 Shantanu Singh
Shantanu Singh Akhilendra Kumar Maurya*
Akhilendra Kumar Maurya*- Biochemistry and Molecular Biology Laboratory, Centre for Advanced Study in Zoology, Institute of Science, Banaras Hindu University, Varanasi, India
T-cell lymphoma is a hematologic neoplasm derived from the lymphoid lineage. It belongs to a diverse group of malignant disorders, mostly affecting the young population worldwide, that vary with respect to molecular features as well as genetic and clinical complexities. Cancer cells rewire the cellular metabolism, persuading it to meet new demands of growth and proliferation. Furthermore, the metabolic alterations and heterogeneity are aberrantly driven in cancer by a combination of genetic and non-genetic factors, including the tumor microenvironment. New insight into cancer metabolism highlights the importance of nutrient supply to tumor development and therapeutic responses. Importantly, oxidative stress due to an imbalance in the redox status of reactive species via exogenous and/or endogenous factors is closely related to multiple aspects of cancer. This alters the signaling pathways governed through the multiple intracellular signal transduction and transcription factors, leading to tumor progression. These oncogenic signaling molecules are regulated through different redox sensors, including nuclear factor-erythroid 2 related factor 2 (Nrf2), phase-II antioxidant enzyme, and NQO1 (NADPH quinone oxidoreductase (1). The existing understanding of the molecular mechanisms of T-cell lymphoma regulation through the cross-talk of redox sensors under the influence of metabolic vulnerability is not well explored. This review highlights the role of the redox dynamics, orchestra of signaling, and genetic regulation involved in T-cell lymphoma progression in addition to the challenges to their etiology, treatment, and clinical response in light of recent updates.
Introduction
Genetic mutations, which are fuel for cancer, have been linked to different human malignancies. Every day, hundreds of cells undergo mutation in the human body, but they do not all go through tumor formation. However, under adverse conditions, adaptive changes may cause cells to behave abnormally, changing survival, growth, and division patterns, which potentially disturbs cellular and social control (1). The multiple rewirings of the cell’s metabolic, genetic, and signaling pathways resulting in a very disruptive destiny for the cell’s social environment. Lymphoid cells, a critical component of the immune system, have been identified as emerging players associated with the progression of multiple forms of cancer, including lymphoma. Lymphoma is a malignant disease consisting of a large heterogeneous group of lymphocytic cells that reside within the lymph nodes, spleen, thymus, bone marrow, and other parts of the body. Broadly, it is classified into Hodgkin’s and non-Hodgkin’s lymphoma (2). Hodgkin’s lymphoma is named after Dr. Thomas Hodgkin who discovered this disease in 1832. Hodgkin’s lymphoma is classified on the basis of the presence of a specific type of cell called a ‘Reed-Sternberg cell’, which is absent in its parallel subtype non-Hodgkin’s lymphoma (2). According to the ‘5th edition of WHO classification of lymphocyte neoplasm 2022’, tumor heterogeneity represents around 115 different types of lymphoid descendants (3). Non-Hodgkin’s lymphoma is classified as abnormal clonal proliferation of T cells and B cells that lack Reed-Sternberg cells, where the majority of them (80–90%) arise from B-type lymphocytes and the rest (10–20%) originate from T lymphocyte or natural killer cells (4). Lymphocytes have a definite life span; however, under certain circumstances they start dividing, causing them to aggregate in lymph nodes, which results in swelling of the lymphatic system. Various immune-suppressed pathologies, including HIV, receiving high doses of chemotherapy, organ transplantation, stem cell therapy, and even infection with Epstein bar virus and Helicobactor pylori bacteria, may cause a predisposition to non-Hodgkin’s lymphoma (5). This review focused on types and subtypes of lymphoma, specifically T-cell Lymphoma, and their dynamic regulation under the influence of redox status, metabolic tuning, and signaling pathways.
T-cell lymphoma
The human body responds to foreign invaders with the help of lymphoid cells and the lymphoid system, including all cells of adaptive and innate immunity. T-cell lymphoma is hematologic neoplasm derived from the lymphoid lineage, which normally governs immune responses. Abnormality in this immune regulation has been reported in post-thymic or activated T lymphocytes. As per the report of the American Cancer Society, T-cell lymphoma accounts for about 10% of all lymphoma. WHO classified T-cell lymphoma into 39 types and 11 different categories (3). Different types of T-cell Lymphoma are listed in Table 1. Based on the involvement of bone marrow, it can be recognized as either lymphoma and/or acute lymphoblastic leukemia. It starts at the thymus site of T-cell maturation and develops into a tumor at the mediastinum and can spread to almost any part of the body, including the liver, bone marrow, gastrointestinal tract, spleen, skin, and brain (6).
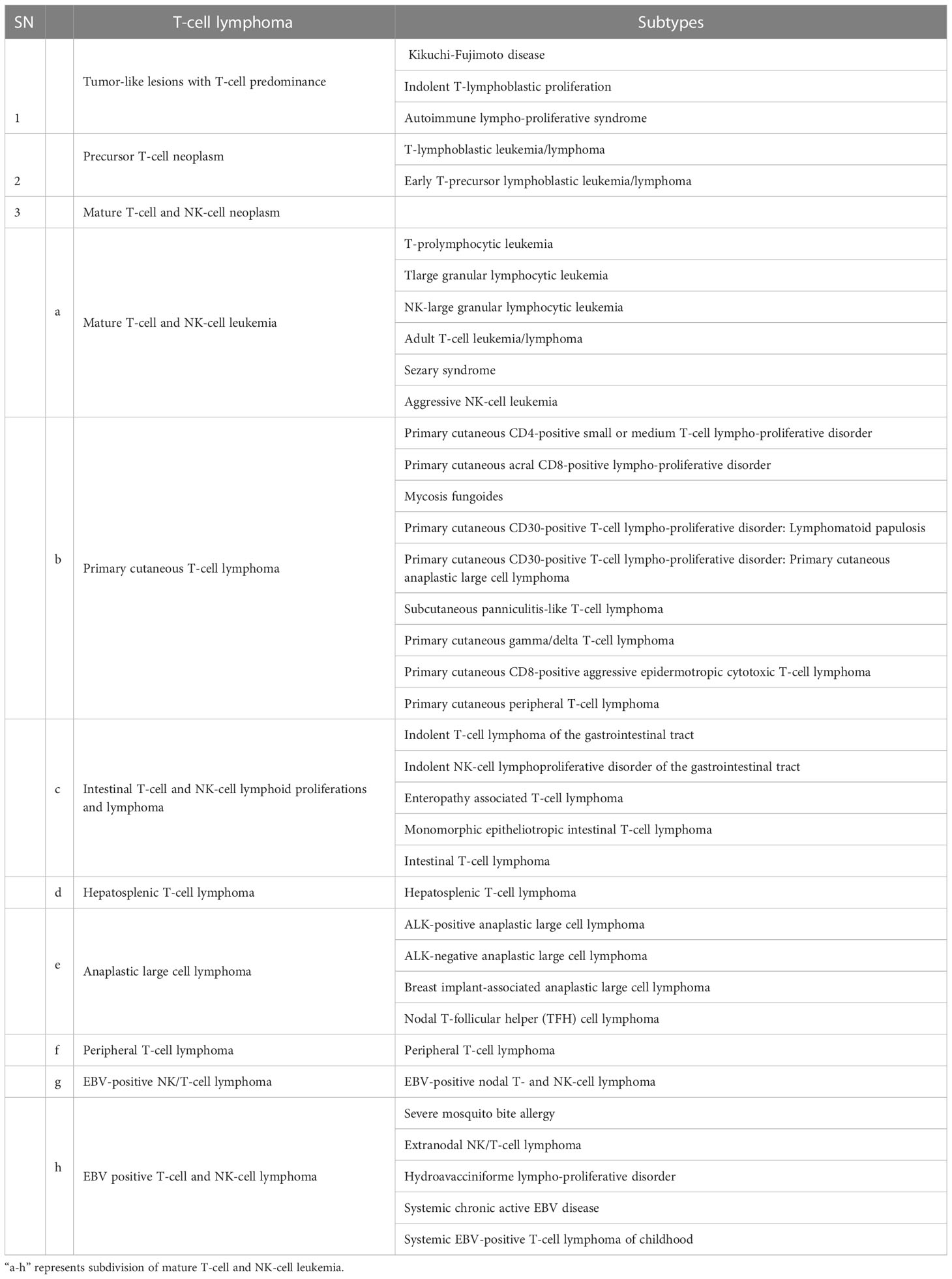
Table 1 T-cell Lymphoma types.
Epithelial–mesenchymal transition (EMT) is initiated in a malignant T-cell lymphoma, which allows epithelial cells to be converted into mesenchymal cells and moves them into distinct locations through a process known as metastasis. Chemotherapy is the most preferred treatment option, being preferable over radiation and surgery. However, a plethora of side effects, including chemotherapy-induced peripheral neuropathy (CIPN) and immunosuppression, can be seen in many parts of the body, including the brain, where it may display lifelong persistence (7–9). The antioxidant defense system is crucial in maintaining cellular homeostasis. An imbalanced redox status due to high metabolic demand and the microenvironment of T-cells could be targeted for treatment.
Redox status and antioxidant defense system
Healthy cells routinely encounter immense amount of stress due to environmental (xenobiotic exposure, microbial exposure, etc.) and cellular (metabolic, immune function, etc.) responses in form of reactive oxygen species (ROS). These ROS are collectively defined under two subtypes, free radical species, including superoxide anions (O2•−), hydroxyl radicals (HO•), peroxyl (RO2•), and alkoxyl (RO•), and non-radical species, such as hydrogen peroxide (H2O2) (10). Accumulation of ROS cumulatively builds up in the cell in the form of oxidative stress. Oxidative stresses are imbalances between oxidants and antioxidants, leading to modification of biochemical properties of biomolecules in cells, which is balanced by an intracellular antioxidant defense system (11). Modulation of redox states contributed to multistage carcinogenesis either by a direct mechanism involving damage of DNA or indirectly by modulating cellular signal transduction (12, 13). ROS affect the function of multiple cellular proteins either by stabilizing them or modifying them in such a way that they can easily interact with their activator for their downstream target where they cause prolonged or constitutive activation of growth factors, cytokines, etc. NF-κB is a potent target of ROS that activates multiple transcription factors associated with cell cycle progression and survival (12). Elevated oxidative stress has been reported in many types of cancer cells where the redox changes have significant consequences. The application of antioxidants on T-cell lymphoma for therapeutic response has been extensively studied.
Models for T-cell lymphoma study
For future drug development and etiological investigation there is a need for a suitable model that can mimic maximum possible properties of problems from the preclinical to clinical grade. A wide range of preclinical models are being studied in research ranging from primary cultures, cell lines, xenografting, and patient derived xenografting (PDX) to more advanced organoid cultures including the patient-derived organoid (PDO). Due to the diversity and/or heterogeneity found in T-cell malignancy a wide variety of in vivo as well as in vitro models have been established (13, 14). Various animal models used in T-cell lymphoma are listed in Table 2. Our elucidation of T cells in this review is mainly dominated by a transplantable T-cell lymphoma model also known as Dalton’s lymphoma. It has shown maximum association with a range of clinical markers of T-cell lymphoma. A wide range of markers, including regulation of CD3+, massive depletion of immature CD4+, CD8+ and mature CD4+, CD8-, and CD8- along with impaired regulation of immunoregulatory cytokines such as IFN-ϒ, IL-10 and IL-2 for T-cell lymphoma, are listed in Table 3.
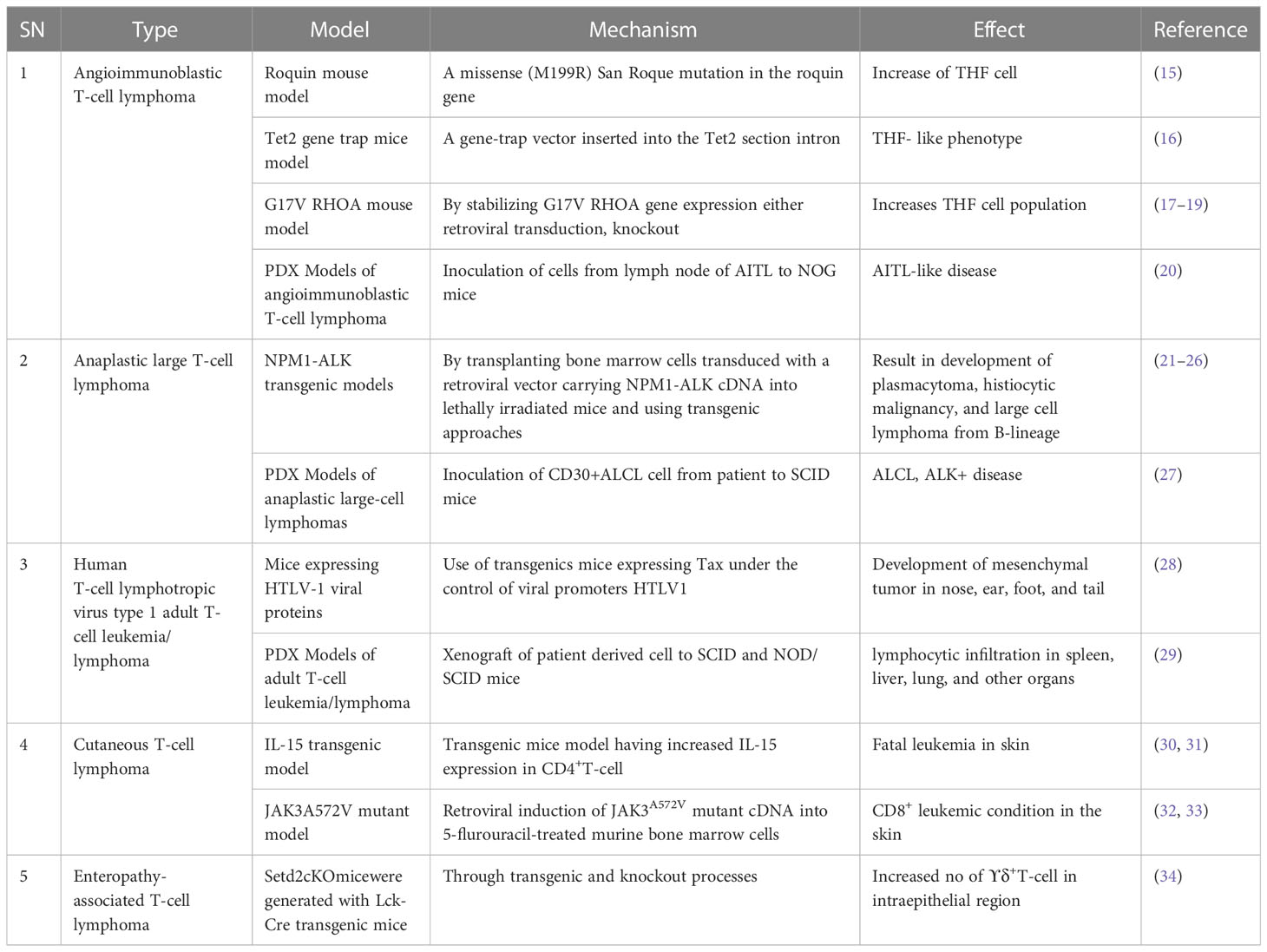
Table 2 T-cell lymphoma model.
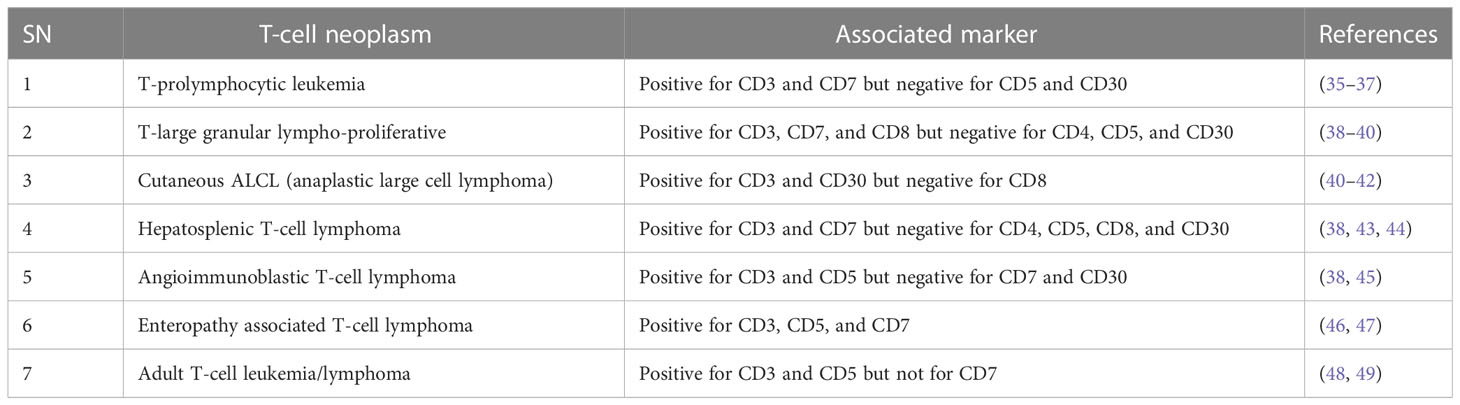
Table 3 T-cell lymphomas and their associated markers.
Regulation of T-cell lymphoma by ROS
ROS governs multiple cellular responses and are collectively referred to as “redox messengers” (10). These messengers further initiate a cascade of events in a small cytoplasmic pool to regulate different cellular functional proteins. Here, we describe some of such signaling events that contribute to progression of T-cell lymphoma in response to these messengers while highlighting signaling molecules involved in it.
Nrf2-NQO1 signaling
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a leucine zipper transcription factor with 7-Neh (Nrf2 ECH homology) domains, each with a different function. Nrf2 is found in every cell type with basal level of activation and its level is regulated by 26s proteasomal degradation mediated by E3 ubiquitin ligase (Keap1-Cul3-Rbx) (50). There are around 200 genes with ARE (antioxidant response element) sequences which are regulated by Nrf2 (51, 52).
Cells are armored with intricate defense system to counter oxidative stress through the Keap1-Nrf2-ARE pathway (53). The Nrf2 protein is reported to be upregulated during oxidative stress, and knockout of Nrf2 results in severe deficiency in the coordination of the gene regulatory program with increased susceptibility to oxidative damage (54). Nrf2 is reported to be a double-edged sword with a dual role in controlling cancer (55). The role of Nrf2 in cancer is a little paradoxical, however, in T-cell lymphoma it is delimited to its traditional pathway where any deterioration in this pathway leads to carcinogenesis and restabilizing it results in the enhancement of lymphoma. Multiple drivers have been reported to activate or stabilize the expression of Nrf2. PKC (Protein kinase C) is one such sensor that stabilizes the translocation of Nrf2 from the cytosol to nucleus by phosphorylating it at the ser-40 position and facilitating its release from its cytosolic anchor Keap1 (56). Nrf2 protects the cell from oxidative damage by preventing cellular damage and halting cell cycle progression. Overexpression of PKC promotes tumor progression (57). Other accessory proteins such as ERK2 and GSK3β have also been reported to regulate Nrf2 activity (58–66).
Genomic response by antioxidant signaling inducers suggest NAD(P)H:quinone oxidoreductase 1 (NQO1) and glutathione S-transferase (GST) are the two main target genes activated through Nrf2 in T-cell lymphoma (67, 68). NQO1 is a phase two antioxidant enzymes; it catalyzes the reduction of several environmental electrophonic contaminants and endogenous compounds, including quinones and nitro compounds (69). NQO1 under the regulation of Nrf2 plays a dual role in protection against carcinogenesis. Nrf2-dependent induction of NQO1 downregulated lipopolysaccharide (LPS)-induced expression of inflammatory cytokines, thereby impairing the inflammatory responses (70). It has also reported to induce apoptosis through p53. p53 is a tumor suppressor protein transcribed in response to cellular alteration like DNA damage and accumulates inside the cell to limit cell proliferation by initiating cell cycle arrest and apoptosis (71). Glutathione s-transferase (GST) is the second important target of the antioxidant-induced downstream response of Nrf2, which regulates metabolic detoxification of various electrophilic xenobiotic by forming a complex with them. Several isoforms of GST are reported to be expressed in a tissue-specific manner (72). The GST null phenotype is associated with an increased rate of cancer development (73). Nrf2 activation and its role in tumor suppression is depicted in Figure 1. Curcumin, an antioxidant, has shown anti-carcinogenic properties by elevating GSTα, GSTµ, and GSTπ activity (74).
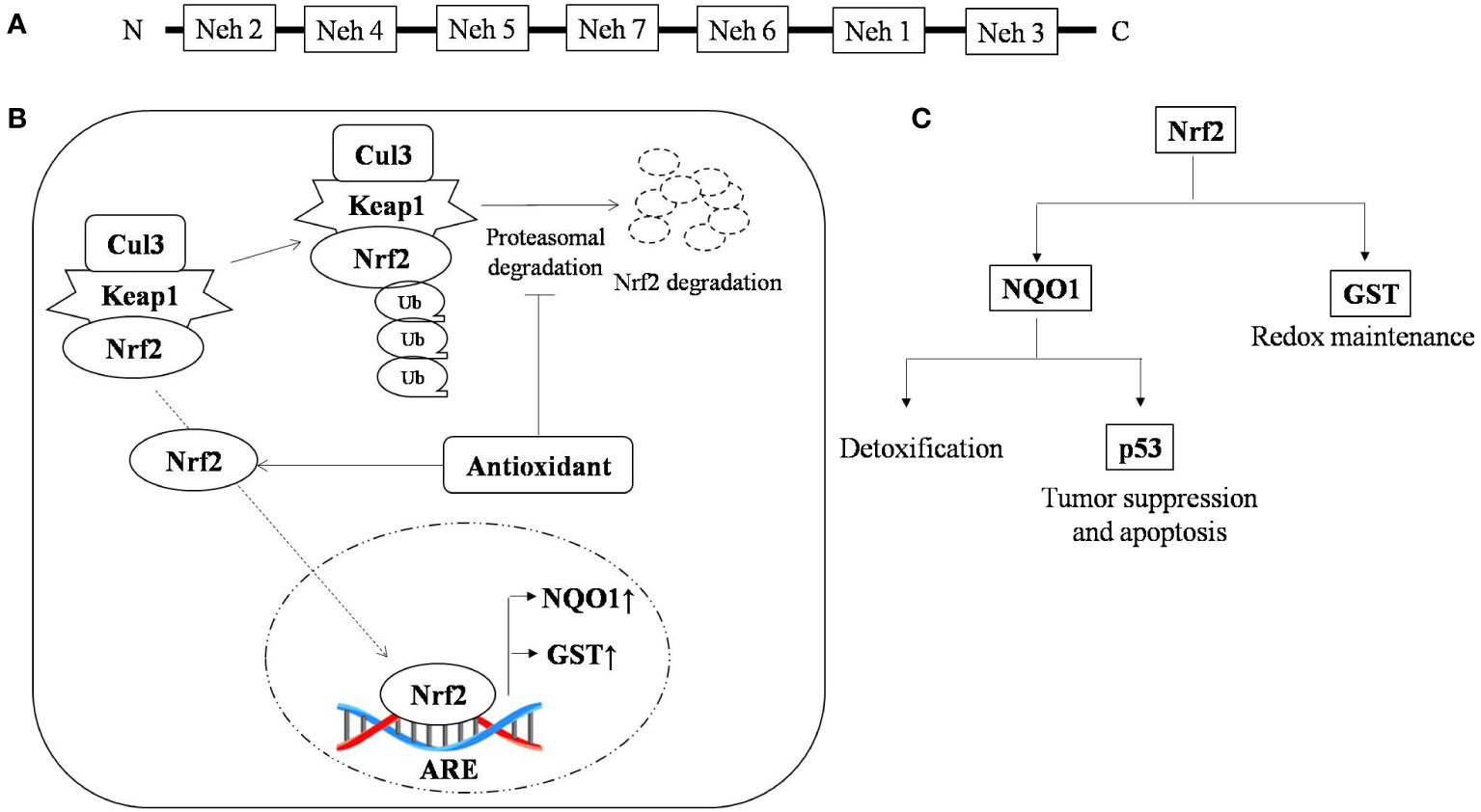
Figure 1 Nrf2 activation and the role of antioxidants in tumor suppression (A) Typical representation of the Nrf2 domain that regulate specific ARE genes; (B) Antioxidant stabilizes Nrf-2 by interfering in its proteasomal degradation pathway, resulting in enhanced binding of Nrf-2 with its promoter sequence, which causes upregulation of antioxidant scavengers NQO1 and GST and (C) Nrf-2 promotes the expression of the antioxidant scavengers, namely NQO1 and GST, where GST is a thiol compound regulating redox status and NQO1 promotes detoxification and expression of p53, which under stress regulates cell cycle and apoptosis. ↑, up-regulation; ↓, down-regulation.
NF-κB signaling
NF-κB represents a group of proteins that are very sensitive to any changes in the cellular redox profile and has a key role in cancer progression (75, 76). Normally, it remains sequestered in the cytoplasm along with inhibitory subunit Ikβ. ROS have been reported as both activators and inhibitory stimuli for NF-κB depending on the type of modification and site of action (77–80). NF-kB regulates multiple proteins, including COX2, VEGE-A, TNF-α, and IL-6. The COX2 protein has been described as one of the major players linking inflammation to cancer through different signaling pathways (81, 82). Vascular endothelial growth factor-A (VEGF-A), another positive downstream of NF-κB, promotes angiogenesis by activating its receptor VEGF-R1 in endothelial cells of micro vessels in lymphoma (83). A schematic representation of the regulation of T-cell lymphoma by NF-κB is depicted in Figure 2. NF-κB has also been found to regulate inflammatory cytokines in a positive feedback mechanism such as TNF-α and IL-6 in T-cell malignant transformation (84).
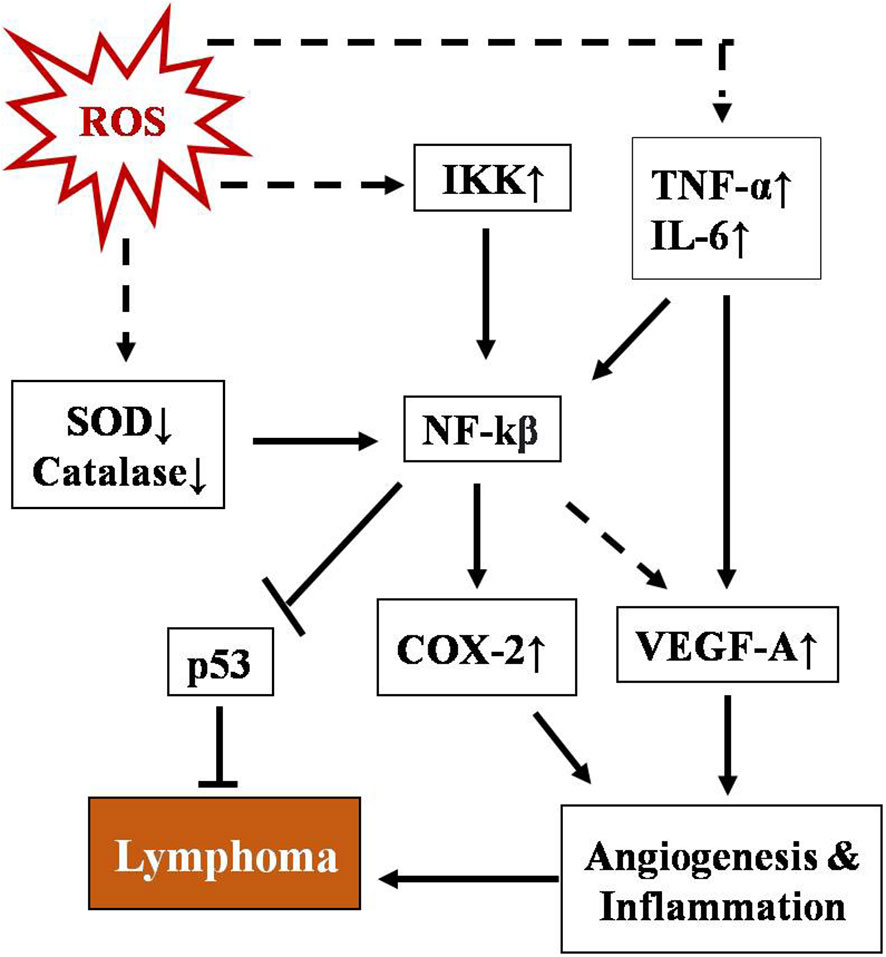
Figure 2 Schematic representation of regulation of T-cell lymphoma by NF-κB. NF-kB activation involves a phosphorylation cascade leading to degradation of its inhibitor IKβ. ROS regulate NF-κB through multiple cellular agents, namely SOD↓, catalase↓, Ikk↑, TNFα, and IL-6↑ (where ↑& ↓represent upregulation and downregulation respectively). Activated NF-κB leads to upregulation of COX-2 and VEGF-A and downregulates p53, promoting angiogenesis and inflammation which ultimately lead to lymphoma.
PI3K-AKT signaling
PI3K signaling acts as master regulator in controlling cell survival, proliferation, and growth. It catalyzes the conversion of phosphatidyl inositol (4, 5)-bisphosphate (PIP2) to phosphatidylinositol (3–5) - triphosphate (PIP3), and this PIP3 initiates the localization of AKT to membrane and its subsequent activation by PDK1.ROS accumulation leads to phosphorylation of PI3K on tyrosine residues of a regulatory subunit (p85α) and thereby results in disassembly of it from its catalytic counterpart p110α (85–87). The p110α triggers rapid transformation of PIP2 to PIP3. AKT activation resulting in cell transformation and generation of cancer phenotypes (88). AKT promotes cell survival by inhibiting proapoptotic protein BAD by phosphorylating it and preventing its interaction with BCL-xl (85, 89–91). Regulation of T-cell lymphoma by the PI3k-AKT signaling pathway is depicted in Figure 3.
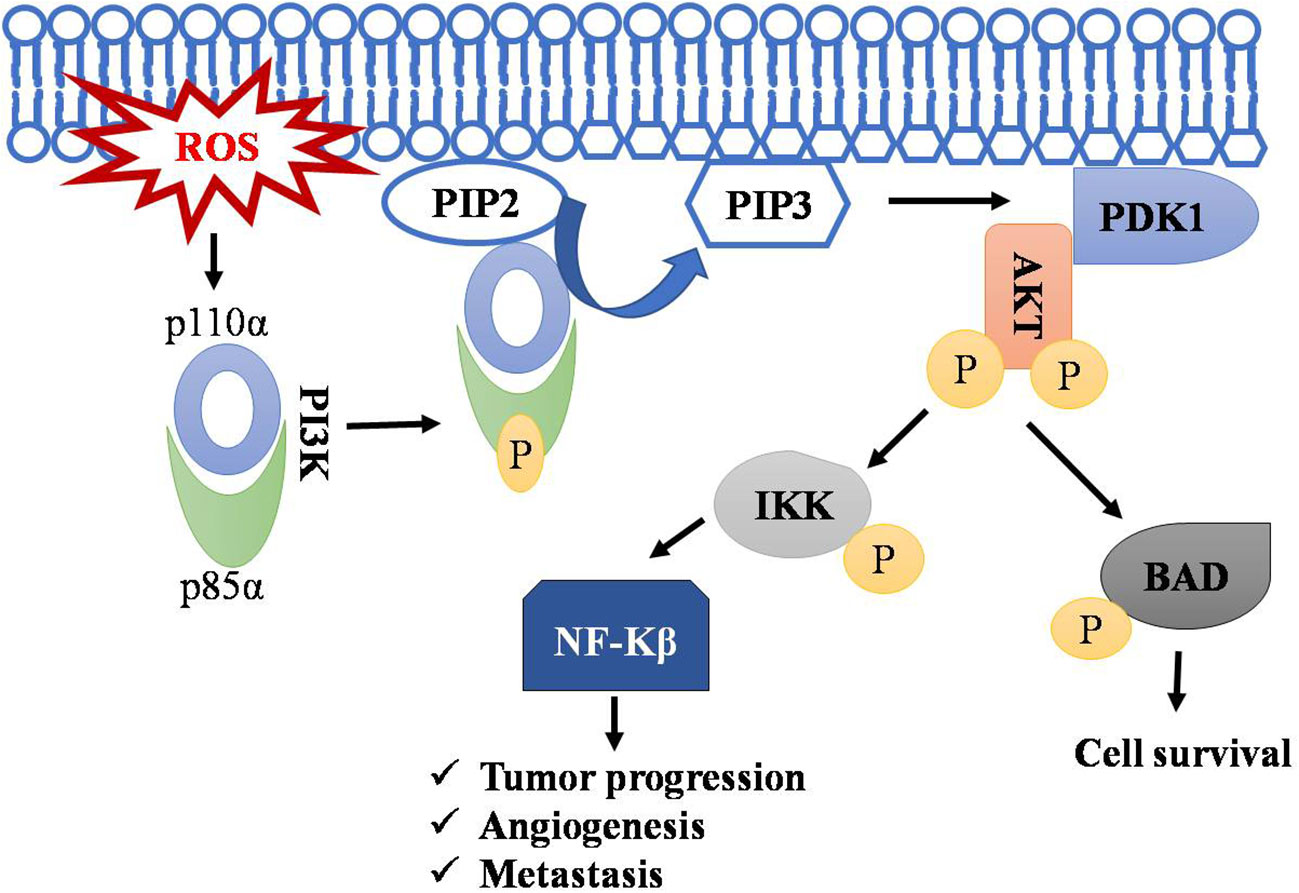
Figure 3 Regulation of T-cell lymphoma by PI3k-AKT pathway. Accumulation of ROS leads to the phosphorylation of PI3K resulting in disassembly of its regulatory subunit (p85α) and catalytic subunit (p110α). The catalytic subunit now phosphorylates PIP2 into PIP3. PIP3 serve as a docking site for AKT, where PDK1 activates AKT by phosphorylation. AKT phosphorylates IKK and BAD, leading to NF-kB activation and thus to cell survival, angiogenesis, and metastasis. Symbol "✔" represents just a separate process. Arrow represents the sequential progression of signaling.
Conclusion and future perspective
From increasing experimental evidence, the role of ROS in tumorigenesis and its progression by regulating and/or altering the cellular environment has become apparent, stabilizing it as a potent hallmark of cancer. Although we have discussed some of the pathway, their complete regulation machinery is unknown and needs more scientific exploration. Changes in cellular redox are stabilized to contribute toward cancer pathogenesis but specific redox levels and their origin are still matters of research. Associated signaling pathways could reveal the hurdle of tumor progression by providing a target for drug development and help us better understand the etiology of cancer progression.
Author contributions
S.S.: writing-original draft and writing- review and editing. A.K.M.: conception and design, writing-original draft, writing-review and editing, administrative, technical, and material support. All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Acknowledgments
Research was supported by Institution of Eminence (IOE), Banaras Hindu University, Varanasi, India. SS thanks UGC, India for their Junior Research Fellowship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, et al. Molecular biology of the cell. 4th edition. New York: Garland Science (2002).
3. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IB de O, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumors: Lymphoid neoplasms. Leukemia (2022) 36(7):1720–48.
4. A clinical evaluation of the international lymphoma study group classification of non-hodgkin's lymphoma. Blood (1997) 89(11):3909–18.
7. Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans (2018) 47(19):6645–53.
8. Hu LY, Mi WL, Wu GC, Wang YQ, Mao-Ying QL. Prevention and treatment for chemotherapy-induced peripheral neuropathy: Therapies based on CIPN mechanisms. CurrNeuropharmacol (2019) . 17(2):184–96.
9. Roufaiel MNKN, Wells JW, Steptoe RJ. Impaired T-cell function in b-cell lymphoma: A direct consequence of events at the immunological synapse. Front Immunol (2015) 6:258.
10. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free RadicBiol Med (2010) 48(6):749–62.
11. Auten RL, Davis JM. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr Res (2009) 66(2):121–7.
12. Karin M, Greten FR. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat Rev Immunol (2005) 5(10):749–59.
13. Visconti R, Grieco D. New insights on oxidative stress in cancer. CurrOpin Drug DiscovDevel. (2009) 12(2):240–5.
14. Hattori K, Shrestha R, Sakamoto T, Kusakabe M, Sakata-Yanagimoto M. Animal models of T-cell lymphoma. Haematol Rep (2006) 2(13):47–55.
15. Ellyard JI, Chia T, Rodriguez-Pinilla SM, Martin JL, Hu X, Navarro-Gonzalez M, et al. Heterozygosity for roquinsan leads to angioimmunoblastic T-cell lymphoma-like tumor in mice. Blood. (2012) 120(4):812–21.
16. Muto H, Sakata-Yanagimoto M, Nagae G, Shiozawa Y, Miyake Y, Yoshida K, et al. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J (2014) 4(12):e264.
17. Cortes JR, Ambesi-Impiombato A, Couronné L, Quinn SA, Kim CS, da Silva Almeida AC, et al. RHOA G17V induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell (2018) 33(2):259–273.e7.
18. Ng SY, Brown L, Stevenson K, DeSouza T, Aster JC, Louissaint A, et al. RhoA G17V is sufficient to induce autoimmunity and promotes T-cell lymphomagenesis in mice. Blood (2018) 132(9):935–47.
19. Nguyen TB, Sakata-Yanagimoto M, Fujisawa M, Nuhat ST, Miyoshi H, Nannya Y, et al. Dasatinib is an effective treatment for angioimmunoblastic T-cell lymphoma. Cancer Res (2020) 80(9):1875–84.
20. Sato F, Ishida T, Ito A, Mori F, Masaki A, Takino H, et al. Angioimmunoblastic T-cell lymphoma mice model. Leuk Res (2013) 37(1):21–7.
21. Kuefer MU, Look AT, Pulford K, Behm FG, Pattengale PK, Mason DY, et al. Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood (1997) 90(8):2901–10.
22. Miething C, Grundler R, Fend F, Hoepfl J, Mugler C, von Schilling C, et al. The oncogenic fusion protein nucleophosmin–anaplastic lymphoma kinase (NPM–ALK) induces two distinct malignant phenotypes in a murine retroviral transplantation model. Oncogene (2003) 22(30):4642–7.
23. Miething C, Grundler R, Mugler C, Peschel C, Duyster J. A new method of retroviral lineage specific expression utilizing the cre/lox system induces a T-lymphoid malignancy in a mouse model of ALCL. Blood. (2004) 104(11):348.
24. Chiarle R, Gong JZ, Guasparri I, Pesci A, Cai J, Liu J, et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood (2003) 101(5):1919–27.
25. Turner SD, Tooze R, Maclennan K, Alexander DR. Vav-promoter regulated oncogenic fusion protein NPM-ALK in transgenic mice causes b-cell lymphomas with hyperactive jun kinase. Oncogene (2003) 22(49):7750–61.
26. Turner SD, Merz H, Yeung D, Alexander DR. CD2 promoter regulated nucleophosmin-anaplastic lymphoma kinase in transgenic mice causes b lymphoid malignancy. Anticancer Res (2006) 26(5A):3275–9.
27. Pfeifer W, Levi E, Petrogiannis-Haliotis T, Lehmann L, Wang Z, Kadin ME. A murine xenograft model for human CD30+ anaplastic large cell lymphoma. successful growth inhibition with an anti-CD30 antibody (HeFi-1). Am J Pathol (1999) 155(4):1353–9.
28. Nerenberg M, Hinrichs SH, Reynolds RK, Khoury G, Jay G. The tat gene of human T-lymphotropic virus type 1 induces mesenchymal tumors in transgenic mice. Science (1987) 237(4820):1324–9.
29. Kawano N, Ishikawa F, Shimoda K, Yasukawa M, Nagafuji K, Miyamoto T, et al. Efficient engraftment of primary adult T-cell leukemia cells in newborn NOD/SCID/beta2-microglobulin(null) mice. Leukemia (2005) 19(8):1384–90.
30. Mishra A, la Perle K, Kwiatkowski S, Sullivan LA, Sams GH, Johns J, et al. Mechanism, consequences, and therapeutic targeting of abnormal IL15 signaling in cutaneous T-cell lymphoma. Cancer Discovery (2016) 6(9):986–1005.
31. Fehniger TA, Suzuki K, Ponnappan A, VanDeusen JB, Cooper MA, Florea SM, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype Cd8+ T cells. J Exp Med (2001) 193(2):219–32.
32. Cornejo MG, Kharas MG, Werneck MB, le BS, SA M, Ball B, et al. Constitutive JAK3 activation induces lymphoproliferative syndromes in murine bone marrow transplantation models. Blood (2009) 113(12):2746–54.
33. Rivera-Munoz P, Laurent AP, Siret A, Lopez CK, Ignacimouttou C, Cornejo MG, et al. Partial trisomy 21 contributes to T-cell malignancies induced by JAK3-activating mutations in murine models. Blood Adv (2018) 2(13):1616–27.
34. Moffitt AB, Ondrejka SL, McKinney M, Rempel RE, Goodlad JR, Teh CH, et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med (2017) 214(5):1371–86.
35. Das DK, Pathan SK, Joneja M, Al-Musawi FA, John B, Mirza KR. T-Cell prolymphocytic leukemia (T-PLL) with overlapping cytomorphological features with T-CLL and T-ALL: a case initially diagnosed by fine-needle aspiration cytology and immunocytochemistry. DiagnCytopathol (2013) 41(4):360–5.
36. Nahmod KA, Thakral B, Aakash FNU, Iyer SP, Medeiros LJ, Quesada AE. From the archives of MD Anderson cancer center: Aleukemic T-prolymphocytic leukemia, a rare presentation and review of the literature. Ann DiagnPathol (2023) 62.
37. Rosenquist R, Porwit A. The chronic lymphoid leukemias. Blood Bone Marrow Pathology: Expert Consult (2011), 407–18.
38. Raj Kumar K. Dalton’s lymphoma as a murine model for understanding the progression and development of T-cell lymphoma and its role in drug discovery. Int J Immunother Cancer Res (2017), 1–6.
39. Shi M, Olteanu H, Jevremovic D, He R, Viswanatha D, Corley H, et al. T-Cell clones of uncertain significance are highly prevalent and show close resemblance to T-cell large granular lymphocytic leukemia. implications for laboratory diagnostics. Modern Pathol (2020) 33(10):2046–57.
40. Booken N, Goerdt S, Klemke CD. Clinical spectrum of primary cutaneous CD30-positive anaplastic large cell lymphoma: An analysis of the Mannheim cutaneous lymphoma registry. J Dtsch Dermatol Ges (2012) 10(5):331–9.
41. Krishnan J, Tomaszewski M -M, Kao GF. Primary cutaneous CD30-positive anaplastic large cell lymphoma. report of 27 cases. J Cutan Pathol (1993) 20(3):193–202.
42. Al-Rohil RN, Torres-Cabala CA, Patel A, Tetzlaff MT, Ivan D, Nagarajan P, et al. Loss of CD30 expression after treatment with brentuximab vedotin in a patient with anaplastic large cell lymphoma: A novel finding. J Cutan Pathol (2016) 43(12):1161–6.
43. Patkar N, Nair S, Alex AA, Parihar M, Manipadam MT, Arora N, et al. Clinicopathological features of hepatosplenic T cell lymphoma: A single centre experience from India. Leuk Lymphoma (2012) 53(4):609–15.
44. Cooke CB, Krenacs L, Stetler-Stevenson M, Greiner TC, Raffeld M, Kingma DW, et al. Hepatosplenic T-cell lymphoma: A distinct clinicopathologic entity of cytotoxic gamma delta T-cell origin. Blood (1996) 88(11):4265–74.
45. Chen W, Kesler MV, Karandikar NJ, McKenna RW, Kroft SH. Flow cytometric features of angioimmunoblastic T-cell lymphoma. Cytometry B Clin Cytom. (2006) 70B(3):142–8.
46. Kakugawa Y, Terasaka S, Watanabe T, Tanaka S, Taniguchi H, Saito Y. Enteropathy-associated T-cell lymphoma in small intestine detected by capsule endoscopy. Leuk Lymphoma (2012) 53(8):1623–4.
47. CecilieAlfsen G, Beiske K, Bell H, Marton PF. Low-grade intestinal lymphoma of intraepithelial T lymphocyties with concomitant enteropathy-associated T cell lymphoma: Case report suggesting a possible histogenetic relationship. Hum Pathol (1989) 20(9):909–13.
48. Yokote T, Akioka T, Oka S, Hara S, Kobayashi K, Nakajima H, et al. Flow cytometric immunophenotyping of adult T-cell Leukemia/Lymphoma using CD3 gating. Am J Clin Pathol (2005) 124(2):199–204.
49. Dahmoush L, Hijazi Y, Barnes E, Stetler-Stevenson M, Abati A. Adult T-cell leukemia/lymphoma. Cancer Cytopathol (2002) 96(2):110–6.
50. Li R, Jia Z, Zhu H. Regulation of Nrf2 signaling. React Oxyg Species (Apex). (2019) 8(24):312–22.
51. Kotlo KU, Yehiely F, Efimova E, Harasty H, Hesabi B, Shchors K, et al. Nrf2 is an inhibitor of the fas pathway as identified by achilles’ heel method, a new function-based approach to gene identification in human cells. Oncogene. (2003) 22(6):797–806.
52. Muiswinkel F, Kuiperij H. The Nrf2-ARE signalling pathway: Promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Target -CNS Neurological Disord (2005) 4(3):267–81.
53. Lu MC, Ji JA, Jiang ZY, You QD. The Keap1–Nrf2–ARE pathway as a potential preventive and therapeutic target: An update. Med Res Rev (2016) 36(5):924–63.
54. Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, et al. The Nrf2–ARE pathway. Ann N Y Acad Sci (2008) 1147(1):61–9.
55. Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res (2008) 58(5–6):262–70.
56. Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at ser-40 by protein kinase c regulates antioxidant response element-mediated transcription. J Biol Chem (2002) 277(45):42769–74.
57. Mishra S, Vinayak M. Ellagic acid inhibits PKC signaling by improving antioxidant defense system in murine T cell lymphoma. Mol Biol Rep (2014) 41(7):4187–97.
58. Armagan G, Sevgili E, Gürkan FT, Köse FA, Bilgiç T, Dagci T, et al. Regulation of the Nrf2 pathway by glycogen synthase kinase-3β in MPP+-induced cell damage. Molecules (2019) 24(7):1–20.
59. Wu X, Stenson M, Abeykoon J, Nowakowski K, Zhang L, Lawson J, et al. Targeting glycogen synthase kinase 3 for therapeutic benefit in lymphoma. Blood (2019) 134(4):363–73.
60. Aschner M, Culbreth M. GSK-3β, a double-edged sword in Nrf2 regulation: Implications for neurological dysfunction and disease. F1000Res (2018) 7:1–7.
61. Dimou A, Syrigos KN. The role of GSK3β in T lymphocytes in the tumor microenvironment. Front Oncol (2020) 10.
62. Khan NM, Haseeb A, Ansari MY, Devarapalli P, Haynie S, Haqqi TM. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human osteoarthritis chondrocytes. Free Radic Biol Med (2017) 106:288–301.
63. Zhang B, Zhao J, Wang Z, Xu L, Liu A, Du G. DL0410 attenuates oxidative stress and neuroinflammation via BDNF/TrkB/ERK/CREB and Nrf2/HO-1 activation. Int Immunopharmacol (2020) 86:106729.
64. van der Zwet JCG, Buijs-Gladdines JGCAM, Cordo’ V, Debets DO, Smits WK, Chen Z, et al. MAPK-ERK is a central pathway in T-cell acute lymphoblastic leukemia that drives steroid resistance. Leukemia (2021) 35(12):3394–405.
65. Salaroglio IC, Mungo E, Gazzano E, Kopecka J, Riganti C. ERK is a pivotal player of chemo immune-resistance in cancer. Int J Mol Sci (2019) 20(10):2505.
66. Liou JT, Lin CS, Liao YC, Ho LJ, Yang SP, Lai JH. JNK/AP-1 activation contributes to tetrandrine resistance in T-cell acute lymphoblastic leukaemia. Acta Pharmacol Sin (2017) 38(8):1–13.
67. Surh YJ, Na HK. NF-κB and Nrf2 as prime molecular targets for chemoprevention and cytoprotection with anti-inflammatory and antioxidant phytochemicals. Genes Nutr (2008) 2(4):313–7.
68. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/Small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. BiochemBiophys Res Commun (1997) 236(2):313–22.
69. Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. NAD(P)H: Quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact (2000) 129(1–2):77–97.
70. Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-induced expression of NAD (P) h: Quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol (2008) 181(10):6730–7.
72. Wu B, Dong D. Human cytosolic glutathione transferases: Structure, function, and drug discovery. Trends Pharmacol Sci (2012) 33(12):656–68.
73. McIlwain CC, Townsend DM, Tew KD. Glutathione s-transferase polymorphisms: Cancer incidence and therapy. Oncogene (2006) 25(11):1639–48.
74. Das L, Vinayak M. Long term effect of curcumin in restoration of tumor suppressor p53 and phase-II antioxidant enzymes via activation of Nrf2 signalling and modulation of inflammation in prevention of cancer. PloS One (2015) 10(4):e0124000.
75. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active b-cell-receptor signalling in diffuse large b-cell lymphoma. Nature (2010) 463(7277):88–92.
76. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large b-cell lymphoma. Nature (2009) 459(7247):717–21.
77. Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-κB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal (2005) 7(3-4):395–403.
78. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. . Cell Res (2011) 21(1):103–15.
79. Das L, Vinayak M. Curcumin modulates glycolytic metabolism and inflammatory cytokines via nrf 2 in dalton’s lymphoma ascites cells In vivo. Anticancer Agents Med Chem (2019) 18(12):1779–91.
80. Gilmore TD. Introduction to NF-κB: Players, pathways, perspectives. Oncogene (2006) 25(51):6680–4.
81. Harris RE. Cyclooxygenase-2 (COX-2) and the inflammogenesis of cancer. Subcell Biochem (2007) 42:93–126.
82. Kopp KLM, Kauczok CS, Lauenborg B, Krejsgaard T, Eriksen KW, Zhang Q, et al. COX-2-dependent PGE2 acts as a growth factor in mycosis fungoides (MF). Leukemia (2010) 24(6):1179–85.
83. Zhao WL, Mourah S, Mounier N, Leboeuf C, Daneshpouy ME, Legrès L, et al. Vascular endothelial growth factor-a is expressed both on lymphoma cells and endothelial cells in angioimmunoblastic T-cell lymphoma and related to lymphoma progression. Lab Invest (2004) 84(11):1512–9.
84. Parakkal D, Sifuentes H, Semer R, Ehrenpreis ED. Hepatosplenic T-cell lymphoma in patients receiving TNF-α inhibitor therapy. Eur J Gastroenterol Hepatol (2011) 23(12):1150–6.
85. Maurya AK, Vinayak M. Quercetin attenuates cell survival, inflammation, and angiogenesis via modulation of AKT signaling in murine T-cell lymphoma. Nutr Cancer (2017) 69(3):470–80.
86. Cohen B, Yoakim M, Piwnica-Worms H, Roberts TM, Schaffhausen BS. Tyrosine phosphorylation is a signal for the trafficking of pp85, an 85-kDa phosphorylated polypeptide associated with phosphatidylinositol kinase activity. Proc Natl Acad Sci (1990) 87(12):4458–62.
87. Xiao ZP, Peng ZY, Peng MJ, Yan WB, Ouyang YZ, Zhu HL, et al. Flavonoids health benefits and their molecular mechanism. Mini-Reviews Medicinal Chem (2011) 11(2):169–77.
88. Maurya AK, Vinayak M. Quercetin regresses dalton’s lymphoma growth via suppression of PI3K/AKT signaling leading to upregulation of p53 and decrease in energy metabolism. Nutr Cancer (2015) 67(2):354–63.
89. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell (1997) 91(2):231–41.
90. Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ (2009) 16(3):368–77.
Keywords: T-cell lymphoma, redox status, cancer metabolism, Nrf2, NF-κB, signaling pathways
Citation: Singh S and Maurya AK (2023) Junction of the redox dynamic, orchestra of signaling, and altered metabolism in regulation of T- cell lymphoma. Front. Oncol. 13:1108729. doi: 10.3389/fonc.2023.1108729
Received: 26 November 2022; Accepted: 21 March 2023;
Published: 19 May 2023.
Edited by:
Santosh Kumar, National Institute of Technology Rourkela, IndiaReviewed by:
Yiqing Li, Department of Hematology, Sun Yat-sen University, ChinaRaj Kumar Koiri, Dr. Harisingh Gour Central University, India
Copyright © 2023 Singh and Maurya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akhilendra Kumar Maurya, YWtoaWxlbmRyYWt1bWFyQGJodS5hYy5pbg==