 Mengmeng Li
Mengmeng Li Ruyue Xing1
Ruyue Xing1 Chao Shi
Chao Shi Huijuan Wang
Huijuan Wang- 1Department of Medical Oncology, The Affiliated Cancer Hospital of Zhengzhou University and Henan Caner Hospital, Zhengzhou, China
- 2Department of Molecular Pathology, The Affiliated Cancer Hospital of Zhengzhou University and Henan Cancer Hospital, Zhengzhou, China
Epithelioid inflammatory myofibroblastic sarcoma (EIMS) is an aggressive variant of inflammatory myofibroblastic tumor (IMT) and has a poor prognosis. EIMS is characterized by epithelioid morphology, neutrophilic infiltrate and specific fusion partners of anaplastic lymphoma kinase (ALK). Despite no standard therapy for EIMS, ALK tyrosine kinase inhibitors (TKIs) are recommended for these tumors. The present case describes an abdominal mass that presented in a 31-year-old male. The patient suffered from recurrence and multiple metastases 2 months after surgery. Ensartinib was administered and RANBP2-ALK fusion was detected. A partial response has been observed for 4 months and there has been no recurrence. This study provided a successful case with sustained response of targeted therapy.
Introduction
Inflammatory myofibroblastic tumor (IMT) is as an intermediate soft tissue tumor composed of myofibroblast-differentiated spindle cells along with numerous inflammatory cells, plasma cells, and/or lymphocytes (1). Epithelioid inflammatory myofibroblastic sarcoma (EIMS) is a rare subtype of IMT that is characterized by aggressiveness, rapid local recurrence, earliest metastasis, and fatality (2, 3). EIMS differs from the conventional spindle-cell IMT in that it consists mostly of round-to-epithelioid cells, with a loose or myxoid stroma that is infiltrated with abundant neutrophils (4–7). Among 11 cases, all tumors were located in the abdomen and most originated in the mesentery or omentum (2). In addition to abdomen, several cases with extra-abdominal sites of EIMS have been reported, including liver (8), lung (7, 9), pericardium (10), ovary (11), cutaneous (12), stomach (13), groin (14) and central nervous system (15). A variety of gene partners have been observed in IMT including NPM, TMP3/4, CARS, CLTC, EML4, DCTN1, SEC31L1, ATIC and FN1 (5), with more prevalent fusions of RANBP2 (2), RRBP1 (16) and EML4 (5) in EIMS. Furthermore, G1269A has been reported as a secondary mutation after resistance to crizotinib (17). As described herein, a patient with RANBP2-ALK EIMS in the greater omentum benefited from an ALK TKI.
Case description
An intermittent abdominal pain and abdominal distention were reported by a 31-year-old Chinese male. Enhanced computed tomography (CT) scans of the abdomen revealed an abdominal mass that was suspected to be gastric stromal tumor (Figure 1 and Table 1). Biopsy showed a loose tissue composed of spindle cells, hollow cells and small blood vessels. Fluorescence in situ hybridization (FISH) was negative for CHOP and MDM2, excluding the presence of liposarcoma. Abdominal tumor resection plus partial colectomy was then performed in March 2022, removing the greater omentum tumor as well as partial transverse colon and ascending colon with a volume of 12 × 10 × 10 cm. The histopathological results revealed the lesion contained both epithelioid and spindle cells with enlarged nuclei and infiltrating inflammatory cells, primarily plasma cells, eosinophils, neutrophils, lymphocytes (Figures 2A, B). EIMS usually expressed vimentin and desmin positively, the Expression situation of EMA, CD30 and SMA was inconsistent. EIMS can be distinguished by IHC from soft tissue tumors with epithelioid cell morphology and tumors with significant mucoid background. Such as anaplastic large cell lymphoma (ALCL) and EIMS were positive for SMA, CD30 and ALK and negative for EMA (29). But no desmin was found in ALCL. Follicular dendritic cell sarcoma (FDCS): Immunohistochemical expression of CD21, CD23, or CD35 was positive, but no ALK,Desmin,WT-l,orD2-40 (30). Extragastrointestinal stromal tumor (EGIST) CD117, CD34 and Dog-1 were positive, and ALK;CKDesmin were negative (31). Therefore, the following markers were selected for immunohistochemistry, and the results were as follows. ALK, Vimentin, Desmin, SAM, Ki-67, CD30, CD31, Catenin and H3K27Me3 were positive, while negative for Cytokeratin (CK), CD34, CD117, S-100, SOX-10, Dog-1, Muc-4, EMA and ERG (Table 2). Antibody clone of ALK-IHC is ALK p80(5A4), and Positive immunohistochemical results of ALK, CD30, desmin, SAM and Vimentin are shown in turn in this Figures 2C–G. Approximately 18% of the tumor cells showed a rearrangement of ALK by FISH (Figure 2H). These findings are in line with those of EIMS. Adjuvant therapy was not administered to the patient following surgery.
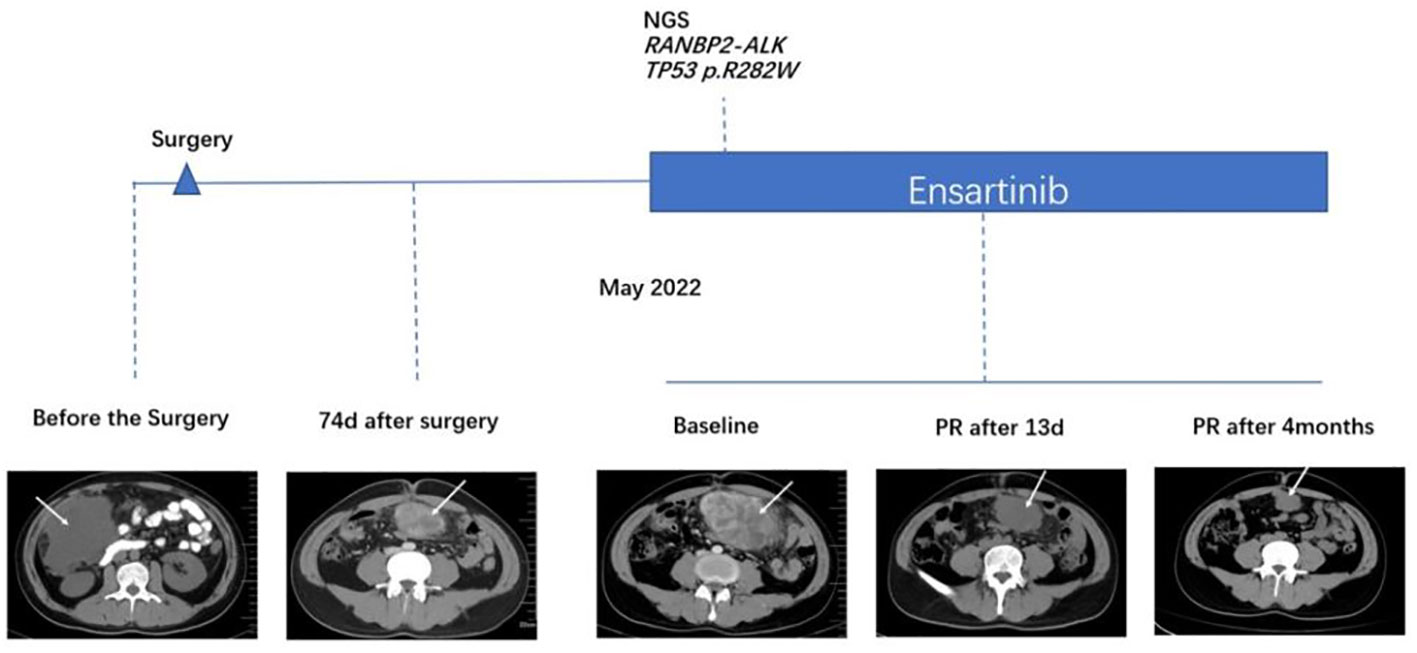
Figure 1 Comparison of computed tomography images before and after treatment with ensartinib.
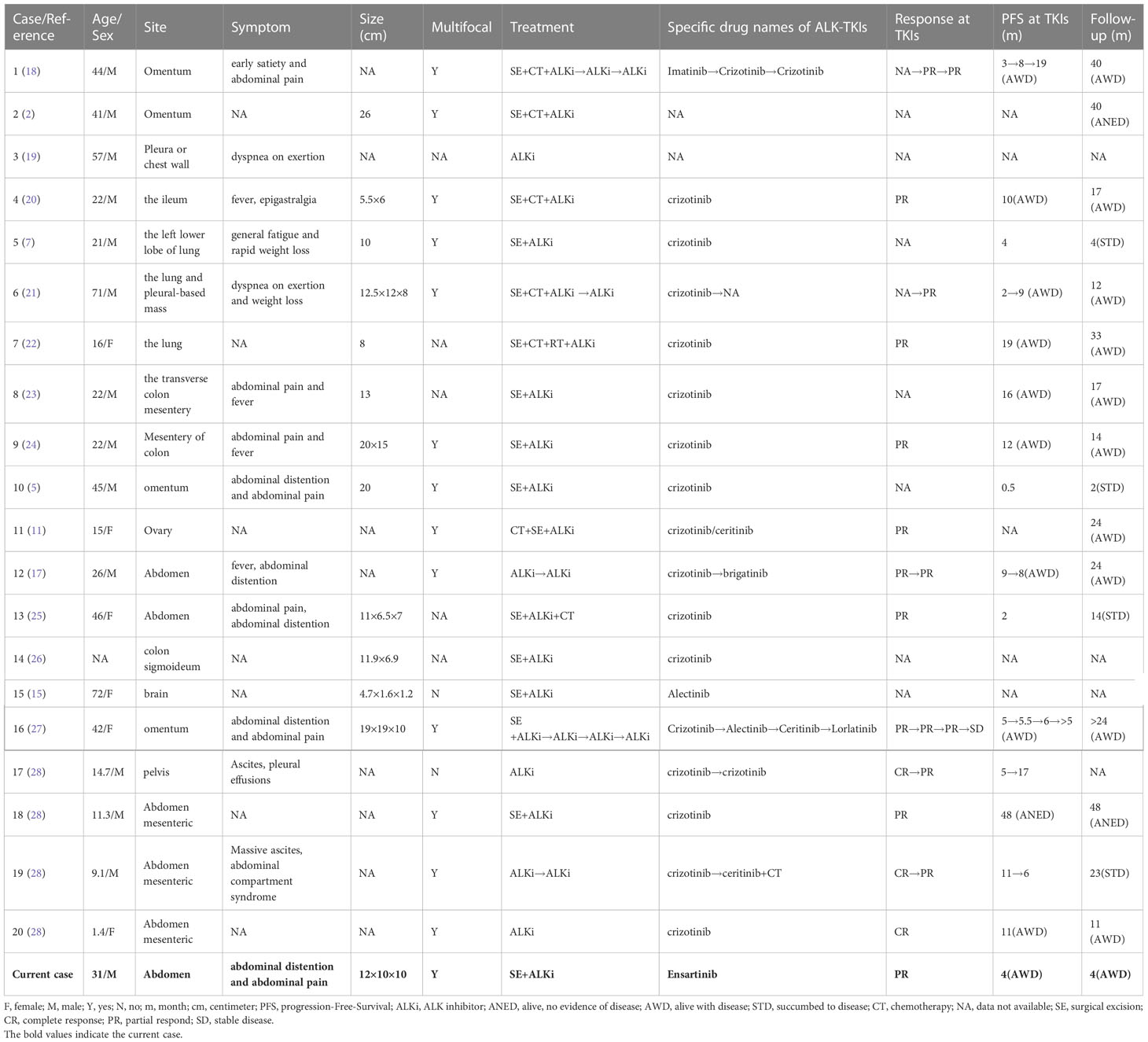
Table 1 Clinicopathological characteristics of EIMS in reported cases and our case.
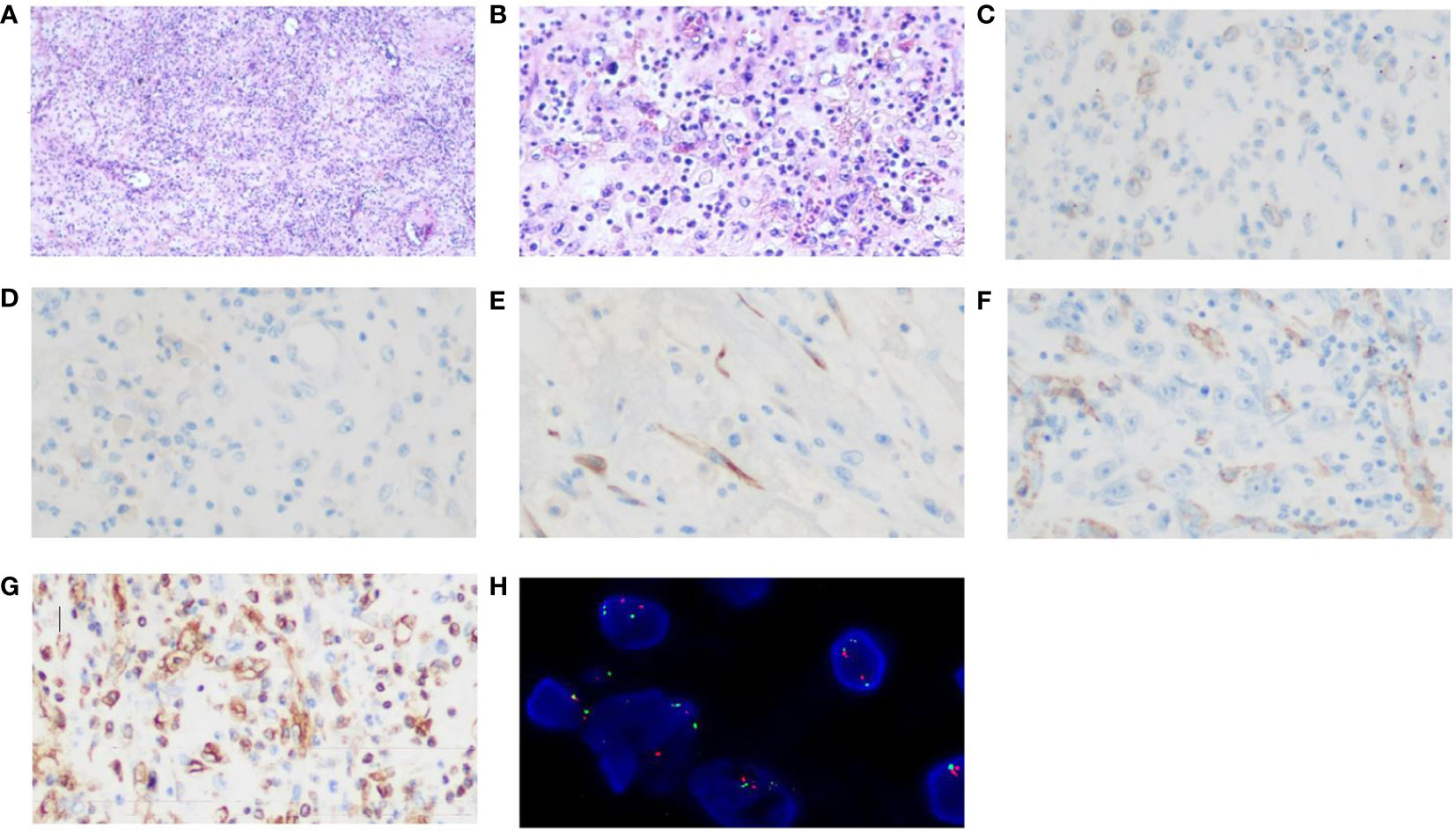
Figure 2 Histopathological examination with haematoxylin and eosin staining and Immunohistochemistry and FISH images. The lesion consisted of both epithelioid and spindle cells with enlarged nucleolus and inflammatory cells infiltration (A) (magnification, x40). (B) (magnification, x200). (C) ALK positivity (magnification, x400); (D)Focal CD30 weakly positive (magnification, x400); (E)Desmin positivity (magnification, x400) (F)SMA positivity (magnification, x400) (G) Vimentin positivity (magnification, x400); (H) A FISH image of ALK rearrangement.
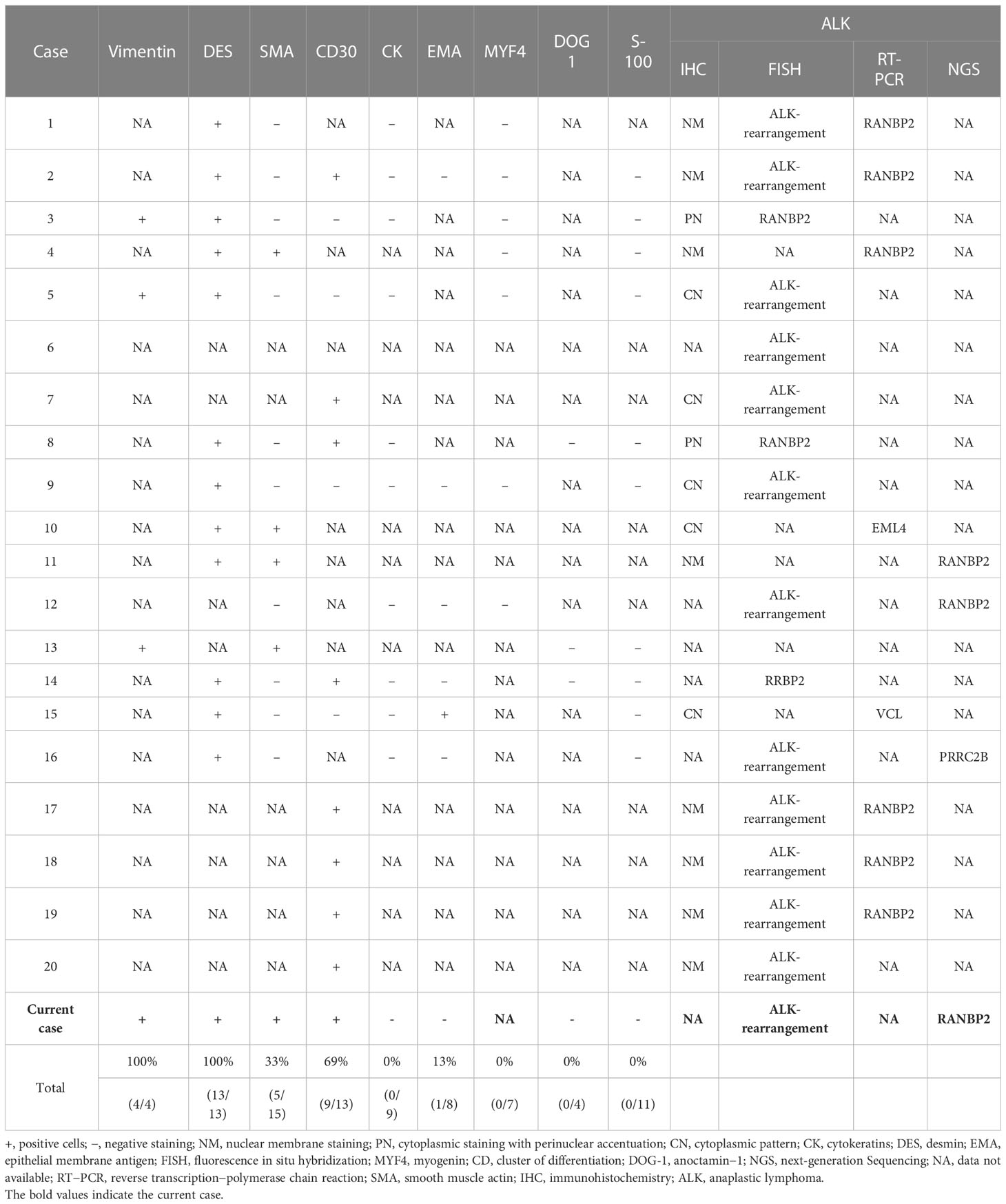
Table 2 Immunohistochemical and genetic characteristics in reported cases and our case.
The patient presented to our hospital with abdominal distension 74 days after surgery. CT showed new soft tissue mass with peritoneal metastasis in lower abdomen and pelvis, indicating tumor recurrence. The tumor was significantly enlarged 14 days later by CT scan. The therapeutic course and radiological examinations of the patient were summarized in Figure 1. The patient was subsequently treated with ensartinib (225 mg, QD). The CT revealed notable tumor shrinkage 13 days after ensartinib treatment. In September 29, 2022, 4 months after initiation of ensartinib, this patient still achieved partial response (PR) according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (Figure 1). Next-generation sequencing (NGS) was employed using a 1021-gene panel (Burning Rock, Guangzhou, China). NGS identified RANBP2-ALK fusion (EX18:EX20) with mutational abundance of 2.3% and a TP53 p.R282W mutation with a mutational abundance of 3.2% using the biopsy specimen of the abdominal mass (data not shown). As we prepared the manuscript, the patient remained PR and continued to receive ensartinib without any significant adverse events.
Discussion
EIMS is first named in 2011 (2) and is a more aggressive subtype of IMT and characterized by epithelioid-to-round cell morphology and prominent inflammatory infiltrate. EIMSs can occur across a wide age range (4 months to 76 years), with a male and intra-abdominal predominance (15). Recently, IMTs associated with EML4-ALK have been classified as EIMS (5).
EIMS is essentially a histopathological diagnosis. Even with auxiliary detection, it is difficult to make a definitive diagnosis of EIMS on a small biopsy due to its genetic overlap with other ALK-positive tumors. To date, more than 40 cases of EIMS have been reported, among which 20 cases (2, 5, 7, 11, 15, 17–28) as well as present case with the complete clinicopathological, immunohistochemical and genetic characteristics were summarized in Table 1, 2. These 21 cases were composed of 14 (67%) adults (21-72 years old) and 6 (29%) children or adolescents. The median age was 24 years old. Fourteen (67%) patients were male while six (29%) were female. In 16 patients, the tumors were found in abdominal cavity (omentum, mesenterium, ileum, colon, ovary, etc.), 4 in pleural cavity and 1 in brain, which consisted with previous reports that EIMS had a male and intra-abdominal predilection. In contrast, conventional spindle-cell IMT was slightly more prevalent among females. The EIMS exhibits distinctive morphological characteristics including loosely arranged round or epithelioid neoplastic cells with vesicular nuclei, prominent nucleoli and myxoid stroma surround by amphophilic to eosinophilic cytoplasm. Neutrophil-rich inflammatory infiltrates are a striking characteristic of EIMS. Almost all tumors contained a spot of spindle cell component.
According to immunohistochemical results in 15 cases with EIMS, 8 (53%) revealed a unique nuclear membrane staining pattern for ALK (Table 2). Nevertheless, cytoplasmic or perinuclear staining of ALK was observed in 7 of 15 cases (47%). All cases (13/13) exhibited strong expression of desmin, another diagnostic immunophenotype. Besides, the tumor displayed variable expression of CD30 (69%, 9/13), alpha smooth muscle actin (33%, 5/15) and epithelial membrane antigen (13%, 1/8). Moreover, all cases were negative for cytokeratin (0/9), myogenin (0/7), anoctamin−1 (0/4) and S-100 (0/11). Of note, FISH assay, PCR assay or NGS can contribute to the diagnosis of EIMS. It has been confirmed that ALK rearrangement is present in 16 cases by FISH, 8 cases by PCR and 4 cases by NGS.
RANBP2-ALK and RRBP1-ALK fusions were the most reported driver mutation of EIMS (16, 32). In previous studies as well as our case with EIMS, nuclear membrane or perinuclear staining pattern for RANBP2-ALK fusion was detected, and almost all cases containing RANBP2-ALK fusion exhibited aggressive behavior (2, 12, 18, 20, 33–35). Despite of the unclear biological function, the chimeric RANPB2-ALK gene was reported to promote cell growth and proliferation regardless of cytokine in vitro (36, 37). There was a specificity for RRBP1-ALK in EMIS with cytoplasmic ALK expression and clinically aggressive progression, suggesting that RRBP1-ALK may exert relapsed oncogenic role in clinically aggressive EIMS (16). Recently, PRRC2B-ALK fusion was also considered as the main oncogenic driver of the EIMS (27).
There is no clear consensus on the best treatment for EIMS. The main treatment option remains surgical resection. Postoperative adjuvant therapy has not yet been identified due to the limited available experiences. In terms of rapid recurrence, postoperative chemotherapy or radiotherapy appeared to exert finite effect (2, 18, 20, 35). Crizotinib has been applied to treat EIMS in several cases and showed favorable efficacy (2, 18–20, 38). In our summarized cases, 10 patients continued to live with disease with follow-up of 11-40 months. Among 21 patients, 17 received crizotinib, apart from 2 unknown TKIs, 1 ensartinib and 1 alectinib. Fifteen of 17 patients who received crizotinib had survival information, with a median PFS of 9 months and mean PFS of 10.8 months. Furthermore, a case with PRRC2B-ALK fusion showed durable clinical response to sequential use of ALK TKIs (crizotinib, alectinib, ceritinib and lorlatinib) (27). The development of drug resistance to ALK TKIs is a major issue. A previous case revealed that R1192P was found as a resistance mutation to crizotinib (27), suggesting that the application of NGS was important to identify actionable mutations and resistance mechanisms. This may contribute to molecular targeted therapies for EIMS with ALK gene arrangement. Considering the broader coverage of targets and stronger tissue penetration, we speculated that patients may have more benefits from the direct second- or third-generation ALK TKIs compared with the sequential treatment of ALK TKIs. Therefore, in our own case, the patient received ensartinib (a second-generation ALK TKI) post relapse. After four months of treatment, the patient felt well with PR, and follow-up CT scan showed that the residual tumor was partially shrank.
Recently, a diffuse positive signal was observed in EIMS for programmed death-ligand 1 (PD-L1) (4), providing a possible inmmunomodulatory therapies targeting the PD-1/PD-L1 pathway. Moreover, CD30 appeared to commonly express in EIMS in our summarized cases (Table 1). The survival time was prolonged by ALK and CD30 combination therapies (39). With increasing evidence of EIMS, molecular mechanisms will be clear and potential treatments will be developed in the future.
In addition to RANBP1-ALK fusion, TP53 p.R282W mutation was also identified in our case. Mutant TP53 is closely related to the occurrence, development and prognosis of tumor (40, 41). Besides, mutations in TP53 independently promoted metastasis, decreased TKI responses and shorten overall survival in ALK-positive lung adenocarcinoma (42). However, the molecular pathological importance of the TP53 mutation in IMT has not been elucidated thus far. A previous study demonstrated that abnormal TP53 staining patterns were detected in only approximately 7% of IMT, with TP53 missense mutations occurring in 13% of cases, suggesting that TP53 mutation in IMT was an infrequent event and may not attribute to its pathogenesis (43). Recently, a study demonstrated that ORR of ensartinib was high regardless of TP53 mutation status (44). In our case, ensartinib also showed favorable efficacy.
Conclusion
In conclusion, we described a typical EIMS case with a round or epithelioid morphology of cells, accompanied by a high relapse and a poor prognosis. To the best of our knowledge, our report is the first case to investigate the efficacy of ensartinib for EIMS. The clinical management and results of the patients were introduced in detail in our case; besides, the pathological and genetic characteristics of the tumors were reviewed. By analyzing the efficacy of ALK TKIs in EIMS in previous literature, we found that ALK TKIs are effective for EIMS treatment. However, given the lack of the clear resistance mechanisms, further research is needed. Detection of ALK rearrangement is essential for correct diagnosis of EIMS and provides a fundamental basis for ALK TKI therapy.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Henan Cancer Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
>HW contributed to the conception of the study. ML and RX integrated all information and wrote the main manuscript. CW supervised the writing process. CS and JH provided critical guidance. All authors contributed to the article and approved the submitted version.
Funding
Our work was supported by the National Natural Science Foundation of China (No. 82102849).
Acknowledgments
All the authors express gratitude to the patient and the members of their research group for useful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alshammari HK, Alzamami HF, Ashoor M, Almarzouq WF, Kussaibi H. A rare presentation of inflammatory myofibroblastic tumor in the nasolabial fold. Case Rep Otolaryngol (2019) 2019:3257697. doi: 10.1155/2019/3257697
2. Mariño-Enríquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD, et al. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear alk. Am J Surg Pathol (2011) 35(1):135–44. doi: 10.1097/PAS.0b013e318200cfd5
3. Lee C, Li C-F, Huang H-Y, Zhu M-J, Mariño-Enríquez A, Lee C-T, et alAlk oncoproteins in atypical inflammatory myofibroblastic tumours: Novel Rrbp1-alk fusions in epithelioid inflammatory myofibroblastic sarcoma. J Pathol (2017) 242(2):260. doi: 10.1002/path.4910
4. Du X, Gao Y, Zhao H, Li B, Xue W, Wang D. Clinicopathological analysis of epithelioid inflammatory myofibroblastic sarcoma. Oncol Lett (2018) 15(6):9317–26. doi: 10.3892/ol.2018.8530
5. Jiang Q, Tong HX, Hou YY, Zhang Y, Li JL, Zhou YH, et al. Identification of Eml4-alk as an alternative fusion gene in epithelioid inflammatory myofibroblastic sarcoma. Orphanet J Rare Dis (2017) 12(1):97. doi: 10.1186/s13023-017-0647-8
6. Telugu RB, Prabhu AJ, Kalappurayil NB, Mathai J, Gnanamuthu BR, Manipadam MT. Clinicopathological study of 18 cases of inflammatory myofibroblastic tumors with reference to alk-1 expression: 5-year experience in a tertiary care center. J Pathol Transl Med (2017) 51(3):255–63. doi: 10.4132/jptm.2017.01.12
7. Fu X, Jiang J, Tian XY, Li Z. Pulmonary epithelioid inflammatory myofibroblastic sarcoma with multiple bone metastases: Case report and review of literature. Diagn Pathol (2015) 10:106. doi: 10.1186/s13000-015-0358-1
8. Chen ST, Lee JC. An inflammatory myofibroblastic tumor in liver with alk and Ranbp2 gene rearrangement: Combination of distinct morphologic, immunohistochemical, and genetic features. Hum Pathol (2008) 39(12):1854–8. doi: 10.1016/j.humpath.2008.04.016
9. Singh P, Nambirajan A, Gaur MK, Raj R, Kumar S, Malik PS, et al. Primary pulmonary epithelioid inflammatory myofibroblastic sarcoma: A rare entity and a literature review. J Pathol Transl Med (2022) 56(4):231–7. doi: 10.4132/jptm.2022.05.08
10. Azad M, Oye M, Torrente N, Mirsaeidi M. Pericardial epithelioid inflammatory myofibroblastic sarcoma: An atypical presentation. Cureus (2022) 14(7):e26827. doi: 10.7759/cureus.26827
11. Fang H, Langstraat CL, Visscher DW, Folpe AL, Schoolmeester JK. Epithelioid inflammatory myofibroblastic sarcoma of the ovary with Ranb2-alk fusion: Report of a case. Int J Gynecol Pathol (2018) 37(5):468–72. doi: 10.1097/pgp.0000000000000431
12. Gadeyne L, Creytens D, Dekeyser S, van der Meulen J, Haspeslagh M. Primary cutaneous epithelioid inflammatory myofibroblastic sarcoma harboring Ranbp2-alk fusion: Report of an exceptional case. Am J Dermatopathol (2022) 44(4):302–5. doi: 10.1097/dad.0000000000002096
13. Xu P, Shen P, Jin Y, Wang L, Wu W. Epithelioid inflammatory myofibroblastic sarcoma of stomach: Diagnostic pitfalls and clinical characteristics. Int J Clin Exp Pathol (2019) 12(5):1738–44.
14. Zilla ML, Khoshnoodi P, Bailey NG, Herradura A, Lee SJ, John I. Epithelioid inflammatory myofibroblastic sarcomas are not exclusive to ventral cavity sites. Histopathology (2022) 80(3):610–2. doi: 10.1111/his.14582
15. Chopra S, Maloney N, Wang WL. Epithelioid inflammatory myofibroblastic sarcoma with vcl-alk fusion of central nervous system: Case report and brief review of the literature. Brain Tumor Pathol (2022) 39(1):35–42. doi: 10.1007/s10014-021-00416-z
16. Lee JC, Li CF, Huang HY, Zhu MJ, Mariño-Enríquez A, Lee CT, et al. Alk oncoproteins in atypical inflammatory myofibroblastic tumours: Novel Rrbp1-alk fusions in epithelioid inflammatory myofibroblastic sarcoma. J Pathol (2017) 241(3):316–23. doi: 10.1002/path.4836
17. Xu X, Li H, Peng K, Yu Y, Chen L, Fang Y, et al. Alk-G1269a mutation in epithelioid inflammatory myofibroblastic sarcoma after progression on crizotinib: A case report. Oncol Lett (2019) 17(2):2370–6. doi: 10.3892/ol.2018.9865
18. Butrynski JE, D'Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, et al. Crizotinib in alk-rearranged inflammatory myofibroblastic tumor. N Engl J Med (2010) 363(18):1727–33. doi: 10.1056/NEJMoa1007056
19. Kozu Y, Isaka M, Ohde Y, Takeuchi K, Nakajima T. Epithelioid inflammatory myofibroblastic sarcoma arising in the pleural cavity. Gen Thorac Cardiovasc Surg (2014) 62(3):191–4. doi: 10.1007/s11748-013-0204-x
20. Kimbara S, Takeda K, Fukushima H, Inoue T, Okada H, Shibata Y, et al. A case report of epithelioid inflammatory myofibroblastic sarcoma with Ranbp2-alk fusion gene treated with the alk inhibitor, crizotinib. Jpn J Clin Oncol (2014) 44(9):868–71. doi: 10.1093/jjco/hyu069
21. Sarmiento DE, Clevenger JA, Masters GA, Bauer TL, Nam BT. Epithelioid inflammatory myofibroblastic sarcoma: A case report. J Thorac Dis (2015) 7(10):E513–6. doi: 10.3978/j.issn.2072-1439.2015.10.55
22. Lee JC, Wu JM, Liau JY, Huang HY, Lo CY, Jan IS, et al. Cytopathologic features of epithelioid inflammatory myofibroblastic sarcoma with correlation of histopathology, immunohistochemistry, and molecular cytogenetic analysis. Cancer Cytopathol (2015) 123(8):495–504. doi: 10.1002/cncy.21558
23. Liu Q, Kan Y, Zhao Y, He H, Kong L. Epithelioid inflammatory myofibroblastic sarcoma treated with alk inhibitor: A case report and review of literature. Int J Clin Exp Pathol (2015) 8(11):15328–32.
24. Yu L, Liu J, Lao IW, Luo Z, Wang J. Epithelioid inflammatory myofibroblastic sarcoma: A clinicopathological, immunohistochemical and molecular cytogenetic analysis of five additional cases and review of the literature. Diagn Pathol (2016) 11(1):67. doi: 10.1186/s13000-016-0517-z
25. Zhang S, Wang Z. A case report on epithelioid inflammatory myofibroblastic sarcoma in the abdominal cavity. Int J Clin Exp Pathol (2019) 12(10):3934–9.
26. Liu D, Luo R, Tang H, Li T. Sigmoid epithelioid inflammatory myofibroblastic sarcoma with high white blood cell count: A case report. Asian J Surg (2020) 43(8):838–9. doi: 10.1016/j.asjsur.2020.03.010
27. Wang Z, Geng Y, Yuan LY, Wang MM, Ye CY, Sun L, et al. Durable clinical response to alk tyrosine kinase inhibitors in epithelioid inflammatory myofibroblastic sarcoma harboring Prrc2b-alk rearrangement: A case report. Front Oncol (2022) 12:761558. doi: 10.3389/fonc.2022.761558
28. Trahair T, Gifford AJ, Fordham A, Mayoh C, Fadia M, Lukeis R, et al. Crizotinib and surgery for long-term disease control in children and adolescents with alk-positive inflammatory myofibroblastic tumors. JCO Precis Oncol (2019) 3:1–11. doi: 10.1200/PO.18.00297
29. Shustov A, Soma L. Anaplastic Large cell lymphoma: Contemporary concepts and optimal management. Cancer Treat Res (2019) 176:127–44. doi: 10.1007/978-3-319-99716-2_6
30. Facchetti F, Simbeni M, Lorenzi L. Follicular dendritic cell sarcoma. Pathologica (2021) 113(5):316–29. doi: 10.32074/1591-951x-331
31. Zhang W, Peng Z, Xu L. Extragastrointestinal stromal tumor arising in the rectovaginal septum: Report of an unusual case with literature review. Gynecol Oncol (2009) 113(3):399–401. doi: 10.1016/j.ygyno.2009.02.019
32. Hallin M, Thway K. Epithelioid inflammatory myofibroblastic sarcoma. Int J Surg Pathol (2019) 27(1):69–71. doi: 10.1177/1066896918767557
33. Cook JR, Dehner LP, Collins MH, Ma Z, Morris SW, Coffin CM, et al. Anaplastic lymphoma kinase (Alk) expression in the inflammatory myofibroblastic tumor: A comparative immunohistochemical study. Am J Surg Pathol (2001) 25(11):1364–71. doi: 10.1097/00000478-200111000-00003
34. Li J, Yin WH, Takeuchi K, Guan H, Huang YH, Chan JK. Inflammatory myofibroblastic tumor with Ranbp2 and alk gene rearrangement: A report of two cases and literature review. Diagn Pathol (2013) 8:147. doi: 10.1186/1746-1596-8-147
35. Ma Z, Hill DA, Collins MH, Morris SW, Sumegi J, Zhou M, et al. Fusion of alk to the ran-binding protein 2 (Ranbp2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer (2003) 37(1):98–105. doi: 10.1002/gcc.10177
36. Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma-associated F1174l alk mutation causes resistance to an alk kinase inhibitor in alk-translocated cancers. Cancer Res (2010) 70(24):10038–43. doi: 10.1158/0008-5472.Can-10-2956
37. Röttgers S, Gombert M, Teigler-Schlegel A, Busch K, Gamerdinger U, Slany R, et al. Alk fusion genes in children with atypical myeloproliferative leukemia. Leukemia (2010) 24(6):1197–200. doi: 10.1038/leu.2010.18
38. Tothova Z, Wagner AJ. Anaplastic lymphoma kinase-directed therapy in inflammatory myofibroblastic tumors. Curr Opin Oncol (2012) 24(4):409–13. doi: 10.1097/CCO.0b013e328354c155
39. Fordham AM, Xie J, Gifford AJ, Wadham C, Morgan LT, Mould EVA, et al. Cd30 and alk combination therapy has high therapeutic potency in Ranbp2-Alk-Rearranged epithelioid inflammatory myofibroblastic sarcoma. Br J Cancer (2020) 123(7):1101–13. doi: 10.1038/s41416-020-0996-2
40. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature (2014) 505(7484):495–501. doi: 10.1038/nature12912
41. Zhou G, Liu Z, Myers JN. Tp53 mutations in head and neck squamous cell carcinoma and their impact on disease progression and treatment response. J Cell Biochem (2016) 117(12):2682–92. doi: 10.1002/jcb.25592
42. Christopoulos P, Kirchner M, Bozorgmehr F, Endris V, Elsayed M, Budczies J, et al. Identification of a highly lethal V3(+) Tp53(+) subset in alk(+) lung adenocarcinoma. Int J Cancer (2019) 144(1):190–9. doi: 10.1002/ijc.31893
43. Yamamoto H, Oda Y, Saito T, Sakamoto A, Miyajima K, Tamiya S, et al. P53 mutation and Mdm2 amplification in inflammatory myofibroblastic tumours. Histopathology (2003) 42(5):431–9. doi: 10.1046/j.1365-2559.2003.01611.x
Keywords: epithelioid inflammatory myofibroblastic sarcoma, inflammatory myofibroblastic tumor, RANBP2-ALK, ensartinib, fluorescence in situ hybridization
Citation: Li M, Xing R, Huang J, Shi C, Wei C and Wang H (2023) Case report: Epithelioid inflammatory myofibroblastic sarcoma treated with an ALK TKI ensartinib. Front. Oncol. 13:1084456. doi: 10.3389/fonc.2023.1084456
Received: 30 October 2022; Accepted: 09 March 2023;
Published: 22 March 2023.
Edited by:
Marla Weetall, PTC Therapeutics, United StatesReviewed by:
Zuoyu Liang, Sichuan University, ChinaErin B. Dickerson, University of Minnesota Twin Cities, United States
Copyright © 2023 Li, Xing, Huang, Shi, Wei and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huijuan Wang, MTg2Mzg1NjE1ODhAMTYzLmNvbQ==