 Danting Wang1†
Danting Wang1† Zhigang Chen
Zhigang Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 12 September 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.984560
This article is part of the Research TopicFrom Metabolism Reprogramming to Cancer Cell Fate: Ferroptosis, Apoptosis, Pyroptosis and SenescenceView all 6 articles
Aberrant lipid metabolism is nonnegligible for tumor cells to adapt to the tumor microenvironment (TME). It plays a significant role in the amount and function of immune cells, including tumor-associated macrophages, T cells, dendritic cells and marrow-derived suppressor cells. It is well-known that the immune response in TME is suppressed and lipid metabolism is closely involved in this process. Immunotherapy, containing anti-PD1/PDL1 therapy and adoptive T cell therapy, is a crucial clinical cancer therapeutic strategy nowadays, but they display a low-sensibility in certain cancers. In this review, we mainly discussed the importance of lipid metabolism in the formation of immunosuppressive TME, and explored the effectiveness and sensitivity of immunotherapy treatment by regulating the lipid metabolism.
Lipids are hydrophobic molecules including phospholipids, monoglycerides, diacylglycerides, triglycerides and sterols, which play the function of participating in forming biological membranes, providing energy, storing energy and mediating cellular signal transduction. Among them fatty acids (FAs), defined as a diverse class of molecules consisting of hydrocarbon chains varying in length and saturation, indispensably involve in the lipid metabolism which includes exogenous lipid uptake, de novo lipid synthesis and lipolysis (1). Fatty acid oxidation (FAO, also called beta-oxidation), shortens fatty acids by two carbons in each cycle and generates NADH, FADH2 and acetyl CoA through its series of cyclic reactions. And the end product of FAO pathway, acetyl CoA, can be ultimately converted back to fatty acids through the catalysis of various enzymes in the process of FA synthesis (FAS). Besides, cholesterol, which deemed as an indispensable part of biological membranes and the substrate for steroid hormones, is also derived from acetyl CoA and its synthesis is inseparable from many enzymes like Acyl coenzyme A-cholesterol acyltransferase (ACAT) (2).The above lipid metabolic processes are of great significance and studies revealed that lipid metabolism has undergone a variety of metabolic reprogramming in tumor cells to promote tumor growth, proliferation and metastasis (3, 4), and the diversity of lipid types and their different roles in tumor cells lead to the complex mechanism alterations. Therefore, targeting lipid metabolism is a promising strategy for cancer treatment. (5).
It is inadequate for tumor cells to rely solely on glucose metabolism for energy. Therefore, in order to meet the need of rapid growth and proliferation, tumor cells promote lipid metabolism through a series of mechanisms. Firstly, higher expression of enzymes related to lipid uptake such as CD36 enables tumor cells to compete with other cells for lipids from TME. Secondly, transcription factors related to lipid oxidation enzymes have been found to be expressed at high frequency. Thirdly, tumor cells have the ability to synthesize lipids independently. In addition to providing energy, lipids also serve as signal transduction molecules and raw materials of cellular structure in tumor cells (6). Furthermore, cholesterol and phospholipid biosynthesis are also very active in tumor cells, they maintain the integrity and fluidity of cell membranes to ensure the survival of tumor cells, and affect the function of receptors on the membrane to enable tumor cells' drug-resistance and better adaptation to the TME (7).
In the hypoxic and acidic TME, dysfunctional immune cells including tumor-associated macrophages (TAMs), tumor infiltrating T cells (TILs), dendritic cells (DC), myeloid-derived suppressor cells (MDSC), and natural killer cells (NK) lead to muted immune responses and further promote tumor progression (8). There is also a fierce competition for the deficient nutrients between immune cells and tumor cells in the TME, during which process their lipid metabolic reprogramming are of great significance and closely involved in the immunosuppressive outcome (9, 10). Pro-tumor and anti-tumor immune cells have different levels of lipid metabolism activity, the former, such as M2-macrophages and Treg cells, tend to have more active lipid metabolism, while the latter prefer to acquire energy through glucose metabolism (11). This difference in lipid metabolism is regulated by many factors, especially some transcriptional regulators such as peroxisome proliferator-activated receptor (PPAR) and sterol regulatory element-binding proteins (SREBP) (12). These lipid metabolism reprogramming in immune cells together leads to the formation of the immunosuppressive microenvironment.
Immunotherapy including adoptive cell therapy, immune checkpoint blockade (ICB), tumor-specific vaccines and small-molecule immune drugs has shown certain therapeutic effects. As one of the representatives of immunotherapy, programmed cell death protein 1 (PD-1) antibodies can inactivate tumor cells’ checkpoint and unleash cytotoxic T cells’ anti-tumor effects, but still major patients do not show complete responses (13). At present, tumor immunotherapy still faces many challenges, and it has a beneficial effect on the minority of patients (14). The immunotherapy resistance and post-immunotherapy cancer progression are two main problems accounting for that low-sensitivity (15, 16). Since lipid metabolism plays an important role in the function of immune cells, there are many experiments to improve the function of immune cells by regulating lipid metabolism, so that immunotherapy may be achieve better efficacy. Our review summarizes and analyzes existing studies with clinical application value, providing useful references for researchers’ follow-up research. For more precise targeted treatment, future research needs to focus on the sensitivity of regulating the same lipid metabolism target in different types of immune cells. Combining lipid metabolism regulation with immunotherapy may lead to new breakthroughs in immunotherapy.
As a feature of many malignant tumors, lipid metabolic reprogramming, especially the dynamic balance between FAS and FAO, plays a significant role in tumor progression (1) (Figure 1).
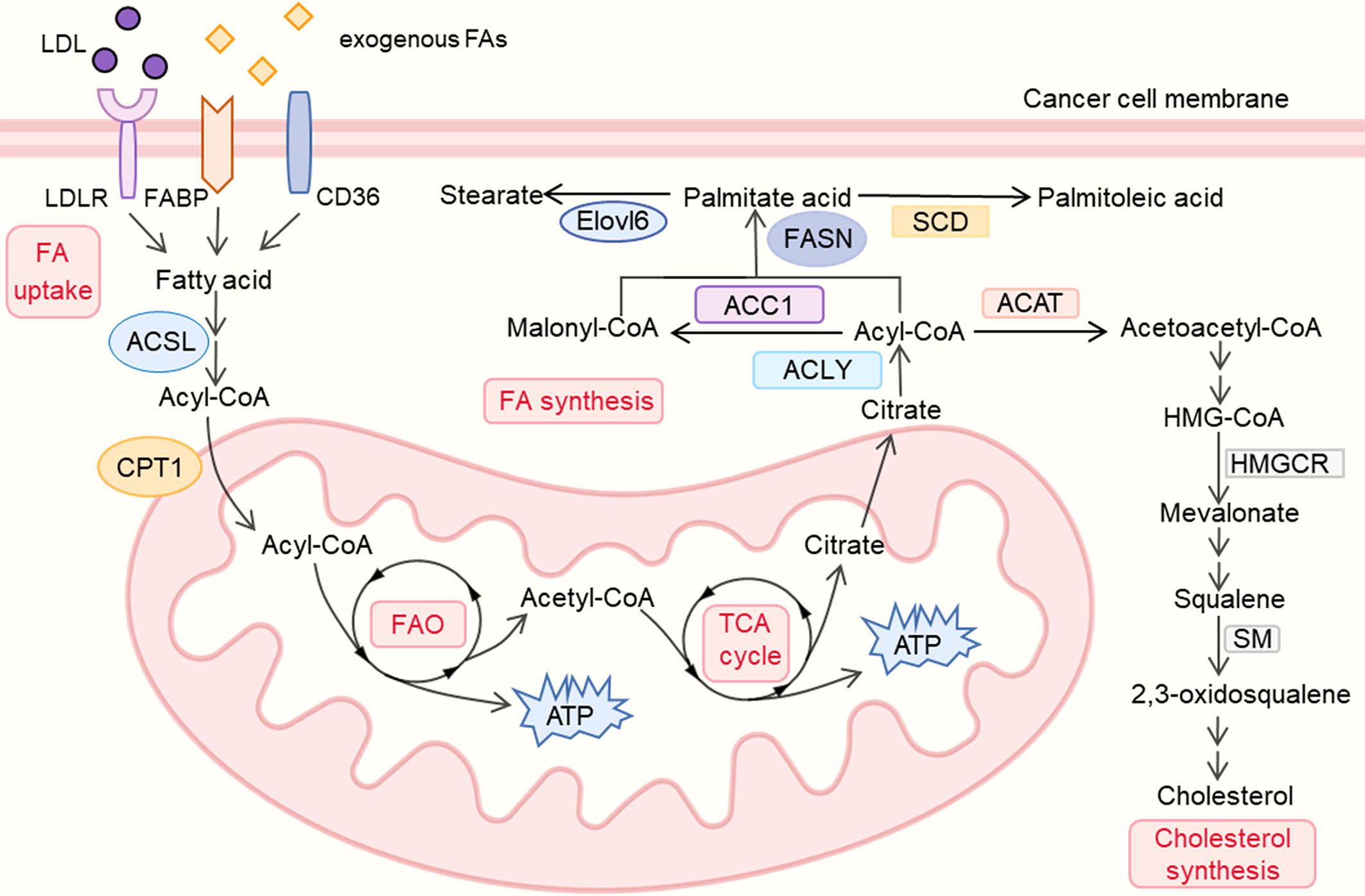
Figure 1 lipid metabolism reprogramming in cancer cells. LDL, low density lipoprotein; LDLR, low density lipoprotein receptor; FA, fatty acid; FABP, fatty acid binding protein; ACSL, Acyl-CoA synthetase long chain; CPT, carnitine palmitoyl transferase; FAO, fatty acid oxidation; TCA, tricarboxylic acid cycle; ACLY, ATP–citrate lyase; ACC, Acetyl-CoA carboxylase; FASN, fatty-acid synthase; SCD, Stearoyl-CoA desaturase; Elovl6, Elongation of long-chain fatty acids family member 6; ACAT, Acyl coenzyme A-cholesterol acyltransferase; HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A reductase; SM, squalene monooxygenase.
Studies have shown that lipogenesis plays an indispensable part in the growth of numerous tumors (17). On the one hand, lipids from the extracellular milieu are crucial for tumor cells. Studies found that tumors can acquire exogenous FAs like palmitic acid which was proved to promote metastasis in carcinoma (18). The upregulation of multiple FA protein transporters, particularly low-density lipoprotein receptors (LDLRs), CD36 (fatty acid translocase) and fatty acid-binding proteins (FABPs), were found in various tumors’ plasma membranes. Low density lipoproteins (LDLs) can be taken in via the LDLRs on the membrane, and the high expression of LDLRs promotes the LDL cholesterol-mediated breast cancer growth (19), which is also associated with the poor prognosis in small cell lung cancer (SCLC) and ovarian cancer (OC) (20, 21). High expression of CD36, which has been correlated with poor survival for patients with gastric, ovarian and breast cancer, could enhance the uptake of diet-derived FAs and fuel tumors’ growth and metastasis (22, 23). Besides, elevated FAs imported via CD36 were also testified to accelerate tumors’ epithelial-mesenchymal transition (EMT) (24), while inhibition of CD36 could impair the metastasis of human oral cancers and prevent the development of the adipocyte-induced malignant phenotypes in ovarian cancer (18, 25). Additionally, FABPs also play an instrumental role in cancer by affecting fatty acid metabolism, high expression of epidermal FABP (E-FABP) in the TAMs could enhance the production of interferon-beta (IFN-β) against the growth of tumor (26). The above evidence indicated the diversity of FA protein transporters’ functions in tumor therapy and suggested potential treatments such as dietary regulation in further study.
Apart from enhancing uptake of exogenous FAs, tumors also exhibit a high level of de novo FA synthesis which alters cellular lipid composition and fuels tumor growth (27). This process involves the upregulation of many enzymes as follows. Acetyl–coenzyme A (CoA), as the main substrate of lipid synthesis, can be converted from citrate by ATP–citrate lyase (ACLY). Since the production of citrate could be derived from glucose (22), ACLY is also regarded as a key enzyme linking glycolysis and lipid synthesis. High ACLY expression was found in gastric adenocarcinoma patients, correlated with advanced stages and lymph node metastasis (28), ACLY also shows a strong association with poor prognosis in various cancers like ovarian cancer (29). On the contrary, the ACLY-targeted treatments exhibit tumor- suppressive effects. ACLY inhibitor SB-204990 could not only suppress lipid synthesis and tumor progression but also show a stronger effect in combination therapy with cisplatin by inhibiting the PI3K-AKT pathway and activating the AMPK-ROS pathway (29, 30).
Acetyl-CoA carboxylase (ACC), which catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, was also found highly expressed in multiple tumors like breast cancer (31), ACC includes two isoforms, the cytosolic ACC1 is present in lipogenic tissues and is critical for FA synthesis, while ACC2 is bound in the mitochondrial outer membrane, mainly presenting in lipid oxidizing tissues like skeletal muscles. ACC1 promotes the FAO‐mediated human hepatocellular carcinoma (HCC) survival under conditions of metabolic stress and also contributes to the metastasis and recurrence of breast cancer (32, 33). While using RNA interference (RNAi) to silence the ACC-α gene could inhibit cell proliferation and induce apoptosis of prostate cancer cells (34). Unlike ACC1, ACC2 plays a role in inhibiting lipid degradation and was found inhibited in various cancers, its expression is also negatively correlated with tumor size and clinical stages of lung adenocarcinoma patients (35, 36). Prolyl hydroxylase domain protein 3 (PHD3) could also repress FAO and inhibit leukemia cell proliferation by activating ACC2 (37). The evidence revealed that both ACC1 and ACC2 could be potential targets in the treatment of cancer.
Fatty-acid synthase (FASN) is a multi-enzyme protein complex which can convert one acetyl-CoA molecule and seven malonyl-CoA molecules into one palmitate acid (C16: 0). FASN was found highly expressed in multiple cancers and closely related to poor prognosis, and notably, many studies have revealed that the downregulation of FASN can repress tumor progression via diverse mechanisms (3). Inhibition of FASN could not only lead to the accumulation of malonyl-CoA, which further represses tumor cells’ FAO and ultimately blocks the cell-cycle, but also impair the correct localization and/or functioning of EGFR and ERBB2 (3, 38). Besides, the apoptosis of tumor cells is also attributed to the inhibition of FASN which causes the downregulation of Akt and the starvation of phosphatidylcholine (3, 39). In terms of combination therapies, FASN inhibition with TVB-3166 or TVB-3664 could inhibit tubulin palmitoylation and show stronger anti-tumor effects with the combination of taxane drugs (40). And another selective FASN inhibitor, Fasnall, also reduces tumor volumes and extends survival when combined with carboplatin in the MMTV-Neu model of HER2+ breast cancer (41). AS a druggable target for cancer treatment, FASN has shown efficacy in diverse tumors, and its relevant metabolic treatments have broad prospects.
Palmitate acid (16:0) produced in de novo FA synthesis can be desaturated to palmitoleic acid (16:1) by Stearoyl-CoA desaturase (SCD). Since the desaturation driven by SCD1 could enhance tumor intrinsic antioxidants and anti-ferroptotic resources for survival and regrowth, suppression of SCD1 was also found to show anti-tumor effects (42). Researchers revealed the association between high expression of SCD1 and lung adenocarcinoma patients’ late stage, which provides SCD1 as a novel potential therapeutic target to suppress tumors (43). Apart from being desaturated by SCDs, palmitate acid could also be elongated by the Elongation of long-chain fatty acids family member 6 (Elovl6) which is also associated with poor prognosis in patients with HCC and breast cancer (44, 45). Knockdown of Elovl6 could also reduce HCC cell proliferation and Akt activation, which further represses tumor growth, and could be used as a potential therapeutic approach (44). High expression of various enzymes which enhances de novo lipid synthesis in tumor cell is also deemed as predictors or biomarkers, and the downregulation of these enzymes shows its anti-tumor effects in different cancers, which reveals the significance of drugs targeting lipid synthesis in tumor therapy.
Tumor cells also exhibit a high level of FAO to meet their demands for energy for rapid proliferation. Since the acidosis in the TME causes the reduction of glucose-derived acetyl-CoA, FAO (also called beta-oxidation), which converts long-chain FAs into acetyl-CoA and meanwhile generates NADH and FADH2 for electron transport chain, plays an essential role in the energy production of tumors (1). Overexpression of FAO enzymes can also be found in multiple tumors like breast cancer, colorectal cancer and gastric cancer (46–48). As the rate-limiting enzyme in FAO, carnitine palmitoyl transferase 1 (CPT1) can transport acetyl-CoA to the mitochondrial matrix and CPT1 includes three isozymes who have different distributions and close relations with tumor. CPT1A is widely distributed in liver, pancreas, brain and blood, and it is also highly correlated with poor prognosis in acute myeloid leukemia (AML) or ovarian cancer (22). CPT1A could also support Castration-Resistant Prostate Cancer (CRPC) by supplying acetyl groups for histone acetylation and CPT1A variant 2 is also found to promote tumor invasion and metastasis (49, 50) Besides, knockdown of CPT1A could repress xenograft tumor initiation through inhibiting adipocytes’ tumor-promoting effects, which indicates CPT1A-dependent FAO as a target for anti-cancer therapeutics (51). CPT1B is selectively present in brown adipose tissue, muscle and heart, JAK/STAT3 induces the expression of CPT1B and further enhances FAO activity to promote breast cancer chemoresistance and stem cells self-renewal, while inhibiting the STAT3-CPT1B-FAO pathway could recover tumor’ sensitivity to chemotherapy (52). Unlike CPT1A and CPT1B, CPT1C is primarily present in the brain and has oncogenic potential (53). Research found CPT1C was remarkably reduced in senescent cells, and silencing of CPT1C could also cause cellular senescence in tumor and suppress xenograft tumor growth (54). CPT1C is also deemed as a contributor to tumor cell metabolic transformation and rapamycin resistance, which is significant for the growth of tumor cells under the conditions of metabolic stress (53).
Acyl-CoA synthetase long chain 3 (ACSL3), an upstream enzyme of CPT1, which converts free FAs into fatty acyl-CoAs, is also crucial for the proliferation of KRAS-driven cancer cells (55). Studies found that ACSL3 is upregulated in human pancreatic ductal adenocarcinoma (PDAC), and the deletion of ACSL3 could delay PDAC progression and reduce fibrosis in mice (56). Apart from energy production, FAO also generates nicotinamide adenine dinucleotide phosphate (NADPH) which is derived from IDH (isocitrate dehydrogenase)-mediated isocitrate oxidation to maintain homeostasis, which is also regarded as a potential therapeutic target (4). Using etomoxir to decrease NADPH level could result in cell death in human glioblastoma cells accompanied by oxidative stress (57). Taken together, the up-regulation of FAS and FAO levels in tumor cells meets the energy need of their rapid proliferation, many enzymes play significant roles in this process, and the down-regulation or inhibitors of those enzymes have been found to have anti-tumor effects with multiple mechanisms. Still some enzymes whose correlation with tumor and potential therapeutic effects remain to be confirmed, but the metabolic regulation related to lipid metabolism is no doubt playing an indispensable role in this field.
Multiple studies suggested that lipids other than fatty acids, such as cholesterol, also play a key role in tumor progression, and the inhibition of cholesterol synthesis is detrimental to cancer cells (6, 58). Cholesterol biosynthesis begins with the formation of acetoacetyl-CoA by the condensation of two molecules of acetyl coenzyme A through Acyl co-enzyme A-cholesterol acyltransferase (ACAT). And the inhibition of ACAT could reduce the formation of lipid droplets (LD) which further suppresses glioblastoma growth (59). Meanwhile, ACAT inhibitor such as avasimibe, was also proved to induce cell cycle arrest and apoptosis, which is regarded as a potential therapy in the treatment of glioblastoma (60).
As the rate-limiting enzyme in cholesterol biosynthesis, 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) which reduces 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) to mevalonate was also found up-regulated in various cancers. Its overexpression induces statin resistance of cancer cells and accelerates tumor migration, while HMGCR knockdown could restore the statin sensitivity of tumor cells like breast cancer and CRPC (61, 62). In addition, HMGCR inhibitor simvastatin could also robustly enhances cancer vaccinations and synergizes with anti-PD-1 antibodies, which shows the broad therapeutic perspectives of drugs targeting cholesterol pathways in combination with other anti-tumor treatments (63). Apart from HMGCR, squalene monooxygenase (SM, also known as squalene epoxidase) which converts squalene to 2,3(S)-oxidosqualene is deemed as the second rate-limiting enzyme in cholesterol biosynthesis and its increased expression was also found associated with poor prognosis in several tumors like prostate cancer, pancreatic adenocarcinoma (PAAD) and head and neck squamous cell carcinoma (HNSCC) (64–66). While inhibition of SM could lead to the accumulation of squalene which further suppresses SCLC proliferation (67), and its pharmacological blockage also inhibits the invasion of CRPC (68). The above evidence reveals the potential value of various enzymes in cholesterol metabolism in tumor therapy, suggesting new ideas for tumor treatment.
When it comes to phospholipid metabolism, the extracellular choline and ethanolamine are transported into tumor cells to be catalyzed by a series of enzymes such as ethanolamine kinase (ETNK), choline kinase (ChK), cytidylyltransferase (CT), diacylglycerol phosphotransferase (PT), and respectively converted into phosphatidylcholine (PtdCho) and phosphatidylethanolamine (PtdEth) (69, 70). Metabolic remodeling of phospholipids can promote tumor progression and drug resistance. In glioblastoma (GBM), overexpressed polymerase 1 and transcript release factors (PTRF) lead to increased activity of cytoplasmic phospholipase A2 (cPLA2), and PTRFs can also induce decreased infiltration of CD8+ T cells. Ultimately, they promote the proliferation of tumor cells (71). Since phospholipid and cholesterol are the main raw materials for forming plasma membrane, their composition changes affect the activity and function of drug efflux pump on plasma membrane, and make tumor cells have stronger drug resistance to some extent (7).
Cancer-associated fibroblasts (CAF) are the major components of tumor stromal cells in TME. CAF-derived exosomes contain a variety of metabolites, including lipids, amino acids, and TCA cycle products (72), which play a major role in regulating various metabolic reprogramming in the tumor microenvironment, also including lipid metabolism (73). These products enter the TME and serve as an important source of extracellular lipids uptake by tumor cells and immune cells. Studies have shown that FAs or lysophosphatidylcholine (LPCs) overexpressed in fibroblasts can be released into TME, which subsequently are partially taken up by tumor cells and promote tumor growth through intracellular lipid metabolism reprogramming pathways (74, 75). Similar experiments have shown that CAF transport proteins and lipids to tumor cells in a one-way process (76). Moreover, CAFs induce the increased expression of fatty acid transport protein 1 (FATP1) in breast cancer cells and accelerate lipid uptake (77). CAFs in lung induce high expression of SCD1 in tumor cells through PI3K/Akt/mTOR pathway, which enhances the ability of tumor cells to metastasize and may be related to lung metastasis of colon cancer or breast cancer (74, 78). Meanwhile, CAFs contribute to promoting immune evasion and are the core source of immunosuppressive molecules. The resistance to ICB is also strictly controlled by CAFs (79). There is evidence proved that stromal cells targeting recombinant IL-2 in combination with ICB can be more effective in immunotherapy than ICB alone (80).
TAMs have distinct characteristics from conventional macrophages. They infiltrate and settle down in the TME. Abundant studies have proved that TAMs have prominent pro-tumor activities, such as tumorigenesis, angiogenesis, spread and metastasis, drug-resistance and tumor immune evasion (81). TAMs are the most abundant immune cells in TME (82). The classically (M1) and alternatively (M2) activated macrophages phenotype are two different TAMs states of polarized activation (83). M2 macrophages are regarded as the main components of TAMs, which exhibit a tumor-promoting effect and are dedicated to forming an immunosuppressive microenvironment, while M1 macrophages show a tumor-inhibiting effect (84) (Figure 2). Consistently, the more amounts of M2 macrophages or total TAMs, the worse prognosis for patients with advanced gastric cancer (AGC) and hepatocellular carcinoma (85, 86). Lipid metabolism is closely involved in the selection of different polarization states in TAMs and ultimately leads to opposite effects in tumor (87).
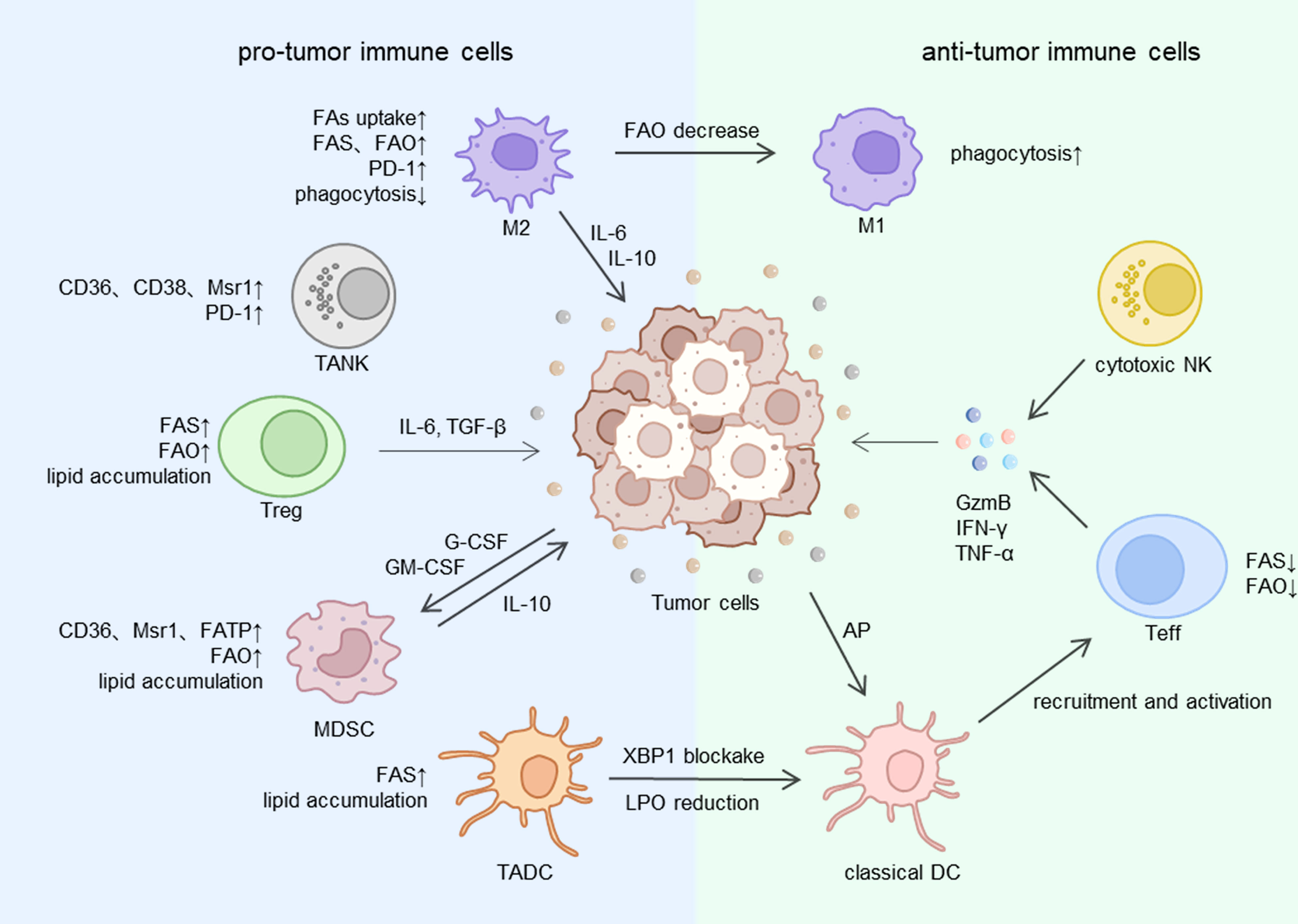
Figure 2 The lipid metabolism of immune cells in TME. FA, fatty acid; FAS, fatty acid synthesis; FAO, fatty acid oxidation; Msr1, macrophage scavenger receptor 1; FATP, fatty acid transport protein; XBP1, X-box binding protein 1; LPO, lipid peroxidation; AP, antigen presentation; IL, interleukin; TGF, tumor growth factor; G-CSF, granulocyte colony stimulating factor; GM-CSF, granulocyte macrophage colony stimulating factor; GzmB, granzyme B; IFN, interferon; TNF, tumor necrosis factor; M1, M1 macrophage; M2, M2 macrophage; NK, natural killer cell; TANK, tumor-associated natural killer cells; Teff, effective T cell; Treg, regulatory T cell; MDSC, myeloid-derived suppressor cell; DC,dendritic cell; TADC, tumor-associated dendritic cell.
Several processes of lipid metabolism like FA biosynthesis, uptake and storage are enhanced in TAMs (88). Free fatty acids are derived from extracellular uptake, de novo synthesis or release of LD from the cellular fatty acid pool. Research has indicated that the immunosuppressive phenotype of TAMs is induced by long-chain fatty acid metabolism, specifically unsaturated fatty acids (UFA) (89). Mechanically, to obtain enough energy and ensure the cellular survival, TAMs overexpress scavenger receptor CD36 and accelerate FAs transportation to accumulate lipid droplets and utilize FAO, enhanced FAO leads to a high rate of oxidative phosphorylation and STAT6 signaling pathway, which finally regulate gene transcription to determine the TAMs function (90). Given that CD36 is expressed extensively in other pro-tumor immune cells like regulatory T cells, the inhibition of CD36 may be a potential adjunct strategy to immunotherapy.
So, what factors will regulate FAO in TAMs? PPAR family has a strong link with FA metabolism in TAMs. PPARα, PPARδ and PPARγ are three major members of the PPAR family and they are involved in respective aspects of FA metabolism (91). S100A4 is a deciding factor for the subsets of TAMs: M2 macrophages display S100A4+ and M1 macrophages display S100A4-. S100A4 controls the upregulation of PPAR-γ during TAMs M2-like polarization, and then PPAR-γ upregulates CD36 to enhance FA uptake and FAO (92). In hepatocellular carcinoma, the deficiency of receptor-interacting protein kinase 3 (RIPK3) inhibits the caspase-1 mediated PPAR cleavage, and the reduction of PPAR clearance results in an increase in FAO and polarization of M2 TAMs (93). Intriguingly, the inhibition of caspase-1 could suppress tumor progression by promoting FAO and exhausting lipid droplets (94). The mechanism behind this contradiction still needs us to explore further. Moreover, another nuclear receptor liver X receptor (LXR), regarded as a cholesterol sensor and contributor to the loss of cholesterol intracellularly (95), play a pivotal role in TAMs gene expression. Activating LXR disrupts the mechanisms of TAMs to maintain immunosuppressive effects (96). M2 and M1 macrophages can be switched to each other in particular conditions. When changing the amounts of FAs or blocking FAO, M2 macrophages can be induced to transform into M1 type polarization (97) and the cancer progression can be prevented.
Compared to M2-like macrophages, M1-like macrophages express less PD-1 and escape from the effects of programmed cell death 1 ligand (PD-L1) (98). Therefore, we assume the combination of anti-PD-1 therapy towards malignant tumors with targeting lipid metabolism of TAMs might selectively strike M2-like macrophages without impairing M1-like macrophages to strengthen the effectiveness and sensibility of treatment.
CD8+ and CD4+ cells are two types of TILs that are familiar to us and they play different roles in TME. The CD8+ clusters include effector T cells (Teff) that kill tumor cells directly, and CD4+ clusters include regulatory T cells (Treg), T follicular helper cells (Tfh) and T helper cells 1, 2, and 17 (Th1, Th2, Th17) (99), the function of Treg significantly contributed to the formation of immunosuppressive microenvironment (100, 101). Intratumor Tregs were found to be at high levels in some malignant tumors (102), one of the reasons is that both FAO and FAS are enhanced in Treg cells, thus Treg cells accumulate lipids and gain abundant energy to adapt to the undernourished environment in TME (103). By contrast, Teff tends to utilize glycolysis for more energy need (11), which will place Teff at a disadvantage in the hypoxia microenvironment (Figure 2).
Since activated T cells exhibit a desperate requirement for a high rate of FAO to maintain energy supply, the intracellular FAs content plays a crucial part in maintaining the levels of FAO in cells (104). FAs are known to be the essential materials for cells, and their deficiency in Teff inhibits cell proliferation and signal transduction so that the quantity of Teffs in tumor is low and cancer cells can’t be prevented efficiently. Another pro-tumor effect of FAs is that FAs catalyzed by ACC1 will fuel the differentiation of Tregs and Th17 (105). Tregs and Th17 have the synergistic effects in tumor-promoting with IL-6 and TGF-β secreted themselves (106). SREBP, a family of membrane-bound transcriptional regulators in ER, are mainly involved in FA and cholesterol synthesis. Three subsets are existing in human cells: SREBR1a, SREBP1c and SREBP2 (12). FAS is especially manipulated by SREBP1c (107). Studies showed that SREBP activity is simulated in intracellular Treg cells (108) and inhibited in Teff cells (109). To summarize, the levels of FA synthesis (modulated by SREBP1c) and FA oxidation (modulated by PPAR) are both elevated in Tregs to achieve a better immunosuppressive effect in tumor. And evidence showed that a high number of Tregs is associated with shorter overall survival and poor prognosis in several tumors like breast cancer or bladder cancer (110, 111).
The role of Th17 cells in tumors is like a coin with two sides, on one side they impair the immune response to the tumor, and on another side, they mediate anti-tumor immunity (112). More specifically, Th17 cells are divided into two categories according to whether they express CD5L (a regulator of lipid metabolism): nonpathogenic Th17 cells (CD5L+) and pathogenic Th17 cells (CD5L-). The former express immunosuppressive genes, while the latter display pro-inflammation genes (113). The deletion of CD5L remarkably reverses the function of Th17 cells by controlling lipid metabolism. For instance, CD5L deficiency leads to an elevated level of cholesterol ester and saturated fatty acid (SFA), and eventually induces nonpathogenic Th17 into pathogenic Th17 (114). Obviously, lipids act as important mediators in determining the phenotype and function of Th17 cells. In conclusion, changing lipid metabolism in T cell lines targeting genes, transcriptional regulators, or SFA content may become a potential strategy.
The regulation of lipid metabolism in T cells interacts with PD-1/PD-L1 expression. PD-1 is the major contributor to hinder tumor immune surveillance. High expression of PD-1 on cell surface is partly responsible for the depletion of CD8+ cells (115). Activation of PD-1 upregulates the expression of CPT1A and fatty triglyceride lipase (ATGL) so that T cells prefer to utilize FAO rather than glycolysis for energy supply (116, 117). We can learn that PD-1 expression reinforce FAO in Teffs. Interestingly, there is a study declaring that fenofibrate, a drug increases FAO of Teff cells, can reverse the poor effects of PD-1 blockade treatment (118). Therefore, we hypothesize that the enhancement of FAO at different levels might increase the PD-1 antibodies binding sites by increasing the expression of PD-1. The exact mechanism needs to be confirmed by abundant experiments in the future.
DCs are the strongest antigen-presenting cells (119). DCs are composed of four main subtypes: classical DCs 1 (cDC1s), classical DCs 2 (cDC2s), plasmacytoid DCs (pDCs) and monocyte-derived DCs (moDCs). In addition to antigen presentation, another pivotal role of DC cells is to trigger the activation of cytotoxic T cells. cDC1s mediate anti-tumor immunity by activating cytotoxic CD8+ T cells, cDC2s activate anti-tumor CD4+ T cells but this process is restricted by Tregs or other immunosuppressive factors (120).
Even though the primary function of DC cells is inducing anti-tumor immune response in TME, a series of research has proved that the capability of anti-tumor of tumor-associated DC cells (TADCs) was destroyed via lipid accumulation (Figure 2). The augmentation of fatty acid synthesis relying on the ER and Golgi body serves as a key regulatory factor of TADC activation (121). High-rated FAS will eventually result in excessive lipids in the cytoplasm of TADCs. Abnormal lipid accumulation prevents the process of antigen-presenting and then weakens the tumor-inhibiting effects (120).
Exposure to the tumor environment results in several metabolic changes in TADCs. Firstly, TADCs overexpress the scavenging receptor macrophage scavenger receptor 1 (Msr1) that facilitates the perpetual uptake of fatty acids and cholesterol in TME (122). Excessive lipids are a sign of ER stress and oxidation damage because increased reactive oxygen species (ROS) mediate lipid peroxidation (123). Afterwards, the activation and cross-presentation to kill tumor cells are impaired. We have already mentioned there are many subtypes of DC cells, so grandly they possess distinct functions and metabolic pathways. It would explain why the phenomenon above was observed in cDC1 cells and cDC2 cells, but not in pDC cells (119). Furthermore, the mechanism of lipid peroxidation in TADCs to the ER stress response is mediated by the inositol-requiring protein 1α (IRE-1α) and its targeting X-box binding protein 1 (XBP1). XBP1 is regarded as the fuel of tumorigenesis and tumor cell progression. In ovarian cancer and triple-negative breast cancer, XBP1 is activated by the lipid peroxidation byproducts and aggregated lipids to promote the growth and dissemination of cancer cells (124). Therefore, the authors have proved that blockade or silencing of XBP1 makes TADCs regain the capability of immunostimulatory response and restriction towards tumor.
This brings us to the next question, what is the mechanism of excessive lipids driving these functional defects of TADCs? The pivotal step of TADCs to exhibit anti-tumor effects is the cross-presentation of tumoral antigens. Studies have established that electrophilic oxidatively truncated (ox-tr) lipids, one of the components of lipid bodies (LB), mediate the aggregation of heat shock protein 70 (HSP70) on the surface of large lipid droplets and then result in the deficiency of cross-presentation. It means that TADCs are unable to transport peptide MHC class I (pMHC) complexes to the cell surface as normal (125). In conclusion, modulating cross-presentation by reducing intracellular lipid aggregation of TADCs might become a potential therapeutic pathway. And there are experiments that exactly did so, deletion of Atg5 (a key autophagy gene) in dendritic cells increased CD36 expression and lipid absorption, therefore, blocking CD36 in TADCs by regulating Atg5 gene expression is a promising method to make a dent in lipids surplus (126, 127).
MDSCs have a powerful contribution to the formation of the immunosuppressive microenvironment of tumor (128) (Figure 2), which is controlled by several key factors like arginase-1 (ARG-1), COX2, TGFβ, iNOS, IL10 et al. But the detailed mechanisms are not completely same in two categories of MDSCs: polymorphonuclear (PMN-MDSCs) and monocytic (M-MDSCs) (129). In advanced gastric cancer, high content of MDSCs can be a conspicuous symbol of the awful prognosis (130). Similar to TADCs, lipid accumulation in cells also has a close relationship with MDSCs to achieve the immunosuppressive microenvironment. Exposure to G-CSF and GM-CSF secreted from tumor cells activates the STAT signal pathway and then elevates FA uptake and FAO in tumor infiltrating MDSCs (131). Continuous contact with exogenous UFAs like oleate but not free FAs synthesized within cells causes a regulatory phenotype induction in MDSCs and suppresses cytotoxic T cells’ proliferation and immune response (132). Exogenous lipids transporters like CD36, Msr1 and FATP are highly expressed on the surface of MDSCs to uptake lipids into cells and lead to a lipid-overloaded situation. Thus CD36 deficiency can diminish the useless lipid accumulation and immunosuppressive effect (9, 10). What’s more, previous studies have shown that FAO maintains the synthesis and secretion of tumor-promoting enzymes and cytokines, which is indispensable to the suppressed function of MDSCs. Thus, blockade of FAO wakens the Immune response tolerance mechanisms in MDSCs, prevents tumor progression, and even boosts adoptive T cells immunotherapy (131).
As a kind of cell easily underappreciated in tumor immunosuppressive environment, tumor-associated natural killer (TANK) cells have the important prognosis pertinence of the interaction with lipid metabolism and tumor cells (133)(Figure 2). In normal circumstances, NK cells express inhibitory receptors (KIRs, killer-cell immunoglobulin-like receptors) and activating receptors (NCRs, natural cytotoxicity receptors) on the surface, an appropriate proportion of them enables NK cells to monitor tumor cells in circulation through recognizing damage-associated molecular patterns (DAMPs) and pathogen-associated patterns (PAMPs) (134). However, studies in gastrointestinal cancer showed when NK cells infiltrate in the TME, their anti-tumor function is damaged on account of the unbalance between inhibitory and activating receptors (135). And the more advanced the tumor stage is, the more completely the function of TANK cells is destroyed.
One of the reasons accounts for the defects in TANK cells’ function and amount is that the immune checkpoint PD-1 high-express on both intratumoral TANK cells and circulating NK cells (136). It is possible that anti-PD-1 therapy can stop the decline of NK cells. Another reason is that oxidative stress caused by lipid peroxidation disrupts glucose metabolism and homeostasis. Nrf2 is the major antioxidant factor to protect TANKs from oxidative stress. Nrf2 agonist RTA-408 recovers TANK cells’ glycolysis and function, maintaining the effective lethality against tumor (137). Interestingly, with perioperative immunology becoming an emerging field recently, researchers suggested that tumor resection caused a postoperative lipid accumulation in NK cells in mice having beared colorectal cancer. This phenomenon is partially caused by gene expression of scavenger receptors like CD36, CD38 and Msr1 (138). These changes of NK cells attenuate the removal of residual tumor cells and increase the possibility of tumor recurrence. Targeting these changes may have an auxiliary effect after operations.
Immunotherapy is defined as a kind of cancer treatment strategy that enhances or restores immune function in the body through a variety of methods, including immune checkpoint blockade, adoptive T cell therapy, cancer vaccines and immune stimulants (139). In our review, we mainly talk about anti-PD-1 treatment which plays an important role in tumor immunotherapy. PD-1 is a well-known kind of inhibitory immune checkpoints. A series of research demonstrated PD-1 is found to be highly expressed in most immune cells including TAMs, T cells, DCs, NK cells and so on (98, 140–142). When PD-1 on Teffs is combined with PD-L1 expressed on tumor cells, the process will initiate the programmed cell death of T cells, thus tumor cells attack the anti-tumor immune response of activated T cells, achieving immune evasion. This is one of the main mechanisms for the formation of tumor immunosuppressive microenvironment. At present, PD-1/PD-L1 inhibitors have shown promising therapeutic effects in various cancers.
Low-response to immunotherapy is the biggest obstacle of immunotherapy and also the difficulty that must be broken through to realize the progress of immunotherapy. This low sensitivity is largely determined by the tumor cells themselves, such as low antigenicity, activation of the WNT pathway or MAPK pathway, inactivation of PTEN protein and the function of CDK proteins (143, 144). The regulation of lipid metabolism sometimes improves immunotherapy efficacy (Table 1) by indirectly influencing these processes. For example, one reason for low-sensitivity is the inactivation of the IFN-γ pathway in tumor cells. Teffs inhibit tumor cells by releasing IFN-γ, and tumor cells reduce Teffs by upregulating PD-L1 through IFN-γR-JAK-STAT pathway (157). Targeted lipid metabolism therapy promotes Teffs IFN-γ production, thereby promoting anti-PD-L1 therapy (153).
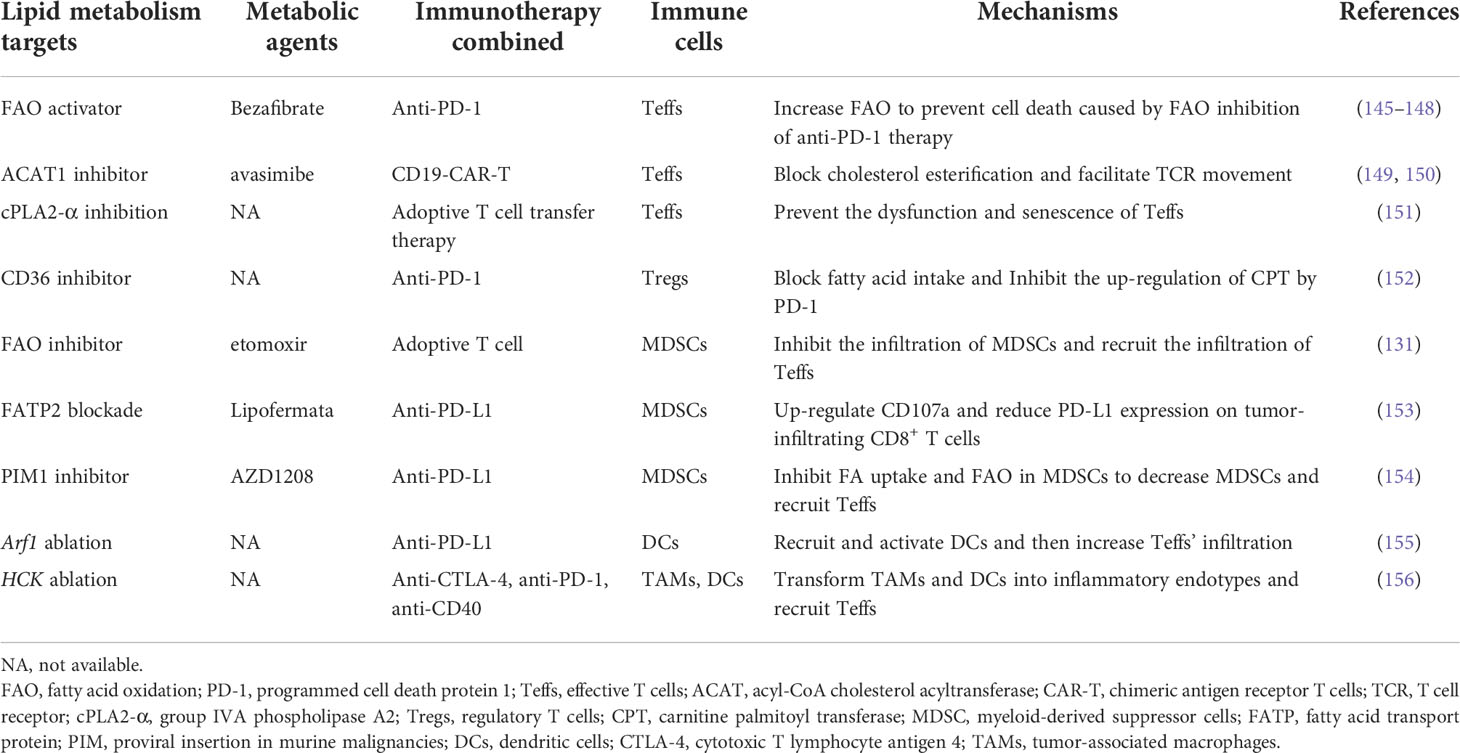
Table 1 The combination of lipid metabolism targeted therapy and immunotherapy.
Moreover, lipid metabolites regulate immune checkpoints in various aspects. A large number of toxic lipid metabolism intermediates and by-products are produced and retained during high-efficiency lipid metabolism in various immune cells, which affect gene expression, transcription, modification and activation of immune checkpoints (158). De novo fatty acid synthesis promotes PD-1 expression in mature Tregs by protein geranylgeranylation and elevates PD-L1 expression in breast cancer by PD-L1 palmitoylation (108, 159). Considering that anti-PD-1 therapy still performs poorly in several patients and lipids play a pivotal role in immune cells’ function, the combination of lipid metabolism targeted therapy and PD-1 inhibitors is a novel idea to break through the bottlenecks in immunotherapy. Increasing research has proved that anti-PD-1 drugs can combine the following lipid metabolism targets at different levels to enhance anti-tumor immunity.
Treg cells are one of the most important members of intratumor immune cells with immunosuppressive effects. So blockade of lipid metabolism in Treg cells is the focus of research nowadays. Diverse patterns inhibiting Treg cells improve anti-tumor responses and prevent tumor metastasis (160, 161), but non-selectively suppression of T cells can lead to accidental damage of cytotoxic T cells and autoimmune caused by healthy Treg cell defects (152). Therefore, how to accurately kill Treg cells in tumors is a problem that needs to be solved at present. This prompts us to seek different biomarkers or signaling pathways between intratumoral Treg cells and normal T cells.
CD36 expression is up-regulated on the surface of both Tregs and cytotoxic T cells. Blocking CD36 effectively suppresses tumor growth (162) and prevents Tregs’ dysfunction. In a recent experiment, inhibiting CD36 in intratumor Tregs by genetic ablating or using CD36 monoclonal antibodies effectively retards tumor growth and has an auxiliary effect for anti-PD-1 treatment (152). CD36 deletion decreased several immune regulatory receptors’ expression but not PD-1, it could explain why the therapeutic effect of PD-1 was not impaired. Surprisingly, the body does not develop a severe autoimmune response in this way. Other studies about CD36 showed the over-expression of CD36 in intratumor effector T cells expose them to oxidative damage and ferroptosis, and the combination of anti-PD-1 therapy with inhibitors of CD36 or ferroptosis will produce greater immunotherapeutic benefits (163). The main reason why CD36 plays different roles in Tregs and effector T cells is that CD8+ T cells tend to rely on glycolysis for energy, while CD4+ T cells tend to obtain energy through FAO and OXPHOS in mitochondria (97). Therefore, Tregs promote mitochondrial adaptation to survive, but effector T cells suffer oxidative damage due to lipid accumulation (162).
We have mentioned above that FAS depends dreadfully on SREBP cleavage active protein (SCAP)/SREBP signaling pathway. Selectively block of this pathway to reduce FAS in Treg cells delays tumor growth and development in B16 melanoma mice. Mechanically, IFN-γ will be produced in quantity after deleting SCAP, and some processes like mevalonate metabolism and protein geranylgeranylation in over-expression of PD-1 rely on SCAP/SREBP pathway (108).
In addition to the dysfunction of Treg cells, the decreased and senescent cytotoxic T cell is another reason for tumor immune evasion. Senescent and dysfunctional T cells exhibit aberrant lipid metabolism and accumulation of lipids intracellularly. Scientists found elevated expression of group IVA phospholipase A2 (cPLA2-α) in both tumor cells and Treg cells, which is closely related to aging and lipid metabolism reprogramming in T cells via MAPK and STAT signaling pathways. Furthermore, cPLA2α inhibition prevents T cells from senescence and enhances immunotherapy such as adoptive T cell transfer therapy (151). Besides, using activator of FAO like bezafibrate could become an adjuvant for anti-PD-1 therapy. And bezafibrate achieves that mainly by facilitating the expression of CXCL9, CXCL10 from tumor cells and CXCR3 on intratumor Teffs (145–147).
There have also been studies on drugs that target lipid metabolism in other immune cells. The quantity of MDSCs is positively correlated with the degree of malignancy of tumor and the poor sensitivity of anti-PD-1 therapy. Combination of 5-fluorouracil and oxaliplatin decreases the amount of infiltrating MDSCs and then recruits CD8+ T cells in TME. However, there exists a disadvantage that PD-L1 expression on tumor cells is facilitated at the same time in this way (164). So it is necessary to explore how to reduce the side effects when diminishing MDSCs. The mechanism behind the connection between MDSCs and ICB resistance is that the gene expressions of CD8+ T cells in sensitive and insensitive patients are very different. CD8+ T cells in sensitive patients express effective and stimulating genes like Fasl, Cd28, G2mb, Tnfsf4, lcos Prf1, while in insensitive patients they express inhibitory and exhausted genes like Pdcd1. Blockade of PIM1 (a serine/threonine kinase) with drugs or genetic deletion inhibits FA uptake and FAO in MDSCs through PPARγ-mediated pathway, which finally decreases the quantity of MDSCs, recruits CD8+ T cells and ameliorate ICB resistance (154).
The removal of Arf1 (a mediator of lipid metabolism to maintain the abundance of tumor stem cells) induces DC cell activation and activated DC cells increase CD8+ T cells’ infiltration to kill tumor cells. Mechanically, Arf1 ablation in tumor stem cells induces mitochondrial disorder and the release of DAMPs, which recruit and activate DCs at tumor sites. Therefore Arf1 ablation and PD-1 blockade have a synergistic effect (155). Besides, another study has demonstrated that genetic ablation of myeloid-specific hematopoietic cell kinase (HCK) boosts the therapeutic effect of anti-CTLA-4, anti-PD-1 and anti-CD40 immunotherapy. The deficiency of HCK transforms TAMs and DCs into inflammatory endotypes and then recruits CD8+ T cells (156).
Nowadays, ferroptosis in tumor cells is an emerging field for tumor treatment strategy. Ferroptosis, an iron-dependant programmed cell death, is induced by lipid peroxides retention and ROS (165). PKCβII-ACSL4 pathway is the main contributor to inducing tumor ferroptosis through sensing and exaggerating lipid peroxides. Ferroptosis is partially associated with immunotherapy and patients with high ACSL4 display a high sensitivity to anti-PD-1 therapy. So enhanced PKCβII-ACSL4 pathway can improve the efficacy of immunotherapy by promoting ferroptosis (166). Consistently, there are a series of experiments showing that manipulating ferroptosis in tumor cells, such as low dose arachidonic acid (AA), inhibition of CAMKK2 or AMPK-NRF2 pathway, inhibition of ALG3 et al. (167–169), can boost the efficacy of anti-PD1 therapy.
In general, FAS and FAO are involved in many aspects of tumor cells and immune cells, including energy acquisition, raw materials of membrane structure, regulation of signaling pathways, formation of the immunosuppressive microenvironment and mediating ferroptosis. Manipulation of lipid metabolism at different levels, such as regulating enzymes, transcription factors or gene expression involved in lipid metabolism, can play a synergistic role with ICB-induced anti-tumor immunotherapy. The common mechanism of most combination therapy is the recruitment and activation of CD8+ T cells. Moreover, some lipid metabolism-related genes can predict the prognosis of anti-PD-1 therapy (170), which may alleviate the problem of low response rates. In order to figure out the mechanisms or pathways involved and improve the effectiveness and sensitivity of immunotherapy, abundant experiments are needed to be warranted.
The combination of lipid metabolism targets in either immune cells or cancer cells therapy and ICB-induced immunotherapy exhibits the potential to be a novel cancer treatment strategy. This idea was put forward based on two facts. One is that the inhibition of PD-1/PD-L1 therapy encounters the low-response rate in certain patients, which is closely linked with the exhaustion of CD8+ T cells, that’s why the major mechanism for many combination therapies is recruitment and activation of Teffs. And the other one is that lipid metabolism reprogramming not only plays a vital role in immune cells and the formation of the immunosuppressive microenvironment, but is also involved in the expression of PD-1. Lipid metabolism reprogramming plays contradictory roles in the immune microenvironment of tumors. In situations of intense competition for nutrients, immune cells use lipids as fuel to mount immune responses. Lipids and cholesterol regulate different immune cells cytotoxic effects, for example, cholesterol enhances cytotoxic effects by restoring the IFN-γ production of iNKT cells. However, Excessive lipid accumulation and LD formation damage T cells or DC cells and induce macrophages and MDSC cells to differentiate into pro-tumor subtypes (171). Although various preclinical trials of this combination therapy achieve convincing and satisfactory results, the systematic and comprehensive mechanisms of lipid metabolism in coordinating anti-tumor or pro-tumor effects of distinct immune cells are still not fully validated. Further, the pathway through which alteration of lipid metabolism reverses the poor sensitivity of anti-PD-1 also requires to be explored through more related experiments. Take FAO as an example, increased FAO is a double-edged sword. On one side, enhanced FAO promotes the expression of PD-1 and polarization of M2, which accelerates tumor progression. On the other side, treatment increasing FAO boosts the effect of anti-PD-1 therapy. So how to balance the level of lipid metabolism manipulation, the specific mechanism and strategy deserve further studies, this may become the novel breakthrough encountering the obstacles of immunotherapy.
ZC and DW conceived the concept, DW and QY wrote the manuscript, ZC and HG revised this review. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81972598), Health Commission of Zhejiang Province (WKJ-ZJ-1803), Natural Science Foundation of Zhejiang Province (LQ20H160064, LY19H160004) and Fundamental Research Funds for the Central Universities (2020FZZX003-02-12, 2021FZ203-02-08).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Maan M, Peters JM, Dutta M, Patterson AD. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun (2018) 504(3):582–9. doi: 10.1016/j.bbrc.2018.02.097
2. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol (2020) 21(4):225–45. doi: 10.1038/s41580-019-0190-7
3. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
4. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer (2013) 13(4):227–32. doi: 10.1038/nrc3483
5. Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol (2014) 24(8):472–8. doi: 10.1016/j.tcb.2014.06.001
6. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab (2020) 31(1):62–76. doi: 10.1016/j.cmet.2019.11.010
7. Kopecka J, Trouillas P, Gasparovic AC, Gazzano E, Assaraf YG, Riganti C. Phospholipids and cholesterol: inducers of cancer multidrug resistance and therapeutic targets. Drug Resist Update (2020) 49:100670. doi: 10.1016/j.drup.2019.100670
8. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
9. Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, et al. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol (2019) 10:1399. doi: 10.3389/fimmu.2019.01399
10. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology (2017) 6(10):e1344804. doi: 10.1080/2162402X.2017.1344804
11. Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair cd8 t cells infiltrating human renal cell carcinoma. JCI Insight (2017) 2(12):e93411. doi: 10.1172/jci.insight.93411
12. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond) (2018) 38(1):27. doi: 10.1186/s40880-018-0301-4
13. Sharpe AH, Pauken KE. The diverse functions of the pd1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
14. Del Paggio JC. Immunotherapy: cancer immunotherapy and the value of cure. Nat Rev Clin Oncol (2018) 15(5):268–70. doi: 10.1038/nrclinonc.2018.27
15. Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer (2016) 16(2):121–6. doi: 10.1038/nrc.2016.2
16. Varade J, Magadan S, Gonzalez-Fernandez A. Human immunology and immunotherapy: main achievements and challenges. Cell Mol Immunol (2021) 18(4):805–28. doi: 10.1038/s41423-020-00530-6
17. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer (2016) 16(11):732–49. doi: 10.1038/nrc.2016.89
18. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor cd36. Nature (2017) 541(7635):41–5. doi: 10.1038/nature20791
19. Gallagher EJ, Zelenko Z, Neel BA, Antoniou IM, Rajan L, Kase N, et al. Elevated tumor ldlr expression accelerates ldl cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene (2017) 36(46):6462–71. doi: 10.1038/onc.2017.247
20. Zhou T, Zhan J, Fang W, Zhao Y, Yang Y, Hou X, et al. Serum low-density lipoprotein and low-density lipoprotein expression level at diagnosis are favorable prognostic factors in patients with small-cell lung cancer (sclc). BMC Cancer (2017) 17(1):269. doi: 10.1186/s12885-017-3239-z
21. Huang X, Wei X, Qiao S, Zhang X, Li R, Hu S, et al. Low density lipoprotein receptor (ldlr) and 3-hydroxy-3-methylglutaryl coenzyme a reductase (hmgcr) expression are associated with platinum-resistance and prognosis in ovarian carcinoma patients. Cancer Manag Res (2021) 13:9015–24. doi: 10.2147/CMAR.S337873
22. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med (2021) 218(1):e20201606. doi: 10.1084/jem.20201606
23. Jiang M, Wu N, Xu B, Chu Y, Li X, Su S, et al. Fatty acid-induced cd36 expression via o-glcnacylation drives gastric cancer metastasis. Theranostics (2019) 9(18):5359–73. doi: 10.7150/thno.34024
24. Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via cd36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep (2015) 5:14752. doi: 10.1038/srep14752
25. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-induced cd36 expression drives ovarian cancer progression and metastasis. Oncogene (2018) 37(17):2285–301. doi: 10.1038/s41388-017-0093-z
26. Zhang Y, Sun Y, Rao E, Yan F, Li Q, Zhang Y, et al. Fatty acid-binding protein e-fabp restricts tumor growth by promoting ifn-beta responses in tumor-associated macrophages. Cancer Res (2014) 74(11):2986–98. doi: 10.1158/0008-5472.CAN-13-2689
27. Hilvo M, Denkert C, Lehtinen L, Muller B, Brockmoller S, Seppanen-Laakso T, et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res (2011) 71(9):3236–45. doi: 10.1158/0008-5472.CAN-10-3894
28. Qian X, Hu J, Zhao J, Chen H. Atp citrate lyase expression is associated with advanced stage and prognosis in gastric adenocarcinoma. Int J Clin Exp Med (2015) 8(5):7855–60.
29. Wei X, Shi J, Lin Q, Ma X, Pang Y, Mao H, et al. Targeting acly attenuates tumor growth and acquired cisplatin resistance in ovarian cancer by inhibiting the pi3k-akt pathway and activating the ampk-ros pathway. Front Oncol (2021) 11:642229. doi: 10.3389/fonc.2021.642229
30. Zhang C, Liu J, Huang G, Zhao Y, Yue X, Wu H, et al. Cullin3-klhl25 ubiquitin ligase targets acly for degradation to inhibit lipid synthesis and tumor progression. Genes Dev (2016) 30(17):1956–70. doi: 10.1101/gad.283283.116
31. Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell (2006) 10(6):529–41. doi: 10.1016/j.ccr.2006.10.009
32. Wang MD, Wu H, Fu GB, Zhang HL, Zhou X, Tang L, et al. Acetyl-coenzyme a carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology (2016) 63(4):1272–86. doi: 10.1002/hep.28415
33. Rios Garcia M, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A, et al. Acetyl-coa carboxylase 1-dependent protein acetylation controls breast cancer metastasis and recurrence. Cell Metab (2017) 26(6):842–55 e5. doi: 10.1016/j.cmet.2017.09.018
34. Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. Rna interference-mediated silencing of the acetyl-coa-carboxylase-alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res (2005) 65(15):6719–25. doi: 10.1158/0008-5472.CAN-05-0571
35. Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of fatty acid oxidation as a therapy for myc-overexpressing triple-negative breast cancer. Nat Med (2016) 22(4):427–32. doi: 10.1038/nm.4055
36. Yu FY, Xu Q, Wei QY, Mo HY, Zhong QH, Zhao XY, et al. Acc2 is under-expressed in lung adenocarcinoma and predicts poor clinical outcomes. J Cancer Res Clin Oncol (2022). doi: 10.1007/s00432-021-03910-1
37. German NJ, Yoon H, Yusuf RZ, Murphy JP, Finley LW, Laurent G, et al. Phd3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of acc2. Mol Cell (2016) 63(6):1006–20. doi: 10.1016/j.molcel.2016.08.014
38. Nagy P, Vereb G, Sebestyen Z, Horvath G, Lockett SJ, Damjanovich S, et al. Lipid rafts and the local density of erbb proteins influence the biological role of homo- and heteroassociations of Erbb2. J Cell Sci (2002) 115(Pt 22):4251–62. doi: 10.1242/jcs.00118
39. Bandyopadhyay S, Zhan R, Wang Y, Pai SK, Hirota S, Hosobe S, et al. Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Cancer Res (2006) 66(11):5934–40. doi: 10.1158/0008-5472.CAN-05-3197
40. Heuer TS, Ventura R, Mordec K, Lai J, Fridlib M, Buckley D, et al. Fasn inhibition and taxane treatment combine to enhance anti-tumor efficacy in diverse xenograft tumor models through disruption of tubulin palmitoylation and microtubule organization and fasn inhibition-mediated effects on oncogenic signaling and gene expression. EBioMedicine (2017) 16:51–62. doi: 10.1016/j.ebiom.2016.12.012
41. Alwarawrah Y, Hughes P, Loiselle D, Carlson DA, Darr DB, Jordan JL, et al. Fasnall, a selective fasn inhibitor, shows potent anti-tumor activity in the mmtv-neu model of her2(+) breast cancer. Cell Chem Biol (2016) 23(6):678–88. doi: 10.1016/j.chembiol.2016.04.011
42. Luis G, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, et al. Tumor resistance to ferroptosis driven by stearoyl-coa desaturase-1 (scd1) in cancer cells and fatty acid biding protein-4 (fabp4) in tumor microenvironment promote tumor recurrence. Redox Biol (2021) 43:102006. doi: 10.1016/j.redox.2021.102006
43. Huang J, Fan XX, He J, Pan H, Li RZ, Huang L, et al. Scd1 is associated with tumor promotion, late stage and poor survival in lung adenocarcinoma. Oncotarget (2016) 7(26):39970–9. doi: 10.18632/oncotarget.9461
44. Su YC, Feng YH, Wu HT, Huang YS, Tung CL, Wu P, et al. Elovl6 is a negative clinical predictor for liver cancer and knockdown of elovl6 reduces murine liver cancer progression. Sci Rep (2018) 8(1):6586. doi: 10.1038/s41598-018-24633-3
45. Feng YH, Chen WY, Kuo YH, Tung CL, Tsao CJ, Shiau AL, et al. Elovl6 is a poor prognostic predictor in breast cancer. Oncol Lett (2016) 12(1):207–12. doi: 10.3892/ol.2016.4587
46. Han S, Wei R, Zhang X, Jiang N, Fan M, Huang JH, et al. Cpt1a/2-mediated fao enhancement-a metabolic target in radioresistant breast cancer. Front Oncol (2019) 9:1201. doi: 10.3389/fonc.2019.01201
47. Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, He MM, et al. Cpt1a-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene (2018) 37(46):6025–40. doi: 10.1038/s41388-018-0384-z
48. Wang M, Yu W, Cao X, Gu H, Huang J, Wu C, et al. Exosomal cd44 transmits lymph node metastatic capacity between gastric cancer cells via yap-cpt1a-mediated fao reprogramming. Front Oncol (2022) 12:860175. doi: 10.3389/fonc.2022.860175
49. Joshi M, Stoykova GE, Salzmann-Sullivan M, Dzieciatkowska M, Liebman LN, Deep G, et al. Cpt1a supports castration-resistant prostate cancer in androgen-deprived conditions. Cells (2019) 8(10):1115. doi: 10.3390/cells8101115
50. Pucci S, Zonetti MJ, Fisco T, Polidoro C, Bocchinfuso G, Palleschi A, et al. Carnitine palmitoyl transferase-1a (cpt1a): a new tumor specific target in human breast cancer. Oncotarget (2016) 7(15):19982–96. doi: 10.18632/oncotarget.6964
51. Xiong X, Wen YA, Fairchild R, Zaytseva YY, Weiss HL, Evers BM, et al. Upregulation of cpt1a is essential for the tumor-promoting effect of adipocytes in colon cancer. Cell Death Dis (2020) 11(9):736. doi: 10.1038/s41419-020-02936-6
52. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. Jak/Stat3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab (2018) 27(6):1357. doi: 10.1016/j.cmet.2018.04.018
53. Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1c promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev (2011) 25(10):1041–51. doi: 10.1101/gad.1987211
54. Wang Y, Chen Y, Guan L, Zhang H, Huang Y, Johnson CH, et al. Carnitine palmitoyltransferase 1c regulates cancer cell senescence through mitochondria-associated metabolic reprograming. Cell Death Differ (2018) 25(4):735–48. doi: 10.1038/s41418-017-0013-3
55. Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S, Mitsche M, et al. Fatty acid oxidation mediated by acyl-coa synthetase long chain 3 is required for mutant kras lung tumorigenesis. Cell Rep (2016) 16(6):1614–28. doi: 10.1016/j.celrep.2016.07.009
56. Rossi Sebastiano M, Pozzato C, Saliakoura M, Yang Z, Peng RW, Galie M, et al. Acsl3-Pai-1 signaling axis mediates tumor-stroma cross-talk promoting pancreatic cancer progression. Sci Adv (2020) 6(44):eabb9200. doi: 10.1126/sciadv.abb9200
57. Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs nadph production and increases reactive oxygen species resulting in atp depletion and cell death in human glioblastoma cells. Biochim Biophys Acta (2011) 1807(6):726–34. doi: 10.1016/j.bbabio.2010.10.022
58. Xi Y, Yani Z, Jing M, Yinhang W, Xiaohui H, Jing Z, et al. Mechanisms of induction of tumors by cholesterol and potential therapeutic prospects. BioMed Pharmacother (2021) 144:112277. doi: 10.1016/j.biopha.2021.112277
59. Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, et al. Inhibition of soat1 suppresses glioblastoma growth via blocking srebp-1-mediated lipogenesis. Clin Cancer Res (2016) 22(21):5337–48. doi: 10.1158/1078-0432.CCR-15-2973
60. Bemlih S, Poirier MD, El Andaloussi A. Acyl-coenzyme a: cholesterol acyltransferase inhibitor avasimibe affect survival and proliferation of glioma tumor cell lines. Cancer Biol Ther (2010) 9(12):1025–32. doi: 10.4161/cbt.9.12.11875
61. Gobel A, Breining D, Rauner M, Hofbauer LC, Rachner TD. Induction of 3-hydroxy-3-methylglutaryl-coa reductase mediates statin resistance in breast cancer cells. Cell Death Dis (2019) 10(2):91. doi: 10.1038/s41419-019-1322-x
62. Kong Y, Cheng L, Mao F, Zhang Z, Zhang Y, Farah E, et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (Crpc). J Biol Chem (2018) 293(37):14328–41. doi: 10.1074/jbc.RA118.004442
63. Xia Y, Xie Y, Yu Z, Xiao H, Jiang G, Zhou X, et al. The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell (2018) 175(4):1059–73 e21. doi: 10.1016/j.cell.2018.08.070
64. You W, Ke J, Chen Y, Cai Z, Huang ZP, Hu P, et al. Sqle, a key enzyme in cholesterol metabolism, correlates with tumor immune infiltration and immunotherapy outcome of pancreatic adenocarcinoma. Front Immunol (2022) 13:864244. doi: 10.3389/fimmu.2022.864244
65. Stopsack KH, Gerke TA, Sinnott JA, Penney KL, Tyekucheva S, Sesso HD, et al. Cholesterol metabolism and prostate cancer lethality. Cancer Res (2016) 76(16):4785–90. doi: 10.1158/0008-5472.CAN-16-0903
66. Li J, Yang T, Wang Q, Li Y, Wu H, Zhang M, et al. Upregulation of sqle contributes to poor survival in head and neck squamous cell carcinoma. Int J Biol Sci (2022) 18(9):3576–91. doi: 10.7150/ijbs.68216
67. Nagaraja R, Olaharski A, Narayanaswamy R, Mahoney C, Pirman D, Gross S, et al. Preclinical toxicology profile of squalene epoxidase inhibitors. Toxicol Appl Pharmacol (2020) 401:115103. doi: 10.1016/j.taap.2020.115103
68. Shangguan X, Ma Z, Yu M, Ding J, Xue W, Qi J. Squalene epoxidase metabolic dependency is a targetable vulnerability in castration-resistant prostate cancer. Cancer Res (2022) 3822.2021. doi: 10.1158/0008-5472.CAN-21-3822
69. Cheng M, Bhujwalla ZM, Glunde K. Targeting phospholipid metabolism in cancer. Front Oncol (2016) 6:266. doi: 10.3389/fonc.2016.00266
70. Iorio E, Mezzanzanica D, Alberti P, Spadaro F, Ramoni C, D’Ascenzo S, et al. Alterations of choline phospholipid metabolism in ovarian tumor progression. Cancer Res (2005) 65(20):9369–76. doi: 10.1158/0008-5472.CAN-05-1146
71. Yi K, Zhan Q, Wang Q, Tan Y, Fang C, Wang Y, et al. Ptrf/Cavin-1 remodels phospholipid metabolism to promote tumor proliferation and suppress immune responses in glioblastoma by stabilizing cpla2. Neuro Oncol (2021) 23(3):387–99. doi: 10.1093/neuonc/noaa255
72. Li C, Teixeira AF, Zhu HJ, Ten Dijke P. Cancer associated-fibroblast-derived exosomes in cancer progression. Mol Cancer (2021) 20(1):154. doi: 10.1186/s12943-021-01463-y
73. Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics (2021) 11(17):8322–36. doi: 10.7150/thno.62378
74. Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X, et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis (2020) 11(4):267. doi: 10.1038/s41419-020-2434-z
75. Radhakrishnan R, Ha JH, Jayaraman M, Liu J, Moxley KM, Isidoro C, et al. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett (2019) 442:464–74. doi: 10.1016/j.canlet.2018.11.023
76. Santi A, Caselli A, Ranaldi F, Paoli P, Mugnaioni C, Michelucci E, et al. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim Biophys Acta (2015) 1853(12):3211–23. doi: 10.1016/j.bbamcr.2015.09.013
77. Lopes-Coelho F, Andre S, Felix A, Serpa J. Breast cancer metabolic cross-talk: fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol Cell Endocrinol (2018) 462(Pt B):93–106. doi: 10.1016/j.mce.2017.01.031
78. Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: from cancer progression to treatment. Prog Lipid Res (2020) 80:101055. doi: 10.1016/j.plipres.2020.101055
79. De Jaeghere EA, Denys HG, De Wever O. Fibroblasts fuel immune escape in the tumor microenvironment. Trends Cancer (2019) 5(11):704–23. doi: 10.1016/j.trecan.2019.09.009
80. Hutmacher C, Gonzalo Nunez N, Liuzzi AR, Becher B, Neri D. Targeted delivery of il2 to the tumor stroma potentiates the action of immune checkpoint inhibitors by preferential activation of nk and cd8(+) t cells. Cancer Immunol Res (2019) 7(4):572–83. doi: 10.1158/2326-6066.CIR-18-0566
81. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discovery (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
82. Lopez-Yrigoyen M, Cassetta L, Pollard JW. Macrophage targeting in cancer. Ann N Y Acad Sci (2021) 1499(1):18–41. doi: 10.1111/nyas.14377
83. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11(10):889–96. doi: 10.1038/ni.1937
84. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci (2021) 22(13):6995. doi: 10.3390/ijms22136995
85. Zhao R, Wan Q, Wang Y, Wu Y, Xiao S, Li Q, et al. M1-like tams are required for the efficacy of pd-l1/pd-1 blockades in gastric cancer. Oncoimmunology (2020) 10(1):1862520. doi: 10.1080/2162402X.2020.1862520
86. Ding T, Xu J, Wang F, Shi M, Zhang Y, Li SP, et al. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum Pathol (2009) 40(3):381–9. doi: 10.1016/j.humpath.2008.08.011
87. Cao S, Saw PE, Shen Q, Li R, Liu Y, Xu X. Reduction-responsive rnai nanoplatform to reprogram tumor lipid metabolism and repolarize macrophage for combination pancreatic cancer therapy. Biomaterials (2022) 280:121264. doi: 10.1016/j.biomaterials.2021.121264
88. Netea-Maier RT, Smit JWA, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: a mutual relationship. Cancer Lett (2018) 413:102–9. doi: 10.1016/j.canlet.2017.10.037
89. Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C, et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med (2019) 11(11):e10698. doi: 10.15252/emmm.201910698
90. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res (2020) 80(7):1438–50. doi: 10.1158/0008-5472.CAN-19-2994
91. Zeng W, Yin X, Jiang Y, Jin L, Liang W. Pparalpha at the crossroad of metabolic-immune regulation in cancer. FEBS J (2021). doi: 10.1111/febs.16181
92. Liu S, Zhang H, Li Y, Zhang Y, Bian Y, Zeng Y, et al. S100a4 enhances protumor macrophage polarization by control of ppar-gamma-dependent induction of fatty acid oxidation. J Immunother Cancer (2021) 9(6):e002548. doi: 10.1136/jitc-2021-002548
93. Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, et al. Ripk3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res (2020) 8(5):710–21. doi: 10.1158/2326-6066.CIR-19-0261
94. Niu Z, Shi Q, Zhang W, Shu Y, Yang N, Chen B, et al. Caspase-1 cleaves ppargamma for potentiating the pro-tumor action of tams. Nat Commun (2017) 8(1):766. doi: 10.1038/s41467-017-00523-6
95. Piccinin E, Cariello M, Moschetta A. Lipid metabolism in colon cancer: role of liver x receptor (lxr) and stearoyl-coa desaturase 1 (scd1). Mol Aspects Med (2021) 78:100933. doi: 10.1016/j.mam.2020.100933
96. Carbo JM, Leon TE, Font-Diaz J, de la Rosa JV, Castrillo A, Picard FR, et al. Pharmacologic activation of lxr alters the expression profile of tumor-associated macrophages and the abundance of regulatory t cells in the tumor microenvironment. Cancer Res (2021) 81(4):968–85. doi: 10.1158/0008-5472.CAN-19-3360
97. Cui MY, Yi X, Zhu DX, Wu J. Aberrant lipid metabolism reprogramming and immune microenvironment for gastric cancer: a literature review. Transl Cancer Res (2021) 10(8):3829–42. doi: 10.21037/tcr-21-655
98. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. Pd-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
99. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific t cells following pd-1 blockade. Nat Med (2019) 25(8):1251–9. doi: 10.1038/s41591-019-0522-3
100. Wang H, Franco F, Ho PC. Metabolic regulation of tregs in cancer: opportunities for immunotherapy. Trends Cancer (2017) 3(8):583–92. doi: 10.1016/j.trecan.2017.06.005
101. Wang L, Simons DL, Lu X, Tu TY, Solomon S, Wang R, et al. Connecting blood and intratumoral treg cell activity in predicting future relapse in breast cancer. Nat Immunol (2019) 20(9):1220–30. doi: 10.1038/s41590-019-0429-7
102. Chakraborty S, Zappasodi R. To go or not to go?-targeting tregs traveling in tumors. Cancer Res (2021) 81(11):2817–9. doi: 10.1158/0008-5472.CAN-21-1203
103. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci U.S.A. (2018) 115(28):E6546–E55. doi: 10.1073/pnas.1720113115
104. Buck MD, O’Sullivan D, Pearce EL. T Cell metabolism drives immunity. J Exp Med (2015) 212(9):1345–60. doi: 10.1084/jem.20151159
105. O’Sullivan D, Pearce EL. Fatty acid synthesis tips the th17-treg cell balance. Nat Med (2014) 20(11):1235–6. doi: 10.1038/nm.3744
106. Corral-Jara KF, Rosas da Silva G, Fierro NA, Soumelis V. Modeling the th17 and tregs paradigm: implications for cancer immunotherapy. Front Cell Dev Biol (2021) 9:675099. doi: 10.3389/fcell.2021.675099
107. Wen YA, Xiong X, Zaytseva YY, Napier DL, Vallee E, Li AT, et al. Downregulation of srebp inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis (2018) 9(3):265. doi: 10.1038/s41419-018-0330-6
108. Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G, et al. Lipid signalling enforces functional specialization of treg cells in tumours. Nature (2021) 591(7849):306–11. doi: 10.1038/s41586-021-03235-6
109. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector t cells and adaptive immunity. Nat Immunol (2013) 14(5):489–99. doi: 10.1038/ni.2570
110. Nunez NG, Tosello Boari J, Ramos RN, Richer W, Cagnard N, Anderfuhren CD, et al. Tumor invasion in draining lymph nodes is associated with treg accumulation in breast cancer patients. Nat Commun (2020) 11(1):3272. doi: 10.1038/s41467-020-17046-2
111. Maeda S, Murakami K, Inoue A, Yonezawa T, Matsuki N. Ccr4 blockade depletes regulatory t cells and prolongs survival in a canine model of bladder cancer. Cancer Immunol Res (2019) 7(7):1175–87. doi: 10.1158/2326-6066.CIR-18-0751
112. He S, Cai T, Yuan J, Zheng X, Yang W. Lipid metabolism in tumor-infiltrating t cells. Adv Exp Med Biol (2021) 1316:149–67. doi: 10.1007/978-981-33-6785-2_10
113. Davis FP, Kanno Y, O’Shea JJ. A metabolic switch for th17 pathogenicity. Cell (2015) 163(6):1308–10. doi: 10.1016/j.cell.2015.11.033
114. Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, et al. Cd5l/Aim regulates lipid biosynthesis and restrains th17 cell pathogenicity. Cell (2015) 163(6):1413–27. doi: 10.1016/j.cell.2015.10.068
115. Huang A, Peng D, Guo H, Ben Y, Zuo X, Wu F, et al. A human programmed death-ligand 1-expressing mouse tumor model for evaluating the therapeutic efficacy of anti-human pd-l1 antibodies. Sci Rep (2017) 7:42687. doi: 10.1038/srep42687
116. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. Pd-1 alters t-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
117. Zhao Y, Shao Q, Peng G. Exhaustion and senescence: two crucial dysfunctional states of t cells in the tumor microenvironment. Cell Mol Immunol (2020) 17(1):27–35. doi: 10.1038/s41423-019-0344-8
118. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing Cd8(+) t cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32(3):377–91 e9. doi: 10.1016/j.ccell.2017.08.004
119. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16(8):880–6. doi: 10.1038/nm.2172
120. Giovanelli P, Sandoval TA, Cubillos-Ruiz JR. Dendritic cell metabolism and function in tumors. Trends Immunol (2019) 40(8):699–718. doi: 10.1016/j.it.2019.06.004
121. Peng X, He Y, Huang J, Tao Y, Liu S. Metabolism of dendritic cells in tumor microenvironment: for immunotherapy. Front Immunol (2021) 12:613492. doi: 10.3389/fimmu.2021.613492
122. Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol (2004) 4(12):941–52. doi: 10.1038/nri1498
123. Lu Y, Shi Y, Luo Z, Guo X, Jiang M, Li X, et al. Reactivation of dysfunctional dendritic cells by a stress-relieving nanosystem resets anti-tumor immune landscape. Nano Today (2022) 43:101416. doi: 10.1016/j.nantod.2022.101416
124. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. Er stress sensor xbp1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell (2015) 161(7):1527–38. doi: 10.1016/j.cell.2015.05.025
125. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun (2017) 8(1):2122. doi: 10.1038/s41467-017-02186-9
126. Oh DS, Lee HK. Autophagy protein atg5 regulates cd36 expression and anti-tumor mhc class ii antigen presentation in dendritic cells. Autophagy (2019) 15(12):2091–106. doi: 10.1080/15548627.2019.1596493
127. Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for atg5 in antigen presentation by dendritic cells. Immunity (2010) 32(2):227–39. doi: 10.1016/j.immuni.2009.12.006
128. Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res (2020) 8(11):1440–51. doi: 10.1158/2326-6066.CIR-20-0111
129. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
130. Choi HS, Ha SY, Kim HM, Ahn SM, Kang MS, Kim KM, et al. The prognostic effects of tumor infiltrating regulatory t cells and myeloid derived suppressor cells assessed by multicolor flow cytometry in gastric cancer patients. Oncotarget (2016) 7(7):7940–51. doi: 10.18632/oncotarget.6958
131. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res (2015) 3(11):1236–47. doi: 10.1158/2326-6066.CIR-15-0036
132. Wu H, Weidinger C, Schmidt F, Keye J, Friedrich M, Yerinde C, et al. Oleate but not stearate induces the regulatory phenotype of myeloid suppressor cells. Sci Rep (2017) 7(1):7498. doi: 10.1038/s41598-017-07685-9
133. Chen Y, Sui M. Lipid metabolism in tumor-associated natural killer cells. Adv Exp Med Biol (2021) 1316:71–85. doi: 10.1007/978-981-33-6785-2_5
134. Raskov H, Orhan A, Salanti A, Gaggar S, Gogenur I. Natural killer cells in cancer and cancer immunotherapy. Cancer Lett (2021) 520:233–42. doi: 10.1016/j.canlet.2021.07.032
135. Wang F, Lau JKC, Yu J. The role of natural killer cell in gastrointestinal cancer: killer or helper. Oncogene (2021) 40(4):717–30. doi: 10.1038/s41388-020-01561-z
136. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on nk cells inhibits nk-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene (2017) 36(44):6143–53. doi: 10.1038/onc.2017.209
137. Poznanski SM, Singh K, Ritchie TM, Aguiar JA, Fan IY, Portillo AL, et al. Metabolic flexibility determines human nk cell functional fate in the tumor microenvironment. Cell Metab (2021) 33(6):1205–20.e5. doi: 10.1016/j.cmet.2021.03.023
138. Niavarani SR, Lawson C, Bakos O, Boudaud M, Batenchuk C, Rouleau S, et al. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer (2019) 19(1):823. doi: 10.1186/s12885-019-6045-y
139. Lizee G, Overwijk WW, Radvanyi L, Gao J, Sharma P, Hwu P. Harnessing the power of the immune system to target cancer. Annu Rev Med (2013) 64:71–90. doi: 10.1146/annurev-med-112311-083918
140. Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, et al. Tgfbeta1-mediated smad3 enhances pd-1 expression on antigen-specific t cells in cancer. Cancer Discovery (2016) 6(12):1366–81. doi: 10.1158/2159-8290.CD-15-1347
141. Lim TS, Chew V, Sieow JL, Goh S, Yeong JP, Soon AL, et al. Pd-1 expression on dendritic cells suppresses cd8(+) t cell function and antitumor immunity. Oncoimmunology (2016) 5(3):e1085146. doi: 10.1080/2162402X.2015.1085146
142. Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L. The immune checkpoint pd-1 in natural killer cells: expression, function and targeting in tumour immunotherapy. Cancers (Basel) (2020) 12(11):3285. doi: 10.3390/cancers12113285
143. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol (2020) 20(1):25–39. doi: 10.1038/s41577-019-0218-4
144. Sherr CJ, Beach D, Shapiro GI. Targeting cdk4 and cdk6: from discovery to therapy. Cancer Discovery (2016) 6(4):353–67. doi: 10.1158/2159-8290.CD-15-0894
145. Wan H, Xu B, Zhu N, Ren B. Pgc-1alpha activator-induced fatty acid oxidation in tumor-infiltrating ctls enhances effects of pd-1 blockade therapy in lung cancer. Tumori (2020) 106(1):55–63. doi: 10.1177/0300891619868287
146. Chowdhury PS, Chamoto K, Kumar A, Honjo T. Ppar-induced fatty acid oxidation in t cells increases the number of tumor-reactive cd8(+) t cells and facilitates anti-pd-1 therapy. Cancer Immunol Res (2018) 6(11):1375–87. doi: 10.1158/2326-6066.CIR-18-0095
147. Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. Stat3 activation-induced fatty acid oxidation in cd8(+) t effector cells is critical for obesity-promoted breast tumor growth. Cell Metab (2020) 31(1):148–61 e5. doi: 10.1016/j.cmet.2019.10.013
148. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the Antitumour Response of Cd8(+) T Cells by Modulating Cholesterol Metabolism. Nature (2016) 531(7596):651–5. doi: 10.1038/nature17412
149. Zhao L, Li J, Liu Y, Kang L, Chen H, Jin Y, et al. Cholesterol Esterification Enzyme Inhibition Enhances Antitumor Effects of Human Chimeric Antigen Receptors Modified T Cells. J Immunother (2018) 41(2):45–52. doi: 10.1097/CJI.0000000000000207
150. Li M, Yang Y, Wei J, Cun X, Lu Z, Qiu Y, et al. Enhanced Chemo-Immunotherapy against Melanoma by Inhibition of Cholesterol Esterification in Cd8(+) T Cells. Nanomedicine (2018) 14(8):2541–50. doi: 10.1016/j.nano.2018.08.008
151. Liu X, Hartman CL, Li L, Albert CJ, Si F, Gao A, et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci Transl Med (2021) 13(587):eaaz6314. doi: 10.1126/scitranslmed.aaz6314
152. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. Cd36-mediated metabolic adaptation supports regulatory t cell survival and function in tumors. Nat Immunol (2020) 21(3):298–308. doi: 10.1038/s41590-019-0589-5
153. Adeshakin AO, Liu W, Adeshakin FO, Afolabi LO, Zhang M, Zhang G, et al. Regulation of ros in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-pd-l1 tumor immunotherapy. Cell Immunol (2021) 362:104286. doi: 10.1016/j.cellimm.2021.104286
154. Xin G, Chen Y, Topchyan P, Kasmani MY, Burns R, Volberding PJ, et al. Targeting pim1-mediated metabolism in myeloid suppressor cells to treat cancer. Cancer Immunol Res (2021) 9(4):454–69. doi: 10.1158/2326-6066.CIR-20-0433
155. Wang G, Xu J, Zhao J, Yin W, Liu D, Chen W, et al. Arf1-mediated lipid metabolism sustains cancer cells and its ablation induces anti-tumor immune responses in mice. Nat Commun (2020) 11(1):220. doi: 10.1038/s41467-019-14046-9
156. Poh AR, Love CG, Chisanga D, Steer JH, Baloyan D, Chopin M, et al. Therapeutic inhibition of the src-kinase hck facilitates T cell tumor infiltration and improves response to immunotherapy. Sci Adv (2022) 8(25):eabl7882. doi: 10.1126/sciadv.abl7882
157. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of ifn-gamma pathway genes in tumor cells as a mechanism of resistance to anti-ctla-4 therapy. Cell (2016) 167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069
158. Liao Q, Zhou Y, Xia L, Cao D. Lipid metabolism and immune checkpoints. Adv Exp Med Biol (2021) 1316:191–211. doi: 10.1007/978-981-33-6785-2_12
159. Wang Y, Wang Y, Ren Y, Zhang Q, Yi P, Cheng C. Metabolic modulation of immune checkpoints and novel therapeutic strategies in cancer. Semin Cancer Biol (2022) S1044–579X(22):00031–1. doi: 10.1016/j.semcancer.2022.02.010
160. Xiong Y, Wang L, Di Giorgio E, Akimova T, Beier UH, Han R, et al. Inhibiting the coregulator corest impairs foxp3+ treg function and promotes antitumor immunity. J Clin Invest (2020) 130(4):1830–42. doi: 10.1172/JCI131375
161. Ji D, Song C, Li Y, Xia J, Wu Y, Jia J, et al. Combination of radiotherapy and suppression of tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J Immunother Cancer (2020) 8(2):e000826. doi: 10.1136/jitc-2020-000826
162. Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor cd36 promotes lipid peroxidation and dysfunction in cd8(+) t cells in tumors. Immunity (2021) 54(7):1561–77.e7. doi: 10.1016/j.immuni.2021.05.003
163. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. Cd36-mediated ferroptosis dampens intratumoral cd8(+) t cell effector function and impairs their antitumor ability. Cell Metab (2021) 33(5):1001–12.e5. doi: 10.1016/j.cmet.2021.02.015
164. Kim W, Chu TH, Nienhuser H, Jiang Z, Del Portillo A, Remotti HE, et al. Pd-1 signaling promotes tumor-infiltrating myeloid-derived suppressor cells and gastric tumorigenesis in mice. Gastroenterology (2021) 160(3):781–96. doi: 10.1053/j.gastro.2020.10.036
165. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science (2020) 368(6486):85–9. doi: 10.1126/science.aaw9872
166. Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, et al. Pkcbetaii phosphorylates acsl4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol (2022) 24(1):88–98. doi: 10.1038/s41556-021-00818-3
167. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. Cd8(+) t cells and fatty acids orchestrate tumor ferroptosis and immunity via acsl4. Cancer Cell (2022) 40(4):365–78 e6. doi: 10.1016/j.ccell.2022.02.003
168. Wang S, Yi X, Wu Z, Guo S, Dai W, Wang H, et al. Camkk2 defines ferroptosis sensitivity of melanoma cells by regulating ampknrf2 pathway. J Invest Dermatol (2022) 142(1):189–200.e8. doi: 10.1016/j.jid.2021.05.025
169. Liu P, Lin C, Liu Z, Zhu C, Lin Z, Xu D, et al. Inhibition of alg3 stimulates cancer cell immunogenic ferroptosis to potentiate immunotherapy. Cell Mol Life Sci (2022) 79(7):352. doi: 10.1007/s00018-022-04365-4
170. Zhang K, Qian Y, Quan X, Zhu T, Qian B. A novel signature of lipid metabolism-related gene predicts prognosis and response to immunotherapy in lung adenocarcinoma. Front Cell Dev Biol (2022) 10:730132. doi: 10.3389/fcell.2022.730132
Keywords: lipid metabolism, tumor, immune cells, immunotherapy, microenvironment
Citation: Wang D, Ye Q, Gu H and Chen Z (2022) The role of lipid metabolism in tumor immune microenvironment and potential therapeutic strategies. Front. Oncol. 12:984560. doi: 10.3389/fonc.2022.984560
Received: 02 July 2022; Accepted: 01 August 2022;
Published: 12 September 2022.
Edited by:
Haineng Xu, University of Pennsylvania, United StatesReviewed by:
Xianyi Cai, Huazhong University of Science and Technology, ChinaCopyright © 2022 Wang, Ye, Gu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhigang Chen, Y2hlbnpoaWdhbmdAemp1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.