 Leqiang Zhang
Leqiang Zhang Ning Chang4†
Ning Chang4† Linlin Sui
Linlin Sui Wei Chen
Wei Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 29 August 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.987499
This article is part of the Research Topic Linking Cellular Metabolism to Hematological Malignancies, volume II View all 11 articles
Hematological malignancies are one of the most lethal illnesses that seriously threaten human life and health. Lipids are important constituents of various biological membranes and substances for energy storage and cell signaling. Furthermore, lipids are critical in the normal physiological activities of cells. In the process of the lethal transformation of hematological malignancies, lipid metabolism reprogramming meets the material and energy requirements of rapidly proliferating and dividing tumor cells. A large number of studies have shown that dysregulated lipid metabolism, commonly occurs in hematological malignancies, mediating the proliferation, growth, migration, invasion, apoptosis, drug resistance and immune escape of tumor cells. Targeting the lipid metabolism pathway of hematological malignancies has become an effective therapeutic approach. This article reviews the oncogenic mechanisms of lipid metabolism reprogramming in hematological malignancies, including fatty acid, cholesterol and phospholipid metabolism, thereby offering an insight into targeting lipid metabolism in the treatment of hematological malignancies.
Hematological malignancies are a collection of malignant tumors that aberrant hematological cells or immune cells are blocked in differentiation and proliferate indefinitely, leading to the dysfunction of biological organisms (1). There are three main types of hematological malignancies: leukemia, multiple myeloma (MM), and lymphoma (2). Hematological malignancies are one of the most lethal illnesses that seriously threaten human life and health with a high mortality rate. According to the World Cancer Report 2020 released by the World Health Organization, the new number of non-Hodgkin’s lymphoma, leukemia, MM and Hodgkin’s lymphoma in 2020 were 544,352, 474,519, 176,404 and 83,087, respectively, accounting for 6.6% of the total number of patients. The corresponding number of deaths were 259,793, 311,594, 117,077 and 23,376 respectively, accounting for 7.1% of the total number of patients (3). Due to its particularity, hematological malignancies cannot be surgically removed like solid tumors, and its clinical first-line treatment options mainly include chemotherapy, radiotherapy and hematopoietic stem cell transplantation (4). Although the traditional first-line therapies have a certain effect, the overall efficacy is not optimistic due to the relapse/refractory caused by the occurrence of primary/secondary drug resistance (5). With the deepening of research, new cancer therapies have brought dawn to relapsed/refractory patients, including CAR-T cell therapies, ADC drugs and immune checkpoint inhibitors (6). The U.S. Food and Drug Administration approved anti-CD19 CAR-T cell therapy Tisagenlecleucel for the treatment of B-cell acute lymphoblastic leukemia (ALL) (7), ADC drug Loncastuximab Tesirine-Lpyl for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL) (8) and PD-1 inhibitor Pembrolizumab for the treatment of Hodgkin lymphoma (9). However, even under novel therapies, there are still a large number of patients with poor clinical prognosis. Therefore, it is still a work with clinical application value and important scientific significance to study the oncogenic mechanism and find new therapeutic targets in order to develop new therapeutic methods (10).
It has been widely reported that cell metabolism affects tumor cell proliferation, apoptosis, migration, invasion, chemical resistance and immune escape (11–14). There is a close relationship between cellular metabolism and functional output, once the metabolic pathway is abnormal, leading to abnormal cell function and disease progression (15). Compared with normal cells, tumor cells undergo metabolic reprogramming due to their excessive proliferation, growth, migration and metastasis requiring faster and more energy, and tumor cell metabolic reprogramming has been identified as a new marker of cancer (16). Clinical observations found that lipid metabolism reprogramming often predicts poorer prognosis in cancer patients (17). A large number of lipid droplets that store lipids and cholesterol can be detected in tumor cells, high lipid droplets and high cholesterol esters are also considered indicators of cancer aggressiveness in tumor cells (18). De novo lipid synthesis pathway and uptake of exogenous lipids are often enhanced in rapidly dividing and energy-consuming tumor cells, such as malignant plasma cells from obese myeloma patients with high expression of acetyl-CoA synthase 2 (19). Acetyl-CoA synthase 2 is a key precursor for the de novo synthesis of fatty acids (FAs). Fatty acid synthase (FASN) is also upregulated in various hematological malignancies (20). The fatty acid transporter protein (FATP), which mediates cellular uptake of FAs, is expressed at high levels on both the cell surface and the intracellular space of patients with MM (21). Lipids are not only important components of organelles and energy substances, but also signaling molecules that are crucial for maintaining cellular homeostasis (22). Lymphoma-derived exosomes promote tumorigenesis by increasing lipid metabolism in recipient cells through surface phospholipase A2 (23). Lysophosphatidic acid (LPA)-mediated activation of the MEK1/2-ERK1/2 signaling pathway increases oxidative phosphorylation in the mitochondria of MM cells, which in turn produces large amounts of NAD+ and ATP. It impairs the activity of proteasome inhibitors and enhances protein folding in the endoplasmic reticulum (ER), thereby conferring resistance to proteasome inhibitors in MM (24). In this review, we discuss the lipid metabolism reprogramming and its oncogenic mechanisms in hematological malignancies, including FA metabolism, cholesterol metabolism, phospholipid metabolism and lipid-related signaling pathways.
FAs are essential molecules in the entire lipid metabolism, not only involved in the synthesis of biological membranes and secondary signaling molecules, but also substrates for mitochondrial ATP and NADH synthesis, eicosanoid production and post-translational protein–lipid modifications of signaling proteins (25, 26). As early as 1924, Warburg proposed that even under sufficient oxygen conditions, tumor cells also prefer the low-utilization form of glycolysis for energy production (27). Even if tumor cells use a large amount of carbohydrates, it is challenging to meet the needs of energy substances, so lipid metabolism is also required for energy. Tumor cells increase lipid metabolism and energy supply mainly by enhancing the de novo synthesis pathway of endogenous FAs, exogenous FAs uptake and lipid mobilization (28). De novo FA synthesis mainly depends on two key rate-limiting enzymes, acetyl-CoA carboxylase (ACC) carboxylates acetyl-CoA to malonyl-CoA, and FASN converts acetyl-CoA and malonyl-CoA Conversion of acyl-CoA to long-chain FAs (Figure 1) (29). It has been reported that the FA de novo synthesis pathway is generally up-regulated in tumor cells, and FASN overexpression has been an independent prognostic marker for the aggressive clinical course of tumor cells (30). MYC+ BCL-2+ DLBCL with high expression of FASN has the characteristics of high invasion and poor prognosis (31). Up-regulated FASN can promote the growth, metastasis, invasion and anti-apoptosis of DLBCL through the pERK/BCL-2 signaling pathway, and FASN inhibition can cause cell growth arrest and apoptosis (32). Overexpressed FASN can also regulate the PI3K/Akt signaling pathway in DLBCL, prompting p70-S6 kinase to phosphorylate USP11. The phosphorylated USP11 mediates eIF4B deubiquitination to increase its stability, and eIF4B promotes key oncogenes biosynthesis, ultimately driving the development of lymphoma (33). PARK2 can also be phosphorylated by mTORC1 to lose its ubiquitination activity, thereby blocking the ubiquitination and degradation of eIF4B protein (Figure 1) (34). Meanwhile, up-regulation of FASN has been reported in acute myeloid leukemia (AML) (35), mantle cell lymphoma (MCL) (36), and MM (37).
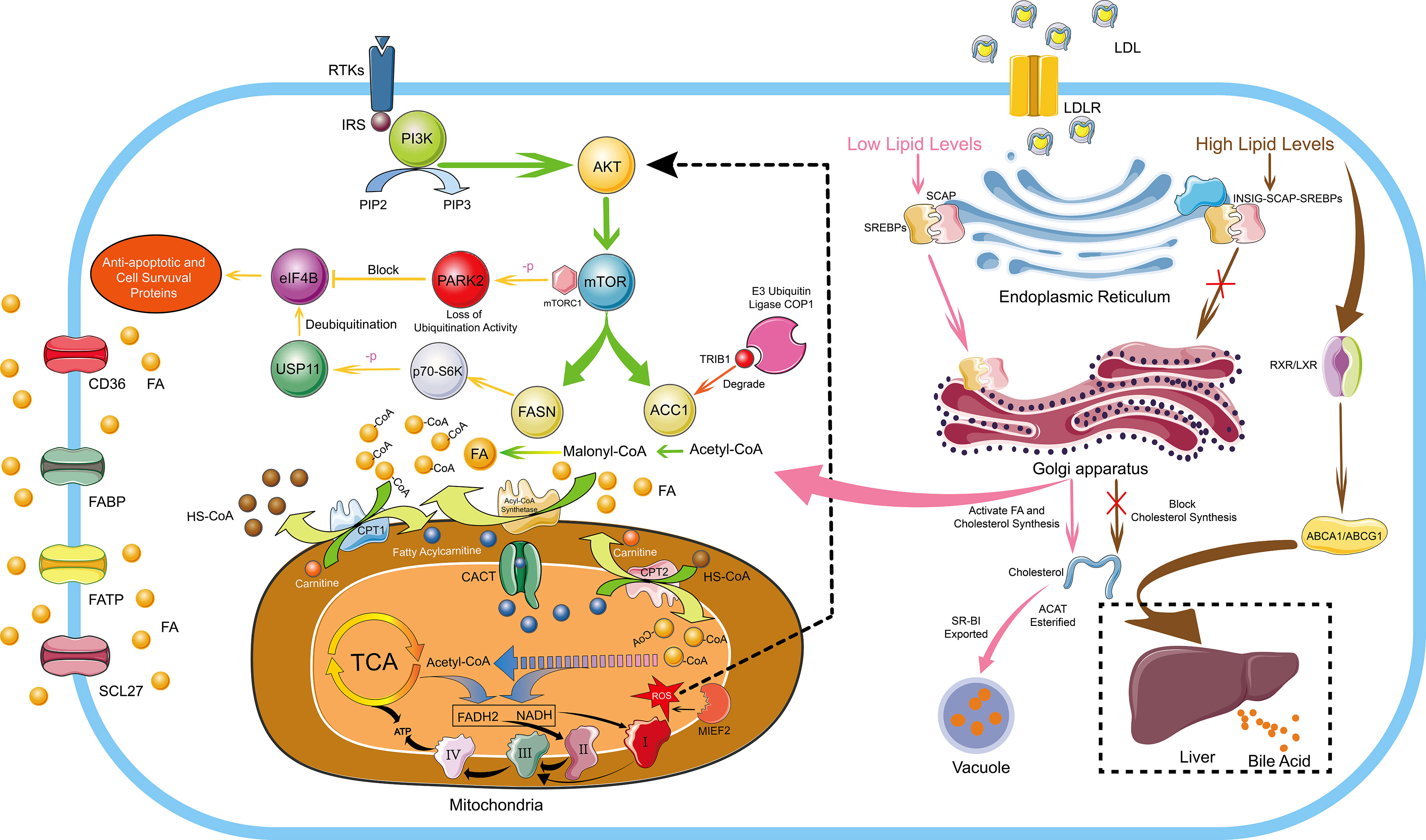
Figure 1 FA And Cholesterol Metabolism in Hematological Malignancies. The de novo synthesis of FA is regulated by the PI3K/Akt signaling pathway. De novo FA synthesis mainly depends on two key rate-limiting enzymes, ACC carboxylates acetyl-CoA to malonyl-CoA, and FASN converts acetyl-CoA and malonyl-CoA Conversion of acyl-CoA to long-chain FA. ACC and FASN are regulated by mTOR, which is a downstream target of the PI3K/Akt signaling pathway. Overexpressed FASN can prompt p70-S6 kinase to phosphorylate USP11 and the phosphorylated USP11 mediates eIF4B deubiquitination to increase its stability. eIF4B mediates the expression of anti-apoptotic and cell survival proteins, ultimately driving the development of lymphoma. In addition, PARK2 can also be phosphorylated by mTORC1 to lose its ubiquitination activity, thereby blocking the ubiquitination and degradation of eIF4B protein. The E3 ubiquitin ligase COP1 binds to ACC1 through Trib1 and causes ACC1 ubiquitination and degradation to inhibit FA synthesis. Cells uptake exogenous FAs mainly through CD36, FATP, FABP and SCL27. Endogenous FAs and exogenous FAs enter the mitochondria through the transmembrane mechanism, and generate a large amount of ATP through FA β-oxidation, the TCA cycle and the electron transport chains. Moreover, MIEF2, a key regulator of mitochondrial fission, can stimulate the production of mitochondrial ROS and activate the AKT/mTOR signaling pathway to further enhance FA synthesis. Cholesterol homeostasis is mainly regulated by SREBPs and LXRs. In conditions of low cholesterol levels, SCAP-SREBP2 can be smoothly transferred from the ER to the Golgi apparatus to activate the de novo synthesis pathway of cholesterol. In conditions of high cholesterol levels, INSIG, SCAP and SREBP2 form a stable trimolecular complex to block the export and activation of SREBP2, and finally inhibiting the de novo cholesterol synthetic route. Moreover, by activating LXR-RXR to express ABCA1 and ABCG1, excessive cholesterol is transported to the liver and excreted in the form of bile acids. Furthermore, excessive cholesterol is rapidly esterified and exported under the action of ACAT and SR-BI to form vacuoles containing cholesterol ester derivatives. I, II, III and IV represent complex I, complex II, complex III and complex IV in the electron transport chains, respectively.
There is a lack of reports about ACC1 in hematological malignancies, only one recent paper mentioned the function of ACC1 to suppress tumors. The E3 ubiquitin ligase COP1 binds to ACC1 through Trib1 and causes ACC1 ubiquitination and degradation to inactivate its biological activity, which leads metabolic reprogramming to support the energy requirements of leukemia progression. A general downregulation of ACC1 can be observed in AML. However, stabilizing the biological activity of ACC1 protein can increase intracellular ROS levels and NADPH consumption, thereby inhibiting leukemia progression, which may be caused by the material and energy competition conflict between ACC1-mediated FA synthesis and tumor cell proliferation (38). As an effective strategy for the treatment of patients with AML and endemic Burkitt lymphoma, the drug combination of bezafibrate and medroxyprogesterone acetate can effectively reduce the expression of FASN and stearoyl-CoA desaturase 1. However, ACC1, which is also a key enzyme in lipid synthesis, did not show a significant change in expression (39). The role of ACC1 remains unknown in hematological malignancies. Interestingly, ACC1 promotes tumor cell growth in some solid tumors. MIEF2, a key regulator of mitochondrial fission, stimulates the production of mitochondrial ROS and activates the AKT/mTOR signaling pathway. The result causes the upregulation of ACC1, FASN, SREBP1/2, SCD1, HMGCS1 and HMGCR to increase lipid synthesis, ultimately promoting the growth and metastasis of ovarian cancer cells (40). Overexpression of the long non-coding RNA (lncRNA, CTD-2245E15) in lung cancer can also regulate ACC1 and pyruvate carboxylase to promote lung cancer development (41).
Tumor cells enhance the uptake of exogenous FAs, mainly through the CD36, FATP, lipid chaperone FA binding protein (FABP) and solute carrier protein family 27 (SCL27). Up-regulation of CD36 has been reported in hematological malignancies such as AML (42), chronic lymphocytic leukemia (CLL) (43), MM (44), DLBCL (30), and MCL (45). Sudjit Luanpitpong (45) used Synchrotron-Based Fourier Transform Infrared Spectroscopy of Single Cells to detect significant increases in total lipids and lipid esters in MCL resistant to Bortezomib (BZ). BZ is a protease inhibitor that leads to accumulation of misfolded and unfolded proteins, mainly by inhibiting protein degradation, ultimately causing the ER stress response. Subsequent Oil Red O staining detection revealed significant lipid droplets accumulation in BZ-resistant MCL cells. Detection of lipid metabolism-related targets revealed increased expression of CD36 protein responsible for exogenous FAs uptake, and CD36 inhibited apoptosis in MCL, which was associated with BZ-resistance. It has been reported that BTK inhibitors can inhibit lipid droplet accumulation in MCL (46). Moreover, CD36 is associated with tumor invasion and metastasis and is a prognostic biomarker for various types of cancer (47–49). Apolipoprotein C2, which is highly expressed in AML, can interact with CD36 to activate LYN-ERK signaling and enhance the metabolic activity of leukemia cells (42). CD36 was also found to promote FAs uptake by activating STAT3 in CLL (43). Up-regulation of CD36 is also one of the reasons why tumor cells develop drug resistance (50). Exogenous interleukin 6 (IL-6) mediates the up-regulation of CD36 by activating STAT3, promoting the uptake of FAs and causing chemotherapy resistance (51). CD36 also induces lipid peroxidation and ferroptosis with concomitant reduction of cytotoxic cytokines and impaired antitumor capacity (52). In addition to the drug resistance of tumor cells caused by increased lipid synthesis, decreased lipid synthesis can also lead to drug resistance. BZ exerts its anti-tumor effect through ER stress caused by the protein accumulation (53), because the ER is the site of lipid synthesis, and BZ also causes lipid accumulation (45). In BZ-resistant MM, it was found that the expression of SREBP1 and its downstream target FA elongase ELOVL6 are reduced, resulting in inhibiting lipid synthesis, thereby reducing the accumulation on the ER and ultimately causing BZ-resistance (54). In addition to CD36 mediating cellular uptake of FAs, FATP also mediates cellular uptake of FAs. FATP is expressed at high levels on the cell surface and intracellular space in MM. Furthermore, MM cells can induce lipolysis of bone marrow adipocytes, and the decomposed free fatty acids (FFA) are taken up by adjacent MM cells through FATP (21). FABP and SCL27 were also observed to be up-regulated in tumor cells, increasing the uptake of exogenous FAs of tumor cells (55–58). Glycoprotein prostaglandin D2 synthase (PTGDS) has dual roles in prostaglandin metabolism and lipid transport. More interestingly, PTGDS exhibits different functions in different tumor cells. PTGDS promotes DLBCL progression by regulating tumor cell viability, proliferation, cell cycle, apoptosis and invasion through MYH9 stimulated Wnt-β-catenin-STAT3 signaling pathway (59). However, PTGDS showed antitumor effect in testicular cancer (60), gastric cancer (61) and breast cancer (62).
Fatty acid oxidation (FAO) provides energy mainly through FA β-oxidation. In order to successfully carry out FAO, FAs first need to enter the mitochondria. Firstly, long-chain FAs need to generate fatty acyl-CoA under the action of fatty acyl-CoA synthase. Fatty acyl-CoA is converted to fatty acylcarnitine under the action of CPT1, which is then transported into the mitochondrial matrix by carnitine/acylcarnitine translocase (CACT) on the inner mitochondrial membrane, and fatty acylcarnitine entering the mitochondrial matrix are reconverted to fatty acyl-CoA by CPT2. The fatty acyl-CoA that smoothly enters the mitochondria repeats the cycle of dehydrogenation, water addition, dehydrogenation, and thiolysis, and finally decomposes the fatty acyl-CoA into acetyl-CoA, accompanied by the generation of a large amount of NADH and FADH2. These substances eventually enter the TCA cycle and the electron transport chains to be oxidized to generate a large amount of ATP for cellular physiological activities (Figure 1) (28). FAO is dysregulated in a variety of malignancies, and it mediates tumor cell proliferation, survival, drug resistance, metastatic progression, immunosuppression and tumor-promoting microenvironment (63). As an enzyme involved in FAO, HADHB is commonly overexpressed in malignant lymphomas and is a poor prognosis predictor in DLBCL, and high expression of HADHB promotes the proliferation and growth of malignant lymphomas (64). HADHA, which forms a heterodimer together with HADHB, is also widely up-regulated in malignant lymphomas, and down-regulation of HADHA can cause G0/G1 cell cycle arrest (65). Acyl-CoA oxidase 1 (ACOX1), a key rate-limiting enzyme in FAO, is overexpressed in malignant lymphomas and confers resistance to the anthracycline antibiotic doxorubicin, mainly by reducing doxorubicin-induced activation of caspase-9 and caspase-3 and reduction of mitochondrial membrane potential. Simultaneously, ACOX1 can also destabilize the tumor suppressor gene family p73 protein and inhibit its expression (66).
Lipid metabolism can affect the immune system in tumor cells, causing immune evasion and promoting tumor growth. Natural killer (NK) cells play an important role in the prevention of hematological malignancies. However, FAs, both in lymphoma cells and in the tumor microenvironment, can reprogram lipid metabolism in NK cells and inhibit the production of cytokines such as IFN-γ, making NK cells lose their immune function to tumor cells (67). The prostaglandin PGD 2, a lipid compound of the eicosanoid family, is abundantly generated under the catalysis of cyclooxygenase overexpressed in tumor cells. PGD 2 can stimulate innate lymphocytes ILC2 to overexpress IL-5, which subsequently promotes the proliferation of Tregs cells. Tregs cells can be involved in immunosuppression, such as inhibition of T effector cell proliferation and production restriction inflammatory response factor IL-10, ultimately promoting the proliferation of hematological stem and progenitor cells (68). Tumor-associated macrophages (TAMs) up-regulate CD36 to uptake lipids resulting in lipid accumulation. Excess lipids provide a large amount of energy through FAO and lead to activation of STAT6, which is accompanied by TAMs differentiation and cancer promotion (69).
Since FA biosynthesis, uptake, and oxidation are significantly enhanced in various types of tumor cells, inhibiting FA mobilization has become a promising antitumor strategy in tumor cells. FASN, a key rate-limiting enzyme in the de novo synthesis pathway of endogenous FAs, is associated with multidrug resistance in tumor cells (70) and is an effective target for the treatment of malignant tumors. Clinically, glucocorticoids such as prednisone and dexamethasone can inhibit the expression of FASN and thereby inhibit the proliferation and growth of tumor cells (71). At the same time, the study found that ginger extract can inhibit the expression of FASN, and combined use with dexamethasone can enhance the drug sensitivity of ALL cells to dexamethasone (72). The combination of bezafibrate and medroxyprogesterone acetate (39), orlistat (73), N-phenylmaleimide (74) and methyl jasmonate (75) can all regulate the expression of FASN to inhibit the growth of tumor cells. In leukemia cells, FABP4 regulates DNMT1 expression through the IL-6/STAT3 axis and DNMT1 controls FABP4 through VEGF signaling, thereby forming a mutually reinforcing positive feedback regulation that ultimately promotes AML aggressiveness. The selective inhibitor BMS309403 can cause FABP4 dysfunction, which in turn promotes the downregulation of DNMT1. Subsequent induction of global DNA methylation and re-expression of tumor suppressor genes ultimately induce AML cell differentiation and inhibit AML progression (76).
Cholesterol is an important substance for cell function, and cholesterol homeostasis is essential for the normal physiological activities of the body (77). Cholesterol homeostasis is mainly regulated by two transcription factor families, sterol regulator element binding proteins (SREBPs) and liver X receptors (LXRs). SREBPs mediate lipid synthesis and LXRs mediate cholesterol transport. Andrea Brendolan has made a detailed summary of cholesterol homeostasis regulation. In brief, in conditions of low cholesterol levels, SREBP2 is escorted to the Golgi apparatus by SREBP cleavage activator protein (SCAP), where a series of biological reactions activate cholesterol synthesis, and LXRs are in an inhibited state at this time. In conditions of high cholesterol levels, INSIG, SCAP and SREBP2 form a stable trimolecular complex in the ER, thereby blocking the export and activation of SREBP2 and finally blocking the de novo cholesterol synthetic route. Moreover, excess oxysterols or desmosterol bind and activate the LXR/RXR heterodimer, which in turn activates specific LXR target genes, such as ATP-binding cassette transporters A1 and G1 (ABCA1 and ASCG1), allowing excess cholesterol to be transported to the liver and excreted as bile acids (Figure 1) (78).
The mechanisms that maintain cholesterol homeostasis are disrupted in tumor cells due to their addiction to cholesterol. It has been reported that a large amount of cholesterol is widely present in malignant tumor cells (79). In human hepatocellular carcinoma cells, the stability of the INSIG, SCAP, and SREBP2 trimolecular complex is destabilized by cascade phosphorylation of the AKT-PCK1-INSIG axis. SCAP-SREBP complex is translated to the Golgi apparatus to activate cholesterol synthesis and ultimately promotes the proliferation and growth of tumor cells (80). Up-regulation of LDLR, SREBP2 and nuclear PBR are detected in CLL, which also explains that hypocholesterolemia in lymphocytic leukemia patients is due to over-uptake of LDL particles from plasma by high LDLR expression (81). At the same time, it has also been found that tumor cells can secrete cytokines through autocrine and paracrine mechanisms to stimulate cellular uptake of LDL in AML (82). In T-cell ALL, the Wnt-β-catenin signaling pathway mediates the oncogenic synergy of Akt and Dlx5 by enhancing cholesterol synthesis (83). Yajie Shen (84) found that SOX9 was highly expressed in the GC-DLBCL with IGH-BCL2+ mutation. Through whole transcriptome analysis and chromatin immunoprecipitation sequencing, it was found that SOX9 could directly bind and transcriptionally activate DHCR24, which is a terminal enzyme in cholesterol biosynthesis that catalyzes the conversion of sterol to cholesterol. Using simvastatin to inhibit cholesterol synthesis, it can effectively inhibit the growth of DLBCL and the progression of lymphoma. Moreover, peroxisome proliferator-activated receptor (PPARδ) is co-expressed with cholesterol synthesis-related genes. The expression level of the key cholesterol synthesis enzyme HMGCR increases nearly 4-fold in malignant B cells with high PPARδ gene expression, and a significant increase in membranous cholesterol was also observed in malignant B cells, indicating changes in cell signal pathways (85). In promyelocytic leukemia (APL) driven by the PML-RAPα oncoprotein, it was found that PML-RAPα can reduce the expression of PPARγ. PML-RAPα and TRIB3 cooperate to destroy the PPARγ/RXR heterodimer to inhibit PPARγ activity, eventually causing abnormal blood lipids in APL (86). Although cholesterol is necessary for maintaining cell homeostasis and cancer cell proliferation, excess free cholesterol is harmful to cells (87). Therefore, cholesterol is rapidly esterified and exported under the action of acetyl-coenzyme A:cholesterol acetyltransferase (ACAT) and scavenger receptor class B member I (SR-BI) to form vacuoles containing cholesterol ester derivatives (Figure 1). A large number of vacuoles have been observed in highly aggressive lymphoma cells, which have been shown to contain lipids by Sudan black positive staining, and up-regulation of molecules related to cholesterol metabolism has also been detected (88).
Cholesterol metabolism reprogramming is also one of the reasons for the drug resistance of tumor cells. HMGCS1, a key enzyme in the mevalonate pathway for cholesterol synthesis, is overexpressed by the upstream regulator GATA1 in patients with relapsed/refractory AML. Activated HMGCS1 protects ER and mitochondria by upregulating the unfolded protein response (UPR) signaling pathway to avoid cell damage caused by RE stress and mitochondrial stress, ultimately endow tumor cells with drug resistance (89). In the tumor microenvironment of MM, there is a large number of oxidatively modified low-density lipoproteins (OxLDLs). These OxLDLs make proteasome inhibitors such as BZ lose their inhibitory and pro-apoptotic effects on the proteasome, and finally make MM patients acquire drug resistance (90). Chemotherapy generally causes drug resistance in tumor cells. After chemotherapy of AML, cellular cholesterol biosynthesis is significantly up-regulated and extracellular vesicles carrying a large number of cholesterol synthesis-related enzymes are secreted, which can be uptake by recipient cells to promote cholesterol synthesis and cell signal transduction, resulting in tumor formation (91).
Because cholesterol affects the occurrence and development of tumors, inhibition of cholesterol synthesis has become a new strategy for the treatment of cancer in recent years (92). In DLBCL, the BCR/SYK/PI3K/AKT signaling pathway regulates the biosynthesis of cholesterol (93). Metformin, a commonly used drug for the treatment of diabetes, can reduce the biosynthesis of cholesterol by blocking the BCR signaling pathway and inhibiting the expression of HMGCS1, thereby inhibiting the growth of DLBCL. Furthermore, limiting the biosynthesis of cholesterol affects the integrity and biological function of the cell membrane and the lipid rafts, further blocking the BCR signaling pathway and the cell activity is severely inhibited (94). Nicotinamide phosphoribosyltransferase inhibitor (KPT-9274) can selectively kill leukemia stem cells (LSC) and is an effective treatment for AML. However, due to the up-regulation of SREBP-regulated genes, LSC developed a certain resistance to KPT-9274 (95). Inhibiting the SREBP signaling pathway with dipyridamole can enhance the drug sensitivity of LSC cells to KPT-9274. In addition, simvastatin, a common statin that reduces plasma cholesterol levels, has been reported to promote apoptosis by inhibiting the miR-19a-3p/HIF-1α axis (96). Conversely, increasing cholesterol biosynthesis can also kill tumor cells. Overactivation of ERK/MAPK signaling pathway using limonoid compounds A1542 and A1543 induces excessive cholesterol biosynthesis in leukemia cells, leading to cholesterol accumulation and programmed apoptosis in leukemia cells (97). In addition, blocking cholesterol efflux by SR-BI inhibitor, resulting in intracellular cholesterol accumulation, can also stimulate ER stress response and eventually lead to apoptosis (88).
Phospholipids, as a large class of lipids, are the main components of biological membranes and important signaling molecules for cell proliferation and growth (98). Sphingosine 1-phosphate (S1P), which is generated by phosphorylation of sphingosine by sphingosine kinase 1 (SK1), is an important lipid metabolite that mediates cellular signal transduction (99). There have been numerous reports that S1P mediates tumor cell proliferation, invasion, angiogenesis, and anti-apoptosis (Figure 2) (100, 101). In addition, overexpression of SK1 also induces malignant transformation and promotes tumor proliferation (102). Michael S. Lee (103) found that S1P is up-regulated in MCL, and S1P can inhibit the activation of NK cells and allow MCL cells to escape immune immunity. Once targeting S1P or inhibiting SK1, the killing effect of NK cells can be restored on MCL cells, and accompanied by the up-regulation of cardiolipin, phosphatidylcholine, phosphatidylethanolamine and sphingomyelin, especially the up-regulation of cardiolipin suggests that cardiolipin induce the activation of NK cells. Furthermore, S1P has also been reported to interact with S1PR3 to promote osteosarcoma proliferation, anti-apoptosis and aerobic glycolysis through the YAP/c-MYC/PGAM1 axis (104).
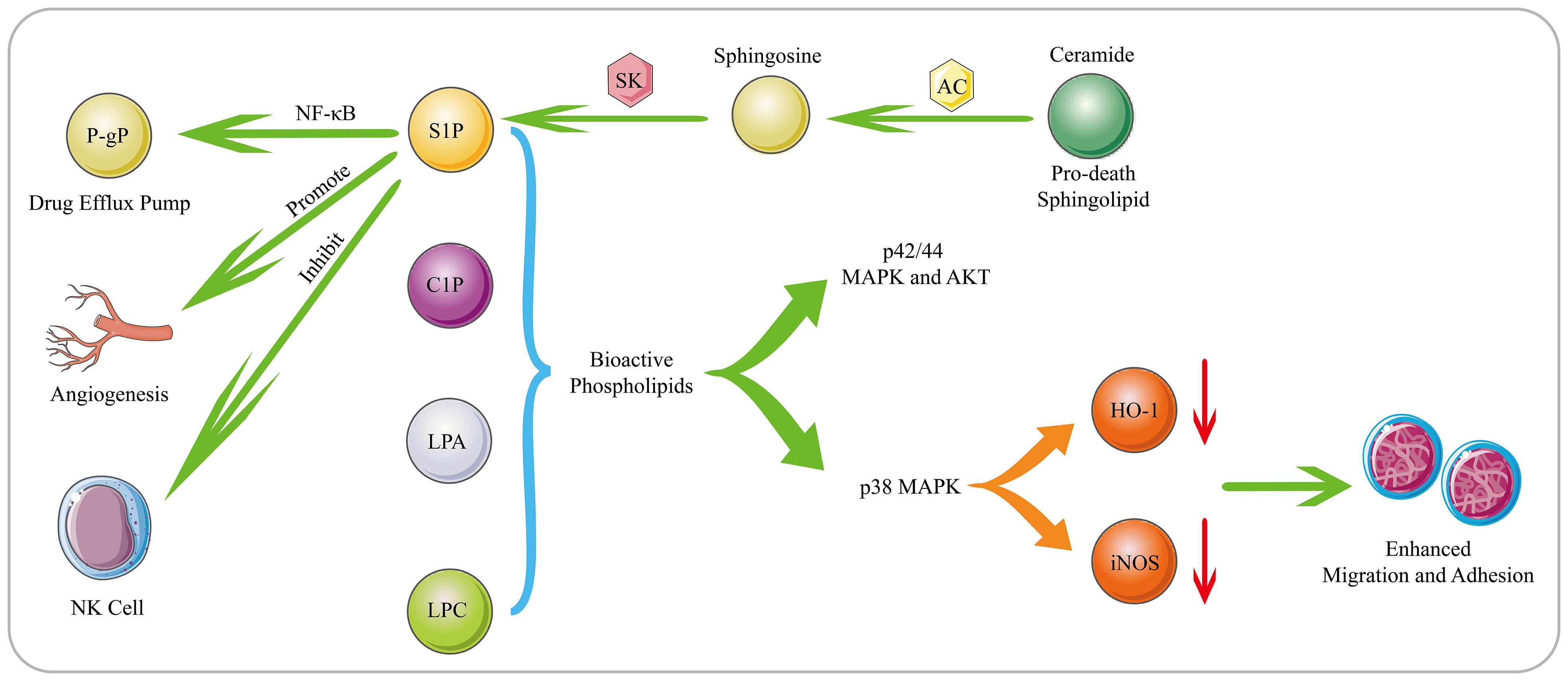
Figure 2 Biological Functions of Bioactive Phospholipids. S1P is generated through phosphorylation of sphingosine by SK. S1P mediates tumor cell proliferation, invasion, angiogenesis, drug resistance and immune escape. Moreover, AC can decompose the pro-death sphingolipid ceramide to generate sphingosine, which subsequently generates S1P. AC and S1P can activate the NF-κB pathway, and mediating the expression of the drug efflux pump P-gp. The bioactive phospholipids such as S1P, C1P, LPC and LPA can stimulate the p42/44 MAPK and AKT signaling pathways. Moreover, as substances that can inhibit the migration of hematological cells, HO-1 and iNOS can be down-regulated by bioactive phospholipids in a p38 MAPK-dependent manner, thereby promoting the migration and adhesion of human leukemia cells.
Phospholipids also play an important role in the occurrence and development of tumor cells and the generation of drug resistance. SK1 is overexpressed in DLBCL, and its downstream product S1P can induce angiogenesis and promote cell proliferation and growth (105). For the increased expression of SK2 in large granular lymphocytic leukemia, inhibiting the expression of SK2 can lead to the degradation of the downstream pro-survival protein Mcl-1, and ultimately induce cell apoptosis (106). In addition to S1P, bioactive phospholipids such as ceramide 1-phosphate (C1P), lysophosphatidylcholine (LPC) and LPA also promote tumor progression. These bioactive phospholipids can stimulate the p42/44 MAPK and AKT signaling pathways. In addition, as substances that can inhibit the migration of hematological cells, HO-1 and iNOS can be down-regulated by bioactive phospholipids in a p38 MAPK-dependent manner, thereby promoting the migration and adhesion of human leukemia cells (107). Lysophospholipase D enzyme converts lysophospholipids into more water-soluble LPA, which activates GPCR-mediated signaling pathways and produces important lipid mediators. They are required to maintain chronic myelogenous leukaemia stem cells function in vivo (108). In MM patients, acid sphingomyelinase (ASM) is significantly up-regulated, which can lead to the increase of ceramide and the decrease of sphingomyelin to cause phospholipid metabolism disorders. The exosomes secreted by MM contain a large amount of ASM, which can make recipient cells resistant to melphalan and BZ (109). Overexpressed Acid ceramidase (AC) in AML can decompose pro-death sphingolipid ceramide to generate sphingosine and FFA, which are converted to S1P by SK1. AC and S1P together stimulate the activation of the NF-κB pathway, which in turn causes the expression of the ATP-binding cassette transporter P-gp. P-gp mediates the excretion of multiple drugs, ultimately conferring resistance to chemotherapeutics in AML (Figure 2) (110).
The rapid growth and continuous invasion of tumor cells require a large amount of energy supply, and metabolic reprogramming is commonly used to meet the material and energy requirements of tumor cells. As an important component of various biological membranes, lipids are also important substances involved in energy storage, production and cell signaling, and play an important role in cell physiological activities (111). Therefore, lipid metabolism reprogramming can be used to meet the material and energy required for rapid proliferation and growth of tumor cells, and lipid metabolism reprogramming has become one of the new markers of cancer (112). In this review, we summarize the proliferation, growth, differentiation, migration, invasion, apoptosis, drug resistance, immune escape and oncogenic mechanisms of tumor cells due to the lipid metabolism reprogramming in hematological malignancies, including FAs, cholesterol, and phospholipids. Tumor cells can increase lipid metabolism by enhancing endogenous lipid de novo synthesis pathway and exogenous lipid uptake, including the overexpression of FASN, ACC1, HMGCR, CD36, FABP and LDLR. Moreover, SREBPs have specific important roles in regulating lipid homeostasis, and SREBPs have three subtypes: SREBP-1a, SREBP-1c and SREBP-2. SREBP-1c mainly regulates the expression of genes required for FA synthesis, while SREBP-1a can regulate FA and cholesterol synthesis, as well as cholesterol uptake. SREBP-2 is relatively specific for the regulation of cholesterol synthesis and uptake (113). SREBPs need to be escorted by SCAP from the ER to the Golgi apparatus to perform their biological functions (114).
Lipid metabolism reprogramming plays an important role in the physiological activities of tumor cells, so targeting the lipid metabolism pathway of tumor cells has become an effective therapeutic approach. For example, inhibition of key rate-limiting enzymes in lipid biosynthesis reduces lipid synthesis, overstimulation of lipid biosynthesis causes ER stress, disruption of mitochondrial oxidative homeostasis causes mitochondrial stress, and blocking lipid-related signaling pathways causes signal pathway dysregulation. However, targeting lipid metabolism reprogramming still faces many challenges for the treatment of hematological malignancies. The main reason is that the relevant mechanisms of lipid metabolism are not fully revealed in hematological malignancies. Therefore, lipid synthesis, storage, utilization and efflux cannot be effectively regulated in hematological malignancies. Ferroptosis, which is a hot research topic in recent years, is also closely related to lipid metabolism. Ferroptosis is a programmed cell death caused by excessive accumulation of iron-dependent lipid peroxidation and reactive oxygen species, and various hematological malignancies are sensitive to ferroptosis. Therefore, ferroptosis is also a promising therapeutic strategy for hematological malignancies. However, what we need to pay attention to is that glucose metabolism, lipid metabolism and amino acid metabolism are interconverted and affect each other, which is a complex connection. These results in that single inhibition of a certain metabolism cannot effectively inhibit the growth of tumor cells because the salvage of other metabolisms is activated. Tumor cells acquire the function of MYC through chromosomal translocations, gene amplifications and single nucleotide polymorphisms, causing a variety of metabolic dysregulations. For example, transporters and enzymes of glycolysis, fatty acid synthesis, glutaminolysis, serine metabolism and mitochondrial metabolism (115). The single inhibition of a downstream metabolic change is not effective in inhibiting cancer development. Therefore, combined inhibition of lipid metabolism, glucose metabolism and amino acid metabolism needs to be considered in clinical applications. In conclusion, understanding the oncogenic mechanism of lipid metabolism and targeting lipid metabolism reprogramming to find new therapeutic targets has important scientific significance and clinical application value. This review provides experience and direction for targeting lipid metabolism reprogramming in the treatment of hematological malignancies.
Conception and design: LZ, LS, and WC. Initial manuscript writing: LZ, NC, LS, and WC. Revision of the manuscript: JL, NC, ZL, and YW. Confirmation of Manuscript: LS and WC. All authors contributed to the article and approved the submitted version.
This review was funded by the National Natural Science Foundation of China (No. 82141113; No. 81901511).
We thank all those who participated in the writing of this article. At the same time, we are very grateful to those who have provided us with excellent advice and encouraged us when we faced challenges.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhang J, Liu Y, Li Q, Xu A, Hu Y, Sun C. Ferroptosis in hematological malignancies and its potential network with abnormal tumor metabolism. BioMed Pharmacother (2022) 148:112747. doi: 10.1016/j.biopha.2022.112747
2. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
3. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
4. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med (2011) 17(9):1086–93. doi: 10.1038/nm.2415
5. Westin J, Sehn LH. Car T cells as a second-line therapy for large b-cell lymphoma: a paradigm shift? Blood (2022) 139(18):2737–46. doi: 10.1182/blood.2022015789
6. Ayyappan S, Maddocks K. Novel and emerging therapies for b cell lymphoma. J Hematol Oncol (2019) 12(1):82–. doi: 10.1186/s13045-019-0752-3
7. Fda approval brings first gene therapy to the united states . Available at: https://www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states (Accessed August 30, 2017).
8. Fda grants accelerated approval to loncastuximab tesirine-lpyl for Large b-cell lymphoma . Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-loncastuximab-tesirine-lpyl-large-b-cell-lymphoma (Accessed August 30, 2017).
9. Shindiapina P, Alinari L. Pembrolizumab and its role in relapsed/refractory classical hodgkin’s lymphoma: evidence to date and clinical utility. Ther Adv Hematol (2018) 9(4):89–105. doi: 10.1177/2040620718761777
10. Lv H, Zhen C, Liu J, Shang P. β-phenethyl isothiocyanate induces cell death in human osteosarcoma through altering iron metabolism, disturbing the redox balance, and activating the mapk signaling pathway. Oxid Med Cell Longev (2020) 2020:5021983–. doi: 10.1155/2020/5021983
11. Han T, Kang D, Ji D, Wang X, Zhan W, Fu M, et al. How does cancer cell metabolism affect tumor migration and invasion? Cell Adh Migr (2013) 7(5):395–403. doi: 10.4161/cam.26345
12. Geng P, Qin W, Xu G. Proline metabolism in cancer. Amino Acids (2021) 53(12):1769–77. doi: 10.1007/s00726-021-03060-1
13. Kikuchi R, Iwai Y, Tsuji T, Watanabe Y, Koyama N, Yamaguchi K, et al. Hypercapnic tumor microenvironment confers chemoresistance to lung cancer cells by reprogramming mitochondrial metabolism in vitro. Free Radic Biol Med (2019) 134:200–14. doi: 10.1016/j.freeradbiomed.2019.01.014
14. Xu W, Liu WR, Xu Y, Tian X, Anwaier A, Su JQ, et al. Hexokinase 3 dysfunction promotes tumorigenesis and immune escape by upregulating monocyte/macrophage infiltration into the clear cell renal cell carcinoma microenvironment. Int J Biol Sci (2021) 17(9):2205–22. doi: 10.7150/ijbs.58295
15. Fajas L. Metabolic control in cancer cells. Ann Endocrinol (Paris) (2013) 74(2):71–3. doi: 10.1016/j.ando.2013.03.021
16. Hönigova K, Navratil J, Peltanova B, Polanska HH, Raudenska M, Masarik M. Metabolic tricks of cancer cells. Biochim Biophys Acta Rev Cancer (2022) 1877(3):188705. doi: 10.1016/j.bbcan.2022.188705
17. Lo Presti C, Fauvelle F, Jacob MC, Mondet J, Mossuz P. The metabolic reprogramming in acute myeloid leukemia patients depends on their genotype and is a prognostic marker. Blood Adv (2021) 5(1):156–66. doi: 10.1182/bloodadvances.2020002981
18. Rozeveld CN, Johnson KM, Zhang L, Razidlo GL. Kras controls pancreatic cancer cell lipid metabolism and invasive potential through the lipase hsl. Cancer Res (2020) 80(22):4932–45. doi: 10.1158/0008-5472.CAN-20-1255
19. Li Z, Liu H, He J, Wang Z, Yin Z, You G, et al. Acetyl-coa synthetase 2: a critical linkage in obesity-induced tumorigenesis in myeloma. Cell Metab (2021) 33(1):78–93.e7. doi: 10.1016/j.cmet.2020.12.011
20. Uddin S, Hussain AR, Ahmed M, Bu R, Ahmed SO, Ajarim D, et al. Inhibition of fatty acid synthase suppresses c-met receptor kinase and induces apoptosis in diffuse large b-cell lymphoma. Mol Cancer Ther (2010) 9(5):1244–55. doi: 10.1158/1535-7163.Mct-09-1061
21. Panaroni C, Fulzele K, Mori T, Siu KT, Onyewadume C, Maebius A, et al. Multiple myeloma cells induce lipolysis in adipocytes and uptake fatty acids through fatty acid transporter proteins. Blood (2022) 139(6):876–88. doi: 10.1182/blood.2021013832
22. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer (2020) 122(1):4–22. doi: 10.1038/s41416-019-0650-z
23. Kudo K, Miki Y, Carreras J, Nakayama S, Nakamoto Y, Ito M, et al. Secreted phospholipase a(2) modifies extracellular vesicles and accelerates b cell lymphoma. Cell Metab (2022) 34(4):615–33.e8. doi: 10.1016/j.cmet.2022.02.011
24. Su P, Xiao L, Ye L, Wang Z, Xiong W, Wang Q, et al. A novel role of lysophosphatidic acid (lpa) in human myeloma resistance to proteasome inhibitors. J Hematol Oncol (2022) 15(1):55. doi: 10.1186/s13045-022-01269-5
25. Fhu CW, Ali A. Fatty acid synthase: an emerging target in cancer. Molecules (2020) 25(17):3935. doi: 10.3390/molecules25173935
26. Hoy AJ, Nagarajan SR, Butler LM. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat Rev Cancer (2021) 21(12):753–66. doi: 10.1038/s41568-021-00388-4
27. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
28. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med (2021) 218(1):e20201606. doi: 10.1084/jem.20201606
29. Jin Z, Chai YD, Hu S. Fatty acid metabolism and cancer. In: Hu S, editor. Cancer metabolomics: methods and applications. Cham: Springer International Publishing (2021). p. 231–41.
30. Danilova OV, Dumont LJ, Levy NB, Lansigan F, Kinlaw WB, Danilov AV, et al. Fasn and Cd36 predict survival in rituximab-treated diffuse large b-cell lymphoma. J Hematop (2013) 6(1):11–8. doi: 10.1007/s12308-012-0166-4
31. Cazzola M. Introduction to a review series: The 2016 revision of the who classification of tumors of hematopoietic and lymphoid tissues. Blood (2016) 127(20):2361–4. doi: 10.1182/blood-2016-03-657379
32. Zhong X, Liu Z, Luo Q, Li J, Zhang W, Shuang Y. Upregulation of fatty acid synthase in myc and bcl-2 double-expressor lymphoma. Oncol Lett (2021) 21(4):245–. doi: 10.3892/ol.2021.12506
33. Kapadia B, Nanaji NM, Bhalla K, Bhandary B, Lapidus R, Beheshti A, et al. Fatty acid synthase induced s6kinase facilitates usp11-eif4b complex formation for sustained oncogenic translation in dlbcl. Nat Commun (2018) 9(1):829. doi: 10.1038/s41467-018-03028-y
34. Kapadia BB, Roychowdhury A, Kayastha F, Nanaji N, Gartenhaus RB. Park2 regulates eif4b-driven lymphomagenesis. Mol Cancer Res (2022) 20(5):735–48. doi: 10.1158/1541-7786.Mcr-21-0729
35. Humbert M, Seiler K, Mosimann S, Rentsch V, Sharma K, Pandey AV, et al. Reducing fasn expression sensitizes acute myeloid leukemia cells to differentiation therapy. Cell Death Differ (2021) 28(8):2465–81. doi: 10.1038/s41418-021-00768-1
36. Gelebart P, Zak Z, Anand M, Belch A, Lai R. Blockade of fatty acid synthase triggers significant apoptosis in mantle cell lymphoma. PloS One (2012) 7(4):e33738. doi: 10.1371/journal.pone.0033738
37. Wang WQ, Zhao XY, Wang HY, Liang Y. Increased fatty acid synthase as a potential therapeutic target in multiple myeloma. J Zhejiang Univ Sci B (2008) 9(6):441–7. doi: 10.1631/jzus.B0740640
38. Ito H, Nakamae I, Kato J-Y, Yoneda-Kato N. Stabilization of fatty acid synthesis enzyme acetyl-coa carboxylase 1 suppresses acute myeloid leukemia development. J Clin Invest (2021) 131(12):e141529. doi: 10.1172/JCI141529
39. Southam AD, Khanim FL, Hayden RE, Constantinou JK, Koczula KM, Michell RH, et al. Drug redeployment to kill leukemia and lymphoma cells by disrupting scd1-mediated synthesis of monounsaturated fatty acids. Cancer Res (2015) 75(12):2530–40. doi: 10.1158/0008-5472.Can-15-0202
40. Zhao S, Cheng L, Shi Y, Li J, Yun Q, Yang H. Mief2 reprograms lipid metabolism to drive progression of ovarian cancer through ros/akt/mtor signaling pathway. Cell Death Dis (2021) 12(1):18–. doi: 10.1038/s41419-020-03336-6
41. Wang C, Meng X, Zhou Y, Yu J, Li Q, Liao Z, et al. Long noncoding rna ctd-2245e15.3 promotes anabolic enzymes acc1 and pc to support non–small cell lung cancer growth. Cancer Res (2021) 81(13):3509–24. doi: 10.1158/0008-5472.CAN-19-3806
42. Zhang T, Yang J, Vaikari VP, Beckford JS, Wu S, Akhtari M, et al. Apolipoprotein C2 - Cd36 promotes leukemia growth and presents a targetable axis in acute myeloid leukemia. Blood Cancer Discov (2020) 1(2):198–213. doi: 10.1158/2643-3230.BCD-19-0077
43. Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, et al. Stat3-activated cd36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget (2018) 9(30):21268–80. doi: 10.18632/oncotarget.25066
44. Kobari L, Auclair M, Piau O, Ferrand N, Zaoui M, Delhommeau F, et al. Circulating cytokines present in multiple myeloma patients inhibit the osteoblastic differentiation of adipose stem cells. Leukemia (2022) 36(2):540–8. doi: 10.1038/s41375-021-01428-6
45. Luanpitpong S, Janan M, Thumanu K, Poohadsuan J, Rodboon N, Klaihmon P, et al. Deciphering the elevated lipid via cd36 in mantle cell lymphoma with bortezomib resistance using synchrotron-based fourier transform infrared spectroscopy of single cells. Cancers (2019) 11(4):576. doi: 10.3390/cancers11040576
46. Liu Z, Liu J, Zhang T, Li L, Zhang S, Jia H, et al. Distinct btk inhibitors differentially induce apoptosis but similarly suppress chemotaxis and lipid accumulation in mantle cell lymphoma. BMC Cancer (2021) 21(1):732. doi: 10.1186/s12885-021-08475-3
47. Luo X, Zheng E, Wei L, Zeng H, Qin H, Zhang X, et al. The fatty acid receptor cd36 promotes hcc progression through activating src/pi3k/akt axis-dependent aerobic glycolysis. Cell Death Dis (2021) 12(4):328. doi: 10.1038/s41419-021-03596-w
48. Sp N, Kang DY, Kim DH, Park JH, Lee HG, Kim HJ, et al. Nobiletin inhibits Cd36-dependent tumor angiogenesis, migration, invasion, and sphere formation through the Cd36/Stat3/Nf-κb signaling axis. Nutrients (2018) 10(6):772. doi: 10.3390/nu10060772
49. Drury J, Rychahou PG, Kelson CO, Geisen ME, Wu Y, He D, et al. Upregulation of Cd36, a fatty acid translocase, promotes colorectal cancer metastasis by increasing Mmp28 and decreasing e-cadherin expression. Cancers (Basel) (2022) 14(1):252. doi: 10.3390/cancers14010252
50. Landberg N, von Palffy S, Askmyr M, Lilljebjörn H, Sandén C, Rissler M, et al. Cd36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica (2018) 103(3):447–55. doi: 10.3324/haematol.2017.169946
51. Zhang Y, Guo H, Zhang Z, Lu W, Zhu J, Shi J. Il-6 promotes chemoresistance via upregulating cd36 mediated fatty acids uptake in acute myeloid leukemia. Exp Cell Res (2022) 415(1):113112. doi: 10.1016/j.yexcr.2022.113112
52. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. Cd36-mediated ferroptosis dampens intratumoral cd8(+) t cell effector function and impairs their antitumor ability. Cell Metab (2021) 33(5):1001–12.e5. doi: 10.1016/j.cmet.2021.02.015
53. Nikesitch N, Lee JM, Ling S, Roberts TL. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin Transl Immunol (2018) 7(1):e1007. doi: 10.1002/cti2.1007
54. Lipchick BC, Utley A, Han Z, Moparthy S, Yun DH, Bianchi-Smiraglia A, et al. The fatty acid elongase elovl6 regulates bortezomib resistance in multiple myeloma. Blood Adv (2021) 5(7):1933–46. doi: 10.1182/bloodadvances.2020002578
55. Konończuk K, Latoch E, Krawczuk-Rybak M, Muszyńska-Rosłan K. Increased levels of adipocyte and epidermal fatty acid-binding proteins in acute lymphoblastic leukemia survivors. J Clin Med (2021) 10(8):1567. doi: 10.3390/jcm10081567
56. Shimizu M, Tachikawa S, Saitoh N, Nakazono K, Yu-Jung L, Suga M, et al. Thalidomide affects limb formation and multiple myeloma related genes in human induced pluripotent stem cells and their mesoderm differentiation. Biochem Biophys Rep (2021) 26:100978. doi: 10.1016/j.bbrep.2021.100978
57. Takahashi-Shishido N, Sugaya M, Morimura S, Suga H, Oka T, Kamijo H, et al. Mycosis fungoides and sézary syndrome tumor cells express epidermal fatty acid-binding protein, whose expression decreases with loss of epidermotropism. J Dermatol (2021) 48(5):685–9. doi: 10.1111/1346-8138.15775
58. Yen MC, Chou SK, Kan JY, Kuo PL, Hou MF, Hsu YL. Solute carrier family 27 member 4 (slc27a4) enhances cell growth, migration, and invasion in breast cancer cells. Int J Mol Sci (2018) 19(11):3434. doi: 10.3390/ijms19113434
59. Hu S, Ren S, Cai Y, Liu J, Han Y, Zhao Y, et al. Glycoprotein ptgds promotes tumorigenesis of diffuse large b-cell lymphoma by myh9-mediated regulation of wnt-β-catenin-stat3 signaling. Cell Death Differ (2022) 29(3):642–56. doi: 10.1038/s41418-021-00880-2
60. Wu CC, Shyu RY, Wang CH, Tsai TC, Wang LK, Chen ML, et al. Involvement of the prostaglandin d2 signal pathway in retinoid-inducible gene 1 (rig1)-mediated suppression of cell invasion in testis cancer cells. Biochim Biophys Acta (2012) 1823(12):2227–36. doi: 10.1016/j.bbamcr.2012.08.013
61. Zhang B, Bie Q, Wu P, Zhang J, You B, Shi H, et al. Pgd2/Ptgdr2 signaling restricts the self-renewal and tumorigenesis of gastric cancer. Stem Cells (2018) 36(7):990–1003. doi: 10.1002/stem.2821
62. Jiang J, Pan W, Xu Y, Ni C, Xue D, Chen Z, et al. Tumour-infiltrating immune cell-based subtyping and signature gene analysis in breast cancer based on gene expression profiles. J Cancer (2020) 11(6):1568–83. doi: 10.7150/jca.37637
63. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang X-Y, et al. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
64. Sekine Y, Yamamoto K, Kurata M, Honda A, Onishi I, Kinowaki Y, et al. Hadhb, a fatty acid beta-oxidation enzyme, is a potential prognostic predictor in malignant lymphoma. Pathology (2022) 54(3):286–93. doi: 10.1016/j.pathol.2021.06.119
65. Yamamoto K, Abe S, Honda A, Hashimoto J, Aizawa Y, Ishibashi S, et al. Fatty acid beta oxidation enzyme hadha is a novel potential therapeutic target in malignant lymphoma. Lab Invest (2020) 100(3):353–62. doi: 10.1038/s41374-019-0318-6
66. Zheng F-M, Chen W-B, Qin T, Lv L-N, Feng B, Lu Y-L, et al. Acox1 destabilizes p73 to suppress intrinsic apoptosis pathway and regulates sensitivity to doxorubicin in lymphoma cells. BMB Rep (2019) 52(9):566–71. doi: 10.5483/BMBRep.2019.52.9.094
67. Kobayashi T, Lam PY, Jiang H, Bednarska K, Gloury R, Murigneux V, et al. Increased lipid metabolism impairs nk cell function and mediates adaptation to the lymphoma environment. Blood (2020) 136(26):3004–17. doi: 10.1182/blood.2020005602
68. Wu L, Lin Q, Ma Z, Chowdhury FA, Mazumder MHH, Du W. Mesenchymal Pgd(2) activates an ilc2-treg axis to promote proliferation of normal and malignant hspcs. Leukemia (2020) 34(11):3028–41. doi: 10.1038/s41375-020-0843-8
69. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res (2020) 80(7):1438–50. doi: 10.1158/0008-5472.CAN-19-2994
70. Wu X, Qin L, Fako V, Zhang JT. Molecular mechanisms of fatty acid synthase (fasn)-mediated resistance to anti-cancer treatments. Adv Biol Regul (2014) 54:214–21. doi: 10.1016/j.jbior.2013.09.004
71. Goossens S, Van Vlierberghe P. Overcoming steroid resistance in t cell acute lymphoblastic leukemia. PloS Med (2016) 13(12):e1002208. doi: 10.1371/journal.pmed.1002208
72. Ghaeidamini Harouni M, Rahgozar S, Rahimi Babasheikhali S, Safavi A, Ghodousi ES. Fatty acid synthase, a novel poor prognostic factor for acute lymphoblastic leukemia which can be targeted by ginger extract. Sci Rep (2020) 10(1):14072–. doi: 10.1038/s41598-020-70839-9
73. Cioccoloni G, Aquino A, Notarnicola M, Caruso MG, Bonmassar E, Zonfrillo M, et al. Fatty acid synthase inhibitor orlistat impairs cell growth and down-regulates pd-l1 expression of a human t-cell leukemia line. J Chemother (2020) 32(1):30–40. doi: 10.1080/1120009x.2019.1694761
74. Rosolen D, Kretzer IF, Winter E, Noldin VF, Rodrigues do Carmo ÍA, Filippin-Monteiro FB, et al. N-phenylmaleimides affect adipogenesis and present antitumor activity through reduction of fasn expression. Chem Biol Interact (2016) 258:10–20. doi: 10.1016/j.cbi.2016.08.005
75. Goel Y, Yadav S, Pandey SK, Temre MK, Maurya BN, Verma A, et al. Tumor decelerating and chemo-potentiating action of methyl jasmonate on a t cell lymphoma in vivo: role of altered regulation of metabolism, cell survival, drug resistance, and intratumoral blood flow. Front Oncol (2021) 11:619351. doi: 10.3389/fonc.2021.619351
76. Yan F, Shen N, Pang JX, Zhao N, Zhang YW, Bode AM, et al. A vicious loop of fatty acid-binding protein 4 and dna methyltransferase 1 promotes acute myeloid leukemia and acts as a therapeutic target. Leukemia (2018) 32(4):865–73. doi: 10.1038/leu.2017.307
77. Shahoei SH, Nelson ER. Nuclear receptors, cholesterol homeostasis and the immune system. J Steroid Biochem Mol Biol (2019) 191:105364. doi: 10.1016/j.jsbmb.2019.04.013
78. Brendolan A, Russo V. Targeting cholesterol homeostasis in hematopoietic malignancies. Blood (2022) 139(2):165–76. doi: 10.1182/blood.2021012788
79. Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab (2020) 2(2):132–41. doi: 10.1038/s42255-020-0174-0
80. Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y, et al. The gluconeogenic enzyme pck1 phosphorylates insig1/2 for lipogenesis. Nature (2020) 580(7804):530–5. doi: 10.1038/s41586-020-2183-2
81. Sankanagoudar S, Singh G, Mahapatra M, Kumar L, Chandra NC. Cholesterol homeostasis in isolated lymphocytes: a differential correlation between male control and chronic lymphocytic leukemia subjects. Asian Pac J Cancer Prev (2017) 18(1):23–30. doi: 10.22034/APJCP.2017.18.1.23
82. Bhuiyan H, Masquelier M, Tatidis L, Gruber A, Paul C, Vitols S. Acute myelogenous leukemia cells secrete factors that stimulate cellular ldl uptake via autocrine and paracrine mechanisms. Lipids (2017) 52(6):523–34. doi: 10.1007/s11745-017-4256-z
83. Tan Y, Sementino E, Liu Z, Cai KQ, Testa JR. Wnt signaling mediates oncogenic synergy between akt and dlx5 in t-cell lymphomagenesis by enhancing cholesterol synthesis. Sci Rep (2020) 10(1):15837–. doi: 10.1038/s41598-020-72822-w
84. Shen Y, Zhou J, Nie K, Cheng S, Chen Z, Wang W, et al. Oncogenic role of the sox9-dhcr24-cholesterol biosynthesis axis in igh-bcl2+ diffuse large b-cell lymphomas. Blood (2022) 139(1):73–86. doi: 10.1182/blood.2021012327
85. Sun L, Shi Y, Wang G, Wang X, Zeng S, Dunn SE, et al. Ppar-delta modulates membrane cholesterol and cytokine signaling in malignant b cells. Leukemia (2018) 32(1):184–93. doi: 10.1038/leu.2017.162
86. Li K, Wang F, Yang Z-N, Cui B, Li P-P, Li Z-Y, et al. Pml-rarα interaction with trib3 impedes pparγ/rxr function and triggers dyslipidemia in acute promyelocytic leukemia. Theranostics (2020) 10(22):10326–40. doi: 10.7150/thno.45924
87. Tippetts TS, Holland WL, Summers SA. Cholesterol - the devil you know; ceramide - the devil you don’t. Trends Pharmacol Sci (2021) 42(12):1082–95. doi: 10.1016/j.tips.2021.10.001
88. Yano H, Fujiwara Y, Horlad H, Pan C, Kai K, Niino D, et al. Blocking cholesterol efflux mechanism is a potential target for antilymphoma therapy. Cancer Sci (2022) 113(6):2129–43. doi: 10.1111/cas.15349
89. Zhou C, Li J, Du J, Jiang X, Xu X, Liu Y, et al. Hmgcs1 drives drug-resistance in acute myeloid leukemia through endoplasmic reticulum-upr-mitochondria axis. BioMed Pharmacother (2021) 137:111378. doi: 10.1016/j.biopha.2021.111378
90. Polusani SR, Cortez V, Esparza J, Nguyen HN, Fan H, Velagaleti GVN, et al. Oxidatively modified low-density lipoproteins are potential mediators of proteasome inhibitor resistance in multiple myeloma. Int J Cancer (2021) 148(12):3032–40. doi: 10.1002/ijc.33497
91. Hong C-S, Jeong E, Boyiadzis M, Whiteside TL. Increased small extracellular vesicle secretion after chemotherapy via upregulation of cholesterol metabolism in acute myeloid leukaemia. J Extracell Vesicles (2020) 9(1):1800979–. doi: 10.1080/20013078.2020.1800979
92. Xu H, Zhou S, Tang Q, Xia H, Bi F. Cholesterol metabolism: new functions and therapeutic approaches in cancer. Biochim Biophys Acta Rev Cancer (2020) 1874(1):188394. doi: 10.1016/j.bbcan.2020.188394
93. Chen L, Monti S, Juszczynski P, Ouyang J, Chapuy B, Neuberg D, et al. Syk inhibition modulates distinct pi3k/akt- dependent survival pathways and cholesterol biosynthesis in diffuse large b cell lymphomas. Cancer Cell (2013) 23(6):826–38. doi: 10.1016/j.ccr.2013.05.002
94. Jiang X-N, Zhang Y, Wang W-G, Sheng D, Zhou X-Y, Li X-Q. Alteration of cholesterol metabolism by metformin is associated with improved outcome in type ii diabetic patients with diffuse large b-cell lymphoma. Front Oncol (2021) 11:608238. doi: 10.3389/fonc.2021.608238
95. Subedi A, Liu Q, Ayyathan DM, Sharon D, Cathelin S, Hosseini M, et al. Nicotinamide phosphoribosyltransferase inhibitors selectively induce apoptosis of aml stem cells by disrupting lipid homeostasis. Cell Stem Cell (2021) 28(10):1851–67.e8. doi: 10.1016/j.stem.2021.06.004
96. Tian H, Qiang T, Wang J, Ji L, Li B. Simvastatin regulates the proliferation, apoptosis, migration and invasion of human acute myeloid leukemia cells via mir-19a-3p/hif-1α axis. Bioengineered (2021) 12(2):11898–908. doi: 10.1080/21655979.2021.1999552
97. Yu F, Gajendran B, Wang N, Sample KM, Liu W, Wang C, et al. Erk activation via a1542/3 limonoids attenuates erythroleukemia through transcriptional stimulation of cholesterol biosynthesis genes. BMC Cancer (2021) 21(1):680–. doi: 10.1186/s12885-021-08402-6
98. Ma Y, Zhang J, Wen L, Lin A. Membrane-lipid associated lncrna: a new regulator in cancer signaling. Cancer Lett (2018) 419:27–9. doi: 10.1016/j.canlet.2018.01.008
99. Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer (2018) 18(1):33–50. doi: 10.1038/nrc.2017.96
100. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol (2008) 9(2):139–50. doi: 10.1038/nrm2329
101. Pyne NJ, El Buri A, Adams DR, Pyne S. Sphingosine 1-phosphate and cancer. Adv Biol Regul (2018) 68:97–106. doi: 10.1016/j.jbior.2017.09.006
102. Selvam SP, Ogretmen B. Sphingosine kinase/sphingosine 1-phosphate signaling in cancer therapeutics and drug resistance. Handb Exp Pharmacol (2013) 216):3–27. doi: 10.1007/978-3-7091-1511-4_1
103. Lee MS, Sun W, Webb TJ. Sphingosine kinase blockade leads to increased natural killer t cell responses to mantle cell lymphoma. Cells (2020) 9(4):1030. doi: 10.3390/cells9041030
104. Shen Y, Zhao S, Wang S, Pan X, Zhang Y, Xu J, et al. S1p/s1pr3 axis promotes aerobic glycolysis by yap/c-myc/pgam1 axis in osteosarcoma. EBioMedicine (2019) 40:210–23. doi: 10.1016/j.ebiom.2018.12.038
105. Lupino L, Perry T, Margielewska S, Hollows R, Ibrahim M, Care M, et al. Sphingosine-1-phosphate signalling drives an angiogenic transcriptional programme in diffuse large b cell lymphoma. Leukemia (2019) 33(12):2884–97. doi: 10.1038/s41375-019-0478-9
106. LeBlanc FR, Pearson JM, Tan S-F, Cheon H, Xing JC, Dunton W, et al. Sphingosine kinase-2 is overexpressed in large granular lymphocyte leukaemia and promotes survival through mcl-1. Br J Haematol (2020) 190(3):405–17. doi: 10.1111/bjh.16530
107. Abdelbaset-Ismail A, Cymer M, Borkowska-Rzeszotek S, Brzeźniakiewicz-Janus K, Rameshwar P, Kakar SS, et al. Bioactive phospholipids enhance migration and adhesion of human leukemic cells by inhibiting heme oxygenase 1 (ho-1) and inducible nitric oxygenase synthase (inos) in a p38 mapk-dependent manner. Stem Cell Rev Rep (2019) 15(1):139–54. doi: 10.1007/s12015-018-9853-6
108. Naka K, Ochiai R, Matsubara E, Kondo C, Yang K-M, Hoshii T, et al. The lysophospholipase d enzyme gdpd3 is required to maintain chronic myelogenous leukaemia stem cells. Nat Commun (2020) 11(1):4681. doi: 10.1038/s41467-020-18491-9
109. Faict S, Oudaert I, D’Auria L, Dehairs J, Maes K, Vlummens P, et al. The transfer of sphingomyelinase contributes to drug resistance in multiple myeloma. Cancers (2019) 11(12):1823. doi: 10.3390/cancers11121823
110. Tan S-F, Dunton W, Liu X, Fox TE, Morad SAF, Desai D, et al. Acid ceramidase promotes drug resistance in acute myeloid leukemia through nf-κb-dependent p-glycoprotein upregulation. J Lipid Res (2019) 60(6):1078–86. doi: 10.1194/jlr.M091876
111. Maan M, Peters JM, Dutta M, Patterson AD. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun (2018) 504(3):582–9. doi: 10.1016/j.bbrc.2018.02.097
112. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J (2012) 279(15):2610–23. doi: 10.1111/j.1742-4658.2012.08644.x
113. Cheng X, Li J, Guo D. Scap/Srebps are central players in lipid metabolism and novel metabolic targets in cancer therapy. Curr Top Med Chem (2018) 18(6):484–93. doi: 10.2174/1568026618666180523104541
114. Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. Srebp transcription factors: Master regulators of lipid homeostasis. Biochimie (2004) 86(11):839–48. doi: 10.1016/j.biochi.2004.09.018
Keywords: lipid metabolism reprogramming, cholesterol, fatty acids, phospholipids, hematological malignancies
Citation: Zhang L, Chang N, Liu J, Liu Z, Wu Y, Sui L and Chen W (2022) Reprogramming lipid metabolism as potential strategy for hematological malignancy therapy. Front. Oncol. 12:987499. doi: 10.3389/fonc.2022.987499
Received: 06 July 2022; Accepted: 01 August 2022;
Published: 29 August 2022.
Edited by:
Hubing Shi, Sichuan University, ChinaCopyright © 2022 Zhang, Chang, Liu, Liu, Wu, Sui and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linlin Sui, bGlubGluMTk4MTA2MjJAc2luYS5jb20=; Wei Chen, Y2hlbncxMjNAYnVhYS5lZHUuY24=
†These authors contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.