 Essam AlShail1†Ahmed Nasser Alahmari1†Anas A. M. Dababo2†Maysoon Alsagob3,4†
Essam AlShail1†Ahmed Nasser Alahmari1†Anas A. M. Dababo2†Maysoon Alsagob3,4† Hindi Al-Hindi2Hala Khalil5Zainab Al Masseri6
Hindi Al-Hindi2Hala Khalil5Zainab Al Masseri6 Razan AlSalamah3Ethar Almohseny3
Razan AlSalamah3Ethar Almohseny3 Amjad Alduhaish7
Amjad Alduhaish7 Dilek Colak8*‡
Dilek Colak8*‡ Namik Kaya3*‡
Namik Kaya3*‡- 1Department of Neurosciences, King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Saudi Arabia
- 2Department of Pathology and Laboratory Medicine, King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Saudi Arabia
- 3Department of Translational Genomics, Center for Genomic Medicine, King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Saudi Arabia
- 4Applied Genomics Technologies Institute, King Abdulaziz City for Science and Technology, Riyadh, Saudi Arabia
- 5Department of Biostatistics, Epidemiology and Scientific Computing, King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Saudi Arabia
- 6Medical Genetics Department, King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Saudi Arabia
- 7Neuroscience Department, King Abdullah Medical City, Mecca, Saudi Arabia
- 8Department of Molecular Oncology, King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Saudi Arabia
Background: Pilocytic Astrocytoma (PA) is the most common pediatric brain tumors. PAs are slow-growing tumors with high survival rates. However, a distinct subgroup of tumors defined as pilomyxoid astrocytoma (PMA) presents unique histological characteristics and have more aggressive clinical course. The studies on genetics of PMA are scarce.
Methods: In this study, we report one of the largest cohort of pediatric patients with pilomyxoid (PMA) and pilocytic astrocytomas (PA) in Saudi population providing a comprehensive clinical picture, retrospective analysis with long-term follow-up, genome-wide copy number changes, and clinical outcome of these pediatric tumors. We examined and compared genome-wide copy number aberrations (CNAs) and the clinical outcome of the patients with PA and PMA.
Results: The median progression free survival for the whole cohort was 156 months and it was 111 months for the PMA, however, not statistically significantly different between the groups (log-rank test, P = 0.726). We have identified 41 CNAs (34 gains and 7 losses) in all tested patients. Our study yielded the previously reported KIAA1549-BRAF Fusion gene in over 88% of the tested patients (89% and 80% in PMA and PA, respectively). Besides the fusion gene, twelve patients had additional genomic CNAs. Furthermore, pathway and gene network analyses of genes in the fusion region revealed alterations in retinoic acid mediated apoptosis and MAPK signaling pathways and key hub genes that may potentially be involved in tumor growth and progression, including BRAF, LUC7L2, MKRN1, RICTOR, TP53, HIPK2, HNF4A, POU5F, and SOX4.
Conclusion: Our study is the first report of a large cohort of patients with PMA and PA in the Saudi population that provides detailed clinical features, genomic copy number changes, and outcome of these pediatric tumors and may help better diagnosis and characterization of PMA.
Introduction
Brain and other central nervous system related tumors are the largest group of childhood cancers and the leading cause of cancer-related death in children. Among these, pilocytic astrocytomas (PAs) are the most frequently encountered tumors with peak incidence in the first decade of life (1, 2). Auspiciously, PAs are not aggressive, typically slow growing brain tumors and have a much better prognosis compared to other more aggressive neoplasms. These tumors are considered being grade I tumors according to the WHO (World Health Organization), and progression to higher grades is quite unusual. Patients with PA have high survival rates and only a small percentage of the patients demonstrate a variable clinical course with decreased length of disease-free survival and higher mortality rates (3). However, a distinct subtype of tumors termed pilomyxoid astrocytomas (PMAs) displays more aggressive clinical course and higher recurrence rate (4). The PMAs prefer the optic-chiasmatic/hypothalamic region, however, there are several studies reporting PMAs in several other locations (5). In addition, some tumors present transitional morphological features with combined pilocytic and pilomyxoid features (mixed) (3). The PMAs were previously considered as WHO grade II tumor (4, 5).
Previous reports have shown that about two thirds of PAs display a recurrent gain at 7q34 (6, 7). This leads to gain of BRAF, with subsequent BRAF activation. Another study indicated that low-grade gliomas have distinct copy number gains at chr6q23 (8). Interestingly, genetic changes may vary among PA, PMA, and IPT (intermediate pilomyxoid tumors). For instance, among 15 patients only 3 had KIAA1549-BRAF-fusion and 2 had BRAFV600E-mutation whereas six patients had wild type BRAF (9). Similarly, it was reported that there was significant difference in the gene expression patterns between PA vs PMA (10). Compared to the general knowledge on PAs, very little is known about the molecular characteristics of PMAs. The molecular mechanisms that may explain PMA’s more aggressive behavior are still lacking. In this study, we examined and compared the clinical features and outcome in a large cohort of the PA and PMA patients, majority of which were PMAs. We also delineated genetic characteristics using genomic arrays and identified copy number changes associated with PMA and PA.
Materials and methods
Tumor samples
We performed a retrospective chart review study of selected patients (n=27) who were histologically determined to have pilomyxoid (PMA) and pilocytic astrocytomas (PA) who were diagnosed between November 1998 and March 2015 at King Faisal Specialist Hospital and Research Centre (KFSHRC). The patients’ charts were reviewed until March 07, 2021. The study was performed under a locally approved IRB (King Faisal Specialist Hospital and Research Centre, Research Advisory Council, Basic Research as well as Ethics Committees, RAC# 2111055) and fulfilled the principles of the Helsinki Declaration, 2013. The histopathological assessment and determination of the tumors were done by neuropathologists at KFSHRC based on microscopic, morphological, and immunohistochemical characteristics of the tumors (5, 11, 12). Among the 27 patients, 17 of them were cytogenetically examined using OncoScan arrays.
DNA isolation
Samples were obtained from the FFPE blocks as several microtome shaves; each is around 5 to 6 μm thick. DNA was extracted using QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) or similar kits based on the Affymetrix’s recommended protocols. DNA was quantified via two different systems Quant-iT (Thermo Fisher, Waltham, Massachusetts, US) and Bioanalyzer (Agilent Technologies, Santa Clara, US). The quality and quantity of the DNA samples were evaluated by assessing the results from both systems.
Genechip OncoScan arrays
OncoScan arrays and related assays were used for detection of chromosomal abnormalities. The array workflow is based on company’s protocols (Affymetrix, Santa Clara, CA, US). The FFPE DNA sample preparation, array hybridization, as well as subsequent reactions such as amplification and labeling were strictly followed according to the assay user guide (P/N 703175). The labeled samples were hybridized to the arrays, washed and scanned using Affymetrix scanner. The data was analyzed using Affymetrix’s Chromosome Analysis Suite (ChAS) using the default settings (high resolution) of the chromosomal abnormality detection.
PCR
We utilized some of the remaining DNA from FFPE samples during DNA amplification. PCR was carried out using the Qiagen HotStar Tag (Life Technologies, USA) according to standard protocols. The products were visually inspected by agarose gel electrophoresis. Afterwards, PCR amplicons were sequenced using the BigDye Terminator reaction mix and run on the Sequencing equipment (Applied Biosystems’ 3730 Sequence Analyzer, Thermo Fisher).
Sanger sequencing
To determine the absence or presence of several known mutations Sanger sequencing (SS) was utilized. Particular mutations, including hotspot mutations in TP53, PIK3CA, KRAS, EGFR, PTEN, BRAF, IDH1, and NRAS, were picked up (Supplementary Table 1) as these are already targeted by OncoScan arrays using molecular inversion probes (MIP). The Sanger sequencing (SS) was carried out as a confirmatory approach to interrogate these mutations. Gene/mutation specific primers (GSP) were used in the PCRs. These primers were modified for SS (M13 universal primer sites were added to the GSP and each primer was used in separate reactions for SS).
Functional pathway and network analysis
We performed gene network and functional analysis using Ingenuity Pathways Analysis (IPA) (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis). We also performed gene ontology (GO) enrichment analysis using Database for Annotation, Visualization and Integrated Discovery (DAVID) (13) and Protein ANalysis Through Evolutionary Relationships (PANTHER™) classification systems (14). The genes affected by copy number changes were mapped in the Ingenuity pathway knowledge base. A right-tailed Fisher’s exact test was used to calculate a p-value determining the probability that the biological function (or pathway) assigned to that data set is explained by chance alone.
Statistical analysis
Comparison of the patients’ characteristics by tumor type (PMA, PA, and others) was performed using non-parametric Kruskal- Wallis test for continuous variables and Fisher’s exact test for categorical variables. Kaplan–Meier survival curves for each tumor type were generated to estimate the progression-free time over the observation period, followed by log-rank test to assess differences between the groups. Statistical analysis was performed using IBM SPSS Statistics Version 26.0. All statistical tests were two-sided and p-value < 0.05 was considered statistically significant.
Results
Clinical features of pilomyxoid and pilocytic astrocytoma patients
We first retrospectively reviewed the clinical and tumor characteristics of 27 pediatric patients who were diagnosed with PMA, PA, angiocentric glioma (AG), and others (intermediate tumor with anaplastic features (IAT). Some representative histopathological images of pilocytic astrocytoma (PA) and pilomyxoid astrocytoma (PMA) are shown in Figure 1A. The median follow-ups for PMA and PA were 60 months and 99 months, respectively. The most common location for PMA was suprasellar area (67%). The clinical and tumor characteristics of the patient cohort that we studied are summarized in Table 1.
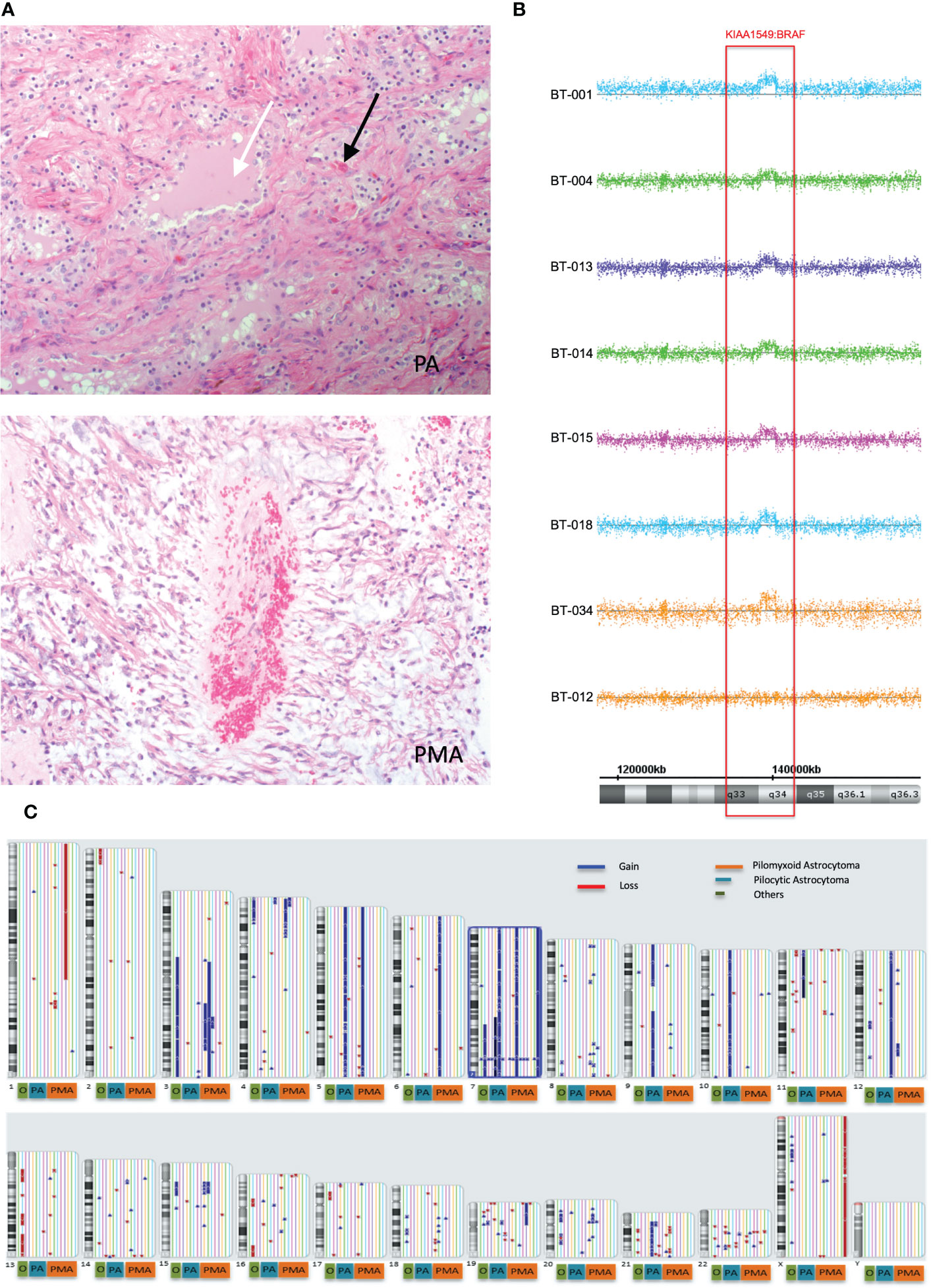
Figure 1 Global copy number analyses of pediatric pilomyxoid and pilocystic astrocytomas. (A) Representative histopathological images of pilocytic astrocytoma (PA) and pilomyxoid astrocytoma (PMA) (original magnification, 200×). The PA shows biphasic pattern with microcystic changes (white arrow) and Rosenthal fibers (black arrows) and the PMA shows perivascular arrangement with myxoid background (bluish color) and the mononmorphic piloid cells without Rosenthal fibers (B) Global copy number analyses of PMA samples, displaying the KIAA1549:BRAF fusion gene. Each sample is color coded. The baseline is represented as a straight line while the region consisting the fusion gene is pointed out by raised probes above the baseline (indicating the gain). The red block covers the region containing the fusion gene/gain. Below the figure, schematic chromosome bands and coordinates on chromosome 7 are illustrated. (C) Global copy number analyses results for all samples tested (PMA, PA and others). Each chromosome and related cytobands are represented in separate boxes. Next to each chromosome displays all samples with different colors. The samples are ordered as PMA, PA, and “others” from right to left next to the chromosome number; with orange, blue and red boxes, respectively, underneath the respective samples. The gains and losses are indicated as blue and red lines/arrow heads, respectively.
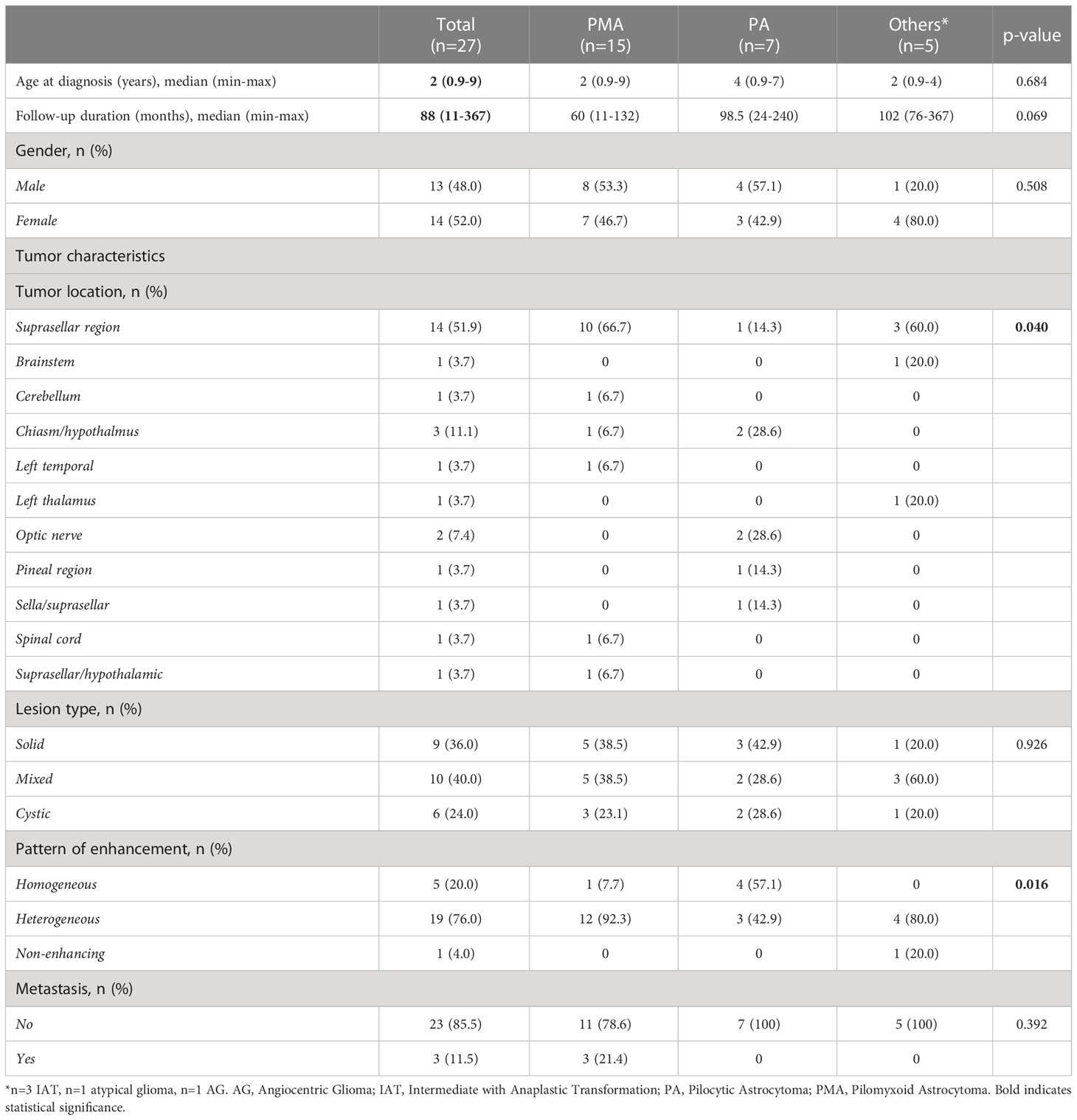
Table 1 Characteristics of patients and tumors by tumor type.
All patients underwent surgeries, with earliest PMA surgery performed in 1998 and all other surgeries were performed from 1998 through 2015, that included unilateral subfrontal, bilateral subfrontal, transcortical transventricular, pterional, temporal or combined subfrontal and pterional approaches. Three-quarters of our patients underwent debulking or subtotal resection (Table 2). Twelve PMA patients (86%) received chemotherapy. Twenty-one patients (80%) received chemotherapy with low-grade glioma protocol consisting of vincristine and carboplatin. The Temozolamide was used in recurrent or resistant cases. Radiation treatment was used for 10 patients (38%). There was only one mortality in a patient with atypical glioma who died after two weeks due to rapid progression of the disease. Three patients had metastases (one to CSF), and all were PMAs.
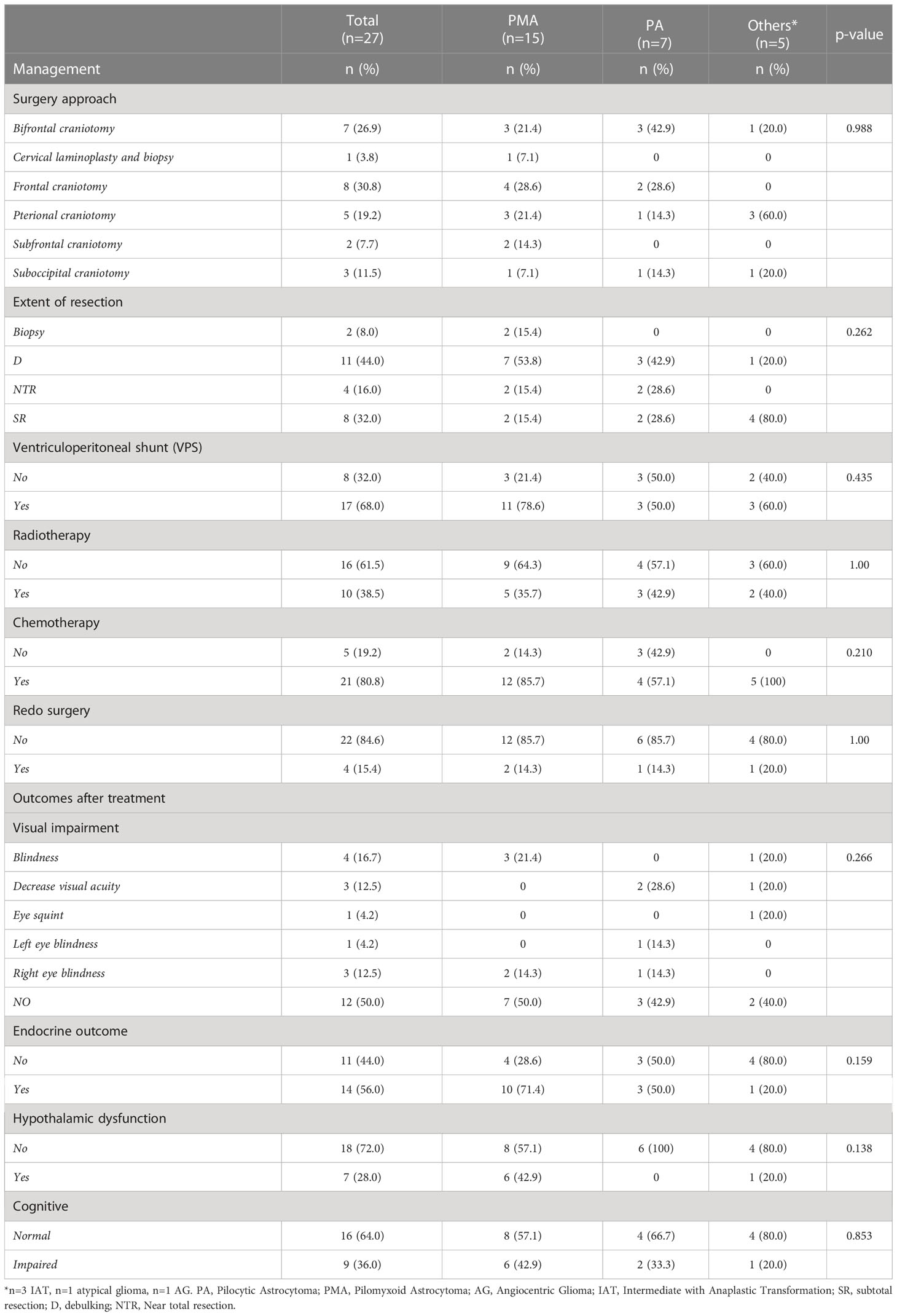
Table 2 Management and outcomes after treatment by tumor type.
Treatment related morbidities among the cohort included visual impairment leading to visual field defect or blindness in 50% (n=12), endocrine deficits in 56% (n=14), such as cortisol deficiency, hypothyroidism, growth hormone deficiency, diabetes insipidus, or panhypopituitarism as well as cognitive impairment in 36% (n=9), such as memory loss or learning difficulties (Table 2). The median progression free survival (PFS) for the whole cohort was 156 months and for PMA it was 111 months that was not statistically significantly different between the groups (log-rank test, P=0.726).
Genomewide copy number analysis of PA and PMAs
We next employed OncoScan® CNV assays in order to identify genome-wide chromosomal aberrations in our 27 tumor samples. The data was analyzed using Affymetrix’s Chromosome Analysis Suite (ChAS) using the default settings. The OncoScan® assays use molecular inversion probes (MIP) for the identification of genomic copy number alterations, loss of heterozygosity (LOH), and somatic mutations. It targets genome-wide chromosomal aberrations with one of the highest resolutions provided by microarrays. The interrogating MIP probes are 40 base pairs and extensively wet-validated using archived FFPE samples of major solid tumor types. It also utilizes MIPs for hotspot mutations in nine genes implicated in cancer (BRAF, KRAS, EGFR, IDH1, IDH2, PTEN, PIK3CA, NRAS and TP53). In our cohort, none of the targeted mutations on the arrays was positively detected. We also performed Sanger sequencing (SS) using standard PCR primers targeting the same mutations (Supplementary Table 1), that also confirmed the OncoScan results.
Genome-wide copy number aberration (CNA) analyses revealed 41 CNAs (34 gains (CNGs) and 7 losses (CNLs)) in all tested patients (n=17) (Figures 1B, C; Table 3). All nine PMAs harbored 20 CNAs (17 gains and 3 losses) and five PAs have 11 CNAs (all gains). Our study yielded the previously reported gain on chromosome 7q34 in over 88% of our patient cohort (89% and 80% in PMA and PA, respectively). The duplication has been previously reported in PA and known as “KIAA1549:BRAF fusion gene” (15, 16). Nearly all samples have similar proximal and distal end breakpoints starting from KIAA1549 gene and reaching to BRAF (Figure 1B). The duplication outspreads nearly 1.87Mb region and comprises 9 pseudogenes, 9 uncharacterized genes, and 10 genes with OMIM ID including KIAA1549 and BRAF at the breakpoints. Other genes that may also have importance in disease pathogenesis include MKRN1, LUC7L2, UBN2, DENND2A, SLC37A3, PARP12, TBXAS1, HIPK2, and C7orf55 (Figure 2).
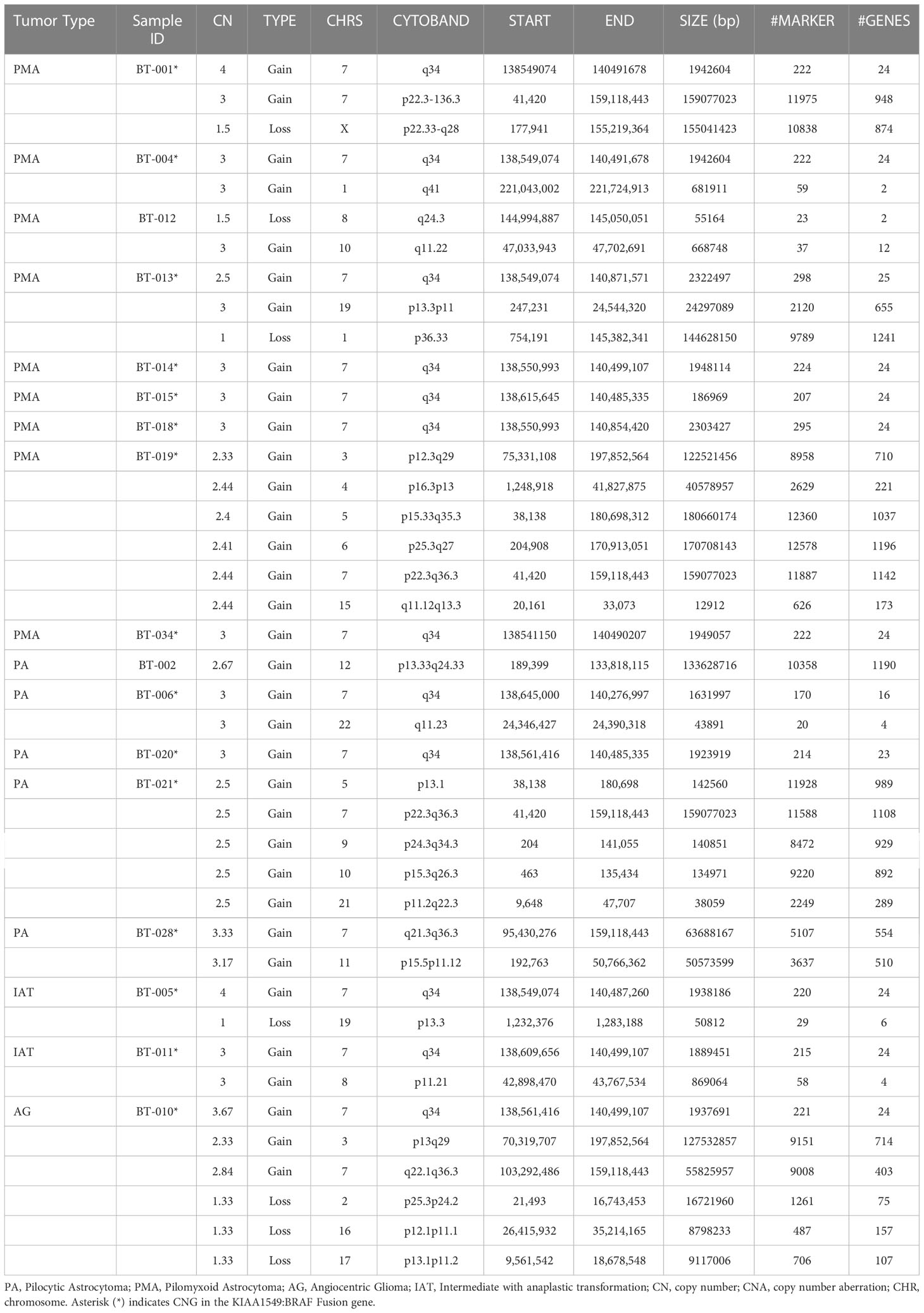
Table 3 Genomic copy number alterations (CNAs) in PA and PMA. CNAs identified in PA/PMA (archived as FFPE) samples using OncoScan arrays.
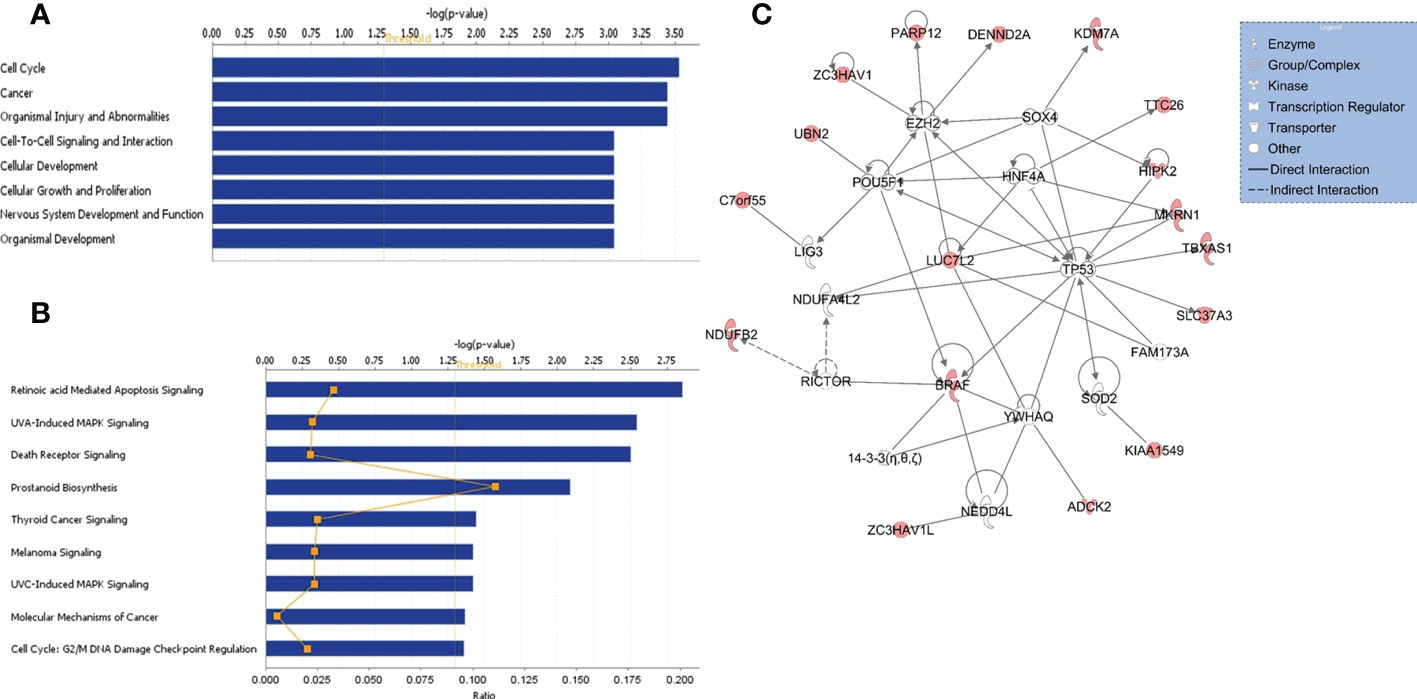
Figure 2 Functional/pathway and network analyses of genes in KIAA1549:BRAF fusion. Enriched biological functions (A) and canonical pathways (B) that are associated with the genes in the fusion region. The significance of function and pathway are shown in X-axis. The P value of 0.05 is indicated as the threshold line. (C) Gene interaction network analysis. Nodes represent genes, with their shape indicating the functional class of the gene product (see legend). The edges indicate biological relationship between the genes, with straight and dashed lines representing direct or indirect gene to gene interactions, respectively. The red color indicate genes in the fusion.
Besides the fusion gene, twelve patients have additional genomic CNAs (Table 3). Briefly, five patients have either partial (n=2) or full (n=3) trisomy of chromosome 7 (Figure S1). Interestingly, only two (BT-001 and BT-010) harbored tetrasomy of the fusion gene region (Figure S2). The partial trisomies in BT-10 and BT-28 are large and cover majority of the q arm. BT-013 (PMA) has a deletion on chromosome 1 (1p36.33q21.1) (Figure S3) and a duplication on chromosome 19 (19p13.3p11) (Figure S4). BT-021 has various large gains of chromosomes 5, 7, 9, 10, and 21 (Figure S5).
Functional, pathway, and gene network analyses
Functional, pathway, and gene network analyses of genes within the KIAA1549:BRAF fusion region were performed using several bioinformatics tools, including DAVID (13) and IPA (QIAGEN Inc.) that indicated enrichment in cancer, cell cycle, cellular development, and nervous system development and function, and significant alterations in retinoic acid mediated apoptosis signaling, MAPK and death receptor signaling pathways (all p values < 0.01) (Figures 2A, B). Furthermore, gene interaction network analysis revealed hub genes that may potentially be involved in PA/PMA, including BRAF, LUC7L2, TP53, MKRN1, RICTOR, HIPK2, HNF4A, POU5F, and SOX4 (Figure 2C).
Discussion
Though PMA shares similar molecular features with PA, the commonest central nervous system (CNS) tumor, however, it presents more aggressive course and higher grade. Histologically, in contrast to PA, PMA does not exhibit calcification, Rosenthal fibers, and eosinophilic granule bodies (4). It mostly appears with angiocentric pattern with small and compact monomorphous piloid cells in myxoid matrix-like background. Although PMA is well characterized histologically, and similarities and differences with PA are recognized at the histological level, the studies on PMAs and differentiating both tumor types genetically are scarce. In this study, we gathered one of the largest cohorts of PA and PMA in Saudi population and investigated genomic copy number changes and outcome of these pediatric tumors.
In one of the earliest studies, Forshew et al. identified the fusion in 23 out of 26 PA samples as well as in singleton PMA (17). Gains on chromosome 5 and 6 were also detected in a few samples. Interestingly, both fusions lack the auto-inhibitory domains of BRAF and RAF1 that were interchanged by in-frame start units of the KIAA1549 and SRGAP3. This replacement creates a constitutive kinase activity. We provided a summary of the previous studies of PAs/PMAs and the identified CNAs using different array platforms (Supplementary Table 2).
We used Affymetrix’s OncoScan arrays that have MIPs targeting selected hot spot mutations in cancers as well as global copy number changes. Interestingly, these arrays did not identify any hot spot mutations in our patient cohort. However, we were able to identify 41 CNAs, including 34 CNGs and 7 CNLs in 17 patients tested (Table 3). Nine PMA samples harbor 20 CNAs (17 CNGs and 3 CNLs) and five PAs have 11 CNGs. Over 88% our patient cohort (89% in PMA and 80% in PA) have the well-defined BRAF fusion. Previous pioneering array studies, mostly done on PA tumors, indicated involvement of KIAA1549-BRAF fusion gene in low-grade brain tumors (15, 16). Alternative approaches such as RNAseq and utilization of droplet-PCR also indicated similar findings in PA (18). Such fusion is predicted to occur owing to tandem duplication events happening especially during homologous recombination of Alu elements (19–21). A large series of pilocytic astrocytoma tumors were screened for likely copy number changes using a custom designed arrays constructed with clones obtained from the Wellcome Trust Sanger Institute. The analysis indicated that 29 samples had duplication between clones RP11-355D18 and RP11-543P6 on chromosome 7. Further analysis confirmed the presence of a newly formed fusion gene between intron 16 of KIAA1549 and intron 8 of BRAF. The fusion gene derives few different transcripts but essentially the fusions retain BRAF’s kinase domain (15). Under normal circumstances, BRAF’s kinase activity is regulated by NH2-terminal region that includes Ras-binding domain and is truncated in the new fusion. Once such truncation removes NH2-terminal of the BRAF, then regulation of fusion gene is not under autoregulation of wild type NH2-terminal but rather it is controlled by newly generated mutant NH2-terminal. Besides the fusion gene, twelve patients in our study had additional genomic CNAs (Table 3).
As over 88% of the samples (15 out of 17) had the gain in KIAA1549-BRAF fusion gene in our study, we investigated the gene interaction network analysis of genes in this region. The analysis indicated alterations in retinoic acid mediated apoptosis signaling, MAPK and death receptor signaling pathways and hub genes, including BRAF, LUC7L2, MKRN1, TP53, HNF4A, RICTOR, POU5F, HIPK2, and SOX4 that may have potentially important role in tumorigenesis and cancer progression (2, 16, 22–25).
This study has potential limitations. We had a relatively small sample size of patients in each comparison group (PA, PMA, and other) and the analysis is based on a retrospective design; hence may have inherent bias. Moreover, though the KFSHRC is a referral hospital and our cohort included local as well as referred patients within Saudi Arabia, the results obtained in this study may not be generalizable to the whole population. Despite these limitations, this study is the first comprehensive study of pediatric patients with PMA and PA in Saudi population providing genome-wide copy number screening, retrospective analysis of clinicopathological features and long-term clinical outcome of these pediatric tumors. In conclusion, our study reports the first comprehensive molecular and clinicopathological analyses of one of the largest cohorts of PMA/PA samples to date in a previously unstudied population to understand the clinical features and genomic makeup of these solid types of tumors and hence may help better diagnosis and characterization of these tumors.
Data availability statement
The dataset generated and analyzed during the current study is available at this link (https://www.dropbox.com/s/fg2vo8ebiwbc8th/Affy-Oncoscan-All-PMA-Data.rar?dl=0).
Ethics statement
This study involved in using archived FFPE samples and was performed under IRB-approved protocols at King Faisal Specialist Hospital and Research Center (KFSHRC’s Research Advisory Council Committees including Basic Research Committee and Research Ethics Committee, RAC# 2111055). All patients were de-identified to the research team and hence the study was exempted from collecting written informed consent for their participation accordance with the institutional requirements.
Author contributions
NK conceived and designed the experiments. NK, EAS, and DC drafted the manuscript. NK, DC, MA, and HK performed the data analyses. AA and MA involved in reviewing and editing manuscript. HA-H and AD review the pathology reports and performed pathology related tests. EAS, HA-H, AD, ANA, AA, EAM, and ZA reviewed the charts, collected clinical data, and prepared the tables related to clinical information. MA and RA performed the experiments. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by King Faisal Specialist Hospital and Research Centre’s seed grants (KFSHRC-RAC# 2120022) for NK and EAS and also for DC (RAC# 2110 006). This research was conducted through intramural funds provided by King Faisal Specialist Hospital and Research Center (KFSHRC, RAC Project # 2111055).
Acknowledgments
We extend our special thanks to KFSHRC Genotyping and Sequencing Core Facilities, and Purchasing Departments for facilitating and expediting our requests. We thank Ibrahim H. Kaya for preparing scientific figures. One of the authors of this study, Dr. Anwar Ul-Haq, passed away due to COVID-19, and we would like to dedicate this work to his memory.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1034292/full#supplementary-material
Abbreviations
PA, Pilocytic Astrocytoma; PMA, Pilomyxoid Astrocytoma; AG, Angiocentric Glioma; FG, Fusion gene referring to KIAA1549:BRAF; IAT, Intermediate with anaplastic transformation; CNA, Copy number aberration; CNG, Copy number gain; CNL, Copy number loss; DAVID, Database for Annotation, Visualization and Integrated Discovery; SR, Subtotal resection; D,Debulking; NTR, Near total resection; IPA, Ingenuity Pathways Analysis.
References
1. Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol (2005) 64:479–89. doi: 10.1093/jnen/64.6.479
2. Roth JJ, Santi M, Rorke-Adams LB, Harding BN, Busse TM, Tooke LS, et al. Diagnostic application of high resolution single nucleotide polymorphism array analysis for children with brain tumors. Cancer Genet (2014) 207:111–23. doi: 10.1016/j.cancergen.2014.03.002
3. Kulac I, Tihan T. Pilomyxoid astrocytomas: a short review. Brain tumor Pathol (2019) 36:52–5. doi: 10.1007/s10014-019-00343-0
4. Ding C, Tihan T. Recent progress in the pathology and genetics of pilocytic and pilomyxoid astrocytomas. Balkan Med J (2019) 36:3–11. doi: 10.4274/balkanmedj.2018.1001
5. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (2007) 114:97–109. doi: 10.1007/s00401-007-0243-4
6. Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol (2008) 67:878–87. doi: 10.1097/NEN.0b013e3181845622
7. Taha H, Yehia M, Mahmoud M, El-Beltagy M, Ghabriel M, El-Naggar S. Incidence of kiaa1549-braf fusion gene in Egyptian pediatric low grade glioma. Clin Transl Med (2015) 4:10. doi: 10.1186/s40169-015-0052-7
8. Tatevossian RG, Tang B, Dalton J, Forshew T, Lawson AR, Ma J, et al. MYB upregulation and genetic aberrations in a subset of pediatric low-grade gliomas. Acta Neuropathol (2010) 120:731–43. doi: 10.1007/s00401-010-0763-1
9. Fadel SA, Von Reppert M, Kazarian E, Omay EZE, Marks A, Linder N, et al. Spectrum of qualitative and quantitative imaging of pilomyxoid, intermediate pilomyxoid and pilocytic astrocytomas in relation to their genetic alterations. Neuroradiology. (2022). doi: 10.1007/s00234-022-03027-3
10. Kleinschmidt-Demasters BK, Donson AM, Vogel H, Foreman NK. Pilomyxoid astrocytoma (PMA) shows significant differences in gene expression vs. pilocytic astrocytoma (PA) and variable tendency toward maturation to PA. Brain Pathol (2015) 25:429–40. doi: 10.1111/bpa.12239
11. Komotar RJ, Mocco J, Carson BS, Sughrue ME, Zacharia BE, Sisti AC, et al. Pilomyxoid astrocytoma: a review. MedGenMed (2004) 6:42.
12. Komotar RJ, Mocco J, Jones JE, Zacharia BE, Tihan T, Feldstein NA, et al. Pilomyxoid astrocytoma: diagnosis, prognosis, and management. Neurosurg Focus (2005) 18:E7. doi: 10.3171/foc.2005.18.6.8
13. Sherman BT, Huang DW, Tan Q, Guo Y, Bour S, Liu D, et al. DAVID knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinf (2007) 8:426. doi: 10.1186/1471-2105-8-426
14. Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res (2003) 31:334–41. doi: 10.1093/nar/gkg115
15. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res (2008) 68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097
16. Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene (2009) 28:2119–23. doi: 10.1038/onc.2009.73
17. Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol (2009) 218:172–81. doi: 10.1002/path.2558
18. Appay R, Fina F, Macagno N, Padovani L, Colin C, Barets D, et al. Duplications of KIAA1549 and BRAF screening by droplet digital PCR from formalin-fixed paraffin-embedded DNA is an accurate alternative for KIAA1549-BRAF fusion detection in pilocytic astrocytomas. Mod Pathol (2018) 31:1490–501. doi: 10.1038/s41379-018-0050-6
19. Kolomietz E, Meyn MS, Pandita A, Squire JA. The role of alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer (2002) 35:97–112. doi: 10.1002/gcc.10111
20. Olesen LH, Aggerholm A, Andersen BL, Nyvold CG, Guldberg P, Norgaard JM, et al. Molecular typing of adult acute myeloid leukaemia: significance of translocations, tandem duplications, methylation, and selective gene expression profiling. Br J Haematol (2005) 131:457–67. doi: 10.1111/j.1365-2141.2005.05791.x
21. O'neil J, Tchinda J, Gutierrez A, Moreau L, Maser RS, Wong KK, et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. J Exp Med (2007) 204:3059–66. doi: 10.1084/jem.20071637
22. Gkountakos A, Pilotto S, Mafficini A, Vicentini C, Simbolo M, Milella M, et al. Unmasking the impact of rictor in cancer: novel insights of mTORC2 complex. Carcinogenesis (2018) 39:971–80. doi: 10.1093/carcin/bgy086
23. Chen P, Duan X, Li X, Li J, Ba Q, Wang H. HIPK2 suppresses tumor growth and progression of hepatocellular carcinoma through promoting the degradation of HIF-1alpha. Oncogene (2020) 39:2863–76. doi: 10.1038/s41388-020-1190-y
24. Basheer AS, Abas F, Othman I, Naidu R. Role of inflammatory mediators, macrophages, and neutrophils in glioma maintenance and progression: Mechanistic understanding and potential therapeutic applications. Cancers (Basel) (2021) 13. doi: 10.3390/cancers13164226
Keywords: pilomyxoid astrocytoma, BRAF, KIAA1549, copy number alteration, pilocytic astrocytoma, genomics, brain tumor, gene network
Citation: AlShail E, Alahmari AN, Dababo AAM, Alsagob M, Al-Hindi H, Khalil H, Al Masseri Z, AlSalamah R, Almohseny E, Alduhaish A, Colak D and Kaya N (2023) A molecular study of pediatric pilomyxoid and pilocytic astrocytomas: Genome-wide copy number screening, retrospective analysis of clinicopathological features and long-term clinical outcome. Front. Oncol. 13:1034292. doi: 10.3389/fonc.2023.1034292
Received: 01 September 2022; Accepted: 13 January 2023;
Published: 13 February 2023.
Edited by:
Jing He, Guangzhou Medical University, ChinaReviewed by:
Prerana Jha, All India Institute of Medical Sciences, IndiaAndrew Michael Donson, University of Colorado Denver, United States
Markus Bookland, Connecticut Children’s Medical Center, United States
Copyright © 2023 AlShail, Alahmari, Dababo, Alsagob, Al-Hindi, Khalil, Al Masseri, AlSalamah, Almohseny, Alduhaish, Colak and Kaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Namik Kaya, bmtheWFAa2ZzaHJjLmVkdS5zYQ==; bmFtaWtrYXlhQGdtYWlsLmNvbQ==; Dilek Colak, ZGtjb2xha0BnbWFpbC5jb20=
†These authors have contributed equally to this work
‡ORCID: Dilek Colak, orcid.org/0000-0001-6485-8768
Namik Kaya, orcid.org/0000-0001-8912-7507