 Kaipeng Jin
Kaipeng Jin Chao Qian
Chao Qian Jinti Lin
Jinti Lin Bing Liu
Bing Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 January 2023
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1099811
This article is part of the Research TopicImmunotherapy and Small Molecule Inhibitors as Combinational Cancer TherapeuticsView all 19 articles
Cyclooxygenases-2 (COX-2) and Prostaglandin E2 (PGE2), which are important in chronic inflammatory diseases, can increase tumor incidence and promote tumor growth and metastasis. PGE2 binds to various prostaglandin E receptors to activate specific downstream signaling pathways such as PKA pathway, β-catenin pathway, NF-κB pathway and PI3K/AKT pathway, all of which play important roles in biological and pathological behavior. Nonsteroidal anti-inflammatory drugs (NSAIDs), which play as COX-2 inhibitors, and EP antagonists are important in anti-tumor immune evasion. The COX-2-PGE2 pathway promotes tumor immune evasion by regulating myeloid-derived suppressor cells, lymphocytes (CD8+ T cells, CD4+ T cells and natural killer cells), and antigen presenting cells (macrophages and dendritic cells). Based on conventional treatment, the addition of COX-2 inhibitors or EP antagonists may enhance immunotherapy response in anti-tumor immune escape. However, there are still a lot of challenges in cancer immunotherapy. In this review, we focus on how the COX-2-PGE2 pathway affects tumor-associated immune cells.
Cyclooxygenases-2 (COX-2) and Prostaglandin E2 (PGE2) are important inflammatory factors, associated with survival, invasion, growth and immune escape of cancer cells. One of the “hallmarks” of cancer is chronic inflammatory disease, which often promotes tumorigenesis and tumor progression (1). For example, inflammatory bowel disease patients had a higher lifetime risk of colon cancer linked to colitis at a younger age than the general population, demonstrating that cancer is easily caused by chronic inflammation (2). Tumor-associated inflammation involves complex interactions between epithelial and mesenchymal cells and in some cases can lead to epigenetic alterations. More broadly, however, chronic inflammation can lead to the production of growth factors that support the development of emerging tumors and cause them to behave as “unhealable wounds” (3). Chronic inflammation can also promote tumor development by facilitating tumor immune escape and establishing an immunosuppressive microenvironment, both of which are cancer-related characteristics. Tumor immune escape occurs through a variety of immunosuppressive mechanisms, such as dysfunctional antigen-presenting cells (APCs), tumor cell resistance to immune attack, decreased cytotoxicity of CD8+ T cells and natural killer (NK) cells, induction of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs), transition of T helper (Th) cells from Th1 to Th2 and Th17, transition of macrophages from M1 to M2 (4).
Numerous studies have shown the importance of nonsteroidal anti-inflammatory drugs (NSAIDs) and EP receptor antagonists in reducing tumor incidence, metastasis and mortality. NSAIDs cause tumor regression and suppress tumor growth by inhibiting the COX-2-PGE2 signaling pathway in several ways: 1. activation of tumor epithelial cells; 2. inhibition of tumor epithelial cell survival and tumor immune surveillance; 3. establishment of tumor-supporting microenvironment; 4. alteration of DNA methylation mechanisms (2). However, the molecular mechanisms underlying the anti-tumor effects of NSAIDs and EP receptor antagonists have not yet been fully understood. More clinical trials and studies are required to explore this further.
In this review, we emphasize our current understanding of COX-2 and PGE2 regulation of tumor immunity. In addition, we are looking into how the COX-2-PGE2 pathway affects tumor-associated immune cells. Given the significance of this pathway in tumor immune escape, we will discuss how to target each component of this pathway as potential strategy for overcoming tumor immune escape while avoiding some serious adverse effects associated with the use of NSAIDs or COX-2 inhibitors.
Cyclooxygenases (COX) include COX-1 and COX-2, also known as prostaglandin G/H synthase-1 and -2, are membrane-bound enzymes that are mainly found on the nuclear membrane and luminal side of the endoplasmic reticulum (5). COX-1 is structurally expressed in many healthy tissues and provides steady-state levels of prostaglandins to perform “housekeeping functions”. In contrast, COX-2 is an inducible isoform produced by prostaglandin-like substances that is typically absent or expressed at low levels in normal organs and tissues, but is overexpressed in many tumor and inflammatory tissues (6). In addition to inducing inflammation, COX-2 can also promote cell survival and proliferation. Therefore, COX-2 may be associated with tumorigenesis and development. Indeed, COX-2 has been found to be highly expressed in different tumors, including breast cancer (7), the melanoma (8), and colorectal cancer (2). Its levels are associated with the development, aggressiveness and prognosis of many tumor entities. Regular use of NSAIDs dramatically decreased the incidence of sporadic colorectal cancer as well as breast, lung, and prostate cancers, according to a new analysis that searched all epidemiological studies (case-control and cohort studies) conducted since 1980 (9). According to one study, genetically removing COX enzymes from mouse melanoma (BRAFV600E or NrasG12D), colorectal cancer (CT26) or breast cancer (4T1) cell lines resulted in dramatic tumor eradication (10). When metastases were assessed 2 weeks after surgical resection of the primary breast cancer tumor, fibroblast-targeted Ptgs2-deficient mice (Ptgs2DFb) were found to develop fewer metastases than WT mice (11). Patients with gastric cancer who have low staging and higher levels of COX-2 expression are at greater risk of dying from the disease. In addition, COX-2 has been identified as an independent prognostic factor for gastric cancer (12). According to the findings mentioned above, COX-2 is thought to be crucial for tumor growth.
COX enzymes convert arachidonic acid to the endogenous peroxidation intermediate PGH2, which is modified by prostaglandin synthase to produce five structurally related prostaglandins, including prostaglandin D2, prostaglandin E2, prostaglandin I2, prostaglandin F2α, and thromboxane A2. The two essential enzymes that catalyze the initial and concluding phases of this synthetic pathway are COX-2 and prostaglandin E2 synthase (PGES), including microsomal PGES (mPGES) and cytosolic PGES. In addition, PGE2 is converted to its inactive form, 15-keto-PGE2 (PGEM), which is then further metabolized to a stable terminal metabolite by the enzyme 15-hydroxyprostaglandin dehydrogenase (15-PGDH). High levels of PGEM increase the risk of gastric and colorectal cancers (13, 14), implying that PGEM may be a biomolecular marker for cancer risk prediction. Prostaglandins control cellular processes by attaching to G protein-coupled receptors on the cell surface. These cell surface receptors are named EP (EP1, EP2, EP3, EP4), DP (DP1, DP2), FP, IP, and TP. PGE2 transmits signals by binding to four receptors, EP1 through EP4 (15) (Figure 1). The expression of each EP receptor and the strength of each EP signal determine the PGE2 signal’s final output. Each EP interacts with its unique G protein to activate particular downstream signaling pathways such as PKA pathway, β-catenin pathway, NF-κB pathway and PI3K/AKT pathway, which have various functions in biological and pathological behavior. Activated EP1 can upregulate the level of intracellular calcium ion concentrations; EP2 and EP4 receptors are associated with cAMP stimulation and PKA signaling through sequential activation of Gas and adenylyl cyclase; EP3 is responsible for downregulating cAMP levels and lead to different cellular responses through different G proteins (16, 17).
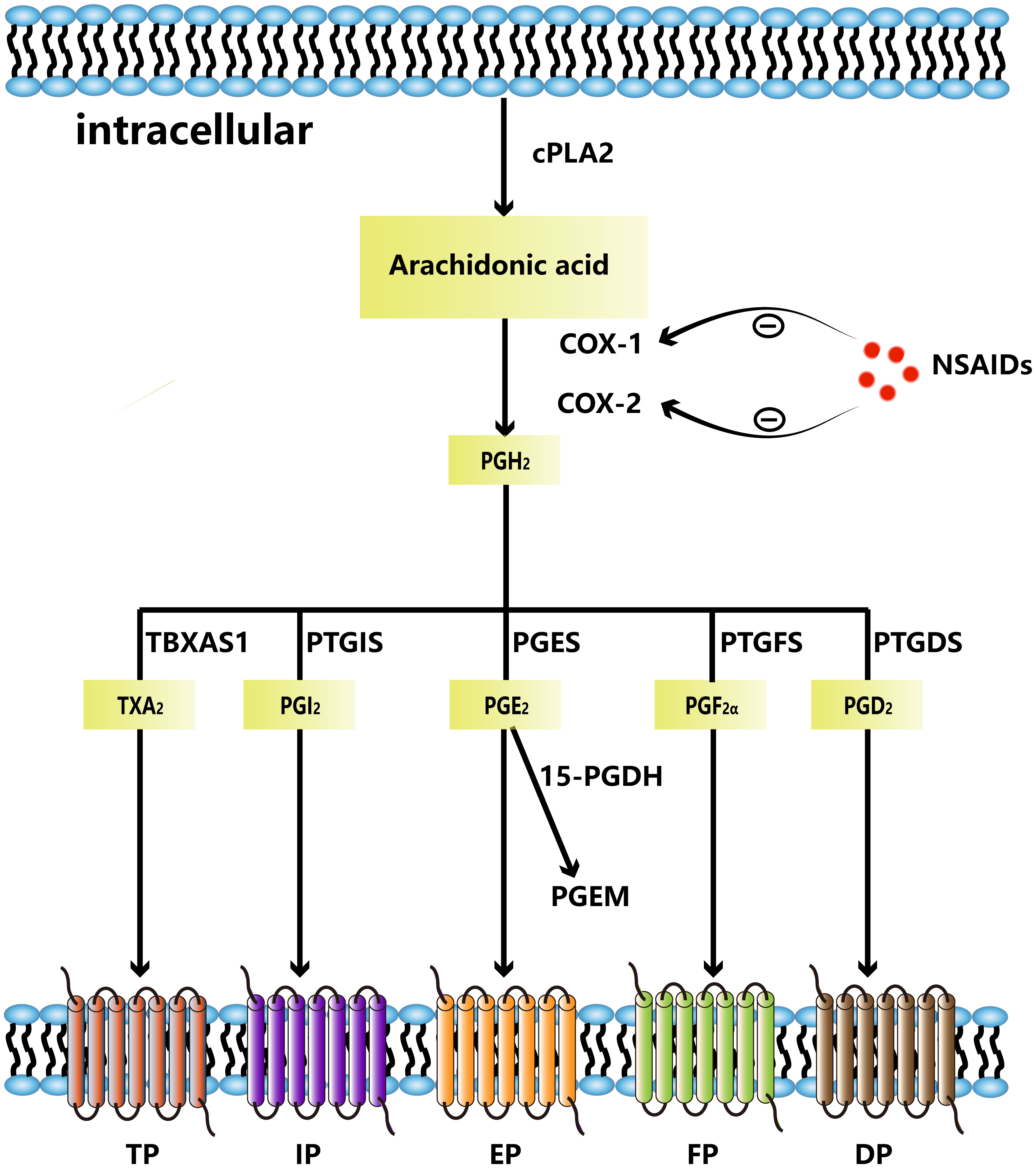
Figure 1 Arachidonic acid is metabolized to PGH2 by COX-1 and COX-2. NSAIDs can inhibit COX-1 and COX-2. PGH2 is metabolized to thromboxane A2(TXA2)by TBXAS1, PGI2 by PTGIS, PGE2 by PGES, PGF2α by PGES, PGD2 by PTGDS, respectively. PGE2 can be metabolized to PGEM by 15-PDGH. TXA2 binds to TP, PGI2 binds to IP, PGE2 binds to EP, PGF2α binds to FP and PGD2 binds to DP.
The most abundant prostaglandin, PGE2, is frequently linked to a poor prognosis in a number of human cancers, including colon, head and neck, lung, and breast cancers (6, 17, 18). In immunologically active hosts, the formation of tumors by mutant BRAFV600E mouse melanoma cells requires the synthesis of prostaglandin E2, which inhibits immunity and fosters tumor inflammation (10). It has been shown that mPGES1 expression increased in human melanoma, and that elevated expression of this protein are linked to reduced patient survival (19). The mPGES-1 gene deletion significantly reduced the risk of developing colon cancer and resulted in reduced multiplicity of distal colon tumors as well as tumor load in mice treated with azoxymethane (AOM) (20). In addition, in ApcMin/+ and AOM mice models, elevated endogenous PGE2, caused by 15-PGDH gene deletion, encouraged the development of colon cancer (21). Taken together, PGE2 is essential in the growth of cancers (Figure 2).
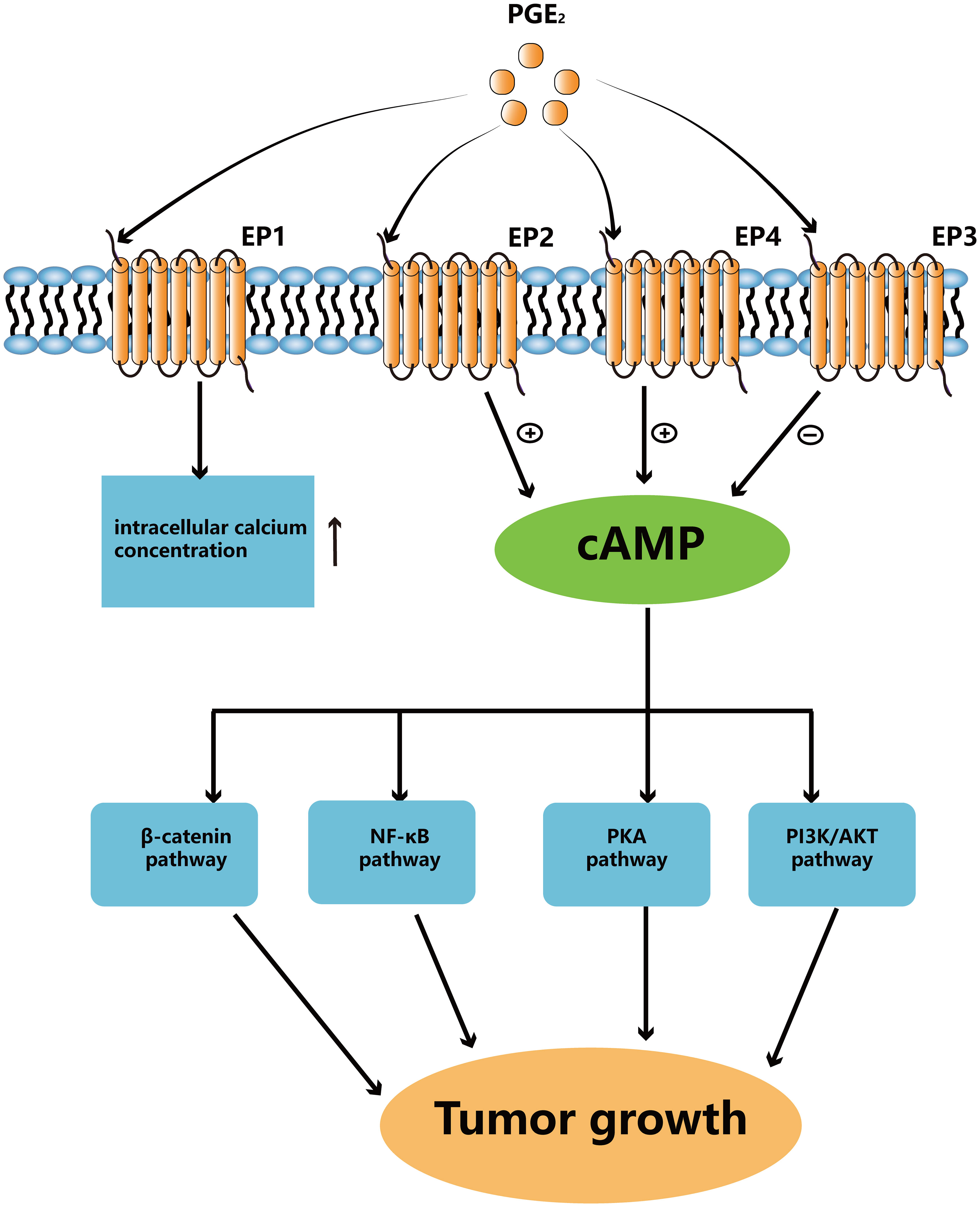
Figure 2 PGE2 binds to different EPs to activate downstream signaling pathways. Activated EP1 can upregulate the level of intracellular calcium ion concentrations. EP2 and EP4 can upregulate cAMP levels to activate different pathways which can promote tumor growth. EP3 is responsible for downregulating cAMP levels.
In addition, numerous evidence has shown that the COX-2-PGE2 pathway promotes tumor development. It has been shown that PGE2 promotes sporadic or colitis-associated colon carcinogenesis via EP1 and EP2 receptor in colon cancer cell lines (22). According to research, numerous tumors are linked to EP1 receptor. For example, selective EP1 antagonist (ONO-8713) can significantly reduce the number of tumor cells in mouse with UVB-induced acute skin inflammation (23). PGE2 upregulates anti-apoptosis protein expression in hepatocellular carcinoma cells via EP1 receptors (24). While EP3 receptors were downregulated, EP1, EP2 and EP4 receptors were all highly increased in COX-2-driven mammary cancers. Downregulation of EP3 receptors suggests a possible protective role against mammary tumors. EP2 and EP4 receptor levels were reduced in the mammary glands of anti-inflammatory pain-treated mice, while EP1 and EP3 levels were not altered (25). According to Fujino et al, EP3 receptors could boost vascular endothelial growth factor receptor-1 signaling and encourage tumor cell metastasis (26). Therefore, it may be debatable how EP3 contributes to carcinogenesis, and more research is required. The role of the EP4 receptor in tumors appears to be more clearly defined than that of other EP receptors. EP4 is a high-affinity EP receptor, and it is considered to be a pro-cancer mediator in many different types of malignancies due to its high expression. In colorectal cancer, deletion of EP4 can attenuates the abnormal AOM-induced crypt (27, 28), and therefore EP4 receptor can be used for prostate cancer immunotherapy. For example, YY001, an antagonist of the EP4 receptor, inhibits prostate cancer growth by modulating the tumor microenvironment (TME), leading to significant tumor regression, long-term survival and long-lasting immune memory (29). Besides, in mouse model, EP4 knockdown suppressed metastasis of oral cancer cells in the lung (30). Collectively, these findings imply a critical function for the PGE2/EP4 signaling pathway in tumor development.
The COX-2-PGE2 pathway induces tumor immune evasion by regulating myeloid-derived suppressor cells (MDSCs), lymphocytes (CD8+ T cells, CD4+ T cells and natural killer cells), and antigen presenting cells (APCs). Understanding the mechanisms of the COX-2-PGE2 pathway may provide a solid foundation for developing new methods to overcome tumor immune escape (Figure 3).
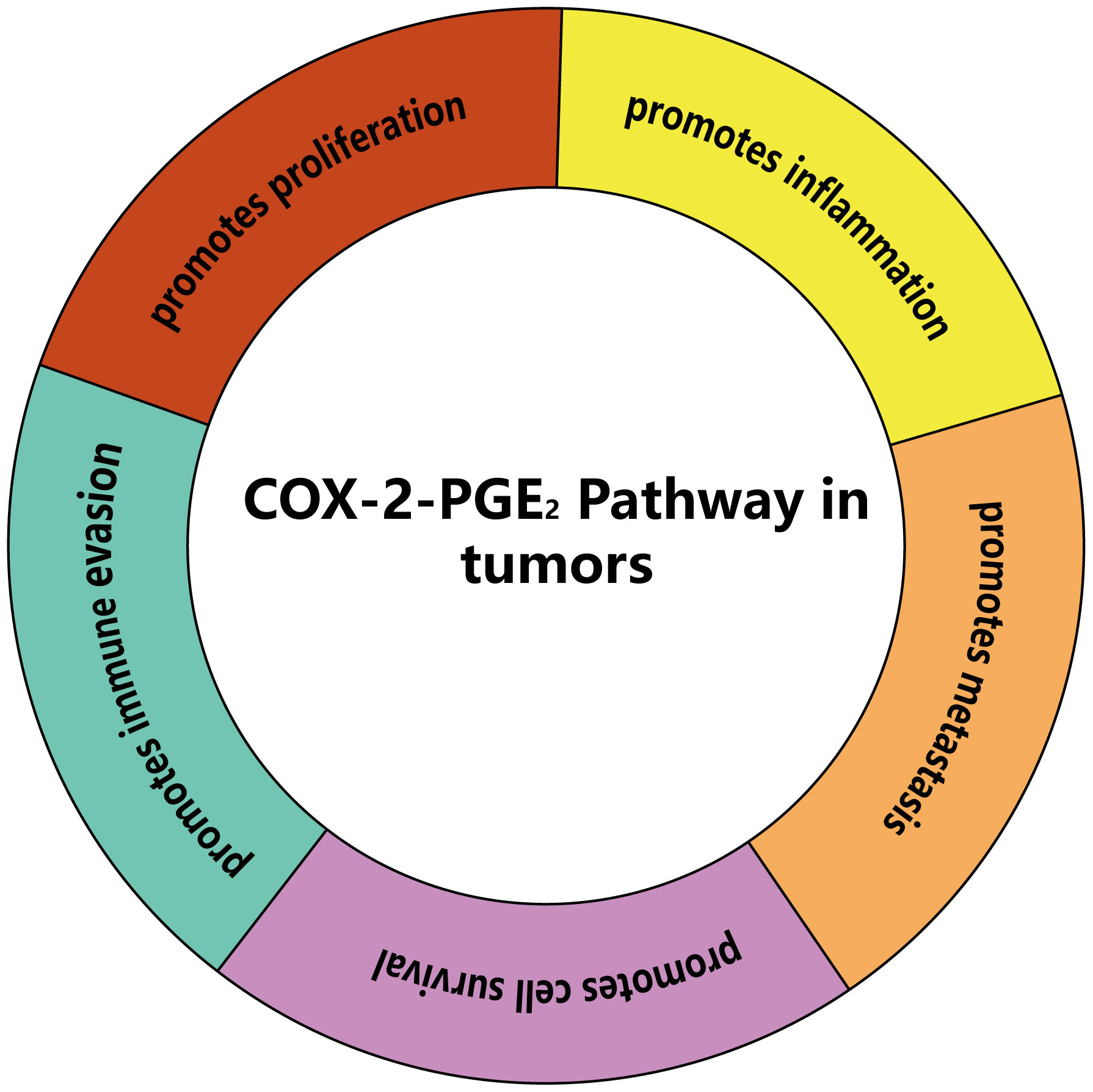
Figure 3 The main effects of COX-2-PGE2 pathway on tumors. COX-2-PGE2 pathway can promote proliferation, inflammation, metastasis, cell survival and immune evasion of tumors.
Granulocyte/polymorphonuclear stem cells and monocyte MDSCs, which come from the granulocyte or monocyte myeloid lineage, respectively, are the two main kinds of MDSCs found in both humans and animals. However, unlike mice, a tiny group of myeloid precursor cells known as “early MDSCs” have been found in humans. These cells have a potent immunosuppressive function and consist mainly of myeloid progenitor cells and precursor cells, accounting for less than 5% of the total number of MDSCs (31). MDSCs mediate immunosuppression by producing arginase and inducible nitric oxide synthase, and TGF-β, IL-10 and COX-2. These substances directly or indirectly enhance Treg activity (32), inhibit cytotoxic activity of NK cells (33), and promote the polarization of macrophages toward M2-like phenotype. They synergistically impair the tumoricidal properties of effector CD8+ T cells, leading to tumor cell evasion of host antitumor immunity (34). MDSCs are significantly linked to a poor clinical course of cancer (35). In mice and patients, the clinical cancer stage and the amount of metastatic tumors are favorably linked with the blood levels of MDSCs (36) (Figure 4).
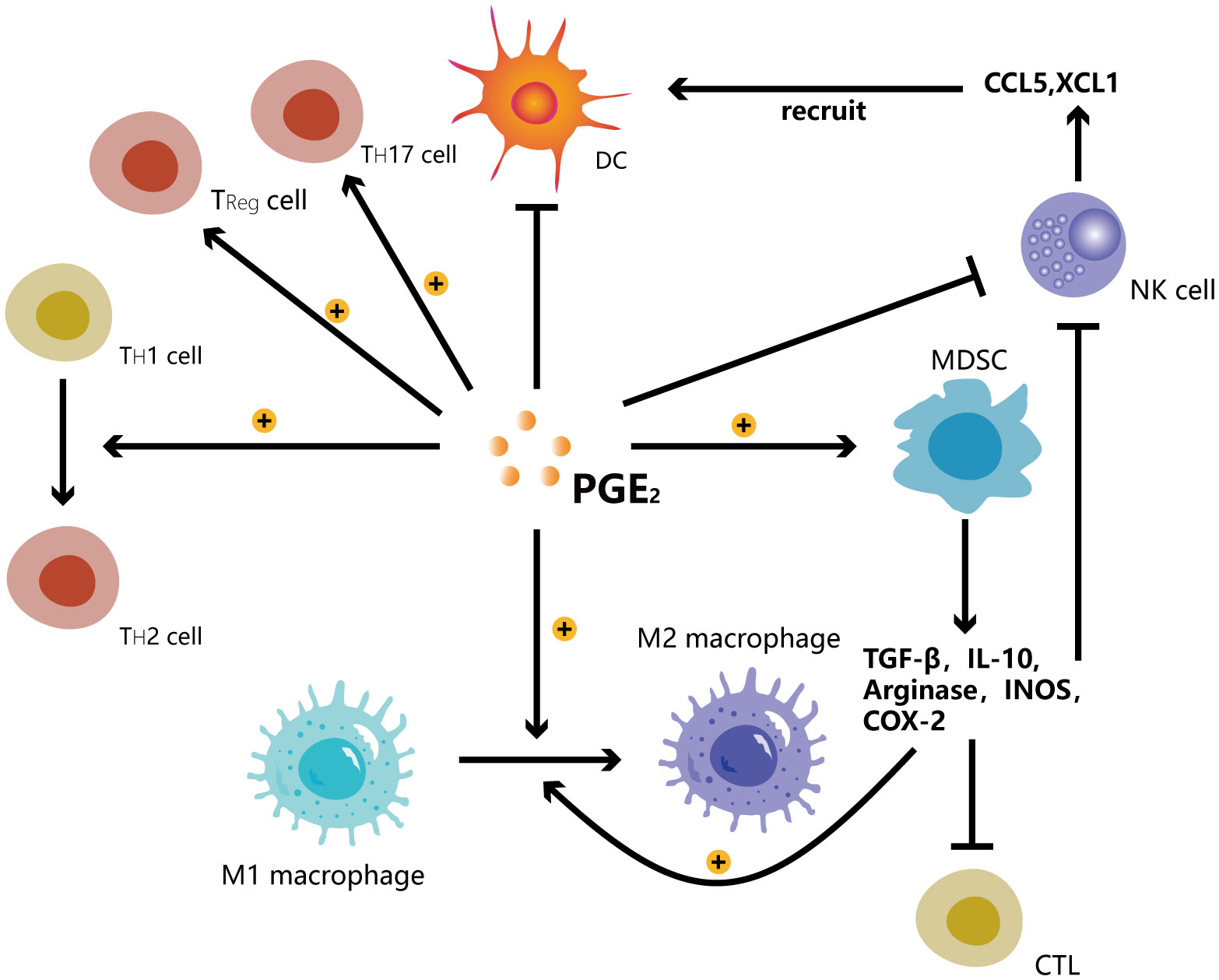
Figure 4 PGE2 promotes the transition of T helper cells from TH1 to TH2 and the shift of macrophages from M1 to M2. PGE2 induces the development of Treg cells, TH17 cells and MDSCs while inhibiting Dendritic cells, NK cells. MDSCs secrete TGF-β, IL-10, arginase, INOS, COX-2 to promote the shift of macrophages from M1 to M2 while inhibiting the functions of cytotoxic lymphocytes (CTL) and NK cells. NK cells can secrete CCL5 and XCL1 chemokines to recruit DCs.
The EP4 antagonist YY001 promotes the proliferation and anti-cancer activities of T lymphocytes while inhibiting myeloid-derived suppressor cells’ development, maturation, and immune-suppressive actions. Additionally, it alters the chemokine profile of tumor cells in vitro and in vivo, which decreases the amounts of MDSCs and T cells in the TME (29). In vitro experiments, PGE2 participated in MDSC-mediated immunosuppression by inducing arginase I expression through EP4 and inhibiting effector T cell activity (37). One study found that treatment of colorectal cancer mice with aspirin or EP antagonists significantly protected mice from tumor formation and reduced aggregation of MDSCs and expression of COX-2/Arg-1 (38). Thus, PGE2 inhibits MDSCs by blocking EP4 receptors, thereby promoting tumor immune evasion. The number of Gr1(+) CD11b(+) immature myeloid suppressor cells was considerably decreased by the celecoxib during the chemoprevention of 1,2-dimethylhydrazine diHCL- (1,2-DMH-) induced colon cancers in Swiss mice (39). The COX-2 pathway promotes glioma formation by directly supporting the phylogeny of MDSCs and their accumulation in TME, where MDSCs limit CTL infiltration (40). Taken together, the COX-2-PGE2 signaling pathway promotes tumor immune evasion by inducing the accumulation of MDSCs cells, which in turn promotes tumor growth and development. However, the regulatory role of COX-2-PGE2 pathway on MDSCs in vivo has not been fully investigated.
Microglia, Kupffer cells, and Langerhans cells in the brain, liver, and epidermis are examples of tissue-specific macrophages. Macrophages possess the ability to remove apoptotic cells, cellular debris and pathogens (41). Similar to the Th1 and Th2 dichotomy, the distinction between M1 and M2 phenotypes supports the idea of macrophage phenotypic heterogeneity. When exposed to lipopolysaccharide and interferon-c, macrophages develop the M1 phenotype and exhibit anticancer properties. Macrophages become polarized to the M2 phenotype in response to Th2 cytokines which promote cell proliferation and tumor formation (42, 43). TAMs have an M2-like phenotype in the majority of cancers (44), which facilitates tumor-associated angiogenesis, promotes tumor cell invasion and migration, and inhibits immune surveillance to promote tumor metastasis (45). Clinical studies have shown that macrophages promote tumorigenesis. In a meta-analysis, more than 80% of studies indicated a link between high macrophage density and poor patient prognosis (46). In primary tumors, TAMs have been shown to inhibit CD8+ T cell recruitment and anti-tumor immunity (47).
Previous studies have pointed out that the COX-2-PGE2 signaling pathway contributes significantly to the polarization of M1 to M2 and tumor immune escape. The majority of the COX-2 seen in both human and mouse intestinal cancers is known to come from TAMs. Celecoxib, a selective COX-2 inhibitor, shifted the TAM phenotype from M2 to M1 in a colorectal cancer animal model in accordance to the decrease in the number of polyps in ApcMin/1 mice (48). While inhibiting the growth of immune-stimulated M1 macrophages, PGE2-binding EP4 promoted development of immune-suppressed M2 macrophages and MDSCs (49). Eruslanov et al. found that in the mouse colon cancer cell line CT26, overexpression of 15-PGDH shifted TAMs from M2-directed TAM to M1-directed macrophages (50). In the GC transgenic model, overexpression of COX-2 and mPGES-1 led to TAM recruitment in gastric tumors (51). These findings collectively imply that COX-2 and PGE2 stimulate tumor development via M2 TAMs. EP1 and EP3 receptors also play an important role in tumor growth. Targeting EP1 receptor can decrease F4/80 (+) macrophage infiltration and inhibit colon cancer growth (52). EP3 signaling in DCs induces a switch of macrophages from pro-inflammatory to pro-reparative phenotype (53).
As a part of innate immunity, NK cells play an important role in tumor immune surveillance and viral infection resistances. In addition to immediately identifying and eliminating tumor cells, NK cells secrete cytokines that promote CTL activation and growth. However, the functional response of NK cells is compromised in tumors (54). In all cancer types, patient survival was substantially correlated with high expression of NK marker genes (55). In cancerous environment, NK cells dysfunction is integral and inevitable, leading to not only the proliferation of tumor cells but also the formation of distant metastases (56). The abundance of peritumoral NK cells is also associated with high pathological complete response rates in neoadjuvant chemotherapy for large and locally advanced breast cancers (57). In the course of infection with chronic lymphocytic choriomeningitis virus, intrinsic NK cell signaling can inhibit the expansion of CD8+ T cells, thereby promoting tumor immune evasion (58).
The COX-2-PGE2 pathway can enhance tumor immune evasion. PGE2 has been reported to inhibit cytotoxic effects and cytokine generation of NK cells in breast cancer as well as thyroid cancer through the EP4 signaling pathway (59, 60). In mouse tumors, NK cell-derived chemokines CCL5 and XCL1 promote the accumulation of dendritic cells (DCs). PGE2-producing tumors inhibit NK cell function, chemokine production, and DCs chemokine receptor expression to promote tumor immune escape (55). Breast tumor cells have been shown to express fewer MHC class I molecules when EP4 signaling is inhibited, which enhances the NK cells’ capacity to fight the tumor (61).
DCs have a unique function in the induction and regulation of innate and acquired immune responses. To direct T cell responses, DCs in the TME collect, prepare, and present tumor-associated antigens on MHC molecules as well as supply co-stimulatory and soluble components (62). In humans, reduced DC numbers and activity affect the prognosis of colorectal cancer patients (63). When patients were stratified based on cDC1-related gene expression, higher cDC1 signaling in tumors was found to be positively correlated with survival (55). However, eight studies have come to the conflicting conclusion that tumor-associated DCs can be a predictor of progressive prognosis in colorectal cancer (64).
PGE2 plays an important role in the control of DCs behaviors include differentiating, producing cytokines, polarizing TH cells, migrating, and maturing (65). In addition, it has been found that inhibiting EP2 and EP4 receptors increases both MHC molecule expression and antigen uptake by lung dendritic cells (11). Tumor-derived prostaglandin analogs inhibited both the aggregation and activation of CD103+ DCs within the tumor, including their capacity to produce IL-12, as well as the expression of type I immune-related markers (10). Secretion of PGE2 may inhibit the capacity of DCs to activate CD8+ T lymphocytes, thus promoting T cell exclusion from TME (41). Human DC surface HLA class II antigen expression levels are downregulated by IL-6 through the functions of COX-2, lysosomal protease, and arginase (66). Furthermore, an in vitro study demonstrated that NSAIDs prevent DCs from presenting MHC-restricted antigens (67). Taken together, the COX-2-PGE2 signaling pathway promotes tumor immune evasion by inhibiting DC cells.
APCs activate naive CD4+ T cells, which then initiate differentiation into various effector T cells during inflammatory and immunological responses to infections and cancer (68). Th1 cells, in general, control more aggressive responses by encouraging cytotoxic immune responses, whereas Th2 cells control less tissue-damaging immune responses (69, 70). As a result of resident tissue cells being stimulated to release chemokines by Th17-associated cytokines, neutrophils and macrophages are drawn to the sites of inflammation. In turn, the recruited cells generate more cytokines and protein hydrolases, exacerbating the immunological reaction (71, 72). TGF-β suppresses TH2-mediated cancer immunity. In a mouse model of breast cancer, induction of TGF-β receptor II gene deletion in CD4+ T cells inhibited tumor growth (73). Higher levels of circulating CD4+ T lymphocytes were linked to smaller tumor sizes in GC patients (74). Th17 cells were found in greater numbers in the peripheral blood and tumor tissue of patients with oral squamous cell carcinoma (66). In conclusion, the formation and progression of tumors are highly correlated with CD4+ T cell activity.
COX-2 deficiency delays mammary carcinogenesis by enhancing type 1 immune responses in breast cancers (75). At high doses, PGE2 induces a shift from a Th1-dominant to a Th2-dominant immune response by downregulating Th1-induced activation of the IL-12 pathway (68). Additionally, PGE2 stimulates IL-23 production and prevents DCs from releasing IL-12 and IL-27, which increases the pathogenic inflammatory Th17 phenotype (76). Although the majority of published studies indicate that PGE2 boosts Th2-type responses, evidence shows that COX-2 has an inhibitory effect on Th2 immune responses. In contrast to Th1-mediated lung inflammation, Jaffar et al. demonstrated that specific inhibition of COX-2 in vivo decreases PGE2 production and causes a considerable rise in Th2-mediated lung inflammation (77). Some studies have shown that PGE2 can promote Th1 differentiation through the EP1 receptor (78). Thus, the results are contradictory and more research is required.
CD8+ cytotoxic T cells, the primary killer cells of pathogens and tumor cells, are essential for the destruction of intracellular infections and malignant cells, and may provide long-term immune protection (79, 80). Exercise alters the metabolism of CD8+ T cells, thereby enhancing their antitumor efficacy (81). In colorectal cancer, low tumor stage, negative nodal stage, longer overall survival, and an inflammatory immune phenotype were all substantially correlated with the density and proportion of proliferating CD8+ cytotoxic T cells (82). These results suggest that CD8+ T cells are capable of suppressing tumor growth.
In ErbB2 transgenic mice, COX-2 (MEC) KO breast cancer tumors contained more CD8+ cytotoxic immune cells (CTL) (7). Some studies found that the amount of CD8+ T cells and the percentage of functional CD8+ T cells were considerably decreased in PTGS2 overexpression tumors (83). These findings suggest that activation of the COX2 enzymes can reduce tumor-infiltrating CD8+ T cells. In addition, effector CD8+ T cells in tumors are inhibited from killing tumors by tumor-producing, PGE2-activated immunosuppressive cells in TME via EP4 receptors (84). During chronic lymphocytic choroidal meningitis virus infection, EP2 and EP4 are upregulated on virus-specific CTL inhibiting CTL survival and function (85). Increased secretion of PGE2 by breast cancer cells also recruits Treg cells into the primary tumor, thereby increasing apoptosis of CD8+ T cells and bone metastasis of cancer cells (86). PGE2 directly inhibits cytotoxic T cell activity and induces regulatory T cell function in vitro through upregulation of CD94 and NKG2A complexes (87). However, more evidence is needed to demonstrate whether the COX-2-PGE2 pathway promotes tumor immune evasion by suppressing CD8+ T cells.
Immunotherapy has grown in popularity as a cancer treatment option in recent years. Given the importance of the COX-2-PGE2 pathway in carcinogenesis and progression, there is a clear opportunity for therapeutic intervention. NSAIDs are a class of drugs that primarily suppress COX enzyme activity, which also lowers prostaglandins production. The use of NSAIDs to inhibit tumor growth has been considered. Combination therapy with NSAIDs and anti-PD-1 monoclonal antibody induces tumor eradication faster than anti-PD-1 alone (10). In addition, an in vivo investigation revealed that NSAIDs improved anti-tumor responses and reversed the imbalance between Th1 and Th2 in the metastatic spread of colorectal cancer (88). Some studies have demonstrated that NSAIDs can reduce cancer-associated mortalities and lower cancer incidence (89). For this reason, the FDA has approved celecoxib, a selective COX-2 inhibitor, for the treatment of population with familial adenomatous polyposis who wish to avoid developing colorectal polyps. In experimental animals, specific COX-2 inhibitors can prevent the development of mammary tumors (90). However, due to the risk of adverse cardiovascular events, celecoxib, along with other NSAIDs (except from aspirin), should not be used for an extended period of time, particularly in patients with a history of atherosclerotic heart disease (91). Excessive bleeding is the most serious side effect of aspirin, and both the risk and mortality rate increase with age (92). One way to prevent these negative consequences is to target only downstream PGE2 signaling. Cell surface inhibitory receptors (such as PD-1) are centrally involved in T cell exhaustion (93). And PGE2 could be one of the inducers of PD-L1 expression (94). We suppose that COX2-PGE2 pathway may result in T cell exhaustion by inducing PD-L1 expression. Emerging data from multiple solid tumor mouse models indicates that EP4 antagonists and PD1/PD-L1 blockade are effective in inhibiting primary tumor growth (49, 95, 96). Therefore, it may be a new potential strategy to combine EP antagonists with PD1/PD-L1 blockade in treating T cell exhaustion. In addition, combination immunotherapy with chimeric antigen receptor T (CAR-T) cells and checkpoint blockade is thought to be the next immunotherapy frontier (97). For instance, combination PD-L1 blockade and CD19 CAR-T cell therapy resulted in better outcomes in patients with heavily B-ALL (98). PGE2 signaling can activate protein kinase A, and then inhibits T-cell receptor activation, which inhibits the antitumor effect of CAR-T cell therapy (99). Thus, we conclude that targeting EP receptors (such as EP2 and EP4) could represent a novel strategy for improving the efficacy of adoptive immunotherapy. Some clinical trials are ongoing to evaluate the efficacy of these inhibitors. A phase I trial showed that EP4 antagonist (E7046) was safe in patients with advanced malignancies (ClinicalTrials.gov, Number NCT02540291). 1 clinical trial is recruiting patients to evaluate whether a dual EP2/EP4 antagonist (TPST-1495) suppresses tumor growth (ClinicalTrials.gov, Number NCT04344795). Although there is abundant EP receptor expression in fibroblasts and inflammatory cells, the role of the COX-2-PGE2 signaling pathway in TME is not fully understood, and more studies are needed to demonstrate this.
In this review, we discuss the role of the COX-2-PGE2 pathway in tumor immune evasion regulation. The idea that efficient therapeutic approaches should involve eliminating tumor cells and suppressing tumor immune evasion is being supported by a growing body of evidence, such as the use of checkpoint inhibitors to target immunosuppressive cells and reactivate immunosuppressive effector T cells. The COX-2-PGE2 signaling pathway may assist tumors in evading immune systems by inducing tumor-associated immune cells aggregation, impaired APC activity, a switch from Th1 to Th2 and Th17 immune responses or by suppressing CD8+ cytotoxic T cell and NK cell functions to promote tumor immune escape. In conclusion, the COX-2-PGE2 pathway not only is an effective target for tumor eradication, but it also suppresses tumor immune escape. Therefore, the addition of cyclooxygenase-2 inhibitors or EP antagonists to standard treatment may enhance the response of immunotherapy in anti-tumor immune escape.
KJ and CQ drafted the manuscript and revised the manuscript. JL and BL were in charge of the whole research conduction and paper writing. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the National Natural Science Foundation of China (NO.81872181), and the Natural Science Foundation of Zhejiang Province (NO. LY21H160034).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hou J, Karin M, Sun B. Targeting cancer-promoting inflammation - have anti-inflammatory therapies come of age? Nat Rev Clin Oncol (2021) 18(5):261–79. doi: 10.1038/s41571-020-00459-9
2. Wang D, DuBois RN. The role of anti-inflammatory drugs in colorectal cancer. Annu Rev Med (2013) 64:131–44. doi: 10.1146/annurev-med-112211-154330
3. Singh R, Mishra MK, Aggarwal H. Inflammation, immunity, and cancer. Mediators Inflamm (2017) 2017:6027305. doi: 10.1155/2017/6027305
4. Wang D, Cabalag CS, Clemons NJ, DuBois RN. Cyclooxygenases and prostaglandins in tumor immunology and microenvironment of gastrointestinal cancer. Gastroenterology (2021) 161(6):1813–29. doi: 10.1053/j.gastro.2021.09.059
5. Tong D, Liu Q, Wang LA, Xie Q, Pang J, Huang Y, et al. The roles of the COX2/PGE2/EP axis in therapeutic resistance. Cancer Metastasis Rev (2018) 37(2-3):355–68. doi: 10.1007/s10555-018-9752-y
6. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer (2010) 10(3):181–93. doi: 10.1038/nrc2809
7. Markosyan N, Chen EP, Evans RA, Ndong V, Vonderheide RH, Smyth EM. Mammary carcinoma cell derived cyclooxygenase 2 suppresses tumor immune surveillance by enhancing intratumoral immune checkpoint activity. Breast Cancer Res (2013) 15(5):R75. doi: 10.1186/bcr3469
8. Botti G, Fratangelo F, Cerrone M, Liguori G, Cantile M, Anniciello AM, et al. COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J Transl Med (2017) 15(1):46. doi: 10.1186/s12967-017-1150-7
9. Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology (2009) 17(2):55–67. doi: 10.1007/s10787-009-8049-8
10. Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell (2015) 162(6):1257–70. doi: 10.1016/j.cell.2015.08.015
11. Gong Z, Li Q, Shi J, Wei J, Li P, Chang CH, et al. Lung fibroblasts facilitate pre-metastatic niche formation by remodeling the local immune microenvironment. Immunity (2022) 55(8):1483–500.e9. doi: 10.1016/j.immuni.2022.07.001
12. Mrena J, Wiksten JP, Thiel A, Kokkola A, Pohjola L, Lundin J, et al. Cyclooxygenase-2 is an independent prognostic factor in gastric cancer and its expression is regulated by the messenger RNA stability factor HuR. Clin Cancer Res (2005) 11(20):7362–8. doi: 10.1158/1078-0432.CCR-05-0764
13. Wang T, Cai H, Zheng W, Michel A, Pawlita M, Milne G, et al. A prospective study of urinary prostaglandin E2 metabolite, helicobacter pylori antibodies, and gastric cancer risk. Clin Infect Dis (2017) 64(10):1380–6. doi: 10.1093/cid/cix106
14. Johnson JC, Schmidt CR, Shrubsole MJ, Billheimer DD, Joshi PR, Morrow JD, et al. Urine PGE-m: A metabolite of prostaglandin E2 as a potential biomarker of advanced colorectal neoplasia. Clin Gastroenterol Hepatol (2006) 4(11):1358–65. doi: 10.1016/j.cgh.2006.07.015
15. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev (1999) 79(4):1193–226. doi: 10.1152/physrev.1999.79.4.1193
16. Sugimoto Y, Narumiya S. Prostaglandin e receptors. J Biol Chem (2007) 282(16):11613–7. doi: 10.1074/jbc.R600038200
17. Reader J, Holt D, Fulton A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev (2011) 30(3-4):449–63. doi: 10.1007/s10555-011-9303-2
18. Hambek M, Baghi M, Wagenblast J, Schmitt J, Baumann H, Knecht R. Inverse correlation between serum PGE2 and T classification in head and neck cancer. Head Neck. (2007) 29(3):244–8. doi: 10.1002/hed.20503
19. Kim SH, Roszik J, Cho SN, Ogata D, Milton DR, Peng W, et al. The COX2 effector microsomal PGE2 synthase 1 is a regulator of immunosuppression in cutaneous melanoma. Clin Cancer Res (2019) 25(5):1650–63. doi: 10.1158/1078-0432.Ccr-18-1163
20. Nakanishi M, Menoret A, Tanaka T, Miyamoto S, Montrose DC, Vella AT, et al. Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev Res (Phila) (2011) 4(8):1198–208. doi: 10.1158/1940-6207.Capr-11-0188
21. Myung SJ, Rerko RM, Yan M, Platzer P, Guda K, Dotson A, et al. 15-hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci USA (2006) 103(32):12098–102. doi: 10.1073/pnas.0603235103
22. Schumacher Y, Aparicio T, Ourabah S, Baraille F, Martin A, Wind P, et al. Dysregulated CRTC1 activity is a novel component of PGE2 signaling that contributes to colon cancer growth. Oncogene (2016) 35(20):2602–14. doi: 10.1038/onc.2015.283
23. Tober KL, Wilgus TA, Kusewitt DF, Thomas-Ahner JM, Maruyama T, Oberyszyn TM. Importance of the EP(1) receptor in cutaneous UVB-induced inflammation and tumor development. J Invest Dermatol (2006) 126(1):205–11. doi: 10.1038/sj.jid.5700014
24. Bai XM, Jiang H, Ding JX, Peng T, Ma J, Wang YH, et al. Prostaglandin E2 upregulates survivin expression via the EP1 receptor in hepatocellular carcinoma cells. Life Sci (2010) 86(5-6):214–23. doi: 10.1016/j.lfs.2009.12.009
25. Chang SH, Liu CH, Conway R, Han DK, Nithipatikom K, Trifan OC, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA (2004) 101(2):591–6. doi: 10.1073/pnas.2535911100
26. Fujino H, Toyomura K, Chen XB, Regan JW, Murayama T. Prostaglandin E2 regulates cellular migration via induction of vascular endothelial growth factor receptor-1 in HCA-7 human colon cancer cells. Biochem Pharmacol (2011) 81(3):379–87. doi: 10.1016/j.bcp.2010.11.001
27. Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, et al. Role of the prostaglandin e receptor subtype EP1 in colon carcinogenesis. Cancer Res (1999) 59(20):5093–6.
28. Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, et al. Involvement of prostaglandin e receptor subtype EP(4) in colon carcinogenesis. Cancer Res (2002) 62(1):28–32.
29. Peng S, Hu P, Xiao YT, Lu W, Guo D, Hu S, et al. Single-cell analysis reveals EP4 as a target for restoring T-cell infiltration and sensitizing prostate cancer to immunotherapy. Clin Cancer Res (2022) 28(3):552–67. doi: 10.1158/1078-0432.CCR-21-0299
30. Osawa K, Umemura M, Nakakaji R, Tanaka R, Islam RM, Nagasako A, et al. Prostaglandin E(2) receptor EP4 regulates cell migration through Orai1. Cancer Sci (2020) 111(1):160–74. doi: 10.1111/cas.14247
31. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
32. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest (2015) 125(9):3356–64. doi: 10.1172/jci80005
33. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res (2014) 20(15):4096–106. doi: 10.1158/1078-0432.Ccr-14-0635
34. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology (2013) 138(2):105–15. doi: 10.1111/imm.12036
35. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21(8):485–98. doi: 10.1038/s41577-020-00490-y
36. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother (2009) 58(1):49–59. doi: 10.1007/s00262-008-0523-4
37. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202(7):931–9. doi: 10.1084/jem.20050715
38. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, et al. A RIPK3-PGE2 circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res (2018) 78(19):5586–99. doi: 10.1158/0008-5472.CAN-17-3962
39. Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol. (2007) 7(2):140–51. doi: 10.1016/j.intimp.2006.09.021
40. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res (2011) 71(7):2664–74. doi: 10.1158/0008-5472.Can-10-3055
41. Hansen M, Andersen MH. The role of dendritic cells in cancer. Semin Immunopathol (2017) 39(3):307–16. doi: 10.1007/s00281-016-0592-y
42. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
43. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
44. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/s1471-4906(02)02302-5
45. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
46. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J Pathol (2002) 196(3):254–65. doi: 10.1002/path.1027
47. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov (2011) 1(1):54–67. doi: 10.1158/2159-8274.Cd-10-0028
48. Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K, et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis (2011) 32(9):1333–9. doi: 10.1093/carcin/bgr128
49. Lu W, Yu W, He J, Liu W, Yang J, Lin X, et al. Reprogramming immunosuppressive myeloid cells facilitates immunotherapy for colorectal cancer. EMBO Mol Med (2021) 13(1):e12798. doi: 10.15252/emmm.202012798
50. Eruslanov E, Kaliberov S, Daurkin I, Kaliberova L, Buchsbaum D, Vieweg J, et al. Altered expression of 15-hydroxyprostaglandin dehydrogenase in tumor-infiltrated CD11b myeloid cells: a mechanism for immune evasion in cancer. J Immunol (2009) 182(12):7548–57. doi: 10.4049/jimmunol.0802358
51. Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. EMBO J (2004) 23(7):1669–78. doi: 10.1038/sj.emboj.7600170
52. O’Callaghan G, Ryan A, Neary P, O’Mahony C, Shanahan F, Houston A. Targeting the EP1 receptor reduces fas ligand expression and increases the antitumor immune response in an in vivo model of colon cancer. Int J Cancer (2013) 133(4):825–34. doi: 10.1002/ijc.28076
53. Nakamoto S, Ito Y, Nishizawa N, Goto T, Kojo K, Kumamoto Y, et al. EP3 signaling in dendritic cells promotes liver repair by inducing IL-13-mediated macrophage differentiation in mice. FASEB J (2020) 34(4):5610–27. doi: 10.1096/fj.201901955R
54. O’Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol (2019) 19(5):282–90. doi: 10.1038/s41577-019-0139-2
55. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172(5):1022–37.e14. doi: 10.1016/j.cell.2018.01.004
56. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol (2016) 16(2):112–23. doi: 10.1038/nri.2015.9
57. Verma C, Kaewkangsadan V, Eremin JM, Cowley GP, Ilyas M, El-Sheemy MA, et al. Natural killer (NK) cell profiles in blood and tumour in women with large and locally advanced breast cancer (LLABC) and their contribution to a pathological complete response (PCR) in the tumour following neoadjuvant chemotherapy (NAC): differential restoration of blood profiles by NAC and surgery. J Transl Med (2015) 13:180. doi: 10.1186/s12967-015-0535-8
58. Chen T, Zhang T, Liu C, Wang C, Ding S, Shao Z, et al. NK cells suppress CD8(+) T cell immunity via NKG2D in severe aplastic anemia. Cell Immunol (2019) 335:6–14. doi: 10.1016/j.cellimm.2018.10.004
59. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E(2) (PGE (2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol Immunother (2011) 60(11):1577–86. doi: 10.1007/s00262-011-1064-9
60. Park A, Lee Y, Kim MS, Kang YJ, Park YJ, Jung H, et al. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of natural killer (NK) cell cytotoxicity and NK cell differentiation. Front Immunol (2018) 9:1859. doi: 10.3389/fimmu.2018.01859
61. Kundu N, Ma X, Holt D, Goloubeva O, Ostrand-Rosenberg S, Fulton AM. Antagonism of the prostaglandin e receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res Treat (2009) 117(2):235–42. doi: 10.1007/s10549-008-0180-5
62. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20(1):7–24. doi: 10.1038/s41577-019-0210-z
63. Della Porta M, Danova M, Rigolin GM, Brugnatelli S, Rovati B, Tronconi C, et al. Dendritic cells and vascular endothelial growth factor in colorectal cancer: Correlations with clinicobiological findings. Oncology (2005) 68(2-3):276–84. doi: 10.1159/000086784
64. Malietzis G, Lee GH, Jenkins JT, Bernardo D, Moorghen M, Knight SC, et al. Prognostic value of the tumour-infiltrating dendritic cells in colorectal cancer: A systematic review. Cell Commun Adhes (2015) 22(1):9–14. doi: 10.3109/15419061.2015.1036859
65. Jia XY, Chang Y, Sun XJ, Dai X, Wei W. The role of prostaglandin E2 receptor signaling of dendritic cells in rheumatoid arthritis. Int Immunopharmacol (2014) 23(1):163–9. doi: 10.1016/j.intimp.2014.08.024
66. Kitamura H, Ohno Y, Toyoshima Y, Ohtake J, Homma S, Kawamura H, et al. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci (2017) 108(10):1947–52. doi: 10.1111/cas.13332
67. Kim HJ, Lee YH, Im SA, Kim K, Lee CK. Cyclooxygenase inhibitors, aspirin and ibuprofen, inhibit MHC-restricted antigen presentation in dendritic cells. Immune Netw (2010) 10(3):92–8. doi: 10.4110/in.2010.10.3.92
68. Li H, Edin ML, Gruzdev A, Cheng J, Bradbury JA, Graves JP, et al. Regulation of T helper cell subsets by cyclooxygenases and their metabolites. Prostaglandins Other Lipid Mediat (2013) 104-105:74–83. doi: 10.1016/j.prostaglandins.2012.11.002
69. Bashyam H. Th1/Th2 cross-regulation and the discovery of IL-10. J Exp Med (2007) 204(2):237. doi: 10.1084/jem.2042fta
70. Endo T, Ogushi F, Sone S. LPS-dependent cyclooxygenase-2 induction in human monocytes is down-regulated by IL-13, but not by IFN-gamma. J Immunol (1996) 156(6):2240–6. doi: 10.4049/jimmunol.156.6.2240
71. Peck A, Mellins ED. Plasticity of T-cell phenotype and function: the T helper type 17 example. Immunology (2010) 129(2):147–53. doi: 10.1111/j.1365-2567.2009.03189.x
72. Peck A, Mellins ED. Breaking old paradigms: Th17 cells in autoimmune arthritis. Clin Immunol (2009) 132(3):295–304. doi: 10.1016/j.clim.2009.03.522
73. Li S, Liu M, Do MH, Chou C, Stamatiades EG, Nixon BG, et al. Cancer immunotherapy via targeted TGF-β signalling blockade in T(H) cells. Nature (2020) 587(7832):121–5. doi: 10.1038/s41586-020-2850-3
74. Li F, Sun Y, Huang J, Xu W, Liu J, Yuan Z. CD4/CD8 + T cells, DC subsets, Foxp3, and IDO expression are predictive indictors of gastric cancer prognosis. Cancer Med (2019) 8(17):7330–44. doi: 10.1002/cam4.2596
75. Markosyan N, Chen EP, Ndong VN, Yao Y, Sterner CJ, Chodosh LA, et al. Deletion of cyclooxygenase 2 in mouse mammary epithelial cells delays breast cancer onset through augmentation of type 1 immune responses in tumors. Carcinogenesis (2011) 32(10):1441–9. doi: 10.1093/carcin/bgr134
76. Khayrullina T, Yen JH, Jing H, Ganea D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J Immunol (2008) 181(1):721–35. doi: 10.4049/jimmunol.181.1.721
77. Jaffar Z, Wan KS, Roberts K. A key role for prostaglandin I2 in limiting lung mucosal Th2, but not Th1, responses to inhaled allergen. J Immunol (2002) 169(10):5997–6004. doi: 10.4049/jimmunol.169.10.5997
78. Nagamachi M, Sakata D, Kabashima K, Furuyashiki T, Murata T, Segi-Nishida E, et al. Facilitation of Th1-mediated immune response by prostaglandin e receptor EP1. J Exp Med (2007) 204(12):2865–74. doi: 10.1084/jem.20070773
79. Raskov H, Orhan A, Christensen JP, Gogenur I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer. (2021) 124(2):359–67. doi: 10.1038/s41416-020-01048-4
80. Reina-Campos M, Scharping NE, Goldrath AW. CD8(+) T cell metabolism in infection and cancer. Nat Rev Immunol (2021) 21(11):718–38. doi: 10.1038/s41577-021-00537-8
81. Rundqvist H, Veliça P, Barbieri L, Gameiro PA, Bargiela D, Gojkovic M, et al. Cytotoxic T-cells mediate exercise-induced reductions in tumor growth. Elife (2020) 9. doi: 10.7554/eLife.59996
82. Blessin NC, Li W, Mandelkow T, Jansen HL, Yang C, Raedler JB, et al. Prognostic role of proliferating CD8(+) cytotoxic tcells in human cancers. Cell Oncol (Dordr) (2021) 44(4):793–803. doi: 10.1007/s13402-021-00601-4
83. Pu D, Yin L, Huang L, Qin C, Zhou Y, Wu Q, et al. Cyclooxygenase-2 inhibitor: A potential combination strategy with immunotherapy in cancer. Front Oncol (2021) 11:637504. doi: 10.3389/fonc.2021.637504
84. Take Y, Koizumi S, Nagahisa A. Prostaglandin e receptor 4 antagonist in cancer immunotherapy: Mechanisms of action. Front Immunol (2020) 11:324. doi: 10.3389/fimmu.2020.00324
85. Chen JH, Perry CJ, Tsui YC, Staron MM, Parish IA, Dominguez CX, et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med (2015) 21(4):327–34. doi: 10.1038/nm.3831
86. Karavitis J, Hix LM, Shi YH, Schultz RF, Khazaie K, Zhang M. Regulation of COX2 expression in mouse mammary tumor cells controls bone metastasis and PGE2-induction of regulatory T cell migration. PloS One (2012) 7(9):e46342. doi: 10.1371/journal.pone.0046342
87. Zeddou M, Greimers R, de Valensart N, Nayjib B, Tasken K, Boniver J, et al. Prostaglandin E2 induces the expression of functional inhibitory CD94/NKG2A receptors in human CD8+ T lymphocytes by a cAMP-dependent protein kinase a type I pathway. Biochem Pharmacol (2005) 70(5):714–24. doi: 10.1016/j.bcp.2005.05.015
88. Yao M, Kargman S, Lam EC, Kelly CR, Zheng Y, Luk P, et al. Inhibition of cyclooxygenase-2 by rofecoxib attenuates the growth and metastatic potential of colorectal carcinoma in mice. Cancer Res (2003) 63(3):586–92.
89. Rothwell PM, Price JF, Fowkes FG, Zanchetti A, Roncaglioni MC. Tognoni G, et al. short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet (2012) 379(9826):1602–12. doi: 10.1016/s0140-6736(11)61720-0
90. Mazhar D, Ang R, Waxman J. COX inhibitors and breast cancer. Br J Cancer (2006) 94(3):346–50. doi: 10.1038/sj.bjc.6602942
91. Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: An international consensus statement. Lancet Oncol (2009) 10(5):501–7. doi: 10.1016/s1470-2045(09)70035-x
92. Cuzick J, Thorat MA, Bosetti C, Brown PH, Burn J, Cook NR, et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann Oncol (2015) 26(1):47–57. doi: 10.1093/annonc/mdu225
94. Goto S, Konnai S, Hirano Y, Kohara J, Okagawa T, Maekawa N, et al. Upregulation of PD-L1 expression by prostaglandin E(2) and the enhancement of IFN-γ by anti-PD-L1 antibody combined with a COX-2 inhibitor in mycoplasma bovis infection. Front Vet Sci (2020) 7:12. doi: 10.3389/fvets.2020.00012
95. Sajiki Y, Konnai S, Cai Z, Takada K, Okagawa T, Maekawa N, et al. Enhanced immunotherapeutic efficacy of anti-PD-L1 antibody in combination with an EP4 antagonist. Immunohorizons (2020) 4(12):837–50. doi: 10.4049/immunohorizons.2000089
96. Wang Y, Cui L, Georgiev P, Singh L, Zheng Y, Yu Y, et al. Combination of EP(4) antagonist MF-766 and anti-PD-1 promotes anti-tumor efficacy by modulating both lymphocytes and myeloid cells. Oncoimmunology (2021) 10(1):1896643. doi: 10.1080/2162402x.2021.1896643
97. Sterner RC, Sterner RM. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
98. Li AM, Hucks GE, Dinofia AM, Seif AE, Teachey DT, Baniewicz D, et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed b-cell acute lymphoblastic leukemia. Blood (2018) 132(Supplement 1):556–. doi: 10.1182/blood-2018-99-112572
Keywords: COX-2, PGE2, COX-2 inhibitors, EP, tumor immune escape
Citation: Jin K, Qian C, Lin J and Liu B (2023) Cyclooxygenase-2-Prostaglandin E2 pathway: A key player in tumor-associated immune cells. Front. Oncol. 13:1099811. doi: 10.3389/fonc.2023.1099811
Received: 16 November 2022; Accepted: 12 January 2023;
Published: 27 January 2023.
Edited by:
Saravanan Rajendrasozhan, University of Hail, Saudi ArabiaReviewed by:
Chiara Focaccetti, Università di Roma Tor Vergata, ItalyCopyright © 2023 Jin, Qian, Lin and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing Liu, bGl1YmluZ3pqdUB6anUuZWR1LmNu; Jinti Lin, MTE5MTgzMzZAemp1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.