
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 21 September 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.973836
This article is part of the Research Topic Proteomic and Metabolic Reprogramming in Myeloma Cells within the Tumor Microenvironment View all 9 articles
 Antonio Giovanni Solimando1,2
Antonio Giovanni Solimando1,2 Eleonora Malerba1
Eleonora Malerba1 Patrizia Leone1
Patrizia Leone1 Marcella Prete3
Marcella Prete3 Carolina Terragna4
Carolina Terragna4 Michele Cavo4
Michele Cavo4 Vito Racanelli3*
Vito Racanelli3*Multiple myeloma (MM) is still an incurable disease, despite considerable improvements in treatment strategies, as resistance to most currently available agents is not uncommon. In this study, data on drug resistance in MM were analyzed and led to the following conclusions: resistance occurs via intrinsic and extrinsic mechanisms, including intraclonal heterogeneity, drug efflux pumps, alterations of drug targets, the inhibition of apoptosis, increased DNA repair and interactions with the bone marrow (BM) microenvironment, cell adhesion, and the release of soluble factors. Since MM involves the BM, interactions in the MM-BM microenvironment were examined as well, with a focus on the cross-talk between BM stromal cells (BMSCs), adipocytes, osteoclasts, osteoblasts, endothelial cells, and immune cells. Given the complex mechanisms that drive MM, next-generation treatment strategies that avoid drug resistance must target both the neoplastic clone and its non-malignant environment. Possible approaches based on recent evidence include: (i) proteasome and histone deacetylases inhibitors that not only target MM but also act on BMSCs and osteoclasts; (ii) novel peptide drug conjugates that target both the MM malignant clone and angiogenesis to unleash an effective anti-MM immune response. Finally, the role of cancer stem cells in MM is unknown but given their roles in the development of solid and hematological malignancies, cancer relapse, and drug resistance, their identification and description are of paramount importance for MM management.
Recent advances in the treatment of MM include the use of novel therapeutic strategies, such as immunomodulatory drugs (thalidomide, lenalidomide, pomalidomide), proteasome inhibitors (PIs: bortezomib, carfilzomib), monoclonal antibodies (MoAbs: daratumumab and elotuzumab), T-cell-based therapies, checkpoint inhibitors and hematopoietic stem cell transplantation. Nonetheless, MM remains an incurable disease (1–3), with 5-year survival rates ranging from 23.1% to 46.7%. For example, in the UK, survival has increased from 6% to 33%, but only 29% of patients will survive more than 10 years (4). Most patients suffer relapse because of the incomplete therapeutic eradication of malignant plasma cells (PCs). In MM, PCs can manifest drug resistance already before treatment or following conventional drug exposure (5). While patients initially respond to modern combination treatment regimens, they inevitably experience serial relapses, with the depth and duration of response following each relapse becoming progressively shorter until their disease becomes refractory (6). How drug-resistant PCs become dominant in MM and are able to persist despite multiple lines of therapy is poorly understood (7). However, elucidation of the mechanisms underlying drug resistance is a prerequisite for MM prevention and for effective therapies.
Drug resistance in MM can involve intrinsic or extrinsic mechanisms (Figure 1) (8, 9). The former includes genetic and epigenetic alterations, as occurs in other solid and hematological malignancies (10–14), the overexpression of drug efflux pumps (15), alteration of drug targets (16), and a dysregulation of intracellular signaling pathways, such as those that mediate apoptosis (17), autophagy, and DNA repair (18). Extrinsic mechanisms are mainly mediated by interactions with the bone marrow (BM) microenvironment (7, 19), cell adhesion to the extracellular matrix (ECM), and other elements of the tumor microenvironment (20), which may induce the production in MM cells of cell cycle inhibitors, anti-apoptotic members of the Bcl-2 family, and ABC drug transporters (21) (18, 22) as well as the release of soluble factors, such as interleukin (IL)-6 and insulin-like growth factor (IGF)-1, by bone marrow stromal cells (BMSCs) that activate the signal transduction pathways leading to drug resistance (23).
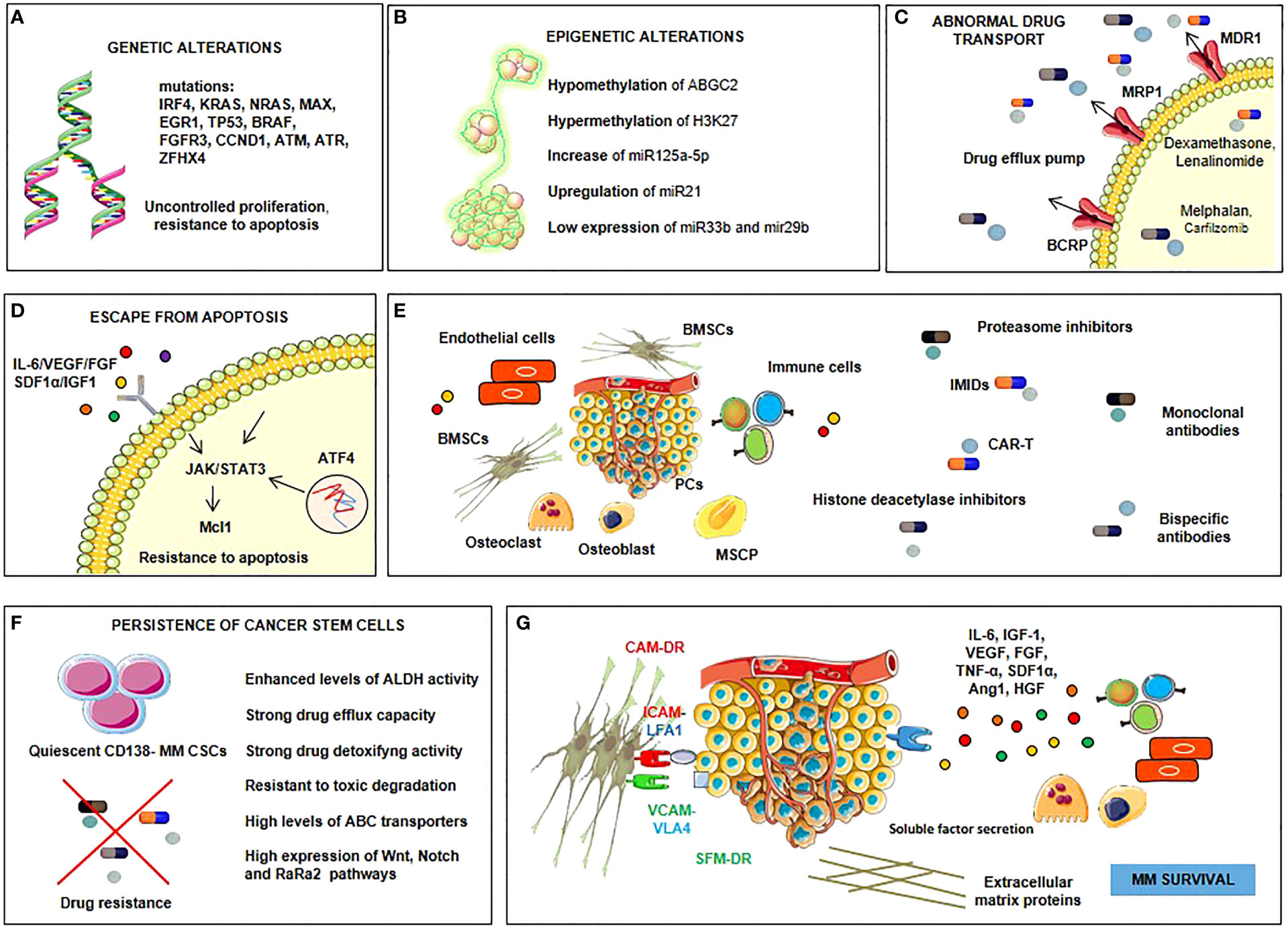
Figure 1 Drug resistance in MM involves intrinsic or extrinsic mechanisms. Intrinsic ones are genetic (A) and epigenetic (B) alterations. Genetic alterations (A) include mutations of a lot of genes such as interferon regulatory factor-4 (IRF-4), KRAS, NRAS, myc-associated factor X (MAX), early growth response protein 1 (EGR1), tumor protein p53 (TP53), BRAF, fibroblast growth factor receptor 3 (FGFR3), cyclin D1 (CCND1), ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related protein (ATR) and zinc finger homeobox 4 (ZFHX4), which procure uncontrolled proliferation and resistance to apoptosis. Epigenetic alterations (B) involve mechanisms of hypomethylation (ATP-binding cassette G2, ABGC2) and hypermethylation (histone 3 lysine 27, H3k27); upregulation of some microRNAs (miRNAs), such as miR125a-5p and miR21, and downregulation of others, such as miR33b and miR29b. Other intrinsic mechanisms are the overexpression of drug efflux pumps and the alteration of drug targets (C), dysregulation of intracellular signaling pathways, such as those that mediate apoptosis (Janus kinase (JAK)-signal transducer and activator of transcription 3 (STAT3)-myeloid cell leukemia sequence 1 (Mcl1), autophagy (activating transcription factor 4, ATF4), and DNA repair (D). Extrinsic mechanisms involve myeloma plasma cell interactions with the BM microenvironment (E), persistence of cancer stem cells (F), soluble factors-mediated drug resistance (SFM-DR) and cell adhesion-mediated drug resistance (CAM-DR) (G) resulting from the production of soluble factors such as interleukin (IL)-6, insulin-like growth factor-1 (IGF-1), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), tumor necrosis factor-alpha (TNF-α), stromal cell-derived factor 1-alpha (SDF1-α), angiopoietin 1 (Ang1), hepatocyte growth factor (HGF) by bone marrow stromal cells (BMSCs) and plasma cells (PCs), and from the overexpression of cell cycle inhibitors, anti-apoptotic members of the B-cell lymphoma-2 (Bcl-2) family and ABC drug transporters in myeloma cells upon direct adhesion with BMSCs.
Myeloma cells reside in the BM, which forms a nutritional niche where they interact with BMSCs via CD44 and other adhesion molecules (24). A vicious cycle is established, driven by the secretion by BMSCs of factors that promote the proliferation of myeloma cells (IL-6, IGF-1, transforming growth factor (TGF)-β, and hepatocyte growth factor (HGF) (25–27). Moreover, due to the median age of disease onset, the BM is enriched with adipose tissue, which secretes tumor-promoting factors such as leptin and adiposin (28). One of the hallmarks of MM is the induction of bone lysis, the consequence of the secretion by malignant clones of osteoclast-activating factors such as MIP1-α and RANKL (29–31). In turn, osteoclasts secrete osteotropic factors, such as osteopontin and IL-6 (32), while the tumor mass produces the osteoblast inhibitors DKK1 and sFRP (33). The interaction of MM cells and osteoclasts is crucial; osteoclasts and MM plasma cells can recruit each other and mutually promote their survival through multiple mechanisms. To better understand these mechanisms, Moreaux et al. have analyzed the osteoclast-gene expression profiling and detected 552 genes overexpressed in osteoclasts compared with other BM cell subpopulations. They have identified genes coding for 4 CCR2-targeting chemokines that are expressed specifically by osteoclasts and genes coding for important MM growth factors such as IGF-1, IL-6, and a proliferation-inducing ligand (APRIL) (34, 35). This suggests that if plasma cell expresses CCR2, it could be attracted by osteoclasts specifically. Indeed, in the bone remodeling compartments CCR2 chemokines as well as plasma cell survival factors were highly concentrated. Furthermore, the bone lesion number in patients with newly diagnosed MM was associated with high CCR2 expression in myeloma plasma cells (34). Thus, targeting the osteoclast/MM plasma cell interaction through MoAb against CCR2 and/or MM growth factors may be a promising therapeutic approach. Importantly, in vitro experiments showed that an anti-CCR2 MoAb blocked osteoclast chemoattractant activity for myeloma cells, but it did not inhibit osteoclast MM cell growth activity. APRIL or IL-6 inhibitors specifically reduced osteoclast-induced MM survival to a partial extent. An anti-IGF-1 receptor MoAb totally suppressed the osteoclast-induced survival of MM plasma cells inhibiting both osteoclast and myeloma cell survival. The exact role of osteoclast in the BM milieu is not fully elucidated. However, the impact in terms of bone involvement prompted a plethora of preclinical and clinical studies aiming to target bone remodeling and, indirectly, tumor progression (29, 36, 37). All targets mentioned above could be potentially valuable in the next-generation immunotherapy for MM patients. For instance, a receptor for APRIL is the B-cell maturation antigen (BCMA), a novel encouraging target for MM immunotherapy. BCMA is a cell biomarker present mainly on CD138pos, CD38pos, CD45neg cells, and it physiologically orchestrates plasma cell homeostasis (38). BCMA pathway highly depends on APRIL and BAFF, boosting MM cell pro-survival, and drug resistance (31, 39).
Furthermore, MM cells also induce neo-angiogenesis in the BM, through their release of the endothelial cell stimulators vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF), which ensures the nutritional support of myeloma growth, including via stromal cell-derived factor (SDF)-1α, angiopoietin (Ang)-1, and HGF (40, 41). Based on these findings, the currently available treatment for MM targets both the malignant cells and the MM niche, through the inclusion of proteasome or HDAC inhibitors, which act on stromal cells, osteoclasts, and angiogenesis (42, 43).
The immunological microenvironment in patients with MM is unique, as are the genetics of the tumor itself. This was recently demonstrated by the use of high-content single-cell techniques (44). The observation that immune activation and exhaustion appear early in the pathogenesis of the preneoplastic phase termed monoclonal gammopathy of undetermined significance (MGUS) (44) suggests that features of the immune response necessary for the long-term stability and persistence of immunity, such as stem-like memory T cells, are critical determinants of immune control. Specific immune response targets may also be relevant, as it may be more effective to therapeutically target clonal rather than subclonal mutations or critical clone features such as stemness (36). Although the spatial elements of the immune response in hematologic malignancies have hardly been investigated, pathologists and radiologists have long recognized that myeloma is multifocal in its growth (hence the name multiple myeloma). Accordingly, understanding the spatial aspects of the immune response as well as the roles played by tissue-resident vs. recruited cells will be crucial in achieving immune control (37, 38).
The evolutionary and ecological context of the tumor must also be considered (39). The former refers to the heterogeneous genetic makeup of the tumor cells as well as changes therein over time. The ecological context is determined by immune cells and the availability of tumor-supporting resources, such as growth hormones and nutrients (39). In MGUS, the evolutionary path is largely established early in the disease, but it is subject to alterations over time. While it is thought that ecological factors, particularly those involving the immune system, are a major predictor of the tumor’s evolutionary trajectory, the respective interactions are likely to be regional. Consequently, the spatial elements of these interactions must be taken into account in the design of therapeutic agents, which must target both the MM cells and the immune niche; combinatorial techniques may thus be required for tumors with a greater rate of genetic change, but in other cases more conservative and sequential tactics may be more effective (36). The immune exhaustion that begins early in MGUS underscores the need to integrate immunologically based prevention with the early detection of the disease (37).
Moreover, the myeloma niche is immunosuppressive, as the activity of myeloid-derived suppressor cells (MDSCs), which secrete IL-10 and TGF-β (45), is enhanced by the production of IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF) by MM cells. MDSCs also inhibit T-cell function, by releasing arginase and inducible nitric oxide synthase, and stimulate neo-angiogenesis by releasing VEGF (46–48).
De Jong et al. presented a single-cell transcriptomic analysis of the hematopoietic and non-hematopoietic BM microenvironment in MM and identified myeloma-specific BM iMSCs able to create an ideal environment for tumor cell proliferation and immune cell recruitment and modulation (49). These cells produced a wide range of inflammatory cytokines and chemokines, such as IL-6, COX2, CXCL2, CXCL8, VEGFA, proteins in the tumor necrosis factor (TNF) pathway, and CCL2, which increases MM cell motility through its association with CCR2 (49). These populations also expressed genes encoding CXCL3, CXCL5, and CD44, the latter of which was proposed as a flow cytometry marker of iMSC, despite the fact that it is not expressed by all iMSCs (50). De Jong et al. identified the expression of IL-1 and TNF receptors on MSCs as determinants of the iMSC phenotype. This was confirmed by the ex vivo activation of MSCs with IL-1 and TNF, which resulted in the iMSC phenotype in both healthy and MM-derived MSCs. The authors examined the interactions between MSC receptors and immune and tumor cells and found that IL-1B was largely expressed by monocytes, and TNF by cytotoxic T and NK cells. In addition to interacting with myeloid cells, iMSCs engaged with proliferating myeloma cells through the CCL2–CCR2 pathway. Interestingly, De Jong et al. revealed an immune cell-mediated stromal inflammation in BM of MM patients persisting even after successful induction therapy, suggesting a potential role of iMSCs in disease relapse (49).
Novel candidate targets that can be used to overcome drug resistance in MM have been identified by dividing the BM microenvironment into the low-oxygen endosteal niche and the high-oxygen vascular niche (51). MM cells reside in the endosteal niche, where the hypoxic environment induces changes in the availability of metabolites, such as amino acids, fatty acids, and nucleotides (52), and induces the enhanced shedding of exosomes (discussed below, 53), both of which contribute to the drug-resistant phenotype (54). Among the significant changes in myeloma cell metabolism in response to hypoxia (55, 56) are those affecting the levels of hexokinase-2 (HK-2) and lactate dehydrogenase A (LDHA, 56). Both have been linked to resistance to bortezomib, a PI shown in an in vivo model to decrease myeloma growth (55, 56). Ikeda et al. focused on KDM3A, another target of hypoxia in myeloma, and found that hypoxia caused an up-regulation of KDM3A that was paralleled by the up-regulation of the downstream protein MALAT1. Thus, according to this result, KDM3A induces MALAT1, which, in turn, induces glycolysis, leading to the proliferation of MM cells (55).
Proteomic profiling was used to examine the changes induced in myeloma cells obtained from patients with active disease. Janker et al. compared cells obtained from healthy donors to MM cells from patients with either a low or a high tumor burden (57). They found that most of the metabolic enzymes were modified in response to hypoxia, thus suggesting that most of the metabolic alterations in MM can be explained by the low oxygen concentration (57). However, studies of agents directed at hypoxia-sensitive targets, such HK2 inhibitors (58), the LDHA-specific inhibitor GSK 2837808A (59), demethylase KDM inhibitors (60), and antisense oligonucleotides (60), are at the preclinical stage. As a proof of concept, Xu et al. used an antisense oligonucleotide against HK2 in a xenograft model of an OPM-2 tumor and reported decreased MM progression (58).
Exosomes are small (100 nm) vesicles that are secreted by most cell types from multivesicular bodies and taken up by endocytosis or micropinocytosis. Importantly, however, they are involved in information transfer, including within the BM environment (31). MM-derived exosomes induce angiogenesis (53, 61), are involved in osteoblast functionality (62, 63) and boost cancer-associated fibroblasts activity in the MM niche (64–66). Roccaro et al. found that exosomes from MSCs of the BM of MM patients stimulate and those from healthy donors reduce the proliferation of myeloma cells, suggesting MM stromal cells as druggable targets (67). These observations were paralleled by in vivo results obtained using silk scaffolds (67). The effect of MSC-derived exosomes in bortezomib resistance was also investigated, with exosomes treatment shown to reduce proteasome inhibition (68). In a proteomic profiling study, Wang et al. compared exosomes from MM cells with those from stromal cells and found the up-regulation of MCP1 and SDF1 in the latter. Furthermore, exosomes from both were shown to be enriched in IL-6 and fibronectin, and exosomal miR15a was shown to be involved in MM suppression (67).
A comparison of the exosomes from the BM MSCs of bortezomib-resistant vs. bortezomib-sensitive MM patients revealed significantly higher levels of PSMA3-AS1, encoding a subunit of the proteasome, in resistant patients (69). Sensitivity could be restored by PSMA3 antisense treatment. These results were corroborated by an in vivo model in which MM mice treated with carfilzomib and PSMA-silencing RNA had reduced tumor development and improved survival (69). Studies of drug-exposure-dependent exosome secretion have shown the increased secretion of exosomes by MM cells treated with melphalan, bortezomib, or carfilzomib, with the respective chemoexosomes containing more heparinase on the surface and enriched with cell cycle proteins (70). The chemoexosomes of MM cells treated with bortezomib and melphalan were also shown to contain acid sphingomyelinase (ASM), which converts sphingomyelin to ceramide (71). The induction of drug resistance by treating bortezomib-sensitive JJN3 cells with bortezomib-resistant U266-cell-derived exosomes suggested that blocking ASM could restore drug sensitivity. Blocking exosome release, such as with the exosome inhibitor GW4869, to modulate osteolysis has been proposed as well (71). In that study, mice were treated with GW4869 in combination with bortezomib (71). Whereas GW4869 alone did not affect MM burden it synergized with bortezomib, whereas with respect to osteolysis GW4869 was effective as a single agent, with its use resulting in enhanced cortico-bone volume and decreased serum collagenase levels (62).
Collectively, exosomes link MM to metabolism and drug resistance, since they contain small molecules and miRNA/lncRNA, inducing metabolic changes in their target cells. They also contain metabolites (such as lactate), metabolic enzymes, and transporters (72). In MM, hypoxia induces changes in metabolism and exosome secretion, which in turn influence MM growth. Taken together, these observations point to exosomes as novel targets to overcome drug resistance.
In MM, the tumor microenvironment induces angiogenesis and hypoxia and alters the ECM as well as MSCs, osteoclasts, osteoblasts, and immune cells (73), representing promising druggable targets as stand-alone or in combinatorial approaches as demonstrated in solid and hematological cancers (74, 75). For example, the co-culture of MM cells with MM MSCs leads to enhanced tumor cell proliferation and adhesion. The gene-expression profile of MSCs from MM patients differs significantly from that of MSCs obtained from patients with multiple myeloma (76). Several vicious cycles are operative within the MM niche since MM cells make use of adhesion molecules and paracrine loops to interact with endothelial cells. Endothelial-multiple myeloma interactions feed into redundant cross-talk, propagating disease progression (77) (22, 27, 78–79) (Figure 2).
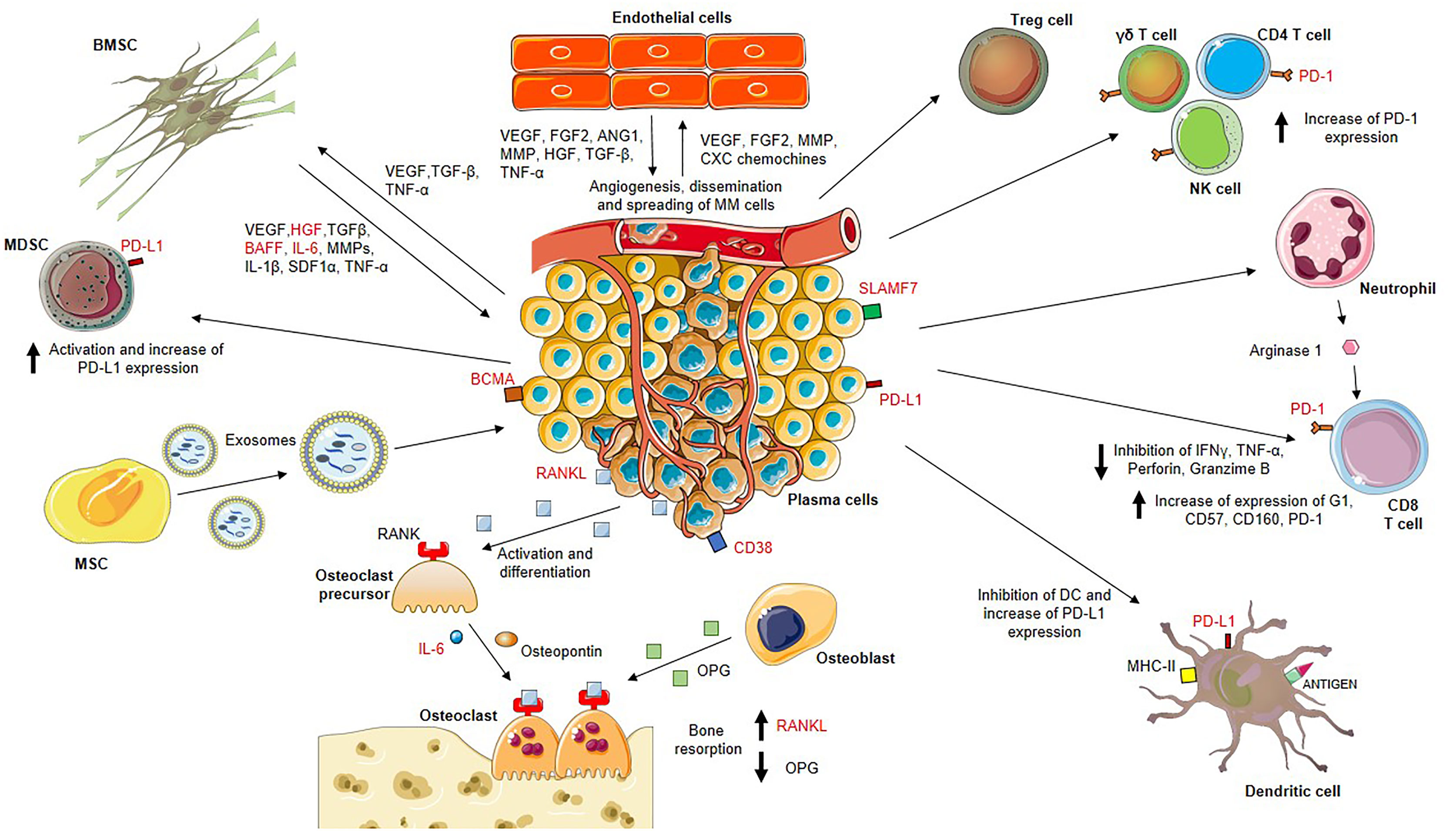
Figure 2 MM BM microenvironment is highly enriched in factors that sustain the proliferation of bone marrow-resident cells including myeloma plasma cells, endothelial cells and bone marrow stromal cells (BMSCs), actors of two important vicious cycles. On one hand, BMSCs release vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), transforming growth factor-β (TGF-β), B cell activating factor (BAFF), interleukin (IL)-6, matrix metalloproteinases (MMPs), IL-1β, stromal cell-derived factor 1 alpha (SDF1α), tumor necrosis factor-α (TNF-α) which stimulate myeloma plasma cell proliferation. On the other hand, myeloma plasma cells produce VEGF, TGF-β, TNF-α sustaining BMSC growth and activation, and endothelial cell stimulators such as VEGF, FGF2, MMP, CXC chemokines promoting neo-angiogenesis. Stimulated endothelial cells also release VEGF, FGF2, angiopoietin 1 (ANG1), MMP, HGF, TGF-β, TNF-α which ensure the nutritional support for myeloma growth. Moreover, MM BM mesenchymal stem cells (MSCs) release exosomes with higher expression of oncogenic proteins, cytokines, protein kinases and microRNAs (miRNAs) that are transferred to myeloma plasma cells resulting in MM growth. Disease progression is also promoted by the strongly immunosuppressive BM microenvironment. MM plasma cells induce activation of T regulatory (Treg) cells and myeloid-derived suppressor cells (MDSCs), and increase of the programmed cell death-1 (PD-1) expression on gamma delta (γδ) T cells, CD4 T cells, natural killer (NK) cells and CD8 T cells. Neutrophils secrete arginase 1 involved in the dysfunction of CD8 T cells due to increased expression of PD-1, G1, CD57, CD160 and decreased secretion of IFNγ, TNF-α, perforin and granzyme (B) Dendritic cells also undergo inactivation and increase levels of inhibitory markers such as programmed death-ligand 1 (PD-L1).
Myeloma plasma cells express and release the receptor activator of nuclear factor kappa beta ligand (RANKL) that binding to its receptor RANK on osteoclast precursors promoting their differentiation into bone-resorbing osteoclasts. Activated osteoclasts secrete osteotropic factors including osteopontin and IL-6, which support bone resorption. The increased osteoclast activity results from the enhanced RANKL production and the reduced osteoprotegerin (OPG) production by osteoblasts.
Potential drug targets are marked in red.
Drug-based attempts to break the lines of communication between MM cells and their environment have demonstrated clinical efficacy, including in overcoming resistance (69, 80) (Table 1). A related strategy is to alter the tumor milieu through druggable targets such as CXCR4, IL-6, IGF-1, VEGF, their cognate receptors, and RANKL (81) in combination therapy to target the cytokines involved in cell adhesion (82) has shown promise whereas monotherapy has been ineffective.

Table 1 Phase III-IV clinical trial aiming to overcome drug resistance in MM, targeting bone marrow microenvironment.
Among the novel anti-MM agents that target the MM niche are alkylators, HDAC inhibitors, PIs, and MEK/ERK and Bcl-2/Mcl-1 inhibitors (83). Alkylators are of interest as they induce tumor cell lysis as well as a more immunogenic niche, by sensitizing MM cells to other immunotherapeutic approaches such as MoAbs. Novel alkylators include melphalan flefenamide (melflufen), which causes the formation of a radical that makes the alkylator inactive and lipophilic. Once inside the cell, the radical is released by the actions of aminopeptidases, which are highly enriched in MM cells, such that high intracellular concentrations of melphalan are obtained (84–87). In the Horizon trial (OP-106) (88), the overall response rate to melflufen was 29%, with the drug shown to also be active in patients with triple refractory disease and those with extramedullary disease. The benefits of an alkylator that may also act in the immune system was evidenced by the ~4-month increase in progression-free survival (PFS). The combination of melflufen with daratumumab and dexamethasone (NCT03481556) may sensitize patients to these MoAbs. Similarly, the alkylator DAC has several immunological effects, involving CD8, NK cells, and MDSCs (89, 90). EDO-S101 is a bendamustine derivative that combines an alkylator with the HDAC inhibitor vorinostat. The latter opens the chromatin, boosting the alkylator’s efficacy. EDO-S101 induces potent DNA damage in MM and may synergize with daratumumab (91).
Kroenke elucidated the mechanism of lenalidomide activity in MM (17, 92) by identifying cereblon (CRBN) as a putative target of the drug and demonstrating that the binding of CRBN to the transcription factors IKZF1 (IKAROS) and IKZF3 (AIOLOS) leads to their proteasome-mediated degradation. The destruction of these factors accounts for lenalidomide’s cytotoxic effect, at it causes the down-regulation of IRF4 and Myc, resulting in an immunomodulating effect that includes an increase in IL-2 and a decrease in TNF (92). Whether IKZF1 and IKZF3 are predictive markers for immunomodulatory drug (IMiD) therapy is thus far unclear (93). In addition to inhibiting the IKAROS-mediated gene expression program (94), lenalidomide inhibits a CRBN-mediated E3 ligase, thus demonstrating pleiotropic, clinically relevant effects in immunomodulation (93, 95). Nonetheless, lenalidomide resistance has been traced to mutations in the CRBN binding region of IKZF1 and IKZF3 (96). This observation prompted intensive research focusing on novel compounds such as iberdomide (CC-220), shown to elicit a 30% response, also in heavily pretreated patients (97), and CC-92480, which is also active against extramedullary disease (98). The biological role of the RAF/RAS pathway in the BM microenvironment (99) led to investigations into the role of RAS in MM. In 2014, Lohr et al. showed that genes encoding RAS family members are the most frequently mutated genes in MM (100), which raised interest in BRAF/MEK targeting (101–104).
Venetoclax is a potent, selective, orally bioavailable, small molecule that inhibits the apoptosis regulator BCL-2 (discussed in detail below). It holds great promise in MM based on its induction of cell death in MM cell lines and primary tumor samples, especially those with t (11, 14) translocation, in which an increased dependency upon BCL-2 for MM cell survival has been determined (105–107). Modulation of the anti-apoptotic pathway by venetoclax, by enhancing the binding of MM cells to the microenvironment, has been proposed (108), and the drug’s activity in t (11, 14) MM (109) and in combination with bortezomib and dexamethasone has been described. In another study, venetoclax improved PFS in patients with t (11, 14) or BCL2high MM, but not in patients with non-t (11, 14) BCL2low MM (110). The finding that MCL-1, an inhibitor of apoptosis, and BCL-2 levels are modulated by the tumor-associated microenvironment (111) has stimulated further investigation focused on MCL-1 inhibition.
Another target in the tumor microenvironment is arginine, which is required for the activation and proliferation of T lymphocytes and NK cells (112) while exerting an immunosuppressive effect. MDSCs and neutrophils produce high levels of arginase (113), which has led to exploration of the therapeutic potential of the arginase inhibitor INCB001158 in combination with daratumumab. Phase I/II clinical trials are being conducted.
Finally, several of the strategies and agents used in the treatment of non-Hodgkin lymphomas (83, 114), such as IMiDs, CELMoDs, naked MoAbs (anti-CD38 or SLAMF-7), ADCs, CAR-Ts, and BiTEs, are state-of-the-art tools being tested for their ability to overcome MM drug-resistance.
An important hallmark of cancer is the evasion of apoptosis. This pathway, which is under the control of BCL-2 family proteins, therefore represents an attractive therapeutic target (115). Programmed cell death is initiated by signaling processes, including the NF-kB, PI3K/AKT, and proteasome pathways (116). IL-6, VEGF, fibroblast growth factor (FGF), SDF1α, and IGF-1 trigger the MAPK/ERK and the JAK/STAT 3 pathways, which have a crucial role in the induction of apoptosis (117–119). Sequestering both BH3 and effector pro-apoptotic proteins blocks apoptosis, as a mechanism of drug resistance that might be overcome using BH3-mimetics. These include the above-discussed BCL-2 inhibitor venetoclax and S64315, AMG176 AZD5991 which target MCL-1 (115). The overexpression of anti-apoptotic BCL-2 proteins accounts for the vulnerability of MM cells to BH3- mimetics. As noted above, venetoclax induces a large response in t (11, 14) MM patients, and specifically those with low-level expression of BCL-XL and MCL-1 resistance factors (109). BH3-mimetics have demonstrated that the many cell lines require MCL-1 for their survival, and MCL-1 dependency was shown to increase in MM relapse (120). Mitsiates et al., demonstrated that TNF–related apoptosis-inducing ligand/Apo2 ligand (TRAIL/Apo2L) causes apoptosis in human MM cells, MM cell lines, and the MM cells of patients with disease either sensitive or resistant to dexamethasone (Dex) and chemotherapy. Doxorubicin and NF-Kβ inhibitors increase TRAIL/Apo2L expression in MM cells, but not in normal B cells, which suggests the targeting of TRAIL/Apo2L as a novel therapeutic strategy in MM (104).
NF-kB family members regulate growth, cell differentiation, and apoptosis in cell lines and tissues but they also play a role in drug resistance and in MM pathogenesis. Studies using the IKK inhibitor PS-1145 have confirmed the role of NF-kB in the growth, survival, and drug resistance of MM cells and therefore raised interest in the targeting of NF-kB in MM (121). Qu et al. found lower NF-kB expression in drug-sensitive than in drug-resistant cells and the up-regulation of NF-kB in the MM cells of patients with disease relapse. The authors showed that NF-kB could be blocked using arsenic trioxide, bortezomib, or Ikβ kinase inhibitors, all of which induced apoptosis in MM cell lines (122).
Activation of the unfolded protein response (UPR) pathway decreases ER stress, inhibits protein synthesis, and increases the transcription of heat shock proteins (HSP) that act as folding chaperones (123, 124). Misfolded proteins are also degraded by proteasomes and autophagy (125). MM cells are characterized by high levels of misfolded or unfolded proteins in the endoplasmic reticulum (ER), which causes their strong dependence on the UPR pathway for survival. Accordingly, MM cells are highly sensitive to PIs such as bortezomib, which inhibits proteasome activity and results in increased levels of misfolded protein in the ER, in turn inducing apoptosis in malignant cells (126). Gambella et al., demonstrated a correlation between higher levels of X-box binding protein 1 (XBP1) and a higher sensitivity to bortezomib (104). Nikesitch et al., showed correlations between ER size, decreased levels of the UPR regulator ATF6, the reduced expression of XBP1 activator, and bortezomib resistance. These observations suggest that decreased UPR activity predicts PI resistance (123).
Another essential survival mechanism for MM cells is autophagy, which helps malignant cells to degrade misfolded proteins and is associated with drug resistance. Milani et al. showed a correlation between the up-regulation of autophagy-inducer activating transcription factor 4 (ATF4) and bortezomib resistance (127). Autophagy thus represents another therapeutic target in MM (128, 129). Indeed, the combination of autophagy inhibitors and bortezomib has been tested in phase I and II clinical trials, with promising results obtained in patients with relapsed refractory disease (130). Carfilzomib in combination with autophagy inhibitors has also shown success in increasing apoptosis levels, both in vitro and in vivo (131).
The heat shock proteins HSP70 and HSP90 play a role in MM survival (132). HSP90 ensures the stability of anti-apoptotic signaling proteins, such as AKT, STAT3, and IL-6 receptors (133). A combination of inhibitors of HSP90 and other drugs has been examined for their ability to activate apoptosis, both in vitro and in vivo (134, 135).
Shadel et al. demonstrated that, in response to chemical stress, mitochondria activate molecular alarm signal triggered by nuclear DNA (nDNA) damage. Previous evidence showed that the response of cells to mitochondrial DNA (mtDNA) changes was similar to the response to an external pathogen, as in both cases, mediators capable of activating cell protection programs are released (136). The authors examined several anti-tumoral drugs, such as doxorubicin, and showed that they damage mtDNA and nDNA, causing the expression of a specific subset of interferon-stimulated genes (ISG), which are usually activated by a virus. The ISGs, including Parp9, remain activated by the unphosphorylated form of ISGF3 (U-ISGF3), which enhances nDNA damage and repair responses and facilitates chemoresistance. mtDNA is therefore a good genotoxic stress sentinel, given its higher sensitivity to the absence of repair (137).
The Bcl-2 inhibitor venetoclax, an inhibitor of BCL2, and compound C, an inhibitor of AMP-activated protein kinase, are more effective in bortezomib and carfilzomib resistant cells, due to their targeting of mitochondria. Moreover, it has been observed a switch in lipid class from lysolipids to sphingomyelins with the accumulation of mono-acylglycerols in PI-resistant cells. A comparison of D609, an inhibitor of sphingomyelin synthase and the sphingomyelin transferase inhibitor tamoxifen on PI-resistant and PI-sensitive cells showed the greater cytotoxicity of D609 in PI-resistant cells than PI-sensitive cells, with the opposite effect determined for tamoxifen. These results suggest the importance of sphingolipid synthesis in PI resistance. The combination of D609 and bortezomib or carfilzomib exhibited synergistic effects in primary MM cells (138). Together, these findings encouraged the study of new compounds that induce cellular oxidative damage or mitochondrial damage. Some of these drugs had acceptable safety profiles but nonetheless promoted the killing of PCs and therefore hold promise as adjuvants in the treatment of MM (139). Song et al., showed that 2-methoxyestradiol increases human MM cell death, by stimulating the production of mitochondrial ROS and raising Ca2+ levels within the cells, due to the increased activation of c-Jun N terminal kinase (JNK) and mitogen-activated protein kinase 4/7 (MKK4/7, 140). Papanikolaou et al. demonstrated the mechanism by which artesunate (ART), a well-tolerated anti-malarial drug, induces apoptosis of MM cell lines by targeting the mitochondria and increasing mitochondrial membrane permeability. In response, the cytosolic and subsequent nuclear translocation of the mitochondrial proteins apoptosis-inducing factor (AIF) and endonuclease G (EndoG) is induced. The nuclear translocation of AIF and EndoG is accompanied by low ROS levels and the increased mitochondrial production of superoxide.
A further target of ART in cancer cells is heme, as the drug reduces heme (bivalent iron) to hemin (trivalent iron). Heme is essential to many protein complexes, such as cytochrome C, a component of the electron transport chain of mitochondria (141). Accordingly, the mitochondria, with their abundance of iron, are an ideal target of ART (141). A recent study showed that the activation of TLR4 signaling in MM cells promotes mitochondrial biogenesis and thus resistance to BTZ. The authors found that TLR4 activation by LPS stimulation increases MM cell mitochondrial mass and enhances the expression of PGC1α, a regulator of mitogenesis, of PRC, a transcriptional cofactor activating genes involved in mitochondrial respiratory function, and of TFAM, a regulator of mitochondrial biogenesis that promotes mtDNA replication and transcription. Tests of the TLR4 inhibitor TAK-242 in combination with bortezomib in human MM cell lines showed higher oxidative stress, due to increases in reactive oxygen and nitrogen species, which in turn caused mitochondrial membrane depolarization and cytochrome c release into the cytosol. This was followed by caspase-9 activation, resulting finally in the overcoming of MM cell resistance (142).
In another study, the authors tested the biological effects of the immunomodulator FTY720 on MM cells and demonstrated potent cytotoxicity against drug-sensitive and drug-resistant MM cell lines as well as against the tumor cells of MM patients whose disease did not respond to conventional drugs. FTY720 triggers the activation of caspase-8, -9 and -3 and causes poly(ADP-ribose) polymerase cleavage. Moreover, it alters mitochondrial membrane potential and causes Bax cleavage, which is followed by the translocation of cytochrome c and Smac/Diablo from the mitochondria to the cytosol, where they activate apoptosis. The addition of FTY720 to the combination of Dex and anti-Fas antibodies improved the response rate and overcame acquired drug resistance (143). These studies show that a reliance solely on diagnostic definitions and the criterion of MRD as the best prognostic marker are insufficient. Rather, investigations of specific compounds and their mechanisms will allow a patient-tailored approach to the treatment of MM (139).
Relapse and tumor progression are mainly caused by a rare population of cancer stem cells (CSCs), which survive treatment and give rise to a PC tumor (144). The characteristics of CSCs include immaturity, quiescence, embryogenic gene expression, drug resistance, and self-renewal (145). MM CSCs differ from normal stem cells in terms of their genetic and epigenetic features, with their phenotype determined by the nature of the mutations (145). In MM, the phenotype of CSCs is not precisely known (146), with some studies claiming that they resemble CD138− B cells (9, 147). Human MM cell lines were shown to contain small subpopulations of CD138− cells with greater clonogenic potential than the corresponding CD138+ cells. CD138− cells were also shown to exhibit stem cell properties, such as enhanced aldehyde dehydrogenase (ALDH) activity (Figure 1F), and were clonogenic both in vitro and in vivo, unlike CD138+ cells. The CD138− PC phenotype includes markers of normal B cells (CD19, CD20 and CD27), suggesting that MM CSCs arise from populations of clonotypic B-cells (148). Another characteristic of CSCs is their resistance to Dex, bortezomib, and lenalidomide, whereas the same drugs inhibit the growth of CD138+ PCs (146). Brandl et al. found a higher proportion of CD138+ cells in proliferating and engrafted cells, and a lower proportion or the absence of CD138 low/CD138− cells, shown to be highly positive for the cell-adhesion molecule JAM-C (junctional adhesion molecule-C, 147). More motile and thus faster disseminating. MM cells are mostly CD138+ at the time of MM diagnosis, but a remarkable percentage of tumor cells lose CD138 expression at relapse. Nonetheless, Kim et al., demonstrated that clonotypic CD138+ PCs share some qualities with CSCs such as self-renewal, tumor-initiating potential, and drug resistance (149). Gao et al., conducted gene expression profiling of CSCs and of MM cells derived from 11 MM patients and found that CD24+ MM cells displayed features of CSCs, including self-renewal and drug resistance, suggesting these cells as a target for MM treatment (9, 146).
Due to their strong drug efflux capacity and drug detoxifying activities, MM CSCs are highly drug-resistant. They also express higher levels of ABC transporters and ATP-binding cassette transporters. These transmembrane proteins use the energy of ATP hydrolysis to transport harmful chemicals across the membrane. One of the key causes of multidrug resistance and chemotherapeutic failure in MM is the ABC transporter-mediated active efflux of drugs (148). The ABCG2 transporter is overexpressed in MM side population (SP) cells, a small subset of tumor cells that can efflux the fluorescent DNA-binding dye Hoechst 33342. ABCG2 inhibition with cyclosporine analogues or verapamil reduces the fraction of CSCs, in turn re-sensitizing MM cells to vincristine, doxorubicin, and Dex (150). A comparison of the expression of ABC transporters in SP cells with that in the main population cells of human MM cell lines showed that ABCG2 expression was significantly higher in SP cells, except in doxorubicin-resistant RPMI-Dox40 SP cells and SP cells of the KMS-11 cell line, which expressed very high levels of ABCB1 (151). SP cells exhibit CSC-like characteristics, such as tumorigenic potential, stem-like gene expression, dye extrusion capacity, and chemoresistance due to the enhanced expression of membrane-bound drug transporters (152). Although previous research suggested that MM SP cells were highly clonogenic and contained CD138− but not CD138+ PCs (148), new evidence suggests that MM SP cells contain both CD138+ and CD138− populations (151). This finding implies that malignant PCs can acquire the SP phenotype, which has consequences for their treatment resistance.
Moreover, contact with BMSCs increases both the number of SP cells and their ability to proliferate. Drugs that target BMSCs and block this stimulatory impact, such as lenalidomide and thalidomide, reduce the percentage of SP cells considerably (151). Both lenalidomide and thalidomide strongly reduce the percentage of SPs, underlining the relevance of the BM microenvironment to cell growth and phenotype acquisition (151).
In addition to ABC transporters, ALDH, a member of the NAD(P)1-dependent enzymes family involved in the metabolic detoxification of aldehydes, including ethanol and cytostatic medications, is another major mediator of drug resistance in MM CSCs (153). ALDH is expressed at high levels in both normal cells and CSCs (154). ALDH1 is also expressed by MM cells that have increased proliferation and tumorigenic features and chromosomal instability linked to treatment resistance (155). Matsui et al. (148) found that ALDH was upregulated in CD138− cells.
In their dynamic and context-dependent phenotypic reprogramming, MM CSCs, like other CSCs, use a variety of pathways, including Wingless (Wnt), Hedgehog (Hh), Notch signaling, and PI3K/Akt/mTOR (156–159). The Wnt pathway promotes cell proliferation, migration, invasion, drug resistance, and the creation of CSCs in MM (154). The expression of Wnt signaling pathway genes is increased in MM cells (160). Peacock et al. (2007) showed that Hh signaling is involved in keeping MM CSCs in an undifferentiated state, allowing clonal expansion. Hh activity is concentrated in CD138−/CD19+ MM CSCs but not in CD138+/CD19- MM PCs, demonstrating that stromal-derived Hh ligands promote MM CSC proliferation without differentiation (161).In CD138+ plasma cells and MM CSCs, overexpression of the retinoic acid receptor alpha 2 (RAR2), results in abnormal Wnt and Hh pathway activation and increased cell self-renewal, proliferation, migration, and drug resistance (160, 162). High-level Notch expression was demonstrated in clonotypic B cells from the BM of MM patients, suggesting that Notch is involved in maintaining MM CSC (163).
MiRNAs have emerged as major CSC regulators, with a distinct expression profile in CSCs and non-CSCs (164). A comparison of SP (side population) cells from MM cell lines and primary MM tumor cells identified ten miRNAs expressed at higher levels and 33 at lower levels. Among the miRNAs in SP cells, miRNA-451 was detected at high levels and shown to play a role in PI3K/Akt/mTOR pathway activation (146).
Contact with BMSCs boosted the number of SP cells and their ability to proliferate. Furthermore, drugs that target BMSCs and block this stimulatory impact, such as lenalidomide and thalidomide, reduced the percentage of SP cells considerably (151). Both lenalidomide and thalidomide strongly reduced the percentage of SP cells when they were co-cultured with BMSCs, underlining the relevance of the BM microenvironment (151).
The identification of more immature pre-plasmablastic cells has been uncovered to be relevant in specific scenarios, such as proteasome inhibitor resistance in MM. According to Orlowski et al., examination of patient samples identified up to five distinct subpopulations of tumor cells, including B-cells, activated B-cells, pre-plasmablasts, plasmablasts, and plasma cells, each of which may respond differently to bortezomib (165). B-cell and pre-plasmablast progenitors were discovered to survive proteasome inhibition and to be highly concentrated in samples resistant to bortezomib, supporting this theory. These results have significant implications for both innate and acquired proteasome inhibitor resistance. First, the authors claim that presumably all patients already have the potential for bortezomib resistance and cross-resistance with other PIs and that this disease phenotype will eventually emerge in step with the loss of bortezomib-sensitive plasmablasts and plasma cells (165). Thus, the identification of the baseline amount of these progenitor cells by whole genome sequencing or gene expression profiling may predict the PIs sensitivity (165). Therefore, promising therapeutic window can be directed to both committed plasma cells and progenitor, or stem cell-like cells. More interestingly, as Leung-Hagesteijn et al. corroborated the insights regarding the MM progenitors by describing a disease’s architecture that mimics the stages of B cell and plasma cell maturation and is a determinant in clinical PI resistance. While maturation defect before the plasmablast stage allows progressive disease on PI treatment in MM, Xbp1s tumor B cells and pre-plasmablasts can be resistant to PI. Indeed, bortezomib resistance is also developed through spliced X-box binding protein 1 (Xbp1) suppression, which reduces immunoglobulin production and consequently endoplasmic reticulum (ER) stress and susceptibility to PI (166). These findings emphasize the tumor progenitor makeup in MM and its contribution to disease progression and drug resistance (167).
Although stem cell properties are limited to a small percentage (possibly < 1%) of the tumor cell population, their eradication along with the bulk of tumor cells is likely to be crucial in obtaining a stable and durable remission, and possibly a cure for MM (9, 146).
Epigenetic modifications regulate normal B-cell development and plasma cell differentiation, and several epigenetic changes are implicated in myeloma pathogenesis and drug resistance (168–170). Alaterre E et al. extended this knowledge by examining the association between treatment response and the differential enrichment of H3K4me3 (an epigenetic modification of the histone H3) on gene promoters in Human multiple myeloma cell lines (HMCLs). Based on this histone alteration, the authors created an epigenetic biomarker predicting HMCL responses to lenalidomide and romidepsin (171). As mentioned earlier, the cereblon complex is a target of IMiDs. In the presence of IMiDs, Cereblon, composed of CUL4, RBX1, DDB1, and CRBN proteins, stimulates the ubiquitination of the B-cell transcription factors IKZF1 and IKZF3 (171, 172). A remarkable difference between lenalidomide-sensitive and -resistant HMCLs in step with H3K4me3 enrichment on the promoter of CUL4B protein was also described, establishing a link between the CUL4B splicing variation 1 and lenalidomide and pomalidomide sensitivity (171).
PIs sensitivity makes no exception. Indeed, with different approaches, De Smetd et al. characterized the role of G9a/GLP complex as a promising epigenetic regulator in MM; the authors showed that G9a/GLP targeting in MM cells induce autophagy-associated apoptosis and makes MM cells more susceptible to PI-based therapy by blocking mTOR signaling and lowering c-MYC levels. Therefore, PI-based therapy for patients with G9a high myeloma can be improved by G9a/GLP targeting (173).
Furthermore, key epigenetic factors that may be possible targets in myeloma include proteins well-known to be crucial in myeloproliferative/myelodysplastic disorders such as IDH2, DNMT3A/B, MMSET (WHSC1/NSD2), SETD2 and the polycomb repressive complex 2 (PRC2) complex (EZH2/PHF19); nonetheless, they are also promising target in MM. From this standpoint, PRC2 core genes were shown to be significantly upregulated in MM cells (174). Herviou et al. tested the impact of EPZ-6438, a selective small molecule inhibitor of EZH2 methyltransferase activity, on the phenotypic and gene expression profile of MM cells. Cell cycle arrest and cell death seem to be impacted due to the activity of polycomb and DNA methylation target genes. DNA methylation of PRC2 target genes mediates resistance to EZH2 inhibitor (171, 174). These results also provide the basis for a synergistic effect with lenalidomide and epigenetic modulators. Of note, in MM cells resistant to EZH2 inhibitor, Herviou L et al. pinpointed a considerable overlap between H3K27me3 and DNA methylation of EPZ-6438 target genes. There is a pathobiological connection between these two epigenetic repression systems. In fact, the PRC2 complex subunit EZH2 is necessary for the methylation of its target gene promoter. These results suggest that PRC2 target genes include essential MM tumor suppressor genes that various epigenetic pathways have repressed. Consequently, the therapeutic potential, since sublethal dosages of DNMTi can make EPZ-6438-resistant MM cell lines sensitive to EZH2 inhibitor (175, 176).
Nonetheless, to date, targeting biomarkers that are both prognostic and predictive is likely to improve outcome for patients with high-risk myeloma fastest but has to be demonstrated in statistically powered trials (177, 178).
Triple refractory MM is the archetype of relapsed or refractory multiple myeloma (RRMM). Data from the MAMMOTH study, which focused on CD38 mAb-refractory myeloma (179), examined resistance to several drugs and found that novel combinations of current drugs might be effective. This was the case for carfilzomib, cyclophosphamide, and Dex, which induced a 52% overall response, a median PFS of 4 months, and a median overall survival (OS) of 12 months (179). As discussed above, melflufen enables aminopeptidase activity inside MM cells but not in non-transformed cells (87). The results of the HORIZON study (88) prompted FDA approval of melflufen, based on promising results in RRMM and in triple refractory MM as well. Selinexor, an inhibitor of nuclear translocation, is another option (180), as it acts on exportin activation, which moves tumor suppressor proteins out into the cytoplasms. Vogl et al. showed that selinexor in combination with Dex was effective in both quad- and penta-refractory disease (181).
Antibody drug conjugates (ADCs) are another novel approach to RRMM management. ADCs are targeted immunotherapies composed by a cytotoxic drug fixed to an antibody scaffold directed against a tumor cell antigen. Upon binding with the tumor cell surface antigen, the ADC is internalized by the tumor cell and processed by the endo-lysosomal system leading to the release of the cancer toxic drug (182). Belantamab mafodotin is the first Food and Drug Administration (FDA)-approved BCMA targeted ADC for treatment of RRMM. It exerts a direct cytotoxic effect through the intracellular release of the monomethyl auristatin E (MMAE) and consequent spindle poisoning and apoptosis. ADCs act also via immunogenic cell death, by releasing, for example, HMGB1 and ATP, leading to the activation and mobilization of an immune effector response that includes T cells, macrophages, and dendritic cells. The DREAMM-2 study demonstrated the efficacy of belantamab mafodotin activity in patients with MM refractory to PIs, IMiDs, and daratumumab (183). In a longer-term follow up, PFS and OS improved in patients with a minimal response or better, but the ocular toxicities and thrombocytopenia must be addressed (184). However, due to the earlier use of novel agents, RRMM is becoming increasingly challenging to manage. The better use of currently available drugs in new combinations should thus be the goal in managing MM. Next-generation approaches should be directed at circumventing key mechanisms of resistance, while eliciting a better response with fewer toxicities. The development of new drugs with novel mechanisms of action must be supported by better predictive tools and immune-microenvironment and biology dissection (185, 186) to ensure that the right drug is delivered to the right patient at the right time (187–189).
Conceptualization, AS, PL and VR. Data curation, EM, AS, MP. Writing, AS, EM, CT and PL. Supervision, PL, MC and VR. Funding, CT, MC, VR. All of the authors reviewed the manuscript, approved the draft submission, and accept responsibility for all aspects of this study. All authors have read and agreed to the published version of the manuscript.
This work was supported by the Italian Association for Cancer Research (AIRC) through an Investigator Grant no. 20441 to VR and Investigator Grant no.22059 to MC. The sponsors of this study are non-profit organizations that support science in general; they had no role in gathering, analyzing, or interpreting the data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Palumbo A, Bringhen S, Ludwig H, Dimopoulos MA, Bladé J, Mateos MV, et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: a report of the European myeloma network (EMN). Blood (2011) 118:4519–29. doi: 10.1182/blood-2011-06-358812
2. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med (2011) 364:1046–60. doi: 10.1056/NEJMra1011442
3. Rajkumar SV. Treatment of multiple myeloma. Nat Rev Clin Oncol (2011) 8:479–91. doi: 10.1038/nrclinonc.2011.63
4. Sant M, Allemani C, Santaquilani M, Knijn A, Marchesi F, Capocaccia R, et al. EUROCARE-4. survival of cancer patients diagnosed in 1995-1999. results and commentary. Eur J Cancer Oxf Engl 1990 (2009) 45:931–91. doi: 10.1016/j.ejca.2008.11.018
5. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer (2007) 7:585–98. doi: 10.1038/nrc2189
6. van de Donk NWCJ, Pawlyn C, Yong KL. Multiple myeloma. Lancet Lond Engl (2021) 397:410–27. doi: 10.1016/S0140-6736(21)00135-5
7. Pinto V, Bergantim R, Caires HR, Seca H, Guimarães JE, Vasconcelos MH. Multiple myeloma: Available therapies and causes of drug resistance. Cancers (2020) 12:E407. doi: 10.3390/cancers12020407
8. Yang W-C, Lin S-F. Mechanisms of drug resistance in relapse and refractory multiple myeloma. BioMed Res Int (2015) 2015:341430. doi: 10.1155/2015/341430
9. Dammacco F, Leone P, Silvestris F, Racanelli V, Vacca A. “Cancer stem cells in multiple myeloma and the development of novel therapeutic strategies.,” Oncogenomics. Elsevier (2019), Chapter 9, p. 121–37. doi: 10.1016/B978-0-12-811785-9.00009-0
10. Leone P, Buonavoglia A, Fasano R, Solimando AG, De Re V, Cicco S, et al. Insights into the regulation of tumor angiogenesis by micro-RNAs. J Clin Med (2019) 8:e2030. doi: 10.3390/jcm8122030
11. Krebs M, Solimando AG, Kalogirou C, Marquardt A, Frank T, Sokolakis I, et al. miR-221-3p regulates VEGFR2 expression in high-risk prostate cancer and represents an escape mechanism from sunitinib. In Vitro. J Clin Med (2020) 9:e670. doi: 10.3390/jcm9030670
12. Desantis V, Saltarella I, Lamanuzzi A, Melaccio A, Solimando AG, Mariggiò MA, et al. MicroRNAs-based nano-strategies as new therapeutic approach in multiple myeloma to overcome disease progression and drug resistance. Int J Mol Sci (2020) 21:e3084. doi: 10.3390/ijms21093084
13. Desantis V, Solimando AG, Saltarella I, Sacco A, Giustini V, Bento M, et al. MicroRNAs as a potential new preventive approach in the transition from asymptomatic to symptomatic multiple myeloma disease. Cancers (2021) 13:3650. doi: 10.3390/cancers13153650
14. Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun (2014) 5:2997. doi: 10.1038/ncomms3997
15. Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell (2013) 23:48–62. doi: 10.1016/j.ccr.2012.12.001
16. Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discovery (2012) 2:214–26. doi: 10.1158/2159-8290.CD-12-0012
17. Choi Y, Zhang J, Murga C, Yu H, Koller E, Monia BP, et al. But not SHIP and SHIP2, suppresses the PI3K/Akt pathway and induces growth inhibition and apoptosis of myeloma cells. Oncogene (2002) 21:5289–300. doi: 10.1038/sj.onc.1205650
18. Gourzones-Dmitriev C, Kassambara A, Sahota S, Rème T, Moreaux J, Bourquard P, et al. DNA Repair pathways in human multiple myeloma: role in oncogenesis and potential targets for treatment. Cell Cycle Georget Tex (2013) 12:2760–73. doi: 10.4161/cc.25951
19. Kawano Y, Moschetta M, Manier S, Glavey S, Görgün GT, Roccaro AM, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev (2015) 263:160–72. doi: 10.1111/imr.12233
20. Furukawa Y, Kikuchi J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int J Hematol (2016) 104:281–92. doi: 10.1007/s12185-016-2048-5
21. Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood (1999) 93:1658–67.
22. Solimando AG, Brandl A, Mattenheimer K, Graf C, Ritz M, Ruckdeschel A, et al. JAM-a as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia (2018) 32:736–43. doi: 10.1038/leu.2017.287
23. Di Marzo L, Desantis V, Solimando AG, Ruggieri S, Annese T, Nico B, et al. Microenvironment drug resistance in multiple myeloma: emerging new players. Oncotarget (2016) 7:60698–711. doi: 10.18632/oncotarget.10849
24. Vacca A, Di Loreto M, Ribatti D, Di Stefano R, Gadaleta-Caldarola G, Iodice G, et al. Bone marrow of patients with active multiple myeloma: angiogenesis and plasma cell adhesion molecules LFA-1, VLA-4, LAM-1, and CD44. Am J Hematol (1995) 50:9–14. doi: 10.1002/ajh.2830500103
25. Vincent T, Mechti N. IL-6 regulates CD44 cell surface expression on human myeloma cells. Leukemia (2004) 18:967–75. doi: 10.1038/sj.leu.2403333
26. Abe M. Targeting the interplay between myeloma cells and the bone marrow microenvironment in myeloma. Int J Hematol (2011) 94:334–43. doi: 10.1007/s12185-011-0949-x
27. Ferrucci A, Moschetta M, Frassanito MA, Berardi S, Catacchio I, Ria R, et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin Cancer Res Off J Am Assoc Cancer Res (2014) 20:5796–807. doi: 10.1158/1078-0432.CCR-14-0847
28. Liu Z, Xu J, He J, Liu H, Lin P, Wan X, et al. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget (2015) 6:34329–41. doi: 10.18632/oncotarget.6020
29. Argentiero A, Solimando AG, Brunetti O, Calabrese A, Pantano F, Iuliani M, et al. Skeletal metastases of unknown primary: Biological landscape and clinical overview. Cancers (2019) 11:E1270. doi: 10.3390/cancers11091270
30. Immune system and bone microenvironment: rationale for targeted cancer therapies - PubMed. Available at pubmed.ncbi.nlm.nih.gov/32064051/ (Accessed April 13, 2022).
31. Solimando AG, Vacca A, Ribatti D. A comprehensive biological and clinical perspective can drive a patient-tailored approach to multiple myeloma: Bridging the gaps between the plasma cell and the neoplastic niche. J Oncol (2020) 2020:6820241. doi: 10.1155/2020/6820241
32. Harmer D, Falank C, Reagan MR. Interleukin-6 interweaves the bone marrow microenvironment, bone loss, and multiple myeloma. Front Endocrinol (2018) 9:788. doi: 10.3389/fendo.2018.00788
33. Giuliani N, Rizzoli V, Roodman GD. Multiple myeloma bone disease: Pathophysiology of osteoblast inhibition. Blood (2006) 108:3992–6. doi: 10.1182/blood-2006-05-026112
34. Moreaux J, Hose D, Kassambara A, Reme T, Moine P, Requirand G, et al. Osteoclast-gene expression profiling reveals osteoclast-derived CCR2 chemokines promoting myeloma cell migration. Blood (2011) 117:1280–90. doi: 10.1182/blood-2010-04-279760
35. Moreaux J, Cremer FW, Reme T, Raab M, Mahtouk K, Kaukel P, et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood (2005) 106:1021–30. doi: 10.1182/blood-2004-11-4512
36. McCachren SS, Dhodapkar KM, Dhodapkar MV. Co-Evolution of immune response in multiple myeloma: Implications for immune prevention. Front Immunol (2021) 12:632564. doi: 10.3389/fimmu.2021.632564
37. Dhodapkar MV, Dhodapkar KM. Tissue-resident memory-like T cells in tumor immunity: Clinical implications. Semin Immunol (2020) 49:101415. doi: 10.1016/j.smim.2020.101415
38. Leone P, Solimando AG, Malerba E, Fasano R, Buonavoglia A, Pappagallo F, et al. Actors on the scene: Immune cells in the myeloma niche. Front Oncol (2020) 10:599098. doi: 10.3389/fonc.2020.599098
39. Maley CC, Aktipis A, Graham TA, Sottoriva A, Boddy AM, Janiszewska M, et al. Classifying the evolutionary and ecological features of neoplasms. Nat Rev Cancer (2017) 17:605–19. doi: 10.1038/nrc.2017.69
40. Bohonowych JE, Gopal U, Isaacs JS. Hsp90 as a gatekeeper of tumor angiogenesis: clinical promise and potential pitfalls. J Oncol (2010) 2010:412985. doi: 10.1155/2010/412985
41. Fuster MM, Wang L. Endothelial heparan sulfate in angiogenesis. Prog Mol Biol Transl Sci (2010) 93:179–212. doi: 10.1016/S1877-1173(10)93009-3
42. Zangari M, Suva LJ. The effects of proteasome inhibitors on bone remodeling in multiple myeloma. Bone (2016) 86:131–8. doi: 10.1016/j.bone.2016.02.019
43. Solimando AG, Summa SD, Vacca A, Ribatti D. Cancer-associated angiogenesis: The endothelial cell as a checkpoint for immunological patrolling. Cancers (2020) 12:e3380. doi: 10.3390/cancers12113380
44. Bailur JK, McCachren SS, Doxie DB, Shrestha M, Pendleton K, Nooka AK, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight (2019) 5:127807. doi: 10.1172/jci.insight.127807
45. Giallongo C, Tibullo D, Parrinello NL, La Cava P, Di Rosa M, Bramanti V, et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget (2016) 7:85764–75. doi: 10.18632/oncotarget.7969
46. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
47. Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Ioannou K, Ziogas AC, et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br J Cancer (2012) 107:1869–75. doi: 10.1038/bjc.2012.468
48. Ribatti D, Solimando AG, Pezzella F. The anti-VEGF(R) drug discovery legacy: Improving attrition rates by breaking the vicious cycle of angiogenesis in cancer. Cancers (2021) 13:3433. doi: 10.3390/cancers13143433
49. de Jong MME, Kellermayer Z, Papazian N, Tahri S, Hofste Op Bruinink D, Hoogenboezem R, et al. The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat Immunol (2021) 22:769–80. doi: 10.1038/s41590-021-00931-3
50. Qian H, Le Blanc K, Sigvardsson M. Primary mesenchymal stem and progenitor cells from bone marrow lack expression of CD44 protein. J Biol Chem (2012) 287:25795–807. doi: 10.1074/jbc.M112.339622
51. Hu J, Van Valckenborgh E, Menu E, De Bruyne E, Vanderkerken K. Understanding the hypoxic niche of multiple myeloma: therapeutic implications and contributions of mouse models. Dis Model Mech (2012) 5:763–71. doi: 10.1242/dmm.008961
52. Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol (2019) 7:4. doi: 10.3389/fcell.2019.00004
53. Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood (2014) 124:3748–57. doi: 10.1182/blood-2014-05-576116
54. Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene (2019) 38:2223–40. doi: 10.1038/s41388-018-0582-8
55. Ikeda S, Abe F, Matsuda Y, Kitadate A, Takahashi N, Tagawa H. Hypoxia-inducible hexokinase-2 enhances anti-apoptotic function via activating autophagy in multiple myeloma. Cancer Sci (2020) 111:4088–101. doi: 10.1111/cas.14614
56. Maiso P, Huynh D, Moschetta M, Sacco A, Aljawai Y, Mishima Y, et al. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res (2015) 75:2071–82. doi: 10.1158/0008-5472.CAN-14-3400
57. Janker L, Mayer RL, Bileck A, Kreutz D, Mader JC, Utpatel K, et al. Metabolic, anti-apoptotic and immune evasion strategies of primary human myeloma cells indicate adaptations to hypoxia. Mol Cell Proteomics MCP (2019) 18:936–53. doi: 10.1074/mcp.RA119.001390
58. Xu S, Zhou T, Doh HM, Trinh KR, Catapang A, Lee JT, et al. An HK2 antisense oligonucleotide induces synthetic lethality in HK1-HK2+ multiple myeloma. Cancer Res (2019) 79:2748–60. doi: 10.1158/0008-5472.CAN-18-2799
59. Massey AJ. Modification of tumour cell metabolism modulates sensitivity to Chk1 inhibitor-induced DNA damage. Sci Rep (2017) 7:40778. doi: 10.1038/srep40778
60. Tumber A, Nuzzi A, Hookway ES, Hatch SB, Velupillai S, Johansson C, et al. Potent and selective KDM5 inhibitor stops cellular demethylation of H3K4me3 at transcription start sites and proliferation of MM1S myeloma cells. Cell Chem Biol (2017) 24:371–80. doi: 10.1016/j.chembiol.2017.02.006
61. Wang J, De Veirman K, Faict S, Frassanito MA, Ribatti D, Vacca A, et al. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol (2016) 239:162–73. doi: 10.1002/path.4712
62. Li B, Xu H, Han H, Song S, Zhang X, Ouyang L, et al. Exosome-mediated transfer of lncRUNX2-AS1 from multiple myeloma cells to MSCs contributes to osteogenesis. Oncogene (2018) 37:5508–19. doi: 10.1038/s41388-018-0359-0
63. Raimondo S, Urzì O, Conigliaro A, Bosco GL, Parisi S, Carlisi M, et al. Extracellular vesicle microRNAs contribute to the osteogenic inhibition of mesenchymal stem cells in multiple myeloma. Cancers (2020) 12:E449. doi: 10.3390/cancers12020449
64. De Veirman K, Wang J, Xu S, Leleu X, Himpe E, Maes K, et al. Induction of miR-146a by multiple myeloma cells in mesenchymal stromal cells stimulates their pro-tumoral activity. Cancer Lett (2016) 377:17–24. doi: 10.1016/j.canlet.2016.04.024
65. Cheng Q, Li X, Liu J, Ye Q, Chen Y, Tan S, et al. Multiple myeloma-derived exosomes regulate the functions of mesenchymal stem cells partially via modulating miR-21 and miR-146a. Stem Cells Int (2017) 2017:9012152. doi: 10.1155/2017/9012152
66. Desantis V, Frassanito MA, Tamma R, Saltarella I, Di Marzo L, Lamanuzzi A, et al. Rhu-epo down-regulates pro-tumorigenic activity of cancer-associated fibroblasts in multiple myeloma. Ann Hematol (2018) 97:1251–8. doi: 10.1007/s00277-018-3293-x
67. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai Y-T, Reagan M, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest (2013) 123:1542–55. doi: 10.1172/JCI66517
68. Wang J, Hendrix A, Hernot S, Lemaire M, De Bruyne E, Van Valckenborgh E, et al. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood (2014) 124:555–66. doi: 10.1182/blood-2014-03-562439
69. Xu H, Han H, Song S, Yi N, Qian C, Qiu Y, et al. Exosome-transmitted PSMA3 and PSMA3-AS1 promote proteasome inhibitor resistance in multiple myeloma. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:1923–35. doi: 10.1158/1078-0432.CCR-18-2363
70. Bandari SK, Purushothaman A, Ramani VC, Brinkley GJ, Chandrashekar DS, Varambally S, et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol J Int Soc Matrix Biol (2018) 65:104–18. doi: 10.1016/j.matbio.2017.09.001
71. Faict S, Oudaert I, D’Auria L, Dehairs J, Maes K, Vlummens P, et al. The transfer of sphingomyelinase contributes to drug resistance in multiple myeloma. Cancers (2019) 11:e1823. doi: 10.3390/cancers11121823
72. Yang E, Wang X, Gong Z, Yu M, Wu H, Zhang D. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct Target Ther (2020) 5:242. doi: 10.1038/s41392-020-00359-5
73. Zavidij O, Haradhvala NJ, Mouhieddine TH, Sklavenitis-Pistofidis R, Cai S, Reidy M, et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer (2020) 1:493–506. doi: 10.1038/s43018-020-0053-3
74. Argentiero A, Solimando AG, Krebs M, Leone P, Susca N, Brunetti O, et al. Anti-angiogenesis and immunotherapy: Novel paradigms to envision tailored approaches in renal cell-carcinoma. J Clin Med (2020) 9:e1594. doi: 10.3390/jcm9051594
75. Rao L, Giannico D, Leone P, Solimando AG, Maiorano E, Caporusso C, et al. HB-EGF-EGFR signaling in bone marrow endothelial cells mediates angiogenesis associated with multiple myeloma. Cancers (2020) 12:173–91. doi: 10.3390/cancers12010173
76. Garayoa M, Garcia JL, Santamaria C, Garcia-Gomez A, Blanco JF, Pandiella A, et al. Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia (2009) 23:1515–27. doi: 10.1038/leu.2009.65
77. Moschetta M, Basile A, Ferrucci A, Frassanito MA, Rao L, Ria R, et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin Cancer Res Off J Am Assoc Cancer Res (2013) 19:4371–82. doi: 10.1158/1078-0432.CCR-13-0039
78. Solimando AG, Da Vià MC, Leone P, Borrelli P, Croci GA, Tabares P, et al. Halting the vicious cycle within the multiple myeloma ecosystem: blocking JAM-a on bone marrow endothelial cells restores angiogenic homeostasis and suppresses tumor progression. Haematologica (2021) 106:1943–56. doi: 10.3324/haematol.2019.239913
79. Leone P, Di Lernia G, Solimando AG, Cicco S, Saltarella I, Lamanuzzi A, et al. Bone marrow endothelial cells sustain a tumor-specific CD8+ T cell subset with suppressive function in myeloma patients. Oncoimmunology (2019) 8:e1486949. doi: 10.1080/2162402X.2018.1486949
80. Rao L, De Veirman K, Giannico D, Saltarella I, Desantis V, Frassanito MA, et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin® protein MP0250: a preclinical study. Oncotarget (2018) 9:13366–81. doi: 10.18632/oncotarget.24351
81. Suzuki K, Nishiwaki K, Yano S. Treatment strategies considering micro-environment and clonal evolution in multiple myeloma. Cancers (2021) 13:E215. doi: 10.3390/cancers13020215
82. Solimando AG, Da Via’ MC, Leone P, Croci G, Borrelli P, Tabares Gaviria P, et al. Adhesion-mediated multiple myeloma (MM) disease progression: Junctional adhesion molecule a enhances angiogenesis and multiple myeloma dissemination and predicts poor survival. Blood (2019) 134:855–5. doi: 10.1182/blood-2019-126674
83. Ho M, Goh CY, Patel A, Staunton S, O’Connor R, Godeau M, et al. Role of the bone marrow milieu in multiple myeloma progression and therapeutic resistance. Clin Lymphoma Myeloma Leuk (2020) 20:e752–68. doi: 10.1016/j.clml.2020.05.026
84. Wickström M, Haglund C, Lindman H, Nygren P, Larsson R, Gullbo J. The novel alkylating prodrug J1: diagnosis directed activity profile ex vivo and combination analyses. vitro. Invest New Drugs (2008) 26:195–204. doi: 10.1007/s10637-007-9092-1
85. Di Martino MT, Gullà A, Cantafio MEG, Lionetti M, Leone E, Amodio N, et al. In vitro and in vivo anti-tumor activity of miR-221/222 inhibitors in multiple myeloma. Oncotarget (2013) 4:242–55. doi: 10.18632/oncotarget.820
86. Ray A, Ravillah D, Das DS, Song Y, Nordström E, Gullbo J, et al. A novel alkylating agent melflufen induces irreversible DNA damage and cytotoxicity in multiple myeloma cells. Br J Haematol (2016) 174:397–409. doi: 10.1111/bjh.14065
87. Wickström M, Nygren P, Larsson R, Harmenberg J, Lindberg J, Sjöberg P, et al. Melflufen - a peptidase-potentiated alkylating agent in clinical trials. Oncotarget (2017) 8:66641–55. doi: 10.18632/oncotarget.18420
88. Richardson PG, Oriol A, Larocca A, Bladé J, Cavo M, Rodriguez-Otero P, et al. Melflufen and dexamethasone in heavily pretreated relapsed and refractory multiple myeloma. J Clin Oncol Off J Am Soc Clin Oncol (2021) 39:757–67. doi: 10.1200/JCO.20.02259
89. Kroesen M, Gielen P, Brok IC, Armandari I, Hoogerbrugge PM, Adema GJ. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget (2014) 5:6558–72. doi: 10.18632/oncotarget.2289
90. Shen L, Orillion A, Pili R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics (2016) 8:415–28. doi: 10.2217/epi.15.118
91. López-Iglesias A-A, Herrero AB, Chesi M, San-Segundo L, González-Méndez L, Hernández-García S, et al. Preclinical anti-myeloma activity of EDO-S101, a new bendamustine-derived molecule with added HDACi activity, through potent DNA damage induction and impairment of DNA repair. J Hematol OncolJ Hematol Oncol (2017) 10:127. doi: 10.1186/s13045-017-0495-y
92. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science (2014) 343:301–5. doi: 10.1126/science.1244851
93. Zhu YX, Braggio E, Shi C-X, Kortuem KM, Bruins LA, Schmidt JE, et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood (2014) 124:536–45. doi: 10.1182/blood-2014-02-557819
94. Bjorklund CC, Lu L, Kang J, Hagner PR, Havens CG, Amatangelo M, et al. Rate of CRL4(CRBN) substrate ikaros and aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-myc and IRF4. Blood Cancer J (2015) 5:e354. doi: 10.1038/bcj.2015.66
95. Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia (2012) 26:2326–35. doi: 10.1038/leu.2012.119
96. Barrio S, Munawar U, Zhu YX, Giesen N, Shi C-X, Viá MD, et al. IKZF1/3 and CRL4CRBN E3 ubiquitin ligase mutations and resistance to immunomodulatory drugs in multiple myeloma. Haematologica (2020) 105:e237–41. doi: 10.3324/haematol.2019.217943
97. Bjorklund CC, Kang J, Amatangelo M, Polonskaia A, Katz M, Chiu H, et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia (2020) 34:1197–201. doi: 10.1038/s41375-019-0620-8
98. Hansen JD, Correa M, Nagy MA, Alexander M, Plantevin V, Grant V, et al. Discovery of CRBN E3 ligase modulator CC-92480 for the treatment of relapsed and refractory multiple myeloma. J Med Chem (2020) 63:6648–76. doi: 10.1021/acs.jmedchem.9b01928
99. Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol (2014) 11:385–400. doi: 10.1038/nrclinonc.2014.83
100. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell (2014) 25:91–101. doi: 10.1016/j.ccr.2013.12.015
101. Andrulis M, Lehners N, Capper D, Penzel R, Heining C, Huellein J, et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discovery (2013) 3:862–9. doi: 10.1158/2159-8290.CD-13-0014
102. Raab MS, Lehners N, Xu J, Ho AD, Schirmacher P, Goldschmidt H, et al. Spatially divergent clonal evolution in multiple myeloma: overcoming resistance to BRAF inhibition. Blood (2016) 127:2155–7. doi: 10.1182/blood-2015-12-686782
103. Da Vià MC, Solimando AG, Garitano-Trojaola A, Barrio S, Munawar U, Strifler S, et al. CIC mutation as a molecular mechanism of acquired resistance to combined BRAF-MEK inhibition in extramedullary multiple myeloma with central nervous system involvement. Oncologist (2020) 25:112–8. doi: 10.1634/theoncologist.2019-0356
104. Slavcev M, Spinelli A, Absalon E, Masterson T, Heuck C, Lam A, et al. Results of a time and motion survey regarding subcutaneous versus intravenous administration of daratumumab in patients with relapsed or refractory multiple myeloma. Clin Outcomes Res CEOR (2021) 13:465–73. doi: 10.2147/CEOR.S302682
105. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med (2013) 19:202–8. doi: 10.1038/nm.3048
106. Touzeau C, Maciag P, Amiot M, Moreau P. Targeting bcl-2 for the treatment of multiple myeloma. Leukemia (2018) 32:1899–907. doi: 10.1038/s41375-018-0223-9
107. Punnoose EA, Leverson JD, Peale F, Boghaert ER, Belmont LD, Tan N, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther (2016) 15:1132–44. doi: 10.1158/1535-7163.MCT-15-0730
108. Gupta VA, Matulis SM, Conage-Pough JE, Nooka AK, Kaufman JL, Lonial S, et al. Bone marrow microenvironment-derived signals induce mcl-1 dependence in multiple myeloma. Blood (2017) 129:1969–79. doi: 10.1182/blood-2016-10-745059
109. Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood (2017) 130:2401–9. doi: 10.1182/blood-2017-06-788786
110. Kumar SK, Harrison SJ, Cavo M, de la Rubia J, Popat R, Gasparetto C, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol (2020) 21:1630–42. doi: 10.1016/S1470-2045(20)30525-8
111. Algarín EM, Quwaider D, Campos-Laborie FJ, Díaz-Tejedor A, Mogollón P, Vuelta E, et al. Stroma-mediated resistance to S63845 and venetoclax through MCL-1 and BCL-2 expression changes induced by miR-193b-3p and miR-21-5p dysregulation in multiple myeloma. Cells (2021) 10:559. doi: 10.3390/cells10030559
112. Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol (2009) 158:638–51. doi: 10.1111/j.1476-5381.2009.00291.x
113. Romano A, Parrinello NL, La Cava P, Tibullo D, Giallongo C, Camiolo G, et al. PMN-MDSC and arginase are increased in myeloma and may contribute to resistance to therapy. Expert Rev Mol Diagn (2018) 18:675–83. doi: 10.1080/14737159.2018.1470929
114. Solimando AG, Ribatti D, Vacca A, Einsele H. Targeting b-cell non Hodgkin lymphoma: New and old tricks. Leuk Res (2016) 42:93–104. doi: 10.1016/j.leukres.2015.11.001
115. Seiller C, Maiga S, Touzeau C, Bellanger C, Kervoëlen C, Descamps G, et al. Dual targeting of BCL2 and MCL1 rescues myeloma cells resistant to BCL2 and MCL1 inhibitors associated with the formation of BAX/BAK hetero-complexes. Cell Death Dis (2020) 11:316. doi: 10.1038/s41419-020-2505-1
116. Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin (2005) 55:178–94. doi: 10.3322/canjclin.55.3.178
117. Yang HH, Ma MH, Vescio RA, Berenson JR. Overcoming drug resistance in multiple myeloma: the emergence of therapeutic approaches to induce apoptosis. J Clin Oncol Off J Am Soc Clin Oncol (2003) 21:4239–47. doi: 10.1200/JCO.2003.06.001
118. Lentzsch S, Chatterjee M, Gries M, Bommert K, Gollasch H, Dörken B, et al. PI3-K/AKT/FKHR and MAPK signaling cascades are redundantly stimulated by a variety of cytokines and contribute independently to proliferation and survival of multiple myeloma cells. Leukemia (2004) 18:1883–90. doi: 10.1038/sj.leu.2403486
119. Ge NL, Rudikoff S. Insulin-like growth factor I is a dual effector of multiple myeloma cell growth. Blood (2000) 96:2856–61. doi: 10.1182/blood.V96.8.2856
120. Gomez-Bougie P, Maiga S, Tessoulin B, Bourcier J, Bonnet A, Rodriguez MS, et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood (2018) 132:2656–69. doi: 10.1182/blood-2018-03-836718
121. Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, et al. NF-kappa b as a therapeutic target in multiple myeloma. J Biol Chem (2002) 277:16639–47. doi: 10.1074/jbc.M200360200
122. Qu X, Du J, Zhang C, Fu W, Xi H, Zou J, et al. Arsenic trioxide exerts antimyeloma effects by inhibiting activity in the cytoplasmic substrates of histone deacetylase 6. PloS One (2012) 7:e32215. doi: 10.1371/journal.pone.0032215
123. Nikesitch N, Ling SCW. Molecular mechanisms in multiple myeloma drug resistance. J Clin Pathol (2016) 69:97–101. doi: 10.1136/jclinpath-2015-203414
124. Benbrook DM, Long A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp Oncol (2012) 34:286–97.
125. Vincenz L, Jäger R, O’Dwyer M, Samali A. Endoplasmic reticulum stress and the unfolded protein response: targeting the Achilles heel of multiple myeloma. Mol Cancer Ther (2013) 12:831–43. doi: 10.1158/1535-7163.MCT-12-0782
126. Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood (2006) 107:4907–16. doi: 10.1182/blood-2005-08-3531
127. Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with bortezomib. Cancer Res (2009) 69:4415–23. doi: 10.1158/0008-5472.CAN-08-2839
128. Lamanuzzi A, Saltarella I, Desantis V, Frassanito MA, Leone P, Racanelli V, et al. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget (2018) 9:20563–77. doi: 10.18632/oncotarget.25003
129. Di Lernia G, Leone P, Solimando AG, Buonavoglia A, Saltarella I, Ria R, et al. Bortezomib treatment modulates autophagy in multiple myeloma. J Clin Med (2020) 9:552–66. doi: 10.3390/jcm9020552
130. Vogl DT, Stadtmauer EA, Tan K-S, Heitjan DF, Davis LE, Pontiggia L, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy (2014) 10:1380–90. doi: 10.4161/auto.29264
131. Jarauta V, Jaime P, Gonzalo O, de Miguel D, Ramírez-Labrada A, Martínez-Lostao L, et al. Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells. Vitro vivo. Cancer Lett (2016) 382:1–10. doi: 10.1016/j.canlet.2016.08.019
132. Chatterjee M, Jain S, Stühmer T, Andrulis M, Ungethüm U, Kuban R-J, et al. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood (2007) 109:720–8. doi: 10.1182/blood-2006-05-024372
133. Khong T, Spencer A. Targeting HSP 90 induces apoptosis and inhibits critical survival and proliferation pathways in multiple myeloma. Mol Cancer Ther (2011) 10:1909–17. doi: 10.1158/1535-7163.MCT-11-0174
134. Ishii T, Seike T, Nakashima T, Juliger S, Maharaj L, Soga S, et al. Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J (2012) 2:e68. doi: 10.1038/bcj.2012.13
135. Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Kung AL, Davies FE, et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood (2006) 107:1092–100. doi: 10.1182/blood-2005-03-1158
136. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol (2017) 17:363–75. doi: 10.1038/nri.2017.21
137. Wu Z, Oeck S, West AP, Mangalhara KC, Sainz AG, Newman LE, et al. Mitochondrial DNA stress signalling protects the nuclear genome. Nat Metab (2019) 1:1209–18. doi: 10.1038/s42255-019-0150-8
138. Besse L, Besse A, Mendez-Lopez M, Vasickova K, Sedlackova M, Vanhara P, et al. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mitochondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica (2019) 104:e415–9. doi: 10.3324/haematol.2018.207704
139. Innao V, Rizzo V, Allegra AG, Musolino C, Allegra A. Promising anti-mitochondrial agents for overcoming acquired drug resistance in multiple myeloma. Cells (2021) 10:439. doi: 10.3390/cells10020439
140. Song IS, Kim HK, Lee SR, Jeong SH, Kim N, Ko KS, et al. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int J Cancer (2013) 133:1357–67. doi: 10.1002/ijc.28149
141. Papanikolaou X, Johnson S, Garg T, Tian E, Tytarenko R, Zhang Q, et al. Artesunate overcomes drug resistance in multiple myeloma by inducing mitochondrial stress and non-caspase apoptosis. Oncotarget (2014) 5:4118–28. doi: 10.18632/oncotarget.1847
142. Giallongo C, Tibullo D, Puglisi F, Barbato A, Vicario N, Cambria D, et al. Inhibition of TLR4 signaling affects mitochondrial fitness and overcomes bortezomib resistance in myeloma plasma cells. Cancers (2020) 12:E1999. doi: 10.3390/cancers12081999
143. Yasui H, Hideshima T, Raje N, Roccaro AM, Shiraishi N, Kumar S, et al. FTY720 induces apoptosis in multiple myeloma cells and overcomes drug resistance. Cancer Res (2005) 65:7478–84. doi: 10.1158/0008-5472.CAN-05-0850
144. Gao M, Kong Y, Yang G, Gao L, Shi J. Multiple myeloma cancer stem cells. Oncotarget (2016) 7:35466–77. doi: 10.18632/oncotarget.8154
145. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CHM, Jones DL, et al. Cancer stem cells–perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res (2006) 66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126
146. Ghosh N, Matsui W. Cancer stem cells in multiple myeloma. Cancer Lett (2009) 277:1–7. doi: 10.1016/j.canlet.2008.08.005
147. Brandl A, Solimando AG, Mokhtari Z, Tabares P, Medler J, Manz H, et al. Junctional adhesion molecule c expression specifies a CD138low/neg multiple myeloma cell population in mice and humans. Blood Adv (2022) 6:2195–206. doi: 10.1182/bloodadvances.2021004354
148. Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res (2008) 68:190–7. doi: 10.1158/0008-5472.CAN-07-3096
149. Kim D, Park CY, Medeiros BC, Weissman IL. CD19-CD45 low/- CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia (2012) 26:2530–7. doi: 10.1038/leu.2012.140
150. Dalton WS, Crowley JJ, Salmon SS, Grogan TM, Laufman LR, Weiss GR, et al. A phase III randomized study of oral verapamil as a chemosensitizer to reverse drug resistance in patients with refractory myeloma. A Southwest Oncol Group study. Cancer (1995) 75:815–20. doi: 10.1002/1097-0142(19950201)75:3<815::aid-cncr2820750311>3.0.co;2-r
151. Jakubikova J, Cervi D, Ooi M, Kim K, Nahar S, Klippel S, et al. Anti-tumor activity and signaling events triggered by the isothiocyanates, sulforaphane and phenethyl isothiocyanate, in multiple myeloma. Haematologica (2011) 96:1170–9. doi: 10.3324/haematol.2010.029363
152. Challen GA, Little MH. A side order of stem cells: the SP phenotype. Stem Cells Dayt Ohio (2006) 24:3–12. doi: 10.1634/stemcells.2005-0116
153. Vasiliou V, Pappa A, Estey T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug Metab Rev (2004) 36:279–99. doi: 10.1081/dmr-120034001
154. Clark DW, Palle K. Aldehyde dehydrogenases in cancer stem cells: potential as therapeutic targets. Ann Transl Med (2016) 4:518. doi: 10.21037/atm.2016.11.82
155. Zhou W, Yang Y, Gu Z, Wang H, Xia J, Wu X, et al. ALDH1 activity identifies tumor-initiating cells and links to chromosomal instability signatures in multiple myeloma. Leukemia (2014) 28:1155–8. doi: 10.1038/leu.2013.383
156. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature (2005) 434:843–50. doi: 10.1038/nature03319
157. Ruiz i Altaba A, Sánchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer (2002) 2:361–72. doi: 10.1038/nrc796
158. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting wnt, notch, and hedgehog pathways. Nat Rev Clin Oncol (2011) 8:97–106. doi: 10.1038/nrclinonc.2010.196
159. Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of notch in stem cells and cancer. Cancer Lett (2009) 279:8–12. doi: 10.1016/j.canlet.2008.09.030
160. Sukhdeo K, Mani M, Zhang Y, Dutta J, Yasui H, Rooney MD, et al. Targeting the beta-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc Natl Acad Sci U.S.A. (2007) 104:7516–21. doi: 10.1073/pnas.0610299104
161. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U.S.A. (2007) 104:4048–53. doi: 10.1073/pnas.0611682104
162. Derksen PWB, Tjin E, Meijer HP, Klok MD, MacGillavry HD, van Oers MHJ, et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc Natl Acad Sci U.S.A. (2004) 101:6122–7. doi: 10.1073/pnas.0305855101
163. Boucher K, Parquet N, Widen R, Shain K, Baz R, Alsina M, et al. Stemness of b-cell progenitors in multiple myeloma bone marrow. Clin Cancer Res Off J Am Assoc Cancer Res (2012) 18:6155–68. doi: 10.1158/1078-0432.CCR-12-0531
164. Chhabra R, Saini N. MicroRNAs in cancer stem cells: current status and future directions. Tumour Biol J Int Soc Oncodevelopment Biol Med (2014) 35:8395–405. doi: 10.1007/s13277-014-2264-7
165. Orlowski RZ. Why proteasome inhibitors cannot ERADicate multiple myeloma. Cancer Cell (2013) 24:275–7. doi: 10.1016/j.ccr.2013.08.014
166. Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, et al. Xbp1s-negative tumor b cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell (2013) 24:289–304. doi: 10.1016/j.ccr.2013.08.009
167. Solimando AG, Da Vià MC, Bolli N, Steinbrunn T. The route of the malignant plasma cell in its survival niche: Exploring “Multiple myelomas”. Cancers (2022) 14:3271. doi: 10.3390/cancers14133271
168. Desantis V, Saltarella I, Lamanuzzi A, Melaccio A, Solimando AG, Mariggiò MA, et al. MicroRNAs-based nano-strategies as new therapeutic approach in multiple myeloma to overcome disease progression and drug resistance. Int J Mol Sci (2020) 21:3084–3101. doi: 10.3390/ijms21093084
169. Walker BA, Wardell CP, Chiecchio L, Smith EM, Boyd KD, Neri A, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood (2011) 117:553–62. doi: 10.1182/blood-2010-04-279539
170. Kaiser MF, Walker BA, Hockley SL, Begum DB, Wardell CP, Gonzalez D, et al. TC classification-based predictor for multiple myeloma using multiplexed real-time quantitative PCR. Leukemia (2013) 27:1754–7. doi: 10.1038/leu.2013.12
171. Alaterre E, Ovejero S, Herviou L, de Boussac H, Papadopoulos G, Kulis M, et al. Comprehensive characterization of the epigenetic landscape in multiple myeloma. Theranostics (2022) 12:1715–29. doi: 10.7150/thno.54453
172. Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature (2014) 512:49–53. doi: 10.1038/nature13527
173. De Smedt E, Devin J, Muylaert C, Robert N, Requirand G, Vlummens P, et al. G9a/GLP targeting in MM promotes autophagy-associated apoptosis and boosts proteasome inhibitor-mediated cell death. Blood Adv (2021) 5:2325–38. doi: 10.1182/bloodadvances.2020003217
174. Herviou L, Kassambara A, Boireau S, Robert N, Requirand G, Müller-Tidow C, et al. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin Epigenet (2018) 10:121. doi: 10.1186/s13148-018-0554-4
175. Kassambara A, Rème T, Jourdan M, Fest T, Hose D, Tarte K, et al. GenomicScape: An easy-to-Use web tool for gene expression data analysis. application to investigate the molecular events in the differentiation of b cells into plasma cells. PloS Comput Biol (2015) 11::e1004077. doi: 10.1371/journal.pcbi.1004077
176. Herviou L, Ovejero S, Izard F, Karmous-Gadacha O, Gourzones C, Bellanger C, et al. Targeting the methyltransferase SETD8 impairs tumor cell survival and overcomes drug resistance independently of p53 status in multiple myeloma. Clin Epigenet (2021) 13:174. doi: 10.1186/s13148-021-01160-z
177. Desantis V, Solimando AG, Saltarella I, Sacco A, Giustini V, Bento M, et al. MicroRNAs as a potential new preventive approach in the transition from asymptomatic to symptomatic multiple myeloma disease. Cancers (2021) 13:3650. doi: 10.3390/cancers13153650
178. Smith EM, Boyd K, Davies FE. The potential role of epigenetic therapy in multiple myeloma. Br J Haematol (2010) 148:702–13. doi: 10.1111/j.1365-2141.2009.07976.x
179. Pennipede D, Mohyuddin GR, Hawkins R, Ganguly S, Shune L, Ahmed N, et al. Carfilzomib, cyclophosphamide, and dexamethasone (KCd) for the treatment of triple-class relapsed/refractory multiple myeloma (RRMM). Eur J Haematol (2021) 107:602–8. doi: 10.1111/ejh.13697
180. Theodoropoulos N, Lancman G, Chari A. Targeting nuclear export proteins in multiple myeloma therapy. Target Oncol (2020) 15:697–708. doi: 10.1007/s11523-020-00758-2
181. Vogl DT, Dingli D, Cornell RF, Huff CA, Jagannath S, Bhutani D, et al. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J Clin Oncol Off J Am Soc Clin Oncol (2018) 36:859–66. doi: 10.1200/JCO.2017.75.5207
182. Hartley-Brown M, Richardson P. Antibody-drug conjugate therapies in multiple myeloma–what’s next on the horizon? Explor Target Anti-Tumor Ther (2022), 3(1):1–10. doi: 10.37349/etat.2022.00067
183. Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol (2020) 21:207–21. doi: 10.1016/S1470-2045(19)30788-0
184. Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Longer term outcomes with single-agent belantamab mafodotin in patients with relapsed or refractory multiple myeloma: 13-month follow-up from the pivotal DREAMM-2 study. Cancer (2021) 127:4198–212. doi: 10.1002/cncr.33809
185. Gnoni A, Brunetti O, Longo V, Calabrese A, Argentiero A, Calbi R, et al. Immune system and bone microenvironment: rationale for targeted cancer therapies. Oncotarget (2020) 11:480–7. doi: 10.18632/oncotarget.27439
186. Kumar SK, Rajkumar SV. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat Rev Clin Oncol (2018) 15:409–21. doi: 10.1038/s41571-018-0018-y
187. Solimando AG, Da Vià MC, Cicco S, Leone P, Di Lernia G, Giannico D, et al. High-risk multiple myeloma: Integrated clinical and omics approach dissects the neoplastic clone and the tumor microenvironment. J Clin Med (2019) 8:997–1023. doi: 10.3390/jcm8070997
188. Davies FE, Pawlyn C, Usmani SZ, San-Miguel JF, Einsele H, Boyle EM, et al. Perspectives on the risk-stratified treatment of multiple myeloma. Blood Cancer Discovery (2022), 3(4):273–84. doi: 10.1158/2643-3230.BCD-21-0205
Keywords: multiple myeloma, drug resistance, bone marrow microenvironment, monoclonal gammopathy of undetermined significance, therapeutic targets
Citation: Solimando AG, Malerba E, Leone P, Prete M, Terragna C, Cavo M and Racanelli V (2022) Drug resistance in multiple myeloma: Soldiers and weapons in the bone marrow niche. Front. Oncol. 12:973836. doi: 10.3389/fonc.2022.973836
Received: 20 June 2022; Accepted: 05 September 2022;
Published: 21 September 2022.
Edited by:
Annamaria Gulla, Dana–Farber Cancer Institute, United StatesReviewed by:
Claudio Cerchione, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalyCopyright © 2022 Solimando, Malerba, Leone, Prete, Terragna, Cavo and Racanelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vito Racanelli, dml0by5yYWNhbmVsbGkxQHVuaWJhLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.