
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 01 November 2022
Sec. Gastrointestinal Cancers: Colorectal Cancer
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.982744
This article is part of the Research TopicGut Microbiota and Chemotherapy Resistance of Colorectal CancerView all 6 articles
 Leitao Sun1†
Leitao Sun1† Zhenzheng Zhu2†
Zhenzheng Zhu2† Xinru Jia2†
Xinru Jia2† Xiangchang Ying2
Xiangchang Ying2 Binbin Wang1
Binbin Wang1 Peipei Wang2*Shuo Zhang3*
Peipei Wang2*Shuo Zhang3* Jieru Yu4*
Jieru Yu4*Metastasis of colorectal cancer is deemed to be closely related to the changes in the human gut microbiome. The purpose of our study is to distinguish the differences in gut microbiota between colorectal cancer with and without metastases. Firstly, this study recruited colorectal cancer patients who met the established inclusion and exclusion criteria in the Oncology Department of Zhejiang Hospital of Traditional Chinese Medicine from February 2019 to June 2019. Fresh stool samples from healthy volunteers, non-metastatic patients, and metastatic patients were collected for 16S rRNA gene sequencing, to analyze the diversity and abundance of intestinal microorganisms in each group. The results showed that the microbial composition of the control group was more aplenty than the experimental group, while the difference also happened in the Tumor and the metastases group. At the phylum level, the abundance of Bacteroidetes significantly declined in the Tumor and the metastases group, compared with the control group. At the class level, Bacilli increased in experimental groups, while its abundance in the Tumor group was significantly higher than that in the metastases group. At the order level, the Tumor group had the highest abundance of Lactobacillales, followed by the metastases group and the control group had the lowest abundance. Overall, our study showed that the composition of the flora changed with the occurrence of metastasis in colorectal cancer. Therefore, the analysis of gut microbiota can serve as a supplement biological basis for the diagnosis and treatment of metastatic colorectal cancer which may offer the potential to develop non-invasive diagnostic tests.
Colorectal cancer (CRC) is not only considered the third most common malignancy in the world (1), but also the second most deadly cancer (2). Meanwhile, over recent years, its incidence has continued to rise (3). According to estimation, there will be 3.2 million new cases globally by 2040 (1). About 20% of CRC patients have metastases at diagnosis while another 20% who develop metastases at the time of follow-up need systemic treatment (4). It is named Metastatic Colorectal Cancer (MCRC). Therefore, the prognosis of colorectal cancer remains poor though surgery, despite radiotherapy and chemotherapy has advanced significantly (5).
Gut microbiota (GM) is the largest microbiome in the human body which involves at least 1,000 different species of bacteria and 100 trillion microbes (6). It plays a vital role in maintaining intestinal stability (7) as well as healthy state by eliminating pathogens and establishing intestinal barriers (8).
In recent decades, it has been proven that the dysregulation of intestinal flora forms biofilm, which leads to damage of intestinal barrier function, further enhances intestinal dysregulation, and promotes the development of colon cancer (9). It is worth emphasizing that there is a complicated relationship between CRC and GM (10). Research has already confirmed that the GM diversity of patients with CRC is lower than healthy people (11). As probiotics, butyrate-producer, Clostridium butyicum, and lactate-producer like S. thermophilus in CRC patients were exhausted (12). Meanwhile, some species of CRC patients’ gut microbiota showed a significant increase. For instance, in cancer tissue, there are more Fusobacterium than adjoining health tissue (13) which will foster tumor proliferation during the development of CRC (14). Meanwhile, the study has shown that gut microbiota could further promote CRC metastasis by interfering with metabolism (15).
As the vast majority of the existing studies have focused on the relationship between CRC and gut microbiota, there is a gap in flora in the colorectal cancer metastasis research field. This study intends to distinguish the differences in gut microbiota between colorectal cancer with and without metastases, to supplement the biological basis for the treatment of metastatic colorectal cancer. The design is shown in Figure 1.
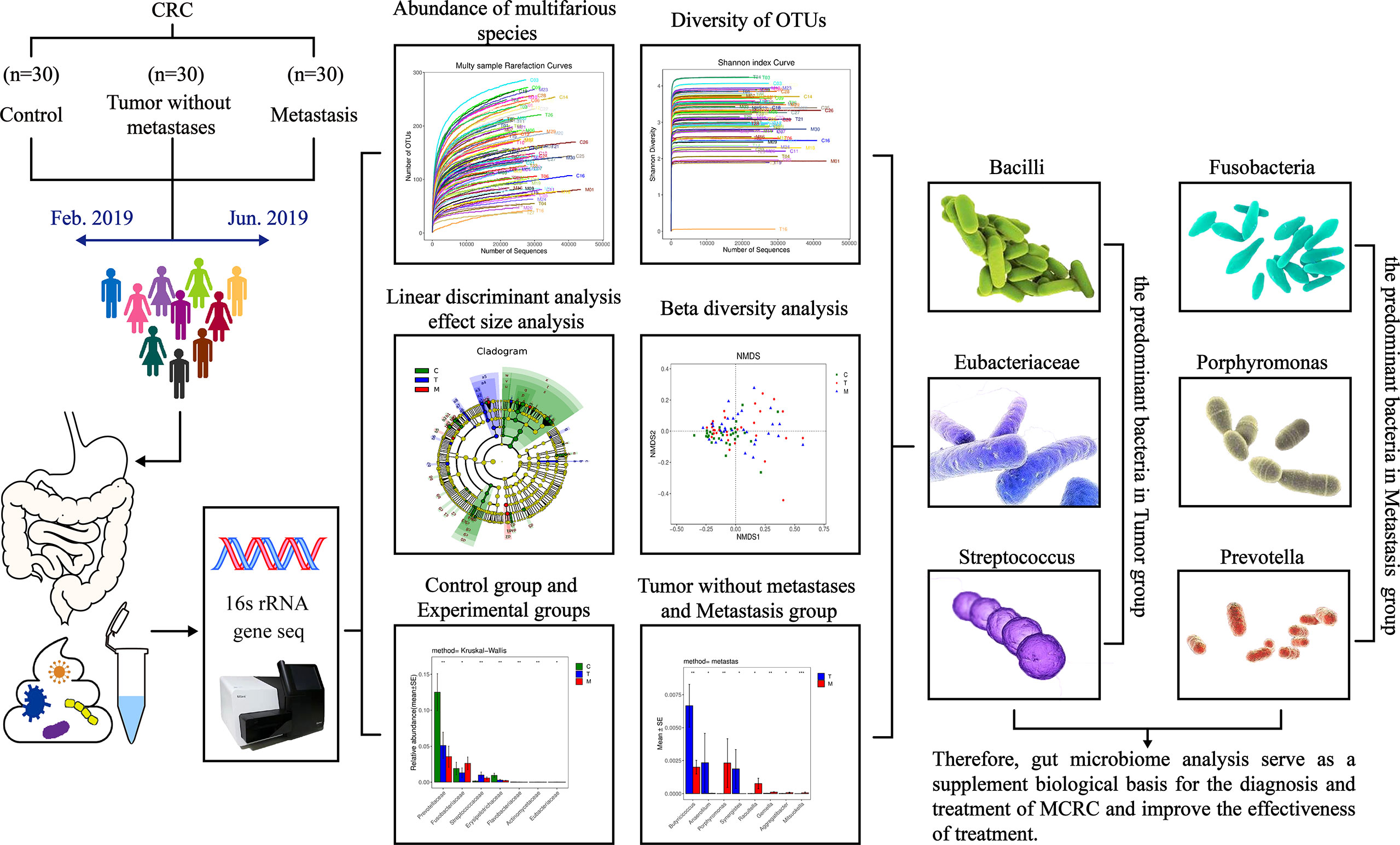
Figure 1 Human gut microbiome in colorectal cancer (CRC) without metastases and metastases.
The collection of samples was carried out in the Oncology Department of Zhejiang Provincial Hospital of Traditional Chinese Medicine between February 2019 and June 2019. The inclusion criteria of the experimental groups were as follows: (a) Individuals who meet the diagnostic criteria and are diagnosed with colorectal cancer by histopathological examination; (b) Individuals with no family history of CRC; (c) People aged between 18 to 75 years old; (d) Individuals who have no diarrhea, vomiting, nausea and other gastrointestinal discomforts during the previous month; (e) Individuals who did not use antibiotics or probiotics in the 4 weeks before taking stool samples. Patients with the following conditions were excluded: (a) Pregnant or breastfeeding women; (b) Individuals with inflammatory, infectious, or other autoimmune diseases; (c) Individuals with fecal discharge through fistulas; (d) Uncooperative people. The research was conducted under the clinical studies rules at home and abroad. All the protocols and procedures of this study were approved by the Zhejiang Province Hospital of TCM Ethics Committee (2020-KL-050-01), and all volunteers signed the informed consent form before participating in the experiment.
All patients had no history of malignant tumors except for the diagnosis of CRC, which was confirmed by pathology and operation. Case classification and grouping were carried out following the eighth edition American Joint Commission on Cancer (AJCC) TNM staging system (16).
The stool samples were collected from CRC patients and healthy volunteers in a clean environment, preserved in an aseptic sampling tube at −80°C. A 200 mg fecal sample was weighed into a 2 ml centrifuge tube. Then, the DNA from the stool samples was extracted by QIAamp DNA Fecal Mini Kit (Qiagen, Germany) according to the instructions. The samples were added ASL and incubated in a water bath at 95°C for 5 min. The process of extracting the final DNA was performed in 50 µL AE. The extracted total DNA was tested for concentration, purity, and integrity by 1% agarose gel electrophoresis and NanoDrop2000 ultramicro spectrophotometer, then stored at 4°C for further analysis.
Three groups were set in the experiment, namely the C (Control) group, the T (Tumor without metastases) group, and the M (Metastases) group.
The hypervariable region of the microbial 16S rRNA gene was amplified by PCR thermocycler. The sequences for the primers were 357F (5’-ACTCCTACGGRAGGCAGCAG-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’). The concrete scheme is as follows: 3 min at 95°C, then 29 cycles of 30 s at 95°C, 30 s at 55°C and 45 s at 72°C, followed by 5 min at 72°C. The PCR reaction was repeated in three times. During the pre-experiment, ten samples were selected randomly to produce the lowest cycle number, and the results showed that most of the samples could be scaled up to the appropriate concentration of the product. Formal experiments were then performed using the TransGen AP221-02, TransStart Fastpfu DNA Polymerase, and 20-μl reaction system. The products of PCR were quantitatively detected with a micro fluorometer based on the results of preliminary sample detection electrophoresis. Afterward, the products were mixed according to the sequencing volume of a single sample.
Paired-end DNA was sequenced by Illumina Miseq. The results of the sequencing were extracted according to the barcode tag complete matching method and transformed into images. Raw fastq files were quality-filtered by Trimmomatic and merged by FLASH. The specific process was as follows: In the 50bp window, if the average quality value was lower than 20, the back-end bases were truncated from the window, and the reads below 50bp after quality control were filtered. According to the overlap relationship between PEreads, the paired reads were merged into one sequence, and the minimum overlap length was 10bp. The maximum mismatch ratio allowed in the overlap region of the spliced sequence was 0.2. Mothur V.1.39.5. (Parameter settings: maxambig=0, maxhomop=8, minlength=200, maxlength=485) was used to filter out the singletons in the spliced long reads (corresponding to a sequence with only one read) to obtain data for subsequent clustering OTUs.
Operational Taxonomy Unit (OTU) is a set of operational definitions used to classify a certain taxonomic unit including domain, kingdom, phylum, class, order, family, genera, and species, which facilitates the analysis in population genetics or phylogenetic research. Clustering was performed at 97% similarity by UPARSE software to obtain representative sequences of OTUs. Chimeras generated by PCR amplification were then removed from the OTU representative sequences by UCHIME software and golddatabase (v20110519). The abundance of each sample at each OTU was obtained by ussearch_global method. Then, the representative sequences of OTUs were aligned with Silva128, Greengene, and RDP databases by mothur (classify.seqs) software for species annotation. The confidence threshold was 0.6.
According to the taxonomic information, the community structure was statistically analyzed at the taxonomic levels of phylum, class, order, family, genus, and species, and the number of sequences at different taxonomic levels was counted for each sample respectively.
The majorizing sequence was randomly selected from the OTU sequence with a similarity of 97%. According to the number of corresponding OTU and the number of the selected majorizing sequences, the rarefaction curve was constructed and drawn R software (version 3.6.3).
Four indices were used to measure alpha diversity. The Chao index and Ace index were calculated to estimate the number of species in the samples, while the Shannon index and the Simpson index reflected the community diversity. Mothur V.1.39.5. and Qiime were used for Alpha diversity analysis.
Based on the results of the taxonomic analysis, the community structure composition of different classification levels was obtained, and the corresponding diagrams were drawn by the R language tool.
Linear discriminant analysis effect size (LEfSe), a software for discovering high-dimensional biomarkers and revealing genomic features, was used to perform linear discriminant analysis on samples with different grouping conditions in line with the classification composition. Then, communities or species that were significantly affected by the differences in sample division were screened by linear discriminant analysis (LDA). The software used in this experiment is lefse (1.0).
Differentially Abundant Features can be evaluated by multiple hypothesis testing and false discovery rate (FDR) analysis of rare frequency data based on the obtained OTU or community abundance. In the metastats software, the samples of the general control group and the experimental group were analyzed and compared at each level to find out the different bacterial species with certain differences in bacterial abundance. Differences were considered statistically significant when the P value was< 0.05.
The difference in the flora data among the C group, the T group, and the M group were evaluated with statistical software (SPSS 22.0). Data were expressed as mean ± standard deviation (). The differences between two independent samples were compared by the T-test, while the differences between groups were analyzed by Kruskal -Wallis and Wilcoxon rank-sum test. Differences were considered statistically significant at p<0.05, significant at p<0.01, and not statistically significant at p>0.05.
This study collected a total of 90 cases, including 30 cases in the C group, 30 cases in the T group, and 30 cases in the M group. There were 47 males and 43 females, aged from 17 to 75 years, with an average of 55.92 ± 10.70 years old. There was no significant difference in any clinical factors such as gender, age, or inflammation location among the individuals. (p>0.05) (Table 1)

Table 1 Characteristics of healthy volunteers and CRC patients.
This study collected a total of 90 cases, including 30 cases in the C group, 30 cases in the T group, and 30 cases in the M group. According to the general principles of systems genetics and population genetics, the fuzzy and repeated base sequences that affected the quality of analysis were eliminated, and 2425308 optimized sequences were accurately obtained.
There were 694 OTUs clustering in the 90 samples in this study, including 199 species, 138 genera, 47 families, 30 orders, 18 classes, 11 phyla, and 1 domain. More data contributed little to the discovery of new OTUs as the curve flattened, suggesting that this study can be conducted with reasonable sample collection and high species richness (Figure 2A). Additionally, the similarity analysis indicated that the differences between the groups were more obvious than the difference within the groups (R=0.058, p<0.01), declaring the amount of data in this study was feasible to reflect most of the information on the microbiota in each group objectively (Figure 2B). Venn diagram (VENN) showed the number of OTUs that were distinct or the same in each set of intestinal tracts. The total number of OTUs in this study was 694, of which the number of OTUs in the general control group was exceedingly higher than that in the experiment groups (619 vs 556,567) (Figure 2C). According to the sample community’s structures, the abundance diversity histogram of the top twelve phylum species suggested that Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia were the predominant flora in most samples and the proportions of them were different in each group (Figure 2D).
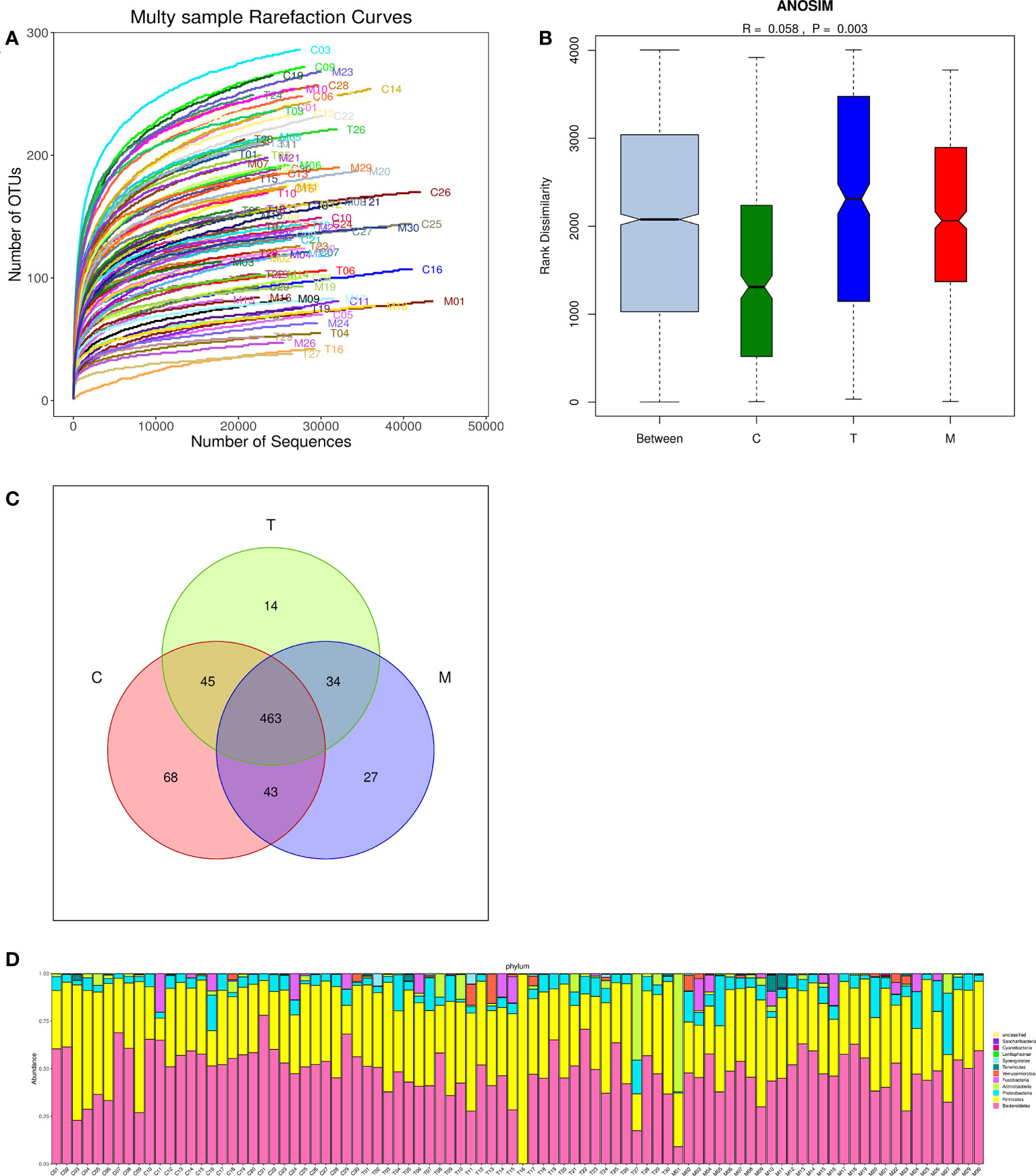
Figure 2 Multiple sparse curves for comparing the abundance of multifarious species (A), Similarity analysis boxplot for identifying the existence of differences between groups (B), Venn diagram for implying the common and specific traits among three groups (C), Histogram of the abundance distribution of species at the phylum level (D).
We performed Kruskal-Wallis test for the Ace, Chao and Shannon index of the three groups and the results showed that the intestinal microbiota diversity of the C group was higher than that of the other two experimental groups (Figures 3A–C). Based on the corresponding OTU number and the selected sequence number, rank-abundance distribution curves showed that the species abundance and uniformity of the T and M group were lower than the C group (Figure 3D). Besides, due to the problems in sample quality and detection process, sequencing results of T16 were meaningless.

Figure 3 Boxplot of the Kruskal -Wallis test for the Ace index (A), Boxplot of the Kruskal -Wallis test for the Chao index (B), Boxplot of the Kruskal -Wallis test for the Shannon index (C), Rank-abundance distribution curves of the general control group and the experimental groups (D). **p < 0.01.
In the NMDS chart, the degree of difference between diverse samples was reflected by the distance between the points. Points represented the 3 groups distributed on the diagram regularly, which indicated that there were significant differences in OTUs types among the three groups. (Figure 4).
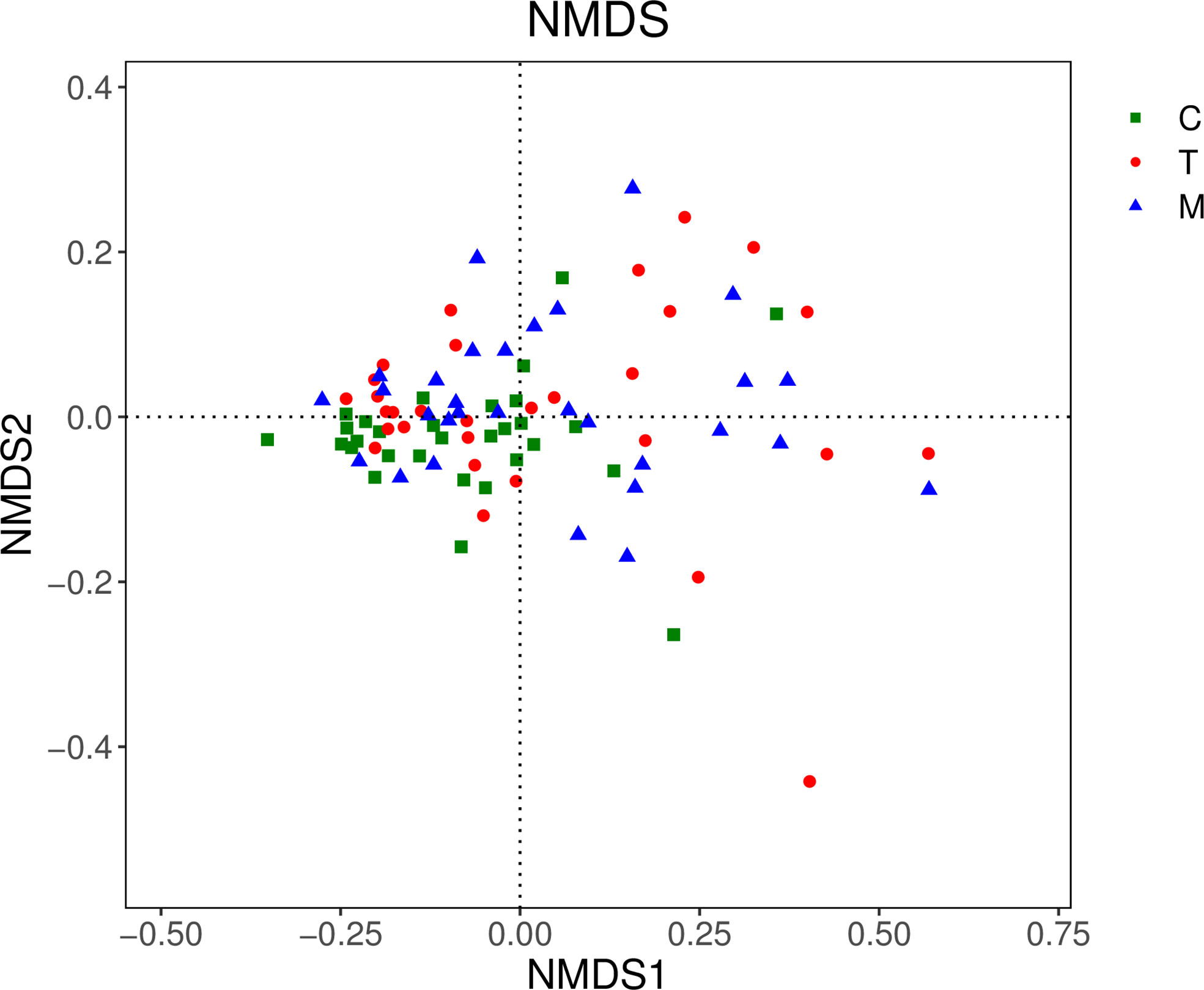
Figure 4 NMDS diagram of the general control group and the experimental groups.
The concentric circles from inside to outside are phylum, class, order, and family, and its nodes correspond to specific flora of different classes. Under the LEfSe diagram (Figure 5), the result indicated the dominant flora of each group: Bacteroidetes on the phylum level in the control group; Bacilli on the class level, Lactobacillales on the order level, Eubacteriaceae on the family level, Streptococcus and Butyricicoccus on the genus level in the tumor without metastases group; Fusobacterium and Lachnoanaerobaculum on the genus level, Porphyromonas_somerae on the species level in the metastases group.
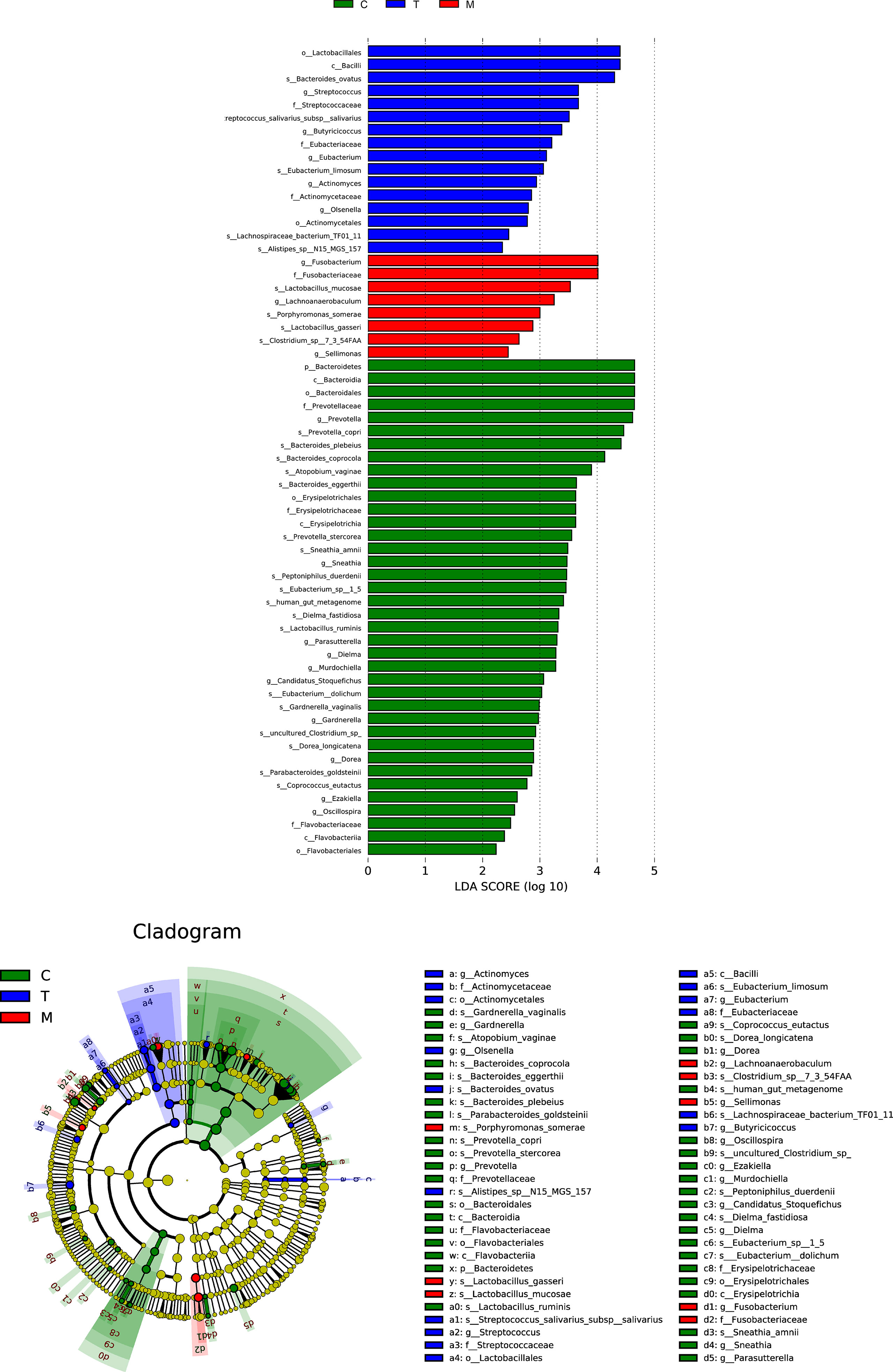
Figure 5 Histogram and cladogram of Linear discriminant analysis effect size (LEfSe) analysis among the general control group and the experimental groups.
Bacteroidetes accounted for 44.0% and 46.8% of the total amount of bacteria in the T and the M group based on the histogram of phylum (Figure 6A), which were lower than 53.2% in the C group (p<0.01). So, Bacteroidetes was the predominant bacteria in the C group.
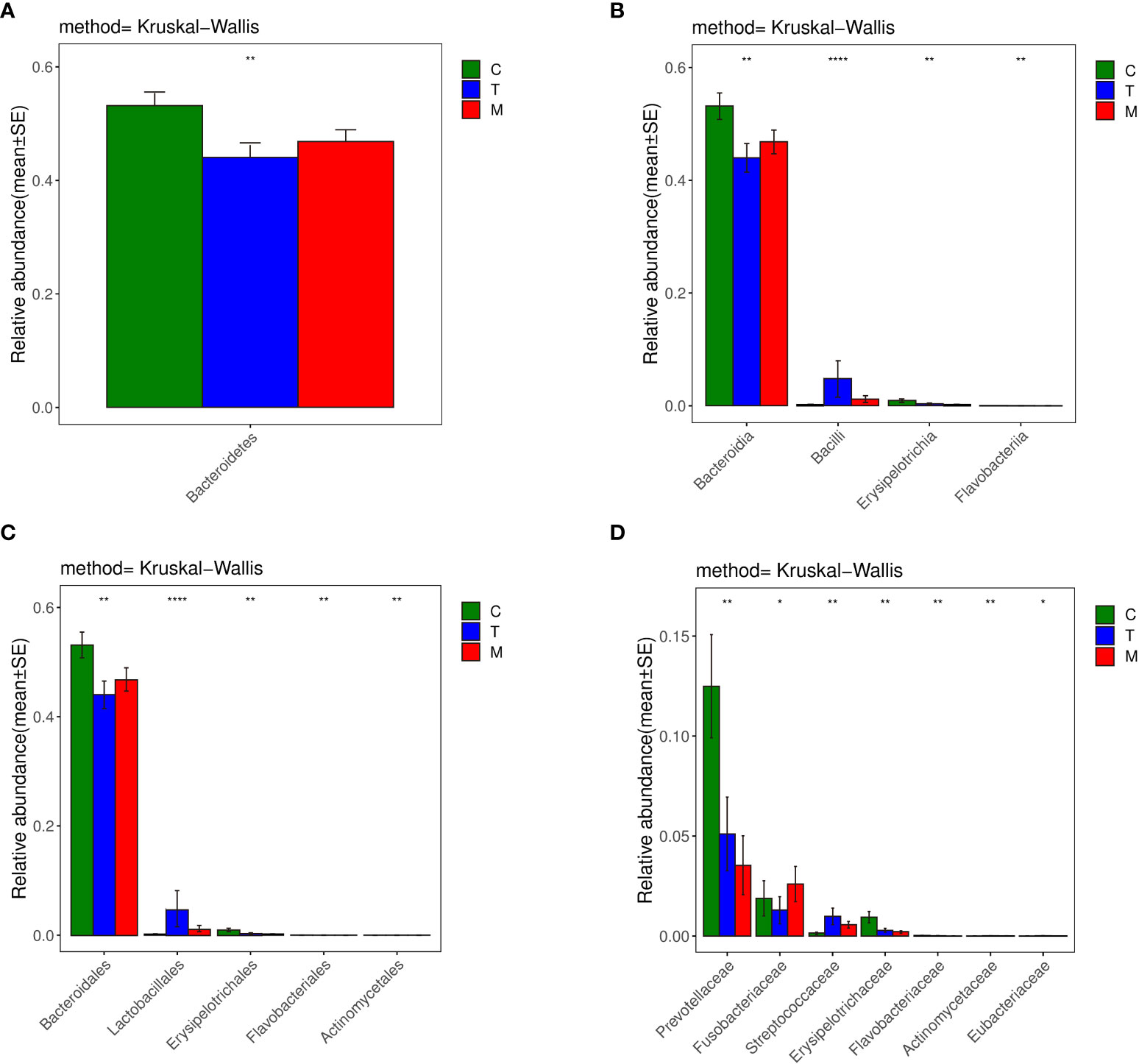
Figure 6 Histograms of differences in the diversity of species analyzed by kruskal_ Wilcox test among three groups at the levels of phylum (A), class (B), order (C), family (D), genus. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Considering the class histograms (Figure 6B), Bacilli accounted for 4.8% and 1.2% of the total number of bacteria in the T and the M group, which were higher than that in the C group, which accounted for 0.2% (p<0.01). Therefore, Bacilli was the predominant bacteria in the T and M group. Erysipelotrichia accounted for 0.3% and 0.2% of the total amount of bacteria in the T and the M group, which were less abundant than that in the C group, which accounted for 0.9% (p<0.01). The abundance of Flavobacteriia in the C group was higher than that in the other two groups (0.02% VS 0.01%, 0.003%) (p<0.01). Thus, the specific bacteria in gut bacterial composition of the C group were Erysipelotrichia and Flavobacteriia.
Based on the histograms of order (Figure 6C), Lactobacillales, belonging to Bacilli, accounted for 4.8% of the total amount of bacteria in the T group, which was higher than that in the C group (0.2%) and M group (1.2%) (p<0.01). Actinomycetales accounted for 0.007% and 0.006% of the total number of bacteria in the T and the M group, which were higher than that in the C group, which accounted for 0.002% (p<0.01). So, Lactobacillales was the specific bacteria in the T group, while Actinomycetales was the specific bacteria in the T and Mgroup.
In terms of the family histogram (Figure 6D), the abundance of Prevotellaceae, which was the specific bacteria in the C group, in the C group was higher than that in the other two groups (12.5% VS 5.1%, 3.5%) (p<0.01).
The species differences between the T group and the M group were analyzed by metastats software. According to the histograms of order (Figure 7A), the differences in Bacillales between the T group and the M group were significant (0.002%<0.01%) (p<0.01). Based on the histograms of the genus (Figure 7B), the differences in Mitsuokella between the two groups were significant (0%<0.006%) (p<0.001). The differences of Butyricicoccus, Gemella and Porphyromonas were both significant (0.7%>0.2%,0.002%<0.01%,0.0002%<0.2%) (p<0.01). The differences of Raoultella, Synergistes, Aggregatibacter and Anaerofilum were statistically significant (0.0008%<0.08%,0.2%>0%,0.0009%<0.007%,0.2%>0.003%) (p<0.05). Considering the species histograms (Figure 7C), the differences of Prevotella_intermedia, Veillonella_magna, Porphyromonas_somerae, Porphyromonas_endodontalis, Prevotella_nigrescens were significant (0%<0.08%,0.08%>0%,0%<0.04%,0%<0.008%,0%<0.009%) (p<0.001). The differences of [Clostridium]_lactatifermentans, Porphyromonas_asaccharolytica, Raoultella_ornithinolytica and Coprococcus_comes between the two groups were statistically significant (0.01%<0.2%,0.0001%<0.2%, 0.0008%<0.08%,0.08%>0.02%) (p<0.05). However, the statistical differences in the histograms of family, class, and phylum between the experimental groups were not evident.
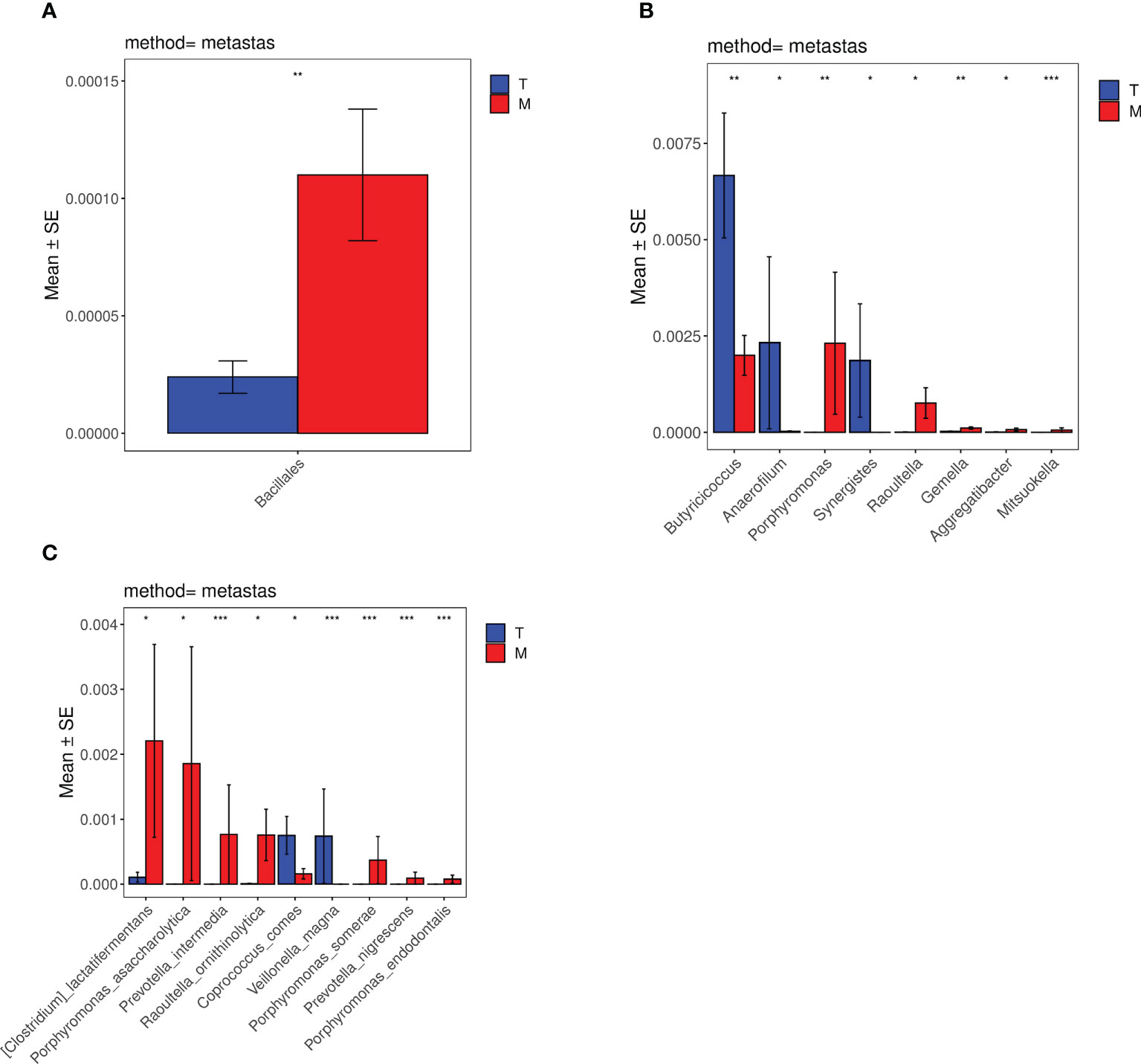
Figure 7 Histograms of distribution differences of species between the tumor without metastases group and the metastases group performed by Metastats software at the levels of order (A), genus (B) and species (C). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Colorectal cancer is one of the common malignancies (17), and the survival of CRC patients is severely influenced by metastasis and recurrence (18). It turns out that the gut microbiota which is associated with the human normal metabolism state (19) plays a significant role in CRC formation and development (20). Yuan N et al. (21) demonstrated that alterations in gut microbiota composition could remodel the liver immune microenvironment by regulating Kupffer cells (KCs) on colorectal cancer, promoting or inhibiting liver metastases. Alice Bertocchi et al. (22) suggested that by breaking the intestinal vascular barrier and forming a premetastatic niche, the bacteria of primary colorectal cancer spread to the liver, thereby promoting the metastasis of colorectal cancer.
There is growing evidence that the gut microbiota diversity of patients with CRC decreased (23). Likewise, the results in our study showed that the diversity of microbial communities in the control group was higher than those in the tumor without metastases group and the metastases group. Comparing the taxonomic groups between healthy volunteers and patients in CRC, our study showed that Bacteroidetes was significantly more abundant in the control group than that in the CRC group, which was the most predominant phylum in healthy individuals. Actinomycetales and Bacilli were also enriched in the CRC group, while Erysipelotrichia and Flavobacteriia were less abundant in the CRC group. The changes and differences in the gut microbiota abundance between healthy people and CRC patients in our experiments were similar to the common experimental results at home and abroad (24–27), and a very small part of the difference may be caused by the influence of the treatment plan, relatively sample size and geographical factors like diet and climate.
Comparing the microbial communities between the tumor without metastases group and the metastases group, the results of our study showed the enrichment of Fusobacterium, Porphyromonas, Raoultella, Lachnoanaerobaculum, and Ezakiella, and the depletion of Butyricicoccus and Succinatimonas in CRC patients with metastases (p<0.05). In previous studies, the higher proportions of Fusobacterium (28), Fusobacterium nucleatum (29), Porphyromonas asaccharolytica, and Porphyromonas gingivalis (30), and the lower proportions of Lachnospira multipara (31), Prevotellaceae, and Butyricicoccus (32) were commonly observed in CRC patients with metastasis. In contrast, the differences observed in our study of Fusobacterium, Porphyromonas, and Butyricicoccus were similar to the previous results, while we also did detect meaningful differences in Raoultella, Lachnoanaerobaculum, Ezakiella, and Succinatimonas. In the tumor without metastases group, the predominant bacterium on the class level was Bacilli; on the family level was Eubacteriaceae; on the genus level was Streptococcus, Butyricicoccus, Synergistes, and Anaerofilum; on the species level was Coprococcus_comes and Veillonella_magna. The differences of them were statistically significant (p<0.05). Among our results, Bacilli and Streptococcus were the same as the former investigation (33–35), and we also obtained valid data of Butyricicoccus, Veillonella_magna, Synergistes, Anaerofilum, Coprococcus_comes, and Eubacteriaceae. Nevertheless, we did not detect meaningful data on Escherichia.
Gut microbiota can promote the proliferation and metastasis of tumor in multiple ways. Porphyromonas gingivalis is related to the occurrence and development of various of tumors (36–38). Mu W et al. (39) found that Porphyromonas gingivalis could promote the proliferation of colorectal cancer cells by activating the MAPK/ERK signaling pathway. In addition, Porphyromonas gingivalis was found to promote the metastasis and malignant progression of lung cancer through long-term colonization of lung cancer cells (40). Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans could initiate the Toll-like receptor (TLR) signaling pathways, and the activation of this pathway produced tumor-promoting effects (41, 42). Raoultella belongs to the Enterobacteriaceae family. The secretome of Enterobacteriaceae enhanced the growth of colorectal cancer cells (43). Besides, Rubinstein MR et al. (44) found that when the benign cells become cancerous and expressed elevated levels of Annexin A1, Fusobacterium nucleatum would be activated and stimulated the growth of colorectal cancer cells by the Wnt/ß‐catenin signaling. Our results showed enrichment of Porphyromonas, Aggregatibacter, Raoultella, and Fusobacterium in the metastases group, so we suggested that their enrichment may promote proliferation and metastasis of colorectal cancer cells and may serve as diagnostic markers for CRC progression.
Gut microbiota contribute to transformation and tumor progression by participating in metabolism and its metabolites. The most important metabolites in the gut microbiota are short-chain fatty acids (SCFAs), of which butyrate is an important member and has been found to have multiple beneficial effects on colon cancer (45). Hu S et al. (46) found that butyrate inhibited miR-92a transcription by reducing c-Myc, ultimately reducing cancer cell proliferation and stimulating apoptosis. Butyrate could also deactivate Akt/ERK signaling in histone deacetylase dependent manner, which impeded CRC cell metastasis and invasion (47). Chang S C et al. (48) found that Butyricicoccus pullicaecorum, a gut butyrate-producing bacterium, reduced the progression of 1,2-dimethylhydrazine-associated colorectal cancer by regulating short-chain fatty acid transporter and its receptor. Butyricicoccus could also downregulate the expression of PLAC8, which may be associated with CRC recurrence, and induce apoptosis in PLAC8-overexpressing cells (49). Therefore, we speculated that the reduction of Butyricicoccus is one of the diagnostic markers of colorectal cancer metastasis, and our experimental results showed that Butyricicoccus in CRC with metastasis was less abundant than that in CRC without metastasis, which supported this speculation. In addition, Ternes D et al. (50) found that formate, a metabolite of Fusobacterium nucleatum, drove CRC tumor invasion by triggering AhR signaling. Li R et al. (51) detected larger primary tumors, more liver metastatic foci, and higher LPS release in intestinal dysbiosis, which was brought about by the excessive administration of Escherichia coli. LPS increased the expression of CTSK in colorectal cancer cells, which promoted the invasion and metastasis of CRC cells by stimulating the secretion of cytokines by M2 TAMs and led to a poor prognosis. The abundance of Prevotella, Mitsuokella, and Fusobacterium was associated with Trimethylamine N-oxide (TMAO), a compound derived from diet and metabolism by the gut microbiome (52). TMAO is involved in a number of genetic pathways with an apparent association to carcinomas, especially colon cancer (53) and it has been found to exert oncogenic effects by promoting cell proliferation and angiogenesis in colorectal cancer (54).
Gut microbiota can alter the tumor microenvironment through inflammatory or immune responses and finally promote the occurrence and development of malignant tumor. Aggregatibacter actinomycetemcomitans, which was a rarely detected periodontopathic bacteria, was identified as being potentially associated with esophageal cancer, pancreatic cancer and precancerous gastric lesions (55). It produced several virulence factors such as cytolethal distending toxin (CDT) and leukotoxin (LtxA) to subvert the host immune response. CDT secreted from Aggregatibacter was pro-inflammatory and could promote carcinogenesis by creating a pro-inflammatory and or growth factor-rich microenvironment (56). LtxA could specifically target and kill activated white blood cells (WBCs) by binding to lymphocyte function-associated antigen-1 (LFA-1) (57). Therefore, Aggregatibacter may promote cancer progression by suppressing the host’s immune response, which was consistent with our finding of increased numbers in the metastatic group. Proença M A et al. (58) found that Fusobacterium nucleatum increased the expression of inflammatory mediators through possible miRNA-mediated activation of TLR2/TLR4. Therefore, the development of CRC was promoted through the immune responses to inflammatory stresses. In addition, Engevik MA et al. (59) found that outer membrane vesicles (OMVs) secreted from Fusobacterium nucleatum could promote proinflammatory cytokine production. Kostic A D et al. (60) also found that Fusobacterium nucleatum could increase tumor multiplicity and selectively recruited tumor-infiltrating myeloid cells in the mouse model, which generated a pro-inflammatory microenvironment and promoted CRC progression. Porphyromonas gingivalis promoted CRC’s formation and development (61) through inflammation of gut tissue (62). It plays a major role in the PD-L1 up-regulation in colon carcinoma cells, which could induce chronic inflammation and activate mechanisms of immune evasion (63). Our experimental results showed that Aggregatibacter, Fusobacterium nucleatum, and Porphyromonas gingivalis were the predominant bacteria in the metastases group, and combined with their above-mentioned functions and mechanisms, we suggested that this gut microbiota may serve as a supplement biological basis for the diagnosis of metastatic colorectal cancer.
Furthermore, gut microbes are closely related to the structure of the systemic immune system (64), the differences of intestinal microbiota in colorectal cancer patients before and after metastasis have influence on chemoradiotherapy and immunotherapy. Eubacterium_limosum which was the predominant flora in T group could enhance ICIs by inducing IFN-γ CD8 T cells (65). In the T group, the abundance of Proteobacteria_eggerthii was also higher than that in the M group, which was same with the result after CCRT that the abundance of Proteobacteria increased (66). After chemoradiotherapy, the Bacteroidetes abundance of individuals with short PFS was lower than that of individuals with long PFS, while the lower ratio may be associated with the progression and recurrence of colorectal cancer (67). Moreover, enrichment of Firmicutes could improve the sensitivity of ICI immunotherapy (68) and Bacteroides could enhance the anti‐CTLA‐4 therapy (69) and the anti-PD-1 immunotherapy (70). Clinically, it was also found that the abundance of Bacteroides vulgatus in anti-PD-1 blockade responders was higher than that in non-responders (64). Bacteroides played a role by upregulating systemic MDSCs and inducing Th1 mediated immune responses (71).The above results were consistent with our results. In our study, Flavobacteriia belonging to Bacteroidetes and Erysipelotrichia belonging to Firmicutes in the M group were lower than that in the T group. Therefore, according to our results combined with previous studies, we hypothesized that Eubacterium_limosum, Proteobacteria, Bacteroidetes and Firmicutes may optimize the effects of chemoradiotherapy and immunotherapy and slow the metastatic progression of colon cancer. Meanwhile Further imbalance of gut microbiota biodiversity can affect the treatment of therapy and promote the development of colorectal cancer. FOLFOX scheme is a chemotherapy regimen based on oxaliplatin and 5-FU which is one of the most commonly used chemotherapy regimens for patients diagnosed with metastatic colorectal cancer, whereas chemoresistance is the main problems in treatment (72). Study had shown that Prevotella and 3-Oxo not only promoted the development of malignant tumors, but also reversed the anticancer effect of FOLFOX (73). Prevotella, as an important carrier of drug resistance genes, was one of the dominant bacteria in the transfer group (67).
Consequently, by improving the structure of intestinal flora in vivo, colorectal cancer metastasis can be slowed down and the effects of radiotherapy, chemotherapy and immunotherapy can be enhanced. Spencer et al. (74) showed that modulation of gut microbes through dietary fiber and probiotics could enhance the effect of cancer immunotherapy. Fusobacterium was enriched in M group, but through the colonization of specific exogenous probiotics, its content in feces of patients could be reduced, thus promoting the effect of immunotherapy (75). On the contrary, Lactobacillus_Mucosae was also the dominant bacterium in M group, but study had shown that Lactobacillus could improve the efficiency of immunotherapy by enhancing PD-1/PD-L1 or CTLA-4 blockade (76). On the other hand, the use of immunotherapy to optimize the intestinal flora in vivo is also a breakthrough direction in the treatment of colorectal cancer in the future. It is generally believed that Treg/Th17 balance plays a fundamental role in stabilizing the homeostasis of intestinal microecology (77) and Treg cells participate in the development and progression of tumors by inhibiting anti-tumor immunity (78). TLR2/TLR4 can inhibit intestinal inflammation caused by Fusobacterium nucleatum in vivo by activating and inducing Tregs (79).
Despite the novel and meaningful findings, there are still some limitations in our study. Due to improper operation during collection, transportation, or storage, the sequencing data of the T16 was insufficient to reflect the vast majority of bacterial diversity information in the sample, and its analysis results were quite different from those in the same group. As for the differences in the abundance of Bacteroidetes (27), Firmicutes (32), and Actinobacteria (34) between healthy people and CRC patients, there are few studies with opposite results. The occurrence of this phenomenon may be related to the geographic location, diet habits, and age characteristics of the patients, the activity, the extent and treatment of the CRC, and the difference in DNA extraction methods. Therefore, larger sample sizes, more groups, and further experimental studies are still required.
The microbiome has received extensive attention in recent years. In the annual report of ASCO (80, 81), the work of identifying biomarkers relevant to immunotherapies that predicted initial response, long-term disease control, adverse events, resistance, and the microenvironment of potentially malignant lesions that were associated with progression to invasive disease were recognized as priority focus areas. And in the 2021 annual meeting special issue of CSCO, gut microbiota was thought of as a predictive biomarker for the occurrence and development of irAEs. It was expected to reverse the destruction of intestinal flora homeostasis and blocked its impact on the human body through a series of methods such as intestinal flora transplantation, probiotic intervention, and targeted drugs for specific intestinal flora. Although there have been lots of studies on the relationship between the gut microbiome and colorectal cancer, studies on the association of gut microbiome with colorectal cancer metastasis are still limited. Therefore, our study innovatively discovered the difference among healthy people, patients in colorectal cancer with and without metastases, which leads to the exploration of the pathogenesis and development of colorectal cancer from the perspective of gut microbiota, contributing to understanding the tumorigenesis of colorectal cancer from microbiological perspective. We hope that this study may offer some novel perspective for the precise treatment of CRC by targeting specific microbiota, which may help to realize the personalized colorectal cancer treatment mediated by the gut microbiota.
In summary, our study suggests the decreased diversity of gut microbiota in CRC patients and the differences in composition, abundance and predominant bacteria of flora between the non-metastatic patients and metastatic patients. Synergistes, Anaerofilum, Coprococcus_comes, Eubacteriaceae, Bacilli, Streptococcus, Butyricicoccus, and Veillonella_magna are the predominant bacteria in the tumor without metastases group, while Fusobacteria, Porphyromonas, Prevotella, Mitsuokella, Gemella, Raoultella, Aggregatibacter, and [Clostridium]_lactatifermentans are the predominant bacteria in the metastases group. Therefore, gut microbiome analysis may offer the potential to develop non-invasive diagnostic tests, serve as a supplement biological basis for the diagnosis and treatment of metastatic colorectal cancer and improve the effectiveness of treatment.
All the protocols and procedures of this study were approved by the Zhejiang Province Hospital of TCM Ethics Committee (2020-KL-050-01), and all volunteers signed the informed consent form before participating in the experiment.
LS, ZZ, XJ: These authors have contributed equally to this work and share first authorship. PW, SZ, JY: These authors are responsible for corresponding. All authors contributed to the article and approved the submitted version.
This study was supported by Natural Science Foundation of Zhejiang Province (LQ22H270008); China Postdoctoral Science Foundation (2021M702928); The Postgraduate Scientific Research Fund of Zhejiang Chinese Medical University (2021YKJ01); Young Elite Scientists Sponsorship Program by CACM (2021-QNRC2-B13).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.982744/full#supplementary-material
1. Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol (2021) 14(10):101174. doi: 10.1016/j.tranon.2021.101174
2. Talaat IM, Elemam NM, Saber-Ayad M. Complement system: An immunotherapy target in colorectal cancer. Front Immunol (2022) 13:810993. doi: 10.3389/fimmu.2022.810993
3. Si H, Yang Q, Hu H, Ding C, Wang H, Lin X. Colorectal cancer occurrence and treatment based on changes in intestinal flora. Semin Cancer Biol (2021) 70:3–10. doi: 10.1016/j.semcancer.2020.05.004
4. Lai E, Liscia N, Donisi C, Mariani S, Tolu S, Pretta A, et al. Molecular-Biology-Driven treatment for metastatic colorectal cancer. Cancers (Basel) (2020) 12(5):1214. doi: 10.3390/cancers12051214
5. Hu HF, Xu WW, Li YJ, He Y, Zhang WX, Liao L, et al. Anti-allergic drug azelastine suppresses colon tumorigenesis by directly targeting ARF1 to inhibit IQGAP1-ERK-Drp1-mediated mitochondrial fission. Theranostics (2021) 11(4):1828–44. doi: 10.7150/thno.48698
6. Duttaroy AK. Role of gut microbiota and their metabolites on atherosclerosis, hypertension and human blood platelet function: A review. Nutrients (2021) 13(1):144. doi: 10.3390/nu13010144
7. Zhang L, Ocansey DKW, Liu L, Olovo CV, Zhang X, Qian H, et al. Implications of lymphatic alterations in the pathogenesis and treatment of inflammatory bowel disease. BioMed Pharmacother (2021) 140:111752. doi: 10.1016/j.biopha.2021.111752
8. Wang G, Huang S, Wang Y, Cai S, Yu H, Liu H, et al. Bridging intestinal immunity and gut microbiota by metabolites. Cell Mol Life Sci (2019) 76(20):3917–37. doi: 10.1007/s00018-019-03190-6
9. Chew SS, Tan LT, Law JW, Pusparajah P, Goh BH, Ab Mutalib NS, et al. Targeting gut microbial biofilms-a key to hinder colon carcinogenesis? Cancers (Basel) (2020) 12(8):2272. doi: 10.3390/cancers12082272
10. Garrett WS. Cancer and the microbiota. Science (2015) 348(6230):80–6. doi: 10.1126/science.aaa4972
11. Wang P, Ding S, Sun L, Feng Y, Guo K, Zhu Y, et al. Characteristics and differences of gut microbiota in patients with different traditional Chinese medicine syndromes of colorectal cancer and normal population. J Cancer (2020) 11(24):7357–67. doi: 10.7150/jca.50318
12. Cheng Y, Ling Z, Li L. The intestinal microbiota and colorectal cancer. Front Immunol (2020) 11:615056. doi: 10.3389/fimmu.2020.615056
13. Gao Y, Bi D, Xie R, Li M, Guo J, Liu H, et al. Fusobacterium nucleatum enhances the efficacy of PD-L1 blockade in colorectal cancer. Signal Transduct Target Ther (2021) 6(1):398. doi: 10.1038/s41392-021-00795-x
14. Velikova T, Krastev B, Lozenov S, Gencheva R, Peshevska-Sekulovska M, Nikolaev G, et al. Antibiotic-related changes in microbiome: The hidden villain behind colorectal carcinoma immunotherapy failure. Int J Mol Sci (2021) 22(4):1754. doi: 10.3390/ijms22041754
15. Nenkov M, Ma Y, Gassler N, Chen Y. Metabolic reprogramming of colorectal cancer cells and the microenvironment: Implication for therapy. Int J Mol Sci (2021) 22(12):6262. doi: 10.3390/ijms22126262
16. Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, et al. The eighth edition AJCC cancer staging manual: Continuing to build a bridge from a population-based to a more "personalized" approach to cancer staging. CA: Cancer J Clin (2017) 67(2):93–9. doi: 10.3322/caac.21388
17. Jiang Z, Tai Q, Xie X, Hou Z, Liu W, Yu Z, et al. EIF4A3-induced circ_0084615 contributes to the progression of colorectal cancer via miR-599/ONECUT2 pathway. J Exp Clin Cancer Res (2021) 40(1):227. doi: 10.1186/s13046-021-02029-y
18. Guo Y, Guo Y, Chen C, Fan D, Wu X, Zhao L, et al. Circ3823 contributes to growth, metastasis and angiogenesis of colorectal cancer: involvement of miR-30c-5p/TCF7 axis. Mol Cancer (2021) 20(1):93. doi: 10.1186/s12943-021-01372-0
19. Pothuraju R, Chaudhary S, Rachagani S, Kaur S, Roy HK, Bouvet M, et al. Mucins, gut microbiota, and postbiotics role in colorectal cancer. Gut Microbes (2021) 13(1):1974795. doi: 10.1080/19490976.2021.1974795
20. Wu Y, Jiao N, Zhu R, Zhang Y, Wu D, Wang AJ, et al. Identification of microbial markers across populations in early detection of colorectal cancer. Nat Commun (2021) 12(1):3063. doi: 10.1038/s41467-021-23265-y
21. Yuan N, Li X, Wang M, Zhang Z, Qiao L, Gao Y, et al. Gut microbiota alteration influences colorectal cancer metastasis to the liver by remodeling the liver immune microenvironment. Gut liver (2022) 16(4):575–88. doi: 10.5009/gnl210177
22. Bertocchi A, Carloni S, Ravenda PS, Bertalot G, Spadoni I, Lo Cascio A, et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell (2021) 39(5):708–24 e11. doi: 10.1016/j.ccell.2021.03.004
23. Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med (2019) 25(6):968–76. doi: 10.1038/s41591-019-0458-7
24. Yang Y, Misra BB, Liang L, Bi D, Weng W, Wu W, et al. Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics (2019) 9(14):4101–14. doi: 10.7150/thno.35186
25. Zhang YK, Zhang Q, Wang YL, Zhang WY, Hu HQ, Wu HY, et al. A comparison study of age and colorectal cancer-related gut bacteria. Front Cell Infect Microbiol (2021) 11:606490. doi: 10.3389/fcimb.2021.606490
26. Yang J, McDowell A, Kim EK, Seo H, Lee WH, Moon CM, et al. Development of a colorectal cancer diagnostic model and dietary risk assessment through gut microbiome analysis. Exp Mol Med (2019) 51(10):1–15. doi: 10.1038/s12276-019-0313-4
27. Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst (2013) 105(24):1907–11. doi: 10.1093/jnci/djt300
28. Amitay EL, Krilaviciute A, Brenner H. Systematic review: Gut microbiota in fecal samples and detection of colorectal neoplasms. Gut Microbes (2018) 9(4):293–307. doi: 10.1080/19490976.2018.1445957
29. Chen S, Su T, Zhang Y, Lee A, He J, Ge Q, et al. Fusobacterium nucleatum promotes colorectal cancer metastasis by modulating KRT7-AS/KRT7. Gut Microbes (2020) 11(3):511–25. doi: 10.1080/19490976.2019.1695494
30. Gao R, Zhu Y, Kong C, Xia K, Li H, Zhu Y, et al. Alterations, interactions, and diagnostic potential of gut bacteria and viruses in colorectal cancer. Front Cell Infect Microbiol (2021) 11:657867. doi: 10.3389/fcimb.2021.657867
31. Mizutani S, Yamada T, Yachida S. Significance of the gut microbiome in multistep colorectal carcinogenesis. Cancer Sci (2020) 111(3):766–73. doi: 10.1111/cas.14298
32. Park J, Kim NE, Yoon H, Shin CM, Kim N, Lee DH, et al. Fecal microbiota and gut microbe-derived extracellular vesicles in colorectal cancer. Front Oncol (2021) 11:650026. doi: 10.3389/fonc.2021.650026
33. Marongiu L, Landry JJM, Rausch T, Abba ML, Delecluse S, Delecluse HJ, et al. Metagenomic analysis of primary colorectal carcinomas and their metastases identifies potential microbial risk factors. Mol Oncol (2021) 15(12):3363–84. doi: 10.1002/1878-0261.13070
34. Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J (2012) 6(2):320–9. doi: 10.1038/ismej.2011.109
35. Zhang Y, Yu X, Yu E, Wang N, Cai Q, Shuai Q, et al. Changes in gut microbiota and plasma inflammatory factors across the stages of colorectal tumorigenesis: a case-control study. BMC Microbiol (2018) 18(1):92. doi: 10.1186/s12866-018-1232-6
36. Gao S, Liu Y, Duan X, Liu K, Mohammed M, Gu Z, et al. Porphyromonas gingivalis infection exacerbates oesophageal cancer and promotes resistance to neoadjuvant chemotherapy. Br J Cancer (2021) 125(3):433–44. doi: 10.1038/s41416-021-01419-5
37. Utispan K, Pugdee K, Koontongkaew S. Porphyromonas gingivalis lipopolysaccharide-induced macrophages modulate proliferation and invasion of head and neck cancer cell lines. Biomed pharmacother = Biomed pharmacother (2018) 101:988–95. doi: 10.1016/j.biopha.2018.03.033
38. Yuan X, Liu Y, Kong J, Gu B, Qi Y, Wang X, et al. Different frequencies of porphyromonas gingivalis infection in cancers of the upper digestive tract. Cancer Lett (2017) 404:1–7. doi: 10.1016/j.canlet.2017.07.003
39. Mu W, Jia Y, Chen X, Li H, Wang Z, Cheng B. Intracellular porphyromonas gingivalis promotes the proliferation of colorectal cancer cells via the MAPK/ERK signaling pathway. Front Cell infect Microbiol (2020) 10:584798. doi: 10.3389/fcimb.2020.584798
40. Liu Y, Yuan X, Chen K, Zhou F, Yang H, Yang H, et al. Clinical significance and prognostic value of porphyromonas gingivalis infection in lung cancer. Trans Oncol (2021) 14(1):100972. doi: 10.1016/j.tranon.2020.100972
41. Abu N, Rus Bakarurraini NAA, Nasir SN. Extracellular vesicles and DAMPs in cancer: A mini-review. Front Immunol (2021) 12:740548. doi: 10.3389/fimmu.2021.740548
42. Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med (2015) 212(12):2077–94. doi: 10.1084/jem.20142162
43. Taddese R, Garza DR, Ruiter LN, de Jonge MI, Belzer C, Aalvink S, et al. Growth rate alterations of human colorectal cancer cells by 157 gut bacteria. Gut Microbes (2020) 12(1):1–20. doi: 10.1080/19490976.2020.1799733
44. Rubinstein MR, Baik JE, Lagana SM, Han RP, Raab WJ, Sahoo D, et al. Fusobacterium nucleatum promotes colorectal cancer by inducing wnt/β-catenin modulator annexin A1. EMBO Rep (2019) 20(4):e47638. doi: 10.15252/embr.201847638
45. McNabney SM, Henagan TM. Short chain fatty acids in the colon and peripheral tissues: A focus on butyrate, colon cancer, obesity and insulin resistance. Nutrients (2017) 9(12):1348. doi: 10.3390/nu9121348
46. Hu S, Liu L, Chang EB, Wang JY, Raufman JP. Butyrate inhibits pro-proliferative miR-92a by diminishing c-myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol Cancer (2015) 14:180. doi: 10.1186/s12943-015-0450-x
47. Li Q, Ding C, Meng T, Lu W, Liu W, Hao H, et al. Butyrate suppresses motility of colorectal cancer cells via deactivating Akt/ERK signaling in histone deacetylase dependent manner. J Pharmacol Sci (2017) 135(4):148–55. doi: 10.1016/j.jphs.2017.11.004
48. Chang SC, Shen MH, Liu CY, Pu CM, Hu JM, Huang CJ. A gut butyrate-producing bacterium butyricicoccus pullicaecorum regulates short-chain fatty acid transporter and receptor to reduce the progression of 1,2-dimethylhydrazine-associated colorectal cancer. Oncol Lett (2020) 20(6):327. doi: 10.3892/ol.2020.12190
49. Huang CC, Shen MH, Chen SK, Yang SH, Liu CY, Guo JW, et al. Gut butyrate-producing organisms correlate to placenta specific 8 protein: Importance to colorectal cancer progression. J adv Res (2020) 22:7–20. doi: 10.1016/j.jare.2019.11.005
50. Ternes D, Tsenkova M, Pozdeev VI, Meyers M, Koncina E, Atatri S, et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat Metab (2022) 4(4):458–75. doi: 10.1038/s42255-022-00558-0
51. Li R, Zhou R, Wang H, Li W, Pan M, Yao X, et al. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ (2019) 26(11):2447–63. doi: 10.1038/s41418-019-0312-y
52. Fu BC, Hullar MAJ, Randolph TW, Franke AA, Monroe KR, Cheng I, et al. Associations of plasma trimethylamine n-oxide, choline, carnitine, and betaine with inflammatory and cardiometabolic risk biomarkers and the fecal microbiome in the multiethnic cohort adiposity phenotype study. Am J Clin Nutr (2020) 111(6):1226–34. doi: 10.1093/ajcn/nqaa015
53. Xu R, Wang Q, Li L. A genome-wide systems analysis reveals strong link between colorectal cancer and trimethylamine n-oxide (TMAO), a gut microbial metabolite of dietary meat and fat. BMC Genomics (2015) 16 Suppl 7(Suppl 7):S4. doi: 10.1186/1471-2164-16-S7-S4
54. Yang S, Dai H, Lu Y, Li R, Gao C, Pan S. Trimethylamine n-oxide promotes cell proliferation and angiogenesis in colorectal cancer. J Immunol Res (2022) 2022:7043856. doi: 10.1155/2022/7043856
55. Kawasaki M, Ikeda Y, Ikeda E, Takahashi M, Tanaka D, Nakajima Y, et al. Oral infectious bacteria in dental plaque and saliva as risk factors in patients with esophageal cancer. Cancer (2021) 127(4):512–9. doi: 10.1002/cncr.33316
56. Faïs T, Delmas J, Serres A, Bonnet R, Dalmasso G. Impact of CDT toxin on human diseases. Toxins (2016) 8(7):220. doi: 10.3390/toxins8070220
57. Prince DJ, Patel D, Kachlany SC. Leukotoxin (LtxA/Leukothera) induces ATP expulsion via pannexin-1 channels and subsequent cell death in malignant lymphocytes. Sci Rep (2021) 11(1):18086. doi: 10.1038/s41598-021-97545-4
58. Proença MA, Biselli JM, Succi M, Severino FE, Berardinelli GN, Caetano A, et al. Relationship between fusobacterium nucleatum, inflammatory mediators and microRNAs in colorectal carcinogenesis. World J Gastroenterol (2018) 24(47):5351–65. doi: 10.3748/wjg.v24.i47.5351
59. Engevik MA, Danhof HA, Ruan W, Engevik AC, Chang-Graham AL, Engevik KA, et al. Fusobacterium nucleatum secretes outer membrane vesicles and promotes intestinal inflammation. mBio (2021) 12(2):e02706–20. doi: 10.1128/mBio.02706-20
60. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe (2013) 14(2):207–15. doi: 10.1016/j.chom.2013.07.007
61. Lichtenstern CR, Ngu RK, Shalapour S, Karin M. Immunotherapy, inflammation and colorectal cancer. Cells (2020) 9(3):618. doi: 10.3390/cells9030618
62. Hamamoto Y, Ouhara K, Munenaga S, Shoji M, Ozawa T, Hisatsune J, et al. Effect of porphyromonas gingivalis infection on gut dysbiosis and resultant arthritis exacerbation in mouse model. Arthritis Res Ther (2020) 22(1):249. doi: 10.1186/s13075-020-02348-z
63. Adel-Khattab D, Groeger S, Domann E, Chakraborty T, Lochnit G, Meyle J. Porphyromonas gingivalis induced up-regulation of PD-L1 in colon carcinoma cells. Mol Oral Microbiol (2021) 36(3):172–81. doi: 10.1111/omi.12332
64. Huang J, Liu D, Wang Y, Liu L, Li J, Yuan J, et al. Ginseng polysaccharides alter the gut microbiota and kynurenine/tryptophan ratio, potentiating the antitumour effect of antiprogrammed cell death 1/programmed cell death ligand 1 (anti-PD-1/PD-L1) immunotherapy. Gut (2022) 71(4):734–45. doi: 10.1136/gutjnl-2020-321031
65. Huang J, Jiang Z, Wang Y, Fan X, Cai J, Yao X, et al. Modulation of gut microbiota to overcome resistance to immune checkpoint blockade in cancer immunotherapy. Curr Opin Pharmacol (2020) 54:1–10. doi: 10.1016/j.coph.2020.06.004
66. Qiu B, Xi Y, Liu F, Li Y, Xie X, Guo J, et al. Gut microbiome is associated with the response to chemoradiotherapy in patients with non-small cell lung cancer. Int J Radiat Oncol Biol Phys (2022) S0360-3016(22)00749-0. doi: 10.1016/j.ijrobp.2022.07.032
67. Xi Y, Liu F, Qiu B, Li Y, Xie X, Guo J, et al. Analysis of gut microbiota signature and microbe-disease progression associations in locally advanced non-small cell lung cancer patients treated with concurrent chemoradiotherapy. Front Cell Infect Microbiol (2022) 12:892401. doi: 10.3389/fcimb.2022.892401
68. Lu Y, Yuan X, Wang M, He Z, Li H, Wang J, et al. Gut microbiota influence immunotherapy responses: Mechanisms and therapeutic strategies. J Hematol Oncol (2022) 15(1):47. doi: 10.1186/s13045-022-01273-9
69. Rezasoltani S, Yadegar A, Asadzadeh Aghdaei H, Reza Zali M. Modulatory effects of gut microbiome in cancer immunotherapy: A novel paradigm for blockade of immune checkpoint inhibitors. Cancer Med (2021) 10(3):1141–54. doi: 10.1002/cam4.3694
70. Wu J, Wang S, Zheng B, Qiu X, Wang H, Chen L. Modulation of gut microbiota to enhance effect of checkpoint inhibitor immunotherapy. Front Immunol (2021) 12:669150. doi: 10.3389/fimmu.2021.669150
71. Yi M, Jiao D, Qin S, Chu Q, Li A, Wu K. Manipulating gut microbiota composition to enhance the therapeutic effect of cancer immunotherapy. Integr Cancer Ther (2019) 18:1534735419876351. doi: 10.1177/1534735419876351
72. Escalante PI, Quinones LA, Contreras HR. Epithelial-mesenchymal transition and MicroRNAs in colorectal cancer chemoresistance to FOLFOX. Pharmaceutics (2021) 13(1):75. doi: 10.3390/pharmaceutics13010075
73. Hou XY, Zhang P, Du HZ, Gao YQ, Sun RQ, Qin SY, et al. Prevotella contributes to individual response of FOLFOX in colon cancer. Clin Transl Med (2021) 11(9):e512. doi: 10.1002/ctm2.512
74. Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science (2021) 374(6575):1632–40. doi: 10.1126/science.aaz7015
75. Lau HCH, Sung JJ, Yu J. Gut microbiota: impacts on gastrointestinal cancer immunotherapy. Gut Microbes (2021) 13(1):1–21. doi: 10.1080/19490976.2020.1869504
76. Kazmierczak-Siedlecka K, Roviello G, Catalano M, Polom K. Gut microbiota modulation in the context of immune-related aspects of lactobacillus spp. and bifidobacterium spp. in gastrointestinal cancers. Nutrients (2021) 13(8):2674. doi: 10.3390/nu13082674
77. Liu YJ, Tang B, Wang FC, Tang L, Lei YY, Luo Y, et al. Parthenolide ameliorates colon inflammation through regulating Treg/Th17 balance in a gut microbiota-dependent manner. Theranostics (2020) 10(12):5225–41. doi: 10.7150/thno.43716
78. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
79. Zhang W, Cheng C, Han Q, Chen Y, Guo J, Wu Q, et al. Flos abelmoschus manihot extract attenuates DSS-induced colitis by regulating gut microbiota and Th17/Treg balance. BioMed Pharmacother (2019) 117:109162. doi: 10.1016/j.biopha.2019.109162
80. Pal SK, Miller MJ, Agarwal N, Chang SM, Chavez-MacGregor M, Cohen E, et al. Clinical cancer advances 2019: Annual report on progress against cancer from the American society of clinical oncology. J Clin Oncol (2019) 37(10):834–49. doi: 10.1200/JCO.18.02037
Keywords: metastasis, metastatic colorectal cancer, gut microbiota, biomarkers, colorectal cancer
Citation: Sun L, Zhu Z, Jia X, Ying X, Wang B, Wang P, Zhang S and Yu J (2022) The difference of human gut microbiome in colorectal cancer with and without metastases. Front. Oncol. 12:982744. doi: 10.3389/fonc.2022.982744
Received: 30 June 2022; Accepted: 30 September 2022;
Published: 01 November 2022.
Edited by:
Shaobin Hou, University of Hawaii at Manoa, United StatesReviewed by:
Satoshi Hayakawa, Nihon University, JapanCopyright © 2022 Sun, Zhu, Jia, Ying, Wang, Wang, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jieru Yu, amllcnV5dUB6Y211LmVkdS5jbg==; Shuo Zhang, emhhbmdzaHVvdGNtQDE2My5jb20=; Peipei Wang, cGVpcGVpLndhbmdAemNtdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.