 Shenghua Zhuo1†
Shenghua Zhuo1† Liangwang Yang1†
Liangwang Yang1† Shenbo Chen1†
Shenbo Chen1† Caiying Tang1
Caiying Tang1 Weicheng Li1Zhenzhong Gao1*
Weicheng Li1Zhenzhong Gao1* Jigao Feng2*
Jigao Feng2* Kun Yang1*
Kun Yang1*- 1Department of Neurosurgery, First Affiliated Hospital of Hainan Medical University, Haikou, China
- 2Department of Neurosurgery, Second Affiliated Hospital of Hainan Medical University, Haikou, China
It is widely thought that the tumor microenvironment (TME) provides the “soil” for malignant tumors to survive. Prior to metastasis, the interaction at the host site between factors secreted by primary tumors, bone-marrow-derived cells, with stromal components initiates and establishes a pre-metastatic niche (PMN) characterized by immunosuppression, inflammation, angiogenesis and vascular permeability, as well as lymphangiogenesis, reprogramming and organotropism. Ferroptosis is a non-apoptotic cell death characterized by iron-dependent lipid peroxidation and metabolic constraints. Ferroptotic cancer cells release various signal molecules into the TME to either suppress or promote tumor progression. This review highlights the important role played by ferroptosis in PMN, focusing on the relationship between ferroptosis and PMN characteristics, and discusses future research directions.
Introduction
It is well-established that tumor cells spread from the primary to other single or multiple sites nearby or far away via blood vessels and lymphatic vessels by separation, migration, invasion and adhesion (1). Notably, tumor metastasis is difficult to cure once it occurs. Metastasis accounts for about 90% of all cancer-related mortality (2). Therefore, early detection, accurate diagnosis and personalized treatment strategies before the onset of metastasis are warranted to cure malignant tumors.
As early as 1889, British surgeon Stephen Paget put forward the “seed” and “soil” hypothesis, whereby cancer cells are analogous to the “seed” and metastatic sites to the “soil” (3). As the “soil” of tumor metastasis, the complex tumor microenvironment (TME) at the metastatic site is closely related to the malignant behavior of tumor cells. Primary tumor cells can create a microenvironment that promotes colonization and growth before metastasis. Prior to metastasis, the metastatic site forms a pre-metastatic niche (PMN) with immunosuppression, inflammation, angiogenesis and vascular permeability, as well as lymphangiogenesis, reprogramming and organotropism characteristics. Functionally, the PMN promotes tumor cell metastasis through four stages, namely priming, licensing, initiation, and progression, to facilitate tumor cell colonization and growth (4). During this spatial time course, tumor cells recruit tumor-mobilized bone marrow-derived cells and various inhibitory and regulatory immune cells by secreting multiple tumor-derived secreted factors (TDSFs) and extracellular vesicles (EVs). Moreover, they interact with the host matrix to form PMN characterized by immunosuppression and extracellular matrix (ECM) remodeling, a phenomenon that enables circulating tumor cells (CTCs) to not only evade the body’s surveillance and pursuit but also take root, sprout and grow in the “fertile soil” (5).
In 2012, researchers discovered that certain lethal compounds could induce a new mode of cell death. This type of cell death, termed ferroptosis, manifests as iron-dependent and reactive oxygen species (ROS) accumulation and is morphologically, genetically, and biochemically distinct from necrosis, apoptosis, and autophagy (6). An increasing body of evidence suggests that ferroptosis is not only closely linked to cancer-acquired drug resistance and immune evasion but also has dual effects (7, 8).
Epithelial-to-mesenchymal transition (EMT) in tumors refers to the dynamic changes in cellular organization from an epithelial to a mesenchymal phenotype, which cause functional changes in cell migration and invasion. Previous studies have shown that EMT plays an irreplaceable role in tumor metastasis (2), can also lead to metastatic spreading of tumors and confer drug resistance to clinical treatment (9). Interestingly, EMT signaling is closely related to ferroptosis (7). Ferroptosis induced via inhibition of the lipid peroxidase signaling pathway is a feature in therapy-resistant cancer cells of various mesenchymal cell states (10). This pathway, which centers around the metabolism of long-chain polyunsaturated fatty acids (PUFAs), is regulated by the EMT-inducible transcription factor zinc finger E-box homeobox 1 (ZEB1). Moreover, glutathione-dependent antioxidant enzyme glutathione peroxidase 4 (GPX4) dissipates reactive peroxides produced by the metabolism of PUFAs and prevents ferroptotic cell death (10). Combination treatment, comprising the ferroptosis inducers (FINs) beta-elemene and cetuximab have been reported to sensitize KRAS-mutated colorectal cancer cells by inhibiting EMT and lymph node (LN) metastasis and induce ferroptosis (11). Furthermore, epigenetic reprogramming of EMT is associated with ferroptosis (12). Although the role of ferroptosis in PMN is currently unclear, there appears to be a link between ferroptosis and PMN. This review focuses on the relationship between ferroptosis and PMN characteristics (including immunosuppression, inflammation, angiogenesis and vascular permeability, as well as lymphangiogenesis, reprogramming and organotropism), and highlights potential areas and directions for future research.
Immunosuppression
Cancer cells metastasize by evading immune surveillance. The primary tumor either forms TME or PMN through recruitment and expansion of immunosuppressive cell populations. It also transforms the phenotype and function of normal immune cells, from a potential immune response to a tumor-promoting state. Notably, tumor-promoting immune cells not only enhance immune evasion by suppressing antitumor immune responses, but also promote tumor cell invasion by inducing an immunosuppressive microenvironment, promoting EMT, and inducing vasculature at the primary tumor or metastatic site to establish PMN. Collectively, these phenomena eventually lead to tumor metastasis (13).
Cell death can be divided into two types, namely tolerogenic cell death (TCD) and immunogenic cell death (ICD), depending on whether an immune response is induced. In TCD, phagocytes efficiently eliminate cellular debris, via endocytosis, critical for preventing inflammatory and autoimmune diseases. ICD entails the release of intracellular molecules or they are exposed to dead or dying cells, a phenomenon that elicits adaptive immunity that subsequently induces immune responses to tumor-associated antigens (14). Among them, damage-associated molecular patterns (DAMPs) and cytokines released during cell death are critical for initiating immune responses (15, 16). The high mobility group box 1 protein (HMGB1), an essential protein required for cancer cell immunogenicity, is a crucial DAMP that promotes antigen presentation by dendritic cells (DCs) to T cells (17). Cancer cells with compound-induced ferroptosis were found to release HMGB1 in an autophagy-dependent manner (18). Remarkably, ferroptosis was recently identified as a type of ICD. Additional evidence showed that the GPX4 inhibitor RSL3 induced early ferroptotic cancer cells was ICD and was accompanied by the production of DAMPs, such as purine adenosine triphosphate (ATP) and HMGB1. Functionally, these components not only trigger in vitro maturation of DCs but also elicit vaccination-like effects in immunocompetent mice (19). In breast cancer, patrolling monocytes establish early interactions with metastatic tumor cells, clear tumor material in blood vessels, and promote recruitment as well as activation of natural killer cells (NK cells), thereby inhibiting tumor cells metastasis (20, 21). Studies have associated hypoxic features in the TME with PMN formation, while TDSFs generated by hypoxia in primary tumors can recruit immunosuppressive cells and reduce NK cells cytotoxicity in PMN (22). Peripheral blood mononuclear cells (PBMCs) have the potential to differentiate into various types of immune cells, under physiological and pathological conditions, while erastin (a FIN) plays an immunomodulatory function, where it promotes proliferation and differentiation of human PBMCs into B and NK cells (23). Another study showed that interferon γ (IFNγ), which is mainly produced by T lymphocytes or NK cells, sensitizes hepatocellular carcinoma cells to ferroptosis by activating the JAK/STAT signaling inhibitory system xc-activation, and promoting lipid peroxidation associated with mitochondrial damage (24). Interestingly, activation of CD8+ T cell activation has been documented to induce ferroptosis in cancer cells. Notably, activation of CD8+ T cells by nivolumab-based immunotherapy mediated downregulation of solute carrier family 3 membrane 2 (SLC3A2) and solute carrier family 7 membrane 11 (SLC7A11) by releasing IFNγ. This phenomenon inhibited uptake of cystine by tumor cells and promoted occurrence of lipid peroxidation and ferroptosis in cancer cells (25). IFNγ from CD8+ T cells and arachidonic acid (AA) from the tumor microenvironment mediate tumorigenic ferroptosis through acyl-CoA synthetase long-chain family member 4 (ACSL4) while targeting tumor ACSL4 reportedly improved immune checkpoint blockade treatment sensitivity (26). On the other hand, exposure to immune-activated cells to ferroptosis may impair their antitumor ability. Notably, conditional deletion of Gpx4 induced ferroptosis by lipid peroxidation in mouse T cells (27), while CD36 reportedly mediated AA uptake by CD8+ T cells, thereby causing lipid peroxidation and ferroptosis, as well as impairing the antitumor function of CD8+ T cells (28). DCs selectively induced by RSL3 undergo ferroptosis and cannot induce CD8+ T cells to produce IFNγ (29). Impaired toxicity of NK cells isolated from the ovarian cancer microenvironment and peripheral blood NK cells exposed to the tumor-derived ascites microenvironment showed cell morphology consistent with ferroptosis (30). Antitumor activity of NK cells could be restored by activation of the nuclear factor E2-related factor 2 (NRF2) antioxidant pathway (30, 31).
There is ample evidence that myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) suppress antitumor immune responses in PMN (32, 33). MDSCs can be induced to differentiate into TAMs under inflammatory stress and hypoxic niche (33). As optimal partners of tumor cells, MDSCs are a population of immature myeloid cells that emerge and accumulate in PMN, where they support tumor cell colonization into PMN during metastasis (34). Induction of ferroptosis in MDSCs may be an effective therapy to inhibit the accumulation of MDSCs during cancer immunotherapy (35). The chemotherapeutic drug gemcitabine (Gem), a FIN, can effectively deplete MDSCs. For instance, a previous study showed that Gem nanoparticles could promote anti-melanoma immunity, effectively deplete MDSCs and regulatory T cells (Tregs), and polarize TAMs to antitumor M1 phenotype, and also improve CD8+ T cell immune response, thereby inhibiting tumor growth (36). Furthermore, activating transcription factor 4 (ATF4) reportedly upregulated expression of heat-shock 70kDa protein 5 (HSPA5) in pancreatic ductal adenocarcinoma (37). HSPA5 binds to GPX4 and avoids GPX4 protein degradation and lipid peroxidation, thus limiting Gem’s anticancer activity. However, promoting ferroptosis was found to enhance Gem sensitivity (37). Similarly, combining FINs and apoptosis activators was shown to significantly improve the cytotoxic effect of Gem in pancreatic cancer (38). Notably, N-acylsphingosine amidohydrolase 2 (ASAH2) suppresses the p53 pathway to protect MDSCs from ferroptosis by destabilizing p53 protein in TME (35).
Macrophages are a vital factor in the progression of cancer metastasis, owing to their effect on PMN formation and CTCs adhesion, as well as extravasation, and colonization (39). Studies have shown that tumor-derived exosomes promote tumor metastasis by stimulating macrophage development in PMN to an immunosuppressive phenotype through glycolytic-dominant metabolic reprogramming (40). ICD induced triggered by FINs has been reported to polarize TAMs to a pro-immune antitumor phenotype, thereby enabling ferroptosis and immune regulation to act synergistically (41, 42). Additional evidence has shown that macrophages can efficiently clear ferroptotic cancer cells by phagocytosis (43), while ferric citrate effectively induces ferroptosis in macrophages (44). Furthermore, different subsets of macrophages have various sensitivities to ferroptosis. Particularly, the M1 type exhibits higher levels of resistance to drug-induced ferroptosis compares to the M2 type, while tumor suppressor M1-type macrophages are more resistant to FINs than tumor-promoting M2-type macrophages. This phenomenon may be attributed to the higher inducible nitric oxide synthase (iNOS) content in M1 than M2 macrophages. The lower iNOS content makes M2 macrophages produce less NO free radicals, a phenomenon that reduces their inhibitory effect on lipid peroxidation, thus rendering them sensitive to ferroptosis (45). Interestingly, different B cell subsets also differ in their sensitivity to ferroptosis. For instance, B1 and marginal zone B cells display higher lipid metabolism and are more sensitive to lipid peroxidation as well as ferroptosis compared to follicular B2 counterparts (46). Tumor-evoked regulatory B cells (tBregs), which are phenotypically similar to activated but less proliferative mature B2 cells, reportedly promote breast cancer metastasis by converting quiescent CD4+ T cells into Tregs (47). Moreover, tBregs can also directly activate the regulatory functions of MDSCs-derived monocytes and granulocyte subsets, while activated MDSCs have been shown to improve production of ROS and NO, and more effectively inhibit CD4+ T and CD8+ T cells, thereby promoting tumor metastasis (48). Tregs activation and suppression of antitumor immunity are maintained by GPX4 by preventing lipid peroxidation and ferroptosis (49). Taken together, findings from studies demonstrate that ferroptosis is closely associated with various immune factors in PMN, especially the complex crosstalk between ferroptosis and tumor cells and immune cells. The sensitivity of different immune cells and tumor types to FINs appears to determine the fate of PMN formation; nonetheless, further research explorations are needed to answer this question (Figure 1).
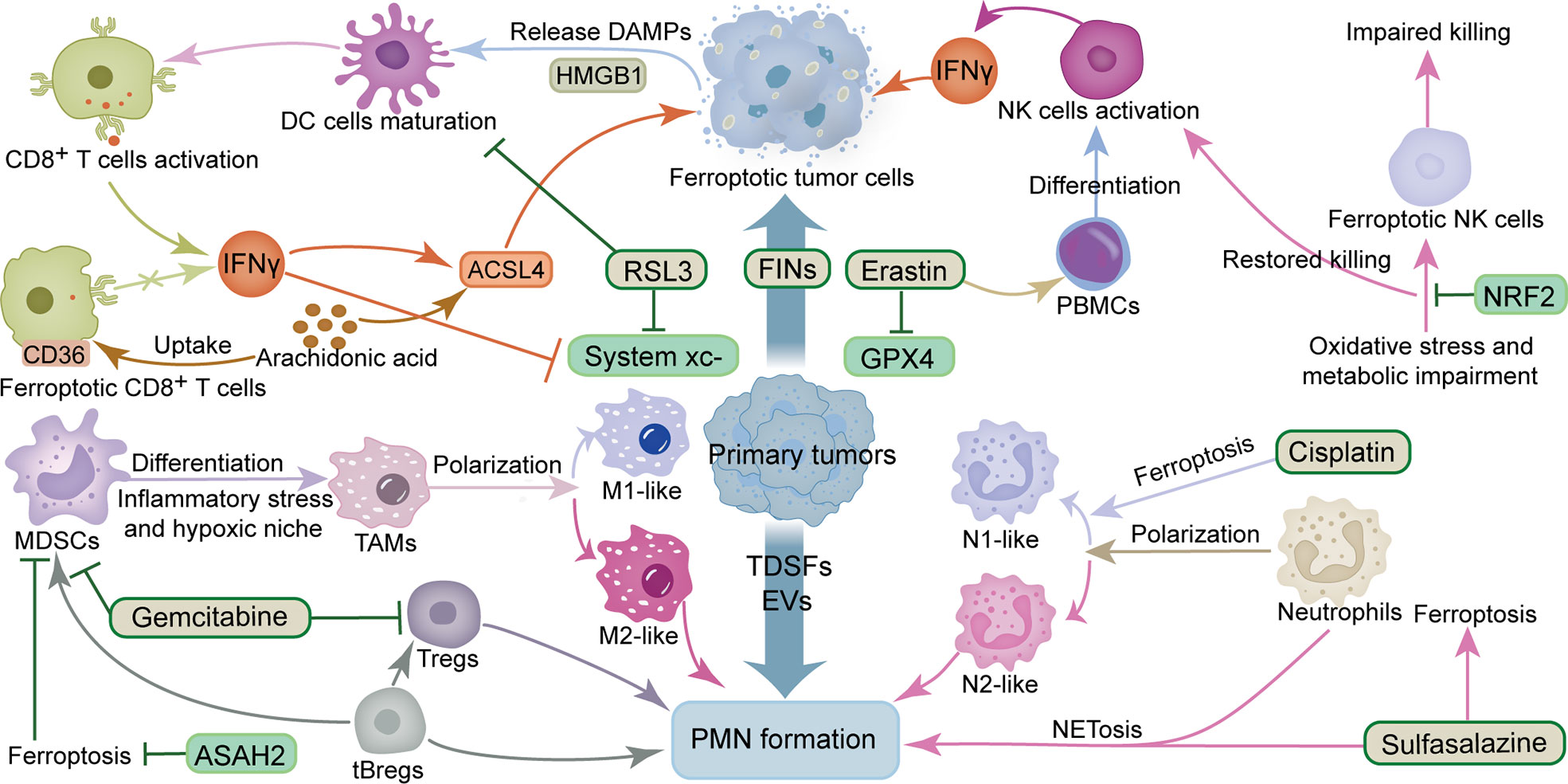
Figure 1 Crosstalk between ferroptosis and immunosuppression and inflammation in PMN. The ingredients that promote the formation of PMN include TDSFs, EVs, MDSCs, M2-like TAMs, tBregs, Tregs and N2-like neutrophils. The components that inhibit the formation of PMN include IFNγ, DC cells, NK cells, M1-like TAMs, N1-like neutrophils and CD8+ T cells. The ingredients that promote ferroptosis include RSL3, erastin, gemcitabine, sulfasalazine, cisplatin, IFNγ, ACSL4 and arachidonic acid. The components that inhibit ferroptosis include GPX4 and system xc-. ACSL4, acyl-CoA synthetase long-chain family member 4; ASAH2, N-acylsphingosine amidohydrolase 2; DAMPs, damage-associated molecular patterns; DC cells, dendritic cells; EVs, extracellular vesicles; FINs, ferroptosis inducers; GPX4, glutathione peroxidase 4; HMGB1, high mobility group box 1 protein; IFNγ, interferon γ; MDSCs, myeloid-derived suppressor cells; NK cells, natural killer cells; NRF2, nuclear factor E2-related factor 2; PBMCs, Peripheral blood mononuclear cells; PMN, pre-metastatic niche; TAMs, tumor-associated macrophages; tBregs, tumor-evoked regulatory B cells; TDSFs, tumor-derived secreted factors; Tregs, regulatory T cells.
Inflammation
Overwhelming evidence substantiates that chronic inflammation transforms susceptible cells into tumors, subsequently driving tumor development and metastasis (50, 51). Establishing an inflammatory microenvironment at the host site facilitates the seeding, survival and proliferation of CTCs in PMN (4). In addition, balancing ROS production and antioxidant defenses is critical for tumor cell survival and growth, owing to the fact that high ROS levels cause cytotoxicity. In contrast, low or moderate ROS levels have been implicated in DNA damage and mutation, as well as development of inflammatory responses and ultimately carcinogenesis (52). Accordingly, ferroptosis plays an essential role in the inflammatory process, representing a new therapeutic target in many inflammatory diseases (53, 54).
The crosstalk between chronic inflammation and various immune and inflammatory cells promotes tumor cell metastasis (55). In peri-tumor-associated inflammatory cells, GPX4 deletion in myeloid cells mediated an increase in ROS production, which was accompanied by secretion of excess H2O2, transforming intestinal epithelial cells (IECs) by triggering DNA mutations (56). H2O2 also induces secretion of chemokines and cytokines by IECs through the tumor necrosis factor α (TNFα) autocrine loop to recruit myeloid cells and promote tumor invasion (56). Neutrophils in TME can display opposing phenotypes: tumor-fighting effector cells (N1) and tumor-promoting effector cells (N2). Studies have shown that neutrophil recruitment is critical for PMN formation. In fact, neutrophils can spontaneously migrate during tumor progression (57). Chronic nicotine exposure has been shown to recruit tumor-promoting N2-type neutrophils, induce pulmonary PMN formation, and promote tumor cell colonization as well as metastatic growth (58). A previous study found that primary tumor-derived exosomes could activate lung epithelial cell Toll-like receptor 3 (TLR3) to initiate neutrophil recruitment and pulmonary PMN formation (59). An antitumor effect was observed in N1-polarized neutrophils induced by cisplatin-mediated non-small cell lung cancer ferroptosis (60). As found in another study, platinum prodrug nanoparticles induced ferroptosis by concurrent chemoradiotherapy to inhibit tumor recurrence and metastasis (61). Moreover, neutrophil infiltration in glioblastoma necrotic sites induces ferroptosis (62). There is ample evidence suggesting that sulfasalazine, an inhibitor of system xc-, can induce tumor ferroptosis (63, 64). Interestingly, sulfasalazine-induced accumulation of oxidized phospholipids in activated neutrophils could promote neutrophil extracellular traps-induced specific death mode NETosis (65, 66). Notably, NETosis is well-established to promote tumor growth and metastasis (67–69), suggesting that complex linkages between cell death pathways influence disease progression. Given the complex crosstalk between different forms of cell death (70), it is essential to fully understand and elucidate which form of cell death dominates in a given physiological or pathological setting.
Pro-inflammatory cytokines secreted by tumor cells or stromal cells, such as interleukin 6 (IL6), IL1β and C-C motif chemokine ligand 2 (CCL2), can induce phenotypic changes in tumor cells, recruit bone marrow-derived cells and form an inflammatory environment. Collectively, they constitute PMN and facilitate colonization of metastatic cells (71). Studies have shown that chronic inflammatory cytokines can also work synergistically with immunosuppressive cells, such as TAMs, to build an immunosuppressive microenvironment (72). Ferroptotic cells have been reported to induce aggregation and chemotaxis of macrophages through CCL2 and secrete cytokines, such as IL6 and IL1β, thereby triggering inflammation and enhancing tissue damage (73). In addition, ferroptosis-related genes play a crucial role in formation of inflammation, and ruptured ferroptotic cells can cause necrotizing inflammation and release pro-inflammatory DAMPs. Moreover, GPX4 controls lipoxygenase (LOX) and prostaglandin internalization through peroxide elasticity activity of prostaglandin-endoperoxide synthase enzymes involved in ferroptosis at multiple levels (74). As DAMPs, modulating HMGB1 activity in inflammation, immune response and tissue repair could generate invaluable treatment strategies for various diseases, including cancer (75). A previous study found that LPS could induce acetylation of HMGB1 in colon cancer cells, thereby promoting its interaction with GPX4, and regulating ROS levels and inflammation (76).
Macrophages are “gatekeepers” whose diversity and heterogeneity are critical in maintaining iron homeostasis (77). M1 macrophages, also known as inflammatory macrophages, can be activated by IFNγ, TNFα, HMGB1 and lipopolysaccharide (LPS), whereas M2 macrophages, also known as anti-inflammatory macrophages, can be activated by IL4, IL10, IL13, colony-stimulating factor 1 (CSF1) and transforming growth factor β (TGFβ) (78). Both have different phenotypes and functions (33). In contrast to the iron-chelating phenotype of inflammatory macrophages induced by pro-inflammatory cytokines and DAMPs, anti-inflammatory macrophages and lymphocytes exhibit an iron-releasing phenotype that not only contributes but also distributes iron in the TME (79). In lung cancer, iron-loaded TAMs enhance ROS production and pro-inflammatory cytokines (TNFα and IL6) to induce tumor cell death (80). Macrophage polarization is highly plastic, and its phenotype is primarily influenced by its current microenvironment, independent of the previous polarization state (81). TAMs actively release iron to the TME in hyper-inflamed tumors, thereby enriching the TME with iron and promoting both cancer development and immune escape. Notably, excess iron predisposes macrophages to a pro-inflammatory phenotype (79). Interestingly, M2 macrophages are more sensitive to ferroptosis (45). Therefore, harnessing the selective sensitization of macrophages to ferroptosis may improve a tumor’s inflammatory microenvironment. Given the complex crosstalk between ferroptosis and iron metabolism with tumor inflammation and immune cells, it is critical to investigate the impact of immune cell heterogeneity and plasticity, such as TAMs, on iron homeostasis as well as the balance in the inflammatory microenvironment. Inducing iron overload to promote ferroptosis and remodel the inflammatory microenvironment represents a potential therapeutic strategy to improve PMN formation (Figure 1).
Angiogenesis and vascular permeability
Multiple factors have been shown to affect angiogenesis in the TME, including various peritumoral cells, ECM, tumor metabolism, and tumor-derived extracellular vesicles (82). Increased angiogenesis and permeability in PMN can reportedly promote metastasis (4). At the pre-metastatic stage, primary tumors upregulate angiopoietin 2 (ANGPT2), matrix metalloproteinase 3 (MMP3) and MMP10 in the lung, increasing pulmonary vascular permeability and extravasation of CTCs, thereby promoting lung metastasis (83). A previous study found that TAMs-derived vascular endothelial growth factor (VEGF) improved vascular permeability, thereby promoting cancer cell invasion and metastasis (84). Another study showed that cancer-derived exosomal miR-25-3p participates in PMN formation and promotes colorectal cancer metastasis by inducing angiogenesis and permeability (85). Notably, ferroptosis has been closely associated with regulation of angiogenesis and permeability. As a crucial regulator of angiogenesis, ATF4 overexpression reportedly conferred resistance to sorafenib and erastin in glioma cells. Moreover, ATF4-induced angiogenesis was effectively inhibited by erastin and RSL3 (86), while another study demonstrated that GPX4 acts as an essential regulator of tumor angiogenesis and vascular maturation by controlling the activity of 12/15-LOX (87). Dysregulation of glutamate transport can enhance the function of Tregs and promote anti-VEGF treatment resistance in glioblastoma. Notably, blocking and eliminating Tregs using CD25 before anti-VEGF treatment reportedly restored production of IFNγ by CD8+ T cells, thereby improving the antitumor response of anti-VEGF therapy (88). Dihydroartemisinin, a semi-synthetic derivative of artemisinin, has been reported to exhibit antitumor activity in head and neck cancer cells by inhibiting angiogenesis, and also inducing ferroptosis and apoptosis (89). Apatinib, an anti-angiogenic drug for the treatment of metastatic gastric cancer, is a highly selective inhibitor of VEGFR2 that induces ferroptosis by reducing cellular glutathione levels and increasing levels of lipid peroxidation (90). Conversely, the application of erastin reportedly activated vascular endothelial cells to a leakier pro-metastatic phenotype, thereby promoting breast cancer cell adhesion and transendothelial migration (91). As the main characteristics of malignant tumor PMN, angiogenesis and vascular permeability represent essential steps that precede metastasis. Although FINs can inhibit angiogenesis, FINs may also increase the permeability of vascular endothelial cells, thereby triggering entry of tumor cells into blood vessels to facilitate hematogenous metastasis. To date, however, it is not known whether ferroptosis can alter this characteristic and interfere with PMN formation, necessitating further research explorations. In addition, the effects of FINs on this characteristic may be heterogeneous in different malignancies (Figure 2).
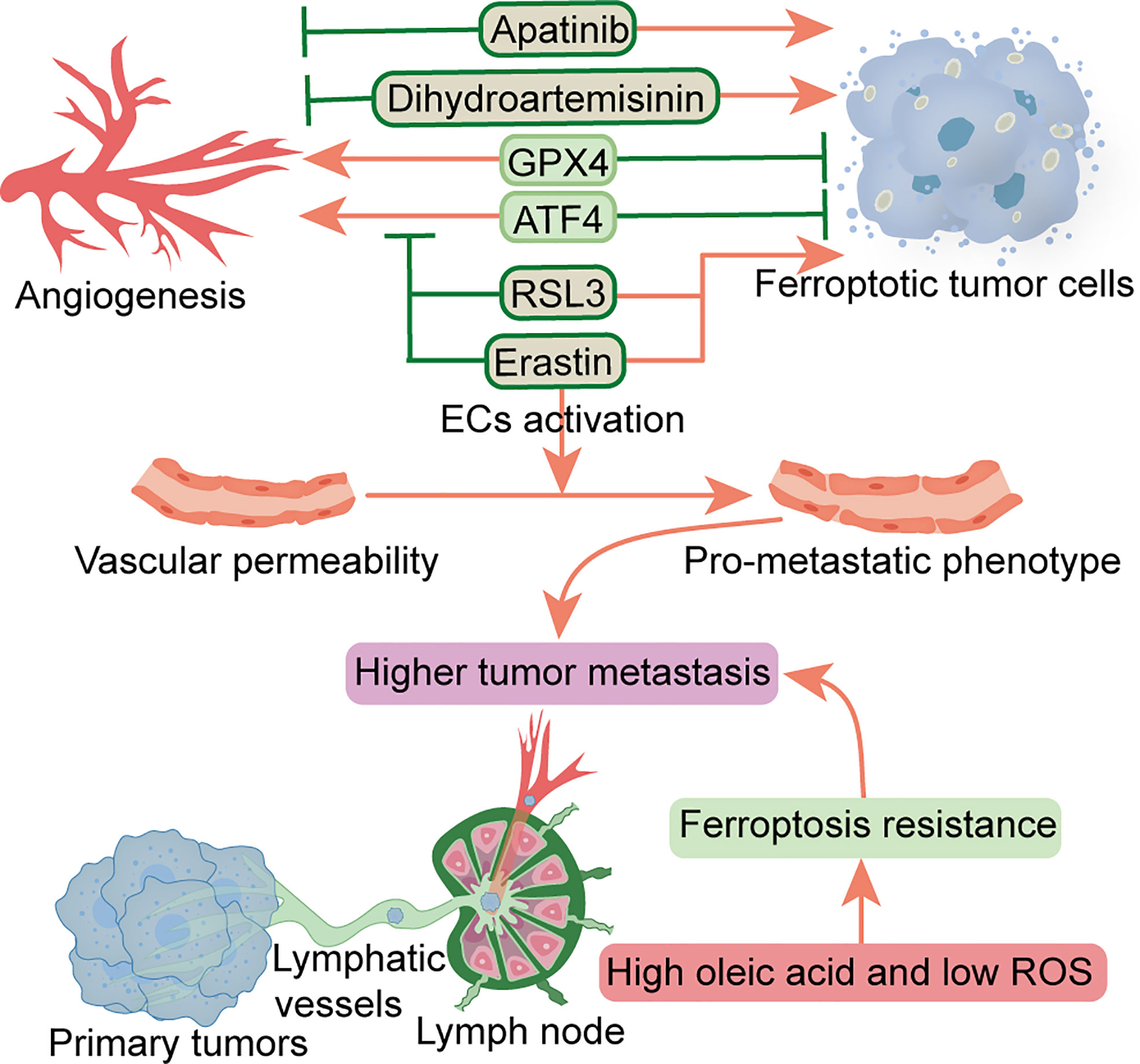
Figure 2 Relationship between ferroptosis and angiogenesis and vascular permeability and lymphangiogenesis. Factors that inhibit angiogenesis and promote ferroptosis include RSL3, erastin, dihydroartemisinin and apatinib. Erastin can also promote vascular permeability. ATF4 and GPX4 can promote angiogenesis and inhibit ferroptosis. Metastatic tumor cells escape ferroptosis through lymphatic vessels in low ROS and high oleic acid lymph node microenvironment. ATF4, transcription factor 4; ECs, endothelial cells; GPX4, glutathione peroxidase 4; ROS, reactive oxygen species.
Lymphangiogenesis
Tertiary lymphoid structures, composed of LNs and lymphatic vessels, are involved in PMN formation and tumor metastasis (92). Before metastatic spread, the primary tumor promotes lymphangiogenesis and high endothelial venule remodeling by secreting soluble factors or releasing EVs transported by lymphatic vessels, forming PMN in the LNs, leading to subsequent survival and growth of metastatic cancer cells (93). It has been established that lymphangiogenic growth factors VEGFC and VEGFD, secreted by a primary tumor, can promote lymphatic metastasis (94). VEGFD can down-regulate the 15-hydroxyprostaglandin dehydrogenase (HPGD) that degrades prostaglandins, and cause secretion of a large number of prostaglandins from the collecting lymphatic endothelial cells. Consequently, this promotes the entry of tumor cells into primary lymphatic vessels and induces collecting lymphatic vessels’ expansion to facilitate the transfer of tumor cells to distant metastases (94). Additional studies have shown that specific lymphoid tissue and immune response signatures contribute to PMN formation in sentinel LN in early-stage cervical cancer (95), while fatty acid oxidation (FAO) is a driver of LN metastasis (96). It is well-recognized that the LN microenvironment is rich in lipids, while fatty acids are a preferential energy source for LN metastatic tumor cells. Notably, LN metastasis requires tumor cells to undergo a metabolic transition to FAO (97). In pre-metastatic LN, melanoma-derived EVs fuse with subcapsular sinus CD169+ macrophages to avoid immune recognition, while the destruction of subcapsular sinus CD169+ macrophages allows EVs to enter the LN cortex to interact with B cells and activate pro-inflammatory cells tumor B cell immunity (98). Communication between EVs and LN was found to contribute to PMN formation and suppression of tumor immunity (99). Interestingly, melanoma cells experience less oxidative stress, owing to higher glutathione and oleic acid levels coupled with less free iron in the lymphatic microenvironment. Among them, tumor cells are protected from ferroptosis, a phenomenon that improves the ability to form metastatic tumors (100) (Figure 2). Additionally, as a vital feature of TME, the hypoxic environment affects the phenotype of various types of cells around the tumor cells (101). Moreover, angiogenesis and lymphangiogenesis are dependent on a hypoxic environment (102), while hypoxia reportedly improves resistance of malignant mesothelioma cells to ferroptosis (103). Similarly, studies have also shown that mitochondrial ferritin (FT) and FT heavy chain synergistically protect macrophages from RSL3-induced ferroptosis under hypoxic conditions (104). Therefore, the role of ferroptosis in angiogenesis and vascular permeability, as well as lymphangiogenesis in PMN warrants further study in the hypoxic environment. Future studies are expected to determine whether induction of ferroptosis can potentiate efficacy of anti-angiogenic or lymphangiogenic therapy for inhibition of tumor metastasis.
Reprogramming
Stromal and metabolic reprogramming has been associated with PMN-promoted tumor metastasis (4). Notably, tumor stroma not only promotes cancer growth, survival, and invasion but also modifies the behavior of stromal cells, including fibroblasts and immune cells, to drive metastasis and resist therapy (105). As an important part of TME stromal cells, cancer-associated fibroblasts (CAFs) are the “architects” of matrix remodeling and are closely related to the poor prognosis of solid tumors (106). Previous studies have shown that CAFs can interact with tumor and immune cells by releasing various regulatory factors, synthesizing and remodeling the ECM, as well as inducing EMT and drug resistance (107). CAF-derived EVs reportedly induced lung PMN formation in mice, thereby improving the lung metastatic ability of salivary gland adenoid cystic carcinoma (108). Disseminated breast cancer cells induce an inflammatory phenotype in lung fibroblasts, thereby creating a microenvironment that supports metastasis (109). Another study found that CAF-derived exosomal miR-522 not only inhibited ferroptosis but also promoted acquired cisplatin and paclitaxel resistance in gastric cancer (110). Collectively, these studies substantiate that CAFs and PMN formation is closely associated with ferroptosis, suggesting the importance of CAFs in matrix reprogramming. It is unknown whether CAFs undergo ferroptosis and inhibit PMN formation, nor is it clear whether targeting CAFs can enhance the sensitivity of tumor cells to ferroptosis. Accordingly, further research is essential to elucidate these questions.
Metabolic reprogramming is a hallmark of malignancy, while metabolic properties and preferences of tumors change during cancer progression (111). Generally, metastatic cancer cells selectively and dynamically adjust their metabolism at each step in the metastatic cascade. To adapt to a new environment, for survival and growth, many metastatic tumors exhibit metabolic profiles that differ from those at the primary site (112). Several review articles have shown that metabolic pathways, such as glucose metabolism, lipid metabolism, iron metabolism, amino acid and glutathione metabolism, are closely related to ferroptosis (113–115). AMP-activated protein kinase (AMPK), a central regulator of energy homeostasis and cellular metabolism, has been shown to play a tumor-promoting or antitumor effect under different circumstances. Notably, AMPK activation reportedly endows tumor cells with a growth advantage by modulating their metabolic plasticity, thereby adapting to metabolic stress (116). Mitochondrial ROS is a physiological activator of AMPK. Notably, AMPK negative feedback limits ROS production, while mitochondrial ROS-activated AMPK can regulate the stability of hypoxia-inducible factor 1α (HIF1α) (117). Similarly, AMPK plays a dual role in regulation of ferroptosis, mainly by blocking system xc- activation (118). In contrast, energy stress-mediated AMPK activation inhibits ferroptosis (119). Moreover, ROS also plays a dual role in cancer prevention and treatment (120). The relationship between metabolic homeostasis and ferroptosis is not static, and critical metabolic regulators may play opposite roles in differential tumor progression. Complex network communication between reprogramming and ferroptosis can promote PMN formation, and how tumor cells avoid ferroptosis through reprogramming deserves further study. Given that ferroptosis is closely associated with many metabolic pathways, exploiting metabolic fragility might be critical in unraveling therapeutic loopholes for inducing ferroptosis to inhibit tumor PMN formation (Figure 3).
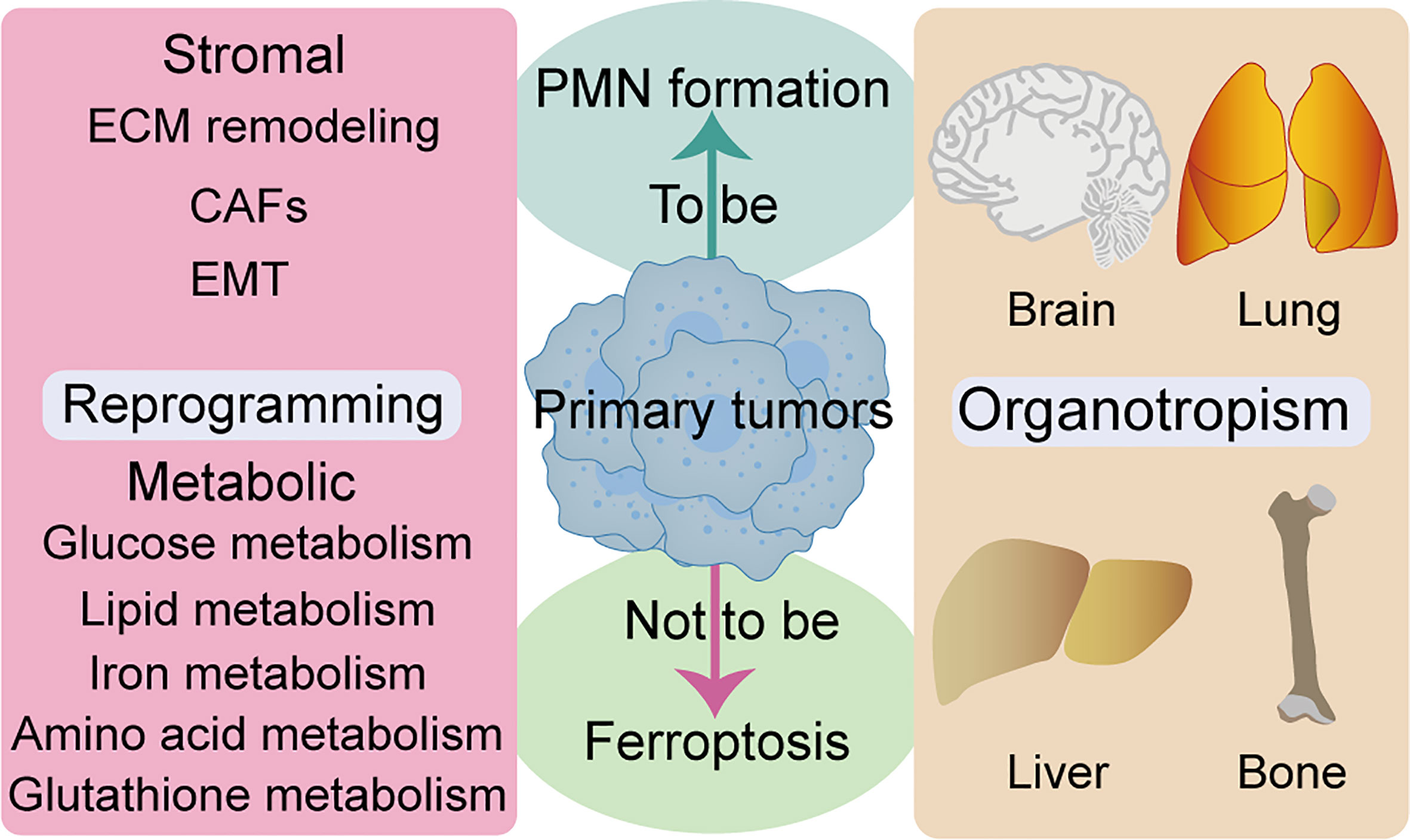
Figure 3 Relationship between ferroptosis and reprogramming and organotropism. ECM remodeling, CAFs, and EMT are closely associated to stromal reprogramming and ferroptosis. Multiple metabolic pathways are closely related to ferroptosis, such as glucose metabolism, lipid metabolism, iron metabolism, amino acid and glutathione metabolism. Several organs are the host parts of metastasis and are prone to ferroptosis, such as brain, lung, liver and bone. CAFs, cancer-associated fibroblasts; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition.
Organotropism
Features of organotropism may be innately associated with PMN, owing to the fact that some cancer types tend to metastasize to specific organs with selective microenvironments (4). Intrinsic properties of tumors, coupled with unique characteristics of host organs, circulation patterns, and interactions between tumor cells and the host microenvironment, jointly determine organ-specific metastatic behavior. Studies have shown that major metastatic target organs, such as bone, liver, lung, and brain, among others, have their specific PMN (121) (Figure 3). Moreover, researchers have documented the role of ferroptosis in many cases of pathological cell death, especially in tissues such as the liver and brain (122). Notably, iron-rich tumors, such as hepatocellular carcinoma and non-small cell lung cancer, maybe particularly responsive to FINs (7). Furthermore, different ferroptosis regulators have distinct gene expression levels throughout the tumor. SLC7A11 inhibitors have been reported to be particularly effective against certain types of cancer that overexpress this target, such as esophageal cancer and glioblastoma (7). Although no studies have demonstrated the relationship between ferroptosis with organotropism during PMN formation, iron-rich organs (more nutrient-rich “soil”) may be more susceptible to PMN formation. Moreover, ferroptosis may have particular effects on prevention and treatment of organotypic metastases in certain tumors, such as breast and lung cancer brain metastases, due to tissue specificity of ferroptosis and the high sensitivity of specific types of tumors to FINs.
Conclusions and perspectives
Formation and maturation of PMN guarantee successful extravasation, colonization and growth of metastatic tumor cells. This complex spatiotemporal process relies on interactions between tumor cells at the primary site and the colonization site microenvironment. Notably, vascular stabilization and immune regulation represent promising pathways to deconstruct the complexity of PMN (123). This review addresses the potential link between ferroptosis and PMN, harnessing our knowledge of PMN characteristics and information from published articles. Although no direct and precise links have so far been established, this information provides a new frontier for discovery of interventions in PMN formation and maturation via ferroptosis to overcome cancer metastasis. In the future, more in-depth studies are needed to identify and characterize the key molecules as well as mechanisms underlying ferroptosis in PMN, as such information will guide development of safe and effective anticancer therapies. This review did not cover various topics, such as tumor cell dormancy characteristics, inter-tumor and intra-tumor heterogeneity, and cancer stem cells. Indeed, epigenetic regulation plays an integral role in malignant tumor progression (124), while ferroptosis mediates various epigenetic regulators and metabolic changes. Studies have shown that epigenetic regulation of ferroptosis may serve as a novel anticancer therapeutic strategy (12, 125). Future studies, seeking to elucidate the role of ferroptosis in PMN, should focus on epigenetic regulation.
In summary, the aforementioned six PMN characteristics not only comprehensively function from different aspects to ensure colonization and metastasis of tumor cells, but also are closely related to ferroptosis, suggesting that ferroptosis may play an essential role in PMN and modulation of ferroptosis to predict or treat tumor metastasis is an attractive strategy. However, its toxic and side effects should be borne in mind, including an elevated risk of neurodegeneration and acute tissue damage, or ineffective therapeutic effects leading to tumor progression. Therefore, researchers need to conduct sufficient preclinical studies to ascertain its safety and efficacy.
Future studies are expected to not only elucidate the mechanism underlying ferroptosis in PMN but also identify and distinguish the effect of ferroptosis on different PMN characteristics during tumor metastasis. Such research endeavors are expected to employ advanced technologies, such as organoid technology, single-cell and spatial transcriptome sequencing platforms, and nanomedicine approaches, to study the effect of ferroptosis on the interaction between tumor cells and peritumoral stromal cells and immune cells, as well as the initiation of ferroptosis in PMN priming, licensing, initiation, and progression roles in various spatiotemporal stages. Findings from these explorations will provide valuable insights to guide future development and utilization of new prevention and treatment strategies against cancer metastasis.
Author contributions
KY, JF, and ZG conceived and designed the study. SZ, LY, and SC provided equal contributions to research article writing. CT and WL revised the manuscript. The final manuscript has been read and approved by all authors.
Funding
This work was supported by Finance science and technology project of hainan province (Grant No. ZDYF2022SHFZ088, ZDYF2019129), the National Nature Science Foundation of China (Grant No. 82060456), the Innovative Research Project of Hainan Graduate Students (Grant No. Qhyb2021-58) and project supported by Hainan Province Clinical Medical Center.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sin B (2015) 5:402–18. doi: 10.1016/j.apsb.2015.07.005
2. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (2011) 331:1559–64. doi: 10.1126/science.1203543
3. Paget S. The distribution of secondary growths in cancer of the breast. Lancet (1889) 133:571–3. doi: 10.1016/S0140-6736(00)49915-0
4. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell (2016) 30:668–81. doi: 10.1016/j.ccell.2016.09.011
5. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer (2017) 17:302–17. doi: 10.1038/nrc.2017.6
6. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
7. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
8. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer (2019) 19:405–14. doi: 10.1038/s41568-019-0149-1
9. Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2020) 21:341–52. doi: 10.1038/s41580-020-0237-9
10. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547:453–7. doi: 10.1038/nature23007
11. Chen P, Li X, Zhang R, Liu S, Xiang Y, Zhang M, et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics (2020) 10:5107–19. doi: 10.7150/thno.44705
12. Lee J, You JH, Kim MS, Roh JL. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol (2020) 37:101697. doi: 10.1016/j.redox.2020.101697
13. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med (Berl) (2016) 94:509–22. doi: 10.1007/s00109-015-1376-x
14. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol (2009) 9:353–63. doi: 10.1038/nri2545
15. Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer (2020) 8:e000337. doi: 10.1136/jitc-2019-000337
16. Garg AD, Agostinis P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol Rev (2017) 280:126–48. doi: 10.1111/imr.12574
17. Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ (2014) 21:69–78. doi: 10.1038/cdd.2013.72
18. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun (2019) 510:278–83. doi: 10.1016/j.bbrc.2019.01.090
19. Efimova I, Catanzaro E, van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer (2020) 8:e001369. doi: 10.1136/jitc-2020-001369
20. Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, et al. Patrolling monocytes control tumor metastasis to the lung. Science (2015) 350:985–90. doi: 10.1126/science.aac9407
21. Narasimhan PB, Eggert T, Zhu YP, Marcovecchio P, Meyer MA, Wu R, et al. Patrolling monocytes control NK cell expression of activating and stimulatory receptors to curtail lung metastases. J Immunol (2020) 204:192–8. doi: 10.4049/jimmunol.1900998
22. Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res (2012) 72:3906–11. doi: 10.1158/0008-5472.CAN-11-3873
23. Wang D, Xie N, Gao W, Kang R, Tang D. The ferroptosis inducer erastin promotes proliferation and differentiation in human peripheral blood mononuclear cells. Biochem Biophys Res Commun (2018) 503:1689–95. doi: 10.1016/j.bbrc.2018.07.100
24. Kong R, Wang N, Han W, Bao W, Lu J. IFNγ-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J Leukoc Biol (2021) 110:301–14. doi: 10.1002/jlb.3ma1220-815rrr
25. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
26. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell (2022) 40:365–78.e6. doi: 10.1016/j.ccell.2022.02.003
27. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T Cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med (2015) 212:555–68. doi: 10.1084/jem.20140857
28. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab (2021) 33:1001–12.e5. doi: 10.1016/j.cmet.2021.02.015
29. Han L, Bai L, Qu C, Dai E, Liu J, Kang R, et al. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys Res Commun (2021) 576:33–9. doi: 10.1016/j.bbrc.2021.08.082
30. Poznanski SM, Singh K, Ritchie TM, Aguiar JA, Fan IY, Portillo AL, et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab (2021) 33:1205–20.e5. doi: 10.1016/j.cmet.2021.03.023
31. Renken S, Nakajima T, Magalhaes I, Mattsson J, Lundqvist A, Arnér ESJ, et al. Targeting of Nrf2 improves antitumoral responses by human NK cells, TIL and CAR T cells during oxidative stress. J Immunother Cancer (2022) 10:e004458. doi: 10.1136/jitc-2021-004458
32. Wang Y, Ding Y, Guo N, Wang S. MDSCs: Key criminals of tumor pre-metastatic niche formation. Front Immunol (2019) 10:172. doi: 10.3389/fimmu.2019.00172
33. Nasrollahzadeh E, Razi S, Keshavarz-Fathi M, Mazzone M, Rezaei N. Pro-tumorigenic functions of macrophages at the primary, invasive and metastatic tumor site. Cancer Immunol Immunother (2020) 69:1673–97. doi: 10.1007/s00262-020-02616-6
34. Trovato R, Cane S, Petrova V, Sartoris S, Ugel S, De Sanctis F. The engagement between MDSCs and metastases: Partners in crime. Front Oncol (2020) 10:165. doi: 10.3389/fonc.2020.00165
35. Zhu H, Klement JD, Lu C, Redd PS, Yang D, Smith AD, et al. Asah2 represses the p53-Hmox1 axis to protect myeloid-derived suppressor cells from ferroptosis. J Immunol (2021) 206:1395–404. doi: 10.4049/jimmunol.2000500
36. Zhang Y, Bush X, Yan B, Chen JA. Gemcitabine nanoparticles promote antitumor immunity against melanoma. Biomaterials (2019) 189:48–59. doi: 10.1016/j.biomaterials.2018.10.022
37. Zhu S, Zhang Q, Sun X, Zeh HJ, Lotze MT 3rd, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res (2017) 77:2064–77. doi: 10.1158/0008-5472.CAN-16-1979
38. Ye Z, Zhuo Q, Hu Q, Xu X, Mengqi L, Zhang Z, et al. FBW7-NRA41-SCD1 axis synchronously regulates apoptosis and ferroptosis in pancreatic cancer cells. Redox Biol (2021) 38:101807. doi: 10.1016/j.redox.2020.101807
39. Doak GR, Schwertfeger KL, Wood DK. Distant relations: Macrophage functions in the metastatic niche. Trends Cancer (2018) 4:445–59. doi: 10.1016/j.trecan.2018.03.011
40. Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C, et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab (2021) 33:2040–58.e10. doi: 10.1016/j.cmet.2021.09.002
41. Li Z, Rong L. Cascade reaction-mediated efficient ferroptosis synergizes with immunomodulation for high-performance cancer therapy. Biomater Sci (2020) 8:6272–85. doi: 10.1039/d0bm01168a
42. Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated tumor cell-derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv (2020) 6:eaay9789. doi: 10.1126/sciadv.aay9789
43. Luo X, Gong HB, Gao HY, Wu YP, Sun WY, Li ZQ, et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ (2021) 28:1971–89. doi: 10.1038/s41418-020-00719-2
44. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology (2017) 66:449–65. doi: 10.1002/hep.29117
45. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol (2020) 16:278–90. doi: 10.1038/s41589-019-0462-8
46. Muri J, Thut H, Bornkamm GW, Kopf M. B1 and marginal zone b cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep (2019) 29:2731–44:e4. doi: 10.1016/j.celrep.2019.10.070
47. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-evoked regulatory b cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res (2011) 71:3505–15. doi: 10.1158/0008-5472.CAN-10-4316
48. Bodogai M, Moritoh K, Lee-Chang C, Hollander CM, Sherman-Baust CA, Wersto RP, et al. Immunosuppressive and prometastatic functions of myeloid-derived suppressive cells rely upon education from tumor-associated b cells. Cancer Res (2015) 75:3456–65. doi: 10.1158/0008-5472.CAN-14-3077
49. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain treg cell activation and suppression of antitumor immunity. Cell Rep (2021) 35:109235. doi: 10.1016/j.celrep.2021.109235
50. Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer (2007) 121:2381–6. doi: 10.1002/ijc.23192
51. Solinas G, Marchesi F, Garlanda C, Mantovani A, Allavena P. Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev (2010) 29:243–8. doi: 10.1007/s10555-010-9227-2
52. Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev (2009) 61:290–302. doi: 10.1016/j.addr.2009.02.005
53. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. BioMed Pharmacother (2020) 127:110108. doi: 10.1016/j.biopha.2020.110108
54. Mao H, Zhao Y, Li H, Lei L. Ferroptosis as an emerging target in inflammatory diseases. Prog Biophys Mol Biol (2020) 155:20–8. doi: 10.1016/j.pbiomolbio.2020.04.001
55. Garner H, de Visser KE. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol (2020) 20:483–97. doi: 10.1038/s41577-019-0271-z
56. Canli O, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M, et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell (2017) 32:869–83:e5. doi: 10.1016/j.ccell.2017.11.004
57. Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol (2018) 19:1236–47. doi: 10.1038/s41590-018-0229-5
58. Tyagi A, Sharma S, Wu K, Wu SY, Xing F, Liu Y, et al. Nicotine promotes breast cancer metastasis by stimulating N2 neutrophils and generating pre-metastatic niche in lung. Nat Commun (2021) 12:474. doi: 10.1038/s41467-020-20733-9
59. Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell (2016) 30:243–56. doi: 10.1016/j.ccell.2016.06.021
60. Zhou Z, Zhao Y, Chen S, Cui G, Fu W, Li S, et al. Cisplatin promotes the efficacy of immune checkpoint inhibitor therapy by inducing ferroptosis and activating neutrophils. Front Pharmacol (2022) 13:870178. doi: 10.3389/fphar.2022.870178
61. Jiang W, Wei L, Chen B, Luo X, Xu P, Cai J, et al. Platinum prodrug nanoparticles inhibiting tumor recurrence and metastasis by concurrent chemoradiotherapy. J nanobiotechnol (2022) 20:129. doi: 10.1186/s12951-022-01322-y
62. Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun (2020) 11:5424. doi: 10.1038/s41467-020-19193-y
63. Gout P, Buckley A, Simms C, Bruchovsky NJL. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia (2001) 15:1633–40. doi: 10.1038/sj.leu.2402238
64. Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. oncotarget (2016) 7:36021–33. doi: 10.18632/oncotarget.8651
65. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
66. Yotsumoto S, Muroi Y, Chiba T, Ohmura R, Yoneyama M, Magarisawa M, et al. Hyperoxidation of ether-linked phospholipids accelerates neutrophil extracellular trap formation. Sci Rep (2017) 7:16026. doi: 10.1038/s41598-017-15668-z
67. Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology (2016) 5:e1134073. doi: 10.1080/2162402X.2015.1134073
68. Cedervall J, Zhang Y, Olsson AK. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res (2016) 76:4311–5. doi: 10.1158/0008-5472.CAN-15-3051
69. De Meo ML, Spicer JD. The role of neutrophil extracellular traps in cancer progression and metastasis. Semin Immunol (2021) 57:101595. doi: 10.1016/j.smim.2022.101595
70. Kist M, Vucic D. Cell death pathways: intricate connections and disease implications. EMBO J (2021) 40:e106700. doi: 10.15252/embj.2020106700
71. Li R, Wen A, Lin J. Pro-inflammatory cytokines in the formation of the pre-metastatic niche. Cancers (Basel) (2020) 12:3752. doi: 10.3390/cancers12123752
72. Farajzadeh Valilou S, Keshavarz-Fathi M, Silvestris N, Argentiero A, Rezaei N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev (2018) 39:46–61. doi: 10.1016/j.cytogfr.2018.01.007
73. Wang Y, Quan F, Cao Q, Lin Y, Yue C, Bi R, et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J Adv Res (2021) 28:231–43. doi: 10.1016/j.jare.2020.07.007
74. Proneth B, Conrad M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ (2019) 26:14–24. doi: 10.1038/s41418-018-0173-9
75. Bianchi ME, Crippa MP, Manfredi AA, Mezzapelle R, Rovere Querini P, Venereau E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol Rev (2017) 280:74–82. doi: 10.1111/imr.12601
76. Yang Y, Yang L, Jiang S, Yang T, Lan J, Lei Y, et al. HMGB1 mediates lipopolysaccharide-induced inflammation via interacting with GPX4 in colon cancer cells. Cancer Cell Int (2020) 20:205. doi: 10.1186/s12935-020-01289-6
77. Pfeifhofer-Obermair C, Tymoszuk P, Petzer V, Weiss G, Nairz M. Iron in the tumor microenvironment-connecting the dots. Front Oncol (2018) 8:549. doi: 10.3389/fonc.2018.00549
78. Li C, Xu X, Wei S, Jiang P, Xue L, Wang J, et al. Tumor-associated macrophages: potential therapeutic strategies and future prospects in cancer. J Immunother Cancer (2021) 9:e001341. doi: 10.1136/jitc-2020-001341
79. Jung M, Mertens C, Tomat E, Brune B. Iron as a central player and promising target in cancer progression. Int J Mol Sci (2019) 20:273. doi: 10.3390/ijms20020273
80. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron induces anti-tumor activity in tumor-associated macrophages. Front Immunol (2017) 8:1479. doi: 10.3389/fimmu.2017.01479
81. Liu SX, Gustafson HH, Jackson DL, Pun SH, Trapnell C. Trajectory analysis quantifies transcriptional plasticity during macrophage polarization. Sci Rep (2020) 10:12273. doi: 10.1038/s41598-020-68766-w
82. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer (2017) 17:457–74. doi: 10.1038/nrc.2017.51
83. Huang Y, Song N, Ding Y, Yuan S, Li X, Cai H, et al. Pulmonary vascular destabilization in the premetastatic phase facilitates lung metastasis. Cancer Res (2009) 69:7529–37. doi: 10.1158/0008-5472.Can-08-4382
84. Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov (2015) 5:932–43. doi: 10.1158/2159-8290.CD-15-0012
85. Zeng Z, Li Y, Pan Y, Lan X, Song F, Sun J, et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat Commun (2018) 9:5395. doi: 10.1038/s41467-018-07810-w
86. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene (2017) 36:5593–608. doi: 10.1038/onc.2017.146
87. Schneider M, Wortmann M, Mandal PK, Arpornchayanon W, Jannasch K, Alves F, et al. Absence of glutathione peroxidase 4 affects tumor angiogenesis through increased 12/15-lipoxygenase activity. Neoplasia (2010) 12:254–63. doi: 10.1593/neo.91782
88. Long Y, Tao H, Karachi A, Grippin AJ, Jin L, Chang YE, et al. Dysregulation of glutamate transport enhances treg function that promotes VEGF blockade resistance in glioblastoma. Cancer Res (2020) 80:499–509. doi: 10.1158/0008-5472.CAN-19-1577
89. Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett (2016) 381:165–75. doi: 10.1016/j.canlet.2016.07.033
90. Zhao L, Peng Y, He S, Li R, Wang Z, Huang J, et al. Apatinib induced ferroptosis by lipid peroxidation in gastric cancer. Gastric Cancer (2021) 24:642–54. doi: 10.1007/s10120-021-01159-8
91. Lopes-Coelho F, Martins F, Hipólito A, Mendes C, Sequeira CO, Pires RF, et al. The activation of endothelial cells relies on a ferroptosis-like mechanism: Novel perspectives in management of angiogenesis and cancer therapy. Front Oncol (2021) 11:656229. doi: 10.3389/fonc.2021.656229
92. Mustapha R, Ng K, Monypenny J, Ng T. Insights into unveiling a potential role of tertiary lymphoid structures in metastasis. Front Mol Biosci (2021) 8:661516. doi: 10.3389/fmolb.2021.661516
93. Gillot L, Baudin L, Rouaud L, Kridelka F, Noël A. The pre-metastatic niche in lymph nodes: formation and characteristics. Cell Mol Life Sci (2021) 78:5987–6002. doi: 10.1007/s00018-021-03873-z
94. Karnezis T, Shayan R, Caesar C, Roufail S, Harris NC, Ardipradja K, et al. VEGF-d promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell (2012) 21:181–95. doi: 10.1016/j.ccr.2011.12.026
95. Balsat C, Blacher S, Herfs M, Van de Velde M, Signolle N, Sauthier P, et al. A specific immune and lymphatic profile characterizes the pre-metastatic state of the sentinel lymph node in patients with early cervical cancer. Oncoimmunology (2017) 6:e1265718. doi: 10.1080/2162402X.2016.1265718
96. Li M, Xian HC, Tang YJ, Liang XH, Tang YL. Fatty acid oxidation: driver of lymph node metastasis. Cancer Cell Int (2021) 21:339. doi: 10.1186/s12935-021-02057-w
97. Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I, et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science (2019) 363:644–9. doi: 10.1126/science.aav0173
98. Pucci F, Garris C, Lai C, Newton A, Pfirschke C, Engblom C, et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-b cell interactions. Science (2016) 352:242–6. doi: 10.1126/science.aaf1328
99. Leary N, Walser S, He Y, Cousin N, Pereira P, Gallo A, et al. Melanoma-derived extracellular vesicles mediate lymphatic remodelling and impair tumour immunity in draining lymph nodes. J extracellular vesicles (2022) 11:e12197. doi: 10.1002/jev2.12197
100. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature (2020) 585:113–8. doi: 10.1038/s41586-020-2623-z
101. LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol (2016) 18:356–65. doi: 10.1038/ncb3330
102. Schito L. Hypoxia-dependent angiogenesis and lymphangiogenesis in cancer. Adv Exp Med Biol (2019) 1136:71–85. doi: 10.1007/978-3-030-12734-3_5
103. Li Z, Jiang L, Chew SH, Hirayama T, Sekido Y, Toyokuni S. Carbonic anhydrase 9 confers resistance to ferroptosis/apoptosis in malignant mesothelioma under hypoxia. Redox Biol (2019) 26:101297. doi: 10.1016/j.redox.2019.101297
104. Fuhrmann DC, Mondorf A, Beifuss J, Jung M, Brune B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol (2020) 36:101670. doi: 10.1016/j.redox.2020.101670
105. Kai F, Drain AP, Weaver VM. The extracellular matrix modulates the metastatic journey. Dev Cell (2019) 49:332–46. doi: 10.1016/j.devcel.2019.03.026
107. Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist Update (2020) 53:100715. doi: 10.1016/j.drup.2020.100715
108. Kong J, Tian H, Zhang F, Zhang Z, Li J, Liu X, et al. Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol Cancer (2019) 18:175. doi: 10.1186/s12943-019-1101-4
109. Pein M, Insua-Rodriguez J, Hongu T, Riedel A, Meier J, Wiedmann L, et al. Metastasis-initiating cells induce and exploit a fibroblast niche to fuel malignant colonization of the lungs. Nat Commun (2020) 11:1494. doi: 10.1038/s41467-020-15188-x
110. Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer (2020) 19:43. doi: 10.1186/s12943-020-01168-8
111. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science (2020) 368:eaaw5473. doi: 10.1126/science.aaw5473
112. Bergers G, Fendt SM. The metabolism of cancer cells during metastasis. Nat Rev Cancer (2021) 21:162–80. doi: 10.1038/s41568-020-00320-2
113. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
114. Yao X, Li W, Fang D, Xiao C, Wu X, Li M, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv Sci (Weinheim Baden-Wurttemberg Germany) (2021) 8:e2100997. doi: 10.1002/advs.202100997
115. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther (2020) 5:108. doi: 10.1038/s41392-020-00216-5
116. Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett (2015) 356:165–70. doi: 10.1016/j.canlet.2014.01.018
117. Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep (2017) 21:1–9. doi: 10.1016/j.celrep.2017.09.026
118. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system xc(-) activity. Curr Biol (2018) 28:2388–99:e5. doi: 10.1016/j.cub.2018.05.094
119. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
120. Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T, et al. The double-edged roles of ROS in cancer prevention and therapy. Theranostics (2021) 11:4839–57. doi: 10.7150/thno.56747
121. Gao Y, Bado I, Wang H, Zhang W, Rosen JM, Zhang XH. Metastasis organotropism: Redefining the congenial soil. Dev Cell (2019) 49:375–91. doi: 10.1016/j.devcel.2019.04.012
122. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci (2016) 73:2195–209. doi: 10.1007/s00018-016-2194-1
123. Wang H, Pan J, Barsky L, Jacob JC, Zheng Y, Gao C, et al. Characteristics of pre-metastatic niche: the landscape of molecular and cellular pathways. Mol biomed (2021) 2:3. doi: 10.1186/s43556-020-00022-z
124. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov (2022) 12:31–46. doi: 10.1158/2159-8290.Cd-21-1059
Keywords: ferroptosis, pre-metastatic niche, tumor, immune escape, therapeutic strategies
Citation: Zhuo S, Yang L, Chen S, Tang C, Li W, Gao Z, Feng J and Yang K (2022) Ferroptosis: A potential opportunity for intervention of pre-metastatic niche. Front. Oncol. 12:980620. doi: 10.3389/fonc.2022.980620
Received: 28 June 2022; Accepted: 23 August 2022;
Published: 09 September 2022.
Edited by:
Zhizhou Shi, Kunming University of Science and Technology, ChinaReviewed by:
Tao Li, First Affiliated Hospital of Anhui Medical University, ChinaArmel Hervé Nwabo Kamdje, University of Garoua, Cameroon
Copyright © 2022 Zhuo, Yang, Chen, Tang, Li, Gao, Feng and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Yang, Y2hieWsxMzc5QDE2My5jb20=; Jigao Feng, ZmVuZ2ppZ2FvQGhhaW5tYy5lZHUuY24=; Zhenzhong Gao, d3VxdWUxMjNAaGFpbm1jLmVkdS5jbg==
†These authors have contributed equally to this work