
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 06 October 2022
Sec. Radiation Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.998388
This article is part of the Research Topic Targeting DNA Damage Response to Enhance Antitumor Innate Immunity in Radiotherapy View all 10 articles
 Lindsey Carlsen1,2,3,4,5
Lindsey Carlsen1,2,3,4,5 Wafik S. El-Deiry1,2,3,4,5,6*
Wafik S. El-Deiry1,2,3,4,5,6*DNA damage response inhibitors are widely used anti-cancer agents that have potent activity against tumor cells with deficiencies in various DNA damage response proteins such as BRCA1/2. Inhibition of other proteins in this pathway including PARP, DNA-PK, WEE1, CHK1/2, ATR, or ATM can sensitize cancer cells to radiotherapy and chemotherapy, and such combinations are currently being tested in clinical trials for treatment of many malignancies including breast, ovarian, rectal, and lung cancer. Unrepaired DNA damage induced by DNA damage response inhibitors alone or in combination with radio- or chemotherapy has a direct cytotoxic effect on cancer cells and can also engage anti-cancer innate and adaptive immune responses. DNA damage-induced immune stimulation occurs by a variety of mechanisms including by the cGAS/STING pathway, STAT1 and downstream TRAIL pathway activation, and direct immune cell activation. Whether or not the relative contribution of these mechanisms varies after treatment with different DNA damage response inhibitors or across cancers with different genetic aberrations in DNA damage response enzymes is not well-characterized, limiting the design of optimal combinations with radio- and chemotherapy. Here, we review how the inhibition of key DNA damage response enzymes including PARP, DNA-PK, WEE1, CHK1/2, ATR, and ATM induces innate and adaptive immune responses alone or in combination with radiotherapy, chemotherapy, and/or immunotherapy. We also discuss current progress in the clinical translation of immunostimulatory DNA-damaging treatment regimens and necessary future directions to optimize the immune-sensitizing potential of DNA damage response inhibitors.
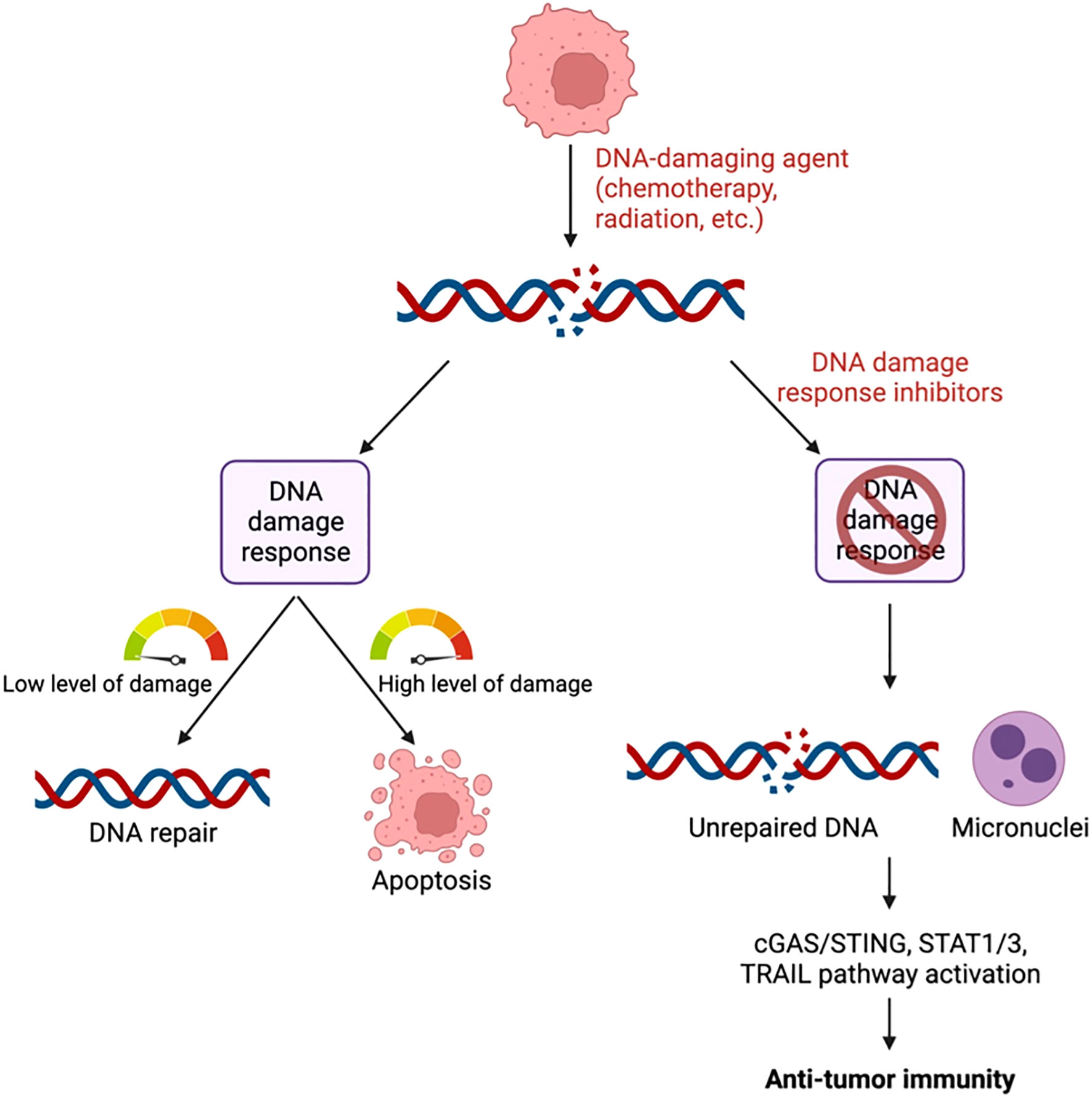
Graphical Abstract
The DNA damage response (DDR) involves several pathways including base excision repair (BER) and nucleotide excision repair (NER) to repair single-stranded DNA breaks as well as homologous recombination (HR) and non-homologous end joining (NHEJ) to repair double-stranded DNA breaks. Activation of these pathways results in cell cycle arrest, DNA repair, senescence, and/or apoptosis depending on the extent of DNA damage (Figure 1) (15). Inhibition of DDR proteins including poly-ADP ribose polymerase (PARP), DNA-dependent protein kinase (DNA-PK), WEE1, checkpoint kinase 1/2 (CHK1/2), ataxia telangiectasia and Rad3 related (ATR), or ataxia telangiectasia mutated (ATM) serine/threonine kinase results in cell cycle progression and accumulation of unrepaired DNA (16). This accumulation eventually leads to cell death and/or DNA leakage into the cytosol in the form of micronuclei (17). DDR inhibitor (DDRi) therapy is used to treat cancer patients with tumors that harbor alterations in DDR proteins such as BRCA1/2. In these tumors, inhibition of additional DDR proteins renders the cell incapable of any type of DNA repair, resulting in cell death (18). This mechanism is known as synthetic lethality, a situation in which inhibition or mutation of two proteins separately is viable, but mutation or inactivation of both is lethal to the cell (19). Even in the absence of DDRi agents, cancer cells with defects in DNA repair pathways tend be more sensitive to anti-cancer therapies (20) including chemotherapy as compared to cells without genetic alterations in these pathways (21, 22). In addition, cells with DNA damage repair defects tend to be sensitive to immunotherapy as a result of enhanced neoantigen generation, upregulation of programmed death ligand 1 (PD-L1), and induction of the cyclic GMP–AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway (23–26).
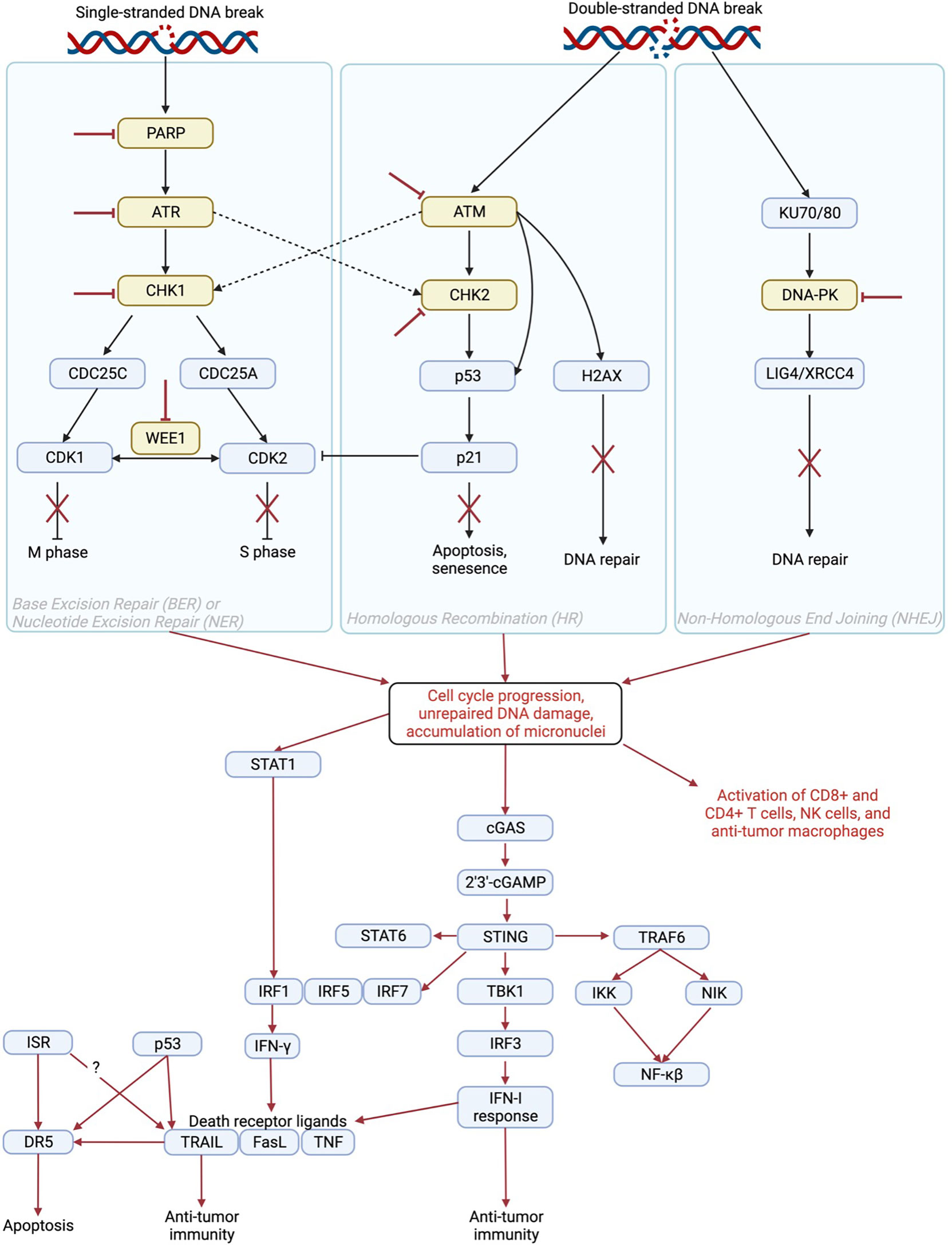
Figure 1 Mechanism of immune activation by inhibition of DNA damage repair proteins. PARP, ATR, CHK1/2, WEE1, ATM, and DNA-PK play roles in DNA repair pathways including base excision repair (BER), nucleotide excision repair (NER), homologous recombination (HR), and non-homologous end joining (NHEJ) to induce cell cycle arrest, apoptosis, senescence, and/or DNA repair. Inhibition of DNA damage repair proteins (red inhibitor lines) results in cell cycle progression, unrepaired DNA damage, and accumulation of cytosolic micronuclei that contain fragments of DNA (1). This results in activation of the STAT1 (2, 3), cGAS/STING (4), and TRAIL pathways as well as direct activation of immune cells (5, 6) to induce an anti-tumor immune response. The STAT1 pathway induces IFN-γ, which can increase levels of death receptor ligands including TRAIL (7), FasL (8), and TNF (9). The cGAS/STING pathway induces an IFN-I response, which also contributes to increased levels of death receptor ligands (10–13) as well as contributes directly to anti-tumor immunity (14). Created in BioRender.com.
DDRi therapy may be used as a single agent or in combination with DNA-damaging agents such as chemotherapy and radiation therapy (RT) (1) (Table 1). Certain PARP inhibitors (PARPi) are FDA-approved to treat breast, prostate, and gynecologic cancers including ovarian cancer (21–23), and there are numerous clinical trials underway to extend their use to other malignancies (Table 1). The WEE1 inhibitor ZN-c3 has been granted fast track designation by the FDA for treatment of patients with uterine serous carcinoma (27, 28) and is included in nine other clinical trials testing its efficacy in various other types of cancer. Additional clinical trials are ongoing to investigate other DDRi therapies including inhibitors of DNA-PK, and CHK1/2, ATR, and ATM.
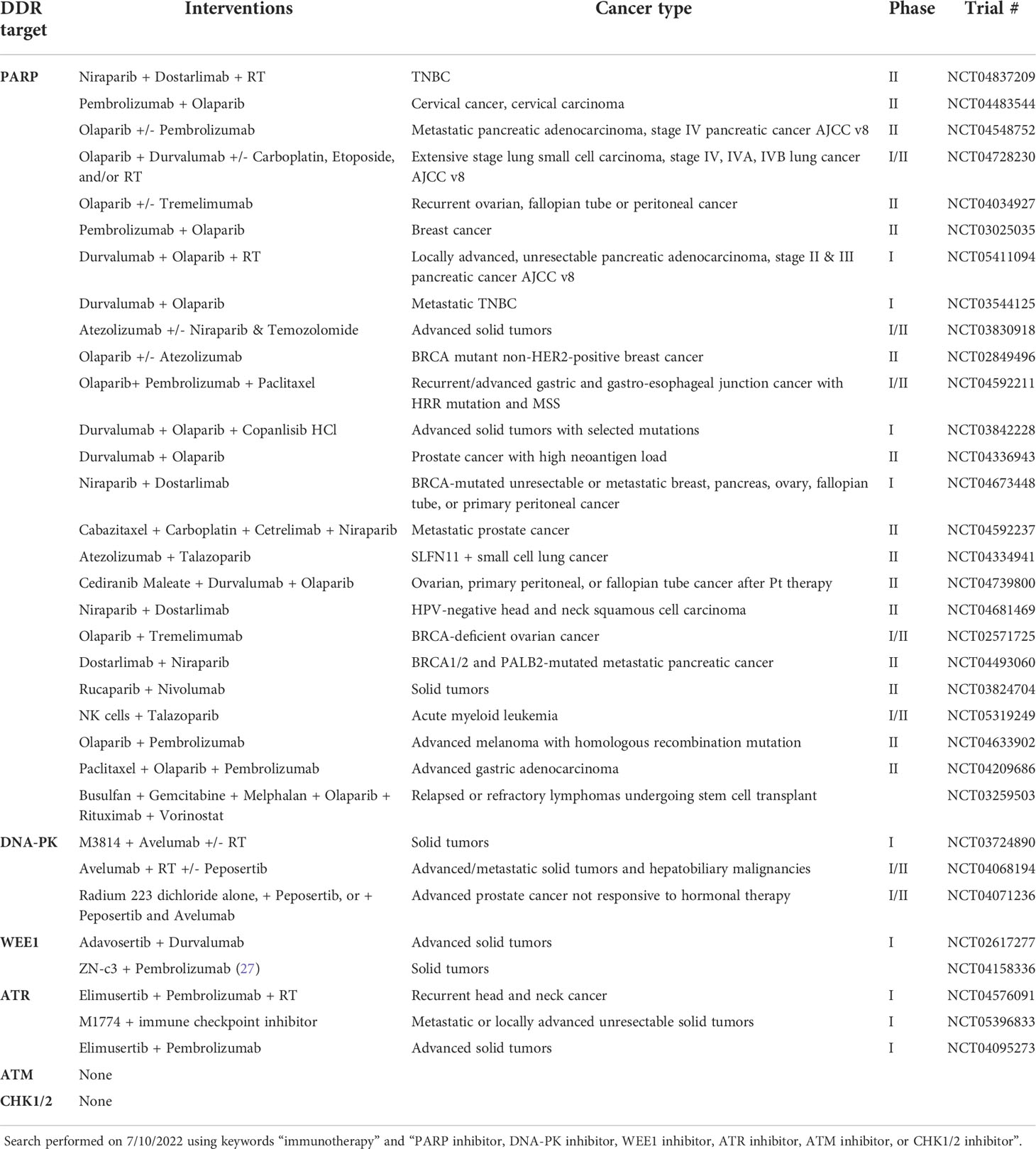
Table 1 Ongoing, completed, and recruiting clinical trials testing the combination of DNA damage inhibitors with immunotherapy in various cancer types.
In addition to inducing cancer cell death by synthetic lethality, it is now well-recognized that DDRi therapy induces innate and adaptive immune responses (29, 30). DDRi-induced immune stimulation primarily occurs via the cGAS/STING pathway (29), but also occurs through signal transducer and activator of transcription 1 (STAT1) pathway activation (2) and direct activation of immune cells including T cells, NK cells, and anti-tumor macrophages (5, 6). As a result of extensive preclinical evidence supporting DDRi-induced immune responses, several clinical trials have been initiated to test the combination of DDRi with immunotherapy, primarily immune checkpoint inhibition (ICI) (Table 1). In this Review, we will discuss the mechanisms of different DDR proteins, their interactions with the immune system, and clinical translation of DDRi + immunotherapy. We also discuss necessary future directions for optimal clinical translation including clarification of variation across different DDRi therapies and across cancer types, as well as the need for a stronger focus on combining DDRi + immunotherapy strategies with DNA-damaging agents such as chemotherapy and RT.
The cGAS/STING pathway is heavily implicated in the immunomodulatory effects of DNA damaging drugs and DDRi therapies. The first step of the pathway involves cGAS interaction with double-stranded DNA in the cytosol (31). These segments of DNA are often referred to as micronuclei (32). Then, cGAMP acts as a second messenger to activate STING, which activates TBK1 to recruit and activate IRF3. IRF3 then translocates to the nucleus to induce transcription of immune-stimulated genes (ISG) and type 1 interferons (IFNs). STING also activates IKK and NIK to mediate the induction of canonical and non-canonical NF-κB-driven inflammatory genes (31).
cGAS/STING-mediated IFN signaling enhances the infiltration of anti-tumor T cells and NK cells into the tumor. Though further study is needed to confirm this mechanism, it is also thought that cytosolic DNA from tumor cells can be transferred to the cytosol of immune cells to induce cGAS/STING signaling and enhance antigen presentation and cross-priming in DCs and T cells, respectively (31). Lastly, c-GAS-STING also promotes the senescence-associated secretory phenotype (SASP), which is characterized by cancer cell secretion of pro-inflammatory cytokines, chemokines, proteases, and growth factors that induce senescence and tumor control (31). However, it is important to mention that SASP can also induce an immunosuppressive TME, promoting cancer progression (33).
It is important to note that the effects of cytosolic DNA on cancer progression are likely dependent on cancer stage. In early stages, cytosolic DNA likely leads to immune surveillance through mechanisms such as the cGAS/STING pathway. In late stages, cancer cells are more likely to have lost functional checkpoints of cell cycle and immune regulation, and therefore cytosolic DNA can induce chronic inflammatory signaling that may be associated with survival and metastasis. Thus, the tumor microenvironment should be carefully monitored during the therapeutic induction of cytosolic DNA accumulation and cGAS/STING pathway activation using DDRi therapy (31).
STING agonists are being investigated to treat many types of cancer either as a single agent or combined with ICI or chemotherapy (34). STING-based therapeutics have yet to be combined with DDRi therapy, though there is rationale for combination to enhance DDRi-induced immune activation (34, 35). STING agonists can activate the cGAS/STING pathway in the absence of cytosolic DNA and therefore circumvent the need for DNA damage to induce the type 1 IFN response (35). Some limitations of targeting cGAS/STING in cancer exist, including evidence of cGAS/STING silencing or loss-of-function mutations in certain tumors (35, 36) and cGAS/STING-driven IL-6-dependent survival of chromosomally instable cancers (37). In these cases, administration of cGAS/STING agonists may have limited to no efficacy or may be pro-tumorigenic. Careful consideration of STING agonist combination therapies and evaluation of patients who may not benefit or be harmed by these therapies is needed prior to clinical translation of cGAS/STING + DDRi combination therapies.
The tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is primarily expressed on the surface of immune cells including NK cells, T cells, NK tumor (NKT) cells, DCs, and macrophages (38). It can also be expressed in soluble form after proteolytic cleavage from the cell surface (39). Both membrane-bound and soluble TRAIL bind to death receptor 5 (DR5) and death receptor 4 (DR4) on cancer cells to induce apoptosis (39). TRAIL is induced by the IFN-γ/STAT1 and cGAS/STING pathways (7, 40–42) that are activated after DDRi therapy. The TRAIL pathway is anti-tumorigenic, as evidenced by the increased susceptibility of TRAIL receptor-deficient mice to chronic inflammation and tumorigenesis (43, 44).
In addition to its role in apoptosis, TRAIL also plays an important role in the anti-cancer immune response. For example, some immune cells kill cancer cells in a TRAIL-dependent manner (38, 45) and targeted delivery of TRAIL to cell surface antigens on T cells may enhance their cytotoxic activity (46). TRAIL-TRAIL receptor interaction on MDSCs can limit their lifespan, supporting an anti-tumor immune microenvironment (47, 48). Due to its ability to induce both apoptosis and anti-tumor immune responses, activation of the TRAIL pathway is a promising clinical strategy (49). Various therapeutic approaches have been considered including TRAIL receptor agonists, DR4/5 agonistic monoclonal antibodies, and different formulations such as PEGylated TRAIL (49). Other exciting new directions are being pursued preclinically such as engineering tumor-homing, TRAIL-expressing mesenchymal stem cells (50, 51). TRAIL-based therapies have been studied extensively in the clinic and some have shown early signs of efficacy in non-Hodgkin’s lymphoma (52) and non-small cell lung cancer (53, 54). Limitations include short half-life, limited induction of receptor clustering, binding to decoy receptors such as DcR1, DcR2, and osteoprotegerin (55, 56), and development of resistance (49). There are some ongoing clinical trials with TRAIL-based therapies such as the TRAIL-receptor agonists ABBV-621 in combination with bortezomib and dexamethasone (NCT04570631) and INBRX-109 alone (NCT04950075) or in combination with DNA damaging agents (NCT03715933) (49). No trials are currently investigating the combination of TRAIL-based therapy with DDRi agents.
Combination therapies may overcome some of these limitations, and various preclinical investigations support the combination of TRAIL-based therapies with DDRi. For example, one study found that PARPi enhanced the efficacy of a DR5 antibody in a pancreatic cancer mouse xenograft model (57). Others found that DNA-PKi potentiates p53-dependent apoptosis after treatment with a DNA damaging agent in AML cells, and that the TRAIL pathway plays a major role in this apoptotic response (58). Others have described upregulation of TRAIL-mediated apoptosis after ATMi treatment of melanoma cells (59). These findings provide rationale for combining TRAIL agonists and DDRi therapy in the clinic to enhance induction of apoptosis. Whether or not these types of combination treatments will enhance the anti-tumor immune response remains to be investigated.
PD-L1 is a ligand that binds programmed cell death protein 1 (PD-1) on activated T cells. Binding of PD-L1 to PD-1 inhibits T cell activity (60) and elevated expression of PD-L1 is a major biomarker of favorable response to immune checkpoint inhibitors (61). Experimental evidence suggests that PARPi agents can upregulate PD-L1 expression by blocking glycogen synthase kinase-3 beta (GSK3β), a regulator of glycogen metabolism, cell cycle, inflammation, and proliferation (62). GSK3β also plays a role in the repair of both single- and double-stranded DNA breaks. PARPi-induced inhibition of GSK3β causes inhibition of DNA damage repair and upregulation of PD-L1 (63–65).
ATR inhibitors, on the other hand, seem to upregulate PD-L1 mRNA but downregulate PD-L1 protein expression (66–68). Further, studies have shown that DNA damage-induced upregulation of PD-L1 by cisplatin or ionizing radiation was suppressed by co-administration with ATRi agents (67). DNA-PK inhibitors seem to upregulate PD-L1 (69) along with WEE1 inhibitors (2) and Chk1/2 inhibitors (70, 71), likely by preventing the repair of double-stranded DNA breaks, which activates STAT1/3 signaling through ATM/ATR/Chk1 kinases, resulting in an upregulation of PD-L1 levels (60).
Poly-ADP ribose polymerase (PARP) is an enzyme that plays a critical role in the DNA repair pathways NER and BER, which repair DNA damage that is caused by therapeutic agents such as alkylating agents and chemotherapy (72). The PARP family contains 17 different proteins, but most studied are PARP1 and PARP2. PARP1 binds to DNA regardless of phosphorylation state and PARP2 preferentially binds phosphorylated DNA breaks, but otherwise these proteins largely function similarly (73). PARP inhibitors (PARPi) lead to death by synthetic lethality in cancer cells with deficiencies in the homologous recombination (HR) DNA repair pathway (74). PARPi is FDA-approved to treat breast cancer, prostate cancer, and gynecologic cancers including ovarian cancer (75–77).
PARP plays an important role in the normal functioning of the immune system. PARP2 contributes to the development of mature CD4+ and CD8+ T cells and in vivo data suggests that dual inhibition of PARP1 and PARP2 leads to a measurable decrease in T cell populations. PARP1 and PARP2 also contribute to normal T cell functioning as demonstrated by experiments in which PARP1/2 inhibition resulted in decreased IL-2 and IFN-γ-secreting T cells. PARP1 is also responsible for marking Foxp3-expressing T regulatory cells (T regs) for degradation. Additionally, PARP1 regulates NFAT, a family of transcription factors that that regulates CD4+ T cell differentiation, but it is unclear if inhibition of PARP1 biases CD4+ cells toward a Th1 or Th2 phenotype (73). Lastly, PARP1 may cooperate with IFI16 to induce noncanonical STING activation in response to chemotherapy-induced DNA damage (31).
Interestingly, in BRCA1-deficient ovarian cancer models, PARP inhibition with olaparib increased CD4+ and CD8+ T cells in the tumor and in circulation, reduced their expression of inhibitory receptors PD-1, Tim-3, and Lag-3, and increased their levels of TNF-alpha and IFN-γ secretion. In dendritic cells, PARPi upregulates costimulatory molecules CD80/CD86 and MHC class II which enhances antigen presentation and interactions with T cells. PARPi may increase expression of cell death receptor ligands and NKG2D ligands, which increases cancer cell sensitivity to NK cell-mediated killing. In macrophages, the impact of PARPi is dependent on factors in the tumor microenvironment including certain cytokines. The DNA damage caused by PARPi leads to cytosolic DNA, activating the cGAS/STING pathway and the type I IFN response (73). PARPi can also increase the amount of DNA in the cytosol, leading to the accumulation of neoantigens (78).
Due to the immune-stimulating properties of PARPi therapy, there is clinical interest in combining PARPi with immunotherapy. Clinical trials testing such combinations are ongoing for ovarian, ovarian, lung, urothelial, prostate, and gastrointestinal cancers (78). The results of these trials have been most promising in ovarian and breast cancer. In ovarian cancer, overall response rates (ORR) ranged from 45-63% and disease control rate (DCR) was 73-81% depending on the patient population. In breast cancer, ORR was 53% and DCR was 47-83% depending on patient population. PARPi alone is effective in patients with prostate cancer and has been combined with IT in several clinical trials. Results of the completed trials have been promising, with 9/17 patients with metastatic castration-resistant prostate cancer (mCRPC) treated with durvalumab and olaparib experiencing a PSA decline of >50% and 4/17 patients experiencing a radiographic response. A combination of pembrolizumab and olaparib in a cohort of patients with wild-type HR proteins had slightly less exciting results, with 7% partial response and 29% DCR. Studies in gastric cancer combining durvalumab and olaparib have reported a 10% ORR and 12-week DCR of 26% (78).
DNA-PK is a serine/threonine protein kinase that plays a critical role in the DNA repair pathways classical NHEJ and HR. DNA-PK inhibitors (DNA-PKi) interfere with its kinase function and sensitize cells to DNA-damaging agents. DNA-PKi can be used as a single agent in some cancers with ATM deficiency by inducing synthetic lethality (79). No DNA-PKi therapies are FDA approved, however there are several ongoing clinical trials involving compounds such as XRD-0394, CC-115, VX-984 (M9831), AZD7648, and M3814 (nedisertib, peposertib, MSC-2490484A) to treat various type of cancer, typically advanced solid tumors (80, 81).
DNA-PK phosphorylates cGAS and suppresses its enzymatic activity. DNA-PK inhibition or deficiency correlates with decreased levels of phosphorylated cGAS and promotes antiviral immune responses (82). Additionally, as DNA-PK is critical to maintaining genomic stability, the loss or inhibition of this kinase may lead to high mutation load secondary to the development of genomic instability. Mutation of the gene encoding DNA-PK protein PRKDC is associated with high mutation load or microsatellite instable (MSI)-high status in The Cancer Genome Atlas pan-cancer cohort. Further, PRKDC knockout and DNA-PKi enhanced the efficacy of ICI (83, 84). The DNA-PKi AZD7648 sensitizes mice with colorectal tumors or melanoma to radiotherapy and induces a tumor control that is dependent on type I IFNs. There are phase I/II clinical trials involving AZD7648 in combination with chemotherapy (NCT03907969) and radiotherapy (NCT04550104) currently ongoing (85). Due to the dependence of AZD7648 on type I IFN responses, it would be interesting to combine this drug with immunomodulatory drugs that enhance the type I IFN response. Additionally, the DNA-PKi peposertib enhanced RT-induced TGFβ/PD-L1-targeted immunotherapy in mice, further supporting the combination of DNA-PKi, RT, and immunotherapy (69).
Three clinical trials are evaluating the combination of the DNA-PKi M3814 combined with the anti-PD-L1 ICI avelumab (86). M3814 has demonstrated monotherapy activity in several tumor cell lines, and M3814 + radiotherapy (RT) combined with avelumab significantly delayed tumor growth as compared to either agent alone + RT in MC38 syngrafts, indicating the benefit of combining DNA-PKi and immunotherapy (87). One trial is investigating M3814 and avelumab +/- radiotherapy for treatment of patients with advanced solid tumors (NCT03724890) (87). Another is investigating avelumab and RT +/- M3814 in advanced solid tumors and hepatobiliary malignancies (NCT04068194) (88). Lastly, one trial is evaluating RT vs. RT + M3814 vs. RT + M3814 + avelumab in patients with advanced prostate cancer that is unresponsive to hormonal therapy (NCT04071236) (89).
The WEE1 kinase family consists of three serine/threonine kinases: WEE1, PKMYT1, and WEE1B (WEE2). WEE1 and PKMYT1 play a crucial role in cell cycle regulation and DNA damage repair, while WEE2 regulates cell cycle progression and largely regulates meiosis. WEE1 and PKMYT1 can act like oncogenes and are a major focus in anti-cancer drug development (90). One WEE1 inhibitor, ZN-c3, has been granted fast track designation by the FDA for treatment of patients with uterine serous carcinoma. Another WEE1 inhibitor adavosertib (AZD1775, MK-1775) is highly developed and has been included in over fifty clinical trials to treat various types of cancer since 2008 (28).
WEE1 overexpression abrogates immune cell killing, for example by protecting cancer cells from granzyme B/TNFα induced cell death. One study found that cancer cells develop resistance to granzyme B/TNFα-mediated cytotoxic T cell killing by activating the G2/M cell cycle checkpoint. Further, they found that administration of WEE1i adavosertib reversed this effect, enhanced T cell killing, and synergized with an anti-PD-1 monoclonal antibody in murine models of oral cavity carcinoma, melanoma and colon adenocarcinoma with various TP53 mutations (91). WEE1 inhibition activates the STING and STAT1 pathways in SCLC and enhances the antitumor immune response to PD-L1 inhibition (2). Like the STING pathway, the STAT1 pathway is a major contributor to the anti-tumor immune response. Along with STAT2, STAT1 induces IFN-regulated genes, enhances antigen presentation, and contributes to an inflammatory, anti-cancer response. It is important to differentiate STAT1 and STAT2 from other STAT family members such as STAT3 and STAT5, which contribute to cancer cell survival, proliferation, and angiogenesis (3). It has also been shown that WEE1 induces anti-tumor immunity by activating endogenous retroviral elements and the dsRNA pathway (92). WEE1i also sensitizes head and neck cancers to natural killer (NK) cell therapies (93).
One ongoing clinical trial is evaluating adavosertib with the anti-PD-L1 ICI durvalumab for treatment of patients with advanced solid tumors (NCT02617277). DCR for the total cohort was 36%, suggesting antitumor activity (94). Notably, adavosertib + immunotherapy has a better safety profile compared to adavosertib + chemotherapy, warranting continued investigation (95). Another actively recruiting trial will test the safety, tolerability, efficacy, pharmacokinetics and pharmacodynamics of ZN-c3 alone and in combination with other drugs including the anti-PD-1 ICI pembrolizumab (NCT04158336) (27). As p53 mutations and overexpression of SKP2 and CUL1 may be biomarkers of a favorable response to WEE1i, additional clinical trials in these patient populations in combination with immunotherapy are needed (27, 91).
ATR and its major downstream effector checkpoint kinase 1 CHK1 play a role in the DNA damage response. In response to single-stranded DNA breaks, ATR activates CHK1 to trigger intra-S and G2/M phase checkpoints. In response to double-stranded DNA breaks, the MRE11/NBS1/RAD5 complex activates ATM and CHK2 to trigger the G1/S-phase checkpoint (96). Because ATR has a broader range of biological functions than CHK1, it is thought that ATRi may have greater toxicity in normal cells. Therefore, the clinical development of CHK1i is more advanced than ATRi (96). There are over twenty CHK1/2 and ten ATR inhibitors in various stages of clinical trials for many different cancer types mostly in combination with chemotherapy but also with RT and histone deacetylase inhibitors (HDACi) (96). No CHK1/2 or ATR inhibitors are FDA-approved yet (97).
One study found that in the leukemia cell line THP-1, CHK1i increased TBK1 but did not increase IRF3 phosphorylation, induce IRF3 or NF-κB reporter activation, nor induce a type 1 IFN response (98). The same group found that in solid tumor cell lines, addition of CHK1i to chemotherapy treatment such as gemcitabine or camptothecin increased the accumulation of cytosolic DNA but decreased the level of chemotherapy-mediated IRF1 and STAT1 phosphorylation. Interestingly, similar results as far as lack of type 1 IFN response were found using ATRi and WEE1i, indicating that context such as cancer type may affect the ability of DDRi to induce the cGAS/STING pathway (99). Another study found that in murine melanoma models, CHK1i induces an immunogenic signaling and increased levels and activity of CD8+ T cells (100). Similarly, others observed that treatment of patients with head and neck squamous cell carcinoma with CHK1i led to an upregulation of transcripts associated with T-cell activation and inflammatory cytokines and chemokines but also T regs (101). Interestingly, others have shown that the combination of CHK1i and ionizing RT increases micronuclei formation and induces an abscopal tumor regression response in a murine melanoma model (102).
Despite the advanced preclinical development of CHK1i alone or in combination with chemotherapy or RT, there are currently no ongoing or completed clinical trials testing the combination of CHK1/2i with immunotherapy. There are three trials that are evaluating ATRi with immunotherapy. One study (NCT04576091) is investigating sensitization to pembrolizumab with the ATRi elimusertib in combination with RT for treatment of patients with recurrent head and neck cancer. As of February 2022, no patients were enrolled in this study. Another trial is currently recruiting patients with advanced solid tumors to evaluate elimusertib + pembrolizumab without RT (NCT04095273). Lastly, one trial will evaluate the combination of ATRi M1774 with immune checkpoint inhibition for treatment of patients with metastatic or locally advanced unresectable solid tumors (NCT05396833). The results of these trials are highly anticipated.
ATM is activated by double stranded breaks in DNA, and cells that are deficient in ATM experience abnormal DNA repair. Activated ATM phosphorylates p53 at serine 15 to activate it and phosphorylates MDM2 to prevent its inhibitory binding to p53. ATM also phosphorylates and activates CHK2, which phosphorylates p53 at another activating site (serine 20). p53 induces p21 to inhibit CDK2/cyclin E to induce arrest at the G1 phase of the cell cycle. Activated ATM also phosphorylates NBS1, which is necessary for RT-induced S phase cell cycle arrest, but the complete mechanism remains to be clarified (103).
In Drosophila models, ATR deficiency causes an innate immune response (104). In murine and human cancer cell lines, ATM deficiency induces ISG expression and tumor infiltration of immune cells in a cGAS/STING-dependent manner. Further investigation revealed this effect was mediated specifically by leakage of mitochondrial DNA rather than nuclear DNA into the cytoplasm. The same group found that ATM expression levels negatively correlate with type 1 IFN gene expression in human tumor tissues and that patients with tumors harboring ATM mutations have a favorable response to ICI (105). Similar findings as far as ATM mutations serving as a biomarker of favorable response to ICI have been made in bladder cancer (106). Other studies in pancreatic cancer have shown that ATMi induces type 1 IFNs in a cGAS/STING-independent manner, but this response was dependent on TBK1 and SRC (107). Despite this preclinical evidence of ATMi-induced immune stimulation, there are no clinical trials testing the combination of ATMi and immunotherapy.
Chemotherapy and radiation therapy are well-known inducers of oxidative stress, a condition in the cell characterized by excess reactive oxygen species and the resulting processes that detoxify the cell and repair oxidative damage (108). Oxidative stress plays a major role in inducing cellular damage after treatment with DNA-damaging agents (109). Oxidative stress increases levels of intracellular Ca2+, induces Fenton reaction DNA lesions, and triggers DNA repair mechanisms (110). BER plays a major role in the cellular response to oxidative DNA damage. During BER, damaged bases are excised, generating apurinic/apyrimidinic (AP) sites. At these sites, apurinic/apyrimidinic endonuclease 2 (APE2, APN2, or APEX2) creates a single-strand break which is then fixed by other DNA repair enzymes. Thus, APE2 plays a critical role in the repair of oxidative damage, and in fact knockdown of APE2 led to increased micronuclei formation in the PANC1 pancreatic cancer cell line (111). Oxidative stress plays many roles in the immune microenvironment of the tumor (112). For example, APE2 is involved in B cell development and immunoglobulin class switch recombination and APE2-knockout mice develop defects in immune responses. BER and ATR pathways, both of which are heavily involved in regulating PD-L1 expression, rely on APE2. APE2 involvement in the response to immunotherapy is likely but has not been investigated. There are no APE2 inhibitors in clinical trials for the treatment of cancer. Future studies should investigate the impact of APE2 and other oxidative damage repair enzymes on the immune response and response to immunotherapy (113).
The clinical applicability of DDRi has been clearly demonstrated for cancers with deficiencies in the DDR pathway. The combination of DDRi with DNA-damaging agents has improved the efficacy of these agents in certain contexts. It is now well-recognized that DDRi compounds stimulate the immune system against cancer and that this effect may be enhanced by combinations with DNA-damaging agents. The cGAS/STING pathway is a major regulator of DDRi-induced immune stimulation, though the STAT1 pathway, TRAIL pathway, and direct activation of anti-cancer immune cells also play important roles. The effects of DDRi on the immune system provide rationale for their combination with immunotherapy such as ICI, as is being tested in various clinical trials.
TRAIL is induced by the IFN-γ/STAT1 and cGAS/STING pathways (7, 40–42) that are activated after DDRi therapy. TRAIL-based therapies have therapeutic potential because of their ability to induce both apoptosis and lasting anti-cancer immunity. There are no FDA-approved TRAIL-based treatments (49), however numerous clinical trials are continuing to investigate new approaches and combination treatments (49, 55, 56). Many preclinical investigations support the combination of TRAIL-based therapies with DDRi (57–59), providing rationale for clinical translation. The addition of ICI to this treatment regimen should also be considered given the heavy involvement of both DDRi and the TRAIL pathway in anti-cancer immune activation.
Less than half (12/33) of the clinical trials that are testing combinations of DDRi and immunotherapy involve combination with a DNA damaging agent such as chemotherapy or radiotherapy (Table 1). Most of the trials that do not include a DNA damaging agent are for treatment of malignancies in which alterations in DDR proteins are common, such as breast cancer and ovarian cancer. While DDRi therapy can induce accumulation of cytosolic DNA and stimulate the immune system in the presence of these alterations, co-administration of DNA damaging agents should be considered to expand the use of this combination therapy to patients without such alterations.
Cancer stem cells are cancer cells with stem-like phenotypes that are slow-cycling and have highly efficient DNA repair, which grants them resistance to chemotherapy and radiotherapy (114). Cancer stem cells present a major challenge as far as overcoming drug resistance and cancer recurrence. Cancer stem cells may also be able to evade the immune system (115), thus immunotherapy alone may not be active against this subset of the tumor. Inhibiting the highly efficient DNA repair processes in cancer stem cells, especially in combination with DNA-damaging agents, may be a promising approach to eliminate this cell population (116). Bulk tumor reduction and elimination of cancer stem cells with the combination of DDRi and DNA damaging agents sequenced with immunotherapy for lasting tumor regression may be a viable treatment option for patients with tumors characterized by cancer stemness. Optimization and validation of treatment dose, timing, and sequencing is necessary in vivo.
Another area in need of further investigation is the differential effects of various DDRi agents on the immune system. Though inhibition of each DDR protein has similar effects in most studies, there seem to be context-specific differences especially for CHK1i. Similar context-dependency may be found with complementary study of other DDR proteins. Further investigation is critical to the application of DDRi + immunotherapy to wider patient populations.
Conceptualization, WE-D and LC. Investigation, WE-D and LC. Writing—original draft preparation, LC. Writing—review and editing, WE-D and LC. Supervision, WE-D. Project administration, WE-D. Funding acquisition, WE-D. All authors have read and agreed to the published version of the manuscript.
This work was funded by the Teymour Alireza P’98, P’00 Family Cancer Research Fund, grant number GFT640730, established by the Alireza Family.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3 related; BER, base excision repair; cGAS, cyclic GMP-AMP synthase; CHK1/2, checkpoint kinase 1/2; DCR, disease control rate; DDR, DNA damage response; DNA-PK, DNA-dependent protein kinase; DR5, death receptor 5; HR, homologous recombination; ICI, immune checkpoint inhibition; IFN, interferon; ISG, interferon stimulated gene; MDSC, myeloid-derived suppressor cell; MSI, microsatellite instable; NER, nucleotide excision repair; NHEJ, non-homologous end joining; NK, natural killer; ORR, overall response rate; PARP, poly-ADP ribose polymerase; PD-1, programmed cell death-1; PD-L1, programmed death-ligand 1; RT, radiation therapy; SASP, senescence-associated secretory phenotype; STAT1, signal transducer and activator of transcription 1; STING, stimulator of interferon genes; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand.
1. Maresca L, Stecca B, Carrassa L. Novel therapeutic approaches with DNA damage response inhibitors for melanoma treatment. Cells (2022) 11(9):1466. doi: 10.3390/cells11091466
2. Taniguchi H, Caeser R, Chavan SS, Zhan YA, Chow A, Manoj P, et al. Wee1 inhibition enhances the antitumor immune response to pd-L1 blockade by the concomitant activation of sting and Stat1 pathways in sclc. Cell Rep (2022) 39(7):110814. doi: 10.1016/j.celrep.2022.110814
3. Owen KL, Brockwell NK, Parker BS. Jak-stat signaling: A double-edged sword of immune regulation and cancer progression. Cancers (Basel) (2019) 11(12):2002. doi: 10.3390/cancers11122002
4. Liao W, Du C, Wang J. The cgas-sting pathway in hematopoiesis and its physiopathological significance. Front Immunol (2020) 11:573915. doi: 10.3389/fimmu.2020.573915
5. Lutfi N, Galindo-Campos MA, Yélamos J. Impact of DNA damage response-targeted therapies on the immune response to tumours. Cancers (Basel) (2021) 13(23):6008. doi: 10.3390/cancers13236008
6. Zhu L, Liu J, Chen J, Zhou Q. The developing landscape of combinatorial therapies of immune checkpoint blockade with DNA damage repair inhibitors for the treatment of breast and ovarian cancers. J Hematol Oncol (2021) 14(1):206. doi: 10.1186/s13045-021-01218-8
7. Miura Y, Tsujioka T, Nishimura Y, Sakaguchi H, Maeda M, Hayashi H, et al. Trail expression up-regulated by interferon-gamma Via phosphorylation of Stat1 induces myeloma cell death. Anticancer Res (2006) 26(6b):4115–24.
8. Xu X, Fu XY, Plate J, Chong AS. Ifn-gamma induces cell growth inhibition by fas-mediated apoptosis: Requirement of Stat1 protein for up-regulation of fas and fasl expression. Cancer Res (1998) 58(13):2832–7.
9. Vila-del Sol V, Punzón C, Fresno M. Ifn-Gamma-Induced tnf-alpha expression is regulated by interferon regulatory factors 1 and 8 in mouse macrophages. J Immunol (2008) 181(7):4461–70. doi: 10.4049/jimmunol.181.7.4461
10. Papageorgiou A, Dinney CP, McConkey DJ. Interferon-alpha induces trail expression and cell death Via an irf-1-Dependent mechanism in human bladder cancer cells. Cancer Biol Ther (2007) 6(6):872–9. doi: 10.4161/cbt.6.6.4088
11. Arbour N, Rastikerdar E, McCrea E, Lapierre Y, Dörr J, Bar-Or A, et al. Upregulation of trail expression on human T lymphocytes by interferon beta and glatiramer acetate. Mult Scler (2005) 11(6):652–7. doi: 10.1191/1352458505ms1222oa
12. Kaser A, Nagata S, Tilg H. Interferon alpha augments activation-induced T cell death by upregulation of fas (Cd95/Apo-1) and fas ligand expression. Cytokine (1999) 11(10):736–43. doi: 10.1006/cyto.1998.0484
13. Yarilina A, Ivashkiv LB. Type I interferon: A new player in tnf signaling. Curr Dir Autoimmun (2010) 11:94–104. doi: 10.1159/000289199
14. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol (2015) 15(7):405–14. doi: 10.1038/nri3845
15. Li LY, Guan YD, Chen XS, Yang JM, Cheng Y. DNA Repair pathways in cancer therapy and resistance. Front Pharmacol (2020) 11:629266. doi: 10.3389/fphar.2020.629266
16. Curtin NJ. Inhibiting the DNA damage response as a therapeutic manoeuvre in cancer. Br J Pharmacol (2013) 169(8):1745–65. doi: 10.1111/bph.12244
17. MacDonald KM, Benguerfi S, Harding SM. Alerting the immune system to DNA damage: Micronuclei as mediators. Essays Biochem (2020) 64(5):753–64. doi: 10.1042/ebc20200016
18. Hengel SR, Spies MA, Spies M. Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chem Biol (2017) 24(9):1101–19. doi: 10.1016/j.chembiol.2017.08.027
19. Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discovery (2020) 19(1):23–38. doi: 10.1038/s41573-019-0046-z
20. Jiang M, Jia K, Wang L, Li W, Chen B, Liu Y, et al. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann Transl Med (2020) 8(24):1685. doi: 10.21037/atm-20-2920
21. Sharma Saha S, Gentles L, Bradbury A, Brecht D, Robinson R, O'Donnell R, et al. Genomic, transcriptomic, and functional alterations in DNA damage response pathways as putative biomarkers of chemotherapy response in ovarian cancer. Cancers (Basel) (2021) 13(6):1420. doi: 10.3390/cancers13061420
22. Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, et al. Somatic Ercc2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discovery (2014) 4(10):1140–53. doi: 10.1158/2159-8290.Cd-14-0623
23. Zhang J, Shih DJH, Lin S-Y. Role of DNA repair defects in predicting immunotherapy response. biomark Res (2020) 8(1):23. doi: 10.1186/s40364-020-00202-7
24. Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from pd-1/Pd-L1 blockade in advanced urothelial cancers. J Clin Oncol (2018) 36(17):1685–94. doi: 10.1200/jco.2017.75.7740
25. Jiang M, Jia K, Wang L, Li W, Chen B, Liu Y, et al. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm Sin B (2021) 11(10):2983–94. doi: 10.1016/j.apsb.2021.01.003
26. Joshi M, Grivas P, Mortazavi A, Monk P, Clinton SK, Sue-Ann Woo M, et al. Alterations of DNA damage response genes correlate with response and overall survival in anti-Pd-1/Pd-L1-Treated advanced urothelial cancer. Cancer Med (2020) 9(24):9365–72. doi: 10.1002/cam4.3552
27. Meng X, Gao JZ, Gomendoza SMT, Li JW, Yang S. Recent advances of Wee1 inhibitors and statins in cancers with P53 mutations. Front Med (Lausanne) (2021) 8:737951. doi: 10.3389/fmed.2021.737951
28. Gorecki L, Andrs M, Korabecny J. Clinical candidates targeting the atr-Chk1-Wee1 axis in cancer. Cancers (Basel) (2021) 13(4):795. doi: 10.3390/cancers13040795
29. Ye Z, Shi Y, Lees-Miller SP, Tainer JA. Function and molecular mechanism of the DNA damage response in immunity and cancer immunotherapy. Front Immunol (2021) 12:797880. doi: 10.3389/fimmu.2021.797880
30. Pilger D, Seymour LW, Jackson SP. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev (2021) 35(9-10):602–18. doi: 10.1101/gad.348314.121
31. Kwon J, Bakhoum SF. The cytosolic DNA-sensing cgas-sting pathway in cancer. Cancer Discovery (2020) 10(1):26–39. doi: 10.1158/2159-8290.Cd-19-0761
32. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. Cgas surveillance of micronuclei links genome instability to innate immunity. Nature (2017) 548(7668):461–5. doi: 10.1038/nature23449
33. Yang J, Liu M, Hong D, Zeng M, Zhang X. The paradoxical role of cellular senescence in cancer. Front Cell Dev Biol (2021) 9:722205. doi: 10.3389/fcell.2021.722205
34. Le Naour J, Zitvogel L, Galluzzi L, Vacchelli E, Kroemer G. Trial watch: Sting agonists in cancer therapy. Oncoimmunology (2020) 9(1):1777624. doi: 10.1080/2162402x.2020.1777624
35. Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, Luke JJ. Sting agonists as cancer therapeutics. Cancers (Basel) (2021) 13(11):2695. doi: 10.3390/cancers13112695
36. Konno H, Yamauchi S, Berglund A, Putney RM, Mulé JJ, Barber GN. Suppression of sting signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene (2018) 37(15):2037–51. doi: 10.1038/s41388-017-0120-0
37. Hong C, Schubert M, Tijhuis AE, Requesens M, Roorda M, van den Brink A, et al. Cgas-sting drives the il-6-Dependent survival of chromosomally instable cancers. Nature (2022) 607(7918):366–73. doi: 10.1038/s41586-022-04847-2
38. Falschlehner C, Schaefer U, Walczak H. Following trail's path in the immune system. Immunology (2009) 127(2):145–54. doi: 10.1111/j.1365-2567.2009.03058.x
39. Kimberley FC, Screaton GR. Following a trail: Update on a ligand and its five receptors. Cell Res (2004) 14(5):359–72. doi: 10.1038/sj.cr.7290236
40. Meissl K, Macho-Maschler S, Müller M, Strobl B. The good and the bad faces of Stat1 in solid tumours. Cytokine (2017) 89:12–20. doi: 10.1016/j.cyto.2015.11.011
41. Shin EC, Ahn JM, Kim CH, Choi Y, Ahn YS, Kim H, et al. Ifn-gamma induces cell death in human hepatoma cells through a Trail/Death receptor-mediated apoptotic pathway. Int J Cancer (2001) 93(2):262–8. doi: 10.1002/ijc.1310
42. Meng RD, El-Deiry WS. P53-independent upregulation of Killer/Dr5 trail receptor expression by glucocorticoids and interferon-gamma. Exp Cell Res (2001) 262(2):154–69. doi: 10.1006/excr.2000.5073
43. Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in tnf-related apoptosis-inducing ligand-deficient mice. J Immunol (2002) 168(3):1356–61. doi: 10.4049/jimmunol.168.3.1356
44. Finnberg N, Klein-Szanto AJ, El-Deiry WS. Trail-r deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest (2008) 118(1):111–23. doi: 10.1172/jci29900
45. Song M, Cubillos-Ruiz JR. Endoplasmic reticulum stress responses in intratumoral immune cells: Implications for cancer immunotherapy. Trends Immunol (2019) 40(2):128–41. doi: 10.1016/j.it.2018.12.001
46. de Bruyn M, Wei Y, Wiersma VR, Samplonius DF, Klip HG, van der Zee AG, et al. Cell surface delivery of trail strongly augments the tumoricidal activity of T cells. Clin Cancer Res (2011) 17(17):5626–37. doi: 10.1158/1078-0432.Ccr-11-0303
47. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23(2):249–62. doi: 10.1016/j.ccr.2013.01.008
48. Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. Er stress regulates myeloid-derived suppressor cell fate through trail-R-Mediated apoptosis. J Clin Invest (2014) 124(6):2626–39. doi: 10.1172/jci74056
49. Ralff MD, El-Deiry WS. Trail pathway targeting therapeutics. Expert Rev Precis Med Drug Dev (2018) 3(3):197–204. doi: 10.1080/23808993.2018.1476062
50. Fakiruddin KS, Ghazalli N, Lim MN, Zakaria Z, Abdullah S. Mesenchymal stem cell expressing trail as targeted therapy against sensitised tumour. Int J Mol Sci (2018) 19(8):2188. doi: 10.3390/ijms19082188
51. Liu M, Hu Y, Chen G. The antitumor effect of gene-engineered exosomes in the treatment of brain metastasis of breast cancer. Front Oncol (2020) 10:1453. doi: 10.3389/fonc.2020.01453
52. Younes A, Vose JM, Zelenetz AD, Smith MR, Burris HA, Ansell SM, et al. A phase 1b/2 trial of mapatumumab in patients with Relapsed/Refractory non-hodgkin's lymphoma. Br J Cancer (2010) 103(12):1783–7. doi: 10.1038/sj.bjc.6605987
53. Ouyang X, Shi M, Jie F, Bai Y, Shen P, Yu Z, et al. Phase iii study of dulanermin (Recombinant human tumor necrosis factor-related apoptosis-inducing Ligand/Apo2 ligand) combined with vinorelbine and cisplatin in patients with advanced non-Small-Cell lung cancer. Invest New Drugs (2018) 36(2):315–22. doi: 10.1007/s10637-017-0536-y
54. Snajdauf M, Havlova K, Vachtenheim J Jr., Ozaniak A, Lischke R, Bartunkova J, et al. The trail in the treatment of human cancer: An update on clinical trials. Front Mol Biosci (2021) 8:628332. doi: 10.3389/fmolb.2021.628332
55. Ashkenazi A. Targeting the extrinsic apoptotic pathway in cancer: Lessons learned and future directions. J Clin Invest (2015) 125(2):487–9. doi: 10.1172/jci80420
56. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol (2020) 17(7):395–417. doi: 10.1038/s41571-020-0341-y
57. Yuan K, Sun Y, Zhou T, McDonald J, Chen Y. Parp-1 regulates resistance of pancreatic cancer to trail therapy. Clin Cancer Res (2013) 19(17):4750–9. doi: 10.1158/1078-0432.Ccr-13-0516
58. Haines E, Nishida Y, Carr MI, Montoya RH, Ostermann LB, Zhang W, et al. DNA-Pk Inhibitor peposertib enhances P53-dependent cytotoxicity of DNA double-strand break inducing therapy in acute leukemia. Sci Rep (2021) 11(1):12148. doi: 10.1038/s41598-021-90500-3
59. Ivanov VN, Zhou H, Partridge MA, Hei TK. Inhibition of ataxia telangiectasia mutated kinase activity enhances trail-mediated apoptosis in human melanoma cells. Cancer Res (2009) 69(8):3510–9. doi: 10.1158/0008-5472.Can-08-3883
60. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling pd-L1 expression in cancer. Mol Cell (2019) 76(3):359–70. doi: 10.1016/j.molcel.2019.09.030
61. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. Pd-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol (2021) 18(6):345–62. doi: 10.1038/s41571-021-00473-5
62. Phukan S, Babu VS, Kannoji A, Hariharan R, Balaji VN. Gsk3beta: Role in therapeutic landscape and development of modulators. Br J Pharmacol (2010) 160(1):1–19. doi: 10.1111/j.1476-5381.2010.00661.x
63. Lin J, Song T, Li C, Mao W. Gsk-3β in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim Biophys Acta Mol Cell Res (2020) 1867(5):118659. doi: 10.1016/j.bbamcr.2020.118659
64. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. Parp inhibitor upregulates pd-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res (2017) 23(14):3711–20. doi: 10.1158/1078-0432.Ccr-16-3215
65. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun (2016) 7:12632. doi: 10.1038/ncomms12632
66. Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA Double-strand break repair pathway regulates pd-L1 expression in cancer cells. Nat Commun (2017) 8(1):1751. doi: 10.1038/s41467-017-01883-9
67. Sun LL, Yang RY, Li CW, Chen MK, Shao B, Hsu JM, et al. Inhibition of atr downregulates pd-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res (2018) 8(7):1307–16.
68. Tang Z, Pilié PG, Geng C, Manyam GC, Yang G, Park S, et al. Atr inhibition induces Cdk1-spop signaling and enhances anti-Pd-L1 cytotoxicity in prostate cancer. Clin Cancer Res (2021) 27(17):4898–909. doi: 10.1158/1078-0432.Ccr-21-1010
69. Carr MI, Chiu LY, Guo Y, Xu C, Lazorchak AS, Yu H, et al. DNA-Pk Inhibitor peposertib amplifies radiation-induced inflammatory micronucleation and enhances Tgfβ/Pd-L1 targeted cancer immunotherapy. Mol Cancer Res (2022) 20(4):568–82. doi: 10.1158/1541-7786.Mcr-21-0612
70. Mouw KW, Konstantinopoulos PA. From checkpoint to checkpoint: DNA damage Atr/Chk1 checkpoint signalling elicits pd-L1 immune checkpoint activation. Br J Cancer (2018) 118(7):933–5. doi: 10.1038/s41416-018-0017-x
71. Wang Z, Zhang X, Li W, Su Q, Huang Z, Zhang X, et al. Atm/Nemo signaling modulates the expression of pd-L1 following docetaxel chemotherapy in prostate cancer. J Immunother Cancer (2021) 9(7):e001758. doi: 10.1136/jitc-2020-001758
72. Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, et al. Review of poly (Adp-ribose) polymerase (Parp) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr (2014) 24(1):15–28. doi: 10.1615/critreveukaryotgeneexpr.2013006875
73. Lee EK, Konstantinopoulos PA. Parp inhibition and immune modulation: Scientific rationale and perspectives for the treatment of gynecologic cancers. Ther Adv Med Oncol (2020) 12:1758835920944116. doi: 10.1177/1758835920944116
74. D'Andrea AD. Mechanisms of parp inhibitor sensitivity and resistance. DNA Repair (Amst) (2018) 71:172–6. doi: 10.1016/j.dnarep.2018.08.021
75. Risdon EN, Chau CH, Price DK, Sartor O, Figg WD. Parp inhibitors and prostate cancer: To infinity and beyond brca. Oncologist (2021) 26(1):e115–e29. doi: 10.1634/theoncologist.2020-0697
76. First parp inhibitor ok'd for breast cancer. Cancer Discovery (2018) 8(3):256–7. doi: 10.1158/2159-8290.Cd-nb2018-008
77. Foo T, George A, Banerjee S. Parp inhibitors in ovarian cancer: An overview of the practice-changing trials. Genes Chromosomes Cancer (2021) 60(5):385–97. doi: 10.1002/gcc.22935
78. Vikas P, Borcherding N, Chennamadhavuni A, Garje R. Therapeutic potential of combining parp inhibitor and immunotherapy in solid tumors. Front Oncol (2020) 10:570. doi: 10.3389/fonc.2020.00570
79. Mohiuddin IS, Kang MH. DNA-Pk As an emerging therapeutic target in cancer. Front Oncol (2019) 9:635. doi: 10.3389/fonc.2019.00635
80. Prakash A, Garcia-Moreno JF, Brown JAL, Bourke E. Clinically applicable inhibitors impacting genome stability. Molecules (2018) 23(5):1166. doi: 10.3390/molecules23051166
81. Topatana W, Juengpanich S, Li S, Cao J, Hu J, Lee J, et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J Hematol Oncol (2020) 13(1):118. doi: 10.1186/s13045-020-00956-5
82. Sun X, Liu T, Zhao J, Xia H, Xie J, Guo Y, et al. DNA-Pk Deficiency potentiates cgas-mediated antiviral innate immunity. Nat Commun (2020) 11(1):6182. doi: 10.1038/s41467-020-19941-0
83. Yang H, Yao F, Marti TM, Schmid RA, Peng RW. Beyond DNA repair: DNA-pkcs in tumor metastasis, metabolism and immunity. Cancers (Basel) (2020) 12(11):3389. doi: 10.3390/cancers12113389
84. Chen Y, Li Y, Guan Y, Huang Y, Lin J, Chen L, et al. Prevalence of prkdc mutations and association with response to immune checkpoint inhibitors in solid tumors. Mol Oncol (2020) 14(9):2096–110. doi: 10.1002/1878-0261.12739
85. Nakamura K, Karmokar A, Farrington PM, James NH, Ramos-Montoya A, Bickerton SJ, et al. Inhibition of DNA-Pk with Azd7648 sensitizes tumor cells to radiotherapy and induces type I ifn-dependent durable tumor control. Clin Cancer Res (2021) 27(15):4353–66. doi: 10.1158/1078-0432.Ccr-20-3701
86. Sun W, Zhang Q, Wang R, Li Y, Sun Y, Yang L. Targeting DNA damage repair for immune checkpoint inhibition: Mechanisms and potential clinical applications. Front Oncol (2021) 11:648687. doi: 10.3389/fonc.2021.648687
87. Bendell JC, Shafique MR, Perez B, Chennoufi S, Beier F, Trang K, et al. Phase 1, open-label, dose-escalation study of M3814 + avelumab ± radiotherapy (Rt) in patients (Pts) with advanced solid tumors. J Clin Oncol (2019) 37(15_suppl):TPS3169–TPS. doi: 10.1200/JCO.2019.37.15_suppl.TPS3169
88. Spencer KR. Testing the combination of new anti-cancer drug peposertib with avelumab and radiation therapy for Advanced/Metastatic solid tumors and hepatobiliary malignancies (2019).
89. Gray H. Radiation medication (Radium-223 dichloride) versus radium-223 dichloride plus radiation enhancing medication (M3814) versus radium-223 dichloride plus M3814 plus avelumab (a type of immunotherapy) for advanced prostate cancer not responsive to hormonal therapy (2019).
90. Ghelli Luserna di Rorà A, Cerchione C, Martinelli G, Simonetti G. A Wee1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J Hematol Oncol (2020) 13(1):126. doi: 10.1186/s13045-020-00959-2
91. Sun L, Moore E, Berman R, Clavijo PE, Saleh A, Chen Z, et al. Wee1 kinase inhibition reverses G2/M cell cycle checkpoint activation to sensitize cancer cells to immunotherapy. OncoImmunology (2018) 7(10):e1488359. doi: 10.1080/2162402X.2018.1488359
92. Guo E, Xiao R, Wu Y, Lu F, Liu C, Yang B, et al. Wee1 inhibition induces anti-tumor immunity by activating erv and the dsrna pathway. J Exp Med (2022) 219(1):e20210789. doi: 10.1084/jem.20210789
93. Friedman J, Morisada M, Sun L, Moore EC, Padget M, Hodge JW, et al. Inhibition of Wee1 kinase and cell cycle checkpoint activation sensitizes head and neck cancers to natural killer cell therapies. J Immunother Cancer (2018) 6(1):59. doi: 10.1186/s40425-018-0374-2
94. Patel MR, Falchook GS, Wang JS-Z, Rodrigo Imedio E, Kumar S, Motlagh P, et al. Open-label, multicenter, phase I study to assess safety and tolerability of adavosertib plus durvalumab in patients with advanced solid tumors. J Clin Oncol (2019) 37(15_suppl):2562. doi: 10.1200/JCO.2019.37.15_suppl.2562
95. Kong A, Mehanna H. Wee1 inhibitor: Clinical development. Curr Oncol Rep (2021) 23(9):107. doi: 10.1007/s11912-021-01098-8
96. Qiu Z, Oleinick NL, Zhang J. Atr/Chk1 inhibitors and cancer therapy. Radiother Oncol (2018) 126(3):450–64. doi: 10.1016/j.radonc.2017.09.043
97. Barnieh FM, Loadman PM, Falconer RA. Progress towards a clinically-successful atr inhibitor for cancer therapy. Curr Res Pharmacol Drug Discovery (2021) 2:100017. doi: 10.1016/j.crphar.2021.100017
98. Brooks T, Wayne J, Massey AJ. Checkpoint kinase 1 (Chk1) inhibition fails to activate the stimulator of interferon genes (Sting) innate immune signalling in a human coculture cancer system. Mol BioMed (2021) 2(1):19. doi: 10.1186/s43556-021-00044-1
99. Wayne J, Brooks T, Landras A, Massey AJ. Targeting DNA damage response pathways to activate the sting innate immune signaling pathway in human cancer cells. FEBS J (2021) 288(15):4507–40. doi: 10.1111/febs.15747
100. Proctor M, Gonzalez Cruz JL, Daignault-Mill SM, Veitch M, Zeng B, Ehmann A, et al. Targeting replication stress using Chk1 inhibitor promotes innate and nkt cell immune responses and tumour regression. Cancers (Basel) (2021) 13(15):3733. doi: 10.3390/cancers13153733
101. Chaudhary R, Slebos R, Song F, McCleary-Sharpe K, Masannat J, Tan AC, et al. Effects of prexasertib, a Chk1 inhibitor, in the immune microenvironment of head and neck squamous cell carcinoma (Hnscc). J Clin Oncol (2020) 38(15_suppl):e18541–e. doi: 10.1200/JCO.2020.38.15_suppl.e18541
102. Chao HH, Karagounis IV, Thomas C, François NB, Facciabene A, Koumenis C, et al. Combination of Chek1/2 inhibition and ionizing radiation results in abscopal tumor response through increased micronuclei formation. Oncogene (2020) 39(22):4344–57. doi: 10.1038/s41388-020-1300-x
103. Kastan MB, Lim DS. The many substrates and functions of atm. Nat Rev Mol Cell Biol (2000) 1(3):179–86. doi: 10.1038/35043058
104. Petersen AJ, Rimkus SA, Wassarman DA. Atm kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in drosophila. Proc Natl Acad Sci USA (2012) 109(11):E656–64. doi: 10.1073/pnas.1110470109
105. Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, et al. Atm inhibition enhances cancer immunotherapy by promoting mtdna leakage and Cgas/Sting activation. J Clin Invest (2021) 131(3):e139333. doi: 10.1172/jci139333
106. Yi R, Lin A, Cao M, Xu A, Luo P, Zhang J. Atm mutations benefit bladder cancer patients treated with immune checkpoint inhibitors by acting on the tumor immune microenvironment. Front Genet (2020) 11:933. doi: 10.3389/fgene.2020.00933
107. Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, et al. Inhibition of atm increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res (2019) 79(15):3940–51. doi: 10.1158/0008-5472.Can-19-0761
108. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic Biol Med (2010) 49(11):1603–16. doi: 10.1016/j.freeradbiomed.2010.09.006
109. Conklin KA. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr Cancer Ther (2004) 3(4):294–300. doi: 10.1177/1534735404270335
110. Davalli P, Marverti G, Lauriola A, D'Arca D. Targeting oxidatively induced DNA damage response in cancer: Opportunities for novel cancer therapies. Oxid Med Cell Longev (2018) 2018:2389523. doi: 10.1155/2018/2389523
111. Hossain MA, Lin Y, Driscoll G, Li J, McMahon A, Matos J, et al. Ape2 is a general regulator of the atr-Chk1 DNA damage response pathway to maintain genome integrity in pancreatic cancer cells. Front Cell Dev Biol (2021) 9:738502. doi: 10.3389/fcell.2021.738502
112. Kotsafti A, Scarpa M, Castagliuolo I, Scarpa M. Reactive oxygen species and antitumor immunity-from surveillance to evasion. Cancers (Basel) (2020) 12(7):1748. doi: 10.3390/cancers12071748
113. Jensen KA, Shi X, Yan S. Genomic alterations and abnormal expression of Ape2 in multiple cancers. Sci Rep (2020) 10(1):3758. doi: 10.1038/s41598-020-60656-5
114. Abad E, Graifer D, Lyakhovich A. DNA Damage response and resistance of cancer stem cells. Cancer Lett (2020) 474:106–17. doi: 10.1016/j.canlet.2020.01.008
115. Lei MML, Lee TKW. Cancer stem cells: Emerging key players in immune evasion of cancers. Front Cell Dev Biol (2021) 9:692940. doi: 10.3389/fcell.2021.692940
Keywords: DNA damage response (DDR), immunotherapy, cGAS/STING, DNA-PK, WEE1, CHK1/2, ATR, ATM
Citation: Carlsen L and El-Deiry WS (2022) Anti-cancer immune responses to DNA damage response inhibitors: Molecular mechanisms and progress toward clinical translation. Front. Oncol. 12:998388. doi: 10.3389/fonc.2022.998388
Received: 19 July 2022; Accepted: 09 September 2022;
Published: 06 October 2022.
Edited by:
Qiang Zhang, University of Michigan, United StatesReviewed by:
Guang Peng, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2022 Carlsen and El-Deiry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wafik S. El-Deiry, d2FmaWtfZWwtZGVpcnlAYnJvd24uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.