
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 15 February 2023
Sec. Molecular and Cellular Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1072802
This article is part of the Research Topic The Role of Helicobacter pylori in Gastric Carcinogenesis View all 6 articles
 Wendy Malespín-Bendaña1*
Wendy Malespín-Bendaña1* Warner Alpízar-Alpízar2,3
Warner Alpízar-Alpízar2,3 Lucía Figueroa-Protti2,4Ledis Reyes5Silvia Molina-Castro1,3Clas Une1
Lucía Figueroa-Protti2,4Ledis Reyes5Silvia Molina-Castro1,3Clas Une1 Vanessa Ramírez-Mayorga1,6
Vanessa Ramírez-Mayorga1,6Introduction: Helicobacter pylori colonizes the gastric mucosa and induces chronic inflammation.
Methods: Using a mouse model of H. pylori-induced gastritis, we evaluated the mRNA and protein expression levels of proinflammatory and proangiogenic factors, as well as the histopathological changes in gastric mucosa in response to infection. Five- to six-week-old female C57BL/6N mice were challenged with H. pylori SS1 strain. Animals were euthanized after 5-, 10-, 20-, 30-, 40- and 50-weeks post infection. mRNA and protein expression of Angpt1, Angpt2, VegfA, Tnf-α, bacterial colonization, inflammatory response and gastric lesions were evaluated.
Results: A robust bacterial colonization was observed in 30 to 50 weeks-infected mice, which was accompanied by immune cell infiltration in the gastric mucosa. Compared to non-infected animals, H. pylori-colonized animals showed an upregulation in the expression of Tnf-A, Angpt2 and VegfA at the mRNA and protein levels. In contrast, Angpt1 mRNA and protein expression was downregulated in H. pylori-colonized mice.
Conclusion: Our data show that H. pylori infection induces the expression of Angpt2, Tnf-A and Vegf-A in murine gastric epithelium. This may contribute to the pathogenesis of H. pylori-associated gastritis, however the significance of this should be further addressed.
Angiogenesis, the process of formation of new blood vessels from the pre-existing, involves proliferation, sprouting and migration of endothelial cells, as well as degradation of the basement membrane. After the re-establishment of cell junctions and coverage of pericytes, the newly formed vessels mature and remain quiescent (1). Importantly, endothelial cells start proliferating to initiate angiogenesis only after stimulation by several growth factors and inflammatory mediators, mainly members of vascular endothelial growth factor (VEGF) family and angiopoietins.
Angiogenesis and tumor promoting inflammation are closely interconnected and are crucial events during cancer progression and metastasis; in fact, they are both considered as hallmarks of cancer (2). Both tumor and stromal cells produce angiogenic factors that ultimately lead to endothelial cell proliferation. These cancer-associated endothelial cells cooperate in the perpetuation of inflammation, which reciprocally promotes angiogenesis by secretion of cytokines, proteases, growth and proangiogenic factors, thus creating a positive feedback loop (1, 2). Aberrant vascular structures can also induce hypoxia, acidosis and DNA damage (3), which contribute to the establishment of a tumor-promoting microenvironment since very early in the carcinogenesis. This could be particularly relevant in cancers in which chronic and persistent inflammation is the main driving force for the malignant transformation, for example in gastric carcinogenesis.
VEGFA is the most potent and ubiquitous member of VEGF family; it is considered as the master inductor of physiological and tumor angiogenesis (4). Secreted by macrophages, lymphocytes, fibroblast and tumor cells, it promotes proliferation, migration and survival of endothelial cells, as well as expression of extracellular matrix proteases (5). Angiopoietin 1 (ANGPT1) and ANGPT2 bind to TIE1-TIE2 (tyrosine kinase with immunoglobulin-like and epidermal growth factor–like domains 1 and 2) receptor complex and α5β1 integrin in endothelial cells, thus activating signaling pathways that lead to blood and lymphatic vessel formation. ANGPT1 is secreted by pericytes and acts in a paracrine manner to keep homeostasis of the mature vasculature. ANGPT2 is mostly produced by endothelial cells, in which it is stored at the cytoplasmatic Weibel-Palade bodies, and released after stimulation, to act in an autocrine way (6). ANGPT2 plays a role as a vessel destabilizing agent that induces permeability and leads to dissociation of cell-cell contacts, allowing the sprouting of new vessels (1, 7). The expression of ANGPT2 in normal tissues is low or absent, but it is upregulated in many cancers in which it is mainly expressed by tumor-associated macrophages (6). In humans, alternative splicing generates a smaller isoform, ANGPT2443, which has been reported as upregulated in tumor cell lines and human tumor tissue (8, 9).
TNF-α is a key mediator in a host’s response against gram-negative bacteria, such as H. pylori. It is produced by lymphocytes and macrophages and promotes leukocyte recruitment. In the context of angiogenesis, TNF-α primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype (10).
Helicobacter pylori colonization of the gastric mucosa induces chronic and persistent inflammation. This is associated with several clinical outcomes, such as peptic ulcer, MALT-lymphoma and gastric cancer (GC). The latter results from a stepwise cascade of preneoplastic lesions known as Correa’s cascade (11, 12). The persistent inflammation mounted in response to this bacterial infection is characterized by the infiltration in the gastric mucosa of neutrophils, lymphocytes and macrophages. Increased permeability of the endothelium is a pivotal event for the extravasation of immune cells, proteins and fluids (13).
Several reports indicate that H. pylori can induce the production of proangiogenic factors in vitro and in vivo (14–18). Specifically, the expression of HIF-1a, VEFGA and the density of CD31+ blood vessels is higher in H. pylori-positive patients, compared to negative persons (19, 20). Those findings suggest that the bacterium is capable of inducing a proangiogenic response after colonizing its host. This could be of pivotal importance during gastric cancer initiation and development. However, the molecules and mechanisms by which H. pylori induces early neovascularization in gastric mucosa, and how this influences the precancerous series of events that precede GC is not completely understood.
The present study, we used a mouse model in order to characterize the expression kinetics of Tnf-A, Angpt1, Angpt2 and VegfA in the gastric mucosa in response to H. pylori infection and to explore the correlation to inflammation, vascularization and gastric pathology.
Thirty-nine C57BL/6N female mice certified as Helicobacter spp-free were acquired from Harlan Laboratories® and housed at the Laboratorio de Ensayos Biológicos (LEBi), University of Costa Rica, at standard temperature (25.5°C ± 1.20°C) and humidity (50–70%), and maintained on a 12-h light/dark cycle (lights off at 6:00 p.m). Water and food (LabDiet 5010.) were provided ad libitum. Mice were distributed into 7 experimental groups, each consisting of 5-7 animals per cage (Figure 1) and were moved to an experimental section one week before they were inoculated with H. pylori. Experimental procedures and methods were carried out in accordance with the guidelines of the Costa Rican Ministry of Science and Technology and were approved by the Institutional Committee for Animal Care and Use of Animals (CICUA) of the University of Costa Rica (permission number CICUA-031-17).
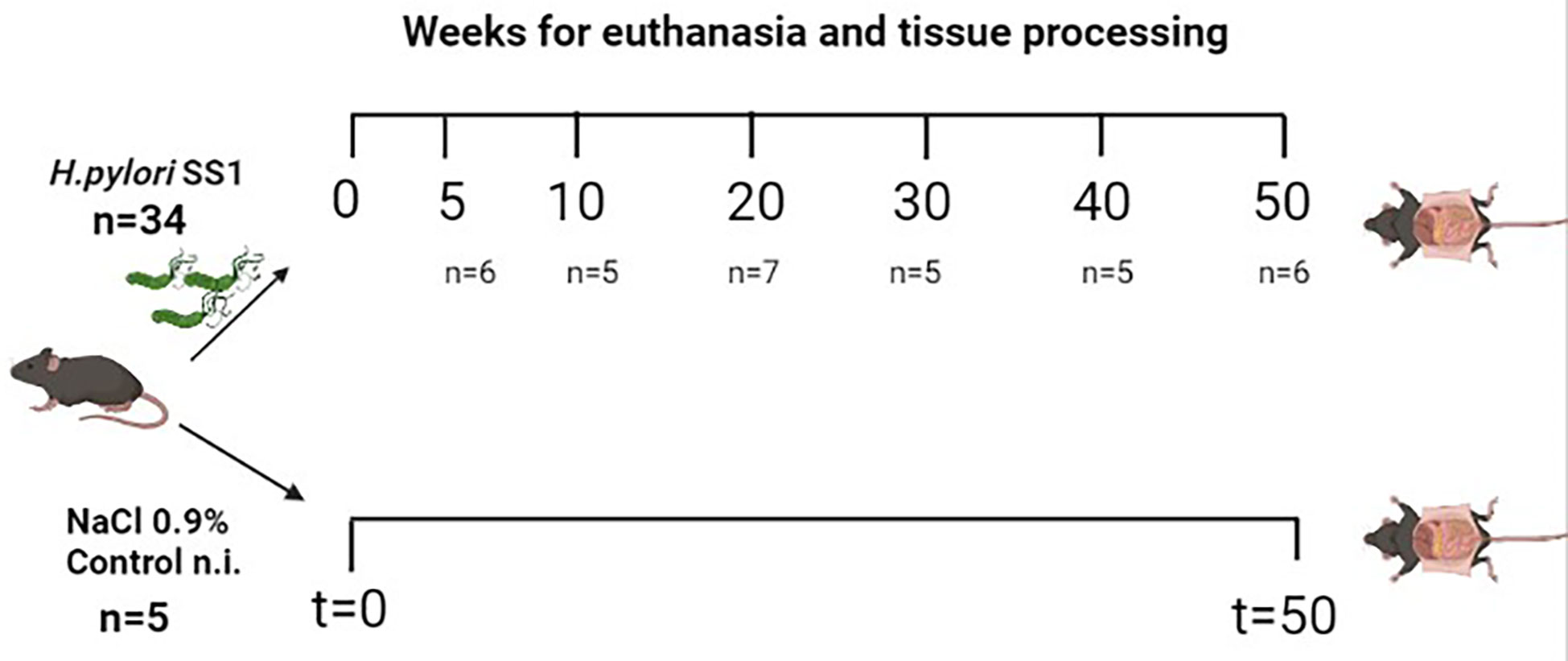
Figure 1 Schematic view of the experimental groups of the mouse model of Helicobacter pylori SS1 infection used in the study. A total of 39 female C57BL/6N mice were divided into 7 groups (n per group indicated between parentheses), 6 of these groups were infected with H. pylori and one group received NaCl 0.9% only (control group, n=5). Mice were euthanized between 5-50 w.p.i. Control non-infected mice were sacrificed at 50 w.p.i. Created with BioRender.com.
The inoculation was performed with SS1 strain (mouse-adapted cagA+, vacA+ strain with no functionality of the cag pathogenicity island; kindly donated by Dr. James Fox, MIT, USA). Bacteria were grown on Skirrow plates and incubated at 37°C for 5-7 days under microaerophilic conditions. The SS1 strain was harvested and mice were inoculated by oral gavage with a single dose of approximately 109 bacteria (CFU) in 0.2 mL of 0.9% NaCl. Control mice received 0.2 mL of the saline solution only. The bacteria used for inoculations were isolated from previously colonized mice from our bioterium.
Infected groups were euthanized by cervical dislocation after 5-, 10-, 20-, 30-, 40- and 50- weeks post-infection (w.p.i.). The control group was euthanized at 50 w.p.i. Entire stomachs were opened along the greater curvature, washed with PBS, and cut longitudinally into four stripes, extending from the squamous forestomach through the duodenum. The stripes were used as follows: one for confirmation of bacterial colonization (culture), one was cut in halves for RNA and protein isolation, and two for immunohistochemistry assays.
The stripe was macerated with saline solution, vortexed 5 s, then 40 μl of the suspension was seeded in both Skirrow and agar-blood BHI plates and incubated at 37°C for 5-7 days under microaerophilic conditions.
The stomach tissue was suspended in RNAlater (Invitrogen) until utilization. Total RNA was isolated with Trizol (Invitrogen) in accordance with the manufacturer’s instructions. RNA from each sample was transcribed into cDNA using High-capacity cDNA reverse transcription kit (ThermoFisher Scientific). A total of 0,5 μl cDNA was used for qRT-PCR amplification using TaqMan Gene Expression Assays (Thermo Fisher Scientific) for Angpt1 (Mm00456503_m1), Angpt2 (Mm00545822_m1), Tnf-A (Mm00443258_m1) and VegfA (Mm00437306_m1) with Gapdh (Mm99999915_g1) as endogenous control, using the StepOne Plus thermal cycler (Applied Biosystems, Foster City, CA, USA) under the following conditions: 95°C for 10 sec, 95°C for 5 sec, and 60°C for 30 sec, run for 40 cycles. Fold-difference for infected vs. non infected mice was estimated by the 2 -ΔΔCt method.
Stomach tissues were macerated and lysed using lysis buffer solution (PBS 1X, 1% Triton X-100, 1% NP-40, proteases and phosphatases cocktail inhibitors, pH 7.4). Following centrifugation at 14 000 g for 30 min, the proteins were quantified using the Protein Assay Dye Reagent Concentrate (BioRad) and 50 µg of protein were separated by 10% SDS−PAGE, prior to being transferred onto a nitrocellulose membrane (BioRad). Following blocking with 5% fat−free milk in PBS buffer in 0.05% Tween 20 for 1 h at room temperature, the membranes were then separately incubated overnight at 4˚C with the following monoclonal antibodies: Rabbit ANGPT1 (1:500; Abcam ab102015), Rabbit ANGPT2 (1:500, Abcam ab8452) and Rabbit GAPDH (1:1000, Abcam, ab9485). The secondary antibodies were applied (dilution 1:10 000, horseradish peroxidase-conjugated anti-rabbit, Sigma-Aldrich A9169) at room temperature for 1 h. Labeled bands were detected by enhanced chemiluminescence (Clarity ECL, BioRad, USA) and analyzed by Chemidoc Imaging System (Bio-Rad). Protein levels were quantified using the Image Lab software, version 4.1 (Bio-Rad, USA).
The stinging protocols were a modification of Alpízar-Alpízar et al. (21). Two stomach stripes were fixed 24 hours in 4% paraformaldehyde and paraffin embedded. Then, 4 μm tissue sections were deparaffinized in xylene and hydrated in a gradual series of ethanol-water dilutions. For H. pylori, sections were pretreated with Proteinase K (10 μg/mL) for 15 min, 37°C. For CD31, Angpt1, Angpt2 and VegfA, sections were pretreated at 98°C for 15 min in 10 mM sodium citrate pH 6.0. In all cases, endogenous peroxidase activity was blocked by incubation in 3% H2O2 solution for 15 min. The primary antibodies were diluted in Antibody Diluent (Dako, code S3022) and incubated overnight at 4°C in Shandon racks (Thermo Shandon, Pittsburg, PA, USA) at the following dilutions: Rabbit-anti H. pylori 1:150 (Dako, code B0471), Rabbit anti-Angpt1 1:500, Rabbit anti-Angpt2 1:1000, Rabbit anti-CD31 1:250, Rabbit anti-VegfA 1:200. All primary antibodies were detected with EnVision reagent anti-rabbit IgG horseradish peroxidase-conjugated polymers (Dako Code: K3468). Each incubation step was followed by washes in TBS containing 0.5% (v/v) Triton X-100. Finally, the reactions were visualized by incubating the sections with Liquid DAB+ substrate chromogen system (Dako, Code S3022) and counterstained with Mayer’s hematoxylin.
H. pylori colonization was evaluated according to the number of gastric glands containing bacteria and their density, as follows: 0: No observed bacteria; 1: Occasional pits and/or glands with individual bacteria; 2: frequent pits and/or glands with individual bacteria; 3, infrequent pits and/or gland with dense bacterial colonies; and 4, frequent pits and/or glands with dense bacterial colonies (22). Sections were stained with hematoxylin and eosin (H&E) for histopathological evaluations of the mouse gastric mucosa describing inflammation, infiltration, metaplasia, and anatomical localization of the lesions, using published guidelines (23): 0, no infiltration, 1, patchy or multifocal small islands of inflammatory cells in the mucosa and/or submucosa; 2, coalescing aggregates of inflammatory cells in submucosa or mucosa; 3, organizing nodules of lymphocytes and other inflammatory cells in submucosa and mucosa; 4, follicles and/or sheets of inflammatory cells extending into or through muscularis propria adventitia. Photomicrographs were captured using the bright field microscope Motic BA400 and Motic Images Plus 3.0 software (China).
Data for gene expression is represented as the mean ± standard deviation for every experimental group. Data for inflammation and H. pylori scores are represented with median values. The association between variables on an ordinal scale is presented by the Spearman rank correlation. Experimental data were compared among the groups using the Kruskal-Wallis test. p-values less than 0.05 were considered significant. ***p < 0.001. Data presented as mean ± SD of each experimental group. All data were analyzed using the GraphPad Prism 9 software.
In the present study, we used a mouse model of H. pylori-induced gastritis to explore the relationship between the bacterium and the induction of proangiogenic and inflammatory mediators in vivo. We evaluated the histopathological changes and lesions induced by the H. pylori infection in the groups of challenged and unchallenged mice. Non infected mice showed a normal mucosal architecture (Figure 2A). Five weeks after colonization, H. pylori-infected mice showed scattered immune cells infiltrating mucosa and submucosa, at antrum, corpus and the squamo-columnar transition. Two of the animals showed patches of mucous metaplasia in corpus (Figure 2B). Ten weeks after challenge, mice showed minor immune cell infiltration at the mucosa, especially at the squamo-columnar and corpus-antrum transitions, with some mucous metaplastic patches (Figure 2C). Twenty-week-colonized mice showed minor immune cell foci in antral mucosa, at the base of the corpus glands and squamo-columnar transition (not shown). Thirty weeks after inoculation, mice presented a stronger and widespread infiltration of immune cells in mucosa and submucosa of corpus and corpus-antrum transition, with hyperplasia and multiple foci of mucous metaplasia (Figure 2D). Forty- and fifty-week challenged animals also showed an abundant and extensive immune cell infiltration in mucosa and submucosa of the corpus, with large areas of hyperplasia and mucous metaplasia (Figures 2E, F).
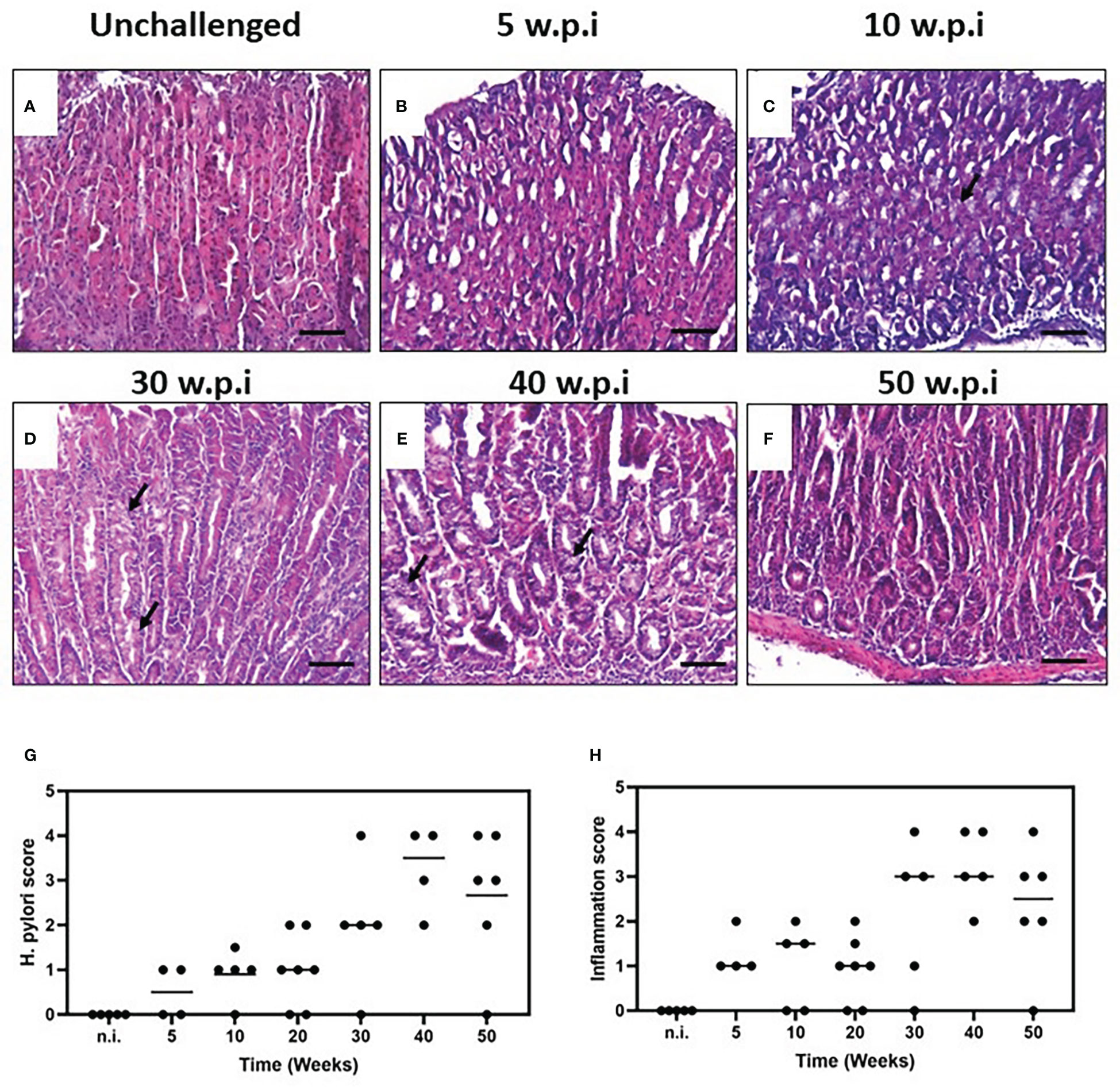
Figure 2 Histopathology of the murine gastric corpus mucosa in unchallenged and H. pylori-challenged mice. Tissue sections from resected gastric mucosa of unchallenged and H. pylori-inoculated mice were stained by H&E. (A) Unchallenged mice show a normal mucosal architecture. (B) Five weeks after H. pylori inoculation, minor multifocal infiltration of immune cells is observed. (C) Minor infiltration and some metaplastic patches are observed ten weeks after challenge. (D) The pathology and infiltration are intensified after thirty weeks of infection, with hyperplasia and multiple foci of mucous metaplasia. (E) Animals infected for forty and fifty (F) weeks also showed an extensive infiltration of immune cells with areas of hyperplasia and metaplasia (black arrows). H. pylori colonization status and histopathological assessment of inflammation. (G) H. pylori score at different time-points. (H). Inflammation score of the murine gastric mucosa with H. pylori infection at different time-points (scored according to the scheme proposed by Wang et al. (22) and Rogers (23), one experiment; n = 5-7 mice per point; black line represents median values). Formalin-fixed, paraffin-embedded gastric tissue were stained with hematoxylin and eosin and examined. Scale bars: 50 μm (20x).
We assessed the H. pylori colonization status by culture and immunohistochemistry for each animal and found H. pylori-positive mice in all time points. In accordance with the bacterial score, the most robust infiltration of immune cells and more severe lesions were found at 30, 40 and 50 w.p.i (Figures 2G, H), p=0.0187).
We assessed the expression at the mRNA and protein level of several proangiogenic factors in the gastric mucosa in our mouse model. In the H. pylori-infected animals Tnf-A, VegfA and Angpt2 mRNAs normalized to Gapdh showed an increased expression in time, showing the lowest ΔCt values at 30 to 50 w.p.i (Figures 3A–C). When comparing unchallenged animals with the 50 w.p.i. group, a significant upregulation was observed for Tnf-A, VegfA and Angpt2 mRNAs expression (Figures 3E–G)
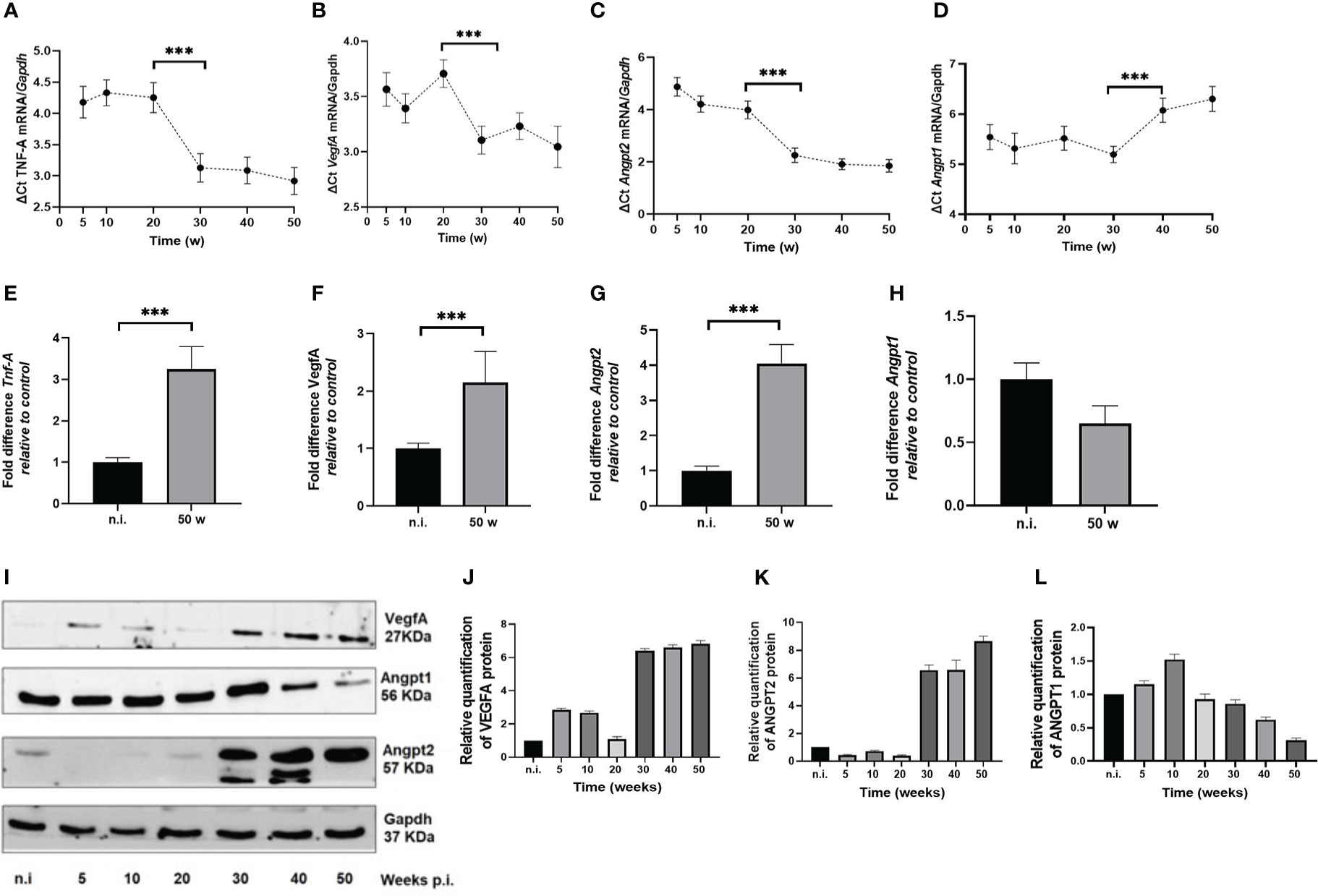
Figure 3 H. pylori-SS1 upregulates the expression of pro-angiogenic factors in murine gastric mucosa. mRNA expression was analyzed by real-time PCR, and represented as Delta (Δ) Ct values. The higher ΔCt values represents the lower expression of genes. (A) Tnf-A, (B) VegfA and (C) Angpt2 mRNAs show a concomitant upregulation in time, becoming more evident from 30 w.p.i, whereas (D) Angpt1 mRNA is downregulated at 40 and 50 w.p.i. When comparing not infected animals with the 50 w.p.i. group, a significant upregulation was observed for (E) Tnf-A, (F) VegfA and (G) Angpt2 mRNAs expression, but a significant deregulation for (H) Angpt1mRNA. (I-L) Western blot and concordant relative quantification of VegfA, Angpt1 and Angpt2 proteins in mouse gastric mucosa of H. pylori-SS1 infected and uninfected mice are shown. VegfA and Angpt2 are upregulated and Angpt1 is downregulated in time. Data presented as mean ± SD of each experimental group. ***p < 0.001.
In H. pylori-infected mice from 5 to 20 w.p.i., there was a slight increment that reached its top level of expression at 30 w.p.i., but at 40 and 50 w.p.i. the mRNA levels were drastically downregulated (Figure 3D). At 50 w.p.i Angpt1 mRNA levels were significantly different from the not infected animals (Figure 3H). When analyzing the expression of Tnf-A, VegfA, Angpt2 and Angpt1 altogether, it is worth nothing that Tnf-A, Vegf-A and Angpt2 mRNAs showed a significant upregulation starting at 30 w.p.i, that coincides with the downregulation of Angpt1 at the 40 and 50 w.p.i. This is important since all these molecules act in concert to trigger the angiogenic switch.
The Western blot analysis showed an upregulation of VegfA and Angpt2 proteins at 30 to 50 w.p.i (Figures 3F, G, I). In contrast, Angpt1 protein was downregulated (Figures 4F, H). Of note, two bands for Angpt2 protein with weights between 50-57 KDa, were detected in 30 and 40 w.p.i mice. This was not observed at 50 w.p.i. (Figure 3F).

Figure 4 H. pylori upregulates Angpt2 expression in murine gastric mucosa. (A, B) IHC staining in blood vessels is positive (brown color) for Angpt1 and negative for Angpt2 in unchallenged mice. (C, D) IHC staining of Angpt1 compared to Angpt2 in lamina propria and blood vessels of infected mice 50 w.p.i. IHC staining with Angpt2 in infected mice 30 w.p.i. compared to 50 w.p.i. mice. Scale bars: (A, B, E, G) ≈ 50 μm; (C, D, F, H) ≈ 100 μm.
We also performed immunohistochemistry to assess the expression pattern and localization of Angpt1 and Angpt2 in murine tissue. H. pylori infection induced Angpt2 expression in gastric epithelial cells. Unchallenged mice showed a strong staining for Angpt1 in blood vessels, but Angpt2 was not detected (Figures 4A, B). Angpt2 immunoreactivity was observed in gastric mucosa of infected mice, with an intense expression in metaplastic areas at the corpus mucosa in 30- and 50-w.p.i. challenged mice (Figures 4D–G), but no signal for Angpt1 (Figure 4C).
Pathological angiogenesis is a process that takes place during chronic inflammation and cancer. It involves activation of endothelial and immune cells that secrete pro-angiogenic factors such as VEGF-A, ANGPT2, TNF-α and MMPs, thus triggering proliferation of quiescent endothelium to make new blood vessels. This has been intensively studied in several cancer types, including GC. Although some evidence from in vitro models and studies in human gastric mucosa suggest that H. pylori infection may be involved in the induction of pro-angiogenic factors as VEGFA and HIF-1 (17–19, 24–26), to the best of our knowledge, no studies have addressed the role of H. pylori infection in the induction of proangiogenic proteins as Angpt1 and Angpt2 in the gastric mucosa. Therefore, we used a mouse model of H. pylori-induced gastritis to explore the kinetics of the expression of Angpt1, Angpt2, Tnf-α and Vegf-A in vivo.
We found a significant upregulation of Angpt2 mRNA and protein, whereas Angpt1 mRNA was downregulated. The ANGPT2/ANGPT1 balance determines the fate of the endothelium. ANGPT2 has opposing role to ANGPT1 since it promotes blood vessel wall destabilization. This is achieved by competitively inhibiting the binding of ANGPT1 to Tie-2 and reducing Tie-2 activation and phosphorylation (1). ANGPT2 is produced by endothelial cells and macrophages, and plays a key role in the promotion of vessel sprouting, pericyte detachment and basement membrane remodeling. ANGPT1 is produced by pericytes and is associated with endothelium quiescence. ANGPT2/ANGPT1 imbalance parallels capillary destabilization; as the inflammatory response mounted against H. pylori infection persists, activated endothelial and infiltrating immune cells produce increasing ANGPT2 levels while ANGPT1 is downregulated, ultimately leading to vessel destabilization (1, 6).
In mouse and human, the Angpt2/ANGPT2 gene encodes a 496 aa protein of 57 KDa. Hypoxia is an inducer of alternative splicing in cancer and endothelial cells that frequently display a temporal and tissue-specific expression (27). In humans, alternative splicing generates the ANGPT2443 isoform lacking exon 2, which produces a smaller (443 aa) 51 KDa protein that is found in activated endothelial cells and macrophages. Reports indicate that functions as a more potent competitive inhibitor of ANGPT1 (8) and is upregulated in cancer cell lines, breast tumor tissues, and canine adrenocortical tumor tissue (27, 28). In the performed Western blots we observed the presence of two bands with weights between 50-57 KDa for the infected mice of groups of 30 and 40 w.p.i. (Figure 3F), using an antibody (ab8452, Abcam) predicted to recognize both isoforms of Angpt2. We hypothesize that the observed bands could be Angpt2443. Nevertheless, as there are no previous reports for the expression of the isoform Angpt2443 in mice in normal or pathological conditions, further transcriptomic and protein assays are necessary to check whether the observation of the present study corresponds to that isoform.
We observed a concomitant upregulation of Tnf-A, Vegf-A, Angpt2 mRNAs at 30, 40 and 50 w.p.i. In vitro studies have revealed that TNF-α upregulates the expression of VEGF-A, ANGPT2, and Tie2, and deregulates ANGPT1 genes at mRNA level (29, 30). After translation, the ANGPT2 protein is stored in Weibel-Palade bodies at endothelial cells. The main factor causing its secretion is TNF-α, which ultimately results in autocrine inhibition of the Tie-2 receptor (31). TNF-α is mainly produced by activated macrophages and T lymphocytes, and is a potent activator of endothelium by inducing vasodilatation, increase of vascular permeability and the recruitment of immune cells (32). TNF-α expression is substantially elevated in response to H. pylori infection thus inhibiting gastric acid secretion, which in turn facilitates H. pylori survival (32). TNF-A transcription is induced by nuclear factor κappa B (NF-κB), which is activated in response H. pylori infection in gastric epithelial cells in vitro and in vivo (15, 33, 34), and upregulates VEGFR in endothelial cells (16).
Besides its role in cancer progression, angiogenesis plays a major role in the multi-step carcinogenesis process due to the fact that the gastric mucosa undergoes important histological changes, which may require gaining access to the vasculature in order to receive an adequate supply of nutrients and oxygen (35). Tuccillo et al. (36) found elevated levels of VEGFA in H. pylori-associated human gastritis. VEGFA and iNOS levels are also high in patients with chronic atrophic gastritis as well as in metaplastic and dysplastic areas (37). These observations, and ours, support the evidence pointing to the role of H. pylori as a promotor of angiogenesis.
The normal gastric epithelium does not express angiogenic factors; those are secreted by endothelial and some immune cells under stimulus. Several in vitro studies, including our own observations (manuscript submitted), show that in co-cultures H. pylori induces the expression of ANGPT2, VEGFA and other angiogenesis-related factors in gastric adenocarcinoma cell lines (17, 26, 38). Atrophic, hyperplastic, metaplastic or dysplastic cells in H. pylori-infected gastric mucosa have accumulated genetic and epigenetic alterations that may change their gene expression programs thus leading to alteration in their phenotypes (39). In this context, it could be possible that the gastric epithelial cells themselves secrete some angiogenic factors, in response to signals from the inflammatory microenvironment, bacterial virulence factors (e.g. CagA, VacA, LPS, urease), or both (Figure 5). In the present study we challenged mice with H. pylori SS1, which has a nonfunctional CagA protein. Therefore, the mechanisms by which the bacterium induces angiogenic responses may be CagA-independent. Other virulence factor, such as urease, may be implicated since it has been demonstrated that this enzyme induces ANGPT2 and other angiogenic factors in human gastric cell lines (17).
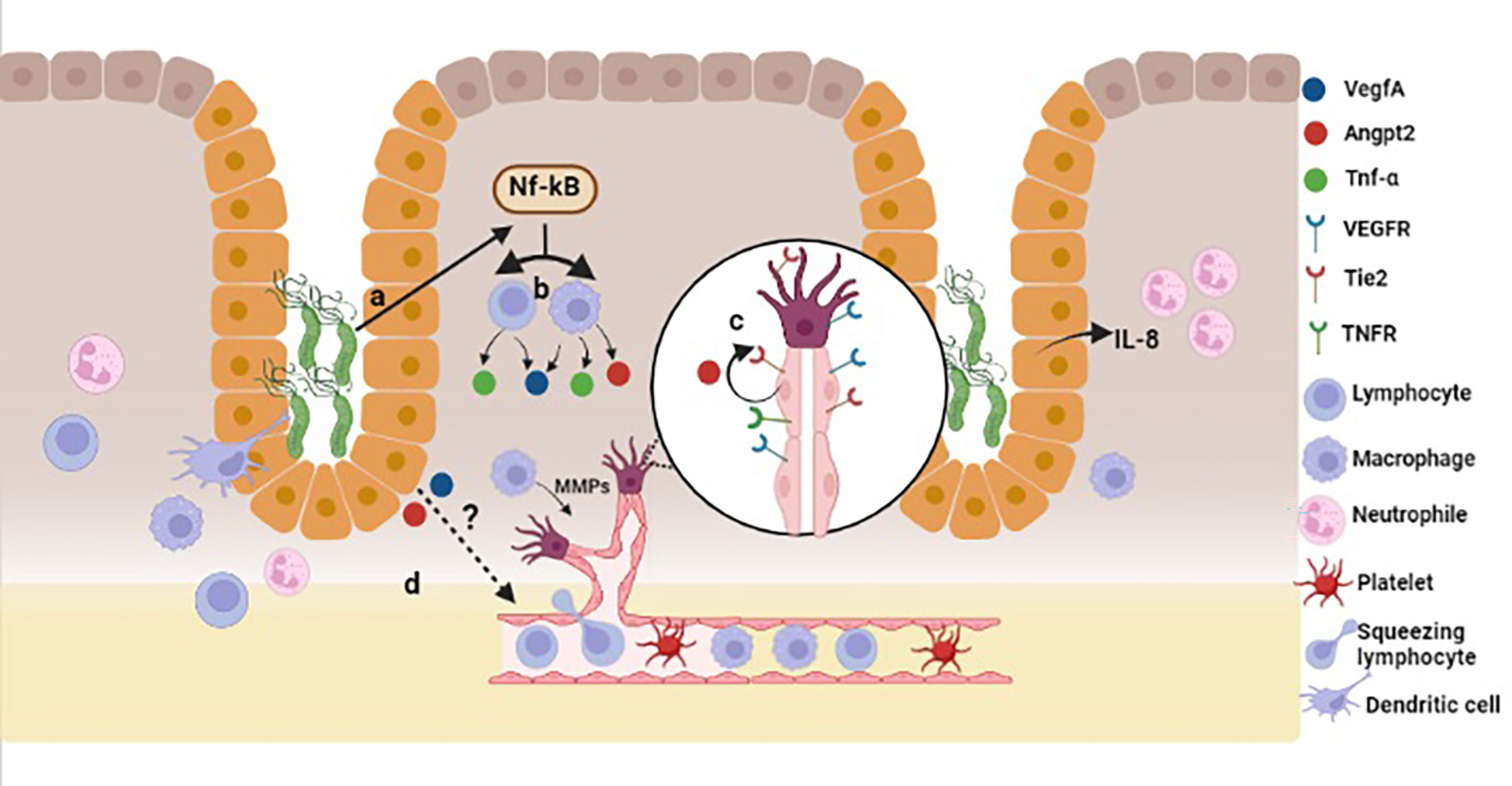
Figure 5 Model for H. pylori-induced angiogenesis in gastric mucosa. In a CagA-independent pathway, H. pylori infection may trigger the activation of NF-κB (A), which in turn induces TNF-α production (B) in lymphocytes and macrophages. TNF-α activates endothelial cells, which promotes Angpt2 release (C) from Weibel-Palade bodies. H. pylori induces the expression of Angpt2 and VegfA from epithelial cells in a CagA-independent pathway, and also the activation and proliferation of endothelial cells (D). Created with BioRender.com.
In conclusion, in this study we have found that H. pylori infection concurrently modulates the expression of several angiogenic mediators in the murine stomach. Specifically, this bacterial infection induces the expression of Angpt2, Tnf-A and VegfA at the mRNA and protein levels. Concomitantly, it downregulates Angpt1 expression in mouse gastric mucosa in a time-dependent manner. Nevertheless, our study has two major limitations; first, we had only one group of non-infected mice for the experiment that was euthanized at 50 weeks, but no control groups for early time points of infected animals. Hence, we show only the evolution of the expression in infected animals, and the comparison with the control group is shown only for the 50 w.p.i. mice. Second; the presented evidence describes the role of H. pylori in the modulation of the studied angiogenic factors, therefore, subsequent mechanistic and functional studies are necessary to establish the impact to H. pylori infection to angiogenesis in vivo.
Datasets are available on request: The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Committee for Animal Care and Use of Animals (CICUA) of the University of Costa Rica (permission number CICUA-031-17).
VR-M, WA-A, WM-B, CU, SM-C and LR designed the study. WM-B, WA-A and LF-P performed the experiments. WM-B, WA-A, VR and SM-C analyzed the data. WM-B, VR and WA-A interpreted the data. WM-B, WA-A and VR wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by: Vicerrectoría de Investigación, Universidad de Costa Rica, Consejo Nacional para Investigaciones Científicas y Tecnológicas (CONICIT, Ministerio de Ciencia y Tecnología, Costa Rica).
Authors wish to thank Dr. James Fox (MIT, USA) for the kind gift of H. pylori SS1, and Elena Vásquez, Eugenio Calvo, Mariana Piedra and Ricardo Martínez for excellent assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fiedler U, Augustin HG. Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol (2006) 27(12):552–8. doi: 10.1016/j.it.2006.10.004
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
3. Kitadai Y. Angiogenesis and lymphangiogenesis of gastric cancer. J Oncol (2010) 2010:468725. doi: 10.1155/2010/468725
4. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell (2011) 146(6):873–87. doi: 10.1016/j.cell.2011.08.039
5. Ucuzian AA, Gassman AA, East AT, Greisler HP. Molecular mediators of angiogenesis. J Burn Care Res (2010) 31(1):158–75. doi: 10.1097/BCR.0b013e3181c7ed82
6. Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discovery (2017) 16(9):635–61. doi: 10.1038/nrd.2016.278
7. Huang H, Bhat A, Woodnutt G, Lappe R. Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer (2010) 10(8):575–85. doi: 10.1038/nrc2894
8. Kim I, Kim JH, Ryu YS, Jung SH, Nah JJ, Koh GY. Characterization and expression of a novel alternatively spliced human angiopoietin-2. J Biol Chem (2000) 275(24):18550–6. doi: 10.1074/jbc.M910084199
9. Kapiainen E, Kihlström MK, Pietilä R, Kaakinen M, Ronkainen VP, Tu H, et al. The amino-terminal oligomerization domain of angiopoietin-2 affects vascular remodeling, mammary gland tumor growth, and lung metastasis in mice. Cancer Res (2021) 81(1):129–43. doi: 10.1158/0008-5472.CAN-19-1904
10. Sainson RC, Johnston DA, Chu HC, Holderfield MT, Nakatsu MN, Crampton SP, et al. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood (2008) 111(10):4997–5007. doi: 10.1182/blood-2007-08-108597
11. Atherton JC. The pathogenesis of helicobacter pylori-induced gastro-duodenal diseases. Annu Rev Pathol (2006) 1:63–96. doi: 10.1146/annurev.pathol.1.110304.100125
12. Correa P, Houghton J. Carcinogenesis of helicobacter pylori. Gastroenterology (2007) 133(2):659–72. doi: 10.1053/j.gastro.2007.06.026
13. Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest (2007) 117(1):60–9. doi: 10.1172/JCI30111
14. Strowski MZ, Cramer T, Schäfer G, Jüttner S, Walduck A, Schipani E, et al. Helicobacter pylori stimulates host vascular endothelial growth factor-a (vegf-a) gene expression via MEK/ERK-dependent activation of Sp1 and Sp3. FASEB J (2004) 18(1):218–20. doi: 10.1096/fj.03-0055fje
15. Wu CY, Wang CJ, Tseng CC, Chen HP, Wu MS, Lin JT, et al. Helicobacter pylori promote gastric cancer cells invasion through a NF-kappaB and COX-2-mediated pathway. World J Gastroenterol (2005) 11(21):3197–203. doi: 10.3748/wjg.v11.i21.3197
16. de Jesus Souza M, de Moraes JA, Da Silva VN, Helal-Neto E, Uberti AF, Scopel-Guerra A, et al. Helicobacter pylori urease induces pro-inflammatory effects and differentiation of human endothelial cells: Cellular and molecular mechanism. Helicobacter (2019) 24(3):e12573. doi: 10.1111/hel.12573
17. Olivera-Severo D, Uberti AF, Marques MS, Pinto MT, Gomez-Lazaro M, Figueiredo C, et al. A new role for helicobacter pylori urease: Contributions to angiogenesis. Front Microbiol (2017) 8:1883. doi: 10.3389/fmicb.2017.01883
18. Pousa ID, Gisbert JP. Gastric angiogenesis and helicobacter pylori infection. Rev Esp Enferm Dig (2006) 98(7):527–41. doi: 10.4321/S1130-01082006000700006
19. Yeo M, Kim DK, Han SU, Lee JE, Kim YB, Cho YK, et al. Novel action of gastric proton pump inhibitor on suppression of helicobacter pylori induced angiogenesis. Gut (2006) 55(1):26–33. doi: 10.1136/gut.2005.067454
20. Lazaraki G, Kountouras J, Metallidis S, Vrettou E, Tzioufa V, Germanidis G, et al. Helicobacter pylori infection upregulates endothelial nitric oxide synthase expression and induces angiogenesis in gastric mucosa of dyspeptic patients. Eur J Gastroenterol Hepatol (2008) 20(5):441–9. doi: 10.1097/MEG.0b013e3282f4c35a
21. Alpízar-Alpízar W, Skindersoe ME, Rasmussen L, Kriegbaum MC, Christensen IJ, Lund IK, et al. Colonization drives urokinase receptor (uPAR) expression in murine gastric epithelium during early pathogenesis. Microorganisms (2020) 8(7):1019. doi: 10.3390/microorganisms8071019
22. Wang TC, Goldenring JR, Dangler C, Ito S, Mueller A, Jeon WK, et al. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with helicobacter felis infection. Gastroenterology (1998) 114(4):675–89. doi: 10.1016/S0016-5085(98)70581-5
23. Rogers AB. Histologic scoring of gastritis and gastric cancer in mouse models. Methods Mol Biol (2012) 921:189–203. doi: 10.1007/978-1-62703-005-2_22
24. Kitadai Y, Sasaki A, Ito M, Tanaka S, Oue N, Yasui W, et al. Helicobacter pylori infection influences expression of genes related to angiogenesis and invasion in human gastric carcinoma cells. Biochem Biophys Res Commun (2003) 311(4):809–14. doi: 10.1016/j.bbrc.2003.10.077
25. Liu N, Zhou N, Chai N, Liu X, Jiang H, Wu Q, et al. Helicobacter pylori promotes angiogenesis depending on wnt/beta-catenin-mediated vascular endothelial growth factor via the cyclooxygenase-2 pathway in gastric cancer. BMC Cancer (2016) 16:321. doi: 10.1186/s12885-016-2351-9
26. Yamac D, Ayyildiz T, Coşkun U, Akyürek N, Dursun A, Seckin S, et al. Cyclooxygenase-2 expression and its association with angiogenesis, helicobacter pylori, and clinicopathologic characteristics of gastric carcinoma. Pathol Res Pract (2008) 204(8):527–36. doi: 10.1016/j.prp.2008.01.002
27. Di Matteo A, Belloni E, Pradella D, Cappelletto A, Volf N, Zacchigna S, et al. Alternative splicing in endothelial cells: novel therapeutic opportunities in cancer angiogenesis. J Exp Clin Cancer Res (2020) 39(1):275. doi: 10.1186/s13046-020-01753-1
28. Kool MM, Galac S, Kooistra HS, Mol JA. Expression of angiogenesis-related genes in canine cortisol-secreting adrenocortical tumors. Domest Anim Endocrinol (2014) 47:73–82. doi: 10.1016/j.domaniend.2013.11.004
29. Yuan J, Fang W, Lin A, Ni Z, Qian J. Angiopoietin-2/Tie2 signaling involved in TNF-α induced peritoneal angiogenesis. Int J Artif Organs (2012) 35(9):655–62. doi: 10.1177/039139881203500905
30. Zhang B, Khalaf H, Sirsjö A, Bengtsson T. Gingipains from the periodontal pathogen porphyromonas gingivalis play a significant role in regulation of angiopoietin 1 and angiopoietin 2 in human aortic smooth muscle cells. Infect Immun (2015) 83(11):4256–65. doi: 10.1128/IAI.00498-15
31. Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med (2006) 12(2):235–9. doi: 10.1038/nm1351
32. Zambon CF, Basso D, Navaglia F, Belluco C, Falda A, Fogar P, et al. Pro- and anti-inflammatory cytokines gene polymorphisms and helicobacter pylori infection: interactions influence outcome. Cytokine (2005) 29(4):141–52. doi: 10.1016/j.cyto.2004.10.013
33. Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-kappa b in gastric epithelial cells. Gastroenterology (1997) 113(4):1099–109. doi: 10.1053/gast.1997.v113.pm9322504
34. Maeda S, Yoshida H, Ogura K, Mitsuno Y, Hirata Y, Yamaji Y, et al. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology (2000) 119(1):97–108. doi: 10.1053/gast.2000.8540
35. Papetti M, Herman IM. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol (2002) 282(5):C947–70. doi: 10.1152/ajpcell.00389.2001
36. Tuccillo C, Cuomo A, Rocco A, Martinelli E, Staibano S, Mascolo M, et al. Vascular endothelial growth factor and neo-angiogenesis in h. pylori gastritis humans J Pathol (2005) 207(3):277–84. doi: 10.1002/path.1844
37. Feng CW, Wang LD, Jiao LH, Liu B, Zheng S, Xie XJ. Expression of p53, inducible nitric oxide synthase and vascular endothelial growth factor in gastric precancerous and cancerous lesions: correlation with clinical features. BMC Cancer (2002) 2:8. doi: 10.1186/1471-2407-2-8
38. Fu YG, Sung JJ, Wu KC, Bai AH, Chan MC, Yu J, et al. Inhibition of gastric cancer cells associated angiogenesis by 15d-prostaglandin J2 through the downregulation of angiopoietin-1. Cancer Lett (2006) 243(2):246–54. doi: 10.1016/j.canlet.2005.11.039
Keywords: Helicobacter pylori, angiogenesis, ANGPT1, ANGPT2, VEGFA, mouse model, TNF-A
Citation: Malespín-Bendaña W, Alpízar-Alpízar W, Figueroa-Protti L, Reyes L, Molina-Castro S, Une C and Ramírez-Mayorga V (2023) Helicobacter pylori infection induces gastric precancerous lesions and persistent expression of Angpt2, Vegf-A and Tnf-A in a mouse model. Front. Oncol. 13:1072802. doi: 10.3389/fonc.2023.1072802
Received: 17 October 2022; Accepted: 26 January 2023;
Published: 15 February 2023.
Edited by:
Javier Torres, Mexican Social Security Institute (IMSS), MexicoReviewed by:
Pirjo Spuul, Tallinn University of Technology, EstoniaCopyright © 2023 Malespín-Bendaña, Alpízar-Alpízar, Figueroa-Protti, Reyes, Molina-Castro, Une and Ramírez-Mayorga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wendy Malespín-Bendaña, d2VuZHkubWFsZXNwaW5AdWNyLmFjLmNy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.