
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 15 August 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.976557
This article is part of the Research TopicHallmark of Cancer: Resisting Cell DeathView all 7 articles
 Zhengyang Zhu1†
Zhengyang Zhu1† Chaoyou Fang1†
Chaoyou Fang1† Houshi Xu1†Ling Yuan1Yichao Du1Yunjia Ni1
Houshi Xu1†Ling Yuan1Yichao Du1Yunjia Ni1 Yuanzhi Xu2
Yuanzhi Xu2 Anwen Shao3,4*
Anwen Shao3,4* Anke Zhang3,4*
Anke Zhang3,4* Meiqing Lou1*
Meiqing Lou1*Glioma is the most common malignant intracranial tumor and exhibits diffuse metastasis and a high recurrence rate. The invasive property of glioma results from cell detachment. Anoikis is a special form of apoptosis that is activated upon cell detachment. Resistance to anoikis has proven to be a protumor factor. Therefore, it is suggested that anoikis resistance commonly occurs in glioma and promotes diffuse invasion. Several factors, such as integrin, E-cadherin, EGFR, IGFR, Trk, TGF-β, the Hippo pathway, NF-κB, eEF-2 kinase, MOB2, hypoxia, acidosis, ROS, Hsp and protective autophagy, have been shown to induce anoikis resistance in glioma. In our present review, we aim to summarize the underlying mechanism of resistance and the therapeutic potential of these molecules.
Glioma is the most common central nervous system (CNS) tumor and has limited therapeutic options and poor overall survival (OS) (1, 2). Glioma comprises approximately 30% of all brain tumors and 80% of all malignant brain tumors in adults (3). According to the 5th edition of the WHO classification of CNS tumors, gliomas are divided into 4 different families (adult-type diffuse gliomas, pediatric-type diffuse low-grade gliomas, pediatric-type diffuse high-grade gliomas, circumscribed astrocytic gliomas) and 17 subfamilies (4). High-grade gliomas tend to infiltrate diffusely, causing extensive areas of necrosis and hypoxia. Furthermore, the diffusive property makes it almost impossible to remove gliomas completely through surgery, leading to frequent recurrence and eventually death. Apart from surgery, radiotherapy (RT) and chemotherapy are also applied in clinical practice. However, high-precision conformal RT, temozolomide (TMZ) and bevacizumab fail to improve OS (5).
The diffusive property of glioma relates to aberrant changes in cell adhesion (6). This suggestion links glioma with a type of programmed cell death called anoikis. Anoikis is a special form of apoptosis activated upon cell detachment. Normal cells live in a microenvironment that is composed of extracellular matrix and various supporting cells. This three-dimensional scaffold provides adherent cells with necessary biochemical and mechanical signals for survival, growth, differentiation and other physiological processes (7). Remodeling of the microenvironment or cell detachment can lead to the activation of anoikis and eventually cell death.
Anoikis is regulated by many factors and pathways in wide range of cancers (8–10). By utilizing these factors, we might inhibit the diffuse infiltration of glioma, thus improving clinical outcomes. Studies have revealed that several regulators take part in anoikis resistance in glioma; these include integrin, E-cadherin, EGFR, IGFR, Trk, TGF-β, the Hippo pathway, NF-κB, eEF-2 kinase, MOB2, hypoxia, acidosis, reactive oxygen species (ROS), heat shock proteins (Hsps) and protective autophagy. Here, we review these regulators that have been reported to induce anoikis resistance in diffuse glioma and their possible utilization in future treatment.
In terms of downstream mechanisms, anoikis is similar to apoptosis, which generally can be converted into the intrinsic pathway and the extrinsic pathway and converges at the activation of Caspases. The intrinsic pathway relies on mitochondrial permeabilization, which can be triggered by cellular signals such as DNA damage and unfolded protein response, followed by Caspase activation (11, 12). Mitochondrial permeabilization is a result of membrane pore formation or mitochondrial swelling. The Bcl-2 protein family controls the formation of pores in the outer mitochondrial membrane (13). In other words, the relative ratio of prosurvival (Bcl-2, BCL-xL) and proapoptotic (Bax, Bak and BH3-only subfamily) proteins of this family determines whether permeabilization occurs. During this process, Bax and Bak translocate to the outer mitochondrial membrane and create pores through oligomerization. BH3-only proteins act as activators and sensitizers. Activators promote the formation of Bax-Bak oligomers, and sensitizers act as competitive inhibitors of Bcl-2, thus neutralizing the inhibitory effect of Bcl-2 on activators and oligomers (14, 15). Permeabilization of mitochondria allows the leakage of several apoptotic proteins, such as cytochrome c (Cyt c), endonuclease G (EndoG), apoptosis-inducing factors (AIFs), second mitochondrial activator of Caspases (Smac) and high temperature requirement protein A2 (HtrA2). Cyt c is the main contributor to the intrinsic pathway, and its leakage leads to the formation of apoptosomes with apoptotic protease activating factor-1 (Apaf-1), thereby activating Caspase-9. EndoG and AIF participate in Caspase-independent DNA fragmentation. Smac and HtrA2 activate Caspase by neutralizing the apoptosis inhibitor IAP. These proteins eventually lead to anoikis via different mechanisms (14).
The extrinsic pathway relies on the ligand binding of death receptors on the cell membrane following the formation of the death-inducing signaling complex (DISC). Tumor necrosis factor receptor (TNFR) subfamily members, such as the first apoptosis signal (Fas) receptor, TNFR1, TNFR2 and TNF-related apoptosis inducing ligand (TRAIL) receptor-1 and -2, are major initiators of this pathway (15, 16). The ligand binding of FAS receptor results in the formation of the death-inducing signaling complex (DISC). DISC recruits and activates Caspase-8 through interaction with Fas-associated death domain protein (FADD). Activated Caspase-8 in turn activates the effectors Caspase-3, Caspase-6, and Caspase-7 (15, 17). TRAIL receptor activates Caspase-8 in a similar way as Fas receptor—via the formation of the Fas/TRAIL DISC (18). TNFR has a more complex way of inducing anoikis. First, upon ligand binding, TNFR1 binds to TNFR1-associated death domain protein (TRADD), while TNFR2 binds to TNFR-associated factor 1 (TRAF1) and TRAF2. The TNFR1-TRADD oligomer then recruits TRAF2 or TRAF5 and cellular inhibitor of apoptosis protein 1 (cIAP1) or cIAP2 to form TNFR1 complex I. Complex I further recruits the linear ubiquitin chain assembly complex (LUBAC) and ubiquitylates receptor-interacting serine/threonine-protein kinase 1 (RIPK1). Then, according to the ubiquitylation status of RIPK1, TNFR1 complex IIa or IIb is formed with RIPK1, FADD, pro-Caspase-8, the long isoform of FLICE-like inhibitory protein (FLIPL) and TRADD (in IIa) or RIPK3 (in IIb). Both TNFR1 complex IIa and IIb are capable of Caspase-8 activation (19).
Despite the differences between the intrinsic and extrinsic pathways, they merge at the point of Caspase activation and are connected by several shared intermediates. For example, Caspase-8, the key downstream molecule of the extrinsic pathway, also activates Bid, which is a BH3-only protein, therefore triggering the intrinsic pathway. Mitochondrial permeabilization is often followed by death receptor activation (15). Thus, anoikis is regulated by both pathways.
Adhesion molecules are first-line mediators in anoikis signaling. They are directly involved in the establishment of cell adhesion and therefore can convey both mechanical and molecular signals of cell detachment. Thus, anomalies in adhesion molecule expression or signaling could result in anoikis resistance. Adhesion molecules such as integrins and E-cadherin have been proven to regulate anoikis resistance in glioma. And their regulation of anoikis resistance is demonstrated in Figure 1.
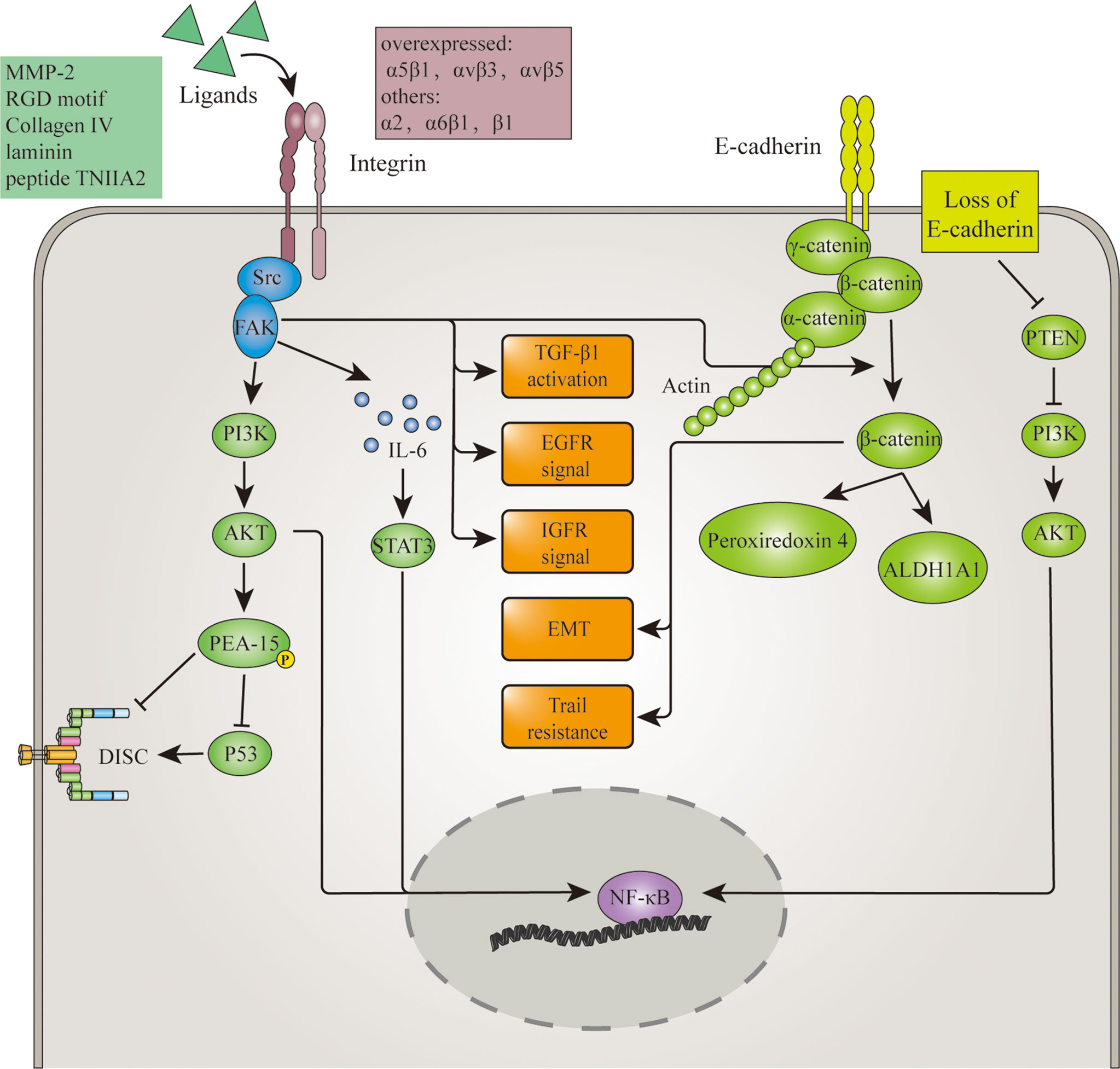
Figure 1 Adhesion molecules involved in anoikis resistance mechanism of glioma. Overexpression of Integrins and loss of E-cadherin in glioma disrupt the initiating signal of anoikis. Upon cell detachment, overexpressed integrins still activate FAK, which in turn activates signalings such as TGFβ, EGFR, IGFR, PI3k/AKT pathway. Then through p53 inhibition, NF-κB activation or other mediators, these pathways lead to anoikis. α2 integrin induced FAK activation also promotes the release of β-catenin from the E-cadherin/β-catenin complex. Along with the loss of E-cadherin and the help of Wnt, this increased the cytoplasmic level of β-catenin. β-catenin thereby promote anoikis resistance through EMT, TRAIL resistance and upregulation of invasion-related protein ALDH1A1 and peroxiredoxin 4. Aside from β-catenin, loss of E-cadherin also downregulates tumor suppressor PTEN and therefore maintains NF-κB activation.
Integrins are transmembrane receptors that facilitate cell–cell and cell-extracellular matrix (ECM) adhesion. They bind fibronectin, vitronectin, collagen, tenascin and laminin, which are directly related to adhesion. Upon ligand binding, integrins activate the receptor tyrosine kinase (RTK) signaling pathway and thereby regulate cellular events such as growth, division, survival, differentiation and programmed death (20, 21). Loss of adhesion can trigger anoikis through this pathway. However, aberrations in the expression of integrins and their ligands often inhibit anoikis during cell detachment.
Integrins are obligate heterodimers composed of α and β subunits. There are 24 integrins in total, and a single cell generally has multiple types of integrins on its surface (21). The α5β1, αvβ3 and αvβ5 integrins are overexpressed in glioma (22). Their binding with the tripeptide sequence Arg-Gly-Asp (RGD) motif in ECM proteins activates the cytosolic tyrosine kinase Src. Src constitutively binds to the integrin β cytoplasmic tail and focal adhesion kinase (FAK) (23). In turn, FAK activation induces anoikis inhibition by activating the transcription factor NF-κB through the PI3K/AKT signaling pathway (24). This inhibition can be reversed by applying the RGD-integrin antagonist 1a-RGD, which downregulates the phosphorylation of FAK (25, 26). The binding of α5β1 integrin with matrix metalloproteinase-2 (MMP-2), which breaks down ECM and induces metastasis, promotes IL-6/Stat3 survival signaling and upregulates NF-κB, leading to anoikis resistance (27). The upregulated binding of α5β1 integrin also inhibits p53-induced apoptosis through the PI3K/AKT pathway, which activates the antiapoptotic function of astrocytic phosphoprotein PEA-15 (28). Moreover, increased binding of MMP-2 with αvβ3, which is upregulated by p21-activated kinase 4 (PAK4), activates the EGFR pathway and induces anoikis resistance (29). αV integrin also participates in TGF-β1 activation, which promotes anoikis resistance in detached glioma cells (30–32). The binding of α6β1 integrin with laminin also enhances glioma proliferation and anoikis resistance, although the involved signaling pathway is not known (33). Similarly, an in vivo study demonstrated the promoting role of the binding of β1-integrin with tenascin-C-derived peptide TNIIA2 in anoikis resistance in GBM without a clear understanding of its subsequent signaling (34).
E-cadherin is a type of calcium-dependent cell adhesion molecule that is important in the formation of adherens junctions to allow cells to adhere to each other. E-cadherin is also referred to as the “suppressor of invasion” because its repression is the major event responsible for dysfunction in cell–cell adhesion. Cell–cell adhesion is achieved through the formation of homodimers in membranes and the binding of the protein complex to the cytoplasmic portion of E-cadherin. E-cadherin anchors to the cytoskeleton via a complex composed of α-, β- and γ-catenin (35). β-Catenin is released upon E-cadherin tension relaxation due to Src-dependent FAK activation, which is integrin related. Then, with the help of Wnt, cytoplasmic β-catenin accumulates and translocates to the nucleus (36–38). Therefore, E-cadherin activates β-catenin upon cell detachment.
A previous study showed that β-catenin can inhibit anoikis resulting from the loss of cell-substrate contact (39, 40). It has also been revealed that ALDH1A1 and peroxiredoxin 4 are direct targets of β-catenin and contribute to glioma invasiveness (41–43). Thus, the E-cadherin/β-catenin/ALDH1A1 and E-cadherin/β-catenin/peroxiredoxin-4 pathways are likely direct promoters of anoikis resistance in glioma.
Furthermore, loss of the E-cadherin/β-catenin complex inhibits anoikis through epithelial-to-mesenchymal transition (EMT). EMT is a process by which epithelial cells lose their cell polarity and cell–cell adhesion and gain migratory and invasive properties to become mesenchymal stem cells (44). Loss of cell–cell contact or E-cadherin upregulates cytoplasmic β-catenin, which, together with transforming growth factor-β (TGF-β), triggers EMT. During EMT, adherens junctions are downregulated, and anoikis is inhibited (38). TRAIL sensitivity is downregulated in this process, which partially explains the common occurrence of TRAIL resistance in glioblastoma (GBM) (45, 46).
Another target protein of E-cadherin is PTEN, a tumor suppressor antagonizing the PI3K-AKT signaling pathway (47). E-cadherin upregulates PTEN expression, which inhibits NF-κB activation through the PI3K/AKT pathway, thus promoting anoikis. In contrast, PTEN mutation in GBM leads to constitutive activation of AKT signaling and anoikis resistance (48). Therefore, loss of E-cadherin promotes anoikis resistance through the downregulation of PTEN.
Cell detachment also affects extracellular signaling molecules and membrane receptors, thereby inducing anoikis through various signaling pathways. On the other hand, aberration in signaling interrupts the anoikis process that normally occurs after cell detachment. Among these signaling pathways, epidermal growth factor receptor (EGFR), insulin-like growth factor receptor (IGFR), tropomyosin receptor kinases (Trk), transforming growth factor-β (TGF-β) and the Hippo pathway have been shown to regulate anoikis resistance in glioma. And these signaling are demonstrated in Figure 2.
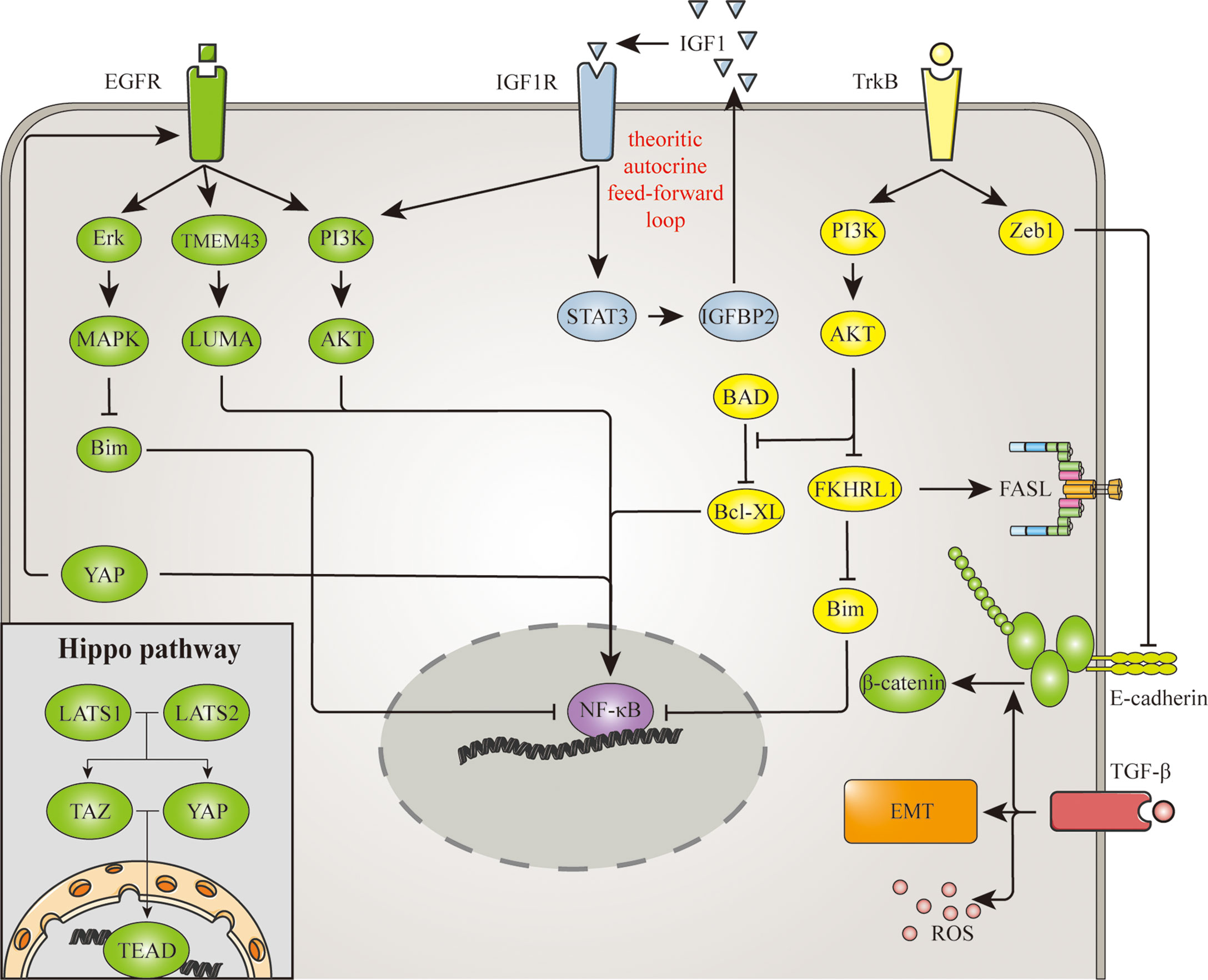
Figure 2 Signaling involved in anoikis resistance mechanism of glioma. EGFR, IGFR, TrkB, TGF-β and the hippo pathway are signaling pathways involved in the anoikis resistance of glioma. NF-κB is a common mediator in these pathways, for it regulates the expression of several anoikis-related proteins. Similarly, the hippo pathway promotes anoikis resistance through its transcription factor TAZ. Apart from transcription factors, EGFR also represses Bim expression through Erk/MAPK pathway. Meanwhile TrkB upregulates Bcl-xl and downregulates FasL and Bim through PI3K/AKT pathway. The signaling of TrkB and TGF-β triggers EMT as well, which promotes cell detachment and anoikis resistance. And there are also EGFR signaling triggered by the hippo pathway, and β-catenin promoted by TGF-β, indicating the connection between adhesion molecules and signaling pathways goes both way.
EGFR is a transmembrane receptor tyrosine kinase for the epidermal growth factor (EGF) family (49). Known ligands of EGFR include EGF, TGF, AREG, EPGN, BTC, EREG and HBEGF. Numerous studies have shown that EGFR has an important role in cancer progression. Its major downstream signaling pathways include the PI3K/AKT pathway, MAPK pathway, PLCγ/PKC and JAK2/STAT3 pathway (50–52). Through these pathways, EGFR can regulate anoikis resistance in gliomas. Furthermore, amplified EGFR was found in 60% of GBMs (53).
Under physiological conditions, Bim, one of the BH3-only proteins, is upregulated via downregulation of EGFR signaling upon cell detachment and then triggers the intrinsic pathway of anoikis. In vitro experiments suggest that overexpression of EGFR inhibits Bim through the MAPK pathway and thus induces anoikis resistance in malignant tumors (54, 55). A study also revealed NF-κB as the downstream target of EGFR in the PI3K/AKT pathway and the inducer of anoikis resistance (56). Similarly, STAT3 was found to maintain FAK activation during detachment (57). Therefore, FAK might in turn promote anoikis resistance through the PI3K/AKT pathway. Apart from the above pathways, the amplification of EGFR also activates NF-κB through TMEM43/LUMA and results in anoikis resistance in glioma (58).
IGFR is a family of transmembrane receptor tyrosine kinases that includes IGF1R, IGF2R, insulin receptor (IR) and hybrid IR/IGF1R. IGFRs bind to their cognate ligands, namely, IGF1, IGF2 and insulin. Six IGF-binding proteins (IGFBP1-6) bind to and carry IGFs in serum. Then, they are cleaved by MMPs in focal adhesions in the ECM, and IGFs are released (59). Activated IGF1R is involved in cell growth and survival control and is crucial for tumor transformation and survival (59). A study showed that cell detachment enhances IGF1R signaling through the collagen IV/α2 integrin/FAK pathway and thus promotes anoikis resistance (60). IGF1R signaling is carried out through the PI3K-Akt pathway and MAPK pathway (61, 62). However, a further study noted that the protective effect of IGF1 against anoikis is transduced via the PI3K-Akt pathway but not the Grb2/Ras/MAPK pathway (63). Furthermore, IGF1R/PI3K/Akt signaling promotes anoikis resistance through activation of NF-κB (64).
IGFBP2 overexpression is common in high-grade glioma and is related to glioma progression (65). Moreover, IR and IGF1R recruit and activate STAT3 via RACK1 mediation and induce anoikis resistance (66). Thus, a theoretic autocrine feed-forward loop model is established in which STAT3 activation leads to the binding of nuclear IGFBP2 to IGF1, resulting in IGF1 secretion that activates IGF1R, which in turn activates STAT3 (67).
Trks are a family of tyrosine kinases that regulate synaptic strength and plasticity in the nervous system (68). The Trk family has three members, each of which binds to different types of growth factors called neurotrophins (NTs). TrkA binds to nerve growth factor (NGF) and is also called a high affinity nerve growth factor receptor. TrkB binds to brain-derived neurotrophic factor (BDNF) and NT-4. TrkC binds to NT-3 (69). Upon ligand binding, Trks can activate effectors, including SHC1, FRS2, SH2B1, SH2B2 and PLCG1 (70). Through these effectors, the PI3K/AKT pathway, MAPK pathway and NF-κB are activated (71, 72).
Trks have been reported to affect tumor behavior. TrkA inhibits tumor growth, invasion and angiogenesis, while TrkB promotes anoikis resistance and metastasis (73). Studies have revealed that TrkB contributes to anoikis resistance through the above signaling pathway and EMT (74, 75). TrkB expression is upregulated in lower grade glioma, proneural GBM and methylated phenotype GBM, and TrkB-induced anoikis resistance has been confirmed in glioma (76, 77). Further study showed the participation of Zeb1, an E-cadherin repressor, in TrkB-induced anoikis resistance (78). Thus, TrkB promotes anoikis resistance through the PI3K/AKT pathway, MAPK pathway, NF-κB activation, EMT and E-cadherin inhibition.
TGF-β is a multifunctional cytokine produced by white blood cells. Activated TGF-β forms a serine/threonine kinase complex and binds to TGF-β receptors (TGFBR1 and TGFBR 2) (79). Upon ligand binding, TGFRBR2 phosphorylates and activates TGFRBR1, which initiates a signaling cascade (80). In this way, TGF-β activates the SMAD pathway, which translocates the Smad complex into the nucleus to induce the transcription of different effectors. There are also non-Smad signaling pathways, including the MAPK and PI3K-AKT pathways, that are initiated in parallel and cooperate with the SMAD pathway (81). Through these pathways, TGF-β exerts both tumor-promoting effects and tumor-suppressing effects.
TGF-β signaling is highly active in high-grade gliomas, which exerts tumor-promoting effects on proliferation, angiogenesis and invasion (82). An in vivo study also proved that TGF-β promotes anoikis resistance upon glioma cell detachment (31). As the most potent inducer of EMT, TGF-β mainly promotes anoikis resistance through this transition and the subsequent loss of cell adhesion (83, 84). TGF-β also promotes anoikis resistance mediated by ROS and β-catenin (85, 86).
The Hippo pathway is a well-known signaling pathway that controls organ size by regulating cell proliferation and apoptosis (87). Large tumor suppressor (LATS) 1 and 2 are starting proteins of the Hippo pathway. LATS1/2 negatively regulate the transcriptional coactivator/corepressor YAP and TAZ. YAP/TAZ then bind to the transcription factor family TEAD and direct gene expression in the nucleus (88). The Hippo pathway is not directly activated by the loss of cell adhesion but by the signaling of other pathways (89). For example, YAP/TAZ are regulated by integrin-SRC signaling, E-cadherin-JUB signaling, growth factor-PI3K-Akt signaling and GPCR signaling (90). Thus, the Hippo pathway induces anoikis by transducing upstream signals.
Under physiological conditions, the Hippo pathway downregulates YAP and induces anoikis (91). However, it is one of the 8 signaling pathways that is frequently dysregulated in cancer (89). Evidence shows that YAP1 upregulation promotes anoikis resistance and EGFR amplification in GBM (92). YAP1 also regulates the glioma phenotype by inhibiting proneural marker OLIG2 expression (93). YAP knockdown increases apoptosis and attenuates GBM metastasis (94, 95). The YAP inhibitor verteporfin has been shown to have significant efficacy in a preclinical GBM model, indicating that the Hippo pathway is a promising therapeutic target for glioma (96).
Cell detachment does not affect cytoplasmic proteins directly. However, proteins such as transcription factors and kinases can act as important mediators of anoikis resistance and are usually regulated by multiple pathways. Nuclear factor-κB (NF-κB), elongation factor-2 kinase (eEF-2 kinase) and Mps one binder kinases 2 (MOB2) are known promotors of glioma.
NF-κB is a primary transcription factor that widely participates in cell proliferation and survival control (97, 98). NF-κB is a dimeric complex formed by RelA, RelB, NF-κB1, NF-κB2 and c-Rel. When bound to DNA, different dimer combinations act as transcriptional activators or repressors (99). NF-κB plays a key role in inducing anoikis resistance and is the downstream effector of many signal-transducing events. Inhibitors of κB (IκBs) block the nuclear entry of NF-κB and keep it in an inactive state. Activation of NF-κB is initiated by the signal-induced degradation of IκB proteins with the help of IκB kinase (IKK) (100).
NF-κB is dysregulated in most malignant tumors and causes aberrant expression of several anoikis-related proteins. The anoikis suppressors Bcl-2, BCL-xL, OPG, IAP, c-FLIP, and RIPK1 were upregulated by NF-κB in vivo (101–103). NF-κB is also reported to counter reactive oxygen species (ROS)-induced anoikis through manganese superoxide dismutase (MnSOD) upregulation (104). High-grade and invasive gliomas often exhibit NF-κB dysregulation. NF-κB promotes the expression of ZEB-1 in glioma, which in turn represses E-cadherin and results in anoikis resistance (105). On the other hand, inhibition of the gene transactivation function of NF-κB by cannabidiol induces cell death in GBM (106).
eEF-2 kinase is a calcium/calmodulin-dependent transferase. eEF-2 kinase phosphorylates eEF-2 and decreases its affinity for ribosomes, leading to retardation of protein elongation and therefore inhibition of protein synthesis (107). It was also discovered that eEF-2 kinase regulates dendritic mRNA translation in neurons (108). eEF-2 kinase is often upregulated in cancer cells and promotes proliferation, survival and migration (109).
eEF-2 kinase is upregulated in glioma (110). Upon detachment, the high expression of eEF-2 kinase induces anoikis resistance and promotes invasion through downregulation of Bim and upregulation of BCL-xL (111, 112). On the other hand, inhibition of eEF-2 kinase promotes anoikis and sensitivity to TMZ (113).
MOB2 regulates the activation of nuclear-Dbf2-related (NDR) kinases, which are associated with apoptosis signaling. MOB2 inhibits FAS-induced NDR activation and consequently suppresses apoptosis (114). In vivo and in vitro studies have shown that MOB2 is downregulated in GBM and that downregulation of MOB2 confers anoikis resistance (115). Further experiments revealed that anoikis resistance is achieved through the integrin/FAK/Akt pathway and the cAMP/PKA pathway (114, 115). Few studies have focused on MOB2 as a tumor suppressor, and its therapeutic value remains to be explored.
The TME is the abnormal environment surrounding tumor cells. The TME has pathogenic properties such as hypoxia, acidosis and lack of nutrition. These properties and factors also regulate anoikis.
Hypoxia is a standard property of the TME. Hypoxia-inducible factors (HIFs) are transcription factors that respond to hypoxia and are commonly upregulated in tumor cells. HIF promotes cell survival by upregulating several genes associated with angiogenesis and metastasis (116, 117). Therefore, it is natural for HIF to play a part in cancer cell survival. An in vivo study showed that HIF-1-induced anoikis resistance can be achieved by repression of the BH3-only proteins Bim and Bmf through the EGFR–Mek–Erk and AKT pathways (118, 119). HIF-1α also promotes anoikis resistance by upregulating ANGPTL4, which activates the FAK/Src/PI3K-Akt/ERK pathway and thereby suppresses Caspase activation (120). HIF-1α functions similarly and promotes anoikis resistance in glioma (121).
Acidosis is another property of the TME and is caused partially by hypoxia. Acidosis has dual effects on tumors: mild acidosis promotes survival, and severe acidosis suppresses survival. A study revealed that acidic pH promotes autocrine TGF-β2 signaling and in turn promotes anoikis resistance (122). In vivo studies also show that low pH induces anoikis resistance in melanoma (pH=6.7) and enhances proliferation in GBM (pH=6.2) (123, 124). However, a very low pH (pH=3.4) immensely increases the surface rigidity of GBM cells by altering the cholesterol and GM3 glycosphingolipid content and causes anoikis instead (124).
ROS are highly reactive byproducts of cell metabolism (125). ROS are present at low and stable levels in normal cells and have roles in signaling and homeostasis (126, 127). However, under cancerous conditions, redox homeostasis is dysregulated, and ROS serve as a double-edged sword. Low levels of ROS facilitate cancer cell survival, while high levels of ROS and therapeutic-induced augmentation of ROS stress kill cancer cells (128, 129).
ROS play important roles in achieving anoikis resistance in cancer. An increase in ROS leads to constitutive inactivation of PTEN, which rescues PI3K-AKT signaling and integrin-mediated anoikis resistance (130). Sustained ROS levels also promote the activation of Src kinases and redox-sensitive transcription factors such as NF-κB and HIF-1α, thereby regulating cell adhesion and anoikis through degradation of Bim and upregulation of BCL-xL (130, 131). On the other hand, high levels of ROS promote anoikis sensitivity upon cell detachment, confirming the dual role of ROS in anoikis (132).
ROS function similarly in glioma. Prohibitin is upregulated in glioma stem-like cells (GSCs) and facilitates therapeutic resistance by regulating ROS (133). GBM cells with low Pax6 expression achieve anoikis resistance. Pax6-overexpressing cells retain higher levels of ROS upon detachment, and their survival is impaired (134). Treatments utilizing ROS stress as a mechanism have been successful. Celastrol induces ROS production and inhibits Akt and mTOR activity, thus triggering cell death in glioma (135). Dihydroartemisinin promotes ROS production, which activates p53 protein and in turn downregulates β-catenin expression, thus inhibiting glioma invasion and anoikis resistance (136).
HSPs are a family of proteins that respond to stress events and function as chaperones (137). HSPs are upregulated by heat shock factors (HSFs) during environmental stress conditions (138). The chaperone function of HSP helps stabilize partially unfolded proteins, assisting protein conformation establishment and preventing unwanted protein aggregation (139).
HSP upregulation is triggered in cancer by hypoxia, starvation and cancer therapy. Therefore, HSPs participate in cancer progression and therapy resistance. For example, HSP27 promotes astrocytoma migration through SPARC-MAPK signaling and is suppressed by PTEN (140). HSP70 and its cochaperone BAG3 induce anoikis resistance in GBM through Bcl-2 overexpression (141).
Autophagy is a cellular process that promotes cell adaptation through the degradation of dysfunctional or unnecessary components. Generally, autophagy is triggered upon stress events such as starvation and infection, therefore promoting cell survival. However, autophagy in cancer can be a double-edged sword. While high-level autophagy has an antitumor effect, low-level autophagy can relieve cancer cells of the pressure from hypoxia, acidosis and lack of nutrients. It also facilitates the degradation of apoptotic mediators, thus interfering with apoptosis or anoikis (142). In short, autophagy can play a protective role in cancer and induce anoikis resistance upon cell detachment.
The mechanism of protective autophagy-induced anoikis resistance is not clear, as various involved pathways have been reported. One review concluded that protective autophagy is induced via integrin-focal adhesion signaling (143). Another in vivo study specified the ATF4/CEMIP/PKCα pathway as a trigger of protective autophagy (144). A study in glioma, however, revealed the MDA-9/Syntenin pathway as the key regulator in maintaining protective autophagy (145). These inducers of protective autophagy can be potential therapeutic targets. However, further investigation is required to fully understand the balance between protective autophagy and anoikis.
The common protocol for glioma contains surgery, radiotherapy, conventional chemotherapy, and target therapy (146). Part of these treatments involve anoikis mechanism and their efficacy are affected by anoikis resistance.
Radiotherapy is a standard measure in glioma management, although gliomas often show high radioresistance. A study in prostate cancer shows that one of the reasons for radiotherapy resistance is the acquirement of anoikis resistance in detached tumor cells through Erk and PI3k-Akt signaling (147). Consistently, AKT inhibitor MK2206 is able to enhance the sensitivity of radiotherapy and inhibit invasion in glioma, which could result from the inhibition of anoikis resistance (148). A recent study identifies an adhesion molecule CD146 as a mediator of radiotherapy resistance through suppression of p53 expression and activation of NF-κB, which could induce anoikis resistance (149). Thus, it is highly possible that anoikis resistance takes part in the resistance of radiotherapy in glioma.
Conventional chemotherapy for glioma includes procarbazine, lomustine, carmustine, vincristine, and temozolomide (146). Among them, TMZ is the most common adjuvant treatment. TMZ is an alkylating agent commonly used in gliomas, especially in astrocytoma and GBM. A recent study deliberately cultures anoikis-resistant astrocytes to form clones and test their resistance to TMZ. After treatment with 0.72mM TMZ, only one out of three of these astrocyte-derived clones show a significant decrease in cell viability, with slowed but not halted colony formation. The unsatisfying response to TMZ of these astrocytes is similar to that of the established GBM cell line U87MG (150). Therefore, anoikis resistance is likely one of the main reasons for TMZ resistance.
Target therapies exceed conventional chemotherapy in preventing drug resistance and often generate a synergistic response. Therefore, target therapies aiming at the anoikis mechanism are an ideal adjuvant treatment for glioma. Target drugs relating to anoikis are concluded in Table 1.
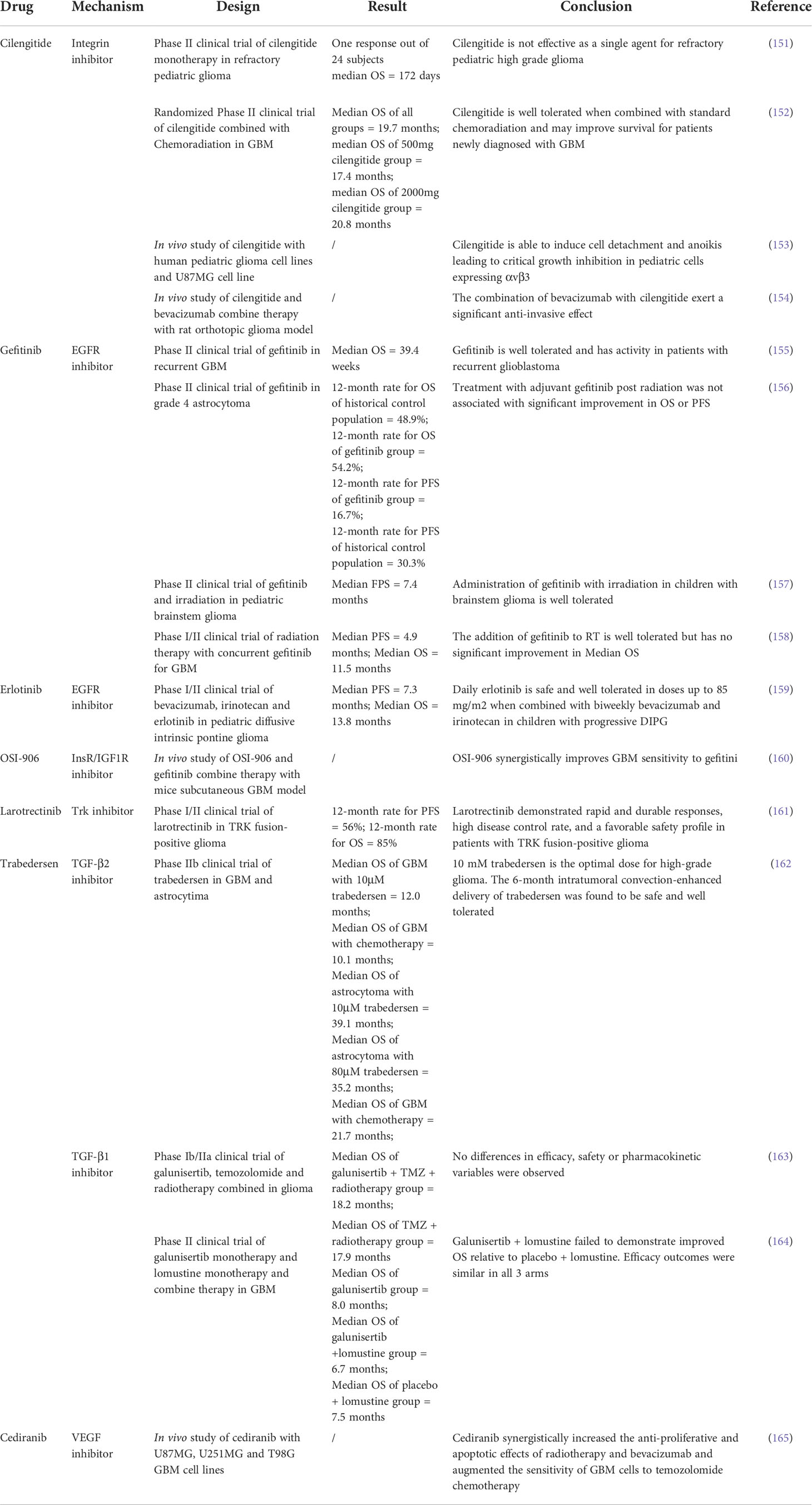
Table 1 Anoikis related.drugs.
Cilengitide, an integrin inhibitor, is able to induce dose-dependent cell detachment of pediatric glioma cells expressing αvβ3 and trigger anoikis. However, the adult glioma cell line U87MG is able to grow despite cilengitide-induced nonadherent conditions. These results indicate that cilengitide cannot reduce anoikis resistance (153). Consistent with this conclusion, only 1 out of 24 subjects has a response in phase 2 clinical trials of cilengitide treating high-grade pediatric gliomas (151). Despite the unsatisfying effect of cilengitide as monotherapy, combined therapy trials with radiation and TMZ, bevacizumab, or other agents have shown promising outcomes (152, 154, 166). Therefore, integrin inhibitors are still reliable adjuvant treatments for glioma treatment.
EGFR inhibitors such as erlotinib and gefitinib have been developed and applied in the clinical treatment of many other cancers. However, despite the common amplification of EGFR in GBM, EGFR inhibitors have been disappointing in single drug clinical trials (155, 156). Nevertheless, combined therapies have shown promising results. Combined treatment of erlotinib with bevacizumab and irinotecan has shown prolonged OS (Median OS = 13.8 months) in children with progressive diffuse intrinsic pontine glioma (159). Adjuvant gefitinib treatment also improved radiotherapy outcome in children (Median PFS =7.4 months) but not in adult (Median PFS = 4.9 months; Median OS = 11.5 months) (157, 158). Thus, EGFR inhibitors are recommended adjuvant treatments in pediatric glioma.
IGF1R inhibitor based treatment has not been successful, and multiple clinical trials of ganitumab have been halted due to lack of benefit (167). However, it is reported that the frequent activation of IR and IGF1R in GBM confers resistance to EGFR inhibitors, and IGF1R has a compensatory effect during EGFR inhibition (160, 168). These finding indicate a promising future for combined treatment with EGFR inhibitor and IGFR inhibitor.
TRK fusion leads to constitutive activation of Trk in all types of gliomas (169). The Trk inhibitors entrectinib and larotrectinib have been approved for the treatment of TRK-fusion glioma (170). While both drugs are still in trial, larotrectinib has demonstrated favorable efficacy in treating glioma (23 responses of 28 subjects; 12-month rate for PFS = 56%; 12-month rate for OS = 85%) (161). Therefore, Trk inhibitors are expected in the future management of TRK-fusion glioma.TGF-β pathway antagonist like trabedersen has been proposed for glioma treatment for a long time (82). A clinical trial of 145 patients shows that trabedersen improves OS in GBM (Median OS 12.0 months vs. 10.1 months) and astrocytoma (Median OS 39.1 months vs. 21.7 months) (162). Another antagonist galunisertib is also trialed in glioma but shows insignificant improvement (Median OS 18.2 months vs. 17.9 months) (Median OS 8.0 months vs. 7.5 months) (163, 164). Many other novel antagonists are developed for cancer treatment, and combined therapies are also expected in the future (171).
HSP is a promising therapeutic target in terms of both monotherapy and combined therapy. A clinical trial shows that immunotherapy targeting HSP96 improves OS in recurrent GBM (Median OS 47 weeks vs. 16 weeks) (172). And an in vivo study shows that silencing HSP27 and HSP72 improves the efficacy of TMZ in glioma (173).
Apart from therapies that target the anoikis mechanism, cediranib, a VEGF inhibitor also affects anoikis resistance in GBM. Study shows that cediranib reduce the survival of anoikis-resistant GBM cells, especially in the T98G cell line, while the U87MG cell line shows intermediate resistance. The study also reveals the synergy effect of cediranib with radiotherapy and TMZ in glioma treatment (165). There has been no clinical trial for cediranib in glioma up till now. However, anti-VEGF drugs like bevacizumab and regorafenib are already in the guideline for glioma management (146).
Currently, anoikis resistance in glioma has not been extensively studied. Nevertheless, the diffuseness of glioma invasion and the high recurrence rate are closely linked to anoikis resistance. Therefore, clarification of the regulation of anoikis resistance at the molecular level could provide a new angle for glioma therapy and prognosis evaluation. In this article, we reviewed current publications on the role of adhesion molecules, signaling pathways, cytoplasmic molecules, the TME and protective autophagy in anoikis resistance and their therapeutic value in glioma. Further studies, however, are still needed to validate the function of these molecules in anoikis resistance and glioma. More research is necessary to explore new directions for targeting anoikis resistance in the treatment of glioma.
ZZ, CF, and HX designed the review and wrote the manuscript. LY and YX conceived the artwork and performed the bibliographical research. AS, AZ, and ML supervised the writing. All the authors revised and approved the final version of the manuscript.
This work was supported by the Natural science foundation of Shanghai (18ZR1430400) and the Zhejiang Provincial Natural Science Foundation of China (LY22H090020).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Huang W, Zhong Z, Luo C, Xiao Y, Li L, Zhang X, et al. The miR-26a/AP-2α/Nanog signaling axis mediates stem cell self-renewal and temozolomide resistance in glioma. Theranostics (2019) 9:5497–516. doi: 10.7150/thno.33800
2. Tong L, Li J, Li Q, Wang X, Medikonda R, Zhao T, et al. ACT001 reduces the expression of PD-L1 by inhibiting the phosphorylation of STAT3 in glioblastoma. Theranostics (2020) 10:5943–56. doi: 10.7150/thno.41498
3. Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genet (2012) 205:613–21. doi: 10.1016/j.cancergen.2012.10.009
4. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
5. Wang H, Xu T, Huang Q, Jin W, Chen J. Immunotherapy for malignant glioma: Current status and future directions. Trends Pharmacol Sci (2020) 41:123–38. doi: 10.1016/j.tips.2019.12.003
6. Gritsenko PG, Atlasy N, Dieteren CEJ, Navis AC, Venhuizen J-H, Veelken C, et al. p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat Cell Biol (2020) 22:97–107. doi: 10.1038/s41556-019-0443-x
7. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci (2010) 123:4195–200. doi: 10.1242/jcs.023820
8. Song J, Liu Y, Liu F, Zhang L, Li G, Yuan C, et al. The 14-3-3σ protein promotes HCC anoikis resistance by inhibiting EGFR degradation and thereby activating the EGFR-dependent ERK1/2 signaling pathway. Theranostics (2021) 11:996–1015. doi: 10.7150/thno.51646
9. Ye G, Yang Q, Lei X, Zhu X, Li F, He J, et al. Nuclear MYH9-induced CTNNB1 transcription, targeted by staurosporin, promotes gastric cancer cell anoikis resistance and metastasis. Theranostics (2020) 10:7545–60. doi: 10.7150/thno.46001
10. Zhao H, Yan G, Zheng L, Zhou Y, Sheng H, Wu L, et al. STIM1 is a metabolic checkpoint regulating the invasion and metastasis of hepatocellular carcinoma. Theranostics (2020) 10:6483–99. doi: 10.7150/thno.44025
11. Rath E, Moschetta A, Haller D. Mitochondrial function - gatekeeper of intestinal epithelial cell homeostasis. Nat Rev Gastroenterol Hepatol (2018) 15:497–516. doi: 10.1038/s41575-018-0021-x
12. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev (2007) 87:99–163. doi: 10.1152/physrev.00013.2006
13. Zhao B, Wang Z, Liang X, Wang X, Lin K, Yuan L, et al. Inhibition of the postsynaptic density protein 95 on the protective effect of ang-(1-7)-Mas on cerebral ischaemia injury. Stroke Vasc Neurol (2022) 84(1):9–19. doi: 10.1136/svn-2021-001396
14. Ola MS, Nawaz M, Ahsan H. Role of bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem (2011) 351:41–58. doi: 10.1007/s11010-010-0709-x
15. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta (2013) 1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026
16. Wajant H. The fas signaling pathway: more than a paradigm. Science (2002) 296:1635–6. doi: 10.1126/science.1071553
17. Liu S, Han S, Dai Q, Li S, Li J. BICAO-induced ischaemia caused depressive-like behaviours and caspase-8/-9-dependent brain regional neural cell apoptosis in mice. Stroke Vasc Neurol (2018) 3:1–8. doi: 10.1136/svn-2017-000109
18. Mandal R, Barron JC, Kostova I, Becker S, Strebhardt K. Caspase-8: The double-edged sword. Biochim Biophys Acta Rev Cancer (2020) 1873:188357. doi: 10.1016/j.bbcan.2020.188357
19. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol (2015) 15:362–74. doi: 10.1038/nri3834
20. Goel HL, Breen M, Zhang J, Das I, Aznavoorian-Cheshire S, Greenberg NM, et al. beta1A integrin expression is required for type 1 insulin-like growth factor receptor mitogenic and transforming activities and localization to focal contacts. Cancer Res (2005) 65:6692–700. doi: 10.1158/0008-5472.CAN-04-4315
21. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell (2002) 110:673–87. doi: 10.1016/S0092-8674(02)00971-6
22. Schnell O, Krebs B, Wagner E, Romagna A, Beer AJ, Grau SJ, et al. Expression of integrin alphavbeta3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature. Brain Pathol (2008) 18:378–86. doi: 10.1111/j.1750-3639.2008.00137.x
23. Zhao J, Guan J-L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev (2009) 28:35–49. doi: 10.1007/s10555-008-9165-4
24. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer (2014) 14:598–610. doi: 10.1038/nrc3792
25. Paolillo M, Galiazzo MC, Daga A, Ciusani E, Serra M, Colombo L, et al. An RGD small-molecule integrin antagonist induces detachment-mediated anoikis in glioma cancer stem cells. Int J Oncol (2018) 53:2683–94. doi: 10.3892/ijo.2018.4583
26. Russo MA, Paolillo M, Sanchez-Hernandez Y, Curti D, Ciusani E, Serra M, et al. A small-molecule RGD-integrin antagonist inhibits cell adhesion, cell migration and induces anoikis in glioblastoma cells. Int J Oncol (2013) 42:83–92. doi: 10.3892/ijo.2012.1708
27. Kesanakurti D, Chetty C, Dinh DH, Gujrati M, Rao JS. Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with alpha5beta1 integrin in glioma. Oncogene (2013) 32:327–40. doi: 10.1038/onc.2012.52
28. Renner G, Janouskova H, Noulet F, Koenig V, Guerin E, Bar S, et al. Integrin alpha5beta1 and p53 convergent pathways in the control of anti-apoptotic proteins PEA-15 and survivin in high-grade glioma. Cell Death Differ (2016) 23:640–53. doi: 10.1038/cdd.2015.131
29. Kesanakurti D, Chetty C, Rajasekhar Maddirela D, Gujrati M, Rao JS. Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell Death Dis (2012) 3:e445. doi: 10.1038/cddis.2012.182
30. Roth P, Silginer M, Goodman SL, Hasenbach K, Thies S, Maurer G, et al. Integrin control of the transforming growth factor-beta pathway in glioblastoma. Brain (2013) 136:564–76. doi: 10.1093/brain/aws351
31. Silginer M, Weller M, Ziegler U, Roth P. Integrin inhibition promotes atypical anoikis in glioma cells. Cell Death Dis (2014) 5:e1012. doi: 10.1038/cddis.2013.543
32. Silginer M, Burghardt I, Gramatzki D, Bunse L, Leske H, Rushing EJ, et al. The aryl hydrocarbon receptor links integrin signaling to the TGF-β pathway. Oncogene (2016) 35:3260–71. doi: 10.1038/onc.2015.387
33. Delamarre E, Taboubi S, Mathieu S, Berenguer C, Rigot V, Lissitzky JC, et al. Expression of integrin alpha6beta1 enhances tumorigenesis in glioma cells. Am J Pathol (2009) 175:844–55. doi: 10.2353/ajpath.2009.080920
34. Fujita M, Sasada M, Iyoda T, Nagai R, Kudo C, Yamamoto T, et al. Anoikis resistance conferred by tenascin-c-derived peptide TNIIIA2 and its disruption by integrin inactivation. Biochem Biophys Res Commun (2021) 536:14–9. doi: 10.1016/j.bbrc.2020.12.050
35. Marie PJ, Hay E, Modrowski D, Revollo L, Mbalaviele G, Civitelli R. Cadherin-mediated cell-cell adhesion and signaling in the skeleton. Calcif Tissue Int (2014) 94:46–54. doi: 10.1007/s00223-013-9733-7
36. Gayrard C, Bernaudin C, Dejardin T, Seiler C, Borghi N. Src- and confinement-dependent FAK activation causes e-cadherin relaxation and beta-catenin activity. J Cell Biol (2018) 217:1063–77. doi: 10.1083/jcb.201706013
37. Minde DP, Anvarian Z, Rüdiger SG, Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer (2011) 10:101. doi: 10.1186/1476-4598-10-101
38. Tian X, Liu Z, Niu B, Zhang J, Tan TK, Lee SR, et al. E-cadherin/beta-catenin complex and the epithelial barrier. J BioMed Biotechnol (2011) 2011:567305. doi: 10.1155/2011/567305
39. Weng Z, Xin M, Pablo L, Grueneberg D, Hagel M, Bain G, et al. Protection against anoikis and down-regulation of cadherin expression by a regulatable beta-catenin protein. J Biol Chem (2002) 277:18677–86. doi: 10.1074/jbc.M105331200
40. Orford K, Orford CC, Byers SW. Exogenous expression of beta-catenin regulates contact inhibition, anchorage-independent growth, anoikis, and radiation-induced cell cycle arrest. J Cell Biol (1999) 146:855–68. doi: 10.1083/jcb.146.4.855
41. Condello S, Morgan CA, Nagdas S, Cao L, Turek J, Hurley TD, et al. β-catenin-regulated ALDH1A1 is a target in ovarian cancer spheroids. Oncogene (2015) 34:2297–308. doi: 10.1038/onc.2014.178
42. Xu S-L, Liu S, Cui W, Shi Y, Liu Q, Duan J-J, et al. Aldehyde dehydrogenase 1A1 circumscribes high invasive glioma cells and predicts poor prognosis. Am J Cancer Res (2015) 5:1471–83.
43. Wang W, Shen X-B, Huang D-B, Jia W, Liu W-B, He Y-F. Peroxiredoxin 4 suppresses anoikis and augments growth and metastasis of hepatocellular carcinoma cells through the β-catenin/ID2 pathway. Cell Oncol (Dordr) (2019) 42:769–81. doi: 10.1007/s13402-019-00460-0
44. Tiwari N, Gheldof A, Tatari M, Christofori G. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol (2012) 22:194–207. doi: 10.1016/j.semcancer.2012.02.013
45. Serrano-Saenz S, Palacios C, Delgado-Bellido D, López-Jiménez L, Garcia-Diaz A, Soto-Serrano Y, et al. PIM kinases mediate resistance of glioblastoma cells to TRAIL by a p62/SQSTM1-dependent mechanism. Cell Death Dis (2019) 10:51. doi: 10.1038/s41419-018-1293-3
46. Peyre L, Meyer M, Hofman P, Roux J. TRAIL receptor-induced features of epithelial-to-mesenchymal transition increase tumour phenotypic heterogeneity: potential cell survival mechanisms. Br J Cancer (2021) 124:91–101. doi: 10.1038/s41416-020-01177-w
47. Costa HA, Leitner MG, Sos ML, Mavrantoni A, Rychkova A, Johnson JR, et al. Discovery and functional characterization of a neomorphic PTEN mutation. Proc Natl Acad Sci (2015) 112:13976–81. doi: 10.1073/pnas.1422504112
48. Edwards LA, Thiessen B, Dragowska WH, Daynard T, Bally MB, Dedhar S. Inhibition of ILK in PTEN-mutant human glioblastomas inhibits PKB/Akt activation, induces apoptosis, and delays tumor growth. Oncogene (2005) 24:3596–605. doi: 10.1038/sj.onc.1208427
49. Kelly DM, Li L, Burgess AI, Poole DL, Duerden JM, Rothwell PM. Associations of blood biomarkers with glomerular filtration rate in patients with TIA and stroke: population-based study. Stroke Vasc Neurol (2021) 6:48–56. doi: 10.1136/svn-2020-000422
50. Kumagai S, Koyama S, Nishikawa H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat Rev Cancer (2021) 21:181–97. doi: 10.1038/s41568-020-00322-0
51. Yarden Y, Shilo BZ. SnapShot: EGFR signaling pathway. Cell (2007) 131:1018. doi: 10.1016/j.cell.2007.11.013
52. Runkle KB, Kharbanda A, Stypulkowski E, Cao XJ, Wang W, Garcia BA, et al. Inhibition of DHHC20-mediated EGFR palmitoylation creates a dependence on EGFR signaling. Mol Cell (2016) 62:385–96. doi: 10.1016/j.molcel.2016.04.003
53. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
54. Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, et al. Integrins and EGFR coordinately regulate the pro-apoptotic protein bim to prevent anoikis. Nat Cell Biol (2003) 5:733–40. doi: 10.1038/ncb1026
55. Buchheit CL, Angarola BL, Steiner A, Weigel KJ, Schafer ZT. Anoikis evasion in inflammatory breast cancer cells is mediated by bim-EL sequestration. Cell Death Differ (2015) 22:1275–86. doi: 10.1038/cdd.2014.209
56. Shao N, Lu Z, Zhang Y, Wang M, Li W, Hu Z, et al. Interleukin-8 upregulates integrin beta3 expression and promotes estrogen receptor-negative breast cancer cell invasion by activating the PI3K/Akt/NF-kappaB pathway. Cancer Lett (2015) 364:165–72. doi: 10.1016/j.canlet.2015.05.009
57. Schlessinger K, Levy DE. Malignant transformation but not normal cell growth depends on signal transducer and activator of transcription 3. Cancer Res (2005) 65:5828–34. doi: 10.1158/0008-5472.CAN-05-0317
58. Jiang C, Zhu Y, Zhou Z, Gumin J, Bengtsson L, Wu W, et al. TMEM43/LUMA is a key signaling component mediating EGFR-induced NF-kappaB activation and tumor progression. Oncogene (2017) 36:2813–23. doi: 10.1038/onc.2016.430
59. Mancarella C, Scotlandi K. IGF system in sarcomas: a crucial pathway with many unknowns to exploit for therapy. J Mol Endocrinol (2018) 61:T45–60. doi: 10.1530/JME-17-0250
60. Burnier JV, Wang N, Michel RP, Hassanain M, Li S, Lu Y, et al. Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene (2011) 30:3766–83. doi: 10.1038/onc.2011.89
61. Valentinis B, Morrione A, Peruzzi F, Prisco M, Reiss K, Baserga R. Anti-apoptotic signaling of the IGF-I receptor in fibroblasts following loss of matrix adhesion. Oncogene (1999) 18:1827–36. doi: 10.1038/sj.onc.1202471
62. Grey A, Chen Q, Xu X, Callon K, Cornish J. Parallel phosphatidylinositol-3 kinase and p42/44 mitogen-activated protein kinase signaling pathways subserve the mitogenic and antiapoptotic actions of insulin-like growth factor I in osteoblastic cells. Endocrinology (2003) 144:4886–93. doi: 10.1210/en.2003-0350
63. Luey BC, May FE. Insulin-like growth factors are essential to prevent anoikis in oestrogen-responsive breast cancer cells: importance of the type I IGF receptor and PI3-kinase/Akt pathway. Mol Cancer (2016) 15:8. doi: 10.1186/s12943-015-0482-2
64. Fernandez S, Genis L, Torres-Aleman I. A phosphatase-independent gain-of-function mutation in PTEN triggers aberrant cell growth in astrocytes through an autocrine IGF-1 loop. Oncogene (2014) 33:4114–22. doi: 10.1038/onc.2013.376
65. Phillips LM, Zhou X, Cogdell DE, Chua CY, Huisinga A, Hess RK, et al. Glioma progression is mediated by an addiction to aberrant IGFBP2 expression and can be blocked using anti-IGFBP2 strategies. J Pathol (2016) 239:355–64. doi: 10.1002/path.4734
66. Zhang W, Zong CS, Hermanto U, Lopez-Bergami P, Ronai Z, Wang LH. RACK1 recruits STAT3 specifically to insulin and insulin-like growth factor 1 receptors for activation, which is important for regulating anchorage-independent growth. Mol Cell Biol (2006) 26:413–24. doi: 10.1128/MCB.26.2.413-424.2006
67. Tan MSY, Sandanaraj E, Chong YK, Lim SW, Koh LWH, Ng WH, et al. A STAT3-based gene signature stratifies glioma patients for targeted therapy. Nat Commun (2019) 10:3601. doi: 10.1038/s41467-019-11614-x
68. Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem (2003) 72:609–42. doi: 10.1146/annurev.biochem.72.121801.161629
69. Segal RA. Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci (2003) 26:299–330. doi: 10.1146/annurev.neuro.26.041002.131421
70. Haniu M, Talvenheimo J, Le J, Katta V, Welcher A, Rohde MF. Extracellular domain of neurotrophin receptor trkB: disulfide structure, n-glycosylation sites, and ligand binding. Arch Biochem Biophys (1995) 322:256–64. doi: 10.1006/abbi.1995.1460
71. Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J. The atypical protein kinase c-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J Biol Chem (2001) 276:7709–12. doi: 10.1074/jbc.C000869200
72. Jing S, Tapley P, Barbacid M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron (1992) 9:1067–79. doi: 10.1016/0896-6273(92)90066-M
73. Thiele CJ, Li Z, McKee AE. On trk–the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin Cancer Res (2009) 15:5962–7. doi: 10.1158/1078-0432.CCR-08-0651
74. Geiger TR, Peeper DS. Critical role for TrkB kinase function in anoikis suppression, tumorigenesis, and metastasis. Cancer Res (2007) 67:6221–9. doi: 10.1158/0008-5472.CAN-07-0121
75. Ricci A, De Vitis C, Noto A, Fattore L, Mariotta S, Cherubini E, et al. TrkB is responsible for EMT transition in malignant pleural effusions derived cultures from adenocarcinoma of the lung. Cell Cycle (2013) 12:1696–703. doi: 10.4161/cc.24759
76. Pinheiro KV, Thomaz A, Souza BK, Metcalfe VA, Freire NH, Brunetto AT, et al. Expression and pharmacological inhibition of TrkB and EGFR in glioblastoma. Mol Biol Rep (2020) 47:6817–28. doi: 10.1007/s11033-020-05739-2
77. Jiang L, Chen S, Zhao D, Yan J, Chen J, Yang C, et al. MNX1 reduces sensitivity to anoikis by activating TrkB in human glioma cells. Mol Med Rep (2018) 18:3271–9. doi: 10.3892/mmr.2018.9329
78. Smit MA, Peeper DS. Zeb1 is required for TrkB-induced epithelial-mesenchymal transition, anoikis resistance and metastasis. Oncogene (2011) 30:3735–44. doi: 10.1038/onc.2011.96
79. Shi A, Li J, Qiu X, Sabbah M, Boroumand S, Huang TC, et al. TGF-β loaded exosome enhances ischemic wound healing in vitro and in vivo. Theranostics (2021) 11:6616–31. doi: 10.7150/thno.57701
80. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol (2012) 13:616–30. doi: 10.1038/nrm3434
81. Zhang YE. Non-smad signaling pathways of the TGF-beta family. Cold Spring Harb Perspect Biol (2017) 9(2):a022129. doi: 10.1101/cshperspect.a022129
82. Joseph JV, Balasubramaniyan V, Walenkamp A, Kruyt FA. TGF-beta as a therapeutic target in high grade gliomas - promises and challenges. Biochem Pharmacol (2013) 85:478–85. doi: 10.1016/j.bcp.2012.11.005
83. Chen N, Balasenthil S, Reuther J, Frayna A, Wang Y, Chandler DS, et al. DEAR1 is a chromosome 1p35 tumor suppressor and master regulator of TGF-beta-driven epithelial-mesenchymal transition. Cancer Discovery (2013) 3:1172–89. doi: 10.1158/2159-8290.CD-12-0499
84. Gupta S, Maitra A. EMT: Matter of life or death? Cell (2016) 164:840–2. doi: 10.1016/j.cell.2016.02.024
85. Feng XX, Luo J, Liu M, Yan W, Zhou ZZ, Xia YJ, et al. Sirtuin 6 promotes transforming growth factor-beta1/H2O2/HOCl-mediated enhancement of hepatocellular carcinoma cell tumorigenicity by suppressing cellular senescence. Cancer Sci (2015) 106:559–66. doi: 10.1111/cas.12632
86. Zhu X, Wang K, Chen Y. Ophiopogonin d suppresses TGF-β1-mediated metastatic behavior of MDA-MB-231 breast carcinoma cells via regulating ITGB1/FAK/Src/AKT/β-catenin/MMP-9 signaling axis. Toxicol In Vitro (2020) 69:104973. doi: 10.1016/j.tiv.2020.104973
87. Wu W, Ziemann M, Huynh K, She G, Pang ZD, Zhang Y, et al. Activation of hippo signaling pathway mediates mitochondria dysfunction and dilated cardiomyopathy in mice. Theranostics (2021) 11:8993–9008. doi: 10.7150/thno.62302
88. Saucedo LJ, Edgar BA. Filling out the hippo pathway. Nat Rev Mol Cell Biol (2007) 8:613–21. doi: 10.1038/nrm2221
89. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell (2018) 173(2):321–37.e10. doi: 10.1016/j.cell.2018.03.035
90. Thompson BJ. YAP/TAZ: Drivers of tumor growth, metastasis, and resistance to therapy. Bioessays (2020) 42:e1900162. doi: 10.1002/bies.201900162
91. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev (2012) 26:54–68. doi: 10.1101/gad.173435.111
92. Vigneswaran K, Boyd NH, Oh SY, Lallani S, Boucher A, Neill SG, et al. YAP/TAZ transcriptional coactivators create therapeutic vulnerability to verteporfin in EGFR-mutant glioblastoma. Clin Cancer Res (2021) 27:1553–69. doi: 10.1158/1078-0432.CCR-20-0018
93. Guichet PO, Masliantsev K, Tachon G, Petropoulos C, Godet J, Larrieu D, et al. Fatal correlation between YAP1 expression and glioma aggressiveness: clinical and molecular evidence. J Pathol (2018) 246:205–16. doi: 10.1002/path.5133
94. Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev (2008) 22:3320–34. doi: 10.1101/gad.1726608
95. Yu OM, Benitez JA, Plouffe SW, Ryback D, Klein A, Smith J, et al. Transcriptional co-activators of RhoA-mediated gene expression, are critical for glioblastoma tumorigenicity. Oncogene (2018) 37:5492–507. doi: 10.1038/s41388-018-0301-5
96. Barrette AM, Ronk H, Joshi T, Mussa Z, Mehrotra M, Bouras A, et al. Anti-invasive efficacy and survival benefit of the YAP-TEAD inhibitor verteporfin in preclinical glioblastoma models. Neuro-oncology (2022) 24:694–707. doi: 10.1093/neuonc/noab244
97. Lu C, Gao S, Xu G. Geraniin inhibits TNF-α-induced impairments of osteogenesis through NF-κB and p38 MAPK signalling pathways in bone marrow stem cells. Stroke Vasc Neurol (2017) 2:47–52. doi: 10.1136/svn-2016-000046
98. Tan Y, Sun R, Liu L, Yang D, Xiang Q, Li L, et al. Tumor suppressor DRD2 facilitates M1 macrophages and restricts NF-κB signaling to trigger pyroptosis in breast cancer. Theranostics (2021) 11:5214–31. doi: 10.7150/thno.58322
99. Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell (1998) 95:749–58. doi: 10.1016/S0092-8674(00)81698-0
100. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol (2000) 18:621–63. doi: 10.1146/annurev.immunol.18.1.621
101. Toruner M, Fernandez-Zapico M, Sha JJ, Pham L, Urrutia R, Egan LJ. Antianoikis effect of nuclear factor-kappaB through up-regulated expression of osteoprotegerin, BCL-2, and IAP-1. J Biol Chem (2006) 281:8686–96. doi: 10.1074/jbc.M512178200
102. Park SH, Riley P.t., Frisch SM. Regulation of anoikis by deleted in breast cancer-1 (DBC1) through NF-kappaB. Apoptosis (2013) 18:949–62. doi: 10.1007/s10495-013-0847-1
103. Hawk MA, Gorsuch CL, Fagan P, Lee C, Kim SE, Hamann JC, et al. RIPK1-mediated induction of mitophagy compromises the viability of extracellular-matrix-detached cells. Nat Cell Biol (2018) 20:272–84. doi: 10.1038/s41556-018-0034-2
104. Kamarajugadda S, Cai Q, Chen H, Nayak S, Zhu J, He M, et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis (2013) 4:e504. doi: 10.1038/cddis.2013.20
105. Edwards LA, Woolard K, Son MJ, Li A, Lee J, Ene C, et al. Effect of brain- and tumor-derived connective tissue growth factor on glioma invasion. J Natl Cancer Inst (2011) 103:1162–78. doi: 10.1093/jnci/djr224
106. Volmar MNM, Cheng J, Alenezi H, Richter S, Haug A, Hassan Z, et al. Cannabidiol converts NF-κB into a tumor suppressor in glioblastoma with defined antioxidative properties. Neuro-oncology (2021) 23:1898–910. doi: 10.1093/neuonc/noab095
107. Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature (1988) 334:170–3. doi: 10.1038/334170a0
108. Heise C, Gardoni F, Culotta L, di Luca M, Verpelli C, Sala C. Elongation factor-2 phosphorylation in dendrites and the regulation of dendritic mRNA translation in neurons. Front Cell Neurosci (2014) 8:35. doi: 10.3389/fncel.2014.00035
109. Ashour AA, Abdel-Aziz A-AH, Mansour AM, Alpay SN, Huo L, Ozpolat B. Targeting elongation factor-2 kinase (eEF-2K) induces apoptosis in human pancreatic cancer cells. Apoptosis an Int J On Programmed Cell Death (2014) 19:241–58. doi: 10.1007/s10495-013-0927-2
110. Wu H, Yang J-M, Jin S, Zhang H, Hait WN. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res (2006) 66:3015–23. doi: 10.1158/0008-5472.CAN-05-1554
111. Zhang L, Zhang Y, Liu XY, Qin ZH, Yang JM. Expression of elongation factor-2 kinase contributes to anoikis resistance and invasion of human glioma cells. Acta Pharmacol Sin (2011) 32:361–7. doi: 10.1038/aps.2010.213
112. Zhang Y, Cheng Y, Zhang L, Ren X, Huber-Keener KJ, Lee S, et al. Inhibition of eEF-2 kinase sensitizes human glioma cells to TRAIL and down-regulates bcl-xL expression. Biochem Biophys Res Commun (2011) 414:129–34. doi: 10.1016/j.bbrc.2011.09.038
113. Liu X-Y, Zhang L, Wu J, Zhou L, Ren Y-J, Yang W-Q, et al. Inhibition of elongation factor-2 kinase augments the antitumor activity of temozolomide against glioma. PloS One (2013) 8:e81345. doi: 10.1371/journal.pone.0081345
114. Kohler RS, Schmitz D, Cornils H, Hemmings BA, Hergovich A. Differential NDR/LATS interactions with the human MOB family reveal a negative role for human MOB2 in the regulation of human NDR kinases. Mol Cell Biol (2010) 30:4507–20. doi: 10.1128/MCB.00150-10
115. Jiang K, Yao G, Hu L, Yan Y, Liu J, Shi J, et al. MOB2 suppresses GBM cell migration and invasion via regulation of FAK/Akt and cAMP/PKA signaling. Cell Death Dis (2020) 11:230. doi: 10.1038/s41419-020-2381-8
116. Benizri E, Ginouvès A, Berra E. The magic of the hypoxia-signaling cascade. Cell Mol Life Sci (2008) 65:1133–49. doi: 10.1007/s00018-008-7472-0
117. Formenti F, Constantin-Teodosiu D, Emmanuel Y, Cheeseman J, Dorrington KL, Edwards LM, et al. Regulation of human metabolism by hypoxia-inducible factor. Proc Natl Acad Sci United States America (2010) 107:12722–7. doi: 10.1073/pnas.1002339107
118. Whelan KA, Caldwell SA, Shahriari KS, Jackson SR, Franchetti LD, Johannes GJ, et al. Hypoxia suppression of bim and bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol Biol Cell (2010) 21:3829–37. doi: 10.1091/mbc.e10-04-0353
119. Whelan KA, Schwab LP, Karakashev SV, Franchetti L, Johannes GJ, Seagroves TN, et al. The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and anoikis resistance. J Biol Chem (2013) 288:15865–77. doi: 10.1074/jbc.M112.426999
120. Baba K, Kitajima Y, Miyake S, Nakamura J, Wakiyama K, Sato H, et al. Hypoxia-induced ANGPTL4 sustains tumour growth and anoikis resistance through different mechanisms in scirrhous gastric cancer cell lines. Sci Rep (2017) 7:11127. doi: 10.1038/s41598-017-11769-x
121. Maurer GD, Brucker DP, Steinbach JP. Loss of cell-matrix contact increases hypoxia-inducible factor-dependent transcriptional activity in glioma cells. Biochem Biophys Res Commun (2019) 515:77–84. doi: 10.1016/j.bbrc.2019.05.115
122. Corbet C, Bastien E, Santiago de Jesus JP, Dierge E, Martherus R, Vander Linden C, et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat Commun (2020) 11(11):454. doi: 10.1073/pnas.1721650115
123. Peppicelli S, Ruzzolini J, Bianchini F, Andreucci E, Nediani C, Laurenzana A, et al. Anoikis resistance as a further trait of acidic-adapted melanoma cells. J Oncol (2019) 2019:8340926. doi: 10.1155/2019/8340926
124. John S, Sivakumar KC, Mishra R. Extracellular proton concentrations impacts LN229 glioblastoma tumor cell fate via differential modulation of surface lipids. Front Oncol (2017) 7:20. doi: 10.3389/fonc.2017.00020
125. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics (2021) 11:1845–63. doi: 10.7150/thno.50905
126. Herb M, Gluschko A, Schramm M. Reactive oxygen species: Not omnipresent but important in many locations. Front Cell Dev Biol (2021) 9:716406. doi: 10.3389/fcell.2021.716406
127. Wan J, Ren H, Wang J, toxicity I. Lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc Neurol (2019) 4:93–5. doi: 10.1136/svn-2018-000205
128. Toler SM, Noe D, Sharma A. Selective enhancement of cellular oxidative stress by chloroquine: implications for the treatment of glioblastoma multiforme. Neurosurg Focus (2006) 21:E10. doi: 10.3171/foc.2006.21.6.1
129. Ramsey MR, Sharpless NE. ROS as a tumour suppressor? Nat Cell Biol (2006) 8:1213–5. doi: 10.1038/ncb1106-1213
130. Parri M, Chiarugi P. Redox molecular machines involved in tumor progression. Antioxid Redox Signal (2013) 19:1828–45. doi: 10.1089/ars.2012.5040
131. Giannoni E, Fiaschi T, Ramponi G, Chiarugi P. Redox regulation of anoikis resistance of metastatic prostate cancer cells: key role for src and EGFR-mediated pro-survival signals. Oncogene (2009) 28:2074–86. doi: 10.1038/onc.2009.77
132. Li S, Mao Y, Zhou T, Luo C, Xie J, Qi W, et al. Manganese superoxide dismutase mediates anoikis resistance and tumor metastasis in nasopharyngeal carcinoma. Oncotarget (2016) 7:32408–20. doi: 10.18632/oncotarget.8717
133. Huang H, Zhang S, Li Y, Liu Z, Mi L, Cai Y, et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat Commun (2021) 12:3720. doi: 10.1038/s41467-021-24108-6
134. Chang JY, Hu Y, Siegel E, Stanley L, Zhou Y-H. PAX6 increases glioma cell susceptibility to detachment and oxidative stress. J Neurooncol 84 (2007) 84(1):9–19. doi: 10.1007/s11060-007-9347-x
135. Liu X, Zhao P, Wang X, Wang L, Zhu Y, Song Y, et al. Celastrol mediates autophagy and apoptosis via the ROS/JNK and Akt/mTOR signaling pathways in glioma cells. J Exp Clin Cancer Res (2019) 38:184. doi: 10.1186/s13046-019-1173-4
136. Que Z, Wang P, Hu Y, Xue Y, Liu X, Qu C, et al. Dihydroartemisin inhibits glioma invasiveness via a ROS to P53 to β-catenin signaling. Pharmacol Res (2017) 119:72–88. doi: 10.1016/j.phrs.2017.01.014
137. Hyun SY, Le HT, Min HY, Pei H, Lim Y, Song I, et al. Evodiamine inhibits both stem cell and non-stem-cell populations in human cancer cells by targeting heat shock protein 70. Theranostics (2021) 11:2932–52. doi: 10.7150/thno.49876
138. Santoro MG. Heat shock factors and the control of the stress response. Biochem Pharmacol (2000) 59:55–63. doi: 10.1016/S0006-2952(99)00299-3
139. Walter S, Buchner J. Molecular chaperones–cellular machines for protein folding. Angew Chem Int Ed Engl (2002) 41:1098–113. doi: 10.1002/1521-3773(20020402)41:7<1098::AID-ANIE1098>3.0.CO;2-9
140. Alam R, Schultz CR, Golembieski WA, Poisson LM, Rempel SA. PTEN suppresses SPARC-induced pMAPKAPK2 and inhibits SPARC-induced Ser78 HSP27 phosphorylation in glioma. Neuro-oncology (2013) 15:451–61. doi: 10.1093/neuonc/nos326
141. Antonietti P, Linder B, Hehlgans S, Mildenberger IC, Burger MC, Fulda S, et al. Interference with the HSF1/HSP70/BAG3 pathway primes glioma cells to matrix detachment and BH3 mimetic-induced apoptosis. Mol Cancer Ther (2017) 16:156–68. doi: 10.1158/1535-7163.MCT-16-0262
142. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol (2007) 8:741–52. doi: 10.1038/nrm2239
143. Vlahakis A, Debnath J. The interconnections between autophagy and integrin-mediated cell adhesion. J Mol Biol (2017) 429:515–30. doi: 10.1016/j.jmb.2016.11.027
144. Yu Y, Liu B, Li X, Lu D, Yang L, Chen L, et al. ATF4/CEMIP/PKCalpha promotes anoikis resistance by enhancing protective autophagy in prostate cancer cells. Cell Death Dis (2022) 13:46. doi: 10.1038/s41419-021-04494-x
145. Talukdar S, Pradhan AK, Bhoopathi P, Shen XN, August LA, Windle JJ, et al. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc Natl Acad Sci USA (2018) 115:5768–73. doi: 10.1073/pnas.1721650115
146. Nabors LB, Portnow J, Ahluwalia M, Baehring J, Brem H, Brem S, et al. Central nervous system cancers, version 3.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2020) 18:1537–70. doi: 10.6004/jnccn.2020.0052
147. Kyjacova L, Hubackova S, Krejcikova K, Strauss R, Hanzlikova H, Dzijak R, et al. Radiotherapy-induced plasticity of prostate cancer mobilizes stem-like non-adherent, erk signaling-dependent cells. Cell Death Differ (2015) 22:898–911. doi: 10.1038/cdd.2014.97
148. Narayan RS, Fedrigo CA, Brands E, Dik R, Stalpers LJ, Baumert BG, et al. The allosteric AKT inhibitor MK2206 shows a synergistic interaction with chemotherapy and radiotherapy in glioblastoma spheroid cultures. BMC Cancer (2017) 17:204. doi: 10.1186/s12885-017-3193-9
149. Liang Y, Voshart D, Paridaen J, Oosterhof N, Liang D, Thiruvalluvan A, et al. CD146 increases stemness and aggressiveness in glioblastoma and activates YAP signaling. Cell Mol Life Sci (2022) 79:398. doi: 10.1007/s00018-022-04420-0
150. da SSR, de SLT, da Silva CT, Goncalves JD, Lerario AM, Marie SKN, et al. Cellular model of malignant transformation of primary human astrocytes induced by Deadhesion/Readhesion cycles. Int J Mol Sci 23 (2022) 23(9):4471. doi: 10.3390/ijms23094471
151. MacDonald TJ, Vezina G, Stewart CF, Turner D, Pierson CR, Chen L, et al. Phase II study of cilengitide in the treatment of refractory or relapsed high-grade gliomas in children: a report from the children's oncology group. Neuro-oncology (2013) 15:1438–44. doi: 10.1093/neuonc/not058
152. Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, et al. A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306). Cancer (2012) 118:5601–7. doi: 10.1002/cncr.27585
153. Leblond P, Dewitte A, Le Tinier F, Bal-Mahieu C, Baroncini M, Sarrazin T, et al. Cilengitide targets pediatric glioma and neuroblastoma cells through cell detachment and anoikis induction. Anticancer Drugs (2013) 24:818–25. doi: 10.1097/CAD.0b013e328362edc5
154. Ishida J, Onishi M, Kurozumi K, Ichikawa T, Fujii K, Shimazu Y, et al. Integrin inhibitor suppresses bevacizumab-induced glioma invasion. Transl Oncol (2014) 7(2):292–302.e1. doi: 10.1016/j.tranon.2014.02.016
155. Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol (2004) 22:133–42. doi: 10.1200/JCO.2004.08.110
156. Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, et al. Phase II evaluation of gefitinib in patients with newly diagnosed grade 4 astrocytoma: Mayo/North central cancer treatment group study N0074. Int J Radiat Oncol Biol Phys (2011) 80:347–53. doi: 10.1016/j.ijrobp.2010.01.070
157. Pollack IF, Stewart CF, Kocak M, Poussaint TY, Broniscer A, Banerjee A, et al. A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: a report from the pediatric brain tumor consortium. Neuro-oncology (2011) 13:290–7. doi: 10.1093/neuonc/noq199
158. Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, et al. RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys (2013) 85:1206–11. doi: 10.1016/j.ijrobp.2012.10.008
159. El-Khouly FE, Veldhuijzen van Zanten SEM, Jansen MHA, Bakker DP, Sanchez Aliaga E, Hendrikse NH, et al. A phase I/II study of bevacizumab, irinotecan and erlotinib in children with progressive diffuse intrinsic pontine glioma. J Neurooncol (2021) 153:263–71. doi: 10.1007/s11060-021-03763-1
160. Ma Y, Tang N, Thompson RC, Mobley BC, Clark SW, Sarkaria JN, et al. InsR/IGF1R pathway mediates resistance to EGFR inhibitors in glioblastoma. Clin Cancer Res (2016) 22:1767–76. doi: 10.1158/1078-0432.CCR-15-1677
161. Doz F, van Tilburg CM, Geoerger B, Hojgaard M, Ora I, Boni V, et al. Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro Oncol (2021) 24(6):997–1007. doi: 10.1093/neuonc/noab274
162. Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol (2011) 13:132–42. doi: 10.1093/neuonc/noq142
163. Wick A, Desjardins A, Suarez C, Forsyth P, Gueorguieva I, Burkholder T, et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Invest New Drugs (2020) 38:1570–9. doi: 10.1007/s10637-020-00910-9
164. Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O, et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol (2016) 18:1146–56. doi: 10.1093/neuonc/now009
165. Momeny M, Shamsaiegahkani S, Kashani B, Hamzehlou S, Esmaeili F, Yousefi H, et al. Cediranib, a pan-inhibitor of vascular endothelial growth factor receptors, inhibits proliferation and enhances therapeutic sensitivity in glioblastoma cells. Life Sci (2021) 287:120100. doi: 10.1016/j.lfs.2021.120100
166. Fujii K, Kurozumi K, Ichikawa T, Onishi M, Shimazu Y, Ishida J, et al. The integrin inhibitor cilengitide enhances the anti-glioma efficacy of vasculostatin-expressing oncolytic virus. Cancer Gene Ther (2013) 20:437–44. doi: 10.1038/cgt.2013.38
167. Allison M. Clinical setbacks reduce IGF-1 inhibitors to cocktail mixers. Nat Biotechnol (2012) 30:906–7. doi: 10.1038/nbt1012-906c
168. Saleem H, Kulsoom Abdul U, Kucukosmanoglu A, Houweling M, Cornelissen FMG, Heiland DH, et al. The TICking clock of EGFR therapy resistance in glioblastoma: Target independence or target compensation. Drug Resist Update (2019) 43:29–37. doi: 10.1016/j.drup.2019.04.002
169. Gambella A, Senetta R, Collemi G, Vallero SG, Monticelli M, Cofano F, et al. NTRK fusions in central nervous system tumors: A rare, but worthy target. Int J Mol Sci (2020) 21(3):753. doi: 10.3390/ijms21030753
170. Perreault S, Chami R, Deyell RJ, El Demellawy D, Ellezam B, Jabado N, et al. Canadian Consensus for biomarker testing and treatment of TRK fusion cancer in pediatric patients. Curr Oncol (2021) 28:346–66. doi: 10.3390/curroncol28010038
171. Kim BG, Malek E, Choi SH, Ignatz-Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the TGF-beta pathway. J Hematol Oncol (2021) 14:55. doi: 10.1186/s13045-021-01053-x
172. Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res an Off J Am Assoc For Cancer Res (2013) 19:205–14. doi: 10.1158/1078-0432.CCR-11-3358
Keywords: anoikis resistance, glioma, molecular pathways, therapeutic targets, apoptosis
Citation: Zhu Z, Fang C, Xu H, Yuan L, Du Y, Ni Y, Xu Y, Shao A, Zhang A and Lou M (2022) Anoikis resistance in diffuse glioma: The potential therapeutic targets in the future. Front. Oncol. 12:976557. doi: 10.3389/fonc.2022.976557
Received: 23 June 2022; Accepted: 25 July 2022;
Published: 15 August 2022.
Edited by:
Qiang Sun, Institute of Biotechnology (CAAS), ChinaReviewed by:
Youwei Ai, Institute of Genetics and Developmental Biology (CAS), ChinaCopyright © 2022 Zhu, Fang, Xu, Yuan, Du, Ni, Xu, Shao, Zhang and Lou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meiqing Lou, TWVpcWluZ19Mb3UyMDIwQDE2My5jb20=; Anke Zhang, dGhlYW5rZUAxNjMuY29t; Anwen Shao, c2hhb2Fud2VuQHpqdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.