
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 24 February 2023
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1100532
This article is part of the Research Topic Improving our Understanding of the Management and Pathogenesis of Rare and Neglected Tumors of the Central and Peripheral Nervous System View all 23 articles
 Giulio Bonomo1,2,3*†‡
Giulio Bonomo1,2,3*†‡ Alessandro Gans1,2†
Alessandro Gans1,2† Elio Mazzapicchi1,2
Elio Mazzapicchi1,2 Emanuele Rubiu1,2
Emanuele Rubiu1,2 Paolo Alimonti1
Paolo Alimonti1 Marica Eoli4
Marica Eoli4 Rosina Paterra4
Rosina Paterra4 Bianca Pollo5Guglielmo Iess1,2
Bianca Pollo5Guglielmo Iess1,2 Francesco Restelli1,2
Francesco Restelli1,2 Jacopo Falco1,2
Jacopo Falco1,2 Francesco Acerbi1
Francesco Acerbi1 Marco Paolo Schiariti1Paolo Ferroli1
Marco Paolo Schiariti1Paolo Ferroli1 Morgan Broggi1
Morgan Broggi1Background: Sporadic Spinal Psammomatous Malignant Melanotic Nerve Sheath Tumor (SSP-MMNST) is a rare subgroup of peripheral nerve sheath tumors arising along the spine. Only a few reports of SSP-MMNST have been described. In this paper, we review the literature on SSP-MMNST focusing on clinical, and diagnostic features, as well as investigating possible pathogenetic mechanisms to better implement therapeutic strategies. We also report an illustrative case of a young female presenting with cervicobrachial pain due to two SSP-MMNSTs arising from C5-6 right spinal roots.
Case description: We report a case of a 28-year-old woman presenting with right arm weakness and dysesthesia. Clinical examination and neuroimaging were performed, and, following surgical removal of both lesions, a histological diagnosis of SSP-MMNST was obtained.
Results: The literature review identified 21 eligible studies assessing 23 patients with SSP-MMNST, with a mean onset age of 41 years and a slight male gender preference. The lumbar district was the most involved spinal segment. Gross-total resection (GTR) was the treatment of choice in all amenable cases, followed in selected cases with residual tumor by adjuvant radiotherapy or chemotherapy. The metastatic and recurrence rates were 31.58% and 36.8%, respectively.
Conclusion: Differently from common schwannomas, MMNST represents a rare disease with known recurrence and metastatization propensity. As reported in our review, SSP-MMNST has a greater recurrence rate when compared to other forms of spinal MMNST, raising questions about the greater aggressiveness of the former. We also found that residual disease is related to a higher risk of systemic disease spreading. This metastatic potential, usually associated with primary lumbar localization, is characterized by a slight male prevalence. Indeed, whenever GTR is unachievable, considering the higher recurrence rate, adjuvant radiation therapy should be taken into consideration.
Malignant melanotic nerve sheath tumor (MMNST) represents a rare variant of nerve sheath neoplasms and accounts for less than 1% of all primary peripheral nerve sheath tumors (1). MMNST predominantly develops from spinal or visceral autonomic nerves (2). Considering its histological and ultrastructural traits of schwannomian differentiation, MMNST was historically considered an atypical variant of schwannoma, hence the definition of “Melanotic schwannoma” (3). However, given its recently uncovered different genetic profile, MMNST now represents a distinct malignant entity, as reported in the 2020 World Health Organization (WHO) classification of soft tissue tumors and in the 2021 WHO classification of central nervous system (CNS) tumors (4, 5). Psammomatous MMNST is an even rarer variant of MMNST, often arising along with the gastrointestinal tract-related nerves or in the extremities (6). The first reported case of MMNST was that by Millar et al. in 1932. The Authors described an uncommon pigmented neural tumor stemming from a sympathetic nerve in the thoracic region (7). The coining of the term would come later, with the 1975 Fu et al. publication (8).
MMNSTs are categorized according to two main groups. The first one is represented by its association with Carney Complex (CC). This entity features autosomal dominant inheritance and is characterized by patchy skin pigmentation, endocrine hyperactivity, and cardiac, mammary, and cutaneous myxomas (9, 10). About 50% of all MMNSTs are associated with CC (9). The second group of MMNSTs includes the presence of round concentric calcifications, known as psammoma bodies. Around 50% of all MMNSTs are psammomatous, and about half of these cases are CC-related (11). Therefore, it is crucial to rule out CC in MMNST patients and consider the possibility of sporadic cases of the disease. Additionally, few reported cases were associated with neurofibromatosis (12).
MMNST displays both malignant and metastatic potential depending on its histology. Microscopically, MMNST features spindle-shaped and epithelioid cells in intertwining fascicles, with a significant concentration of melanin both in the neoplastic cells and in the associated melanophages (1).
Clinical presentation depends on anatomical location. MMNST typically arises along the spinal nerve sheath (13), thus explaining frequent symptoms such as pain and paraesthesia (14–17).
To date, the literature has reported approximately 150 cases of MMNST. In this paper, we review the literature on SSP-MMNST in order to improve the knowledge of this disease, focusing on clinical, and diagnostic data, as well as investigating possible pathogenetic mechanisms to better implement therapeutic strategies. We also report a case of a young female presenting with cervicobrachial pain due to two SSP-MMNSTs arising from C5 and C6 right roots.
A PRISMA flowchart is presented in Figure 1. A primary search returned 449 records. After excluding 192 duplicates, 257 records were screened. After screening titles and abstracts and removing articles without full-text, 85 full-text articles were left and evaluated for eligibility. Given that 64 of them were ineligible for inclusion, we selected 21 full-text papers for the review. We identified 23 cases of SSP-MMNST, including 9 (39.13%) females and 14 (60.87%) males. The male-to-female ratio was 1.56:1, suggesting a slight male prevalence of the disease. The mean age of onset of the disease was 40.39 ± 13.11 years. Spinal localization was as follows: 4 cervical (16.67%), 7 thoracic (29.17%), 9 lumbar (37.5%), and 4 sacral (16.67%). Metastases were reported in 6 cases (4 male, 2 female) with a metastatic rate of 31.58%. Lungs were the main site of metastatization (50%), followed by spinal cord, meninges, bone, chest wall, lymph nodes, and brain parenchyma. In 83.33% of the metastatic cases, the primary tumor site was in the lumbar region, particularly in L4/L5. The mean age of metastatic patients was 37.33 ± 9.09 years. There was no significant difference between the mean age of metastatic and non-metastatic patients (p=0.519). Nonetheless, males appeared slightly more at risk of developing metastasis, with a relative risk (RR) of 1.45 (p=0.6082). Treatment approaches in patients included gross-total resection (GTR) in 17 cases (73.91%), and subtotal resection in 5 cases (22.73%), while one refused the treatment. Patients underwent adjuvant radiotherapy in 4 cases (18.18%), with only one without any recurrence. The disease recurrence rate among the 19 patients with known data was 36.84% (7/19). Of these 7 patients, 4 succumbed to the disease (57.14%), 2 were alive with disease stability (28.57%), and 1 was not reported. Of these 7 patients with disease recurrence, 4 had metastatic locations (57.14%). Finally, 3 of 5 (60%) patients who underwent subtotal resection (STR) developed disease recurrence. On the other hand, only 4 of the 17 patients that underwent GTR suffered a recurrence (23.53%). Considering all 23 patients with SSP-MMNST, the outcome was unknown in 6 patients, 9 were disease-free (52.94%), 6 died, and 2 were still affected by the disease at the time of follow-up. Follow-up was different for each of the cases.
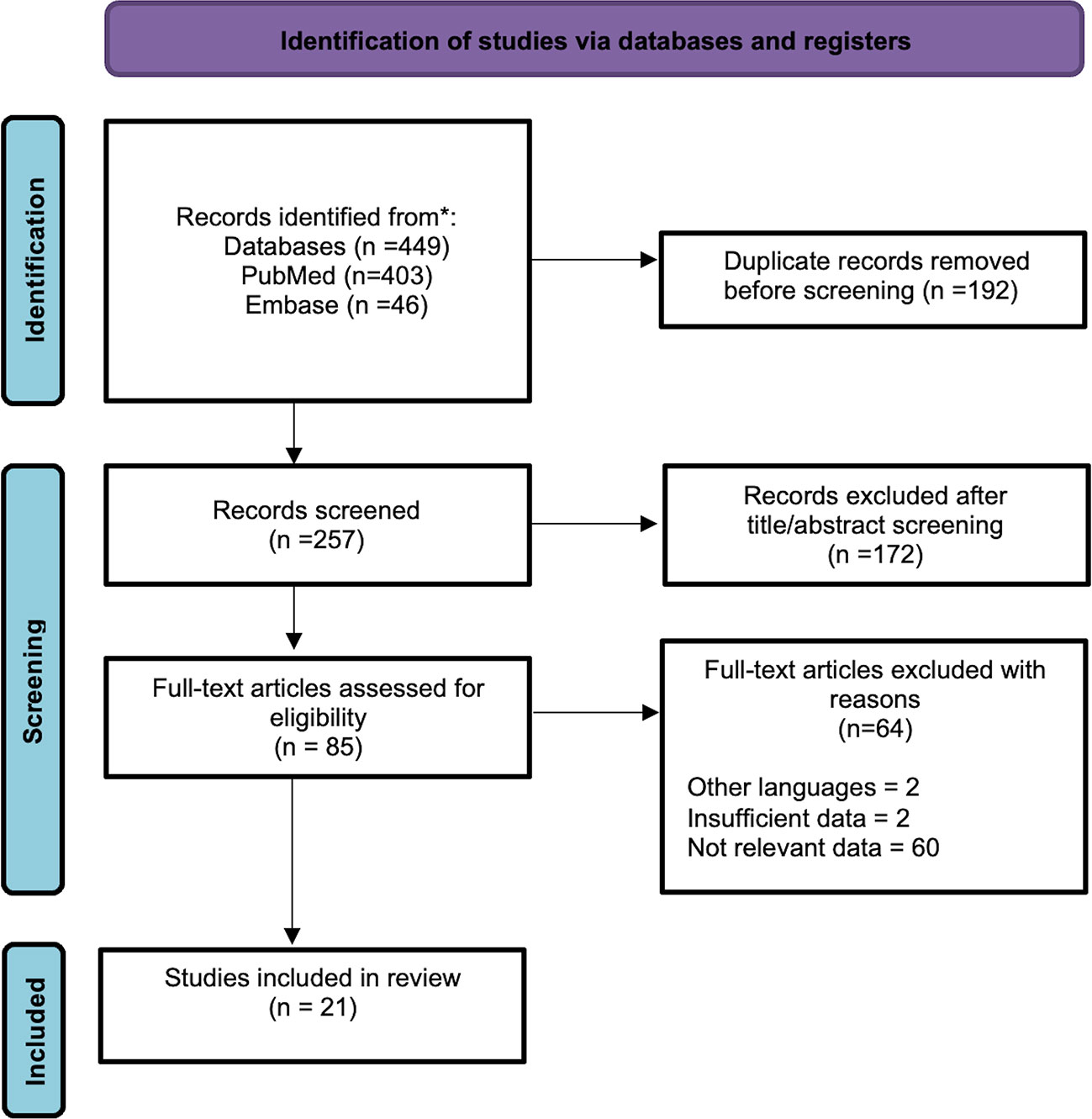
Figure 1 PRISMA flowchart diagram.
A 28-year-old woman presented with a one-year history of cervical pain radiating to the right arm. She had no history of other illnesses, surgeries, related traumas, or a family history of spinal diseases. No skin pigmentary abnormalities were observed. Neurological examination revealed mild weakness in the right arm and dysesthesia in the C5-C6 right dermatome. Electromyography (EMG) testing was performed and did not disclose any abnormality. Spinal cord Magnetic Resonance Imaging (MRI) demonstrated two right intracanal extradural and intra/extra-foraminal lesions at the cervical level: the first lesion measured 20x10mm and was located at the C6-C7 level with paravertebral extension and “dumbbell” shape; the other smaller one was located at C5-C6 level (Figure 2).
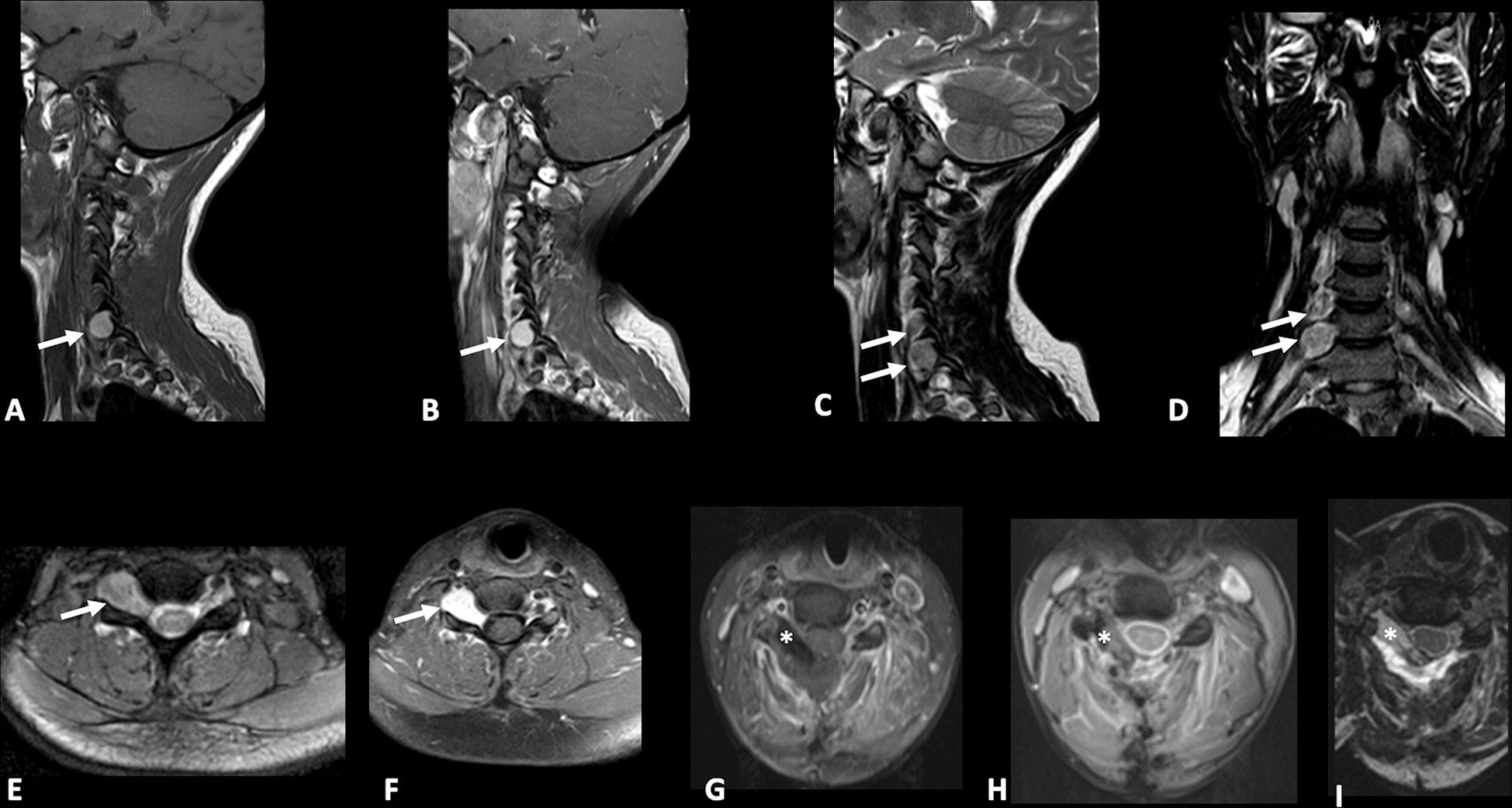
Figure 2 Pre-operative sagittal T1-weighted (A), sagittal (B) and axial (F) post-gadolinium T1-weighted, sagittal (C), coronal (D), axial (E) T2-weighted Nuclear Magnetic Resonance (MRI) images demonstrating the presence of two right sided extradural spinal lesions (arrows) at C5-C7 levels one of which is larger with intraforaminal and paravertebral extension and “dumbbell” shape. Postoperative axial post-gadolinium T1-weighted and fat suppression (G), Gradient echo (GRE) (H), and T2-weighted (I) MRI images demonstrating gross total resection (GTR) of lesions and placement of fat tissue (asterisk) to reinforce dural closure.
The case was discussed by a multidisciplinary team which set the indication for surgical removal of the lesions. The patient gave written informed consent for the procedure. On the day of surgery, the patient was positioned supine, and a laminectomy was performed at the C5-C7 levels. Through a microscopic technique and an extended transcanalar approach, a GTR of intracanal, intra-foraminal, and paravertebral components of the two lesions was performed. Fat tissue was placed to reinforce the dural closure. The postoperative course was uneventful and characterized by strength recovery. On the other hand, dysesthesia slightly improved, but never recovered completely. The patient underwent a postoperative cervical MRI, which confirmed radical excision of both lesions (Figure 2). Surgical specimen underwent histopathological examination, which showed spindle-shaped epithelioid cells with eosinophilic cytoplasm carrying abundant melanin and scattered psammoma bodies. Neoplastic cells featured nuclear polymorphisms and evident nucleoli as well (Figure 3). Immunochemistry showed cellular positivity for “ HMB-45, S100, Synaptophysin, Melan-A and Collagen-IV “, thereby confirming a diagnosis of MNNST. MIB-1 proliferative index was 4-5%. To rule out CC, molecular screening on PRKAR1A gene was performed using Sanger sequencing, and no germline pathogenic variant in PRKAR1A was identified. Concerning the young age of the patient, research on the pathogenic variant in phacomatosis susceptibility genes (NF1, NF2, SPRED1, SMARCB1, LZTR1) and other 24 genes relevant in the pathogenesis of nervous system tumors (TP53, EGFR, PDGFRA, PTEN, PIK3CA, PIK3R, RB1, NOTCH1, CDK4, CDKN2A, CDKN2B, IDH1, IDH2, FUBP1 CIC, TACC3, TERTp, ATRX, DAXX, FGFR3, ACVR1, H3F3A, KIAA1549 BRAF), was performed using a customized gene panel on lymphocyte DNA and tumor tissue. No pathogenic variants of those genes were detected in both tissues. 30 days after the surgical procedure, a whole-body Fluorodeoxyglucose-positron emission tomography/computed tomography (FDG-PET/CT) was performed, showing no residual focal pathology nor metastasis, and an echocardiogram revealed no cardiac myxoma. One year after surgery, the patient was in clinical and radiological remission without any signs of recurrence of the disease as confirmed by a recent one-year FDG-PET/CT.
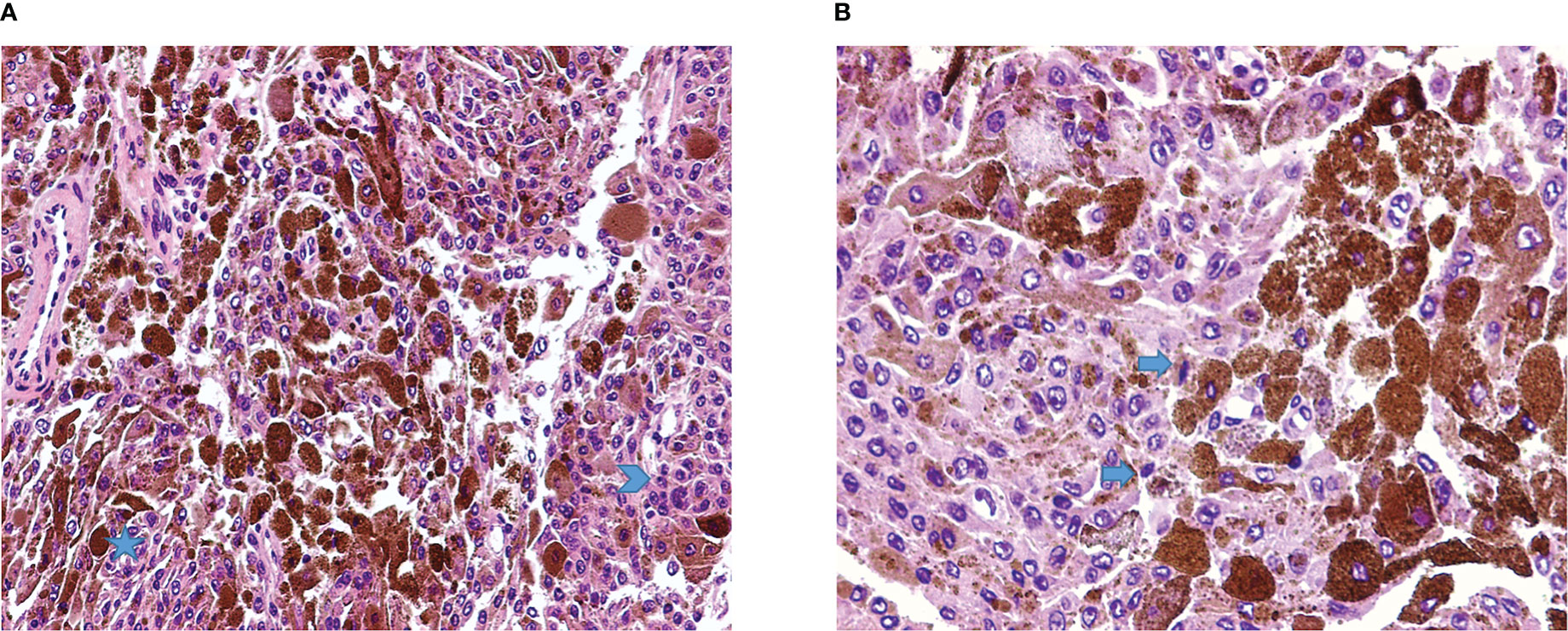
Figure 3 (A) Malignant melanotic nerve sheath tumor (MMNST) composed by pleomorphic cells, with spindle to polygonal shape, organized on fascicles (star) or sheets of roughly syncytial epithelioid cells (arrowhead), and numerous elements with heavy pigment deposits (H&E, magnification 20x). (B) Cellular details show the presence of occasional mitoses (arrows) and brown finely granular melanin pigment (H&E, magnification 40x).
SSP-MMNST is a rare, aggressive, and diagnostically challenging tumor of spinal nerves. We reviewed cases of SSP-MMNST by performing a literature search on PubMed and Embase databases. Their features are summarized in Table 1. It should be noted that some case reports did not include complete clinical, treatment, and genetic data of patients.
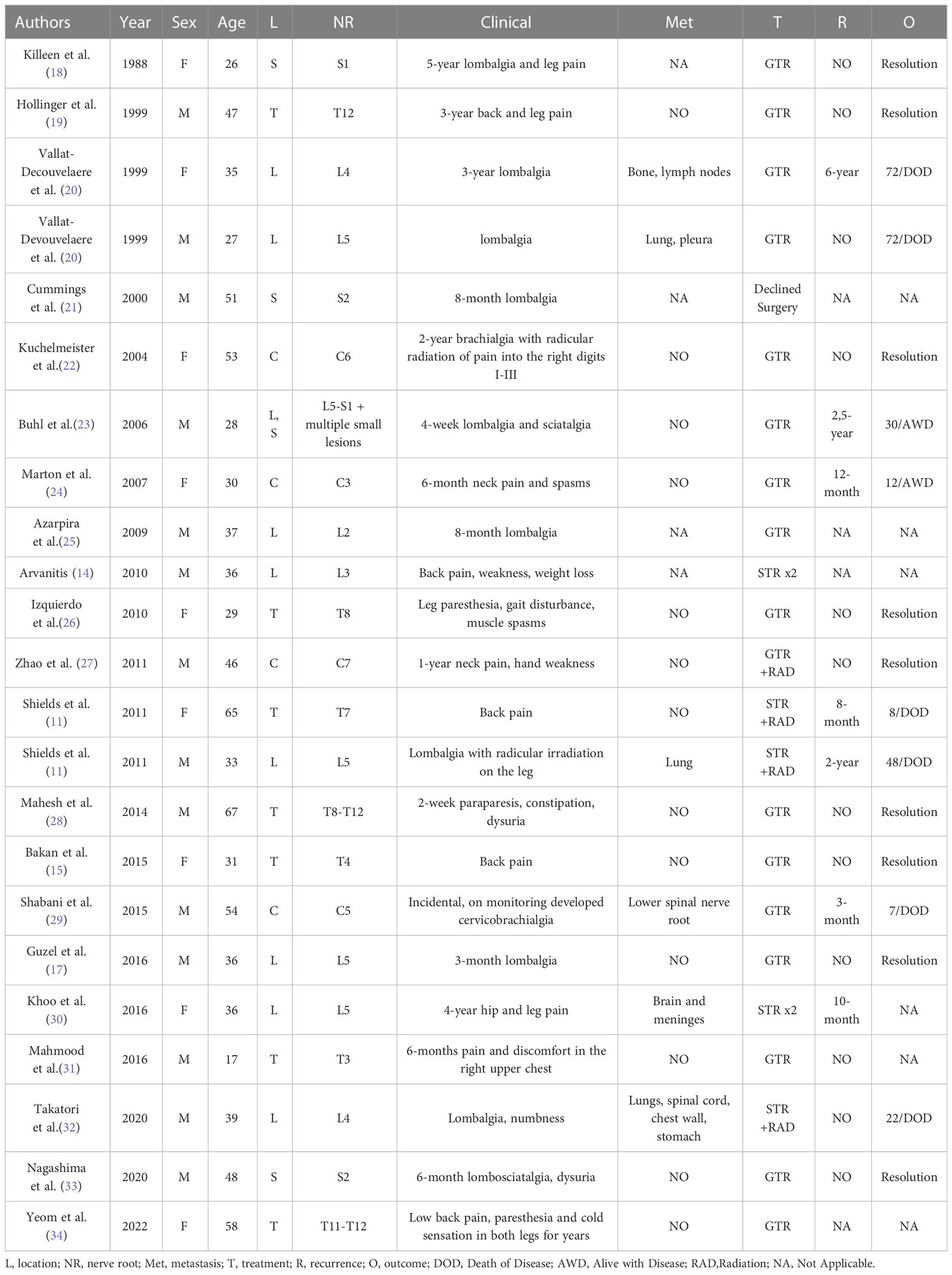
Table 1 Reported cases of sporadic spinal psammomatous melanotic schwannoma.
MMNST, formerly known as Melanotic Schwannoma or Schwannian melanotic tumor, was classified as a malignant lesion in the 2020 WHO classification of soft tissue tumors (4). The neoplasm rarely affects children and most commonly arises in adults, presenting as a sporadic lesion or as part of CC. The mean age of presentation is 38 years and no geographic, racial, ethnic, or gender preferences have been reported so far (35, 36). MMNST development in younger patients may be related to Carney syndrome (24). Psammomatous variants may be seen in patients with CC and arise a decade earlier than isolated sporadic cases, with a prevalence peak in the third decade of life (24, 37). In our analysis, the average disease age of onset for SSP-MMNSTs was 40±13 years (range 17 to 65 years), with slight male prevalence (1.56:1, M:F ratio). This is in contrast with previous studies in which no significant sex predilection was reported.
MMNST generally arises from spinal nerves and sympathetic ganglia as a single lesion (6, 9, 23). Nonetheless, literature reports scarce occurrences in other sites such as sympathetic chains, cranial nerve roots, peripheral nerves, cerebellum, orbit, choroids, soft tissue, alveolar nerves, palate, parotid gland, heart, oral cavity, oesophageal wall, pancreas, trachea, and bones (2, 10, 38–45). Spinal MMNST occurs in the lumbosacral region in 47.2% of cases, in the thoracic tract in 30.5%, and in the cervical region in 22.2%. The tumor may grow both in an intramedullary and extramedullary pattern. Furthermore, the lesion usually moves into the vertebral foramen or paravertebrally mainly affecting the posterior nerve roots (6, 7, 20, 46–48). Our review focused on SSP-MMNST and showed that the lumbar district is the most affected one, with no significant difference between the right and left sides.
The clinical presentation of spinal MMNST is related to its anatomical location. Approximately one-third of cases are asymptomatic (9). On the contrary, symptoms of spinal nerve and spinal cord involvement, which are observed in 35.5% of patients, include radicular and back pain, dysesthesia, progressive sensory and motor deficits, ataxia, and sphincter disorders (38). The peculiar combination of radicular and back pain leads to misdiagnosis, with discopathy being the most frequent one. Mechanical dysfunction of adjacent organs due to compression is reported in 13% of cases (49). Complications vary according to tumor location and may include CC sequelae such as heart failure and stroke (1). Our case was characterized by mild weakness in the right arm and dysesthesia in the C5-C6 right dermatome with progressive deterioration within a year.
Unlike common schwannomas, MMNST is prone to local recurrence and displays metastatic potential (6, 9, 39). Metastases primarily occur in the lung and pleura but may also involve mediastinum, diaphragm, pericardium, endocardium, bone, liver, and spleen (6, 9, 38). In their studies, Torres-Mora et al. and Alexiev et al. showed local recurrence in 15 to 35% of cases and a metastasis rate of 26% to 44% (1, 6).
Our review on SSP-MMNST demonstrated metastasis occurrence in 31.58% of cases, with male prevalence (66.6%) and with no statistically significant difference from the metastatic rate of spinal MMNST (p=0.463) (40). The main metastatic site was lung parenchyma, consistent with previous literature.
In 83% of cases of metastasis, the primitive tumor developed in the lumbar region with a slightly greater risk in males (RR=1.45, p=0.608). The mean age in patients with metastatic disease was lower than non-metastatic ones (37.33 ± 9 vs 42 ± 15 years), with no significant difference.
Comparing whole-body MMNST data (6) with ours on SSP-MMNST, we observed no significant difference in the metastasic rate (44% vs 31.58%, p= 0.563) nor in the recurrence rate (35% vs 36.84%, p = 0.8833). Furthermore, when considering only sporadic spinal MMNSTs (40) and comparing them with our SSP-MMNSTs, we did not find a significant difference in the rate of metastasis (32.7% vs 31.58%, p= 0.9273) nor recurrence (25% vs 36.8%, p= 0.2639). These results seem to support the hypothesis that this histological type has no greater local or distant aggressiveness than sporadic and spinal non-psammomatous cases and whole-body MMNST. We believe that the lack of significant difference between the spinal sporadic MMNST cases and our SSP-MMNST ones may arise from the small sample size available. Nevertheless, the recurrence rate of 36.8% in SSP-MMNST versus 25% in sporadic spinal MMNST appears to be an interesting finding that, if confirmed, could reveal a greater aggressiveness of the former.
Histologically, MMNST generally consists of a single ovoid lesion, and it is rarely multifocal. Adjacent bone erosion may be observed (6, 9, 23, 41). Although being a circumscribed tumor, MMNST does not feature any capsule, in contrast to the typical schwannoma. This characteristic may reflect the potential aggressiveness of the disease. Microscopically, spindle-shaped and plump epithelioid cells appear in intertwined fascicles or nests (1). Melanin accretions in the neoplastic cells and associated melanophages are usually identified. Cytoplasmic pigmentation is highly variable. The pigment is positive for silver Fontana-Masson melanin staining and negative for Prussian blue and periodic acid-Schiff (PAS) staining (50). Rare mitoses are discernible in most lesions. MMNSTs typically lack Verocay bodies, microcysts and thick-walled hyalinized blood vessels (42). The criteria for malignancy in MMNST are not yet fully established. Nonetheless, histological characteristics like large vesicular nuclei with macronucleoli, intense mitotic activity, and necrosis point towards aggressive behavior (6, 38). Immunohistochemical staining for S100, SOX10, HMB-45, Melan-A, p16 and vimentin yields positive results, whereas GFAP, EMA, and CK staining are mostly negative (6, 9, 20, 41–45, 49, 50). All MMNST cases display positive laminin and collagen IV linear and pericellular immunoreactions (50). Aside from our case report, we identified two other MMNST cases with a hemorrhagic presentation, one of which was a heart attack mimicker (27, 40). In our case, the histological features matched with data available in the literature (Figure 3).
MMNST pathogenesis is still poorly understood. Several theories about histogenesis of the tumor have been described. Schwann cells and melanocytes share both their neuroectodermal origin and migration routes. Given this common developmental lineage, Schwann cells may be capable of synthesizing melanin under specific circumstances (18, 51, 52). Further hypotheses point to a melanomatous transformation of neoplastic Schwann cells (53).
Genetically, MMNST features a complex karyotype with recurring 22q band monosomy, variable whole chromosomal gains, and recurrent losses usually affecting chromosomes 1 and 21 and chromosome arm 17p.45 (54). Approximately half of the psammomatous cases are associated with CC, an autosomal dominant genetic disease harboring a chromosome 17 mutation that affects the cAMP-dependent protein kinase type I regulatory subunit alpha (PRKAR1A) gene (9, 42). CC diagnosis requires at least two major clinical findings. However, a single finding is sufficient in cases of positivity for the inactivating PRKAR1A gene mutation (38). MMNST may also develop in neurofibromatosis type I (12, 48). Recent cytogenetic studies demonstrated trisomy 6p at ring chromosome 11 in MMNST, suggesting some similarities with malignant melanoma. However, MMNST lacks the prototypical BRAFV600E mutation (55).
MMNST diagnosis is based on clinical, histopathological, and instrumental findings. The histological diagnosis requires careful differential considerations between neurofibroma, pigmented dermatofibrosarcoma protuberans, melanocytoma, and malignant melanoma.
Our case matched previous literature, with both tumors being positive for HMB-45, S100, Synaptophysin, Melan-A, and Collagen-IV. MIB-1 proliferative index was 4-5%. Phacomatosis susceptibility and CNS tumorigenesis genes were also investigated with negative results. Their negativity allowed us to exclude melanoma, CC, and Neurofibromatosis.
Radiologically, SSP-MMNST is characterized by hyperintensity in T1-weighted sequences, hypointensity in T2-weighted sequences, and homogeneous post-contrast enhancement (Table 2). These features were confirmed in our case (Figure 2). By contrast, schwannomas often present hypointensity in T1 and hyperintensity in T2 (56–58). Melanin displays paramagnetic effects, leading to stable free radicals. Essentially, melanin protons have shorter T1 and T2 relaxation times. Nevertheless, melanin concentration and tumor density are not always consistent. On axial images, the tumor may appear dumbbell-shaped with varying relationships to the medulla and dura. According to the Asazuma et al. classification (59), dumbbell schwannomas are classified into 6 types with our case classified as type IIb (Extradural, inside the spinal canal, intraforaminal and paravertebral).

Table 2 Comparison of Spinal MMNST and SSP-MMNST.
FDG PET/CT provides a unique contribution to: (a) the differential diagnosis of benign and malignant lesions, (b) the detection of covert metastases, (c) the monitoring of treatment response, and (d) the evaluation of MMNST prognosis (6, 9, 39, 60, 61). The patient in our case underwent two full-body FDG-PET-CT scans, the first in the immediate post-operative period and the second at one-year follow-up. In both cases, the scan was negative for focal uptake areas, ruling out disease recurrence and metastasis.
Surgery represents the cornerstone of treatment for MMNST, with GTR being the gold standard for all subtypes. During surgery, a residual tumor capsule may be left to prevent spinal cord injury if the tumor is closely adherent (62, 63).
In our review on SSP-MMNST, the most common approach was GTR in 73.91% of cases. Among these, only 23.53% developed disease recurrence, as opposed to 60% of patients who underwent STR. The total recurrence rate was 36.84%, and 57.14% of that percentage was associated with metastasis. This finding may suggest that recurrent SSP-MMNSTs tend to be more aggressive and, therefore more prone to spread systemically.
Given the limited number of cases, post-surgical management is controversial. No current guidelines for adjuvant treatment in MMNST are available (38, 50). Some studies show a beneficial role of adjuvant radiotherapy in post-operative tumor residuals, reducing the rate of relapse and metastasis during a 24-month follow-up (50). Fractionated radiotherapy is a suitable option for complex MMNST close to susceptible structures, such as the spinal cord, although no significant data on mortality reduction are yet available (38). Indeed, some studies have recommended 54 Gy fractionated radiation therapy applied to the spine (46). Spinal stereotactic radiosurgery may also be considered. Chemotherapy has instead demonstrated low response rates and no mortality benefit (64). In our analysis, radiotherapy was employed in only 18.18% of cases as an adjuvant treatment following STR. Given the metastatic potential of SSP-MMNST, these data seem to suggest that, whenever residual disease is left, adjuvant radiotherapy may be appropriate.
Differently from common schwannomas, MMNST represents a rare disease with known recurrence and metastatization propensity. As reported in our review, SSP-MMNST has a greater recurrence rate when compared to other forms of spinal MMNST, raising questions about its greater aggressiveness. We also found that residual disease is related to a higher risk of systemic spreading. This metastatic potential, usually associated with primary lumbar localization, is characterized by a slight male prevalence. Indeed, whenever GTR is unachievable, considering the higher recurrence rate, adjuvant radiation therapy should be taken into consideration.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Patient signed informed consent regarding publishing his data and photographs.
GB, EM, and MB performed the clinical assessment. GB, AG, EM, ER, PA, and GI critically reviewed the literature and drafted the manuscript. All authors were responsible for important intellectual content. All authors contributed to the article and approved the submitted version.
This work was supported by the Italian Ministry of Health (RRC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
CC, Carney’s complex; CNS, central nervous system; CT, Computed Tomography; EMG, Electromyography; FDG-PET/CT, Fluorodeoxyglucose-Positron Emission Tomography/Computed Tomography; GTR, Gross-Total Resection; MMNST, Malignant Melanotic Nerve Sheath Tumor; MRI, magnetic resonance imaging; PAS, periodic acid-Schiff; SSP-MMNST, Sporadic Spinal Psammomatous Malignant Melanotic Nerve Sheath Tumor; STR, Sub-Total Resection; RR, Relative risk; WHO, World Health Organization.
1. Alexiev BA, Chou PM, Jennings LJ. Pathology of melanotic schwannoma. Arch Pathol Lab Med Allen Press (2018) 142:1517–23. doi: 10.5858/arpa.2017-0162-RA
2. Brierley JD, Asamura H, Eycken EV, Rous B eds. BONE AND SOFT TISSUE TUMOURS. TNM Atlas: Wiley (2021) p. 209–25.
3. Hodson JJ. An intra-osseous tumor combination of biological importance–invasion of a melanotic schwannoma by an adamantinoma. J Pathol Bacteriol (1961) 82:257–66. doi: 10.1002/path.1700820203
4. Sbaraglia M, Bellan E, Dei Tos AP. The 2020 WHO classification of soft tissue tumours: news and perspectives. Pathologica (2020) 113:70–84. doi: 10.32074/1591-951X-213
5. Osborn AG, Louis DN, Poussaint TY, Linscott LL, Salzman KL. The 2021 world health organization classification of tumors of the central nervous system: What neuroradiologists need to know. Am J Neuroradiology (2022) 43:928–37. doi: 10.3174/ajnr.A7462
6. Torres-Mora J, Dry S, Li X, Binder S, Amin M, Folpe AL. Malignant melanotic schwannian tumor: A clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of ‘melanotic schwannoma’. Am J Surg Pathol (2014) 38:94–105. doi: 10.1097/PAS.0b013e3182a0a150
7. Millar WG. A malignant melanotic tumour of ganglion cells arising from a thoracic sympathetic ganglion. J Pathol Bacteriol (1932) 35:351–7. doi: 10.1002/path.1700350305
8. Fu Y-S, Kaye GI, Lattes R. Primary malignant melanocytic tumors of the sympathetic ganglia, with an ultrastructural study of one. Cancer (1975) 36:2029–41. doi: 10.1002/cncr.2820360917
9. Carney JA. Psammomatous melanotic schwannoma. a distinctive, heritable tumor with special associations, including cardiac myxoma and the cushing syndrome. Am J Surg Pathol (1990) 14:206–22. doi: 10.1097/00000478-199003000-00002
10. Aidan Carney J, Gordon H, Carpenter PC, Vittal Shenoy B, Go VLW. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Med (Baltimore) (1985) 64:270–83. doi: 10.1097/00005792-198507000-00007
11. Shields C, Glassman S, LisaBE S, Raque G. Malignant psammomatous melanotic schwannoma of the spine: A component of Carney complex. Surg Neurol Int (2011) 2:136. doi: 10.4103/2152-7806.85609
12. Murakami T, Kiyosawa T, Murata S, Usui K, Ohtsuki M, Nakagawa H. Malignant schwannoma with melanocytic differentiation arising in a patient with neurofibromatosis. Br J Dermatol (2000) 143:1078–82. doi: 10.1046/j.1365-2133.2000.03849.x
13. Greenberg MS. Neurology | Handbook of Neurosurgery [Internet]. karger.com (2019). Available from: https://www.thieme.com/books-main/neurology/product/5411-handbook-of-neurosurgery.
14. Arvanitis LD. Melanotic schwannoma: A case with strong CD34 expression, with histogenetic implications. Pathol Res Pract (2010) 206:716–9. doi: 10.1016/j.prp.2010.02.011
15. Bakan S, Kayadibi Y, Ersen E, Vatankulu B, Ustundag N, Hasiloglu ZI. Primary psammomatous melanotic schwannoma of the spine. Ann Thorac Surg (2015) 99:e141–3. doi: 10.1016/j.athoracsur.2015.02.060
16. Chandran RS, Patil AK, Prabhakar RB, Balachandran K. Melanotic schwannoma of spine: Illustration of two cases with diverse clinical presentation and outcome. Asian J Neurosurg (2018) 13:881–4. doi: 10.4103/ajns.AJNS_353_16
17. Güzel E, Er U, Güzel A, Toktaş Z, Yaplcler Ö. Melanotic schwannoma of the L5 root. Neuroradiol J (2016) 29:219. doi: 10.1177/1971400916638359
18. Killeen RM DCBS. Melanocytic schwannoma. Cancer (1988) 62:174–83. doi: 10.1002/1097-0142(19880701)62:1<174::AID-CNCR2820620127>3.0.CO;2-G
19. Hollinger P, Godoy N, Sturzenegger M. Magnetic resonance imaging findings in isolated spinal psammomatous melanotic schwannoma [4]. J Neurol (1999) 246:1100–2. doi: 10.1007/s004150050522
20. Vallat-Decouvelaere AV, Wassef M, Lot G, Catala M, Moussalam M, Caruel N, et al. Spinal melanotic schwannoma: A tumour with poor prognosis. Histopathology (1999) 35:558–66. doi: 10.1046/j.1365-2559.1999.00786.x
21. Cummings TJ, Liu K, Jordan LK 3rd, Dodd LG. Fine-needle aspiration diagnosis of psammomatous melanotic schwannoma. Diagn Cytopathol (2000) 23(1):55–58. doi: 10.1002/1097-0339(200007)23:1<55::AID-DC13>3.0.CO;2-B
22. Kuchelmeister K, Lotz C, Schönmayr R, Schachenmayr W. April 2004: Woman in her early fifties with a cervical intraspinal and extraspinal mass lesion. Brain pathology (2004) 14(4):453-4–458-9. doi: 10.1111/j.1750-3639.2004.tb00090.x
23. Buhl R, Barth H, Hugo HH, Mautner VF, Mehdorn HM. Intracranial and spinal melanotic schwannoma in the same patient. J Neurooncol (2004) 68:249–54. doi: 10.1023/B:NEON.0000033491.23654.6c
24. Marton E, Feletti A, Orvieto E, Longaitti P. Dumbbell-shaped c-2 psammomatous melanotic malignant schwannoma. case report and review of the literature. J Neurosurg Spine (2007) 6:591–9. doi: 10.3171/spi.2007.6.6.14
25. Azarpira N, Torabineghad S, Sepidbakht S, Rakei M, Bagheri MH. Cytologic findings in pigmented melanotic schwannoma. Acta Cytol (2009) 53(1):113–115. doi: 10.1159/000325096
26. Martinez Izquierdo MA, Lopez-Soto V, Saenz-Santamaria J, Lacruz-Pelea C. Intraoperative cytological findings in two cases of psammomatous melanotic schwannoma. Cytopathology (2011)22:60–2. doi: 10.1111/j.1365-2303.2010.00740.x
27. Zhao QH, Zhi S, Wang Z, Tian JW. Psammomatous melanotic schwannoma with cystic changes from old hemorrhages in the cervical spinal canal: A case report. Orthop Surg (2011) 3:143–6. doi: 10.1111/j.1757-7861.2011.00133.x
28. Mahesh I, Karthikeyan VS, Malathi M. Spotty skin pigmentation and multiple blue naevi as cutaneous markers for spinal melanotic schwannoma. BMJ Case Rep (2014) 2014. doi: 10.1136/bcr-2013-201567
29. Shabani S, Fiore SM, Seidman R, Davis RP. Intraspinal psammomatous melanotic schwannoma not associated with Carney complex: Case report. J Neurosurg Spine (2015) 23:233–8. doi: 10.3171/2014.11.SPINE13990
30. Khoo M, Pressney I, Hargunani R, Tirabosco R. Melanotic schwannoma: An 11-year case series. Skeletal Radiol (2016) 45:29–34. doi: 10.1007/s00256-015-2256-8
31. Mahmood UB, Khan FW, Fatima B, Tariq MU, Fatimi SH. Primary melanotic schwannoma with typical histology. J Coll Physicians Surgeons Pakistan (2016) 26:707–10.
32. Takatori N, Hiyama A, Sakai D, Katoh H, Sato M, Watanabe M. A rare case of intraspinal psammomatous melanotic schwannoma: A case report. spine surg relat res. Japanese Soc Spine Surg Related Res (2020) 4:91. doi: 10.22603/ssrr.2019-0034
33. Nagashima Y, Nishimura Y, Eguchi K, Awaya T, Yoshikawa S, Haimoto S, et al. Intraosseous melanotic schwannoma in the sacrum mimicking primary bone tumor. NMC case rep J. Japan Neurosurgical Soc (2020) 7:107. doi: 10.2176/nmccrj.cr.2019-0238
34. Yeom JA, Song YS, Lee IS, Han IH, Choi KU. Malignant melanotic nerve sheath tumors in the spinal canal of psammomatous and non-psammomatous type: Two case reports. World J Clin Cases (2022) 10:8735–41. doi: 10.12998/wjcc.v10.i24.8735
35. Faria MHG, Dória-Netto RH, Osugue GJ, Queiroz L de S, Chaddad-Neto FE. Melanotic schwannoma of the cervical spine progressing with pulmonary metastasis: Case report. Neurol Med Chir (Tokyo) (2013) 53:712–6. doi: 10.2176/nmc.cr2012-0203
36. Koeller KK, Shih RY. Intradural extramedullary spinal neoplasms: Radiologic-pathologic correlation. Radiographics Radiological Soc North America Inc.; (2019) 39:468–90. doi: 10.1148/rg.2019180200
37. Keskin E, Ekmekci S, Oztekin O, Diniz G. Melanotic schwannomas are rarely seen pigmented tumors with unpredictable prognosis and challenging diagnosis. Case Rep Pathol (2017) 2017:1–4. doi: 10.1155/2017/1807879
38. Siordia JA, Golden TR. Current discoveries and management of psammomatous melanotic schwannoma. J Cancer Tumor Int (2016) 3:1–7. doi: 10.9734/JCTI/2016/23786
39. Kang YE, Jeong JO, Kim KH, Ki CS, Kim HJ. Malignant intercostal psammomatous melanotic schwannoma in a patient with Carney complex. Korean J Intern Med (2018) 33:1256. doi: 10.3904/kjim.2017.139
40. Soyland DJ, Goehner DR, Hoerschgen KM, Gust TD, Vuong SM. Hemorrhagic spinal melanotic schwannoma presenting as acute chest pain: A case report and literature review. Surg Neurol Int (2021) 12:164. doi: 10.25259/SNI_786_2020
41. Kurtkaya-Yapicier O, Scheithauer BW, Woodruff JM. The pathobiologic spectrum of schwannomas. Histol Histopathol (2003) 18(3):925–934. 10.14670/HH-18.925
42. Rodriguez FJ, Stratakis CA, Gareth D. Genetic predisposition to peripheral nerve neoplasia: diagnostic criteria and pathogenesis of neurofibromatoses. Acta Neuropathol (2012) 123(3):349–367. 10.1007/s00401-011-0935-7
43. Chetty R, Vajpeyi R, Penwick JL. Psammomatous melanotic schwannoma presenting as colonic polyps. Virchows Archiv (2007) 451:717–20. doi: 10.1007/s00428-007-0453-0
44. Choi SE, Cha YJ, Kim J, Cha H, Seo J, Kuh SU, et al. A rare case of aggressive melanotic schwannoma occurred in spinal nerve of a 59-Year-Old Male. J Pathol Transl Med (2017) 51:505. doi: 10.4132/jptm.2017.01.04
45. Li B, Chen Q. Melanotic schwannoma of thoracic spinal root mimics metastatic melanoma: A potential pitfall for misdiagnosis. Int J Clin Exp Pathol (2015) 8:8639.
46. Santaguida C, Sabbagh AJ, Guiot MC, Del Maestro RF. Aggressive intramedullary melanotic schwannoma: Case report. Neurosurgery (2004) 55(6):1430. doi: 10.1227/01.NEU.0000143617.25417.68
47. Solomon RA, Handler MS, Sedelli RV, Stein BM. Intramedullary melanotic schwannoma of the cervicomedullary junction. Neurosurgery (1987) 20:36–8. doi: 10.1227/00006123-198701000-00010
48. Zhang HY, Yang GH, Chen HJ, Wei B, Ke Q, Guo H, et al. Clinicopathological, immunohistochemical, and ultrastructural study of 13 cases of melanotic schwannoma. Chin Med J (Engl) (2005) 118:1451–61.
49. Merat R, Szalay-Quinodoz I, Laffitte E, Dermatopathology G--. Psammomatous melanotic schwannoma: A challenging histological diagnosis. karger.com (2015). Available at: https://www.karger.com/Article/Abstract/442708.
50. Gulati HK, Joshi AR, Anand M, Deshmukh SD. Non psammomatous melanocytic schwannoma presenting as a subcutaneous nodule: A rare presentation of a rare lesion. Asian J Neurosurg (2016) 11:317. doi: 10.4103/1793-5482.148789
51. Janzer RC, Makek M. Intraoral malignant melanotic schwannoma. ultrastructural evidence for melanogenesis by schwann’s cells. Arch Pathol Lab Med (1983) 107:298–301.
52. Çulhaci N, Dikicioglu E, Meteoglu I, Bukru S. Multiple melanotic schwannoma. Ann Diagn Pathol (2003) 7(4):254–8. doi: 10.1016/s1092-9134(03)00073-x
53. Mandybur TI. Melanotic nerve sheath tumors. J Neurosurg (1974) 41:187–92. doi: 10.3171/jns.1974.41.2.0187
54. Koelsche C, Hovestadt V, Jones DTW, Capper D, Sturm D, Sahm F, et al. Melanotic tumors of the nervous system are characterized by distinct mutational, chromosomal and epigenomic profiles. Brain Pathol (2015) 25:202–8. doi: 10.1111/bpa.12228
55. Italiano A, Michalak S, Soulié P, Peyron AC, Pedeutour F. Trisomy 6p and ring chromosome 11 in a melanotic schwannoma suggest relation to malignant melanoma rather than conventional schwannoma. Acta Neuropathol (2011) 121:669–70. doi: 10.1007/s00401-011-0820-4
56. Bendszus M, Urbach H, Wolf HK, Schramm J, Solymosi L. Magnetic resonance imaging of intraspinal melanotic schwannoma. Eur Radiol (1998) 8:1197. doi: 10.1007/s003300050534
57. Küsters-Vandevelde HVN, van Engen-Van Grunsven IACH, Küsters B, van Dijk MRCF, Groenen PJTA, Wesseling P, et al. Improved discrimination of melanotic schwannoma from melanocytic lesions by combined morphological and GNAQ mutational analysis. Acta Neuropathol (2010) 120:755. doi: 10.1007/s00401-010-0749-z
58. Solomou G, Dulanka Silva AH, Wong A, Pohl U, Tzerakis N. Extramedullary malignant melanotic schwannoma of the spine: Case report and an up to date systematic review of the literature. Ann Med Surg (2020) 59:217. doi: 10.1016/j.amsu.2020.10.003
59. Asazuma T, Toyama Y, Maruiwa H, Fujimura Y, Hirabayashi K. Surgical strategy for cervical dumbbell tumors based on a three-dimensional classification. Spine (Phila Pa 1976) (2004) 29:E10–4. doi: 10.1097/01.BRS.0000103662.13689.76
60. Shen XZ, Wang W, Luo ZY. 18F-FDG PET/CT imaging for aggressive melanotic schwannoma of the L3 spinal root: A case report. Med NLM (Medline) (2021) 100:e24803. doi: 10.1097/MD.0000000000024803
61. Trufant JW, Brenn T, Fletcher CDM, Virata AR, Cook DL, Bosenberg MW. Melanotic schwannoma arising in association with nevus of ota: 2 cases suggesting a shared mechanism. Am J Dermatopathology (2009) 31:808–13. doi: 10.1097/DAD.0b013e3181accd0e
62. Chen D, Gu W. Subdural extramedullary melanotic schwannoma of the thoracic spinal cord: A case report. Turk Neurosurg (2015) 25:326–31. doi: 10.5137/1019-5149.JTN.8153-13.0
63. Xiao LL, Shun-Dong D. Melanotic schwannoma: Two cases of rare lesions. Pathol Oncol Res (2019) 25:1667–70. doi: 10.1007/s12253-018-0417-5
Keywords: malignant melanotic nerve sheath tumor, spinal, nerve sheath, tumor, psammomatous, sporadic, case report, melanotic schwannoma
Citation: Bonomo G, Gans A, Mazzapicchi E, Rubiu E, Alimonti P, Eoli M, Paterra R, Pollo B, Iess G, Restelli F, Falco J, Acerbi F, Schiariti MP, Ferroli P and Broggi M (2023) Sporadic spinal psammomatous malignant melanotic nerve sheath tumor: A case report and literature review. Front. Oncol. 13:1100532. doi: 10.3389/fonc.2023.1100532
Received: 16 November 2022; Accepted: 06 February 2023;
Published: 24 February 2023.
Edited by:
Pierpaolo Alongi, ARNAS Ospedali Civico Di Cristina Benfratelli, ItalyReviewed by:
Vadim Byvaltsev, Irkutsk State Medical University, RussiaCopyright © 2023 Bonomo, Gans, Mazzapicchi, Rubiu, Alimonti, Eoli, Paterra, Pollo, Iess, Restelli, Falco, Acerbi, Schiariti, Ferroli and Broggi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulio Bonomo, ZG90dC5naXVsaW9ib25vbW9AZ21haWwuY29t
†These authors have contributed equally to this work
‡ORCID: Giulio Bonomo, orcid.org/0000-0002-4749-7929
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.