 Josephine A. Hinneh1,2,3,4
Josephine A. Hinneh1,2,3,4 Joanna L. Gillis1,3Nicole L. Moore1,3
Joanna L. Gillis1,3Nicole L. Moore1,3 Lisa M. Butler1,2,3*Margaret M. Centenera1,2,3*
Lisa M. Butler1,2,3*Margaret M. Centenera1,2,3*- 1South Australian Immunogenomics Cancer Institute and Adelaide Medical School, Adelaide, SA, Australia
- 2Freemason’s Centre for Male Health and Wellbeing, The University of Adelaide, Adelaide, SA, Australia
- 3Precision Cancer Medicine, South Australian Health and Medical Research Institute, Adelaide, SA, Australia
- 4Nagoya University Graduate School of Medicine, Nagoya, Japan
Receptor for hyaluronic acid-mediated motility (RHAMM) is a cell surface receptor for hyaluronic acid that is critical for cell migration and a cell cycle protein involved in microtubule assembly and stability. These functions of RHAMM are required for cellular stress responses and cell cycle progression but are also exploited by tumor cells for malignant progression and metastasis. RHAMM is often overexpressed in tumors and is an independent adverse prognostic factor for a number of cancers such as breast and prostate. Interestingly, pharmacological or genetic inhibition of RHAMM in vitro and in vivo ablates tumor invasiveness and metastatic spread, implicating RHAMM as a potential therapeutic target to restrict tumor growth and improve patient survival. However, RHAMM’s pro-tumor activity is dependent on its subcellular distribution, which complicates the design of RHAMM-directed therapies. An alternative approach is to identify downstream signaling pathways that mediate RHAMM-promoted tumor aggressiveness. Herein, we discuss the pro-tumoral roles of RHAMM and elucidate the corresponding regulators and signaling pathways mediating RHAMM downstream events, with a specific focus on strategies to target the RHAMM signaling network in cancer cells.
Introduction
The development and progression of cancer is characterized by a number of hallmarks that include uncontrolled cellular proliferation and aberrant activation of the host tumor microenvironment (TME) (1). Cell proliferation is normally driven and regulated by the cell cycle machinery, and is essential for cell growth, DNA replication and cell division. However, this activity is usurped by tumor cells for uncontrolled replicative capacity and metastatic spread (2). In fact, genetic or pharmacological manipulation of proteins required for cell cycle progression is currently providing clinically-approved therapeutic targets to limit tumor growth (2, 3). The TME, comprising cellular (stroma, fibroblasts, endothelial cells and infiltrating immune cells) and non-cellular (extracellular matrix) components, is an important determinant of tumor fate and metastatic progression (4, 5). Multiple experimental and clinical studies have indicated that cancer cells control TME factors to facilitate disease progression, aggressiveness and drug resistance (4, 5). Of note, inhibition of components of the TME, typified by the success of anti-PDL1 immunotherapies, has become relevant for the treatment of cancer (6, 7). Accordingly, proteins with dual functions in both cancer cells and the TME are attractive therapeutic targets for cancer management.
One such protein is the Receptor for Hyaluronic Acid Mediated Motility (RHAMM). RHAMM has both extracellular and intracellular functions depending on its cellular location (8, 9). Extracellular RHAMM is a well-characterized hyaluronic acid (HA) receptor that modulates HA-induced cell migration critical for inflammation and wound healing (8, 10, 11). Intracellular RHAMM is a cell cycle protein that regulates mitotic spindle and microtubule formation (9, 12). Importantly, these physiological functions of RHAMM are often dysregulated in cancer for growth advantage and disease progression (11, 13). RHAMM expression in most homeostatic tissues is relatively low and its expression is induced upon inflammatory stimuli (11). In contrast, human tumors have been reported to overexpress extracellular RHAMM, and this overexpression is commonly associated with metastatic and aggressive phenotypes, as well as poorer disease outcomes in prostate, breast and hematological malignancies (13–16). RHAMM overexpression is also an independent prognostic factor in many cancers (17–19).
Given that RHAMM is chronically overexpressed in cancer, and has multiple functional roles in cancer development and progression, it has significant potential as a therapeutic target for cancer. Thus, the discovery of agents that interfere with the expression of RHAMM in tumors could provide novel therapeutic strategies to limit tumor growth. However, the current incomplete understanding of RHAMM functions and its varied cellular distribution restricts the development of effective RHAMM-targeted therapies. Herein, we (i) discuss the importance of RHAMM as a potential oncogene and (ii) examine the regulators and signaling pathways activated by RHAMM, with the aim of highlighting new therapeutic avenues.
Structure and functions of RHAMM
RHAMM (also known as CD168) is a helical glycoprotein encoded by the gene HMMR, located on chromosome 5 (5q33.2-qter) in humans, with 18 exons and 2 start codons as depicted in Figure 1A (10, 20, 21). The full-length human RHAMM protein has a molecular weight of 84kDa and is made up of 725 amino acids (22, 23). Turley et al. first isolated and characterized RHAMM as part of the hyaluronic acid receptor complex (HARC) system from sub-confluent chick heart fibroblast cells (10, 24). Subsequent to this study, several RHAMM variants have been identified and are reported to occur via alternate gene splicing or the use of multiple start codons (10, 21, 23, 25). The best-characterized variant of RHAMM is the intracellular dominant exon 4 deficient variant (v3) (Figure 1A). RHAMM v3 is reported to localize with microtubules and induce oncogenic transformation when overexpressed in fibroblasts (23, 26). Additionally, RHAMM v3 is frequently detectable in cancer cells compared to normal tissues and has been shown to promote metastatic progression of tumors in vivo (23, 27). But why cancer cells express this particular variant is yet to be fully understood.
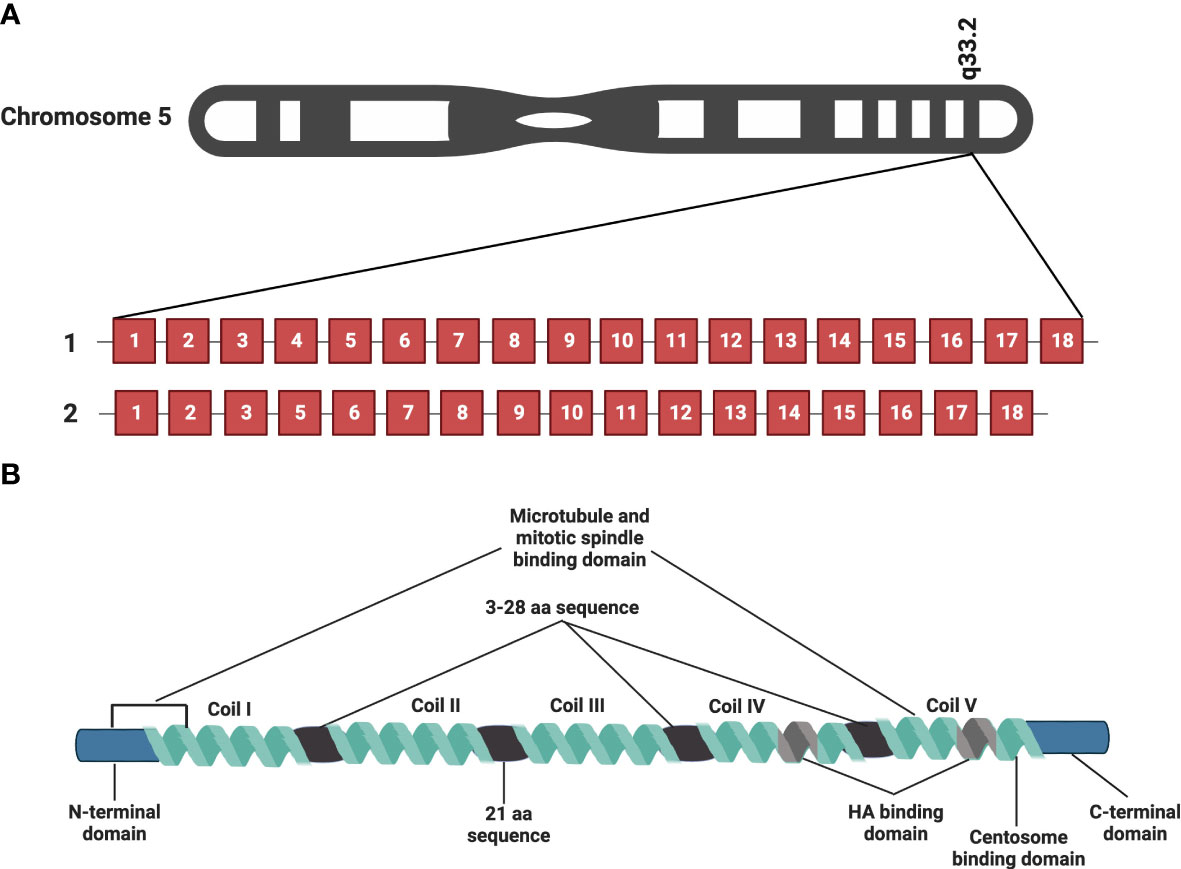
Figure 1 RHAMM gene and protein structure. HMMR, the gene encoding RHAMM is located on chromosome 5 and is made of 18 exons. Figure 1A is a representation of full-length HMMR and the most commonly occurring isoform, the exon 4 deficient variant (v3). Figure 1B is a diagrammatic representation of the protein structure of full length human RHAMM indicating the major functional domains. Five coils make up the supercoiled coiled domain with each separated by a 3-28 amino acid sequence with the exception of coil II and III, which are separated by a 21 amino acid sequence. The leucine-rich basic HA binding domain is located in coil IV and V close to the carboxyl terminal of the protein. Generated with BioRender.com.
RHAMM is a supercoiled-coil hydrophilic protein with three main functional domains, namely the amino-terminal domain (about 163 amino acids), the rod-like middle supercoiled-coil domain (made up of 5 coils) and the carboxyl-terminal domain (about 11 amino acids) (10, 20, 22, 23). Figure 1B is a pictorial description of the RHAMM protein, identifying the key features and various functional domains. The supercoiled-coil domain is a feature responsible for mediating protein-protein interactions (12, 23, 28). RHAMM is known to interact with cell surface receptors such as CD44, and platelet derived growth factor receptor (PDGFR) to mediate cell migration (29–31). RHAMM also interacts with intracellular proteins such breast cancer gene 1 (BRCA1), targeting protein for Xklp2 (TPX2) and extracellular signal-regulated kinase 1/2 (ERK1/2) to regulate mitotic spindle formation and stability (28, 32, 33).
Despite being an HA-binding protein, RHAMM lacks the classic HA-binding motif (proteoglycan tandem repeat) and instead interacts with HA via a basic amino acid-rich domain denoted by B(X7)B, where B is a lysine or arginine and the X is a non-acidic amino acid (aa) located on aa635-646 and 657-666 (10, 20). This region has also been shown to overlap with a carboxyl basic leucine zipper motif responsible for the interaction with centrosomes during mitosis (12). The N-terminal domain (aa 40-59 and 76-90) and exon 4 encoded region localize with microtubules during interphase (12, 23, 26).
Physiologically, RHAMM is transiently upregulated in response to injury in fibroblast cells and it also promotes neural development by enhancing neuroprogenitor cell division (34–36). Consequently, in vivo studies in RHAMM null mice have demonstrated defective wound closure after injury and neurodevelopment abnormalities such as megalocephaly (34–37). In homeostatic adult human tissues, RHAMM is expressed in the testes, thymus and placenta, with very low expression levels in the lungs and pancreas (13, 38).
Extracellular functions of RHAMM
RHAMM as an extracellularly expressed protein is predominantly in the lamellipodia at the leading edge of migrating cells (39, 40). As a well-characterized HA receptor, extracellular RHAMM modulates HA-induced cell migration (8, 10, 24). HA is an extracellular matrix (ECM) glycosaminoglycan, produced by hyaluronan synthases (HAS1-3) and undergoes fragmentation by chemical stress or enzymatic degradation to generate lower molecular weight fragments (11, 41). The size of the HA polymer is often predictive of its physiological function (11). The native high molecular weight HA, the most abundant in normal tissues, provides hydration and resistance to mechanical stress and further exhibits anti-migratory and anti-proliferative effects (11, 41). In contrast, low molecular weight HA (LMWHA), which preferentially interacts with RHAMM, promotes cell proliferation and migration in response to injury or tumor cell death (11, 41). Despite being extracellularly expressed, RHAMM lacks the signal peptide necessary for canonical ER-golgi export, hence it is speculated that RHAMM may be exported by some uncharacterized unconventional pathway(s) (42, 43). Surprisingly, no study to date has evaluated the exact mechanisms by which RHAMM is exported into the extracellular space, therefore studies focused in this direction may help unravel this aspect of RHAMM function and reveal therapeutic opportunities.
RHAMM lacks a transmembrane domain and therefore couples with a number of membrane-spanning receptors such as recepteur d’origine nantais (RON), CD44, PDGFR and transforming growth factor β1 (TGF-β1) to activate intracellular signaling pathways involved in cell migration (24, 29, 44, 45). For example, RHAMM couples with either CD44 in breast cancer cells or PDGFR in fibroblast cells to activate ERK1/2, in mediating cell migration (24, 29, 31, 44). HA and RHAMM interaction (HA : RHAMM) also activates other intracellular signaling pathways such as RHO-ROCK and β-catenin through other unknown cell surface partner proteins (46–51).
Intracellular functions of RHAMM
RHAMM was first identified as a novel microtubule associated protein (MAP) that interacted directly with interphase and mitotic microtubules by Assman and co-workers using confocal microscopy, an observation later confirmed by Maxwell et al. (12, 23, 26). Similar to other MAPs, both full length and RHAMM v3 decorated the entire length of microtubules in multiple cell lines and were also found to localise with centrosomes and interact with other centrosomal proteins during mitosis (12, 23, 26, 28, 32). Functionally, RHAMM regulates mitotic spindle integrity and progression of cells through the G2/M phase of the cell cycle. Consequently, inhibition of RHAMM results in the formation of multipolar spindles and loss of spindle integrity whilst overexpression of RHAMM produces large centrosomes with a resultant metaphase block that leads to cell death (12, 26, 52).
RHAMM interacts with a number of MAPS and centrosomal proteins in regulating microtubule dynamics. RHAMM binds to dynein motor complex, a cargo protein, during mitosis to correctly localize RHAMM to the minus end of spindle poles where it is involved in maintaining spindle stability by crosslinking centrosomal microtubules (Figure 2) (12, 52, 53). Additionally, RHAMM may contribute to microtubule formation indirectly by stabilizing and correctly positioning TPX2 for the effective activation of aurora kinase A (AURKA) at microtubule forming centers as depicted in Figure 2 (28). During cell cycle progression, RHAMM is regulated by the BRCA1-BRAD1 E3 ubiquitin ligase system (54). RHAMM also acts as an adaptor protein to correctly position and direct ERK1/2 to mitotic spindle forming centers and this has been demonstrated to partly modulate RHAMM associated microtubule functions (12, 26, 55–58). Intracellular RHAMM also acts as a transcriptional co-activator to modulate the transcription of target genes involved in migration (59, 60). For example, RHAMM serves as a co-activator for the E2F1 transcription factor to mediate the transcription of fibronectin in melanoma cells (59).
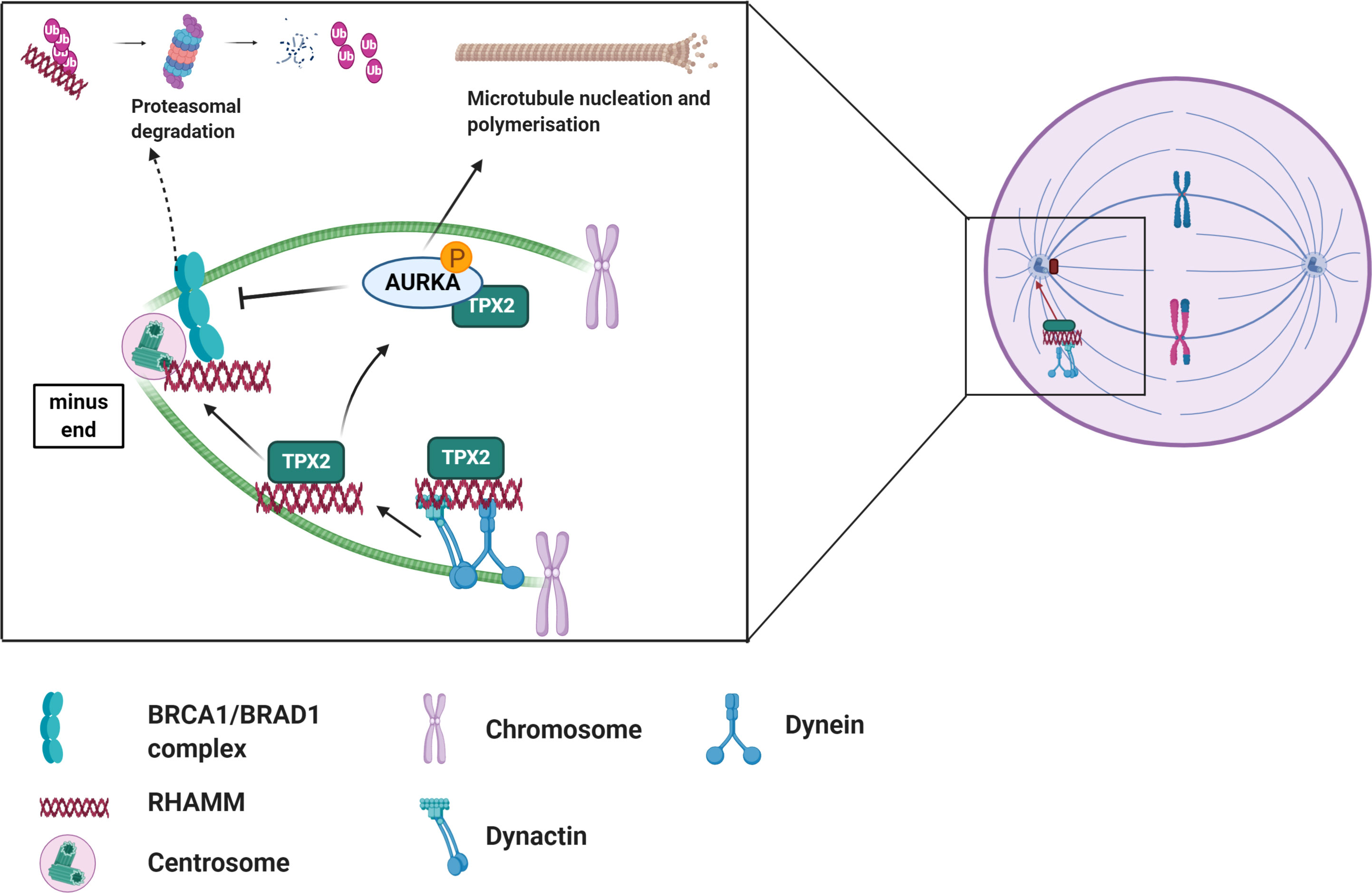
Figure 2 Overview of the role of RHAMM during cell cycle. RHAMM is a cell cycle gene localized at spindle assembly points where it associates with other MAPs such TPX2 to regulate spindle dynamics and microtubule stability. Like most cell cycle proteins, RHAMM mediated microtubule functions are regulated by BRCA1/BRAD1 complex which tags RHAMM for ubiquitination and subsequent proteasomal degradation, an event nullified by activation of AURKA. Generated with BioRender.com.
Whilst RHAMM performs disparate extracellular and intracellular roles, it is becoming more apparent that these roles may be somewhat linked. The observation that HA expression increases during mitosis and the ability of exogenous HA to modify RHAMM : TPX2 binding affinity and alter AURKA activity, further demonstrates that HA : RHAMM interactions may contribute in part to RHAMM intracellular functions (61, 62). A detailed understanding of the RHAMM interactome may yield further insights into the factors linking its intra- versus extracellular actions.
Molecular signaling pathways regulated by RHAMM
Interaction between HA : RHAMM is known to regulate a number of downstream signaling pathways; the most predominant are the ERK1/2 and RHO-ROCK pathways (29, 31, 36, 63). It was first reported that H-ras transformed cells exhibited characteristics similar to RHAMM overexpressing cells, and that loss-of-function mutation of RHAMM resulted in decreased ERK1/2 activation in Ras mutant cells (31, 64). Subsequent studies demonstrated that RHAMM overexpressing cells exhibit high basal ERK1/2 activation while RHAMM null fibroblasts showed reduced ERK1/2 phosphorylation with no change in total ERK1/2 levels (31, 36). RHAMM has been shown to co-localize with ERK1/2 in breast cancer cells, but the exact mechanism by which RHAMM activates ERK1/2 is currently unknown (29).
However, studies have suggested that RHAMM may activate ERK1/2 pathways by partnering with a number of cell surface proteins in response to HA stimulation (26, 62). RHAMM has been shown to co-localize with CD44, another well-characterized receptor for HA, in aggressive MDA-MB-231 breast cancer cells to maintain the activity of ERK1/2 (29). Additionally, RHAMM may co-activate ERK1/2 by partnering with upstream activators such as PDGFR and epidermal growth factor receptor (EGFR) in mesenchymal and fibroma cells respectively to promote cell motility (36, 62). Once activated, ERK1/2 facilitates cell motility by either activating the focal adhesion pathway (FAK) pathway or by mediating transcription of motogenic genes such as MMP9 in a RHAMM dependent manner (46, 47, 60).
HA : RHAMM interaction is also reported to activate the RHO-ROCK signaling pathway in prostate cancer cells, ultimately leading to the phosphorylation of translation initiation factor eIF4E, an observation that was associated with treatment resistance (65). Functionally, HA : RHAMM interaction promoted cell proliferation, metastasis and invasion of androgen independent PC3 prostate cancer cells, and this observation was abrogated by a ROCK inhibitor (50, 65).
Stimulation of RHAMM by HA has also been shown to activate less reported pathways such as β-catenin to up-regulate the expression of the oncogene c-MYC which promoted tumour growth and proliferation in fibrosarcoma cells (51). Figure 3 is a summary of the RHAMM signaling pathway.
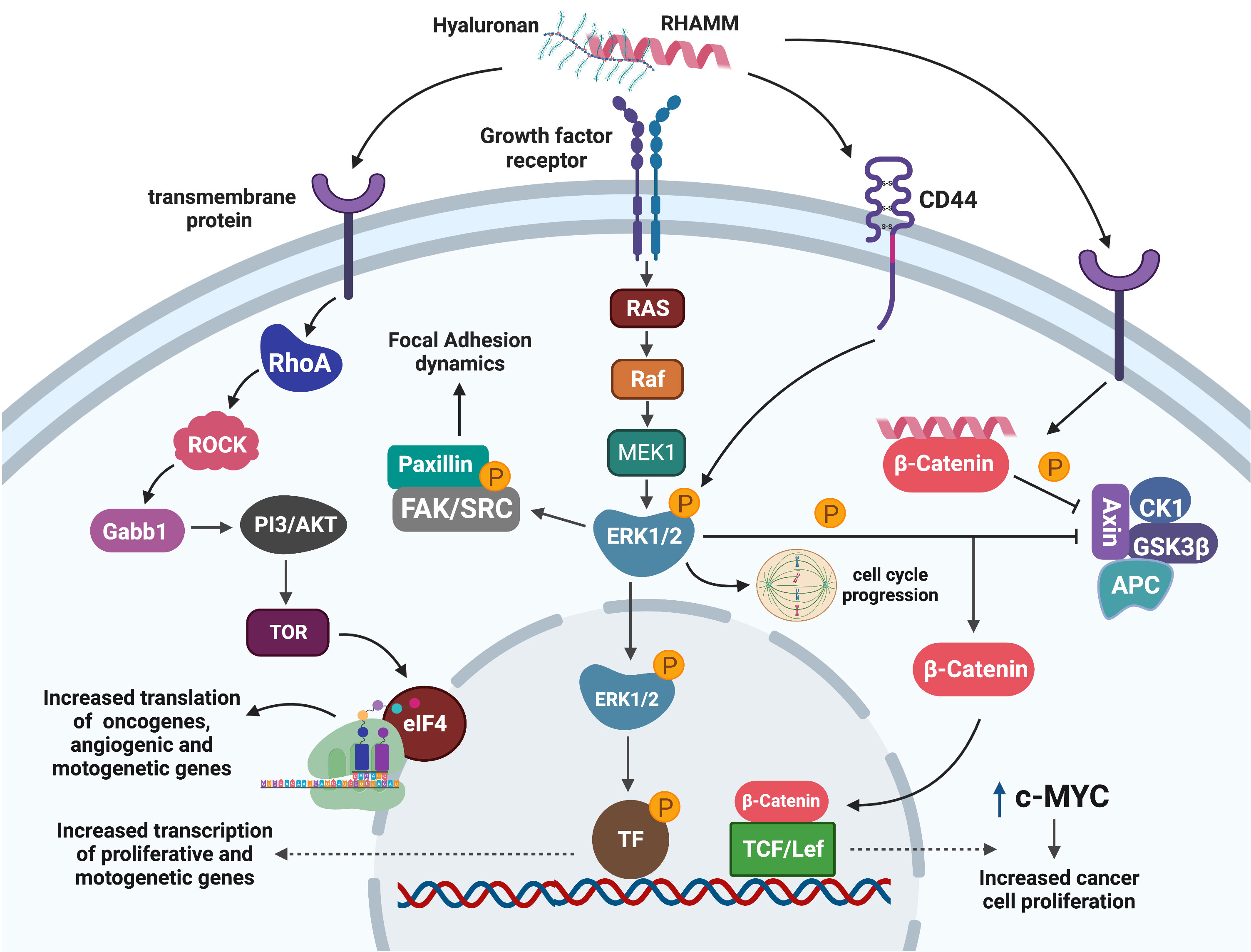
Figure 3 Signaling pathways mediating RHAMM oncogenic effects. Cell surface HA : RHAMM interaction may co-localize with transmembrane receptors/proteins to activate intracellular signaling pathways that result in the phosphorylation of ERK1/2, the key RHAMM modulated protein. pERK1/2 influences cell migration by regulating cell adhesion dynamics through FAK or is translocated into the nucleus to activate unknown transcription factors that enhance the transcription of mitogenic and motogenic genes. Alternatively, pERK1/2 may influence mitotic entry by regulating mitotic spindle formation needed for effective progression through mitosis. Activation of RHAMM may also result in the translation of growth-promoting and motogenic genes through the ROCK-eIF4E pathways. Similarly, RHAMM augments the stability of β-catenin by inhibiting the β -catenin degradation complex and hence promotes nuclear translocation and activation of transcription factors which enhance transcription of the oncogene c-MYC with a resultant increase in cell proliferation. Generated with BioRender.com.
Role of RHAMM in oncogenesis
RHAMM plays an important role in the development and progression of a number of cancers (11). The oncogenic potential of RHAMM is attributed to its intracellular function in cell cycle progression, specifically mitotic spindle formation and stability and extracellular functions in cell migration. In cancer, RHAMM overexpression has been reported in breast, prostate, leukemia, pancreatic, lung cancers, and glioblastoma, with strongest expression in metastatic tumors (38, 64, 66–71). The significance of RHAMM in cancer is documented below and tabulated in Table 1 is a summary of the role of RHAMM reported in various malignancies.
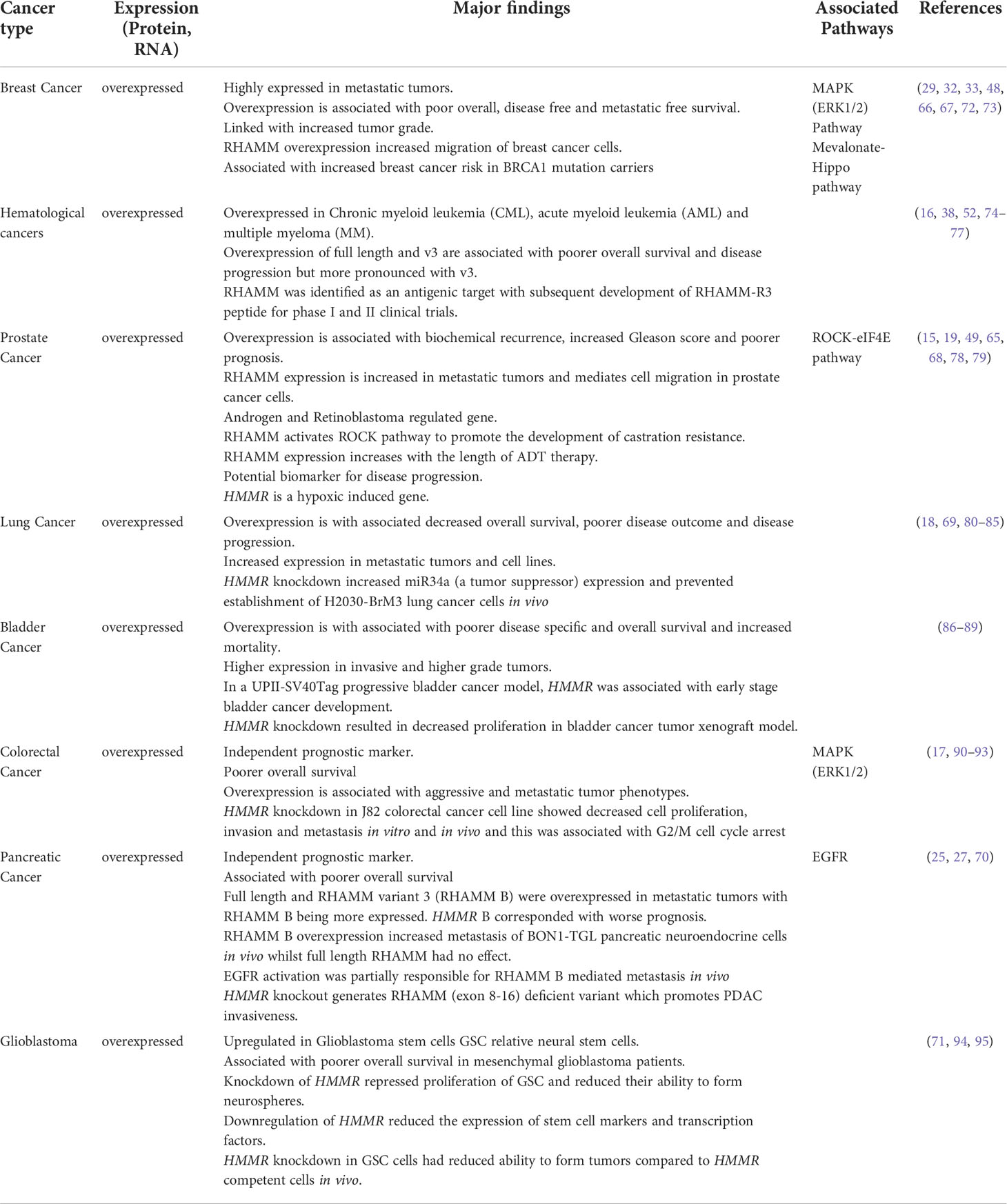
Table 1 A summary of the oncogenic role of RHAMM in various cancers.
Role of RHAMM in cell proliferation
The critical role of RHAMM in the cell cycle means that overexpression or downregulation of RHAMM results in the accumulation of cells at G2/M phase (67, 96–98). Levels of RHAMM mRNA and protein have both been reported to increase during G2/M phase, but the exact mechanism mediating transcriptional upregulation of RHAMM during cell cycle progression remains unknown (98). Multiple independent studies have reported that downregulation of RHAMM suppresses cell proliferation in cancer cells and that overexpression of RHAMM is transforming (64, 97, 99). One mechanism by which RHAMM may promote cell proliferation is by augmenting the activity of M phase promoting factor Cdc2/cyclin B currently known as the Cdk1/cyclin B kinase activity through stabilisation of Cdc2 mRNA levels in an HA dependent manner (80, 97). Cdk1/cyclin B complex regulates mitotic entry by phosphorylating a number of spindle associated proteins such as importin to control effective microtubule formation and chromosomal segregation (100). Therefore, RHAMM overexpression may facilitate premature mitotic entry resulting in genetic alterations that favour tumour growth through an increase in Cdk1/cyclin B activity.
In regulating cell cycle progression, RHAMM is reported to associate with other MAPs to regulate mitotic spindle assembly. RHAMM interacts with TPX2 during mitosis to correctly localize TPX2 to the minus end of microtubules for AURKA phosphorylation and activation (28, 101). Activation of AURKA promotes centrosome nucleation and maturation for mitotic progression (28). Cancer cells overexpressing RHAMM exhibit increased AURKA activation, hence increased mitotic progression and proliferation. As a result, knockdown of RHAMM increased the sensitivity of malignant peripheral nerve sheath tumors to the AURKA inhibitor MLN8237 (102).
Similar to most cell cycle proteins, RHAMM activity is regulated by the BRCA1-BRAD1 E3 ubiquitin ligase complex, which targets it for degradation during cell cycle progression (54). Thus, RHAMM overexpression (in BRCA1 competent cells) has been shown to induce phenotypic changes similar to BRCA1 deficient cells, which are characterized by polyploidy, formation of micronuclei, centrosomal amplification and chromosomal mis-segregation; genomic events associated with DNA damage and mutations (72). In the event of RHAMM overexpression, which is indeed the case for most cancers, cancer cells may mimic BRCA1 deficient phenotypes that facilitate genomic instability and hence promote tumorigenesis (32, 72). Additionally, overexpression of RHAMM is linked to increased AURKA activation, which corresponds to decreased BRCA1 ubiquitin activity, thereby providing a BCRA1 deficient environment necessary for genomic instability (103). A more recent study has demonstrated that loss of BRCA1 and overexpression of HMMR promotes AURKA activation which results in ARPC2-mediated decreased mitotic cortex stability and subsequently genomic instability (104). Thus, RHAMM has been linked with increased breast cancer susceptibility in BRCA1 mutation carriers (32, 67, 104).
Role of RHAMM in cell migration/metastasis
RHAMM influences cell motility in two ways, first by influencing mechanical changes in the cell that impacts unidirectional movements, and second by enhancing epithelial-to-mesenchymal transition (EMT). These functions are linked to RHAMM’s extracellular activity, which is achieved through coupling with transmembrane protein partners to activate intracellular signaling pathways, particularly ERK1/2 (29, 31). The role of ERK1/2 in cell motility has been well documented over the years and reviewed by Tanimura et al. (105).
Relevant to RHAMM is that ERK1/2 regulates the association between FAK and paxillin, a focal adhesion (FA) adaptor protein, by ensuring a phosphorylation/dephosphorylation feedback loop that influences FA assembly and disassembly during cell migration (46, 106, 107). Activation of the ERK1/2 pathway by HA : RHAMM interaction increased tyrosine phosphorylation of FAK with a resultant increase in FA turnover and consequently cell migration (46, 47). In regulating FAK signaling, RHAMM has also been reported to co-activate the E2F1 transcription factor to increase the transcription of fibronectin, which promoted integrin-β1-FAK induced motility in melanoma cells (59). RHAMM induced ERK1/2 activation also exerts control on mechanotaxis by regulating the activity of proteins involved in protrusion and matrix adhesion, therefore aiding detachment and polarized movement (105).
In terms of EMT, RHAMM mediated ERK1/2 activation facilitates that nuclear translocation and activation of yet to be identified transcription factors that mediate the transcription of EMT genes such as MMP9 (60), hence promoting the development of the mesenchymal phenotype (105). Indeed, inhibition of RHAMM using a mimetic peptide, P15-1, coincided with decreased myofibroblast differentiation and expression of mesenchymal markers such as vimentin and N-cadherin, resulting in decreased migration of fibroblast and prostate cancer cells (108). Similarly, inhibition of HA : RHAMM interaction suppresses TGF-β1 (a potent regulator of EMT) mediated cell migration (45, 109, 110). RHAMM-mediated decreased TGF-β1 activity was associated with decreased mesenchymal transformation and ultimately a decrease in cell migration (45, 108). It is still quite unclear how RHAMM modulates TGF-β1 induced cell migration and therefore future studies focused on understanding this phenomenon would provide better insights for effective targeting of this pathway.
HA : RHAMM interactions can also influence cytoskeletal changes of migratory cells by activating the RHO-ROCK pathway to mediate actin polymerization and promotion of unidirectional movement at the leading edge of the cell (50, 65, 111, 112). Overexpression of RHAMM in prostate cancer cells increased phosphorylation of ROCKII and coflin with an accompanying increase in filopodia formation and EMT markers, vimentin and N-cadherin, and a decrease in E- cadherin (50). The RHAMM-ROCK pathway is commonly hyperactivated in castration-resistant prostate cancer and associated with metastatic disease progression (50, 65). Collectively, these findings suggest a critical role for RHAMM in cell migration and more importantly metastasis of cancer cells and further underscores the relevance of targeting RHAMM in advanced cancers.
Regulators of RHAMM expression
RHAMM is transcriptionally regulated by a number of factors that either activate or repress its expression (Figure 4). HA is a potent transcriptional up-regulator of RHAMM in both normal and transformed cells (36, 113, 114). Expression of HMMR was significantly increased when fibroblast, HUVEC and breast cells were stimulated with HA (45, 114, 115). Mechanistically, HA induced RHAMM gene and protein expression by activating CD44/protein kinase Cδ (PKCδ) pathway (115). Activation of the CD44/PKCδ complex induced c-fos and c-jun nuclear translocation, resulting in increased RHAMM transcription, an effect rescued by the inhibition of PKCδ (115).
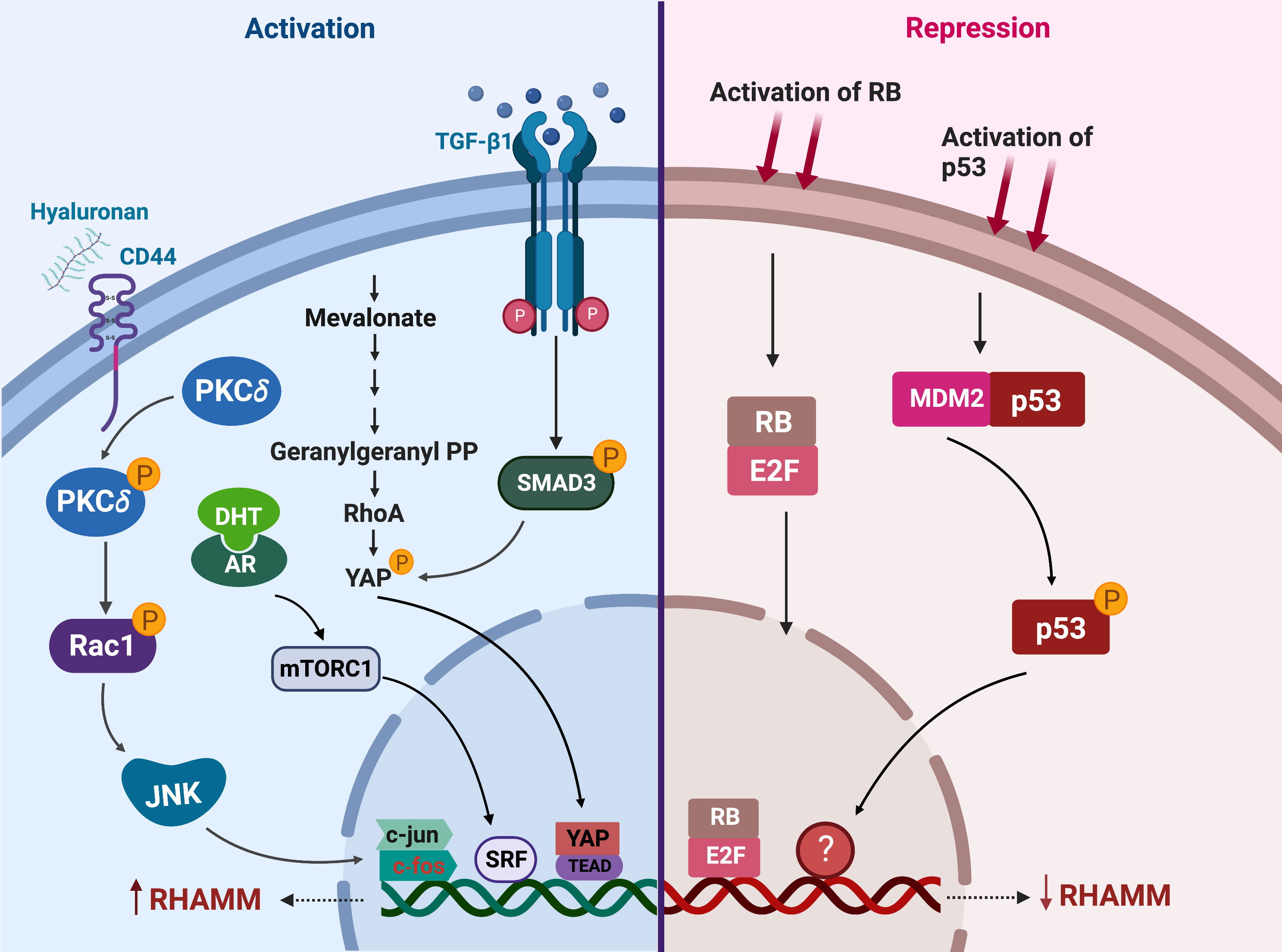
Figure 4 Regulators of RHAMM expression. This figure enumerates the transcriptional regulators of RHAMM activation (on the left) and repression (on the right) in cancer and normal cells. Activation of CD44, mevalonate, TGF-β1 or the androgen receptor (AR) signaling pathways induces transcriptional upregulation of HMMR. On the contrary, p53 and RB activation results in decreased transcription of HMMR. Generated with BioRender.com.
Stimulation of fibroblasts with TGF-β1 markedly increased both HMMR transcription and cell surface expression of RHAMM (45, 109). TGF-β1-induced RHAMM transcription was mediated through a SMAD3-YAP dependent pathway (110). The increase in RHAMM expression enhanced TGF-β1-induced cell migration, and this effect was partially ablated by the addition of RHAMM blocking antibodies (45, 110, 115). Also mediated via YAP, but through prenylation of RhoA, mevalonate treatment of breast cancer cells induced both RHAMM protein and mRNA expression (48). These observations were nullified by the inhibition of YAP or treatment with simvastatin, a mevalonate inhibitor, and this consequently resulted in decreased cell migration in an ERK1/2 driven manner (48).
RHAMM is also transcriptionally up-regulated by DHT treatment in LNCaP prostate cancer cells (49, 116). Whilst the exact mechanism (s) underpinning DHT-induced RHAMM expression is currently unknown, it was reported that this could be a DHT-mediated mTORC1 activation of the SRF transcription factor which directly binds to the promoter region of HMMR in LNCaP cells (116). Further studies are required to fully clarify the direct association between the androgen receptor (AR) and mTOR pathways in regulating HMMR expression given that crosstalk between mTOR and AR is complex and context-dependent (117, 118).
Transcriptional downregulation of RHAMM has been achieved through activation of tumor suppressors p53 and retinoblastoma (RB) (50, 98, 119). Induction of p53 in D53wt ovarian cancer derived cells significantly repressed both full length and v3 RHAMM gene and protein expressions, likely through an indirect mechanism since no putative p53 binding site was observed on the HMMR gene transcript (98). Further studies are needed to provide insights into how p53 represses HMMR expression transcriptionally during cell cycle progression. Similarly, RB regulates RHAMM gene and protein expressions through the E2F1 transcription factor, which binds directly to the promoter region of RHAMM as observed in prostate and lung cancer cells (50). RNA sequencing of RB deficient cells identified HMMR as one of the most significantly upregulated genes promoting cell migration in response to RB-loss (50). Accordingly, drugs that induce or augment p53 or RB activity such as Nutlin-3 and CDK4/CDK6 inhibitors respectively, repressed RHAMM expression (50, 98).
Therapeutic approaches to target RHAMM
Immunological targeting of RHAMM
RHAMM was first identified as a tumor associated antigen in multiple myeloma and chronic myeloid leukemia (CML) and that overexpression was associated with poor prognosis in these patients (16, 38). This discovery stimulated preclinical immunogenic targeting of RHAMM in mouse glioma and melanoma models wherein mice received either dendritic cells pulsed with HMMR mRNA or a DNA-based xenopus RHAMM (xRHAMM), respectively (120, 121). In both studies, RHAMM vaccination significantly reduced tumor burden and increased infiltration of RHAMM specific cytotoxic T cells (120, 121). Following these discoveries, the RHAMM-R3 peptide vaccine was developed and evaluated in Phase I and II clinical trials (122, 123). In the first study, six chronic lymphocytic leukemia patients were vaccinated and about 80% of the patients produced significant numbers of CD8+ T cells with a 20% decrease in white blood cells (122). To further expand the therapeutic scope of the peptide vaccine, Greiner in that same year evaluated a high dose of RHAMM-R3 peptide in patients with acute myeloid leukemia (AML), multiple myeloma and myelodysplastic syndrome, where 4 out of nine patients showed an increase in CD8+ T cells and 3 showed positive clinical response (74). It was later reported that the RHAMM-R3 vaccine when co-administered with chemotherapy or stem cell transplantation in leukemic patients may delay relapse and ensure patients remained in remission (123).
Despite these promising initial clinical reports, RHAMM-R3 peptide is yet to receive clinical approval. To understand the therapeutic limitations of RHAMM-R3 peptides, Snauwaert and co-workers showed that for AML patients, RHAMM expression on normal hematopoietic cells were similar to that of leukemic stem cells and that expression was cell cycle dependent. Thus, they proposed that the lack of specificity of RHAMM expression limits its therapeutic application in AML (124). But for reasons not unique to the RHAMM vaccine, cancer vaccine development in general has encountered substantial setbacks due to immune tolerance and immunosuppressive factors in the tumor microenvironment (125, 126). A potential alternative approach would be to combine RHAMM vaccines with immune checkpoint inhibitors as a mechanism to evade these immunosuppressive factors (127).
Targeting RHAMM signaling
The dependence of extracellular RHAMM on HA for effective signaling coupled with the oncogenic nature of LMWHA (RHAMM specific HA fragments), proffers HA as an effective target for limiting RHAMM downstream signaling activities.
Using phage technology, Tolg and colleagues first identified a number of HA binding peptides, worthy of note is Peptide 15-1 (P15-1), which exhibited substantial anti-migratory effects on fibroblasts in a dose-dependent manner and specifically served as a decoy for LMWHA (108). Subsequent to this discovery, these peptides have been evaluated in a number of preclinical cancer models and have shown significant effects on proliferation, angiogenesis, metastasis and invasion (15, 50, 108, 128). Mechanistically, P15 treatment markedly reduced the expression of TGF-β1, and suppressed the FAK pathway as identified in a pathway analysis from a microarray study of P15-1 treated fibroblasts (108). Additionally, a number of tubulin derived HA targeting peptides have also been developed against RHAMM-specific HA activities (129). A unique feature of these peptides was that they were internalised by breast and prostate cancer cells, suggestive of an inherent ability of these peptides to possibly interfere with RHAMM’s intracellular functions, although this requires experimental substantiation (129). These peptides present a burgeoning area of research worth exploring for the effective development of RHAMM specific anticancer therapies. However, the clinical use of peptides as therapeutics are limited by their unfavourable physicochemical properties (130). Whilst the identification of specific RHAMM therapies have mainly focused on identifying peptides that interfere with RHAMM function, their application have been restricted to in vitro assays with limited in vivo efficacy studies. Currently, the use of peptidomimetics (compounds that mimic a natural peptide in their ability to bind to specific receptors but with improved physicochemical properties) are becoming widely used alternatives in clinical therapeutics (131). Therefore, future studies focused on identifying and evaluating RHAMM peptidomimetics may accelerate the in vivo use of these peptides and potentially facilitate its clinical application.
Alternatively, therapeutic approaches focused on inhibiting HA synthesis 4-Methylubelliferone (4-MU) or HA fragmentation by hyaluronidase (O-sulphated HA) have been explored as anti-cancer therapies (132–134). These HA targeted therapies additionally downregulate RHAMM expression, making them attractive alternatives for modulating RHAMM signaling. These pharmacological agents regulate cancer promoting pathways such PI3K/AKT, EGFR and ERK1/2/MAPK to promote apoptosis and repress proliferation, migration, angiogenesis and invasion of cancer cells (132–134). 4-MU is a clinically approved drug sold under the brand name Hymercromone for use as an anti-spasmodic on the biliary tract in Europe and some parts of Asia (135). Despite multiple studies extensively showing the anti-cancer effects of 4-MU, it is yet to be evaluated in any clinical trials for its anti-cancer effects.
Conclusion and future directions
There has been a growing appreciation of the role of RHAMM in cancer over the last three decades. A plethora of recent studies, incorporating cell and animal models as well as patient samples, have delved into the mechanisms associated with the individual identified roles of RHAMM in an attempt to understand the significance of this protein in cancer. More importantly, efforts have been made to explore the therapeutic potential of targeting RHAMM and to this end, much progress has been made with respect to preclinical evaluations, but the translation of these observations into patients has not yet materialised.
To fully expedite the translation of all these preclinical data into clinical use, it will first of all be important to clearly define the diverse compartmental functions of RHAMM. Whilst a lot of information exists on the intracellular and extracellular functions of RHAMM, much more insight is needed to decipher whether these functions are independent of each other or if there is some form of crosstalk between these identified roles. By employing computational biology approaches to existing transcriptomic and proteomic data, a RHAMM interactome can be developed to identify all RHAMM associated genes and proteins involved in cell cycle and migration and how these proteins are interconnected. This may also help identify functional nodes within these networks which are targetable by drugs. Additionally, different variants of RHAMM have been identified and are reported to play functional roles in oncogenic transformation, but current knowledge about how these isoforms are generated and their specific roles in RHAMM-mediated cancer development and progression are yet to be fully stratified. More in vitro and in vivo studies should be conducted to identify the mechanisms of generating these isoforms and with the aid of omics, proteins and genes associated with these isoforms may be identified to subsequently provide more insight into which RHAMM specific role they regulate.
It has been reported that RHAMM is exported to the extracellular space by yet to be identified mechanisms, where it associates with transmembrane proteins to activate intracellular signaling pathways. Shedding light on these mechanism(s) may provide new therapeutic targets to block the extracellular functions of RHAMM, hence providing treatment alternatives for the management of advanced disease. Also, RHAMM co-localizes with other transmembrane proteins to activate intracellular signaling pathways due to the lack of a transmembrane domain. Although some transmembrane partners have been identified, there are a number yet to be discovered, and even for the identified cell surface partners, the mechanism by which RHAMM modifies their activity is largely unknown. Therefore, identifying and further clarifying these interactions using affinity purification and in silico protein-protein interaction models, may provide mechanistic insights and also reveal indirect therapeutic targets for cancer and other inflammatory related diseases.
Taken together, RHAMM is a vital driver of cancer progression and metastasis, and by addressing some of the knowledge gaps identified in this review article, more specific and effective RHAMM-targeted therapies can be developed for the management of cancer.
Author contributions
Study conception and design, JH, NM, JG, LB, and MC. Drafting of manuscript, JH, NM, and JG. Critical review of the manuscript, LB and MC. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by a grant from Cancer Australia (Project Grant AP1138766). MC was supported by the Freemasons Centre for Male Health and Wellbeing. LB was supported by a Beat Cancer SA Beat Cancer Project Principal Cancer Research Fellowship (PRF1117). JAH is supported by the Beacon of Enlightenment Scholarship from the University of Adelaide
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov (2022) 12(1):31–46. doi: 10.1158/2159-8290.Cd-21-1059
2. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer (2017) 17(2):93–115. doi: 10.1038/nrc.2016.138
3. Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med (2019) 381(4):307–16. doi: 10.1056/NEJMoa1903765
4. Ribeiro Franco PI, Rodrigues AP, de Menezes LB, Miguel Pacheco M. Tumor microenvironment components: Allies of cancer progression. Pathol Res Pract (2020) 216(1):152729. doi: 10.1016/j.prp.2019.152729
5. Li W, Ng JM, Wong CC, Ng EKW, Yu J, et al. Molecular alterations of cancer cell and tumour microenvironment in metastatic gastric cancer. Oncogene (2018) 37(36):4903–20. doi: 10.1038/s41388-018-0341-x
6. Patel MR, Ellerton J, Infante JR, Agrawal M, Gordon M, Aljumaily R, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN solid tumor): Pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol (2018) 19(1):51–64. doi: 10.1016/s1470-2045(17)30900-2
7. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med (2018) 379(23):2220–9. doi: 10.1056/NEJMoa1809064
8. Turley EA, Moore D, Hayden LJ. Characterization of hyaluronate binding proteins isolated from 3T3 and murine sarcoma virus transformed 3T3 cells. Biochemistry (1987) 26(11):2998–3005. doi: 10.1021/bi00385a007
9. Assmann V, Gillett CE, Poulsom R, Ryder K, Hart IR, Hanby AM. The pattern of expression of the microtubule-binding protein RHAMM/IHABP in mammary carcinoma suggests a role in the invasive behaviour of tumour cells. J Pathol (2001) 195:191–6. doi: 10.1002/path.941
10. Hardwick C, Hoare K, Owens R, Hohn HP, Hook M, Moore D, et al. Molecular cloning of a novel hyaluronan receptor that mediates tumor cell motility. J Cell Biol (1992) 117(6):1343–50. doi: 10.1083/jcb.117.6.1343
11. Tolg C, Messam BJ, McCarthy JB, Nelson AC, Turley EA. Hyaluronan functions in wound repair that are captured to fuel breast cancer progression. Biomolecules (2021) 11(11):1551. doi: 10.3390/biom11111551
12. Maxwell CA, Keats J.J, Crainie M, Sun X, Yen T, Shibuya E, et al. RHAMM is a centrosomal protein that interacts with dynein and maintains spindle pole stability. Mol Biol Cell (2003) 14(6):2262–76. doi: 10.1091/mbc.e02-07-0377
13. Chen Y, Chen Z, Du YN. Immunohistochemical analysis of RHAMM expression in normal and neoplastic human tissues: A cell cycle protein with distinctive expression in mitotic cells and testicular germ cells. Oncotarget (2018) 9(30):20941–52. doi: 10.18632/oncotarget.24939
14. Wu W, Chen L, Wang Y, Jin J, Xie X, Zhang J. Hyaluronic acid predicts poor prognosis in breast cancer patients: A protocol for systematic review and meta analysis. Medicine (2020) 99(22):e20438. doi: 10.1097/md.0000000000020438
15. Rizzardi AE, Vogel R.I, Koopmeiners J.S, Forster C.L, Marston L.O, Rosener N.K, et al. Elevated hyaluronan and hyaluronan-mediated motility receptor are associated with biochemical failure in patients with intermediate-grade prostate tumors. Cancer (2014) 120(12):1800–9. doi: 10.1002/cncr.28646
16. Greiner J, Ringhoffer M, Taniguchi M, Schmitt A, Kirchner D, Krähn G, et al. Receptor for hyaluronan acid–mediated motility (RHAMM) is a new immunogenic leukemia-associated antigen in acute and chronic myeloid leukemia. Exp Hematol (2002) 30:1029–35. doi: 10.1016/s0301-472x(02)00874-3
17. Mele V, Soko L, Kölzer VH, Pfaff D, Muraro MG, Keller I, et al. The hyaluronan-mediated motility receptor RHAMM promotes growth, invasiveness and dissemination of colorectal cancer. Oncotarget (2017) 8(14):70617–29. doi: 10.18632/oncotarget.19904
18. Song YJ, Tan J, Gao XH, Wang LX, et al. Integrated analysis reveals key genes with prognostic value in lung adenocarcinoma. Cancer Manag Res (2018) 10:6097–108. doi: 10.2147/CMAR.S168636
19. Rizzardi AE, Rosener NK, Koopmeiners JS, Vogel RSI, Metzger GJ, Forster CL, et al. Evaluation of protein biomarkers of prostate cancer aggressiveness. BMC Cancer (2014) 14(244):1–14. doi: 10.1186/1471-2407-14-244
20. Yang B, Zhang L, Turley EA. Identification of two hyaluronan-binding domains in the hyaluronan receptor RHAMM. J Biol Chem (1993) 268(12):8617–23. doi: 10.1016/S0021-9258(18)52919-7
21. Zhou R, Wu X, Skalli O. The hyaluronan receptor RHAMM/IHABP in astrocytoma cells: expression of a tumor-specific variant and association with microtubules. J Neuro-Oncol (2002) 59:15–26. doi: 10.1023/A:1016373015569
22. Wang C, Entwistle J, Hou G, Li Q, Turley EA. The characterization of a human RHAMM cDNA: Conservation of the hyaluronan-binding domains. Gene (1996) 174:299–306. doi: 10.1016/0378-1119(96)00080-7
23. Assmann V, Marshall JF, Fieber C, Hofmann M, Hart IR. The human hyaluronan receptor RHAMM is expressed as an intracellular protein in breast cancer cells. J Cell Sci (1999) 111:1685–94. doi: 10.1002/path.941
24. Turley EA, Austen L, Vandeligt K, Clary C. Hyaluronan and a cell-associated hyaluronan binding protein regulate the locomotion of ras-transformed cells. J Cell Biol (1991) 112(5):1041–7. doi: 10.1083/jcb.112.5.1041
25. Lin A, Feng J, Chen X, Wang D, Wong M, Zhang G, et al. High levels of truncated RHAMM cooperate with dysfunctional p53 to accelerate the progression of pancreatic cancer. Cancer Lett (2021) 514:79–89. doi: 10.1016/j.canlet.2021.05.011
26. Tolg C, Hamilton SR, Morningstar L, Zhang J, Zhang S, Esguerra KV, et al. RHAMM promotes interphase microtubule instability and mitotic spindle integrity through MEK1/ERK1/2 activity. J Biol Chem (2010) 285(34):26461–74. doi: 10.1074/jbc.M110.121491
27. Choi S, Wang D, Chen X, Tang LH, Verma A, Chen Z, et al. Function and clinical relevance of RHAMM isoforms in pancreatic tumor progression. Mol Cancer (2019) 18(1):92. doi: 10.1186/s12943-019-1018-y
28. Chen H, Mohan P, Jiang J, Nemirovsky O, He D, Fleisch MC, et al. Spatial regulation of aurora a activity during mitotic spindle assembly requires RHAMM to correctly localize TPX2. Cell Cycle (2014) 13(14):2248–61. doi: 10.4161/cc.29270
29. Hamilton SR, Fard SF, Paiwand FF, Tolg C, Veiseh M, Wang C, et al. The hyaluronan receptors CD44 and rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J Biol Chem (2007) 282(22):16667–80. doi: 10.1074/jbc.M702078200
30. Carvalho AM, Costa Soares D, Paulo PMR, Reis RL, Pashkuleva I, et al. Co-Localization and crosstalk between CD44 and RHAMM depend on hyaluronan presentation. Acta Biomater (2021) 119:114–24. doi: 10.1016/j.actbio.2020.10.024
31. Zhang W, Chang MCY, Zylka D, Turley S, Harrison R, Turley EA, et al. The hyaluronan receptor RHAMM regulates extracellular-regulated kinase. J Of Biol Chem (1998) 273(18):11342–8. doi: 10.1074/jbc.273.18.11342
32. Maxwell CA, Benı´tez J, Go´ L, Osorio A, Bonifaci N, Ferna´ndez-Ramires F, et al, et al. Interplay between BRCA1 and RHAMM regulates epithelial apicobasal polarization and may influence risk of breast cancer. PLoS Biol (2011) 9(11):1–18. doi: 10.1371/journal.pbio.1001199
33. Blanco I, Kuchenbaecker K., Cuadras D., Wang X., Barrowdale D., Garibay G.R., et al. Assessing associations between the AURKA-HMMR-TPX2-TUBG1 functional module and breast cancer risk in BRCA1/2 mutation carriers. PLoS One (2015) 10(4):e0120020. doi: 10.1371/journal.pone.0120020
34. Li H, Kroll T, Moll J, Frappart L, Herrlich P, Heuer H, et al. Spindle misorientation of cerebral and cerebellar progenitors is a mechanistic cause of megalencephaly. Stem Cell Rep (2017) 9(4):1071–80. doi: 10.1016/j.stemcr.2017.08.013
35. Cui Z, Liao J, Cheong N, Longoria C, Cao G, DeLisser H.M, et al. The receptor for hyaluronan-mediated motility (CD168) promotes inflammation and fibrosis after acute lung injury. Matrix Biol J Int Soc Matrix Biol (2019) 78–9:255–71. doi: 10.1016/j.matbio.2018.08.002
36. Tolg C, Hamilton S.R, Nakrieko K.A, Kooshesh F, Walton P, McCarthy J.B, et al. Rhamm-/- fibroblasts are defective in CD44-mediated ERK1,2 motogenic signaling, leading to defective skin wound repair. J Cell Biol (2006) 175(6):1017–28. doi: 10.1083/jcb.200511027
37. He Z, Mei L, Connell M, Maxwell CA. Hyaluronan mediated motility receptor (HMMR) encodes an evolutionarily conserved homeostasis, mitosis, and meiosis regulator rather than a hyaluronan receptor. Cells (2020) 9(4):1–14. doi: 10.3390/cells9040819
38. Greiner J, Ringhoffer M, Taniguchi M, Hauser T, Schmitt A, Dohner H, et al. Characterization of several leukemia-associated antigens inducing humoral immune responses in acute and chronic myeloid leukemia. Int J Cancer (2003) 106(2):224–31. doi: 10.1002/ijc.11200
39. Turley EA, Torrance J. Localization of hyaluronate and hyaluronate-binding protein on motile and non-motile fibroblasts. Exp Cell Res (1985) 161(1):17–28. doi: 10.1016/0014-4827(85)90486-0
40. Menko AS, Romisher A, Walker JL. The pro-fibrotic response of mesenchymal leader cells to lens wounding involves hyaluronic acid, its receptor RHAMM, and vimentin. Front Cell Dev Biol (2022) 10:862423. doi: 10.3389/fcell.2022.862423
41. Liu M, Tolg C, Turley E. Dissecting the dual nature of hyaluronan in the tumor microenvironment. Front Immunol (2019) 10:947. doi: 10.3389/fimmu.2019.00947
42. Maxwell CA, McCarthy J, Turley E. Cell-surface and mitotic-spindle RHAMM: moonlighting or dual oncogenic functions? J Cell Sci (2008) 121(Pt 7):925–32. doi: 10.1242/jcs.022038
43. Radisky DC, Hirai Y, Bissell MJ. Delivering the message: epimorphin and mammary epithelial morphogenesis. Trends Cell Biol (2003) 13(8):426–34. doi: 10.1016/s0962-8924(03)00146-6
44. Manzanares D, Monzon ME, Savani RC, Salathe M. Apical oxidative hyaluronan degradation stimulates airway ciliary beating via RHAMM and RON. Am J Respir Cell Mol Biol (2007) 37(2):160–8. doi: 10.1165/rcmb.2006-0413OC
45. Samuel SK, Hurta RAR, Spearman MA, Wright JA, Turley EA, Greenberg AH. TGF-31 stimulation of cell locomotion utilizes the hyaluronan receptor RHAMM and hyaluronan. J Cell Biol (1993) 123:749–58. doi: 10.1083/jcb.123.3.749
46. Kouvidi K, Berdiaki A, Nikitovic D, Katonis P, Afratis N, Hascall VC, et al. Role of receptor for hyaluronic acid-mediated motility (RHAMM) in low molecular weight hyaluronan (LMWHA)-mediated fibrosarcoma cell adhesion. J Biol Chem (2011) 286(44):38509–20. doi: 10.1074/jbc.M111.275875
47. Hall CL, Wang C, Lange LA, Turley EA. Hyaluronan and the hyaluronan receptor RHAMM promote focal adhesion turnover and transient tyrosine kinase activity. J Cell Biol (1994) 126(2):575–88. doi: 10.1083/jcb.126.2.575
48. Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, et al. Interplay of mevalonate and hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci U S A (2014) 111(1):E89–98. doi: 10.1073/pnas.1319190110
49. Lin SL, Chang D, Chiang A, Ying SY. Androgen receptor regulates CD168 expression and signaling in prostate cancer. Carcinogenesis (2008) 29(2):282–90. doi: 10.1093/carcin/bgm259
50. Thangavel C, Boopathi E, Liu Y, Haber A, Ertel A, Bhardwaj A, et al. RB loss promotes prostate cancer metastasis. Cancer Res (2017) 77(4):982–95. doi: 10.1158/0008-5472.CAN-16-1589
51. Kouvidi K, Berdiaki A, Tzardi M, Karousou E, Passi A, Nikitovic D, et al. Receptor for hyaluronic acid- mediated motility (RHAMM) regulates HT1080 fibrosarcoma cell proliferation via a beta-catenin/c-myc signaling axis. Biochim Biophys Acta (2016) 1860(4):814–24. doi: 10.1016/j.bbagen.2016.01.019
52. Maxwell CA, Keats JJ, Belch AR, Pilarski LM, Reiman T. Receptor for hyaluronan-mediated motility correlates with centrosome abnormalities in multiple myeloma and maintains mitotic integrity. Cancer Res (2005) 65(3):850–60. doi: 10.1158/0008-5472.850.65.3
53. Entwistle J, Hall CL, Turley EA. HA receptors: Regulators of signalling to the cytoskeleton. J Cell Biochem (1999) 61:569–77. doi: 10.1002/(sici)1097-4644(19960616)61:4<569::aid-jcb10>3.0.co;2-b
54. Joukov V, Groen AC, Prokhorova T, Gerson R, White E, Rodriguez A, et al. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell (2006) 127(3):539–52. doi: 10.1016/j.cell.2006.08.053
55. Shinohara M, Mikhailov AV, Aguirre-Ghiso JA, Rieder C.L. Extracellular signal-regulated kinase 1/2 activity is not required in mammalian cells during late G2 for timely entry into or exit from mitosis. Mol Biol Cell (2006) 17(12):5227–40. doi: 10.1091/mbc.e06-04-0284
56. Liu X, Yan S, Zhou T, Terada Y, Erikson RL. The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene (2004) 23(3):763–76. doi: 10.1038/sj.onc.1207188
57. Shapiro PS, Vaisberg E, Hunt AJ, Tolwinski NS, Whalen AM, McIntosh JR, et al. Activation of the MKK/ERK pathway during somatic cell mitosis: Direct interactions of active ERK with kinetochores and regulation of the mitotic 3F3/2 phosphoantigen. J Cell Biol (1998) 142(6):1533–45. doi: 10.1083/jcb.142.6.1533
58. Roberts EC, Hammond K, Traish AM, Resing KA, Ahn NG. Identification of G2/M targets for the MAP kinase pathway by functional proteomics. Proteomics (2006) 6(16):4541–53. doi: 10.1002/pmic.200600365
59. Meier C, Spitschak A, Abshagen K, Gupta S, Mor JM, Wolkenhauer O, et al. Association of RHAMM with E2F1 promotes tumour cell extravasation by transcriptional up-regulation of fibronectin. J Pathol (2014) 234(3):351–64. doi: 10.1002/path.4400
60. Tolg C, Liu M, Cousteils K, Telmer P, Alam K, Ma J, et al. Cell-specific expression of the transcriptional regulator RHAMM provides a timing mechanism that controls appropriate wound re-epithelialization. J Biol Chem (2020) 295(16):5427–48. doi: 10.1074/jbc.RA119.010002
61. Evanko SP, Wight TN. Intracellular localization of hyaluronan in proliferating cells. J Histochem Cytochem (1999) 47(10):1331–42. doi: 10.1177/002215549904701013
62. Hatano H, Shigeishi H, Kudo Y, Higashikawa K, Tobiume K, Takata T, et al. RHAMM/ERK interaction induces proliferative activities of cementifying fibroma cells through a mechanism based on the CD44-EGFR. Lab Invest (2011) 91(3):379–91. doi: 10.1038/labinvest.2010.176
63. Wang C, Thor AD, Moore DH, Zhao Y, Kerschmann R, Stern R, et al. The overexpression of RHAMM, a hyaluronan-binding protein that regulates ras signaling, correlates with overexpression of mitogen-activated protein kinase and is a significant parameter in breast cancer progression. Clin Cancer Res (1998) 4:567–76.
64. Hall CL, Yang S, Yang X, Zhang S, Turley M, Samuel S, et al. Overexpression of the hyaluronan receptor RHAMM is transforming and is also required for h-ras transformation. Cell (1995) 62:19–28. doi: 10.1016/0092-8674(95)90048-9
65. Lin SL, Chang D, Ying SY. Hyaluronan stimulates transformation of androgen-independent prostate cancer. Carcinogenesis (2007) 28(2):310–20. doi: 10.1093/carcin/bgl134
66. Yeh MH, Tzeng YJ, Fu TY, You JJ, Chang HT, Ger LP, et al. Extracellular matrix-receptor interaction signaling genes associated with inferior breast cancer survival. Anticancer Res (2018) 38(8):4593–605. doi: 10.21873/anticanres.12764
67. Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet (2007) 39(11):1338–49. doi: 10.1038/ng.2007.2
68. Gust KM, Hofer MD, Perner SR, Kim R, Chinnaiyan AM, Varambally S, et al. RHAMM (CD168) is overexpressed at the protein level and may constitute an immunogenic antigen in advanced prostate cancer disease. Neoplasia (2009) 11(9):956–63. doi: 10.1593/neo.09694
69. He R, Zuo S. A robust 8-gene prognostic signature for early-stage non-small cell lung cancer. Front Oncol (2019) 9:693. doi: 10.3389/fonc.2019.00693
70. Cheng XB, Sato N, Kohi S, Koga A, Hirata K. Receptor for hyaluronic acid-mediated motility is associated with poor survival in pancreatic ductal adenocarcinoma. J Cancer (2015) 6(11):1093–8. doi: 10.7150/jca.12990
71. Tilghman J, Wu H, Sang Y, Shi X, Guerrero-Cazares H, Quinones-Hinojosa A, et al. HMMR maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res (2014) 74(11):3168–79. doi: 10.1158/0008-5472.CAN-13-2103
72. He Z, Kannan N, Nemirovsky O, Chen H, Connell M, Taylor B, et al. BRCA1 controls the cell division axis and governs ploidy and phenotype in human mammary cells. Oncotarget (2017) 8(20):32461–75. doi: 10.18632/oncotarget.15688
73. Wang J, Li D, Shen W, Sun W, Gao R, Jiang P, et al. RHAMM inhibits cell migration via the AKT/GSK3β/Snail axis in luminal a subtype breast cancer. Anat Rec (Hoboken) (2020) 303(9):2344–56. doi: 10.1002/ar.24321
74. Greiner J, Schmitt A, Giannopoulos K, Rojewski M.T, Gotz M, Funk I, et al. High-dose RHAMM-R3 peptide vaccination for patients with acute myeloid leukemia, myelodysplastic syndrome and multiple myeloma. Haematologica (2010) 95(7):1191–7. doi: 10.3324/haematol.2009.014704
75. Greiner J, Li L, Ringhoffer M, Barth TF, Giannopoulos K, Guillaume P, et al. Identification and characterization of epitopes of the receptor for hyaluronic acid-mediated motility (RHAMM/CD168) recognized by CD8+ T cells of HLA-A2-positive patients with acute myeloid leukemia. Blood (2005) 106(3):938–45. doi: 10.1182/blood-2004-12-4787
76. Crainie M, Belch AR, Mant MJ, Pilarski LM. Overexpression of the receptor for hyaluronan-mediated motility (RHAMM) characterizes the malignant clone in multiple myeloma: Identification of three distinct RHAMM variants. Blood (1999) 93(5):1684–96. doi: 10.1182/blood.V93.5.1684
77. Schmitt M, Hückelhoven AG, Hundemer M, Schmitt A, Lipp S, Emde M, et al. Frequency of expression and generation of T-cell responses against antigens on multiple myeloma cells in patients included in the GMMG-MM5 trial. oncotarget (2017) 8(No. 49):84847–62.
78. Korkes F, Castro MG, Zequi Cassio S, Nardi L, Giglio Del A, Pompeo Lima AC. Hyaluronan-mediated motility receptor (RHAMM) immunohistochemical expression and androgen deprivation in normal peritumoral, hyperplasic and neoplastic prostate tissue. BJU Int (2014) 113(5):822–9. doi: 10.1111/bju.12339
79. Gomez CR, Kosari F, Munz JM, Schreiber CA, Knutson GJ, Ida CM, et al. Prognostic value of discs large homolog 7 transcript levels in prostate cancer. PLoS One (2013) 8(12):e82833. doi: 10.1371/journal.pone.0082833
80. Chen F, Zhu XQ, Zheng J, Xu TT, Wu K, Ru CH. RHAMM regulates the growth and migration of lung adenocarcinoma A549 cell line by regulating Cdc2/CyclinB1 and MMP9 genes. Math Biosci Eng (2020) 17(3):2150–63. doi: 10.3934/mbe.2020114
81. Wang K, Zhang T. Prognostic significance of CD168 overexpression in colorectal cancer. Oncol Lett (2016) 12(4):2555–9. doi: 10.3892/ol.2016.4974
82. Xiao Y, Feng M, Ran H, Han X, Li X, et al. Identification of key differentially expressed genes associated with nonsmall cell lung cancer by bioinformatics analyses. Mol Med Rep (2018) 17(5):6379–86. doi: 10.3892/mmr.2018.8726
83. Li Z, Sang M, Tian Z, Liu Z, Lv J, Zhang F, et al. Identification of key biomarkers and potential molecular mechanisms in lung cancer by bioinformatics analysis. Oncol Lett (2019) 18(5):4429–40. doi: 10.3892/ol.2019.10796
84. Stevens LE, Cheung WKC, Adua SJ, Arnal-Estape A, Zhao M, Liu Z, et al. Extracellular matrix receptor expression in subtypes of lung adenocarcinoma potentiates outgrowth of micrometastases. Cancer Res (2017) 77(8):1905–17. doi: 10.1158/0008-5472.CAN-16-1978
85. Li X, Zuo H, Zhang L, Sun Q, Xin Y, Zhang L. Validating HMMR expression and its prognostic significance in lung adenocarcinoma based on data mining and bioinformatics methods. Front Oncol (2021) 11:720302. doi: 10.3389/fonc.2021.720302
86. Yang D, Ma Y, Zhao P, Ma J, He C. Systematic screening of protein-coding gene expression identified HMMR as a potential independent indicator of unfavorable survival in patients with papillary muscle-invasive bladder cancer. BioMed Pharmacother (2019) 120:109433. doi: 10.1016/j.biopha.2019.109433
87. Niedworok C, Kretschmer I, Rock K, Dorp Vom F, Szarvas T, Hess J, et al. The impact of the receptor of hyaluronan-mediated motility (RHAMM) on human urothelial transitional cell cancer of the bladder. PLoS One (2013) 8(9):e75681. doi: 10.1371/journal.pone.0075681
88. Morera DS, Hennig MS, Talukder A, Lokeshwar SD, Wang J, Garcia-Roig M, et al. Hyaluronic acid family in bladder cancer: Potential prognostic biomarkers and therapeutic targets. Br J Cancer (2017) 117(10):1507–17. doi: 10.1038/bjc.2017.318
89. Wang Y, Chen L, Ju L, Qian K, Liu X, Wang X, et al. Novel biomarkers associated with progression and prognosis of bladder cancer identified by Co-expression analysis. Front Oncol (2019) 9:1030. doi: 10.3389/fonc.2019.01030
90. Burren S, Reche K, Blank A, Galvàn J.A, Dawson H, Berger MD, et al. RHAMM in liver metastases of stage IV colorectal cancer with mismatch-repair proficient status correlates with tumor budding, cytotoxic T-cells and PD-1/PD-L1. Pathology-Res Pract (2021) 223:153486. doi: 10.1016/j.prp.2021.153486
91. Tang YP, Yin YX, Xie MZ, Liang XQ, Li JL, Li KZ, et al. Systematic analysis of the clinical significance of hyaluronan-mediated motility receptor in colorectal cancer. Front Mol Biosci (2021) 8:733271. doi: 10.3389/fmolb.2021.733271
92. Koelzer VH, et al. Expression of the hyaluronan-mediated motility receptor RHAMM in tumor budding cells identifies aggressive colorectal cancers. Hum Pathol (2015) 46(11):1573–81. doi: 10.1016/j.humpath.2015.07.010
93. Zlobec I, Terracciano L, Tornillo L, Gunthert U, Vuong T, Jass JR, et al. Role of RHAMM within the hierarchy of well-established prognostic factors in colorectal cancer. Gut (2008) 57(10):1413–9. doi: 10.1136/gut.2007.141192
94. Stangeland B, Mughal AA, Grieg Z, Sandberg CJ, Joel M, Nygård S, et al. Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget (2015) 6(28):26192. doi: 10.18632/oncotarget.4613
95. Hartheimer JS, Park S, Rao SS, Kim Y. Targeting hyaluronan interactions for glioblastoma stem cell therapy. Cancer Microenviron (2019) 12(1):47–56. doi: 10.1007/s12307-019-00224-2
96. Yang CW, Su JY, Tsou AP, Chau GY, Liu HL, Chen CH, et al. Integrative genomics based identification of potential human hepatocarcinogenesis-associated cell cycle regulators: RHAMM as an example. Biochem Biophys Res Commun (2005) 330(2):489–97. doi: 10.1016/j.bbrc.2005.03.005
97. Mohapatra S, Yang X, Wright JA, Turley EA, Greenberg AH, et al. Soluble hyaluronan receptor RHAMM induces mitotic arrest by suppressing Cdc2 and cyclin B1 expression. J Exp Med (1996) 183(4):1663–8. doi: 10.1084/jem.183.4.1663
98. Sohr S, Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle (2008) 7(21):3448–60. doi: 10.4161/cc.7.21.7014
99. Tolg C, Poon R, Fodde R, Turley EA, Alman BA. Genetic deletion of receptor for hyaluronan-mediated motility (Rhamm) attenuates the formation of aggressive fibromatosis (desmoid tumor). Oncogene (2003) 22(44):6873–82. doi: 10.1038/sj.onc.1206811
100. Guo L, Mohd KS, Ren H, Xin G, Jiang Q, Clarke PR, et al. Phosphorylation of importin-alpha1 by CDK1-cyclin B1 controls mitotic spindle assembly. J Cell Sci (2019) 132(18):1–12. doi: 10.1242/jcs.232314
101. Groen AC, Cameron LA, Coughlin M, Miyamoto DT, Mitchison TJ, Ohi R. XRHAMM functions in ran-dependent microtubule nucleation and pole formation during anastral spindle assembly. Curr Biol (2004) 14(20):1801–11. doi: 10.1016/j.cub.2004.10.002
102. Mohan P, Castellsague J, Jiang J, Allen K, Chen H, Nemirovsky O, et al. Genomic imbalance of HMMR/RHAMM regulates the sensitivity and response of malignant peripheral nerve sheath tumour cells to aurora kinase inhibition. Oncotarget (2013) 4(1):80–93. doi: 10.18632/oncotarget.793
103. Sankaran S, Crone DE, Palazzo RE, Parvin JD. Aurora-a kinase regulates breast cancer associated gene 1 inhibition of centrosome-dependent microtubule nucleation. Cancer Res (2007) 67(23):11186–94. doi: 10.1158/0008-5472.CAN-07-2578
104. Mateo F, He Z, Mei L, Garibay GR, Herranz C, García N, et al. Modification of BRCA1-associated breast cancer risk by HMMR overexpression. Nat Commun (2022) 13(1):1895. doi: 10.1038/s41467-022-29335-z
105. Tanimura S, Takeda K. ERK signalling as a regulator of cell motility. J Biochem (2017) 162(3):145–54. doi: 10.1093/jb/mvx048
106. Mousson A, Legrand M, Steffan T, Vauchelles R, Carl P, Gies JP, et al. Inhibiting FAK-paxillin interaction reduces migration and invadopodia-mediated matrix degradation in metastatic melanoma cells. Cancers (Basel) (2021) 13(8):1–20. doi: 10.3390/cancers13081871
107. Hu YL, Lu S, Szeto KW, Sun J, Wang Y, Lasheras JC, et al. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci Rep (2014) 4:6024. doi: 10.1038/srep06024
108. Tolg C, Hamilton SR, Zalinska E, McCulloch L, Amin R, Akentieva N, et al. A RHAMM mimetic peptide blocks hyaluronan signaling and reduces inflammation and fibrogenesis in excisional skin wounds. Am J Pathol (2012) 181(4):1250–70. doi: 10.1016/j.ajpath.2012.06.036
109. Amara FM, Entwistle J, Kuschak TI, Turley EA, Wright JA. Transforming growth factor-b1 stimulates multiple protein interactions at a unique cis-element in the 3*-untranslated region of the hyaluronan receptor RHAMM mRNA. J Of Biol Chem (1996) 271(25):15279–84. doi: 10.1074/jbc.271.25.15279
110. Ye S, Liu Y, Fuller AM, Katti R, Ciotti GE, Chor S, et al. TGFbeta and hippo pathways cooperate to enhance sarcomagenesis and metastasis through the hyaluronan-mediated motility receptor (HMMR). Mol Cancer Res (2020) 18(4):560–73. doi: 10.1158/1541-7786.MCR-19-0877
111. Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res (2010) 8(5):629–42. doi: 10.1158/1541-7786.MCR-10-0139
112. Devreotes P, Horwitz AR. Signaling networks that regulate cell migration. Cold Spring Harb Perspect Biol (2015) 7(8):a005959. doi: 10.1101/cshperspect.a005959
113. Turley EA, Austen L, Moore D, Hoare K. Ras transformed cells express both CD44 and RHAMM hyaluran receptor: Only RHAMM is essensial for hyaluronan-promoted locomotion. Exp Cell Res (1993) 207:277–82. doi: 10.1006/excr.1993.1194
114. Carvalho AM, Costa Soares D, Reis RL, Pashkuleva I. RHAMM expression tunes the response of breast cancer cell lines to hyaluronan. Acta Biomater (2022). doi: 10.1016/j.actbio.2022.05.013
115. Park D, Kim Y, Kim H, Kim K, Lee YS, Choe J, et al. Hyaluronic acid promotes angiogenesis by inducing RHAMM-TGFbeta receptor interaction via CD44-PKCdelta. Mol Cells (2012) 33(6):563–74. doi: 10.1007/s10059-012-2294-1
116. Sun Y, Li Z, Song K. AR-mTOR-SRF axis regulates HMMR expression in human prostate cancer cells. Biomol Ther (Seoul) (2021) 29(6):667–77. doi: 10.4062/biomolther.2021.040
117. Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin d proteins. Cancer Res (2006) 66(15):7783–92. doi: 10.1158/0008-5472.Can-05-4472
118. Zhang J, Wu D, He Y, Li L, Liu S, Lu J, et al. Rapamycin inhibits AR signaling pathway in prostate cancer by interacting with the FK1 domain of FKBP51. Biochem Biophys Rep (2020) 23:100778. doi: 10.1016/j.bbrep.2020.100778
119. Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ (2018) 25(1):114–32. doi: 10.1038/cdd.2017.172
120. Yang HS, Zhang DM, Deng HX, Peng F, Wei YQ. Antitumor and anti-angiogenesis immunity induced by CR-SEREX-identified xenopus RHAMM. Cancer Sci (2010) 101(4):862–8. doi: 10.1111/j.1349-7006.2009.01473.x
121. Amano T, Kajiwara K, Yoshikawa K, Morioka J, Nomura S, Fujisawa H, et al. Antitumor effects of vaccination with dendritic cells transfected with modified receptor for hyaluronan-mediated motility mRNA in a mouse glioma model. J Neurosurgry (2007) 106:638–45. doi: 10.3171/jns.2007.106.4.638
122. Giannopoulos K, Dmoszynska A, Kowal M, Rolinski J, Gostick E, Price DA, et al. Peptide vaccination elicits leukemia-associated antigen-specific cytotoxic CD8+ T-cell responses in patients with chronic lymphocytic leukemia. Leukemia (2010) 24(4):798–805. doi: 10.1038/leu.2010.29
123. Casalegno-Garduno R, Meier C, Schmitt A, Spitschak A, Hilgendorf I, Rohde S, et al. Immune responses to RHAMM in patients with acute myeloid leukemia after chemotherapy and allogeneic stem cell transplantation. Clin Dev Immunol (2012) 2012:146463. doi: 10.1155/2012/146463
124. Snauwaert S, Vanhee S, Goetgeluk G, Verstichel G, Caeneghem Van Y, Velghe I, et al. RHAMM/HMMR (CD168) is not an ideal target antigen for immunotherapy of acute myeloid leukemia. Haematologica (2012) 97(10):1539–47. doi: 10.3324/haematol.2012.065581
125. Paston SJ, Brentville V.A, Symonds P, Durrant L.G. Cancer vaccines, adjuvants, and delivery systems. Front Immunol (2021) 12:627932. doi: 10.3389/fimmu.2021.627932
126. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines (2019) 4(1):7. doi: 10.1038/s41541-019-0103-y
127. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun (2020) 11(1):3801. doi: 10.1038/s41467-020-17670-y
128. Akent'eva NP, Shushanov SS, Kotel'nikov AI. Effects of RHAMM/HMMR-selective peptides on survival of breast cancer cells. Bull Exp Biol Med (2015) 159(5):658–61. doi: 10.1007/s10517-015-3041-3
129. Esguerra KV, Tolg C, Akentieva N, Price M, Cho CF, Lewis JD, et al. Identification, design and synthesis of tubulin-derived peptides as novel hyaluronan mimetic ligands for the receptor for hyaluronan-mediated motility (RHAMM/HMMR). Integr Biol (Camb) (2015) 7(12):1547–60. doi: 10.1039/c5ib00222b
130. Marqus S, Pirogova E, Piva TJ. Evaluation of the use of therapeutic peptides for cancer treatment. J Biomed Sci (2017) 24(1):21. doi: 10.1186/s12929-017-0328-x
131. Mabonga L, Kappo AP. Peptidomimetics: A synthetic tool for inhibiting protein–protein interactions in cancer. Int J Pept Res Ther (2020) 26(1):225–41. doi: 10.1007/s10989-019-09831-5
132. An G, Park S, Lee M, Lim W, Song G. Antiproliferative effect of 4-methylumbelliferone in epithelial ovarian cancer cells is mediated by disruption of intracellular homeostasis and regulation of PI3K/AKT and MAPK signaling. Pharmaceutics (2020) 12(7):1–14. doi: 10.3390/pharmaceutics12070640
133. Lokman NA, Price ZK, Hawkins EK, Macpherson AM, Oehler MK, Ricciardelli C. 4-methylumbelliferone inhibits cancer stem cell activation and overcomes chemoresistance in ovarian cancer. Cancers (Basel) (2019) 11(8). doi: 10.3390/cancers11081187
134. Benitez A, Yates TJ, Lopez LE, Cerwinka WH, Bakkar A, Lokeshwar VB, et al. Targeting hyaluronidase for cancer therapy: Antitumor activity of sulfated hyaluronic acid in prostate cancer cells. Cancer Res (2011) 71(12):4085–95. doi: 10.1158/0008-5472.CAN-10-4610
Keywords: cancer, RHAMM, migration, signaling, microtubules
Citation: Hinneh JA, Gillis JL, Moore NL, Butler LM and Centenera MM (2022) The role of RHAMM in cancer: Exposing novel therapeutic vulnerabilities. Front. Oncol. 12:982231. doi: 10.3389/fonc.2022.982231
Received: 30 June 2022; Accepted: 18 July 2022;
Published: 10 August 2022.
Edited by:
Matiullah Khan, AIMST University, MalaysiaReviewed by:
Sara Pedron, University of Illinois at Urbana-Champaign, United StatesDemitrios Vynios, University of Patras, Greece
Copyright © 2022 Hinneh, Gillis, Moore, Butler and Centenera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa M. Butler, TGlzYS5idXRsZXJAYWRlbGFpZGUuZWR1LmF1; Margaret M. Centenera, TWFyZ2FyZXQuY2VudGVuZXJhQGFkZWxhaWRlLmVkdS5hdQ==