 Ningzhi Zhang
Ningzhi Zhang- 1Department of Medical Oncology, Harbin Medical University Cancer Hospital, Harbin, China
- 2Medical Affairs Department, Harbin Medical University Cancer Hospital, Harbin, China
The role of long non-coding RNA (lncRNA) in human tumors has gradually received increasing attention in recent years. Particularly, the different functions of lncRNAs in different subcellular localizations have been widely investigated. The upregulation of lncRNA small nucleolar RNA host gene 17 (SNHG17) has been observed in various human tumors. Growing evidence has proved that SNHG17 plays a tumor-promoting role in tumorigenesis and development. This paper describes the molecular mechanisms by which SNHG17 contributes to tumor formation and development. The different functions of SNHG17 in various subcellular localizations are also emphasized: its function in the cytoplasm as a competing endogenous RNA (ceRNA), its action in the nucleus as a transcriptional coactivator, and its function through the polycomb repressive complex 2 (PRC2)-dependent epigenetic modifications that regulate transcriptional processes. Finally, the correlation between SNHG17 and human tumors is summarized. Its potential as a novel prognostic and diagnostic biomarker for cancer is explored especially.
1 Introduction
The diversity of cancers increases the complexity of treatment. Hanahan and Weinberg first summarized and classified the functional capabilities acquired by cancer cells that facilitate cell survival, proliferation, and dissemination into six cancer hallmarks in 2000, which include the following: (1) sustaining proliferative signaling, (2) evading growth suppressors, (3) enabling replicative immortality, (4) activating invasion and metastasis, (5) inducing/accessing vasculature, and (6) resisting cell death (1). The following four cancer hallmarks were later added in 2011: deregulating cellular metabolism, avoiding immune destruction, tumor-promoting inflammation, and genome instability/mutation (2). Hanahan (2022) proposed that unlocking phenotypic plasticity, nonmutational epigenetic reprogramming, polymorphic microbiomes, and senescent cells may also be emerging hallmarks of cancer (3). Tumor features in the hallmarks could enable tumors to acquire hallmark capabilities called enabling characteristics, including tumor-promoting inflammation, genome instability/mutation, nonmutational epigenetic reprogramming, and polymorphic microbiomes.
A growing stream of research has focused on the role of non-coding RNAs in tumors. Among them, long non-coding RNAs (lncRNAs) have been found to regulate genomic expression through transcriptional and post-transcriptional levels (4). The function of lncRNA correlates with subcellular localization. LncRNAs can function as competing endogenous RNA (ceRNAs) in the cytoplasm and regulate gene transcription in the nucleus through cis- or trans-regulation (5, 6). This review mainly focuses on the different roles of SNHG17 in various localizations and finds that the effects of SNHG17 emphasized the role of ceRNA in the cytoplasm and regulated transcription in the nucleus as a transcriptional co-activator or through transcriptional repressive chromatin modifications. The vital role of epigenetic modifications has been gaining attention in recent years. Thus, the role of the epigenetic modification function of SNHG17 in promoting tumorigenesis and progression is also discussed.
SNHG is the small nucleolar host gene and belongs to lncRNA. Several SNHG family members, such as SNHG1 (7, 8), SNHG20 (9), SNHG3 (10, 11), SNHG5 (12, 13), and SNHG16 (14), are recently closely associated with tumors. Meanwhile, as the homolog of SNHG, snoRNA has a connection with SNHG. snoRNA plays a role in ribosomal RNA modification, stress response, and tumor development. However, but the relationship between snoRNA and SNHG is unclear. Previous literature describes that SNHG17 in prostate cancer increases the expression of its homolog snoRA71B through a positive feedback loop, which promotes tumor progression (15). The regulatory relationship between SNHG and snoRNA is also investigated in a new insight.
This paper discusses the correlation between SNHG17 and tumors by describing the molecular mechanisms by which SNHG17 contributes to the formation of cellular hallmark capabilities and the enabling characteristics. The paper also focuses on its different functions in various subcellular localizations and finally discusses the potential of SNHG17 as a new prognostic and diagnostic biomarker for cancer.
2 Materials and methods
2.1 Raw data
The data samples of the differentially expressed genes of LUAD after SNHG17 knockdown were downloaded from GSE131543. The immune-related genes were acquired from the ImmPort database.
2.2 GO analyze
GO analyze was performed using Metascape (http://metascape.org/). Terms with p-value cutoff of 0.01, min overlap of 3, and min enrichment of 1.5 were considered. The top 20 enriched terms are displayed in Figure 4B.
2.3 Immune and stromal infiltration analysis
ssGSEA was applied to explore the infiltration degrees of immune cell types in LUAD of the TCGA database using the GSVAR package in R (version 1.34.0). The estimated package was used to generate ImmuneScore, StromalScore, and ESTIMATEScore. R language version 3.6.3 loaded with ggplot2 package (version 3.3.3) was used to demonstrate the correlation between SNHG17 and PD-L1. All the correlation between SNHG17 and others were studied using Spearman correlation analysis.
3 Results
3.1 Association of SNHG17 in the acquisition of hallmark capabilities
3.1.1 Role of SNHG17 in sustaining proliferative signaling
SNHG17 is associated with increased cell proliferation capacity in various types of cancers. The mechanisms that promote the formation of this phenotype include the activation of cyclin-dependent kinases (CDKs), phosphoinositide-3 kinase/protein kinase B (PI3K/Akt) and Wnt/β-catenin signaling pathway, and the inhibition of cyclin-dependent kinase inhibitor (CKI) (Figure 1).
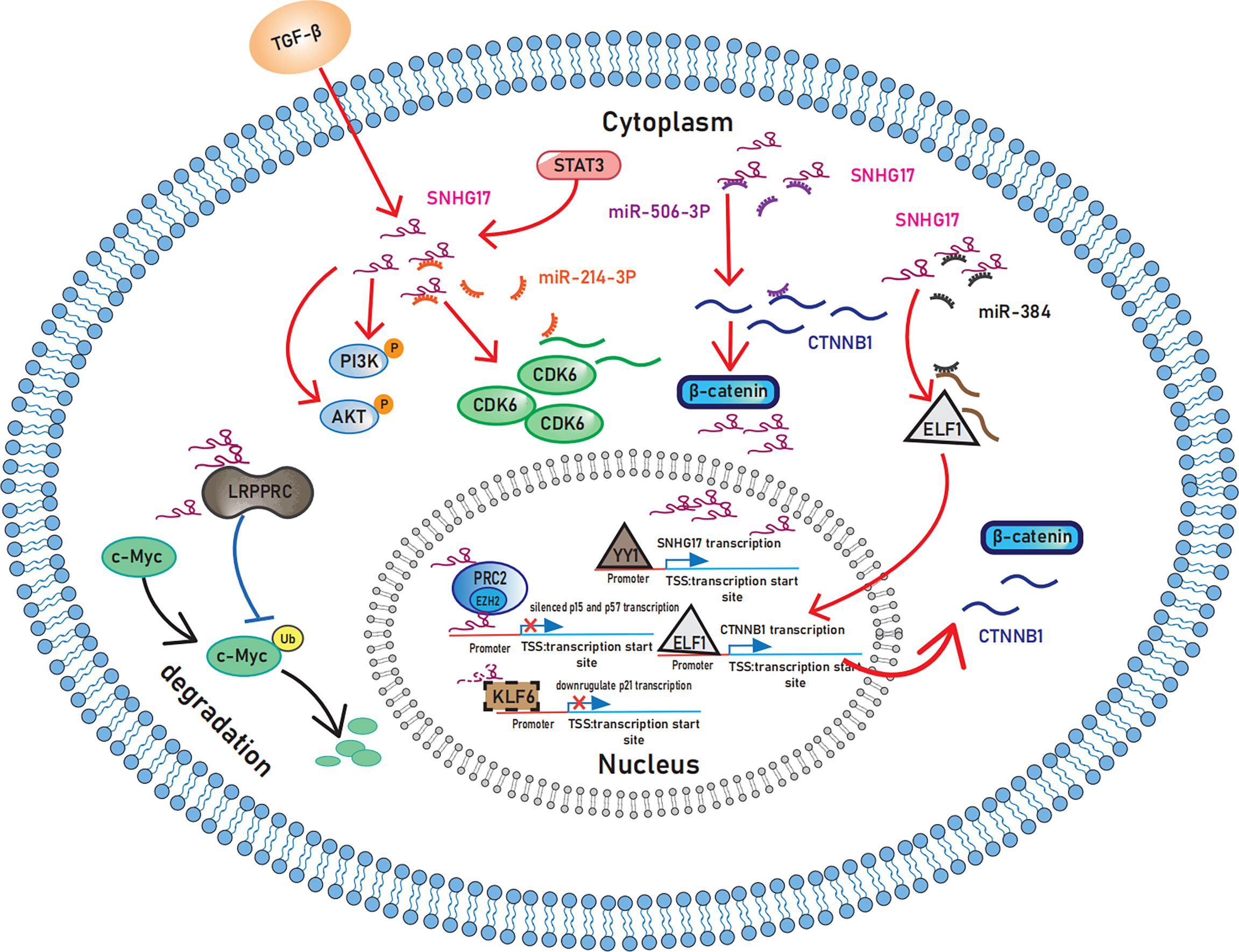
Figure 1 Schematic representation of the mechanisms by which SNHG17 plays in sustaining proliferative signaling. In the cytoplasm, SNHG17 acts as the ceRNA for miR-506-3 and miR-384 to activate the Wnt/β-catenin pathway and enhance the expression of CDK6 by targeting miR-214-3p. Moreover, STAT3 and TGF-β activate the PI3K/AKT signaling pathway by increasing SNHG17 levels. By interacting with LRPPRC, SNHG17 can also reduce c-Myc ubiquitination. In nucleus, SNHG17 targets p57 and p15 by binding to EZH2 and PRC2. p21 may also exist downstream of SNHG17 considering the regulatory relationship between KLF6 and SNHG17. And transcription factor YY1 upregulate the transcription of SNHG17. SNHG17, lncRNA small nucleolar RNA host gene 17; CDK6, Cyclin-dependent kinase 6; STAT3, Signal transducer and activator of transcription 3; TGF-β, Transforming growth factor β; PI3K, Phosphoinositide-3 kinase; AKT, Protein kinase B; LRPPRC, Leucine-rich pentatricopeptide repeat-containing protein; EZH2, enhancers on zeste homolog 2; PRC2, Polycomb repressive complex 2; KLF6: Krupple-like factor 6; YY1, Yin-yang 1.
3.1.1.1 SNHG17 regulates CKI, CDK, and c-Myc
Ma Z et al. (16), first identified the relevance between SNHG17 and tumor cell proliferation in colorectal cancer, and revealed that SNHG17 epigenetically targets p57 by binding to enhancers on zeste homolog 2 (EZH2). As a well-known CKI, p57 plays a vital role in regulating the cell cycle. Briefly, the cell cycle regulatory machinery comprises three major types of proteins: cyclin, CDK, and CKI. The binding of cyclin with CDK promotes cell cycle progression, and CKI hinders this effect by inhibiting the cyclin-CDK complex. The tumor suppressor-like properties of CKI have been reported in recent years (17), and lncRNAs can promote cancer cell proliferation and migration by decreasing CKI expression (18). SNHG17 acts as a suppressor of multiple CKIs in tumors; in addition to p57, SNHG17 can also inhibit p15 and p16, and this CKI inhibitory property contributes to increasing cell proliferation capacity (19, 20). Moreover, RNA sequencing predicts that SNHG17 can interact with the transcription factor krupple-like factor 6 (KLF6) (21). p21 may also exist downstream of SNHG17 considering the regulatory relationship between KLF6 and p21 (22). Furthermore, SNHG17 can enhance the expression of CDK4 and CDK6, promoting cell cycle progression (20, 23).
The role of SNHG17 in the cell cycle also involves the ubiquitin–proteasome system (UPS). As a considerably method for intracellular protein degradation, the UPS is one of the essential mechanisms controlling the levels of MYC protein (24). A recent study in hepatocellular carcinoma found that SNHG17 can reduce c-Myc ubiquitination, increase c-Myc levels, and promote G1/S phase transition (25). SNHG17 can play multiple roles through UPS. For example, in colorectal cancer, SNHG17 competes with the E3 ligase Trim23 to bind Pescadillo (PES1), protecting PES1 from degradation (26). In non-tumor diseases, SNHG17 reduces MST ubiquitination and degradation and regulates the apoptosis of podocytes and Parkin-dependent mitophagy in diabetic nephropathy (27). However, the relationship between SNHG17 and UPS currently needs rigorous experimental data support, and this area of research requires further exploration.
3.1.1.2 SNHG17 regulates PI3K/AKT signaling pathway
PI3K/AKT signaling, one of the critical intracellular pathways, is also involved in the mechanism of how SNHG17 enables tumor cells to obtain the capacity of sustained proliferation. STAT3 and transforming growth factor-β (TGF-β) are located upstream of SNHG17, which contribute to activate the PI3K/AKT signaling pathway by increasing SNHG17 levels (28, 29). In addition, SNHG17 may indirectly activate the PI3K/AKT signaling pathway through its regulatory effects on NETO2 and PES1 (26, 30–32).
3.1.1.3 SNHG17 regulates Wnt/β-catenin signaling pathway
SNHG17 acts as the ceRNA for miR-506-3p and miR-384 in the cytoplasm to exert pro-proliferative effects through the Wnt/β-catenin pathway (33, 34). For the specific mechanism, SNHG17 affects the CTNNB1 gene, which encodes β-catenin by targeting miR-506-3p (33) and miR-384 to act on the transcription factor ELF1 (34) to promote the transcription of the CTNNB1 gene. Moreover, transcription factor yin-yang 1 (YY1) facilitates this upstream regulation of SNHG17 (33). Intriguingly, Wnt ligand secretion mediator (WLS) and stanniocalcin 2 (STC2) are involved in the pro-proliferative effects of SNHG17 as downstream targets (35, 36), which are both closely related to the Wnt/β-catenin signaling pathway (37, 38). However, additional research needs to verify whether SNHG17 can activate the Wnt/β-catenin pathway and thus promote tumor progression through WLS and STC2.
3.1.2 Role of SNHG17 in forming death-resistant phenotype
The forms of cell death include accidental cell death (ACD) and regulatory cell death (RCD). Apoptosis has been widely investigated as the first identified regulatory cell death in tumors; some non-apoptotic regulatory cell deaths, such as autophagic cell death, pyroptosis, and ferroptosis, are also currently gaining attention. Cancer cells can resist cell death through multiple pathways, which is also one of the hallmark capabilities. Interestingly, the upregulation of SNHG17 is associated with increased drug resistance in astrocytoma and prostate cancer (39, 40). Furthermore, several studies have shown the formation mechanisms of the death-resistant phenotype. Herein, several reported mechanisms are introduced, and examples for known modes of action are provided.
3.1.2.1 SNHG17 regulates apoptosis-related proteins
Multiple innate tumor suppressive mechanisms exist in mammals to ensure that cells have normal levels of proliferation, and these mechanisms are activated when cells show aberrant proliferation, leading to apoptosis or senescence (41). Thus, resistance to apoptosis is a barrier that must be breached for tumor formation. The anti-apoptotic effect of SNHG17 on tumor cells has been currently observed in a variety of tumors, including cervical cancer, pancreatic cancer, astrocytoma, hepatocellular carcinoma, oral squamous cell carcinoma, prostate cancer, ovarian cancer, gastric cancer, melanoma, and colorectal cancer (15, 16, 19, 23, 28, 39, 42–45). This anti-apoptotic mechanism was associated with increased anti-apoptotic protein Bcl-2 and the decreased activity of the pro-apoptotic proteins caspase3, caspase8, caspase9, and Bax (33, 35, 36, 40, 46, 47). In addition, RNA-seq analysis performed in non-small cell lung cancer revealed that the genes of the pro-apoptotic proteins BIK and XIAP-associated factor 1 (XAF1) are the downstream targets of SNHG17 (48).
In addition, IGF binding protein 3 (IGFBP3) may act as a bridge linking SNHG17 to p53-dependent apoptosis in colorectal cancer (21). IGFBP3 promotes colorectal cancer progression through p53-dependent apoptosis (49). Notably, Parkin protein was recently found to be regulated by SNHG17 in non-tumor diseases (27). Parkin is a ubiquitin-protein ligase (E3), which can be involved in the regulation of apoptosis by ubiquitinating various apoptosis-related proteins. Notably, studies on whether SNHG17 can regulate apoptosis through Parkin protein in tumors, which may become a director in future SNHG17 investigations, are lacking.
3.1.2.2 SNHG17 regulates autophagy-related protein
Autophagy plays a role in the maintenance of normal cellular homeostasis-like apoptosis. Interestingly, as with TGF-β, cellular autophagy plays a dual role in tumor progression, inhibiting early tumor formation and promoting late tumor progression (50–52). Increased mitochondrial autophagy in tumors leads to high chemo- and radiotherapy resistance by increasing metabolic plasticity in cancer cells (53–55). This section further described cellular metabolism deregulation below. Overall, mitochondrial autophagy has a vital role in tumorigenesis and development.
SNHG17 was screened as an effective autophagy-related lncRNA signature closely associated with prognosis in ovarian cancer and renal clear cell carcinoma (56, 57). Intriguingly, SNHG17 has been shown to reduce Parkin-dependent mitochondrial autophagy by regulating Parkin proteins in non-tumor diseases (27). Parkin is known to act as a mitophagy initiator, and Parkin-dependent mitochondrial autophagy is downregulated in various tumors and has been suggested to belong to a tumor suppressor mechanism (58). Studies on whether SNHG17 can regulate Parkin-dependent mitochondrial autophagy and affect tumor progression, which requires further experimental verification in the future, are still unavailable.
3.1.2.3 SNHG17 is associated with ferroptosis
Ferroptosis is an iron-dependent oxidative cell death (59), and this cell death pathway could be one of the directions for tumor therapy (60). SNHG17 is involved in tumor progression as a ferroptosis-associated lncRNA. Risk assessment and diagnostic models constructed with ferroptosis-associated lncRNAs, such as SNHG17, have shown excellent prognostic and diagnostic values in renal cancer (61). However, further studies are still needed to elucidate the mechanistic role of SNHG17 in ferroptosis.
3.1.3 Role of SNHG17 in accelerating EMT process
Concerning the studies on the relationship between SNHG17 and tumors, the most significant relationship is the promotion of tumor proliferation, invasion, and metastasis. Several studies have shown that the high-level expression of SNHG7 correlates with lymph node metastasis, distant metastasis, and tumor invasion depth in various tumors (19, 29, 42, 44, 46). Spreading to distant organs is recognized to be the most prominent hallmark of cancer cells. A primary biological process that sustains invasion and 1metastasis is the epithelial–mesenchymal transition (EMT), wherein cells gain the mesenchymal phenotypes, such as high migration and invasion and the capability to degrade extracellular matrix.
SNHG17 can have different functions in various subcellular localizations. The best-known capability of SNHG17 lies in its function as ceRNA in the cytoplasm. In esophageal squamous and hepatocellular carcinomas, SNHG17 can promote EMT by targeting miR-338-3p/SOX4 and miR-3180-3p/regulatory factor X-box 1 (RFX1) axes, respectively (62, 63).
The presence of positive feedback regulatory loop SNHG17/miR-339-5p/STAT5A/SNHG17 and SNORA71B in prostate cancer can promote cellular EMT and thus facilitate cancer progress (15). Similarly, in castration-resistant prostate cancer, SNHG17 regulates CD51 and thus promotes tumor EMT via sponge miR-144 (64) (Figure 2).
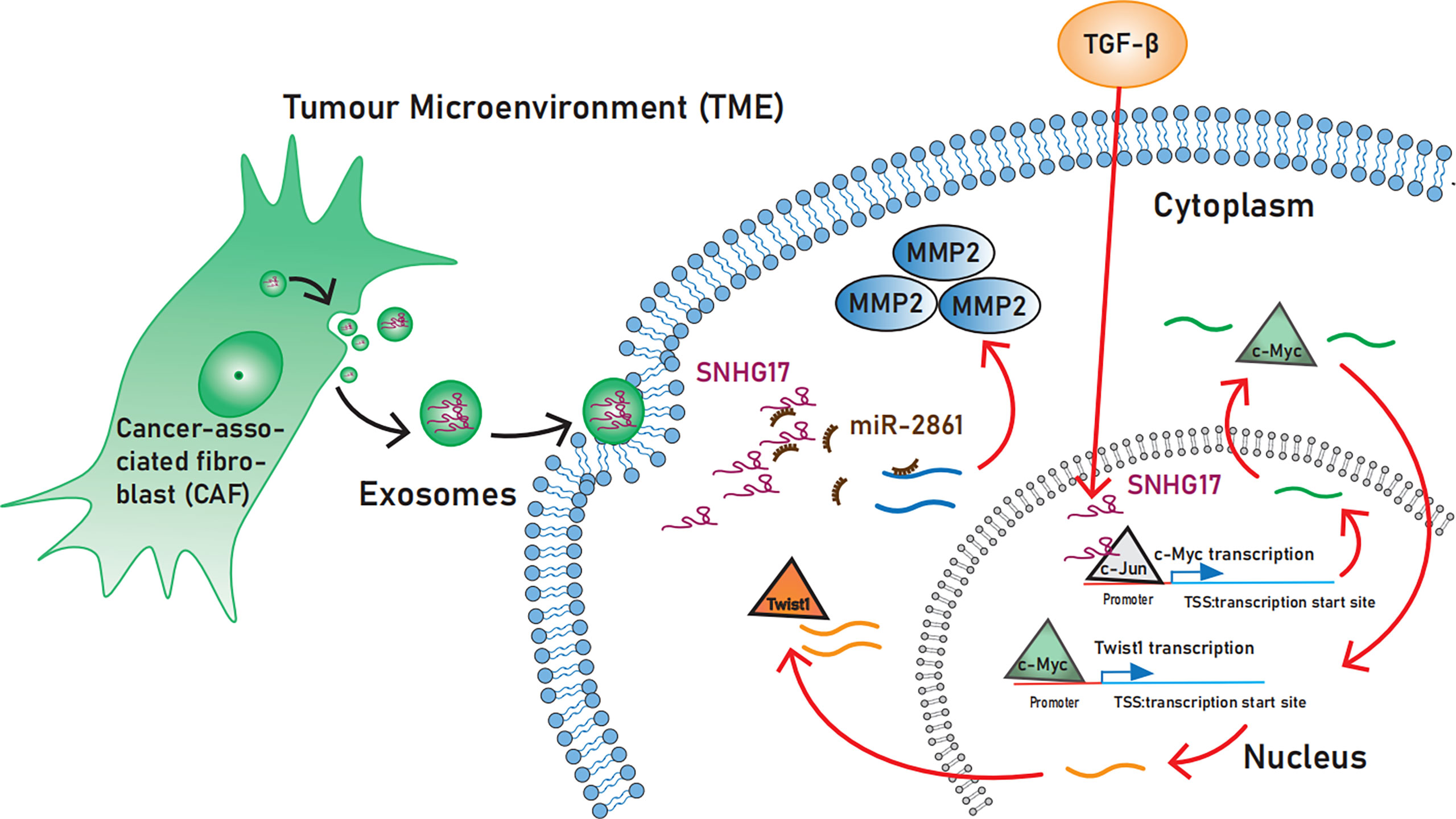
Figure 2 The role SNHG17 plays in activating invasion and metastasis. SNHG17 acts as a transcriptional co-activator by recruiting the transcription factor c-Jun to the c-Myc promoter region, and promotes the expression of the EMT-associated transcription factor Twist1 via c-Myc. In the Extracellular, SNHG17 can be secreted by tumor-associated fibroblasts and target MMP2 in the form of exosomes. EMT, Epithelial-mesenchymal transition; MMP2, Matrix metalloproteinases 2.
3.1.3.1 SNHG17 regulates EMT process via TGF-β, Twist 1 and c-Myc
TGF-β is widely known to play an important role in cancer. In the early stages, TGF-β exerts an oncogenic effect by inhibiting the cell cycle process; in the late stages, it can induce invasion and metastasis and promote EMT (65).
TGF-β-induced SNHG17 hyperactivation promotes EMT. Unlike in the cytoplasm, lncRNAs can regulate the transcriptional process of some oncogenes by binding to transcription factors while in the nucleus (66). Shen S et al. (29), recently demonstrated that SNHG17 is involved in TGF-β1-mediated EMT in esophageal squamous cell carcinoma. Mechanistically, SNHG17 acts in the nucleus by recruiting the transcription factor c-Jun to the c-Myc promoter region, which increases the transcriptional activity of the c-Myc promoter. Furthermore, SNHG17 promotes the expression of the EMT-associated transcription factor Twist1 via c-Myc (29).
3.1.3.2 SNHG17 regulates EMT process via matrix metalloproteinases
Matrix metalloproteinases (MMPs) belong to the family of extracellular proteases and promote tumor progression through tumor microenvironment (TME) regulation. MMPs are not only known as a marker of EMT but also induce EMT. Exosomes can act as tools for cancer cells to regulate the TME and promote proliferation and invasion. As the lncRNA released from tumor-derived exosome, SNHG17 is crucial in promoting tumor migration by increasing MMP2 levels through sponge miR-2861 (47).
3.1.3.3 SNHG17 regulates EMT process via Wnt/β-catenin signaling pathway
The role of the Wnt/β-catenin signaling pathway in EMT is widely recognized. However, the relationship between SNHG17 and Wnt/β-catenin signaling pathway is yet to be investigated. For reference, STC2, a downstream target of miR-361-3p, was shown to promote tumor invasion and migration via the Wnt/β-catenin signaling pathway in vivo experiments (38), while SNHG17 acts as a ceRNA for miR-361-3p (36).
3.1.4 Role of SNHG17 in increasing tumor angiogenesis
The pro-angiogenic effect of SNHG17 can be achieved by sponging miR-23a-3p and thus regulating the chemokine CXCL12 (67). In addition, SNHG17 can target miR-942 to regulate vascular endothelial growth factor (VEGF) expression (43), but the exact mechanism needs further exploration. H2AX, which can be regulated by SNHG17, also plays an essential role in tumor angiogenesis (46, 68). Intriguingly, CDK6 can promote tumor angiogenesis through kinase-independent function; the transcriptional regulator activity of CDK6 provides new ideas for the functional exploration of SNHG17 (23, 69).
3.1.5 Role of SNHG17 in deregulating cellular metabolism
Although no studies in tumors have confirmed the direct effect of SNHG17 on regulating cellular metabolism, a regulatory relationship may exist between SNHG17 and deregulating cellular metabolism according to the mechanism exploration of SNHG17 in some studies.
3.1.5.1 SNHG17 regulates abnormal glucose metabolism
Considering the non-negligible role of cellular metabolism in cancer development, deregulating cellular metabolism was included in the cancer hallmarks as an emerging hallmark by Hanahan and Weinberg (2, 70). Otto Warburg first identified the Warburg effect regarding cancer cells having different energy metabolism from normal cells even under the aerobic environment; cancer cells still prefer to obtain energy through the glycolytic pathway (71, 72). This metabolic mechanism is seemingly counterintuitive because glycolysis is much less efficient than mitochondrial oxidative phosphorylation. However, this particular metabolic is conducive to rapid cell proliferation (73), and metabolic plasticity can act as a survival mechanism of tumors under cancer treatment (53).
Mitochondrial autophagy proves tumors with metabolic plasticity by degrading cellular structures and recycling metabolites in response to environmental stress (54, 55). As described in the resisting cell death section above, SNHG17 reduces mitochondrial autophagy by downregulating Parkin in non-tumor diseases (27), and Parkin plays a role in glucose metabolism and the Warburg effect as a p53 target gene (74). Furthermore, as the E3 ubiquitin ligase, Parkin can promote hypoxia inducible factor-1α (HIF-1α) degradation through ubiquitination and proteasomal degradation (75). HIF-1α is known to act as a hypoxia-inducible transcription factor and can promote the Warburg effect by regulating cellular metabolism (76).
Several transcription factors are involved in establishing the Warburg effect. One factor, namely c-Myc, can confer metabolic advantages to tumor cells by regulating the expression of multiple genes (77, 78). Interestingly, SNHG17 has been demonstrated in several studies to increase c-Myc levels by promoting transcription with inhibition of ubiquitination and degradation (25, 26, 29). c-Myc may also serve as a bridge for SNHG17 to establish the Warburg effect in cancer cells.
3.1.5.2 SNHG17 modulates lipid metabolism
Previous interest in tumor metabolic abnormalities has focused on glucose metabolism. However, recent studies have shown that abnormalities in lipid metabolism are also present in tumors. Reprogramming of lipid metabolism can contribute to tumorigenesis and drug resistance (79–81). Low-density lipoprotein cholesterol (LDL-C) levels are positively associated with increased cancer risk (82). Relevant studies on tumors are lacking. However, SNHG17 was found to be negatively associated with high-density lipoprotein cholesterol (HDL-C) levels in a study on diabetes and may be involved in the formation of type 2 diabetes (83). This study demonstrates a regulatory relationship between SNHG17 and HDL-C and the possible involvement of SNHG17 in tumor progression through lipid metabolism. Moreover, lipid metabolism may become a new direction to explore the tumor-promoting mechanism of SNHG17.
3.1.6 Role of SNHG17 in regulating telomerase activity
The contribution of SNHG17 to achieving replicative immortality has not been fully investigated. However, the telomerase regulatory activity of the SNHG17-associated protein pescadillo (PES1) deserves consideration. Telomeres, which protect the ends of the chromosome, are closely associated with the capability of tumor cells to proliferate indefinitely. Consequently, telomerase, specifically expressed in most tumors, plays an important role and prevents telomere shortening. Telomerase is an attractive target for tumor therapy. Its catalytic core includes telomerase reverse transcriptase and telomerase RNA. SNHG17-related protein PES1 can promote tumor progression in multiple ways. As a key component of telomerase composition, PES1 promotes telomerase assembly by facilitating the direct interaction between telomerase reverse transcriptase and telomerase RNA. The increased expression of PES1 can also lead to enhanced telomerase activity and affect telomere length maintenance (26, 84).
3.1.7 Role of SNHG17 in avoiding immune destruction
Hanahan and Weinberg (2011) introduced immune destruction avoidance as another hallmark capability (2). The extensive crosstalk between the tumor microenvironment (TME) and the tumor promotes the capability of avoiding immune destruction.
Cancer-derived exosomes have been of broad interest as the tumor TME component, which plays a vital role in tumor occurrence and development (85). They have recently been proposed to have the potential to become emerging enabling characteristics (86). SNHG17 can be secreted by tumor-associated fibroblasts in the form of exosomes and targets MMP2 (47). Matrix metalloproteinases 2 (MMP2) promotes immunosuppressive TME formation by reducing anti-tumor-associated immune cells (CD4+ and CD8+ T cells, NK cells, and CD103+ DCs) and increasing M2-like macrophages (87).
3.1.8 Role of SNHG17 in unlocking phenotypic plasticity
According to “Hallmarks of Cancer: New Dimensions” reported by Hanahan in 2022, unlocking phenotypic plasticity may be included in the hallmark capabilities (3). Unlocking phenotypic plasticity is mainly manifested by blocking normal cell differentiation in developmental lineages. Notably, as cell development-related transcription factors involved in this process, SOX2, SOX4, and homeobox A1 (HOXA1) can be regulated by SNHG17 (3, 30, 39, 62, 88).
3.1.9 Role of SNHG17 in regulating senescent cells
Many studies have shown that senescent cells promote tumor proliferation, migration, and other malignant behaviors through senescence-associated secretory phenotype (89–93). Therefore, senescent cells are also classified as potential emerging tumor markers (3). Genomic instability is one of the significant causes of senescence, and the relationship between SNHG17 and genomic instability will be described in the genome instability/mutation section; meanwhile, the senescence-associated secretory phenotype regulator NF-κB is involved in maintaining high levels of SNHG17 expression (94).
3.2 Association of SNHG17 in the development of enabling characteristics
3.2.1 Role of SNHG17 in induces genome instability/mutation
The formation of most cancer hallmarks is associated with genome dysfunction. Genome maintenance systems provide numerous contributions to maintain the stability of the genome.
The impairment of caretaker mechanisms, such as DNA repair defect, aberrant cell cycle regulation, and abnormal telomere DNA maintenance mechanisms, can lead to genome instability (95). As described in the sustaining proliferative signaling section, SNHG17 has been found to inhibit cell cycle protein-dependent kinase inhibitor (CKI) p15, 16, 57, and to increase (16, 19, 20, 96) cell cycle protein-dependent kinase CDK4 and CDK6 expression (20, 23) to involve in aberrant cell cycle regulation.
3.2.1.1 SNHG17 affects the selection of DNA damage repair pathway
Most cancers are characterized by genome instability/mutation, giving cells a selective growth advantage (21, 97). Multiple repair pathways are available after DNA damage; compared with homologous recombination, non-homologous end joining leads to genomic instability and is conducive to tumor development because it cannot guarantee repair accuracy (98). Notably, SNHG17 can promote the non-homologous end joining pathway by targeting non-POU domain-containing octamer-binding protein and acts as a ceRNA to downregulate the expression of the central homologous recombination protein Rad51 (94), which predisposes DNA double-strand breaks to select the non-homologous end joining repair pathway. The role of SNHG17 in DNA repair makes it a potential biomarker for cancer diagnosis and treatment (Figure 3).
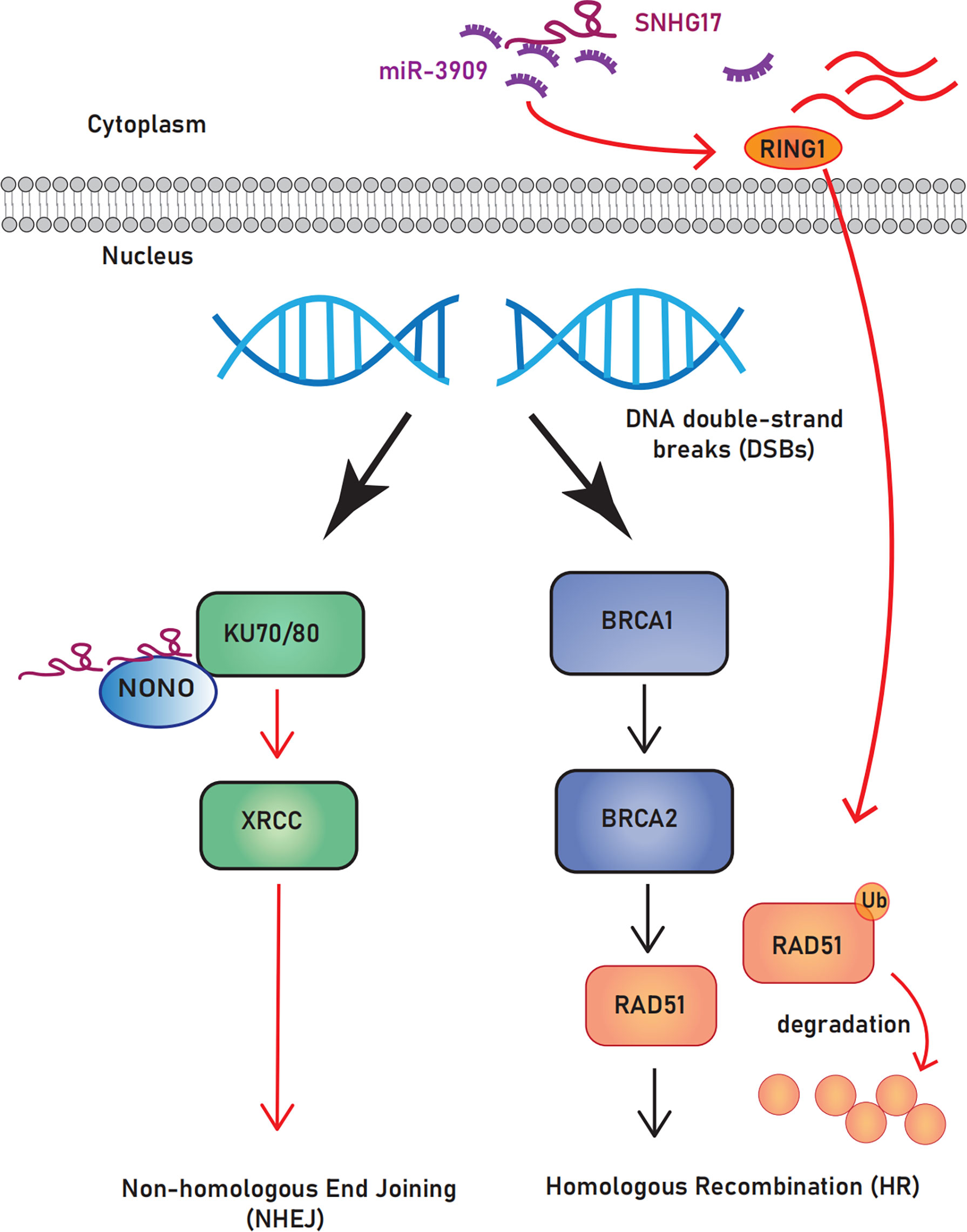
Figure 3 The role of SNHG17 plays in DNA repair.
3.2.1.2 SNHG17 affects telomerase activity
The relationship between SNHG17 and telomerase lacks support from additional rigorous experimental data. For reference, SNHG17 can regulate the ubiquitinate level of PES1 (26), and PES1 can interact with telomerase reverse transcriptase to regulate telomerase activity (84).
3.2.2 Role of SNHG17 in tumor-promoting inflammation
Appropriate inflammation has cancer-suppressing effects on cancers as part of innate immunity, but chronic inflammatory cell infiltration leads to an increased risk of tumor formation. The inflammation that precedes tumor development promotes tumorigenesis by causing genomic instability, recruiting growth factors, and angiogenesis. By contrast, tumor-associated inflammation can lead to immunosuppression addition (99–101).
SNHG17 is a new link between inflammation and cancer. In a study on h. pylori-induced gastric carcinogenesis, SNHG17 was upregulated by the inflammation-associated transcription factor NF-κB; as described in the genome stability/mutation section, SNHG17 consequently affected the selection of the DNA Damage Repair pathway, thereby promoting gastric carcinogenesis (94).
Considering the extensive search for inflammation and immunity in tumor formation and progression, the correlation between SNHG17 and inflammation and immunity was further explored. A set of differentially expressed genes was used after knocking down SNHG17 in lung adenocarcinoma (LUAD) and intersected them with immune-related genes downloaded from the ImmPort database (Figure 4A) (Supplementary Data Sheet 1). Gene ontology (GO) enrichment analysis indicated that these intersected genes mainly focused on receptor ligand activity, inflammatory response, positive regulation of response to external stimulus, positive regulation of cytokine production, cell activation, and the regulation of immune effector process (Figure 4B) (Supplementary Data Sheet 2). SNHG17 was almost mapped to the inflammatory response in the innate immune response. The relationship between SNHG17 expression levels and immune infiltration was then explored. Figure 4 shows the following: SNHG17 had a positive correlation with Th2 and NK CD56 bright cells and a negative correlation with Macrophages, DCs, Neutrophils, Th1 cells, T cells; it was negatively correlated to ESTIMATEScore, ImmuneScore, StromalScore, and PD-L1 (Figures 4C-E) (Supplementary Data Sheet 3–5).
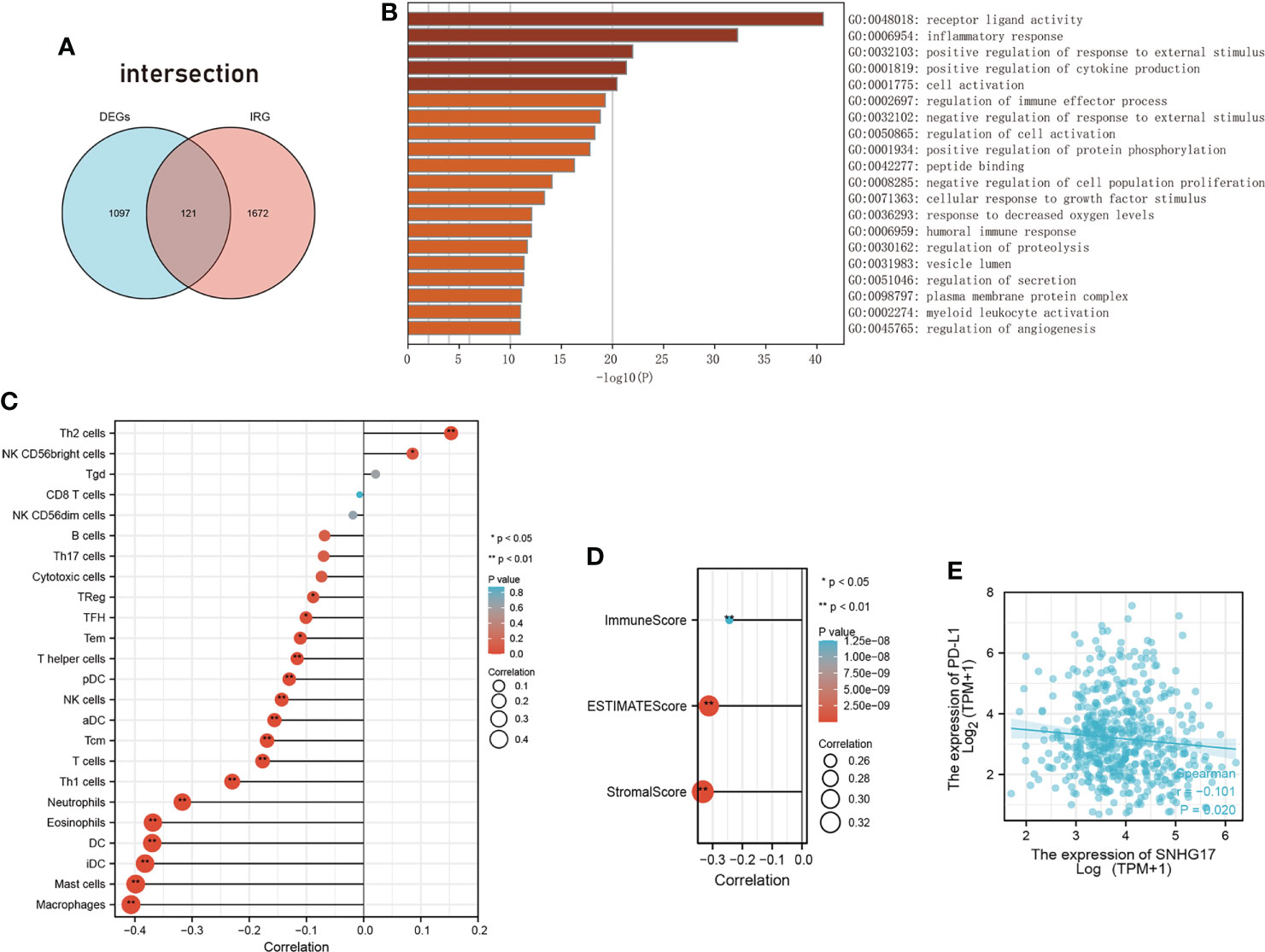
Figure 4 The relationship between SNHG17 and immune. (A) Venn diagram visualizing the overleaping genes between DEGs and IRGs. (B) Summary of enrichment analysis across input the intersected genes of DEGs with IRGs. (C) The correlation between SNHG17 and immune cell infiltration, respectively. (D) Correlation analysis between SNHG17 and immune-related scores (ESTIMATEScore, ImmuneScore, StromalScore). (E) Correlation analysis between SNHG17 and PD-L1 expression. DEGs, Differentially expressed genes; IRGs, Immunity-related genes; PD-L1, Programmed death ligand 1.
In addition, the SNHG17-associated protein MMP2 is involved in the formation of the inflammatory TME through TLR2 and TLR4 (47, 87).
3.2.3 New dimensions: Nonmutational epigenetic reprogramming
Nonmutational epigenetic reprogramming is a possible enabling characteristic newly proposed by Hanahan (3). Epigenetic modifications are prevalent in many cancers and facilitate cancer hallmark formation. SNHG17 is involved in nonmutational epigenetic reprogramming mainly by interacting with the chromatin-associated methyltransferase polycomb repressive complex dependent 2 (16, 19) and blocking ubiquitination and degradation (25, 26).
In addition, as the spliced transcripts of the same primary transcript, SNHG and snoRNA theoretically have the same promoter. A recent study demonstrated a positive feedback loop between SNHG17 and its homolog snoRA71B, promoting tumor progression. Specifically, SNHG17 can promote transcription factor STAT5A expression by targeting miR-339-5p, thereby increasing SNHG17 and cognate snoRA71B levels (15). This finding further confirms the link between SNHG and snoRNA. SnoRNA is known to play an oncogenic role in a variety of tumors (102), and its broadest function is the epigenetic modification of rRNA at the post-transcriptional level through 2’-O-methylation and pseudouridylation. The high relevance to the response to immune therapy and the broad function beyond the ribosome of snoRNA is recently gaining attention. The relationship between SNHG and snoRNA is currently an open question. However, SNHG promotes the expression of its cognate snoRNA through the same promoter positive feedback, thus exerting oncogenic effects through snoRNA. The feedback may become a worthy direction for the subsequent study of SNHG and snoRNA.
3.2.4 Investigating feature: Cancer-derived exosomes
Cancer-derived exosomes can be observed in most cancers, promoting the formation of multiple hallmarks, which is in line with Hanahan’s definition of enabling features (3). Moreover, Kok VC et al. (86), recently raised the possibility of cancer-derived exosomes as an emerging enabling characteristic. Interestingly, SNHG17 can be released by cancer-associated fibroblast as exosomal lncRNA in osteosarcoma to promote tumor proliferation, migration, and apoptosis resistance (47).
4 Conclusion
SNHG17 has been found to be involved in forming cancer hallmarks in numerous ways. SNHG17 acts as ceRNA in the cytoplasm. Meanwhile, SNHG17 in the nucleus can act as a transcriptional co-activator or repress chromatin modifications transcriptionally to increase the malignant progression. These different functions suggest that SNHG17 plays a diverse role in tumorigenesis and progression. Thus, targeting SNHG17 may become a promising strategy for tumor therapy. In addition, SNHG17 can be an independent prognostic factor in hepatocellular carcinoma, renal cell carcinoma, colorectal cancer, gastric cancer, and melanoma (26, 28, 44, 46, 103) and a diagnostic predictor in cervical and gastric cancers (19, 42) (Table 1). Therefore, SNHG17 is an emerging biomarker for cancer diagnosis and prognosis.
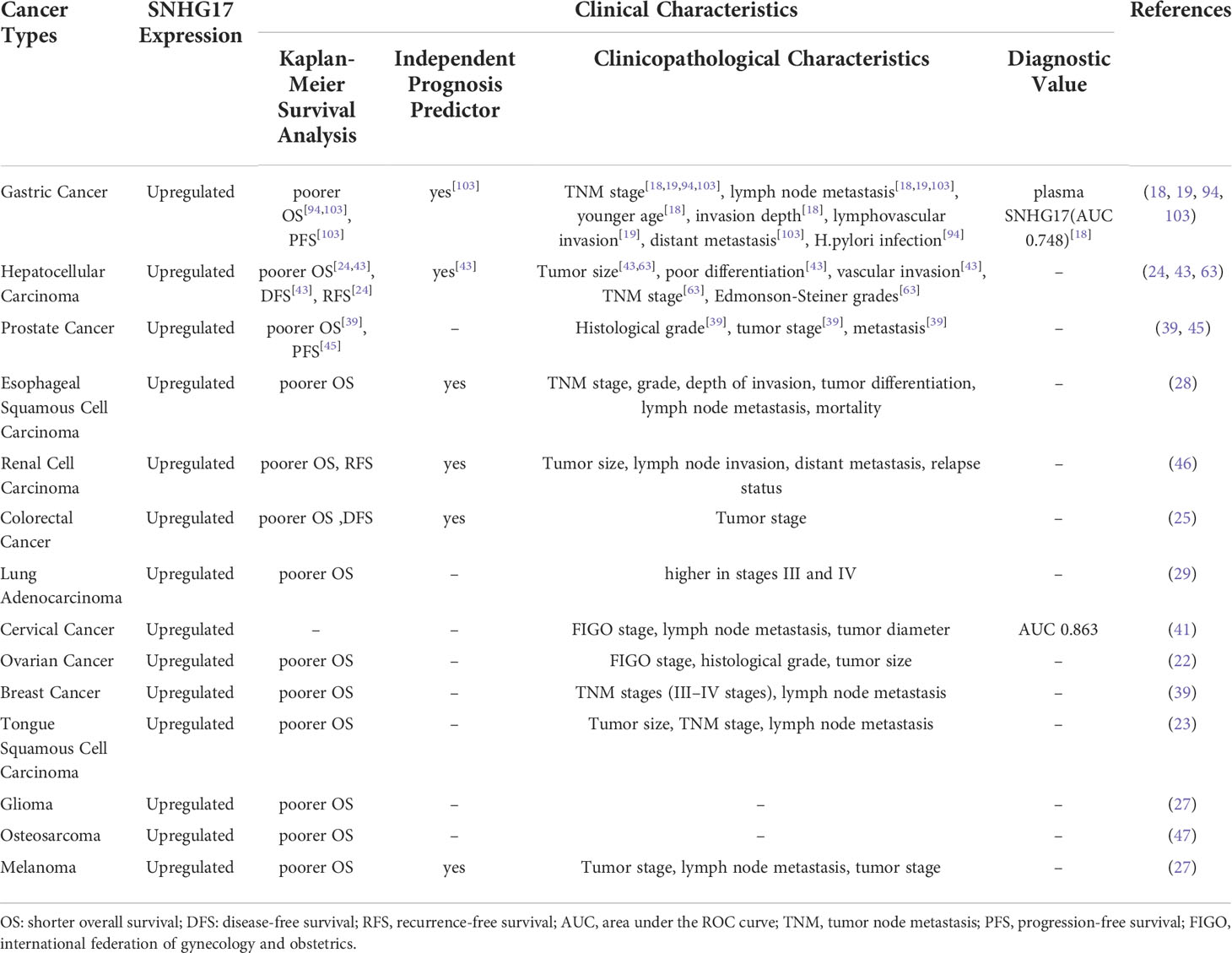
Table 1 Expression and clinical significance of SNHG17 in human cancers.
SNHG17 was highly expressed in many cancers. Mechanistically, YY1 (33), TGF‐β1 (29), STAT3 (23, 28), and two positive feedback loops: SNHG17/miR-339-5p/STAT5A/SNHG17 (15), SNHG17/miR-339-5p/FOSL2/SNHG17 (26) are upstream regulators of SNHG17. This paper describes the mechanisms of SNHG17 that enable cells to gain cancer hallmarks (Figure 5) (Table 2). However, the relevance between SNHG17 and two other hallmarks, namely evading growth suppressors and polymorphic microbiomes, has been excluded due to the limitations of the published literature. Some proposed potential mechanisms of SNHG17 also require further experimental validation, which will be essential in future SNHG17 explorations. Meanwhile, SNHG17 regulation of its homolog snoRA71B through a positive feedback loop provides a new idea for the regulatory relationship between SNHG and snoRNA.

Table 2 The targets and mechanisms underlying the effects of SNHG17.
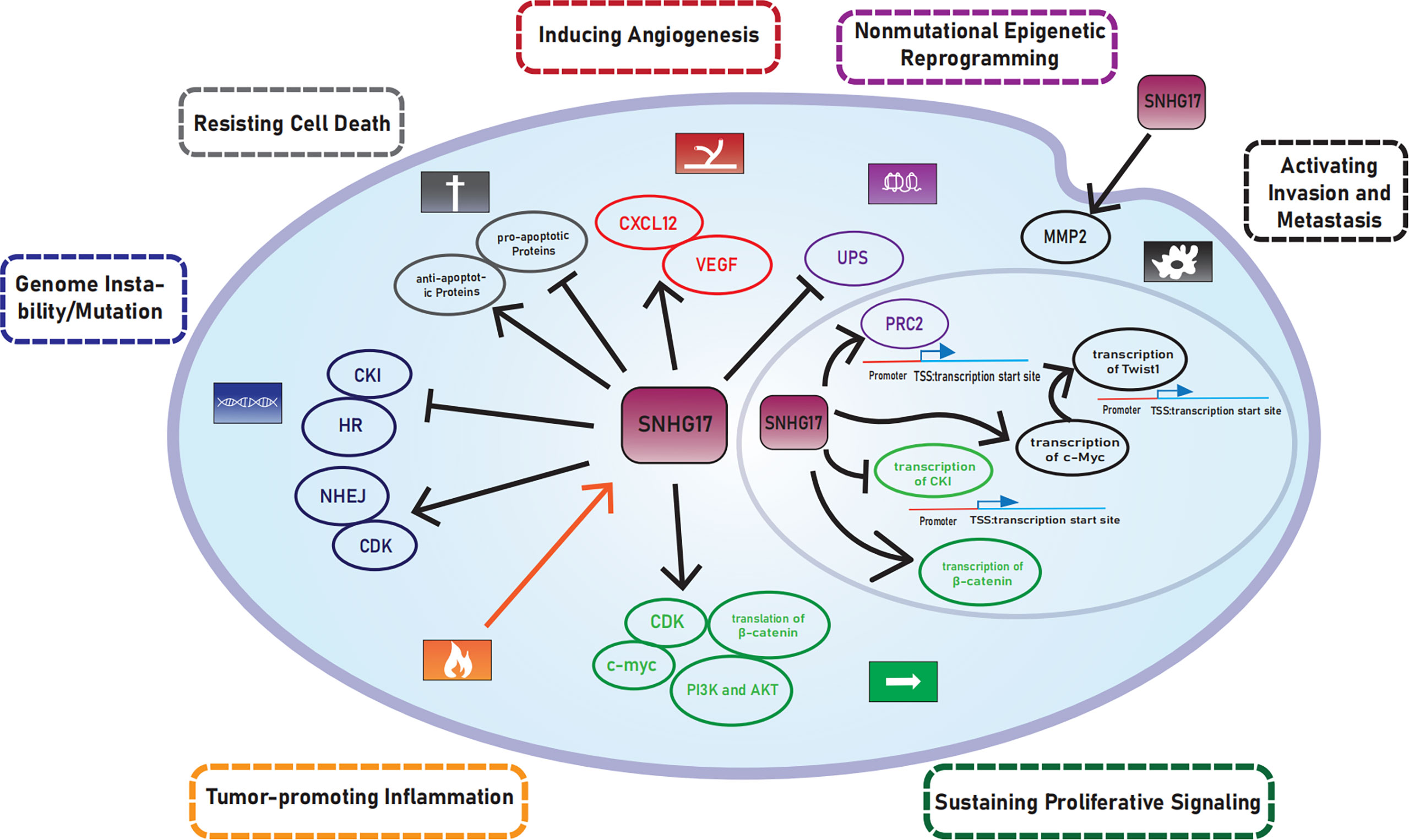
Figure 5 The relationship between SNHG17 and cancer hallmark.
Author contributions
Conceived the idea: NZ, MC. Wrote the majority of the manuscript: NZ. Collected the data, designed the tables and figures: NZ, YS, TW and XX. Critical revisions: MC and XX. All authors read and approved the final manuscript.
Funding
This work was supported by Heilongjiang Postdoctoral Scientific Research Developmental Fund (LBH-Q20040).
Acknowledgments
We are grateful for the help of Heilongjiang Human Resources and Social Security Bureau.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.974939/full#supplementary-material
References
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
3. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
4. Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell (2016) 29(4):452–63. doi: 10.1016/j.ccell.2016.03.010
5. Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol (2021) 220(2). doi: 10.1083/jcb.202009045
6. Statello L, Guo CJ, Chen LL, et al. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol (2021) 22(2):96–118. doi: 10.1038/s41580-020-00315-9
7. Xiao B, Huang Z, Zhou R, et al. The prognostic value of expression of the long noncoding RNA (lncRNA) small nucleolar RNA host gene 1 (SNHG1) in patients with solid malignant tumors: A systematic review and meta-analysis. Med Sci Monit: Int Med J Exp Clin Res (2018) 24:5462–72. doi: 10.12659/MSM.911687
8. Huang L, Jiang X, Wang Z, et al. Small nucleolar RNA host gene 1: a new biomarker and therapeutic target for cancers. (2018) 214(9):1247–52. doi: 10.1016/j.prp.2018.07.033
9. Guan YX, Zhang MZ, Chen XZ, et al. Lnc RNA SNHG20 participated in proliferation, invasion, and migration of breast cancer cells via miR-495. J Cell Biochem (2018) 119(10):7971–81. doi: 10.1002/jcb.26588
10. Huang W, Tian Y, Dong S, et al. The long non-coding RNA SNHG3 functions as a competing endogenous RNA to promote malignant development of colorectal cancer. Oncol Rep (2017) 38(3):1402–10. doi: 10.3892/or.2017.5837
11. Zhang T, Cao C, Wu D, et al. SNHG3 correlates with malignant status and poor prognosis in hepatocellular carcinoma. Tumour Biol: J Int Soc Oncodevelopmental Biol Med (2016) 37(2):2379–85. doi: 10.1007/s13277-015-4052-4
12. He B, Bai Y, Kang W, et al. LncRNA SNHG5 regulates imatinib resistance in chronic myeloid leukemia via acting as a CeRNA against MiR-205-5p. Am J Cancer Res (2017) 7(8):1704–13.
13. Zhao L, Han T, Li Y, et al. The lncRNA SNHG5/miR-32 axis regulates gastric cancer cell proliferation and migration by targeting KLF4. FASEB J: Off Publ Fed Am Soc Exp Biol (2017) 31(3):893–903. doi: 10.1096/fj.201600994R
14. Gong CY, Tang R, Nan W, et al. Role of SNHG16 in human cancer. Clin Chim Acta; Int J Clin Chem (2020) 503:175–80. doi: 10.1016/j.cca.2019.12.023
15. Wu G, Hao C, Qi X, et al. LncRNA SNHG17 aggravated prostate cancer progression through regulating its homolog SNORA71B via a positive feedback loop. Cell Death Dis (2020) 11(5):393. doi: 10.1038/s41419-020-2569-y
16. Ma Z, Gu S, Song M, et al. Long non-coding RNA SNHG17 is an unfavourable prognostic factor and promotes cell proliferation by epigenetically silencing P57 in colorectal cancer. Mol Biosyst (2017) 13(11):2350–61. doi: 10.1039/C7MB00280G
17. Georgakilas AG, Martin OA, Bonner WM. p21: A two-faced genome guardian. Trends Mol Med (2017) 23(4):310–9. doi: 10.1016/j.molmed.2017.02.001
18. Zhang F, Peng H. LncRNA-ANCR regulates the cell growth of osteosarcoma by interacting with EZH2 and affecting the expression of p21 and p27. J Orthopaedic Surg Res (2017) 12(1):103. doi: 10.1186/s13018-017-0599-7
19. Zhang G, Xu Y, Wang S, et al. LncRNA SNHG17 promotes gastric cancer progression by epigenetically silencing of p15 and p57. J Cell Physiol (2019) 234(4):5163–74. doi: 10.1002/jcp.27320
20. Gao C, Wu X, Zhai J, et al. Long non-coding RNA SNHG17 promotes gastric cancer progression by inhibiting P15 and P16. Trans Cancer Res (2019) 8(2):520–31. doi: 10.21037/tcr.2019.04.14
21. Liu J-X, Li W, Li J-T, et al. Screening key long non-coding RNAs in early-stage colon adenocarcinoma by RNA-sequencing. (2018) 10(09):1215–28. doi: 10.2217/epi-2017-0155
22. Cho YG, Choi BJ, Song JW, et al. Aberrant expression of krUppel-like factor 6 protein in colorectal cancers. World J Gastroenterol (2006) 12(14):2250–3. doi: 10.3748/wjg.v12.i14.2250
23. Pan X, Guo Z, Chen Y, et al. STAT3-induced lncRNA SNHG17 exerts oncogenic effects on ovarian cancer through regulating CDK6.Molecular therapy. Nucleic Acids (2020) 22:38–49. doi: 10.1016/j.omtn.2020.08.006
24. Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor myc. Adv Cancer Res (2011) 110:77–106. doi: 10.1016/B978-0-12-386469-7.00004-9
25. Liu JY, Chen YJ, Feng HH, et al. LncRNA SNHG17 interacts with LRPPRC to stabilize c-myc protein and promote G1/S transition and cell proliferation. Cell Death Dis (2021) 12(11):970. doi: 10.1038/s41419-021-04238-x
26. Bian Z, Zhou M, Cui K, et al. SNHG17 promotes colorectal tumorigenesis and metastasis via regulating Trim23-PES1 axis and miR-339-5p-FOSL2-SNHG17 positive feedback loop. J Exp Clin Cancer Res: CR (2021) 40(1):360. doi: 10.1186/s13046-021-02162-8
27. Guo F, Wang W, Song Y, et al. LncRNA SNHG17 knockdown promotes parkin-dependent mitophagy and reduces apoptosis of podocytes through Mst1. Cell Cycle (2020) 19(16):1997–2006. doi: 10.1080/15384101.2020.1783481
28. Gao H, Liu R, Sun X. STAT3-induced upregulation of lncRNA SNHG17 predicts a poor prognosis of melanoma and promotes cell proliferation and metastasis through regulating PI3K-AKT pathway. Eur Rev Med Pharmacol Sci (2019) 23(18):8000–10.
29. Shen S, Liang J, Liang X, et al. SNHG17, as an EMT-related lncRNA, promotes the expression of c-myc by binding to c-jun in esophageal squamous cell carcinoma. Cancer Sci (2022) 113(1):319–33. doi: 10.1111/cas.15184
30. Zhang Z, Yan Y, Zhang B, et al. Long non-coding RNA SNHG17 promotes lung adenocarcinoma progression by targeting the microRNA-193a-5p/NETO2 axis. Oncol Lett (2021) 22(6):818. doi: 10.3892/ol.2021.13079
31. Xu JC, Chen TY, Liao LT, et al. NETO2 promotes esophageal cancer progression by inducing proliferation and metastasis via PI3K/AKT and ERK pathway. Int J Biol Sci (2021) 17(1):259–70. doi: 10.7150/ijbs.53795
32. Wang J, Sun J, Zhang N, et al. PES1 enhances proliferation and tumorigenesis in hepatocellular carcinoma via the PI3K/AKT pathway. (2019) 219:182–9. doi: 10.1016/j.lfs.2018.12.054
33. Li H, Li T, Huang D, et al. Long noncoding RNA SNHG17 induced by YY1 facilitates the glioma progression through targeting miR-506-3p/CTNNB1 axis to activate wnt/β-catenin signaling pathway. Cancer Cell Int (2020) 20:29. doi: 10.1186/s12935-019-1088-3
34. Qiao C, Qiao T, Yang S, et al. SNHG17/miR-384/ELF1 axis promotes cell growth by transcriptional regulation of CTNNB1 to activate wnt/β-catenin pathway in oral squamous cell carcinoma. Cancer Gene Ther (2022) 29(1):122–32. doi: 10.1038/s41417-021-00294-9
35. Li W, Zheng Y, Mao B, et al. SNHG17 upregulates WLS expression to accelerate lung adenocarcinoma progression by sponging miR-485-5p. Biochem Biophys Res Commun (2020) 533(4):1435–41. doi: 10.1016/j.bbrc.2020.09.130
36. Huang F, Li H, Qin Z, et al. SNHG17 serves as an oncogenic lncRNA by regulating the miR-361-3p/STC2 axis in rectal cancer. Front Genet (2021) 12:654686. doi: 10.3389/fgene.2021.654686
37. Lu D, Li Y, Liu QR, et al. Wls promotes the proliferation of breast cancer cells via wnt signaling. Med Oncol (Northwood London England) (2015) 32(5):140. doi: 10.1007/s12032-015-0585-z
38. Li Q, Zhou X, Fang Z, et al. Effect of STC2 gene silencing on colorectal cancer cells. Mol Med Rep (2019) 20(2):977–84. doi: 10.3892/mmr.2019.10332
39. Du F, Hou Q. SNHG17 drives malignant behaviors in astrocytoma by targeting miR-876-5p/ERLIN2 axis. BMC Cancer (2020) 20(1):839. doi: 10.1186/s12885-020-07280-8
40. Zhao H, Dong H, Wang P, et al. Long non-coding RNA SNHG17 enhances the aggressiveness of C4-2 human prostate cancer cells in association with β-catenin signaling. Oncol Lett (2021) 21(6):472. doi: 10.3892/ol.2021.12733
41. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature (2004) 432(7015):307–15. doi: 10.1038/nature03098
42. Cao S, Li H, Li L. LncRNA SNHG17 contributes to the progression of cervical cancer by targeting microRNA-375-3p. Cancer Manag Res (2021) 13:4969–78. doi: 10.2147/CMAR.S312469
43. Zhao L, Ye J, Lu Y, et al. lncRNA SNHG17 promotes pancreatic carcinoma progression via cross-talking with miR-942. Am J Trans Res (2021) 13(3):1037–50.
44. Luo Y, Lin J, Zhang J, et al. LncRNA SNHG17 contributes to proliferation, migration, and poor prognosis of hepatocellular carcinoma. Can J Gastroenterol Hepatol (2021) 2021:9990338. doi: 10.1155/2021/9990338
45. Tong F, Guo J, Miao Z, et al. LncRNA SNHG17 promotes the progression of oral squamous cell carcinoma by modulating miR-375/PAX6 axis. Cancer Biomarkers: Section A Dis Markers (2021) 30(1):1–12. doi: 10.3233/CBM-191070
46. Wu J, Dong G, Liu T, et al. LncRNA SNHG17 promotes tumor progression and predicts poor survival in human renal cell carcinoma via sponging miR-328-3p. Aging (2021) 13(17):21232–50. doi: 10.18632/aging.203440
47. Zhao A, Zhao Z, Liu W, et al. Carcinoma-associated fibroblasts promote the proliferation and metastasis of osteosarcoma by transferring exosomal LncRNA SNHG17. Am J Trans Res (2021) 13(9):10094–111.
48. Xu T, Yan S, Jiang L, et al. Gene amplification-driven long noncoding RNA SNHG17 regulates cell proliferation and migration in human non-Small-Cell lung cancer. molecular therapy. Nucleic Acids (2019) 17:405–13. doi: 10.1016/j.omtn.2019.06.008
49. Williams AC, Collard TJ, Perks CM, et al. Increased p53-dependent apoptosis by the insulin-like growth factor binding protein IGFBP-3 in human colonic adenoma-derived cells. Cancer Res (2000) 60(1):22–7.
50. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res: an Off J Am Assoc Cancer Res (2009) 15(17):5308–16. doi: 10.1158/1078-0432.CCR-07-5023
51. Apel A, Zentgraf H, Büchler MW, et al. Autophagy-a double-edged sword in oncology. Int J Cancer (2009) 125(5):991–5. doi: 10.1002/ijc.24500
52. Rao S, Tortola L, Perlot T, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun (2014) 5:3056. doi: 10.1038/ncomms4056
53. Smith AG, Macleod KF. Autophagy, cancer stem cells and drug resistance. (2019) 247(5):708–18. doi: 10.1002/path.5222
54. Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab (2017) 25(5):1037–43. doi: 10.1016/j.cmet.2017.04.004
55. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell (2016) 61(5):667–76. doi: 10.1016/j.molcel.2016.02.011
56. Meng C, Zhou JQ, Liao YS. Autophagy-related long non-coding RNA signature for ovarian cancer. J Int Med Res (2020) 48(11):300060520970761. doi: 10.1177/0300060520970761
57. Xuan Y, Chen W, Liu K, et al. A risk signature with autophagy-related long noncoding RNAs for predicting the prognosis of clear cell renal cell carcinoma: Based on the TCGA database and bioinformatics. Dis Markers (2021) 2021:8849977. doi: 10.1155/2021/8849977
58. Bernardini JP, Lazarou M, Dewson G. Parkin and mitophagy in cancer. Oncogene (2017) 36(10):1315–27. doi: 10.1038/onc.2016.302
59. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
60. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol (2020) 30(6):478–90. doi: 10.1016/j.tcb.2020.02.009
61. Shu X, Zhang Z, Yao ZY, et al. Identification of five ferroptosis-related LncRNAs as novel prognosis and diagnosis signatures for renal cancer. Front Mol Biosci (2021) 8:763697. doi: 10.3389/fmolb.2021.763697
62. Chen W, Wang L, Li X, et al. LncRNA SNHG17 regulates cell proliferation and invasion by targeting miR-338-3p/SOX4 axis in esophageal squamous cell carcinoma. Cell Death Dis (2021) 12(9):806. doi: 10.1038/s41419-021-04093-w
63. Ma T, Zhou X, Wei H, et al. Long non-coding RNA SNHG17 upregulates RFX1 by sponging miR-3180-3p and promotes cellular function in hepatocellular carcinoma. Front Genet (2020) 11:607636. doi: 10.3389/fgene.2020.607636
64. Bai M, Lei Y, Wang M, et al. Long non-coding RNA SNHG17 promotes cell proliferation and invasion in castration-resistant prostate cancer by targeting the miR-144/CD51 axis. Front Genet (2020) 11:274. doi: 10.3389/fgene.2020.00274
66. Long Y, Wang X, Youmans DT, et al. How do lncRNAs regulate transcription? Sci Adv (2017) 3(9):eaao2110. doi: 10.1126/sciadv.aao2110
67. Liu Y, Li Q, Tang D, et al. SNHG17 promotes the proliferation and migration of colorectal adenocarcinoma cells by modulating CXCL12-mediated angiogenesis. Cancer Cell Int (2020) 20(1):566. doi: 10.1186/s12935-020-01621-0
68. Xiao H, Tong R, Ding C, et al. γ-H2AX promotes hepatocellular carcinoma angiogenesis via EGFR/HIF-1α/VEGF pathways under hypoxic condition. Oncotarget (2015) 6(4):2180–92. doi: 10.18632/oncotarget.2942
69. Kollmann K, Heller G, Schneckenleithner C, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell (2013) 24(2):167–81. doi: 10.1016/j.ccr.2013.07.012
70. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science (2020) 368(6487). doi: 10.1126/science.aaw5473
71. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
72. Warburg O. On respiratory impairment in cancer cells. Science (1956) 124(3215):269–70. doi: 10.1126/science.124.3215.269
73. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
74. Zhang C, Lin M, Wu R, et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the warburg effect. Proc Natl Acad Sci U S A (2011) 108(39):16259–64. doi: 10.1073/pnas.1113884108
75. Liu J, Zhang C, Zhao Y, et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun (2017) 8(1):1823.
76. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene (2010) 29(5):625–34. doi: 10.1038/onc.2009.441
77. Dang CV, Kim JW, Gao P, et al. The interplay between MYC and HIF in cancer. Nat Rev Cancer (2008) 8(1):51–6. doi: 10.1038/nrc2274
78. Dang CV. The interplay between MYC and HIF in the warburg effect, in: Ernst Schering Foundation symposium proceedings, , Vol. 2007. pp. 35–53.
79. Broadfield LA, Pane AA, Talebi A, et al. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev Cell (2021) 56(10):1363–93. doi: 10.1016/j.devcel.2021.04.013
80. Maan M, Peters JM, Dutta M, et al. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun (2018) 504(3):582–9. doi: 10.1016/j.bbrc.2018.02.097
81. Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest (2019) 129(8):3006–17. doi: 10.1172/JCI127201
82. Patel KK, Kashfi K. Lipoproteins and cancer: The role of HDL-c, LDL-c, and cholesterol-lowering drugs. Biochem Pharmacol (2022) 196:114654. doi: 10.1016/j.bcp.2021.114654
83. Mohamadi M, Ghaedi H, Kazerouni F, et al. Deregulation of long noncoding RNA SNHG17 and TTC28-AS1 is associated with type 2 diabetes mellitus. Scand J Clin Lab Invest (2019) 79(7):519–23. doi: 10.1080/00365513.2019.1664760
84. Cheng L, Yuan B, Ying S, et al. PES1 is a critical component of telomerase assembly and regulates cellular senescence. Sci Adv (2019) 5(5):eaav1090. doi: 10.1126/sciadv.aav1090
85. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
86. Kok VC, Yu CC. Cancer-derived exosomes: Their role in cancer biology and biomarker development. Int J Nanomed (2020) 15:8019–36. doi: 10.2147/IJN.S272378
87. Muniz-Bongers LR, McClain CB, Saxena M, et al. MMP2 and TLRs modulate immune responses in the tumor microenvironment. JCI Insight (2021) 6(12). doi: 10.1172/jci.insight.144913
88. Li J, Zeng T, Li W, et al. Long non-coding RNA SNHG1 activates HOXA1 expression via sponging miR-193a-5p in breast cancer progression. Aging (2020) 12(11):10223–34. doi: 10.18632/aging.103123
89. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev (2020) 34(23-24):1565–76. doi: 10.1101/gad.343129.120
90. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer (2019) 19(8):439–53. doi: 10.1038/s41568-019-0156-2
91. Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: Defining a path forward. Cell (2019) 179(4):813–27. doi: 10.1016/j.cell.2019.10.005
92. He S, Sharpless NE. Senescence in health and disease. Cell (2017) 169(6):1000–11. doi: 10.1016/j.cell.2017.05.015
93. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. (2019) 21(1):94–101. doi: 10.1038/s41556-018-0249-2
94. Han T, Jing X, Bao J, et al. H. pylori infection alters repair of DNA double-strand breaks via SNHG17. J Clin Invest (2020) 130(7):3901–18.
95. Levitt NC, Hickson ID. Caretaker tumour suppressor genes that defend genome integrity. Trends Mol Med (2002) 8(4):179–86. doi: 10.1016/S1471-4914(02)02298-0
96. Peng X, Zou R, Pan C, et al. Long non-coding RNA small nucleolar RNA host gene 17 promotes hepatocellular carcinoma progression by inhibiting p57 expression. Ann Clin Lab Sci (2021) 51(6):766–71.
97. Wang C, Qu Y, Wang D, et al. The proangiogenic roles of long non-coding RNAs revealed by RNA-sequencing following oxygen-glucose Deprivation/Re-oxygenation. (2019) 52(4):708–27.
98. Aparicio T, Baer R, Gautier J. DNA Double-strand break repair pathway choice and cancer. DNA Repair (2014) 19:169–75. doi: 10.1016/j.dnarep.2014.03.014
99. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
100. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6(1):24–37. doi: 10.1038/nrc1782
101. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
102. Williams GT, Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer? Nature reviews. Cancer (2012) 12(2):84–8.
Keywords: long non-coding RNA, lncRNA small nucleolar host gene 17 (SNHG17), cancer, cancer hallmark, biomarker
Citation: Zhang N, Sun Y, Wang T, Xu X and Cao M (2022) Enabling factor for cancer hallmark acquisition: Small nucleolar RNA host gene 17. Front. Oncol. 12:974939. doi: 10.3389/fonc.2022.974939
Received: 21 June 2022; Accepted: 19 August 2022;
Published: 14 September 2022.
Edited by:
Brice Laffleur, INSERM UMR1236 Microenvironnement, Différenciation cellulaire, Immunologie et Cancer, FranceReviewed by:
Wenxia Xu, Zhejiang University, ChinaZehua Bian, Affiliated Hospital of Jiangnan University, China
Copyright © 2022 Zhang, Sun, Wang, Xu and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mengru Cao, mengru527@hotmail.com