF1AKMT3, EEF1AKMT3 methylates eEF1A1 on lysine 165 and EEF1AKMT2 methylates eEF1A1 on lysine 318 (109). Finally, the discovery of SETD2 for non-histone substrates is particularly crucial for a more in-depth understanding of its biological role (Figure 2).
3.2.9 Other functions
Recent research has depicted that multiple chromatin remodeling enzymes are genetically inactive in ccRCC. Even though there is emerging evidence that epigenetic changes are important in cancer, only DNA methylation changes have been identified (92). Widespread DNA hypomethylation correlates to the mutation of the H3K36 methyltransferase SETD2 (94).
Patients with polycystic kidney disease (PKD) have a high probability of converting to RCC. However, there is a paucity of knowledge regarding how PKD can develop into RCC, necessitating further research into genetic alterations or the regulation of signaling pathways. Li et al. found that SETD2 deletion can lead to increased activation of the Wnt/β-catenin signaling pathway and promote epithelial-mesenchymal transition and tumor formation. SETD2 plays an essential role in the process of the conversion of PKD to ccRCC (49).
Emerging evidence suggests that exosomal circRNAs might be potential cancer biomarkers (110–112). He et al. reported that circulating exosomal mRNA (emRNA) is a potential diagnostic biomarker of ccRCC; thus, an emRNA-based screening signature could be developed to provide noninvasive indicators for ccRCC (113).
4 Conclusion
SETD2-mediated H3K36me3 enhances transcriptional elongation and is also involved in DNA damage repair and alternative splicing (Figures 2, 3). SETD2 mutations have been identified in ccRCC (41), but further research should focus on the association with the function of SETD2 and ccRCC. Loss of SETD2 in ccRCC is related to decreased autophagy processing, greater levels of general metabolic activity, poorer cancer-specific survival in ccRCC patients, and slower replication fork speed.
As a tumor suppressor, SETD2 may serve as a biomarker to reduce drug resistance to targeted therapy and as a potential therapeutic target to promote individualized treatment and improve patient survival. The TCGA pan-cancer cohort shows that patients with SETD2 mutations have a higher immune-related gene expression and MSI. Clinical data analysis of cancer patients treated with immune checkpoint inhibitors demonstrated that SETD2 mutation is a potential biomarker (114). 5-aza-2’-deoxycytidine (DAC) is used clinically to treat tumors with mutations in chromatin regulators, which competitively inhibits DNA methyltransferase activity and demethylates DNA. H3K36me3 is reduced in SETD2-deficient tumor cells, decreasing the recruitment of DNMT3B and the methylation of DNA, increasing interferon immune responses and the expression of transposable elements, therefore improving the sensitivity to DAC. In wild-type tumors, the number of myeloid-derived immune suppressive cell (MDSC) increased with DAC treatment. In the SETD2-knockdown tumor model, increased CD8+ T cell infiltration and fewer MDSC following combined treatment with DAC and anti-PD-L1. ccRCC with altered SETD2 gene provides preclinical support for a therapeutic target for DAC and anti-PD-L1 (57). A case report about advanced HCC showed that immunotherapy could be effective, leading to long-term survival, and they focused on two mutated genes, SETD2 and LRP1B, to further explore (115). Thus, the hypermutated SETD2 in ccRCC is worthy of attention.
With current innovations in genome engineering and proteomics, the role of SETD2 in normal cells and cancer will be better understood at the molecular level. Nonetheless, it is urgent to explore whether and how SETD2 regulates the molecular mechanisms of recurrence and ccRCC metastasis.
Furthermore, SMYD5 and SETD5 were also demonstrated to catalyze H3K36me3 (7, 116). A growing number of enzymes were initially discovered for methylating additional amino acid residues of histones and other proteins (117), so a reanalysis of known histone methyltransferases is necessary.
In conclusion, the in-depth study of SETD2 during tumor formation and development is warranted for diagnosing, treating, and preventing tumors. It is anticipated that further epigenetics studies will reveal the regulatory pathway of SETD2 expression.
Author contributions
YZ and LJ supervised the review study. MY, YZ, and LJ reviewed the literature and drafted the first draft. GW and KQ provided suggestions to improve the draft. MY, YX, and LJ edited the figures and tables. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (31900902), Hubei Province health and family planning scientific research project (WJ2023M063), and Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2020-PT320-004).
Acknowledgments
We gratefully acknowledge the exceptional assistance in editing diagram by Dr. Yuruo Chen, and we would like to thank Freescience Editorial Team for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AWS, associated with SET; AID, auto-inhibitory domain; ATG, autophagy-related genes; ATR, Ataxia telangiectasia and Rad3 related; CTD, C terminal dоmain; ccRCC, clear cell renal cell carcinoma; CC: coiled-coil; CtIP, C-terminal binding protein interacting protein; DAC, 5-aza-2’-deoxycytidine; DDR, DNA damage response; DSBs, DNA double-strand breaks; DNMT3B, DNA-methyltransferase 3B; emRNA, exosomal mRNA; FH, Fumarate hydratase; GSEA, Gene set enrichment analysis; hnRNP L, heterogeneous nuclear ribonucleoprotein L; H3K36me3: histone H3 lysine36 trimethylation; HR, homologous recombination; HIP1R, HTT-interacting protein 1-related protein; HTT, Huntingtin; LEDGF, Lens epithelium-derived growth factor; MSI, microsatellite instability; MMR, mismatch repair; MRG15, MORF4-related gene on chromosome 15; MDSC, myeloid-derived immune suppressive cell; NHEJ, non-homologous end-joining; PHD, plant homeodomain; PKD, polycystic kidney disease; PTB, polypyrimidine tract-binding protein; PRR, proline-rich region; PWWP, Pro-Trp-Trp-Pro; RCC, renal cell carcinoma; PTECs, renal primary tubular epithelial cells; RPA, replication protein A; PHRF1, ring finger domains 1; RNAPII, RNA polymerase II; RRM2, RNA-recognition motif 2; SAM, S-adenosylmethionine; SDH, Succinate dehydrogenase; SETD2, SET domain-containing 2; SRI, Set2-Rpb1 interacting; SHI, SETD2-hnRNP interaction; ssDNA, single-stranded DNA; VHL, von Hippel-Lindau; ZMYND11, zinc finger MYND-domain containing 11.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
3. WHO. Classification of tumours, urinary and Male genital tumours. Lyon (France: IARC Press (2022).
4. Walton J, Lawson K, Prinos P, Finelli A, Arrowsmith C, Ailles L. PBRM1, SETD2 and BAP1 - the trinity of 3p in clear cell renal cell carcinoma. Nat Rev Urol (2022) 20(2):96–115. doi: 10.1038/s41585-022-00659-1
5. Patard JJ, Pignot G, Escudier B, Eisen T, Bex A, Sternberg C, et al. ICUD-EAU international consultation on kidney cancer 2010: treatment of metastatic disease. Eur Urol (2011) 60(4):684–90. doi: 10.1016/j.eururo.2011.06.017
6. Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol (2012) 13(2):115–26. doi: 10.1038/nrm3274
7. Zhang Y, Fang Y, Tang Y, Han S, Jia J, Wan X, et al. SMYD5 catalyzes histone H3 lysine 36 trimethylation at promoters. Nat Commun (2022) 13(1):3190. doi: 10.1038/s41467-022-30940-1
8. Ho TH, Park IY, Zhao H, Tong P, Champion MD, Yan H, et al. High-resolution profiling of histone h3 lysine 36 trimethylation in metastatic renal cell carcinoma. Oncogene. (2016) 35(12):1565–74. doi: 10.1038/onc.2015.221
9. Wang J, Liu L, Qu Y, Xi W, Xia Y, Bai Q, et al. Prognostic value of SETD2 expression in patients with metastatic renal cell carcinoma treated with tyrosine kinase inhibitors. J Urol (2016) 196(5):1363–70. doi: 10.1016/j.juro.2016.06.010
10. Lam UTF, Tan BKY, Poh JJX, Chen ES. Structural and functional specificity of H3K36 methylation. Epigenet Chromatin (2022) 15(1):17. doi: 10.1186/s13072-022-00446-7
11. Zhang Y, Shan CM, Wang J, Bao K, Tong L, Jia S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep (2017) 7:43906. doi: 10.1038/srep43906
12. Liu Y, Zhang Y, Xue H, Cao M, Bai G, Mu Z, et al. Cryo-EM structure of SETD2/Set2 methyltransferase bound to a nucleosome containing oncohistone mutations. Cell Discovery (2021) 7(1):32. doi: 10.1038/s41421-021-00261-6
13. Li M, Phatnani HP, Guan Z, Sage H, Greenleaf AL, Zhou P. Solution structure of the Set2-Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated c-terminal domain of Rpb1. Proc Natl Acad Sci U S A (2005) 102(49):17636–41. doi: 10.1073/pnas.0506350102
14. Chiang YC, Park IY, Terzo EA, Tripathi DN, Mason FM, Fahey CC, et al. SETD2 haploinsufficiency for microtubule methylation is an early driver of genomic instability in renal cell carcinoma. Cancer Res (2018) 78(12):3135–46. doi: 10.1158/0008-5472.CAN-17-3460
15. Kearns S, Mason FM, Rathmell WK, Park IY, Walker C, Verhey KJ, et al. Molecular determinants for alpha-tubulin methylation by SETD2. J Biol Chem (2021) 297(1):100898. doi: 10.1016/j.jbc.2021.100898
16. Molenaar TM, Malik M, Silva J, Liu NQ, Haarhuis JHI, Ambrosi C, et al. The histone methyltransferase SETD2 negatively regulates cell size. J Cell Sci (2022) 135(19):jcs259856. doi: 10.1242/jcs.259856
17. Gao YG, Yang H, Zhao J, Jiang YJ, Hu HY. Autoinhibitory structure of the WW domain of HYPB/SETD2 regulates its interaction with the proline-rich region of huntingtin. Structure. (2014) 22(3):378–86. doi: 10.1016/j.str.2013.12.005
18. Seervai RNH, Jangid RK, Karki M, Tripathi DN, Jung SY, Kearns SE, et al. The huntingtin-interacting protein SETD2/HYPB is an actin lysine methyltransferase. Sci Adv (2020) 6(40):eabb7854. doi: 10.1126/sciadv.abb7854
19. Molenaar TM, van Leeuwen F. SETD2: from chromatin modifier to multipronged regulator of the genome and beyond. Cell Mol Life Sci (2022) 79(6):346. doi: 10.1007/s00018-022-04352-9
20. Bhattacharya S, Levy MJ, Zhang N, Li H, Florens L, Washburn MP, et al. The methyltransferase SETD2 couples transcription and splicing by engaging mRNA processing factors through its SHI domain. Nat Commun (2021) 12(1):1443. doi: 10.1038/s41467-021-21663-w
21. Bhattacharya S, Wang S, Reddy D, Shen S, Zhang Y, Zhang N, et al. Structural basis of the interaction between SETD2 methyltransferase and hnRNP l paralogs for governing co-transcriptional splicing. Nat Commun (2021) 12(1):6452. doi: 10.1038/s41467-021-26799-3
22. Wang Y, Niu Y, Li B. Balancing acts of SRI and an auto-inhibitory domain specify Set2 function at transcribed chromatin. Nucleic Acids Res (2015) 43(10):4881–92. doi: 10.1093/nar/gkv393
23. Gopalakrishnan R, Marr SK, Kingston RE, Winston F. A conserved genetic interaction between Spt6 and Set2 regulates H3K36 methylation. Nucleic Acids Res (2019) 47(8):3888–903. doi: 10.1093/nar/gkz119
24. Zhu K, Lei PJ, Ju LG, Wang X, Huang K, Yang B, et al. SPOP-containing complex regulates SETD2 stability and H3K36me3-coupled alternative splicing. Nucleic Acids Res (2017) 45(1):92–105. doi: 10.1093/nar/gkw814
25. Bhattacharya S, Workman JL. Regulation of SETD2 stability is important for the fidelity of H3K36me3 deposition. Epigenet Chromatin (2020) 13(1):40. doi: 10.1186/s13072-020-00362-8
26. Bhattacharya S, Lange JJ, Levy M, Florens L, Washburn MP, Workman JL. The disordered regions of the methyltransferase SETD2 govern its function by regulating its proteolysis and phase separation. J Biol Chem (2021) 297(3):101075. doi: 10.1016/j.jbc.2021.101075
27. Kim H, Shim BY, Lee SJ, Lee JY, Lee HJ, Kim IH. Loss of Von hippel-lindau (VHL) tumor suppressor gene function: VHL-HIF pathway and advances in treatments for metastatic renal cell carcinoma (RCC). Int J Mol Sci (2021) 22(18):9795. doi: 10.3390/ijms22189795
28. Li J, Ahn JH, Wang GG. Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell Mol Life Sci (2019) 76(15):2899–916. doi: 10.1007/s00018-019-03144-y
29. Skucha A, Ebner J, Grebien F. Roles of SETD2 in leukemia-transcription, DNA-damage, and beyond. Int J Mol Sci (2019) 20(5):1029. doi: 10.3390/ijms20051029
30. Xiao C, Fan T, Tian H, Zheng Y, Zhou Z, Li S, et al. H3K36 trimethylation-mediated biological functions in cancer. Clin Epigenetics (2021) 13(1):199. doi: 10.1186/s13148-021-01187-2
31. Hacker KE, Fahey CC, Shinsky SA, Chiang YJ, DiFiore JV, Jha DK, et al. Structure/Function analysis of recurrent mutations in SETD2 protein reveals a critical and conserved role for a SET domain residue in maintaining protein stability and histone H3 lys-36 trimethylation. J Biol Chem (2016) 291(40):21283–95. doi: 10.1074/jbc.M116.739375
32. Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, Liu XY, Sturm D, Korshunov A, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol (2013) 125(5):659–69. doi: 10.1007/s00401-013-1095-8
33. Yuan H, Han Y, Wang X, Li N, Liu Q, Yin Y, et al. SETD2 restricts prostate cancer metastasis by integrating EZH2 and AMPK signaling pathways. Cancer Cell (2020) 38(3):350–65.e7. doi: 10.1016/j.ccell.2020.05.022
34. Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet (2014) 46(3):287–93. doi: 10.1038/ng.2894
35. Kanu N, Gronroos E, Martinez P, Burrell RA, Yi Goh X, Bartkova J, et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene. (2015) 34(46):5699–708. doi: 10.1038/onc.2015.24
36. Dong Y, Zhao X, Feng X, Zhou Y, Yan X, Zhang Y, et al. SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints. Leukemia. (2019) 33(11):2585–98. doi: 10.1038/s41375-019-0456-2
37. Hakimi AA, Chen YB, Wren J, Gonen M, Abdel-Wahab O, Heguy A, et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur Urol (2013) 63(5):848–54. doi: 10.1016/j.eururo.2012.09.005
38. Yang S, Zheng X, Lu C, Li GM, Allis CD, Li H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev (2016) 30(14):1611–6. doi: 10.1101/gad.284323.116
39. Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res (2014) 24(2):241–50. doi: 10.1101/gr.158253.113
40. Li F, Mao G, Tong D, Huang J, Gu L, Yang W, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. (2013) 153(3):590–600. doi: 10.1016/j.cell.2013.03.025
41. Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RM, et al. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res (2010) 70(11):4287–91. doi: 10.1158/0008-5472.CAN-10-0120
42. Scelo G, Riazalhosseini Y, Greger L, Letourneau L, Gonzalez-Porta M, Wozniak MB, et al. Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat Commun (2014) 5:5135. doi: 10.1038/ncomms6135
43. Gonzalez-Rodriguez P, Engskog-Vlachos P, Zhang H, Murgoci AN, Zerdes I, Joseph B. SETD2 mutation in renal clear cell carcinoma suppress autophagy via regulation of ATG12. Cell Death Dis (2020) 11(1):69. doi: 10.1038/s41419-020-2266-x
44. Seervai RNH, Grimm SL, Jangid RK, Tripathi DN, Coarfa C, Walker CL. An actin-WHAMM interaction linking SETD2 and autophagy. Biochem Biophys Res Commun (2021) 558:202–8. doi: 10.1016/j.bbrc.2020.09.025
45. Liu J, Hanavan PD, Kras K, Ruiz YW, Castle EP, Lake DF, et al. Loss of SETD2 induces a metabolic switch in renal cell carcinoma cell lines toward enhanced oxidative phosphorylation. J Proteome Res (2019) 18(1):331–40. doi: 10.1021/acs.jproteome.8b00628
46. Li L, Miao W, Huang M, Williams P, Wang Y. Integrated genomic and proteomic analyses reveal novel mechanisms of the methyltransferase SETD2 in renal cell carcinoma development. Mol Cell Proteomics (2019) 18(3):437–47. doi: 10.1074/mcp.RA118.000957
47. Xie Y, Sahin M, Sinha S, Wang Y, Nargund AM, Lyu Y, et al. SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes. Nat Cancer (2022) 3(2):188–202. doi: 10.1038/s43018-021-00316-3
48. Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, et al. Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell (2015) 28(5):557–68. doi: 10.1016/j.ccell.2015.09.015
49. Rao H, Li X, Liu M, Liu J, Feng W, Tang H, et al. Multilevel regulation of beta-catenin activity by SETD2 suppresses the transition from polycystic kidney disease to clear cell renal cell carcinoma. Cancer Res (2021) 81(13):3554–67. doi: 10.1158/0008-5472.CAN-20-3960
50. Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res (2013) 19(12):3259–67. doi: 10.1158/1078-0432.CCR-12-3886
51. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med (2012) 366(10):883–92. doi: 10.1056/NEJMoa1113205
52. Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, et al. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res (2013) 41(5):2881–93. doi: 10.1093/nar/gks1472
53. Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature. (2017) 543(7643):72–7. doi: 10.1038/nature21373
54. Ha SD, Ham S, Kim MY, Kim JH, Jang I, Lee BB, et al. Transcription-dependent targeting of Hda1C to hyperactive genes mediates H4-specific deacetylation in yeast. Nat Commun (2019) 10(1):4270. doi: 10.1038/s41467-019-12077-w
55. Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. (2015) 520(7546):243–7. doi: 10.1038/nature14176
56. Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol (2009) 27(4):361–8. doi: 10.1038/nbt.1533
57. Li HT, Jang HJ, Rohena-Rivera K, Liu M, Gujar H, Kulchycki J, et al. RNA Mis-splicing drives viral mimicry response after DNMTi therapy in SETD2-mutant kidney cancer. Cell Rep (2023) 42(1):112016. doi: 10.1016/j.celrep.2023.112016
58. Jimeno-Gonzalez S, Payan-Bravo L, Munoz-Cabello AM, Guijo M, Gutierrez G, Prado F, et al. Defective histone supply causes changes in RNA polymerase II elongation rate and cotranscriptional pre-mRNA splicing. Proc Natl Acad Sci U S A (2015) 112(48):14840–5. doi: 10.1073/pnas.1506760112
59. Zhang P, Du J, Sun B, Dong X, Xu G, Zhou J, et al. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res (2006) 34(22):6621–8. doi: 10.1093/nar/gkl989
60. Fang D, Gan H, Lee JH, Han J, Wang Z, Riester SM, et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science (2016) 352(6291):1344–8. doi: 10.1126/science.aae0065
61. Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. (2010) 327(5968):996–1000. doi: 10.1126/science.1184208
62. Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep (2014) 7(6):2006–18. doi: 10.1016/j.celrep.2014.05.026
63. Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PloS Genet (2012) 8(5):e1002717. doi: 10.1371/journal.pgen.1002717
64. LeRoy G, Oksuz O, Descostes N, Aoi Y, Ganai RA, Kara HO, et al. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells. Sci Adv (2019) 5(10):eaay3068. doi: 10.1126/sciadv.aay3068
65. Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell (2014) 56(2):298–310. doi: 10.1016/j.molcel.2014.08.022
66. Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature (2014) 508(7495):263–8. doi: 10.1038/nature13045
67. Li J, Kluiver J, Osinga J, Westers H, van Werkhoven MB, Seelen MA, et al. Functional studies on primary tubular epithelial cells indicate a tumor suppressor role of SETD2 in clear cell renal cell carcinoma. Neoplasia. (2016) 18(6):339–46. doi: 10.1016/j.neo.2016.04.005
68. Carvalho S, Vitor AC, Sridhara SC, Martins FB, Raposo AC, Desterro JM, et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife. (2014) 3:e02482. doi: 10.7554/eLife.02482.016
69. Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sorensen CS, et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol (2012) 19(8):803–10. doi: 10.1038/nsmb.2314
70. Marnef A, Cohen S, Legube G. Transcription-coupled DNA double-strand break repair: Active genes need special care. J Mol Biol (2017) 429(9):1277–88. doi: 10.1016/j.jmb.2017.03.024
71. Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol (2014) 21(4):366–74. doi: 10.1038/nsmb.2796
72. Chang CF, Chu PC, Wu PY, Yu MY, Lee JY, Tsai MD, et al. PHRF1 promotes genome integrity by modulating non-homologous end-joining. Cell Death Dis (2015) 6:e1716. doi: 10.1038/cddis.2015.81
73. Fang J, Huang Y, Mao G, Yang S, Rennert G, Gu L, et al. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc Natl Acad Sci U S A (2018) 115(38):9598–603. doi: 10.1073/pnas.1806355115
74. Huang Y, Gu L, Li GM. H3K36me3-mediated mismatch repair preferentially protects actively transcribed genes from mutation. J Biol Chem (2018) 293(20):7811–23. doi: 10.1074/jbc.RA118.002839
75. Jiang A, Song J, Fang X, Fang Y, Wang Z, Liu B, et al. A novel thinking: DDR axis refines the classification of ccRCC with distinctive prognosis, multi omics landscape and management strategy. Front Public Health (2022) 10:1029509. doi: 10.3389/fpubh.2022.1029509
76. Chabanon RM, Rouanne M, Lord CJ, Soria JC, Pasero P, Postel-Vinay S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat Rev Cancer (2021) 21(11):701–17. doi: 10.1038/s41568-021-00386-6
77. Coutts AS, La Thangue NB. Actin nucleation by WH2 domains at the autophagosome. Nat Commun (2015) 6:7888. doi: 10.1038/ncomms8888
78. Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. (2013) 500(7463):468–71. doi: 10.1038/nature12313
79. Artal-Martinez de Narvajas A, Gomez TS, Zhang JS, Mann AO, Taoda Y, Gorman JA, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol (2013) 33(20):3983–93. doi: 10.1128/MCB.00813-13
80. Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol (2014) 15(1):65–74. doi: 10.1038/nrm3716
81. Wei FZ, Cao Z, Wang X, Wang H, Cai MY, Li T, et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy. (2015) 11(12):2309–22. doi: 10.1080/15548627.2015.1117734
82. Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. (2016) 534(7608):553–7. doi: 10.1038/nature18014
83. Baek SH, Kim KI. Epigenetic control of autophagy: Nuclear events gain more attention. Mol Cell (2017) 65(5):781–5. doi: 10.1016/j.molcel.2016.12.027
84. Zhu L, Ding R, Yan H, Zhang J, Lin Z. ZHX2 drives cell growth and migration via activating MEK/ERK signal and induces sunitinib resistance by regulating the autophagy in clear cell renal cell carcinoma. Cell Death Dis (2020) 11(5):337. doi: 10.1038/s41419-020-2541-x
85. Xiao W, Xiong Z, Xiong W, Yuan C, Xiao H, Ruan H, et al. Melatonin/PGC1A/UCP1 promotes tumor slimming and represses tumor progression by initiating autophagy and lipid browning. J Pineal Res (2019) 67(4):e12607. doi: 10.1111/jpi.12607
86. Xu Y, Zhou J, Li L, Yang W, Zhang Z, Zhang K, et al. FTO-mediated autophagy promotes progression of clear cell renal cell carcinoma via regulating SIK2 mRNA stability. Int J Biol Sci (2022) 18(15):5943–62. doi: 10.7150/ijbs.77774
87. González-Rodríguez P, Delorme-Axford E, Bernard A, Keane L, Stratoulias V, Grabert K, et al. SETD2 transcriptional control of ATG14L/S isoforms regulates autophagosome-lysosome fusion. Cell Death Dis (2022) 13(11):953. doi: 10.1038/s41419-022-05381-9
88. White J, Suklabaidya S, Vo MT, Choi YB, Harhaj EW. Multifaceted roles of TAX1BP1 in autophagy. Autophagy. (2023) 19(1):44–53. doi: 10.1080/15548627.2022.2070331
89. Pan M, Yin Y, Hu T, Wang X, Jia T, Sun J, et al. UXT attenuates the CGAS-STING1 signaling by targeting STING1 for autophagic degradation. Autophagy. (2023) 19(2):440–56. doi: 10.1080/15548627.2022.2076192
90. Yamamoto K, Venida A, Perera RM, Kimmelman AC. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy. (2020) 16(8):1524–5. doi: 10.1080/15548627.2020.1769973
91. Wettersten HI, Aboud OA, Lara PN Jr., Weiss RH. Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol (2017) 13(7):410–9. doi: 10.1038/nrneph.2017.59
92. Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: A metabolic disease. Nat Rev Urol (2010) 7(5):277–85. doi: 10.1038/nrurol.2010.47
93. Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell (2016) 29(1):104–16. doi: 10.1016/j.ccell.2015.12.004
94. Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature (2013) 499(7456):43–9. doi: 10.1038/nature12222
95. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet (2013) 45(8):860–7. doi: 10.1038/ng.2699
96. Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet (2014) 46(3):225–33. doi: 10.1038/ng.2891
97. Hsieh JJ, Chen D, Wang PI, Marker M, Redzematovic A, Chen YB, et al. Genomic biomarkers of a randomized trial comparing first-line everolimus and sunitinib in patients with metastatic renal cell carcinoma. Eur Urol (2017) 71(3):405–14. doi: 10.1016/j.eururo.2016.10.007
98. Ho TH, Kapur P, Joseph RW, Serie DJ, Eckel-Passow JE, Tong P, et al. Loss of histone H3 lysine 36 trimethylation is associated with an increased risk of renal cell carcinoma-specific death. Mod Pathol (2016) 29(1):34–42. doi: 10.1038/modpathol.2015.123
99. Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Gruner BM, et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell. (2016) 166(2):328–42. doi: 10.1016/j.cell.2016.05.052
100. Morrow JJ, Bayles I, Funnell APW, Miller TE, Saiakhova A, Lizardo MM, et al. Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat Med (2018) 24(2):176–85. doi: 10.1038/nm.4475
101. Rodrigues P, Patel SA, Harewood L, Olan I, Vojtasova E, Syafruddin SE, et al. NF-kappaB-Dependent lymphoid enhancer Co-option promotes renal carcinoma metastasis. Cancer Discovery (2018) 8(7):850–65. doi: 10.1158/2159-8290.CD-17-1211
102. Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol (2013) 20(1):14–22. doi: 10.1038/nsmb.2461
103. Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol (2015) 16(3):178–89. doi: 10.1038/nrm3941
104. Sharda A, Humphrey TC. The role of histone H3K36me3 writers, readers and erasers in maintaining genome stability. DNA repair (2022) 119:103407. doi: 10.1016/j.dnarep.2022.103407
105. Zhu Q, Yang Q, Lu X, Wang H, Tong L, Li Z, et al. SETD2-mediated H3K14 trimethylation promotes ATR activation and stalled replication fork restart in response to DNA replication stress. Proc Natl Acad Sci U.S.A. (2021) 118(23):e2011278118. doi: 10.1073/pnas.2011278118
106. Santos-Rosa H, Millan-Zambrano G, Han N, Leonardi T, Klimontova M, Nasiscionyte S, et al. Methylation of histone H3 at lysine 37 by Set1 and Set2 prevents spurious DNA replication. Mol Cell (2021) 81(13):2793–807.e8. doi: 10.1016/j.molcel.2021.04.021
107. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, et al. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell. (2017) 170(3):492–506.e14. doi: 10.1016/j.cell.2017.06.042
108. Schuhmacher MK, Beldar S, Khella MS, Bröhm A, Ludwig J, Tempel W, et al. Sequence specificity analysis of the SETD2 protein lysine methyltransferase and discovery of a SETD2 super-substrate. Commun Biol (2020) 3(1):511. doi: 10.1038/s42003-020-01223-6
109. Hapke R, Venton L, Rose KL, Sheng Q, Reddy A, Prather R, et al. SETD2 regulates the methylation of translation elongation factor eEF1A1 in clear cell renal cell carcinoma. Kidney Cancer J Off J Kidney Cancer Assoc (2022) 6(3):179–93. doi: 10.3233/kca-220009
110. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. (2020) 367(6478):eaau6977. doi: 10.1126/science.aau6977
111. Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther (2020) 5(1):145. doi: 10.1038/s41392-020-00261-0
112. Hu W, Liu C, Bi ZY, Zhou Q, Zhang H, Li LL, et al. Comprehensive landscape of extracellular vesicle-derived RNAs in cancer initiation, progression, metastasis and cancer immunology. Mol Cancer (2020) 19(1):102. doi: 10.1186/s12943-020-01199-1
113. He X, Tian F, Guo F, Zhang F, Zhang H, Ji J, et al. Circulating exosomal mRNA signatures for the early diagnosis of clear cell renal cell carcinoma. BMC Med (2022) 20(1):270. doi: 10.1186/s12916-022-02467-1
114. Lu M, Zhao B, Liu M, Wu L, Li Y, Zhai Y, et al. Pan-cancer analysis of SETD2 mutation and its association with the efficacy of immunotherapy. NPJ Precis Oncol (2021) 5(1):51. doi: 10.1038/s41698-021-00193-0
115. Reiter FP, Rau M, Kunzmann V, Kickuth R, Klein I, Neumann O, et al. Profound tumor response to combined CTLA-4 and PD-1 inhibition in systemic fourth line therapy observed in a patient with hepatocellular carcinoma harboring SETD2 and LRP1B mutations. Z Gastroenterol (2023) 61(1):71–5. doi: 10.1055/a-1952-1233
116. Sessa A, Fagnocchi L, Mastrototaro G, Massimino L, Zaghi M, Indrigo M, et al. SETD5 regulates chromatin methylation state and preserves global transcriptional fidelity during brain development and neuronal wiring. Neuron. (2019) 104(2):271–89.e13. doi: 10.1016/j.neuron.2019.07.013
Keywords: SETD2, clear cell renal cell carcinoma (ccRCC), H3K36me3, epigenetic regulation, mutation
Citation: Yu M, Qian K, Wang G, Xiao Y, Zhu Y and Ju L (2023) Histone methyltransferase SETD2: An epigenetic driver in clear cell renal cell carcinoma. Front. Oncol. 13:1114461. doi: 10.3389/fonc.2023.1114461
Received: 02 December 2022; Accepted: 09 March 2023;
Published: 21 March 2023.
Edited by:
Linhui Wang, Second Military Medical University, ChinaReviewed by:
Wei Huang, Capital Medical University, ChinaHaifeng Yang, Thomas Jefferson University, United States
Copyright © 2023 Yu, Qian, Wang, Xiao, Zhu and Ju. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Zhu, emh1eXVhbjE5OTBAd2h1LmVkdS5jbg==; Lingao Ju, anVsaW5nYW8xOTkwQHdodS5lZHUuY24=