 Yuan-rui Xu
Yuan-rui Xu An-long Wang2
An-long Wang2- 1The Second Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China
- 2Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Hangzhou, Zhejiang, China
Patients with chronic obstructive pulmonary disease (COPD), irrespective of their smoking history, are more likely to develop lung cancer than the general population. This is mainly because COPD is characterized by chronic persistent inflammation and hypoxia, which are the risk factors for lung cancer. However, the mechanisms underlying this observation are still unknown. Hypoxia-inducible factor 1-alpha (HIF-1α) plays an important role in the crosstalk that exists between inflammation and hypoxia. Furthermore, HIF-1α is the main regulator of somatic adaptation to hypoxia and is highly expressed in hypoxic environments. In this review, we discuss the molecular aspects of the crosstalk between hypoxia and inflammation, showing that HIF-1α is an important signaling pathway that drives COPD progression to lung cancer. Here, we also provide an overview of HIF-1α and its principal regulatory mechanisms, briefly describe HIF-1α-targeted therapy in lung cancer, and summarize substances that may be used to target HIF-1α at the level of COPD-induced inflammation.
Introduction
Lung cancer is the leading cause of cancer-related death worldwide (1). Although immunotherapy has drastically improved the prognosis of lung cancer patients, its efficacy is constrained to individuals with more than 50% programmed cell death 1 ligand 1 (PD-L1) expression. Accordingly, a global multicenter study of PD-L1 expression in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) reported that only 22% of the patients expressed PD-L1 levels greater than 50% (2). NSCLC is the predominant histological subtype of lung cancer that accounts for 85% of all lung cancer cases (3). Chronic obstructive pulmonary disease (COPD), which is categorized by chronic inflammation and airflow restriction, is prevalent among patients with NSCLC and notably increases mortality. COPD occurs because individuals with COPD and lung cancer are less likely to receive chemotherapy or immunotherapy. However, if the progression of COPD to lung cancer can be delayed, the incidence and mortality rates of lung cancer will be reduced. Therefore, it is crucial to first understand how the recognized risk factors contribute to the early progression of lung cancer in order to achieve this objective.
COPD and lung cancer are related
Smoking is a major risk factor for lung cancer, and patients with COPD are four to six times more likely to develop lung cancer than smokers without COPD (4). Furthermore, COPD may cause lung cancer in nonsmokers (4). Therefore, COPD/emphysema is a risk factor for lung cancer regardless of smoking status (5). In a study, 2,507 patients with COPD were monitored for an average of 60 months. Overall, 215 individuals developed lung cancer at a rate of 16.7 per 1,000 person-years after the follow-up (6). COPD severity is also a critical risk factor for lung cancer. In a large epidemiological study with a 22-year follow-up of 5,402 participants, an increased COPD severity was linked to an elevated risk of lung cancer (7). Moreover, emphysema diagnosed using computed tomography (CT) and airflow restriction determined using spirometry is also associated with lung cancer (8). Accordingly, annual CT screening has been demonstrated to enhance lung cancer diagnosis and decrease mortality, although this is only in individuals with normal or minimally compromised lung function (9).
Approximately 1% of COPD patients develop lung cancer each year (10, 11). According to the latest Global Burden of Disease (GBD) statistics, the number of people with COPD exceeded 1,400,000 in 2018, and the number of people with lung cancer increased by about 400,000 from 2018 to 2019 (12). Therefore, it is likely that about 30% of the new lung cancer population from 2018 to 2019 is related to COPD. It’s a staggeringly theoretical value. However, there are no real-world statistical findings to confirm this figure. In addition, smoking is known to be a common risk factor for COPD and lung cancer. Therefore, this theoretical value is not influenced by COPD alone. However, it has been shown above that the risk of lung cancer is much greater in COPD patients than in the smoking population. Therefore, this theoretical data is sufficient to warn us to pay attention to COPD among the risk factors for lung cancer.
Inflammation and hypoxia link COPD with lung cancer
Numerous mechanisms, including chronic inflammation, aberrant immunology, oxidative stress, epithelial-mesenchymal transition, hypoxia, and genetic variables, have been used to elucidate the association between COPD and lung cancer (13–22). Hypoxia as a carcinogenic etiology of COPD has recently piqued the interest of researchers. Interestingly, a reciprocal relationship exists between hypoxia and inflammation. Hypoxia can aggravate inflammation by activating inflammatory pathways and influencing immune cell functions. Conversely, during inflammation, pathologies such as thrombosis, trauma, compression (interstitial hypertension), and atelectasis (airway obstruction) elevate cells’ metabolic requirements, consequently decreasing oxygen delivery and worsening tissue hypoxia (23). Therefore, hypoxia and inflammation significantly contribute to COPD carcinogenesis, and HIF-1α plays a critical role in both processes. Additionally, hypoxia can increase HIF-1α expression in hypoxic tissues and the serum. Two key angiogenic factors, which include vascular endothelial growth factor-A (VEGF-A) and angiopoietin-2 (Ang-2), are controlled by HIF-1α. Ang-2 promotes vasculature remodeling by inhibiting the associated angiopoietin-1 protein (24). Angiogenesis is also crucial for tumor progression and development. Administering VEGF-specific monoclonal antibodies to mice suppressed tumor development (25). VEGF-A also mediates vascular permeability, which is linked with malignant effusions (26). Furthermore, neo-angiogenesis and VEGF expression in NSCLC are markers of a poor prognosis (27). Surprisingly, VEGF receptor 2 (VEGFR2), VEGF-A and C, matrix metalloproteinase-2 (MMP-2), and MMP-9 were overexpressed in the lungs and tumors of hypoxic mice. VEGF-A and MMP-9 also increase in hypoxic malignancies (28). Angiogenesis is controlled by tie receptors, which are involved in vascular homeostasis, and VEGF receptors are the essential components of angiogenesis. VEGF receptor 1 (VEGFR1) and Tie2 are downregulated in hypoxic mouse tumors, which may cause aberrant vascular leakage linked to tumor invasion (28). In the 3-methylcholanthrene/butylated hydroxytoluene animal model, which is an inflammation-dependent priming-promoting model, chronic persistent alveolar hypoxia increased the expression of epidermal growth factor receptor (EGFR), fibroblast growth factor receptor 2, and platelet-derived growth factor receptor, which are known to promote tumor growth and angiogenesis. However, intermittent hypoxia did not induce the same increase in these growth factor receptors (28).
In addition to VEGF family, the downstream target genes of HIF, which include nuclear factor kappa B (NF-κB) and toll-like receptors, link hypoxia to inflammation (23). Inflammation significantly contributes to an increased susceptibility to lung cancer in patients with COPD. In addition, several NF-κB (key regulators of inflammation) target genes involved in carcinogenesis, including interleukin-6, MMP-9, and cyclooxygenase 2, are also downstream genes of HIF-1α (29). Hypoxia induces NF-κB expression (30). Furthermore, NF-κB stimulates cell proliferation, inhibits programmed cell death, facilitates tumor dissemination, and modifies tumor metabolism in the context of chronic inflammation (29). Moreover, NF-κB can also decrease p53 stability to promote carcinogenesis and can collaborate with HIF-1α to enable the activation of tumor-promoting gene promoters (31). NF-κB also directly regulates the expression of genes encoding MMPs such as MMP-9, leading to extracellular matrix remodeling, which promotes cancer cell distribution in the vicinity (29).
Furthermore, chronically inflamed tissue leads to a hypoxic environment that prevents hypoxia-inducible factor prolyl hydroxylase (PHD), thereby regulating HIF stability. Interleukin-1β and lipopolysaccharide are proinflammatory cytokines that increase the basal transcription rate of HIF messenger RNA probably because NF-κB binds to the HIF-1α promoter and upregulates HIF-1α (32). NF-κB also promotes HIF-1α activation and enhances HIF-1α expression during hypoxia (33). In contrast, PHD and HIF-1α inhibitors (FIH) regulate NF-κB activation by controlling inhibitor of kappa B kinase (IKKβ)complex activity, which is a regulatory component of NF-κB (34) (Table 1 and Figure 1).
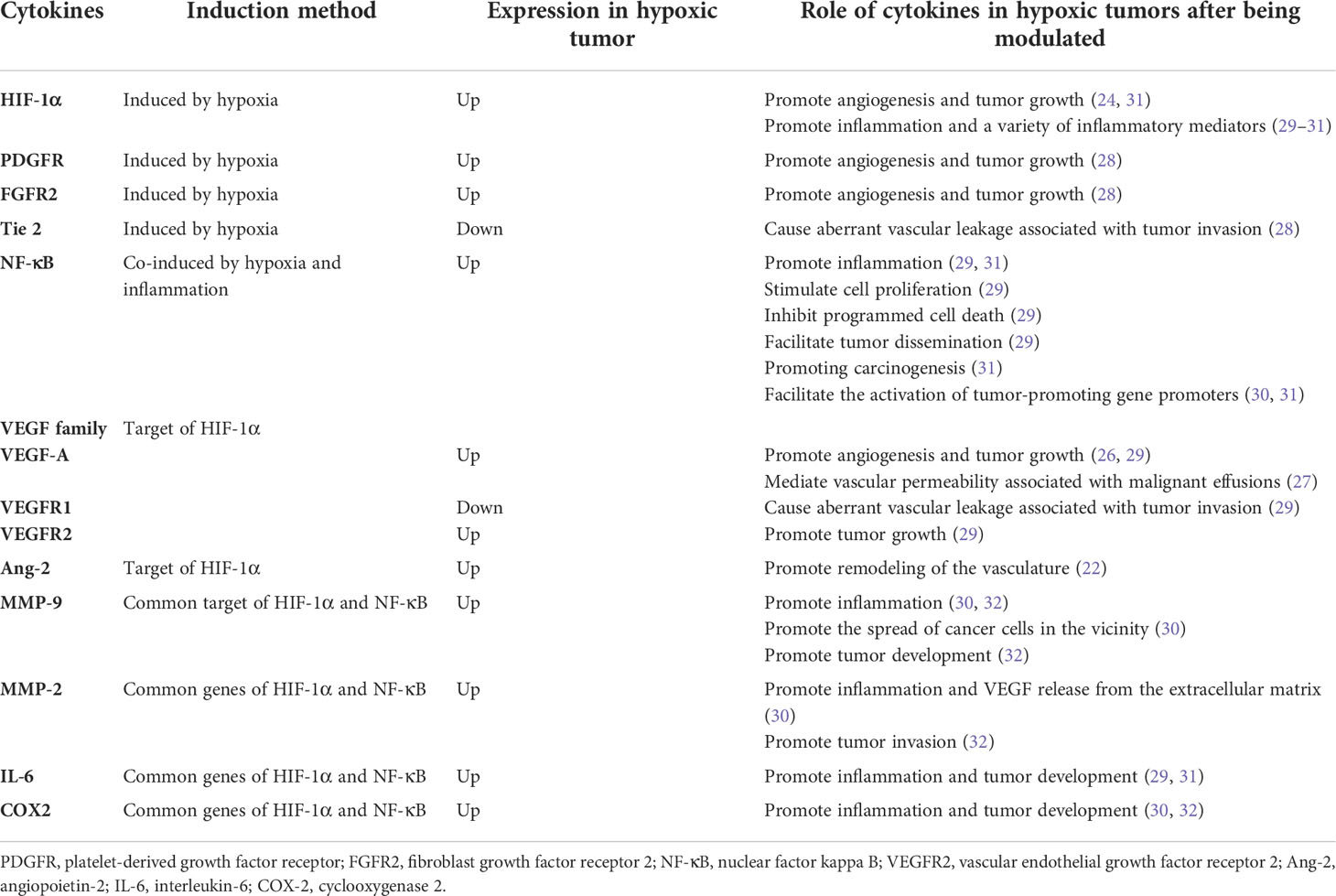
Table 1 Hypoxia and inflammation combined in cancer.
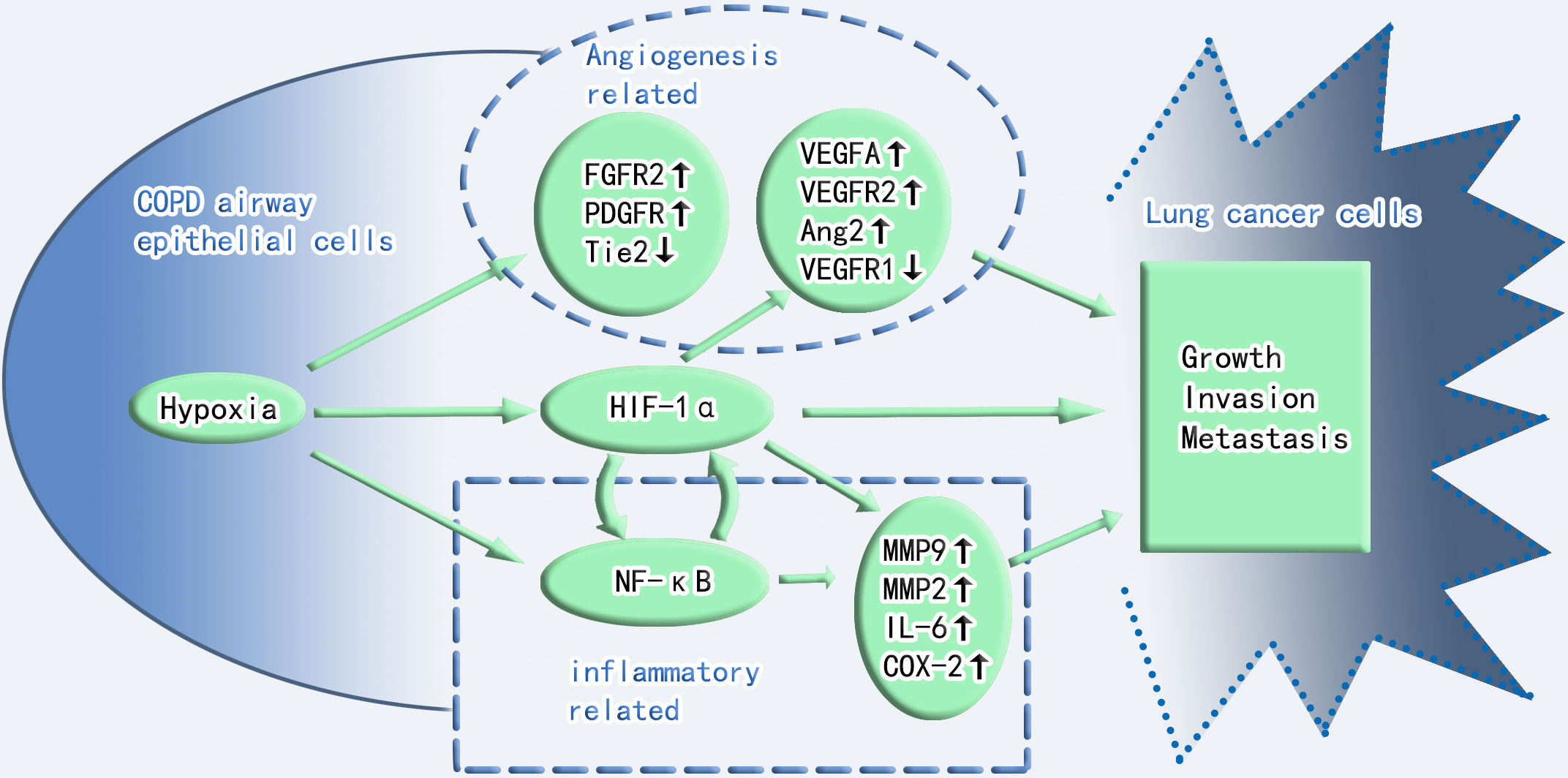
Figure 1 Crosstalk between hypoxia and inflammation in COPD and lung cancer toxicity. Angiogenesis, HIF-1α, and inflammation play important roles in the progression of COPD to lung cancer. Angiogenesis and inflammation cross-talk through HIF-1α. FGFR2, fibroblast growth factor receptor 2; PDGFR, platelet-derived growth factor receptor; Tie2, Tie2 (a member of the receptor tyrosine kinase family and the receptor for angiopoietin); VEGF-A, vascular endothelial growth factor-A; VEGFR2, vascular endothelial growth factor receptor 2; VEGFR1, vascular endothelial growth factor receptor 1; Ang2, angiopoietin-2; NF-κB, nuclear factor kappa B; IL-6, interleukin-6; COX-2, cyclooxygenase 2; MMP9, matrix metalloproteinase-9; MMP2, matrix metalloproteinase-2.
HIF-1
HIF-1 initiation
HIF-1 is essential for the body’s adaptive regulatory response to changes in the oxygen environment and is involved in several physiological and pathological processes in the body, as well as in the origin of many disorders. HIF-1 is a heterodimeric transcription factor with three oxygen-sensitive alpha subunits, HIF-1α, HIF-2α, and HIF-3α, and one constitutive beta subunit, HIF-1β (35). Interestingly, studies on the mammalian HIF-1α have significantly contributed to the knowledge of the structure and regulation of transcription factors. Both HIF-1α and HIF-1β possess a basic helix loop helix (bHLH) domain and two Per-Arnt-Sim (PAS) domains, which include PAS-A and PAS-B (36). Additionally, the bHLH and PAS structural domains mediate the heterodimerization of HIF-1α and HIF-1β. A basal region that precedes the N-terminus of the bHLH domain binds this heterodimer to the hypoxia response element (HRE)-DNA motifs of the target gene promoters (36). The two transactivation domains (TADs) are categorized as follows: NH2-terminal TAD (N-TAD) and COOH-terminal TAD (C-TAD), which are located at the NH2-and COOH terminuses, respectively. N-TAD is found within the oxygen-dependent degradation domain (ODDD), which is constrained to amino acid residues (400–600) of HIF-1α. Conversely, ODDD and C-TAD control the activity and stability of the α-subunit, which will be described in detail below. Furthermore, the amino acid sequence (576–785) of the inhibitory domain (ID) that prevents the transcriptional activity of N-TAD and C-TAD under normoxic conditions separates N-TAD and C-TAD (36). Summarily, HIF-1 signaling begins with its dimerization. HIF-1α migrates to the nucleus and dimerizes with HIF-1β. Subsequently, the dimer binds to the promoters of target genes containing HREs, initiating the transcription of multiple genes, including EGFR (35), which is involved in cellular adaptation to hypoxia, metabolism, and cellular function (37). Furthermore, the ability of HIF-1 to cause hypoxia-induced transactivation significantly depends on its HIF-1α subunit (36) (Table 2).
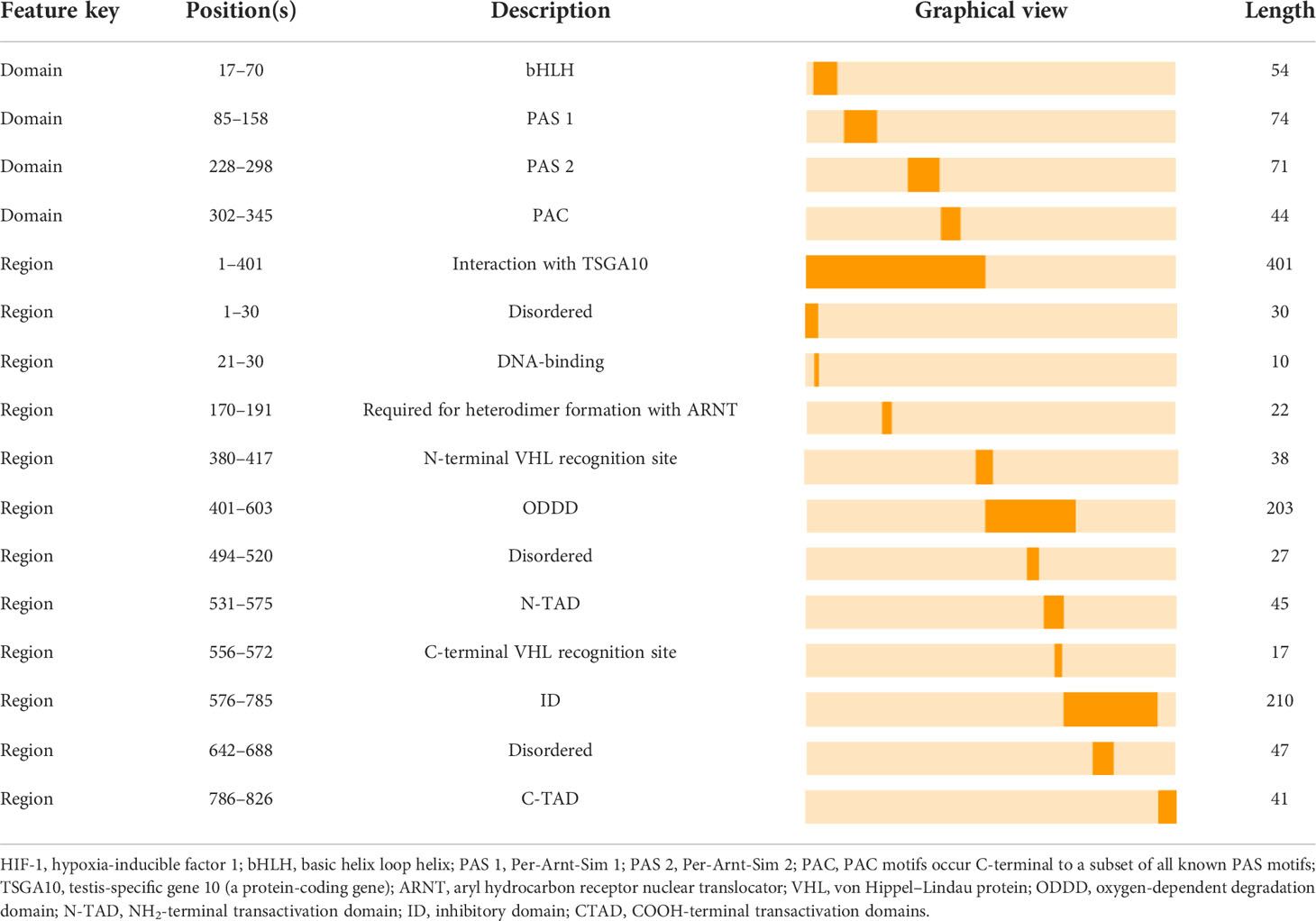
Table 2 The Location of the HIF-1 Partial Functional Domains/Regions.
Hydroxylation regulates HIF-1α
Several environmental factors influence HIF-1 expression. Interestingly, the principal mechanism by which HIF-1 is regulated is hydroxylation, with most of the mechanism occurring in the α-subunit. In resting cells, PHD, which includes PHD1, PHD2, and PHD3, hydroxylates proline residues within the ODDD region of HIF-1α, enabling the ubiquitination of HIF-1α by the von Hippel–Lindau protein (pVHL) E3 ubiquitin ligase (36, 38). Therefore, the PHD and pVHL proteins primarily facilitate HIF-1α degradation. Additionally, since PHD depends on oxygen, the baseline level of HIF-1α remains low under normoxic conditions with a half-life of less than 5 minutes (39). Conversely, hypoxia inhibits PHD and reduces HIF-1 hydroxylation, resulting in HIF-1 accumulation, which translocates to the nucleus and binds to its coactivator (38). Moreover, asparagine hydroxylase, under normal conditions, hydroxylates asparagine residues 803 and 847 within the HIF-1α C-TADs. It prevents HIF-1α from interacting with its coactivator, which is a cyclic adenosine monophosphate response element binding protein-binding Protein/P300 (CBP/p300) (36, 40). In contrast, this FIH function is suppressed under hypoxic conditions, which enables the binding of HIF-1α to its coactivators and boosts the expression of its downstream target genes. Additionally, the signal transducer and activator of transcription 3 enhance HIF-1α activity by blocking VHL from binding to HIF-1α and facilitating the coactivator CBP/p300 to bind to HIF-1α. Additionally, the mammalian target of rapamycin (mTOR) induces HIF-1α binding to its coactivator (41) (Figure 2).
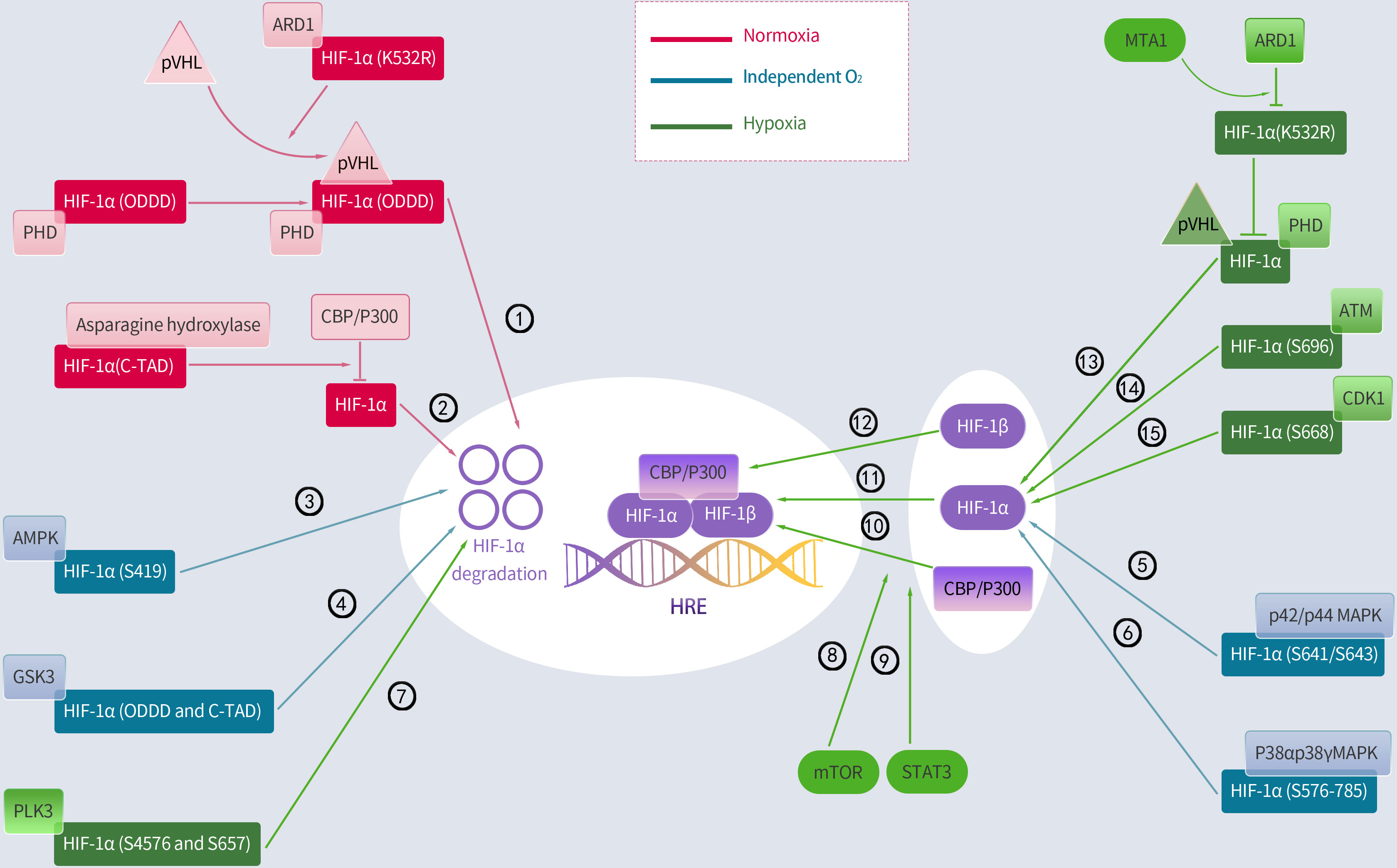
Figure 2 Regulation of HIF-1α by hydroxylation, phosphorylation and acetylation. Activated regulatory pathways under normoxia: ①②; Activation of the following pathway is less associated with oxygen: ③④⑤⑥;Activated regulatory pathways under hypoxia: ⑦⑧⑨⑩⑪⑫⑬⑭⑮. PHD, prolyl hydroxylase; pVHL, Von Hippel-Lindau protein; ARD1, arrest defective-1; CBP/P300, cAMP response element binding protein (CREB)-binding protein and P300; ODDD, oxygen-dependent degradation domain; AMPK, adenosine 5’-monophosphate (AMP)-activated protein kinase; GSK3, glycogen synthase kinase-3; PLK3, polo-like kinase-3; MTA1, metastasis-associated protein 1; ATM, ataxia and telangiectasia mutated; p42/p44 MAPK, mitogen-activated protein kinase; CDK1, cyclin-dependent kinase-1; mTOR, mammalian target of rapamycin; STAT3, signal transducer and activator of transcription 3.
Acetylation regulates HIF-1α
Moreover, lysine acetylation can alter the HIF-1α protein at various sites, resulting in obvious downstream effects. However, these findings remain controversial. A report indicated that the lysine (K532) residue of HIF-1α directly interacts with arrest defective-1 (ARD1: a protein acetyltransferase) to acetylate HIF-1α, thereby enabling HIF-1α binding to pVHL and finally HIF-1α degradation. This study further proved that pVHL-dependent HIF-1α degradation is associated with intra-ODDD acetylation. Metastasis-associated protein 1 (MTA1) is a member of the MTA family that opposes the acetylation function of ARD1 and upregulates the expression of histone deacetylase 1, which induces HIF-1α deacetylation at the K532R site, thereby improving the transcriptional activity and stability of the HIF-1α protein (36). A hypoxic environment notably enhances MTA1 expression (42). However, a report indicates that ARD1 neither acetylates nor destabilizes HIF-1α (43). Similarly, ARD1 inhibition and overexpression had minimal effect on basal HIF-1α levels or hypoxic responses (44). Additionally, the CBP/p300-associated factor acetylates the lysine residue (K674), which elevates HIF-1α levels and promotes HIF-1α binding to P300 (45). In particular, Geng et al. (46) demonstrated that P300 stabilizes HIF-1α by acetylating lysine residues (K709). Summarily, most current studies indicate that HIF-1α is acetylated and deacetylated, leading to its removal and accumulation, respectively (Figure 2).
Phosphorylation regulates HIF-1α
Protein kinases control HIF-1α activity and phosphorylate specific HIF-1α residues under normoxic conditions. Cyclin-dependent kinase-1 (CDK1) phosphorylates HIF-1α at serine 668, increasing the expression of HIF-1α and its target genes and boosting tumor angiogenesis, proliferation, and growth (47). Similarly, ataxia and telangiectasia mutated (ATM) proteins phosphorylate the inhibitory domain residue S696 to stabilize HIF-1α (48). Conversely, polo-like kinase-3 (P3) phosphorylates residues S576 and S657 to destabilize HIF-1α (49). Moreover, glycogen synthase kinase-3 (GSK3) also phosphorylates residues S551, T555, S589, T498 S502, S505, T506, and S510 in ODDD and N-TAD, which enhances HIF-1α degradation (50–52). In the human liver hepatocellular cell lines (HepG2), chronic hypoxia induced an increase in GSK3 activity, which decreased HIF-1α protein levels (52); however, this effect was undetected in other cell types. Additionally, adenosine 5’-monophosphate-activated protein kinase (AMPK) phosphorylates S419 in HIF-1α to downregulate HIF-1α (53). Additionally, two members of the p38 mitogen-activated protein kinase (MAPK) family, p38α and p38γ, can phosphorylate the amino acid 576–785 domain of HIF-1α, thereby contributing to its stability (54). The activation of the p42/p44 MAPK pathway induces HIF-1α by phosphorylating S641/S643 in HIF-1α (55–57).This study further found that p42/p44 MAPK activation was sufficient to promote the transcriptional activity of HIF-1 in the VEGF promoter mutated at a MAPK-sensitive site (SP1/AP2-88-66 site) (57) (Figure 2).
COPD and lung cancer exhibit aberrant HIF-1α expression
HIF-1α corresponds with COPD severity
COPD causes a hypoxic environment for HIF-1α. Numerous studies have proven that HIF-1α is overexpressed in the lungs of patients (58–60). Wang et al. (59) measured HIF-1α serum levels using enzyme-linked immunosorbent assay. They discovered that serum HIF-1α levels were higher in stable patients with COPD than in the general population and that these higher serum levels were positively correlated with the Global Initiative for Chronic Obstructive Lung Disease (GOLD) classification, the United Kingdom modified Medical Research Council (mMRC) score, and medical history. Therefore, this result suggests that increased HIF-1α serum levels are associated with COPD progression. Additionally, Fu et al. (61) assessed HIF-1α expression in the lung tissues of 102 smokers with or without COPD. Patients with COPD had increased levels of HIF-1α and its downstream target genes, including VEGF and VEGFR2, in their lung tissue compared with healthy individuals and nonsmokers without COPD. Furthermore, COPD severity has been associated with HIF-1α expression and downstream target genes. In another study, Zhang et al. (60) investigated the bronchoalveolar lavage fluid from patients with COPD and healthy participants. They found that HIF-1α was overexpressed, and the production of inflammatory markers was elevated in patients with COPD through the activation of the EGFR/PI3K/AKT pathway. Moreover, in a feedback loop, the lung inflammation-induced EGFR/PI3K/AKT pathway upregulates HIF-1α expression, eventually exacerbating COPD.
HIF-1α stimulates tumor development
Increased levels of HIF-1α expression promote tumor growth, whereas a reduction in its activity inhibits tumor growth (62). HIF-1α levels in lung cancer tissue samples are excessively high and play a crucial role in tumor genesis, development, and metastasis (63–65). Infantino et al. (66) demonstrated that HIF-1α contributes to the regulation of glutamine and serine metabolism as well as the one-carbon cycle and fatty acid metabolism in hypoxic cancer cells, thereby reprogramming cancer cell metabolism under hypoxic conditions. Additionally, these changes promote cell survival, proliferation, and tumor growth. Furthermore, HIF-1α overexpression enhanced tumor development, vascularization, and energy metabolism, whereas a decrease in its activity had the opposite impact during in vitro xenograft tests (67–70). Moreover, cigarette smoke extract, which is a prevalent risk factor for COPD and lung cancer, also elevates HIF-1α in a concentration-dependent manner (71). In addition, VEGFR, which is one of the essential target genes downstream of HIF-1α, adapts cancer cells to the hypoxic environment and stimulates the formation of new blood vessels in the tumor, provides nutrients and oxygen to promote tumor growth and metabolism, and presents a major pathway for cancer cells to metastasize to distant organs (72).
HIF-1α links COPD with lung cancer
HIF-1α may increase the risk of cancer in patients with COPD. Although when the gene encoding HIF-1α was disrupted in the lung epithelium of a K-ras mutant mouse model (CC-LR), independent of COPD-like airway inflammation, the number of surface tumors in the mouse lung, tumor angiogenesis, and tumor cell proliferation were considerably reduced (73). However, COPD- and adenocarcinoma-like phenotypes were observed in their offspring when CC-LR mice were bred with transgenic animals overexpressing human HIF-1α in airway epithelial cells. According to the same study, the CC-LR mice with overexpressed HIF-1α in the COPD airway epithelium developed substantial emphysema. Additionally, they possessed an aggressive metastatic phenotype, with increased tumorigenesis, angiogenesis, and cell proliferation. Polosukhin et al. (74) analyzed 55 bronchial biopsies from smokers with COPD and found that HIF-1α contributes to the progression of precancerous epithelial lesions in the airways of smokers with chronic airway inflammation. Furthermore, HIF-1α promotes cigarette smoke exposure-induced malignant transformation of bronchial epithelial cells, which is a process dependent on AKT/NF-κB pathway (75). Moreover, HIF-1α also encourages epithelial-mesenchymal transition and the acquisition of cancer stem cell-like characteristics in the bronchial epithelium (76, 77). Therefore, these findings indicated a link between COPD-associated airway inflammation, HIF-1α, and lung cancer.
Targeting HIF-1α in lung cancer
The mechanism of action of HIF-1α in lung cancer, HIF-1α targets, and HIF-1α pharmacology are of significant interest to researchers. In NSCLC experiments, several substances, such as the HIF-1α inhibitor LW6 (78), chetomin (79), gamma-linolenic acid (80), propofol (81), a novel mycotoxin-derived compound GL331 (82), resveratrol (83), sevoflurane (84), flavanols (85), MiR-199a (86, 87), and connective tissue growth factor (88), possess anticancer properties focusing on cancer cell proliferation and invasion capabilities, by downregulating HIF-1α levels. Interestingly, digoxin inhibits hypoxia-induced VEGF, HIF-1α, and N-myc downstream-regulated genes 1 (NDRG1) in a concentration-dependent manner in A549 cells (89). Topotecan and etoposide inhibited the hypoxia-induced expression of HIF-1α protein in NSCLC cell lines in a dose- and time-dependent manner (90). Conversely, the combination of emodin and cisplatin downregulated the multidrug resistance-1 gene and HIF-1α production in lung tumor cells (91), which restricted cancer cell proliferation, adhesion, migration, and tumor angiogenesis. Additionally, PX-478, which is a potent small-molecule HIF-1α inhibitor, had a substantial anticancer effect in two adenocarcinoma models, PC14-PE6 and NCI-H441, and two small cell lung cancer (SCLC) models, NCI-H187 and NCI-N41 (92). Furthermore, cytokine signaling 3 (SOCS3) suppresses HIF-1α expression, reducing the progression and spread of SCLC (93). Sulforaphane inhibits HIF-1α expression under hypoxic conditions probably because of its H2S-donating characteristics (94, 95). Sphingosine kinase 1 (96) and andrographolide (97) prevent HIF-1α expression in NSCLC cells, although no link with anticancer activity was detected. Moreover, SU5416 and KRN633 are VEGFR tyrosine kinase inhibitors that reduce HIF-1α expression in various cancer cells (excluding lung cancer) (98). Therefore, future studies are necessary to determine whether they have similar effects on lung cancer cells (Table 3).

Table 3 Substances that can target HIF-1α in lung cancer.
Targeting HIF-1α could reduce COPD-related oncogenic effects
The above evidence implies that an increase in HIF-1α levels in COPD leads to the progression of COPD to lung cancer progression. Conversely, inhibiting HIF-1α expression in COPD may delay or block this process. However, some studies have proven that HIF-1α plays a crucial role in the energy metabolism of inflammatory cells and that preventing it causes severe immunodeficiency (24). In a preliminary assessment of HIF interference in inflammatory diseases, the hydroxylase inhibitors dimethyloxalylglycine and FG4497 had a significant protective effect on two models of inflammatory bowel disease (IBD), such as dextran sodium sulfate and 2,4,6-trinitrobenzene sulfonic acid (99, 100). Notably, the respiratory and digestive systems have a common embryonic origin in the primitive foregut. Consequently, their structural similarities may explain their comparable immunological responses to noxious stimuli (101). Furthermore, the closely related IBD and COPD are chronic inflammatory diseases of the digestive and respiratory systems, respectively. Moreover, exposure to cigarette smoke is also a common risk factor for both diseases. Accordingly, both disease states show increased levels of innate lymphocytes and unconventional T cells, such as γδT cells, and myeloid cells, including neutrophils, eosinophils, and macrophages (102). Because VEGF is an important downstream target gene of HIF, we also considered whether blocking its pathway would have a similar effect, and we searched for relevant information. Although evidence that VEGF inhibitors can improve COPD exists, whether they can delay or block the progression of COPD to lung cancer has not been elucidated. Therefore, additional studies are required to clarify this issue. Accordingly, we briefly describe the drugs that can target HIF-1α at the level of COPD, as well as the therapies that can accomplish this effect.
Non-coding RNA
Non-coding RNAs (ncRNAs) are RNAs that do not code for proteins and are categorized into short microRNAs (miRNAs) and long ncRNAs. Targeting ncRNAs is a promising therapeutic strategy since they are involved in various complex pathophysiological processes, such as immune system development and function, immunological diseases, and neurodevelopmental and neurological disorders (103).
Interestingly, miRNAs are prevalent in most somatic tissues. Additionally, they are generally 22 nucleotides long and created by two ribonuclease III proteins, Drosha and Dicer (104), which are essential for gene expression regulation (105), predominantly through translational repression or messenger RNA degradation (106). Additionally, miRNAs control the expression of HIF-1α (107, 108). Notably, Shiro et al. (109) were the first to reveal that individuals with COPD/emphysema had higher lung tissue expression of miR-34a and miR-199-5p. Specifically, they observed that human pulmonary microvascular endothelial cells (HPMVECs) transfected with the miR-199a-5p gene showed a reduced HIF-1α protein expression. Additionally, miR-199-5p expression was elevated in HPMVECs transfected with miR-34a precursor gene. In a recent study, Wu et al. (110) found that miR-125a-5p levels were higher in normal smokers and COPD smokers than in the healthy population. Furthermore, researchers have used smoke to induce COPD in mouse models. They found that miR-125a-5p targeted the 3’-untranslated region of Sp1 mRNA. Sp1 promotes sirtuin 1 (SIRT1) expression, and SIRT1 deacetylates HIF-1α, thereby downregulating HIF-1α on Lys674. Therefore, when miR-125a-5p was knocked down, HIF-1α expression was increased. Similarly, Li et al. (105) conducted a cell culture experiment with lung fibroblasts (MRC-5) and found that miR-186 decreased HIF-1α by directly targeting HIF-1α mRNA. These results indicate that the increased expression of miR-199a-5p, miR-34a, miRNA-186, or miR-125a-5p in the lung tissue of patients with COPD may inhibit HIF-1α production, consequently reducing COPD carcinogenesis. Furthermore, numerous miRNA-targeted therapies, including those for solid tumors, viral hepatitis C, atherosclerosis, myocardial infarction, renal fibrosis, diabetes and its complications, and COPD, have been employed in animal trials and perhaps attained clinical development (111). Despite insufficient studies on the application of miRNAs to lower HIF-1α expression in patients with COPD, this treatment strategy appears beneficial and promising.
Sodium hydrosulfide
Sodium hydrosulfide (NaHS) is an H2S donor. Wu et al. (112) discovered that NaHS reversed HIF-1α accumulation during hypoxia and that this effect persisted even after the degradation of the ubiquitin-proteasome pathway was inhibited. Guan et al. (113) also discovered that NaHS significantly reduced cigarette smoke-induced HIF-1α protein expression and enhanced PHD2 protein levels in a mouse model and in in vitro A549 alveolar epithelial cells. Notably, NaHS reduces inflammation, epithelial cell damage, and apoptosis by blocking HIF-1α signaling. These findings suggest that NaHS may be a novel therapy for COPD. Additionally, NaHS protects against stress-induced lung injury because of its antioxidative, anti-inflammatory, antifibrotic, and antiapoptotic properties (114). Over the past few decades, several H2S-based treatment approaches, including those based on H2S donation and the restriction of H2S generation, have been identified. H2S donors can be obtained through the inhalation of H2S gas, parenteral or enteral injection of H2S salts, or other methods. Furthermore, H2S donors can be rapidly or slowly released, such as NaHS, Na2S, or GY4137, which is a prototype chemical employed in several in vivo and in vitro studies (115). These findings indicate that NaHS may be used as a therapy to prevent the progression of COPD to lung cancer.
Pyrrolidine dithiocarbamate
Pyrrolidine dithiocarbamate (PDTC) is an antioxidant that is believed to be a potent NF-κB inhibitor (116). Briefly, rats were orally administered PDTC, treated by the intratracheal instillation of lipopolysaccharide, and subsequently exposed to cigarette smoke, as described by Jiang et al. (117). They found a decreased HIF-1α expression through NF-κB inhibition in the COPD group caused by PDTC pretreatment. Therefore, PDTC may be used to lower HIF-1α expression in patients in the future. Moreover, PDTC, which is an NF-κB inhibitor, may also be effective for treating COPD.
Lycium barbarum polysaccharides
Lycium barbarum polysaccharides (LBP), comprising glucose, arabinose, galactose, mannose, rhamnose, and xylose, are the primary bioactive components of the Chinese herbal remedy, Lycium barbarum. Interestingly, multiple conditions, such as cancer, aging, fatigue, colitis, stroke, diabetes, Alzheimer’s disease, and glaucoma, have been proven to benefit from LBP (118). Chen et al. (119) demonstrated that oral LBP (5 mg/mL, 100 mL twice daily for 2 weeks) improved COPD symptoms and significantly decreased blood HIF-1α levels. In this study, oral LBP elicited moderate adverse effects, including constipation, dry mouth, inflamed gums, and gastrointestinal reactions. However, these unfavorable reactions to LBP diminished when the patient stopped taking LBP, signifying that the side effects associated with LBP were transient and reversible. Furthermore, animal studies reveal that LBP has no negative or teratogenic effects on sperm (120), although further studies are warranted to validate this claim. LBP may benefit patients with COPD and reduce lung cancer incidence; however, this also requires additional clinical verification.
Conclusion
HIF-1α signaling plays a key role in the progression of COPD to lung cancer and is a potential therapeutic target to delay the progression. Additional insight into the processes that govern HIF-1α expression and activation during COPD inflammatory carcinogenesis would be advantageous in developing COPD-specific HIF-1α therapies.
Author contributions
The structure of the manuscript was decided on by all the authors. Y-RX wrote the first draft of this manuscript. A-LW worked on the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81870028 and 81470241) and the Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents (No. A-2017-CXCR02).
Acknowledgments
The authors thank Yaqing Li for editing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet (London England) (2021) 398(10299):535–54. doi: 10.1016/s0140-6736(21)00312-3
2. Dietel M, Savelov N, Salanova R, Micke P, Bigras G, Hida T, et al. Real-world prevalence of programmed death ligand 1 expression in locally advanced or metastatic non-Small-Cell lung cancer: The global, multicenter express study. Lung Cancer (Amsterdam Netherlands) (2019) 134:174–9. doi: 10.1016/j.lungcan.2019.06.012
3. Gridelli C, Rossi A, Carbone DP, Guarize J, Karachaliou N, Mok T, et al. Non-Small-Cell lung cancer. Nat Rev Dis Primers (2015) 1:15009. doi: 10.1038/nrdp.2015.9
4. Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD. Copd prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur Respir J (2009) 34(2):380–6. doi: 10.1183/09031936.00144208
5. Turner MC, Chen Y, Krewski D, Calle EE, Thun MJ. Chronic obstructive pulmonary disease is associated with lung cancer mortality in a prospective study of never smokers. Am J Respir Crit Care Med (2007) 176(3):285–90. doi: 10.1164/rccm.200612-1792OC
6. de Torres JP, Marin JM, Casanova C, Cote C, Carrizo S, Cordoba-Lanus E, et al. Lung cancer in patients with chronic obstructive pulmonary disease– incidence and predicting factors. Am J Respir Crit Care Med (2011) 184(8):913–9. doi: 10.1164/rccm.201103-0430OC
7. Mannino DM, Aguayo SM, Petty TL, Redd SC. Low lung function and incident lung cancer in the united states: Data from the first national health and nutrition examination survey follow-up. Arch Intern Med (2003) 163(12):1475–80. doi: 10.1001/archinte.163.12.1475
8. Wilson DO, Weissfeld JL, Balkan A, Schragin JG, Fuhrman CR, Fisher SN, et al. Association of radiographic emphysema and airflow obstruction with lung cancer. Am J Respir Crit Care Med (2008) 178(7):738–44. doi: 10.1164/rccm.200803-435OC
9. Young RP, Hopkins RJ. Chronic obstructive pulmonary disease (Copd) and lung cancer screening. Transl Lung Cancer Res (2018) 7(3):347–60. doi: 10.21037/tlcr.2018.05.04
10. Tockman MS, Anthonisen NR, Wright EC, Donithan MG. Airways obstruction and the risk for lung cancer. Ann Internal Med (1987) 106(4):512–8. doi: 10.7326/0003-4819-106-4-512
11. Skillrud DM, Offord KP, Miller RD. Higher risk of lung cancer in chronic obstructive pulmonary disease. A Prospect Matched Controlled Study Ann Internal Med (1986) 105(4):503–7. doi: 10.7326/0003-4819-105-4-503
12. Ihme. (2022). Available at: https://vizhub.healthdata.org/gbd-results/.
13. King PT. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin Transl Med (2015) 4(1):68. doi: 10.1186/s40169-015-0068-z
14. Bozinovski S, Anthony D, McQualter J, Anderson G, Irving L, Steinfort D. Copd and squamous cell lung Cancer:Aberrant inflammation and immunity is the common link. Br J Pharmacol (2016) 173(4):635–48. doi: 10.1111/bph.2016.173.issue-4
15. Wang DC, Shi L, Zhu Z, Gao D, Zhang Y. Genomic mechanisms of transformation from chronic obstructive pulmonary disease to lung cancer. Semin Cancer Biol (2017) 42:52–9. doi: 10.1016/j.semcancer.2016.11.001
16. Shih YM, Chang YJ, Cooke MS, Pan CH, Hu CH, Chao MR, et al. Alkylating and oxidative stresses in smoking and non-smoking patients with copd: Implications for lung carcinogenesis. Free Radic Biol Med (2021) 164:99–106. doi: 10.1016/j.freeradbiomed.2020.12.442
17. Durham AL, Adcock IM. The relationship between copd and lung cancer. Lung Cancer (Amsterdam Netherlands) (2015) 90(2):121–7. doi: 10.1016/j.lungcan.2015.08.017
18. Caramori G, Ruggeri P, Mumby S, Ieni A, Lo Bello F, Chimankar V, et al. Molecular links between copd and lung cancer: New targets for drug discovery? Expert Opin Ther Targets (2019) 23(6):539–53. doi: 10.1080/14728222.2019.1615884
19. Eapen MS, Hansbro PM, Larsson-Callerfelt AK, Jolly MK, Myers S, Sharma P, et al. Chronic obstructive pulmonary disease and lung cancer: Underlying pathophysiology and new therapeutic modalities. Drugs (2018) 78(16):1717–40. doi: 10.1007/s40265-018-1001-8
20. Hou W, Hu S, Li C, Ma H, Wang Q, Meng G, et al. Cigarette smoke induced lung barrier dysfunction, emt, and tissue remodeling: A possible link between copd and lung cancer. BioMed Res Int (2019) 2019:2025636. doi: 10.1155/2019/2025636
21. Houghton AM. Mechanistic links between copd and lung cancer. Nat Rev Cancer (2013) 13(4):233–45. doi: 10.1038/nrc3477
23. Eltzschig HKHypoxia and inflammation. N Engl J Med. (2011) 364(7):656–65. doi: 10.1056/NEJMra0910283
24. Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature (2006) 441(7092):437–43. doi: 10.1038/nature04871
25. Kim KJ, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature (1993) 362(6423):841–4. doi: 10.1038/362841a0
26. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol (2005) 23(5):1011–27. doi: 10.1200/JCO.2005.06.081
27. Bremnes RM, Camps C, Sirera R. Angiogenesis in non-small cell lung cancer: The prognostic impact of neoangiogenesis and the cytokines vegf and bfgf in tumours and blood. Lung Cancer (Amsterdam Netherlands) (2006) 51(2):143–58. doi: 10.1016/j.lungcan.2005.09.005
28. Karoor V, Le M, Merrick D, Fagan KA, Dempsey EC, Miller YE. Alveolar hypoxia promotes murine lung tumor growth through a vegfr-2/Egfr-Dependent mechanism. Cancer Prev Res (Phila) (2012) 5(8):1061–71. doi: 10.1158/1940-6207.CAPR-12-0069-T
29. D'Ignazio L, Batie M, Rocha S. Hypoxia and inflammation in cancer, focus on hif and nf-kappab. Biomedicines (2017) 5(2): 21. doi: 10.3390/biomedicines5020021
30. Palazon A, Goldrath AW, Nizet V, Johnson RS. Hif transcription factors, inflammation, and immunity. Immunity (2014) 41(4):518–28. doi: 10.1016/j.immuni.2014.09.008
31. Balamurugan K. Hif-1 at the crossroads of hypoxia, inflammation, and cancer. Int J Cancer (2016) 138(5):1058–66. doi: 10.1002/ijc.29519
32. Scholz CC, Taylor CT. Targeting the hif pathway in inflammation and immunity. Curr Opin Pharmacol (2013) 13(4):646–53. doi: 10.1016/j.coph.2013.04.009
33. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. Nf-kappab links innate immunity to the hypoxic response through transcriptional regulation of hif-1alpha. Nature (2008) 453(7196):807–11. doi: 10.1038/nature06905
34. Eltzschig HK. Hypoxia and inflammation. N Engl J Med (2011) 364(7):656–65. doi: 10.1056/NEJMra0910283
35. Semenza GL. Hif-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol Med (2002) 8(4 Suppl):S62–7. doi: 10.1016/s1471-4914(02)02317-1
36. Albadari N, Deng S, Li W. The transcriptional factors hif-1 and hif-2 and their novel inhibitors in cancer therapy. Expert Opin Drug Discovery (2019) 14(7):667–82. doi: 10.1080/17460441.2019.1613370
37. Polosukhin VV, Cates JM, Lawson WE, Milstone AP, Matafonov AG, Massion PP, et al. Hypoxia-inducible factor-1 signalling promotes goblet cell hyperplasia in airway epithelium. J Pathol (2011) 224(2):203–11. doi: 10.1002/path.2863
38. Corcoran SE, O'Neill LA. Hif1alpha and metabolic reprogramming in inflammation. J Clin Invest (2016) 126(10):3699–707. doi: 10.1172/JCI84431
39. Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem (1996) 271(50):32253–9. doi: 10.1074/jbc.271.50.32253
40. Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Fih-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev (2002) 16(12):1466–71. doi: 10.1101/gad.991402
41. McGettrick AF, O'Neill LAJ. The role of hif in immunity and inflammation. Cell Metab (2020) 32(4):524–36. doi: 10.1016/j.cmet.2020.08.002
42. Yoo YG, Kong G, Lee MO. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J (2006) 25(6):1231–41. doi: 10.1038/sj.emboj.7601025
43. Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, et al. Interaction between hif-1 alpha (Odd) and Hard1 does not induce acetylation and destabilization of hif-1 alpha. FEBS Lett (2005) 579(28):6428–32. doi: 10.1016/j.febslet.2005.10.036
44. Fisher TS, Etages SD, Hayes L, Crimin K, Li B. Analysis of Ard1 function in hypoxia response using retroviral rna interference. J Biol Chem (2005) 280(18):17749–57. doi: 10.1074/jbc.M412055200
45. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell (2010) 38(6):864–78. doi: 10.1016/j.molcel.2010.05.023
46. Geng H, Liu Q, Xue C, David LL, Beer TM, Thomas GV, et al. Hif1alpha protein stability is increased by acetylation at lysine 709. J Biol Chem (2012) 287(42):35496–505. doi: 10.1074/jbc.M112.400697
47. Warfel NA, Dolloff NG, Dicker DT, Malysz J, El-Deiry WS. Cdk1 stabilizes hif-1alpha Via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle (2013) 12(23):3689–701. doi: 10.4161/cc.26930
48. Cam H, Easton JB, High A, Houghton PJ. Mtorc1 signaling under hypoxic conditions is controlled by atm-dependent phosphorylation of hif-1alpha. Mol Cell (2010) 40(4):509–20. doi: 10.1016/j.molcel.2010.10.030
49. Xu D, Yao Y, Lu L, Costa M, Dai W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of hif-1alpha. J Biol Chem (2010) 285(50):38944–50. doi: 10.1074/jbc.M110.160325
50. Flugel D, Gorlach A, Kietzmann T. Gsk-3beta regulates cell growth, migration, and angiogenesis Via Fbw7 and Usp28-dependent degradation of hif-1alpha. Blood (2012) 119(5):1292–301. doi: 10.1182/blood-2011-08-375014
51. Cassavaugh JM, Hale SA, Wellman TL, Howe AK, Wong C, Lounsbury KM. Negative regulation of hif-1alpha by an Fbw7-mediated degradation pathway during hypoxia. J Cell Biochem (2011) 112(12):3882–90. doi: 10.1002/jcb.23321
52. Flugel D, Gorlach A, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a vhl-independent manner. Mol Cell Biol (2007) 27(9):3253–65. doi: 10.1128/MCB.00015-07
53. Hwang AB, Ryu EA, Artan M, Chang HW, Kabir MH, Nam HJ, et al. Feedback regulation Via ampk and hif-1 mediates ros-dependent longevity in caenorhabditis elegans. Proc Natl Acad Sci U.S.A. (2014) 111(42):E4458–67. doi: 10.1073/pnas.1411199111
54. Kietzmann T, Mennerich D, Dimova EY. Hypoxia-inducible factors (Hifs) and phosphorylation: Impact on stability, localization, and transactivity. Front Cell Dev Biol (2016) 4:11. doi: 10.3389/fcell.2016.00011
55. Mylonis I, Chachami G, Paraskeva E, Simos G. Atypical Crm1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by mapk. J Biol Chem (2008) 283(41):27620–7. doi: 10.1074/jbc.M803081200
56. Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. Mapk signaling up-regulates the activity of hypoxia-inducible factors by its effects on P300. J Biol Chem (2003) 278(16):14013–9. doi: 10.1074/jbc.M209702200
57. Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. P42/P44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (Hif-1alpha) and enhance the transcriptional activity of hif-1. J Biol Chem (1999) 274(46):32631–7. doi: 10.1074/jbc.274.46.32631
58. Shukla SD, Walters EH, Simpson JL, Keely S, Wark PAB, O'Toole RF, et al. Hypoxia-inducible factor and bacterial infections in chronic obstructive pulmonary disease. Respirology (2020) 25(1):53–63. doi: 10.1111/resp.13722
59. Rong B, Liu Y, Li M, Fu T, Gao W, Liu H. Correlation of serum levels of hif-1alpha and il-19 with the disease progression of copd: A retrospective study. Int J Chron Obstruct Pulmon Dis (2018) 13:3791–803. doi: 10.2147/COPD.S177034
60. Zhang H-X, Zhang S-A, Zhang S-M, Wang J-X, Xu Z-Y. Hif-1α promotes inflammatory response of chronic obstructive pulmonary disease by activating egfr or Pi3k or akt pathway. Eur Rev Med Pharmacol Sci (2018) 22(18):6077–84.
61. Fu X, Zhang F. Role of the hif-1 signaling pathway in chronic obstructive pulmonary disease. Exp Ther Med (2018) 16(6):4553–61. doi: 10.3892/etm.2018.6785
62. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene (2010) 29(5):625–34. doi: 10.1038/onc.2009.441
63. Giatromanolaki A, Koukourakis MI, Sivridis E, Turley H, Talks K, Pezzella F, et al. Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non-small cell lung cancer to Angiogenic/Molecular profile of tumours and survival. Br J Cancer (2001) 85(6):881–90. doi: 10.1054/bjoc.2001.2018
64. Koukourakis MI, Papazoglou D, Giatromanolaki A, Panagopoulos I, Maltezos E, Harris AL, et al. C2028t polymorphism in exon 12 and dinucleotide repeat polymorphism in intron 13 of the hif-1alpha gene define hif-1alpha protein expression in non-small cell lung cancer. Lung Cancer (Amsterdam Netherlands) (2006) 53(3):257–62. doi: 10.1016/j.lungcan.2006.05.025
65. Ghattass K, El-Sabban M, Gali-Muhtasib H. Targeting hypoxia for sensitization of tumors to radio- and chemotherapy. Curr Cancer Drug Targets (2013) 13(6):670–85. doi: 10.2174/15680096113139990004
66. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer cell metabolism in hypoxia: Role of hif-1 as key regulator and therapeutic target. Int J Mol Sci (2021) 22(11):5703. doi: 10.3390/ijms22115703
67. Semenza GL. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol Med (2001) 7(8):345–50. doi: 10.1016/s1471-4914(01)02090-1
68. Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, et al. Transcription factor hif-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol (2001) 21(10):3436–44. doi: 10.1128/mcb.21.10.3436-3444.2001
69. Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. Regulation of tumor angiogenesis by P53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev (2000) 14(1):34–44. doi: 10.1101/gad.14.1.34
70. Kung AL, Wang S, Klco JM, Kaelin WG, Livingston DM. Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat Med (2000) 6(12):1335–40. doi: 10.1038/82146
71. Yoo YM, Jung EM, Jeon BH, Tran DN, Jeung EB. Cigarette smoke extraxt influences intracellular calcium concentration in A549 cells. J Physiol Pharmacol (2020) 71(5):679–687. doi: 10.26402/jpp.2020.5.08
72. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Sci (New York NY) (2016) 352(6282):175–80. doi: 10.1126/science.aaf4405
73. de la Garza MM, Cumpian AM, Daliri S, Castro-Pando S, Gong L, Khosravi1 N, et al. Copd-type lung inflammation promotes K-ras mutant lung cancer through epithelial hif-1α mediated tumor angiogenesis and proliferation. Oncotarget (2018) 9(68):32972–83. MJT. doi: 10.18632/oncotarget.26030
74. Polosukhin VV, Lawson WE, Milstone AP, Egunova SM, Kulipanov AG, Tchuvakin SG, et al. Association of progressive structural changes in the bronchial epithelium with subepithelial fibrous remodeling: A potential role for hypoxia. Virchows Arch (2007) 451(4):793–803. doi: 10.1007/s00428-007-0469-5
75. Lu L, Xu H, Yang P, Xue J, Chen C, Sun Q, et al. Involvement of hif-1alpha-Regulated mir-21, acting Via the Akt/Nf-kappab pathway, in malignant transformation of hbe cells induced by cigarette smoke extract. Toxicol Lett (2018) 289:14–21. doi: 10.1016/j.toxlet.2018.02.027
76. Jiang P, Hao S, Xie L, Xiang G, Hu W, Wu Q, et al. Lncrna Neat1 contributes to the acquisition of a tumor like-phenotype induced by pm 2.5 in lung bronchial epithelial cells. Via Hif-1α Activation Environ Sci pollut Res Int (2021) 28(32):43382–93. doi: 10.1007/s11356-021-13735-7
77. Gu X, Zhang J, Shi Y, Shen H, Li Y, Chen Y, et al. Esm1/Hif−1α pathway modulates chronic intermittent Hypoxia−Induced Non−Small−Cell lung cancer proliferation, stemness and Epithelial−Mesenchymal transition. Oncol Rep (2021) 45(3):1226–34. doi: 10.3892/or.2020.7913
78. Sato M, Hirose K, Kashiwakura I, Aoki M, Kawaguchi H, Hatayama Y, et al. Lw6, a hypoxia-inducible factor 1 inhibitor, selectively induces apoptosis in hypoxic cells through depolarization of mitochondria in A549 human lung cancer cells. Mol Med Rep (2015) 12(3):3462–8. doi: 10.3892/mmr.2015.3862
79. Min S, Wang X, Du Q, Gong H, Yang Y, Wang T, et al. Chetomin, a Hsp90/Hif1alpha pathway inhibitor, effectively targets lung cancer stem cells and non-stem cells. Cancer Biol Ther (2020) 21(8):698–708. doi: 10.1080/15384047.2020.1763147
80. Wang Y, Shi J, Gong L. Gamma linolenic acid suppresses hypoxia-induced proliferation and invasion of non-small cell lung cancer cells by inhibition of Hif1alpha. Genes Genomics (2020) 42(8):927–35. doi: 10.1007/s13258-020-00961-5
81. Yang N, Liang Y, Yang P, Ji F. Propofol suppresses lps-induced nuclear accumulation of hif-1alpha and tumor aggressiveness in non-small cell lung cancer. Oncol Rep (2017) 37(5):2611–9. doi: 10.3892/or.2017.5514
82. Chang H, Lee C-C, Tsai S-C, Wang B-W, Lee YuH, Lin S. Gl331 inhibits hif-1alpha expression in a lung cancer model. Biochem Biophys Res Commun (2003) 302(1):95–100. doi: 10.1016/S0006-291X(03)00111-6
83. Jung KH, Lee JH, Thien Quach CH, Paik JY, Oh H, Park JW, et al. Resveratrol suppresses cancer cell glucose uptake by targeting reactive oxygen species-mediated hypoxia-inducible factor-1alpha activation. J Nucl Med (2013) 54(12):2161–7. doi: 10.2967/jnumed.112.115436
84. Liang H, Yang CX, Zhang B, Wang HB, Liu HZ, Lai XH, et al. Sevoflurane suppresses hypoxia-induced growth and metastasis of lung cancer cells Via inhibiting hypoxia-inducible factor-1alpha. J Anesth (2015) 29(6):821–30. doi: 10.1007/s00540-015-2035-7
85. Zhang J, Wang Q, Wang Q, Guo P, Wang Y, Xing Y, et al. Chrysophanol exhibits anti-cancer activities in lung cancer cell through regulating Ros/Hif-1a/Vegf signaling pathway. Naunyn Schmiedebergs Arch Pharmacol (2020) 393(3):469–80. doi: 10.1007/s00210-019-01746-8
86. Wang L-M, Zhang L-L, Wang L-W, Zhu L, MA X-X. Influence of mir-199a on rats with non-small cell lung cancer Via regulating the hif-1α/Vegf signaling pathway. Eur Rev Med Pharmacol Sci (2019) 23(23):10363–9. doi: 10.26355/eurrev_201912_19675
87. Ding G, Huang G, Liu HD, Liang HX, Ni YF, Ding ZH, et al. Mir-199a suppresses the hypoxia-induced proliferation of non-small cell lung cancer cells through targeting Hif1alpha. Mol Cell Biochem (2013) 384(1-2):173–80. doi: 10.1007/s11010-013-1795-3
88. Chang CC, Lin MT, Lin BR, Jeng YM, Chen ST, Chu CY, et al. Effect of connective tissue growth factor on hypoxia-inducible factor 1alpha degradation and tumor angiogenesis. J Natl Cancer Inst (2006) 98(14):984–95. doi: 10.1093/jnci/djj242
89. Wei D, Peng JJ, Gao H, Li H, Li D, Tan Y, et al. Digoxin downregulates Ndrg1 and vegf through the inhibition of hif-1alpha under hypoxic conditions in human lung adenocarcinoma A549 cells. Int J Mol Sci (2013) 14(4):7273–85. doi: 10.3390/ijms14047273
90. Choi YJ, Rho JK, Lee SJ, Jang WS, Lee SS, Kim CH, et al. Hif-1alpha modulation by topoisomerase inhibitors in non-small cell lung cancer cell lines. J Cancer Res Clin Oncol (2009) 135(8):1047–53. doi: 10.1007/s00432-009-0543-2
91. Shrimali D, Shanmugam MK, Kumar AP, Zhang J, Tan BK, Ahn KS, et al. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett (2013) 341(2):139–49. doi: 10.1016/j.canlet.2013.08.023
92. Jacoby JörgJ, Korshunova MV, Williams RR, Furutani K, Takahashi O, Kirkpatrick L, et al. Treatment with hif-1alpha antagonist px-478 inhibits progression and spread of orthotopic human small cell lung cancer and lung adenocarcinoma in mice. J Thorac Oncol (2010) 5(7):940–9. doi: 10.26355/eurrev_201912_19675
93. Wan J, Che Y, Kang N, Wu W. Socs3 blocks hif-1alpha expression to inhibit proliferation and angiogenesis of human small cell lung cancer by downregulating activation of akt, but not Stat3. Mol Med Rep (2015) 12(1):83–92. doi: 10.3892/mmr.2015.3368
94. Yao H, Wang H, Zhang Z, Jiang BH, Luo J, Shi X. Sulforaphane inhibited expression of hypoxia-inducible factor-1alpha in human tongue squamous cancer cells and prostate cancer cells. Int J Cancer (2008) 123(6):1255–61. doi: 10.1002/ijc.23647
95. Pei Y, Wu B, Cao Q, Wu L, Yang G. Hydrogen sulfide mediates the anti-survival effect of sulforaphane on human prostate cancer cells. Toxicol Appl Pharmacol (2011) 257(3):420–8. doi: 10.1016/j.taap.2011.09.026
96. Ader I, Brizuela L, Bouquerel P, Malavaud B, Cuvillier O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res (2008) 68(20):8635–42. doi: 10.1158/0008-5472.CAN-08-0917
97. Lin HH, Tsai CW, Chou FP, Wang CJ, Hsuan SW, Wang CK, et al. Andrographolide down-regulates hypoxia-inducible factor-1alpha in human non-small cell lung cancer A549 cells. Toxicol Appl Pharmacol (2011) 250(3):336–45. doi: 10.1016/j.taap.2010.11.014
98. Ban HS, Uno M, Nakamura H. Suppression of hypoxia-induced hif-1alpha accumulation by vegfr inhibitors: Different profiles of Aal993 versus Su5416 and Krn633. Cancer Lett (2010) 296(1):17–26. doi: 10.1016/j.canlet.2010.03.010
99. Robinson A, Karhausen Jörn, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology (2008) 134(1):145–55. doi: 10.1053/j.gastro.2007.09.033
100. Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology (2008) 134(1):156–65. doi: 10.1053/j.gastro.2007.10.012
101. Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol (2017) 15(1):55–63. doi: 10.1038/nrmicro.2016.142
102. Raftery AL, Tsantikos E, Harris NL, Hibbs ML. Links between inflammatory bowel disease and chronic obstructive pulmonary disease. Front Immunol (2020) 11:2144. doi: 10.3389/fimmu.2020.02144
103. Winkle M, El-Daly SM, Fabbri M, Calin GA. Noncoding rna therapeutics - challenges and potential solutions. Nat Rev Drug Discovery (2021) 20(8):629–51. doi: 10.1038/s41573-021-00219-z
104. Ha M, Kim VN. Regulation of microrna biogenesis. Nat Rev Mol Cell Biol (2014) 15(8):509–24. doi: 10.1038/nrm3838
105. Lin L, Sun J, Wu D, Lin D, Sun D, Li Q, et al. Microrna-186 is associated with hypoxia-inducible factor-1α expression in chronic obstructive pulmonary disease. Mol Genet Genomic Med (2019) 7(3):e531. doi: 10.1002/mgg3.531
106. Correia de Sousa M, Gjorgjieva M, Dolicka D, Sobolewski C, Foti M. Deciphering mirnas' action through mirna editing. Int J Mol Sci (2019) 20(24):6249. doi: 10.3390/ijms20246249
107. Gonsalves CS, Kalra VK. Hypoxia-mediated expression of 5-Lipoxygenase-Activating protein involves hif-1alpha and nf-kappab and micrornas 135a and 199a-5p. J Immunol (2010) 184(7):3878–88. doi: 10.4049/jimmunol.0902594
108. Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of mir-199a derepresses hypoxia-inducible factor-1alpha and sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res (2009) 104(7):879–86. doi: 10.1161/circresaha.108.193102
109. Mizuno S, Bogaard HJ, Gomez-Arroyo J, Alhussaini A, Kraskauskas D, Cool CD, et al. Microrna-199a-5p is associated with hypoxia-inducible factor-1alpha expression in lungs from patients with copd. Chest (2012) 142(3):663–72. doi: 10.1378/chest.11-2746
110. Wu H, Ma H, Wang L, Zhang H, Lu L, Xiao T, et al. Regulation of lung epithelial cell senescence in smoking-induced Copd/Emphysema by micror-125a-5p Via Sp1 mediation of Sirt1/Hif-1a. Int J Biol Sci (2022) 18(2):661–74. doi: 10.7150/ijbs.65861
111. Rupaimoole R, Slack FJ. Microrna therapeutics: Towards a new era for the management of cancer and other diseases. Nat Rev Drug Discovery (2017) 16(3):203–22. doi: 10.1038/nrd.2016.246
112. Wu B, Teng H, Yang G, Wu L, Wang R. Hydrogen sulfide inhibits the translational expression of hypoxia-inducible factor-1alpha. Br J Pharmacol (2012) 167(7):1492–505. doi: 10.1111/j.1476-5381.2012.02113.x
113. Guan R, Wang J, Li D, Li Z, Liu H, Ding M, et al. Hydrogen sulfide inhibits cigarette smoke-induced inflammation and injury in alveolar epithelial cells by suppressing Phd2/Hif-1alpha/Mapk signaling pathway. Int Immunopharmacol (2020) 81:105979. doi: 10.1016/j.intimp.2019.105979
114. Ali FF, Mohammed HH, Elroby Ali DM. Protective effect of hydrogen sulfide against stress-induced lung injury: Involvement of Nrf2, Nfκb/Inos, and hif-1α signaling pathways. Cell Stress chaperones (2022) 27(1):55–70. doi: 10.1007/s12192-021-01248-8
115. Szabo C, Papapetropoulos A, International Union of Basic and Clinical Pharmacology. Cii: Pharmacological modulation of H2s levels: H2s donors and H2s biosynthesis inhibitors. Pharmacol Rev (2017) 69(4):497–564. doi: 10.1124/pr.117.014050
116. Liu SF, Ye X, Malik AB. Inhibition of nf-kappab activation by pyrrolidine dithiocarbamate prevents in vivo expression of proinflammatory genes. Circulation (1999) 100(12):1330–7. doi: 10.1161/01.cir.100.12.1330
117. Jiang H, Zhu Y, Xu H, Sun Y, Li Q. Activation of hypoxia-inducible factor-1alpha Via nuclear factor-kappa b in rats with chronic obstructive pulmonary disease. Acta Biochim Biophys Sin (Shanghai) (2010) 42(7):483–8. doi: 10.1093/abbs/gmq041
118. Cheng J, Zhou ZW, Sheng HP, He LJ, Fan XW, He ZX, et al. An evidence-based update on the pharmacological activities and possible molecular targets of lycium barbarum polysaccharides. Drug design Dev Ther (2015) 9:33–78. doi: 10.2147/dddt.S72892
119. Chen LJ, Xu W, Li YP, Ma LT, Zhang HF, Huang XB, et al. Lycium barbarum polysaccharide inhibited hypoxia-inducible factor 1 in copd patients. Int J Chron Obstruct Pulmon Dis (2020) 15:1997–2004. doi: 10.2147/COPD.S254172
Glossary

Keywords: chronic obstructive pulmonary disease, lung cancer, non-small cell lung cancer, hypoxia-inducible factor 1-alpha, hypoxia-inducible factor 1
Citation: Xu Y-r, Wang A-l and Li Y-q (2022) Hypoxia-inducible factor 1-alpha is a driving mechanism linking chronic obstructive pulmonary disease to lung cancer. Front. Oncol. 12:984525. doi: 10.3389/fonc.2022.984525
Received: 02 July 2022; Accepted: 10 October 2022;
Published: 21 October 2022.
Edited by:
Bikul Das, KaviKrishna Laboratory, IndiaReviewed by:
Saba Khaliq, University of Health Sciences, PakistanDhruv Kumar, University of Petroleum and Energy Studies, India
Copyright © 2022 Xu, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-qing Li, bGlkb2N0b3IwM0AxMjYuY29t