
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 29 August 2022
Sec. Radiation Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.971959
This article is part of the Research TopicTargeting DNA Damage Response to Enhance Antitumor Innate Immunity in RadiotherapyView all 10 articles
 Charleen M. L. Chan Wah Hak1*
Charleen M. L. Chan Wah Hak1* Antonio Rullan2Emmanuel C. Patin2
Antonio Rullan2Emmanuel C. Patin2 Malin Pedersen2Alan A. Melcher1
Malin Pedersen2Alan A. Melcher1 Kevin J. Harrington2
Kevin J. Harrington2Radiotherapy is one of the most effective and frequently used treatments for a wide range of cancers. In addition to its direct anti-cancer cytotoxic effects, ionising radiation can augment the anti-tumour immune response by triggering pro-inflammatory signals, DNA damage-induced immunogenic cell death and innate immune activation. Anti-tumour innate immunity can result from recruitment and stimulation of dendritic cells (DCs) which leads to tumour-specific adaptive T-cell priming and immunostimulatory cell infiltration. Conversely, radiotherapy can also induce immunosuppressive and anti-inflammatory mediators that can confer radioresistance. Targeting the DNA damage response (DDR) concomitantly with radiotherapy is an attractive strategy for overcoming radioresistance, both by enhancing the radiosensitivity of tumour relative to normal tissues, and tipping the scales in favour of an immunostimulatory tumour microenvironment. This two-pronged approach exploits genomic instability to circumvent immune evasion, targeting both hallmarks of cancer. In this review, we describe targetable DDR proteins (PARP (poly[ADP-ribose] polymerase); ATM/ATR (ataxia–telangiectasia mutated and Rad3-related), DNA-PKcs (DNA-dependent protein kinase, catalytic subunit) and Wee1 (Wee1-like protein kinase) and their potential intersections with druggable immunomodulatory signalling pathways, including nucleic acid-sensing mechanisms (Toll-like receptors (TLR); cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) and retinoic acid-inducible gene-I (RIG-I)-like receptors), and how these might be exploited to enhance radiation therapy. We summarise current preclinical advances, recent and ongoing clinical trials and the challenges of therapeutic combinations with existing treatments such as immune checkpoint inhibitors.
Radiotherapy continues to be one of the most effective treatments for a wide range of cancers since its discovery over a century ago. Approximately half of cancer patients receive radiotherapy at some point in their cancer treatment (1), whether in the curative or palliative settings.
Radiotherapy exploits ionising radiation to cause cell death or senescence via DNA damage. Broadly, necrotic or apoptotic cell death occurs depending on cell type, radiotherapy dose and fractionation schedule (2). Cancer cells that evade apoptosis and continue to divide with accumulated DNA damage can die via mitotic catastrophe. Also, excess autophagy can force the cell into apoptotic or necrotic cell death (3, 4). Classically, the response of tumours to conventional fractionated radiotherapy is governed by the principles of the 4 “R”s of radiobiology: repair of sublethal DNA damage after exposure to ionising radiation, redistribution of cells in the cell cycle whereby cells in the G2/M-phase are most radiosensitive and are preferentially killed in comparison to the more radioresistant late S-phase, repopulation of tumour cells and reoxygenation of previously hypoxic tumour areas (5). A 5th “R” of intrinsic radiosensitivity has also postulated by Steel, after observing the varying survival curves of different tumour cell lines following irradiation, which is thought to be independent of their DNA repair capacity (6). Combining agents that can target DNA damage repair pathways, as one of the 4 “R”s, with radiotherapy holds considerable potential to enhance therapeutic outcomes.
In addition to direct cell killing, radiotherapy can induce immunogenic cell death (ICD) and modulate the immune tumour microenvironment to lead to anti-tumour innate immune activation (7). Due to these immunostimulatory effects, there is increased interest in radiotherapy as a promising combinatorial agent with other immuno-oncology agents such as DNA-damage response (DDR)-targeting agents (8). This two-pronged approach exploits two hallmarks of cancer, namely genomic instability and evasion of immune surveillance (9, 10). The DDR sensing and signalling pathway are the collective mechanisms evolved by cells to combat the threat of DNA damage, namely the detection of DNA lesions, signalling of their presence and promotion of DNA repair (11). Promising DDR druggable targets include those within DNA repair pathways and cell cycle checkpoints, as well as damage-associated molecular pattern (DAMP)-sensing receptors which can amplify the DDR-induced immune response when combined with radiotherapy.
Radiotherapy has both immunostimulatory and immunosuppressive effects. The difference in the ability of radiotherapy to initiate pro-immunostimulatory effects and turn immunogenically “cold” (low T-cell infiltrated) tumours “hot” (high T-cell infiltrated) may account for the enhanced response to radiotherapy of some pre-clinical models and clinical cancer histotypes.
As a defence against microbial infection, the innate immune system has evolved pattern-recognition receptors (PRRs) that detect microbial pathogenic molecules known as pathogen-associated molecular patterns (PAMPs). However, these pathways do not exclusively sense foreign molecules. Immune activation can also occur in the absence of microbial infection, instead being triggered by inflammatory signals released from stressed or dying cells collectively known as damage-associated molecular patterns (DAMPs) (12). Radiotherapy-induced cellular stress and ICD can stimulate an immune response through the generation of DAMPs (13) detected by their cognate pattern recognition receptors (PRRs) (14). ICD has been defined as the chronic exposure of DAMPs in the tumour environment (TME), which can induce an innate and adaptive anti-tumour immune response in the host (15).
A characteristic DAMP induced by ICD is the secretion of adenosine triphosphate (ATP) from dying cancer cells into the extracellular space. Extracellular ATP functions as a “find-me” chemoattractant signal for the recruitment and activation of dendritic cells (DCs) (15–17). High-mobility group box-1 (HMGB1), secreted from the nucleus during ICD, binds to Toll-like receptor (TLR-4) and is critical for activating DCs and facilitating antigen processing and presentation to T cells (18). Translocation of calreticulin to the cell surface on dying cells provides an “eat-me” signal to antigen-presenting cells (APCs) and results in their phagocytosing target cells (19). In the context of cancer, ICD leads to release of tumour-associated antigens (TAA) and subsequent priming of a cancer-specific immune response. Another characteristic of ICD is the expression of heat shock proteins (HSP) HSP70 and HSP90 on dying cell membranes that drives cross-presentation of tumour-derived antigens on major histocompatibility complex class I (MHC-I) (15).
Radiotherapy-induced DNA damage can function as a viral mimic through the accumulation of cytosolic DNA or RNA in irradiated cells (20). Cytosolic DNA and RNA activate cyclic GMP-AMP synthase (cGAS)/stimulator of interferon (IFN) genes (STING) and retinoic acid-inducible gene I (RIG-I)/mitochondrial antiviral-signalling protein (MAVS) pathways, respectively (21). These pathways activate complex downstream signalling via interferon regulatory factor 3 (IRF3)/TANK-binding kinase 1 (TBK1) and nuclear factor kappa B (NF-κB) that results in production of Type I IFN and other inflammatory cytokines (e.g. interleukin (IL)-1, tumour necrosis Factor (TNF)-α) (20).
Radiotherapy is a form of ionising radiation that hydrolyses water and forms reactive molecules, such as reactive oxygen species (ROS) and nitric oxide species (NOS), which can directly alter DNA, cellular components, and molecules in the extracellular matrix (ECM) (22). ROS and NOS can be derived both from these direct ionisation events or activated immune cells, and work with other DAMPs to accelerate lymphocyte and DC recruitment. These activated immune cells generate pro-inflammatory cytokines (e.g. TNF-α, IL-1β, IL-6, IL-12) (14, 23, 24), chemokines and growth factors leading to a sustained inflammatory response (22, 25).
Recent data suggest that radiation can enhance cancer cell antigenicity through upregulation of genes involved in DNA damage repair and cellular stress responses (20). Immune cell recruitment is subsequently increased via expression of adhesion molecules (e.g. intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1) and E-selectin) (26) and chemokines (e.g. chemokine (C-X-C motif) ligand 16 (CXCL16)) (27). Within the appropriate inflammatory environment, DCs take up antigens in peripheral tissues and mature and migrate to draining lymph nodes, where they induce activation of naïve T-cells and differentiation into effector T-cells (28). Radiotherapy-induced ICD, as discussed above, increases tumour-associated antigen presentation that can lead to specific tumour-associated antigen T-cell priming, expansion of tumour reactive CD8+ T cells and infiltration into the tumour microenvironment (TME) (29). In summary, inflammatory DAMP signalling generates a favourable environment for activated DCs to process and cross-present tumour-derived antigens from irradiated cells as a “tumour vaccine”, to naïve T cells. These T cells subsequently can be primed and sustain a systemic tumour-specific immune response. The T-cell receptor (TCR) repertoire is also known to be shaped following radiotherapy, including when used in conjunction with immune checkpoint inhibitors (ICI) (30–32).
Whilst pro-inflammatory signalling can lead to a positive anti-tumour effect, cancer cells adapt to survive with mechanisms such as hypoxia resistance and unrestricted proliferation that can result in a state of chronic inflammation and evasion of immune surveillance (33–35). Evasion of immune recognition or immune escape (36) is now a recognised hallmark of cancer (9) and this inclination towards pro-tumour growth is mediated by changes in cytokine signalling (TNF-α, IL-1β, IL-6, IL-10 and TGF-β) (37, 38) and recruitment of TME-immunosuppressive immune cells such as tumour-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs) (39) and regulatory T cells (Tregs) (40, 41).
PD-L1 (programmed death-ligand 1) expression is found to be elevated on tumour cells following irradiation due to interferon gamma (IFN-γ) release from tumour-infiltrating lymphocytes (TILs) (42) and TILs have increased expression of PD-1 (programmed death-1) following ex-vivo irradiation (43). A recent publication found that irradiation of colorectal cancer cells triggered an ATR-mediated DNA repair signalling pathway to upregulate CD47 and PD-L1, through engagement of signal-regulator protein α (SIRPα) and PD-1, respectively, to limit tumour-associated cross-presentation and suppression of innate immune activation (44).
Recruited MDSCs and TAMs can suppress T-cell function through antagonistic cytokine signals (45). Supporting data includes that from a phase I/II clinical trial testing the combination of radiotherapy and a primed DC vaccine in which non-responders had significantly higher baseline tumour levels of MDSCs (46).
Tregs are relatively more radioresistant than other lymphocyte subsets and radiotherapy may increase the infiltration by phenotypically and functionally suppressive Tregs within the TME (40, 41, 47). In several pre-clinical mouse models (B16/F10, RENCA and MC38), Tregs in irradiated tumours expressed higher levels of cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), 4-1BB (CD137, tumour necrosis factor receptor superfamily 9) and Helios compared with Tregs in non-irradiated tumours (47).
Cancer-associated fibroblasts (CAFs) can be the predominant component of the stroma in the TME and facilitate stroma-mediated radioprotection through multiple mechanisms. Following radiotherapy, CAFs can survive through formation of integrin-mediated attachments (48) and radioprotective integrin β-1 signalling (49). CAFs can promote an oxygen-rich, immunosuppressive and pro-inflammatory TME (50–52) resulting in increased tumour growth, invasion and metastasis (53).
Conversion of ATP to adenosine by CD39 and/or CD73 is a mechanism by which tumour cells can escape immune-surveillance by limiting the functionality of multiple potentially protective immune infiltrates, while enhancing the activity of immunosuppressive cell-types (54). CD39 and/or CD73 (over)expression has been found on the surface of tumour cells (55), CAFs (56) MDSCs (57), TAMs (58), Tregs and exhausted conventional CD4+ and CD8+ T cells (59–61).
One of the 4 “R”s of radiobiology is repopulation (5), and tumour repopulation during radiotherapy and chemotherapy is an important cause of treatment failure (62). Some tumours exhibit accelerated tumour repopulation following irradiation by paracrine caspase 3-dependent prostaglandin E2 (PGE2)-mediated signalling (63). Tumour repopulation may also be driven by a small number of cancer stem cells (CSC) which promote tumour growth following an insult, such as radiotherapy (64). Rapid proliferation of cancer cells is generally accepted as a prerequisite for most conventional chemotherapies and radiotherapy to be effective, and any senescent and/or quiescent tumour cells, such as CSCs, may be treatment-resistant (64). The CSC response to therapy may underpin why macroscopic tumour response to (chemo)radiation is not a robust predictor for clinical outcome, since small numbers of these relatively resistant and less immunogenic CSCs may survive to repopulate the tumour (64). However, in vitro pre-clinical data from human breast cancer cell lines (MCF-7 and T47D) have shown that radiotherapy can recruit CSC cells from a quiescent state into the cell cycle (65) and a CSC-druggable target in combination with radiotherapy would be useful.
As we have seen, radiotherapy can trigger key events leading to potent anti-tumour immune responses via production of immunostimulatory cytokines, DC recruitment, and T-cell recruitment and activation. However, these are negatively balanced by the potential for concurrent triggering of immunosuppressive cells within the TME and accelerated tumour cell repopulation. Targeting the DNA-damage response pathway (DDR) is an attractive approach to tip the scales towards maintaining positive immune anti-tumour states, which can be characterised as ‘pro-immunogenic’ and ‘pro-inflammatory’.
Radiotherapy causes cell damage, stress and death through induction of DNA lesions in the form of crosslinking, single-strand breaks (SSBs) and, most significantly, double-strand breaks (DSBs) (66). These processes induce a plethora of intracellular signalling pathways involved in detecting and repairing DNA damage. Targeting both DNA damage repair and DDR’s downstream cytosolic nucleic acid sensing pathways with small molecules in combination with radiotherapy can lead to increased immune activation and anti-tumour efficacy of these treatments (Figure 1).
Radiotherapy induces double-strand breaks (DSBs) in cancer cell DNA, which results in genomic instability, cell cycle arrest, apoptosis or death via mitotic catastrophe (66). In response to radiotherapy, cancer cells can respond to exploit individualised DNA damage repair mechanisms for survival (67). Three primary DNA repair pathways have evolved to process DSB repair and maintain genomic integrity: homologous recombination, non-homologous end-joining (NHEJ) and alternative end-joining (68). Upregulation of these pathways is a mechanism by which cancer cells may acquire radioresistance and, accordingly, radiosensitisation strategies which inhibit radiation-induced DNA damage repair are expected to provide increased cancer control (66). When DNA repair is inhibited in cancer cells, this leads to accumulation of DNA damage, cellular stress and cell death which subsequently increases the likelihood of these cells triggering innate immune pathways and being recognised by anti-tumour immune surveillance.
ATM and ATR are both key mediators of the DSB signalling response that induce cell cycle arrest to facilitate DNA repair (69). In addition, conditions that activate ATM and ATR as part of DDR may also participate in regulating the innate immune system and alert it to potentially ‘dangerous’ tumour cells (70).
In response to DSB, the MRE11-RAD50-Nibrin (NBS1) (MRN) complex assembles at DSB sites to act as a DNA damage sensor that activates and recruits ATM to DSB sites (71). Briefly, when a cell triggers the DDR, ATM initiates a massive signalling cascade with the phosphorylation of hundreds of substrates, including p53 and checkpoint kinase 2 (Chk2). Activated p53 transactivates the expression of p21Cip1/kip1, which inhibits Cyclin Dependent Kinase (CDK) 2 and CDK4/6 to induce G1/S arrest (66). Chk2 in turn phosphorylates and inactivates Cell Division Cycle 25 (CDC25C), maintaining the inhibitory phosphorylation of CDK1 by Wee1-like protein kinase (Wee1) and Myelin Transcription Factor 1 (Myt1) to induce G2/M cell cycle arrest or apoptosis (66, 72). Inhibition of the ATM/Chk2 axis can lead to replication stress and accumulation of cytosolic DNA that subsequently activates the cGAS-STING-mediated innate immune response (73).
ATM was recognised as the defective gene in the inheritable human disorder, ataxia-telangiectasia (A-T) (74), and these patients have characteristic features including genomic instability and profound radiosensitivity (75). Deficiency of ATM-mediated signalling reactions causes sensitisation of cells to radiation (76), which has sparked interest in ATM as a therapeutic target for cancer treatment (69). Inhibition of ATM and ATR have the potential to improve radiotherapy outcomes as they are both key mediators of the DDR (69). Indeed, ATM inhibitors such as caffeine (77), wortmannin (78), CP-466722 (79), KU-55933 (80), KU-60019 (81) and KU-59403 (82) increase cell radiosensitivity (83, 84), particularly in p53 low/deficient and phosphatidylinositol 3-kinase (PI3K) highly-expressing cells (77, 85). In a preclinical study in vivo with KU60019 and radiotherapy, combination treatment enhanced TBK1 activity, type I IFN production, antigen presentation and increased CD8+ TILs; moreover, complete responders had established immunological memory (86) (Table 1). The ATM inhibitor (AZD1390) and radiotherapy is being investigated in a phase I clinical trial in brain cancer (NCT03423628). A dual ATM and DNA-PKc inhibitor (XRD-0394) and radiotherapy phase I trial is also recruiting (NCT05002140) (Table 2).

Table 1 Preclinical RT and DDR combination studies.
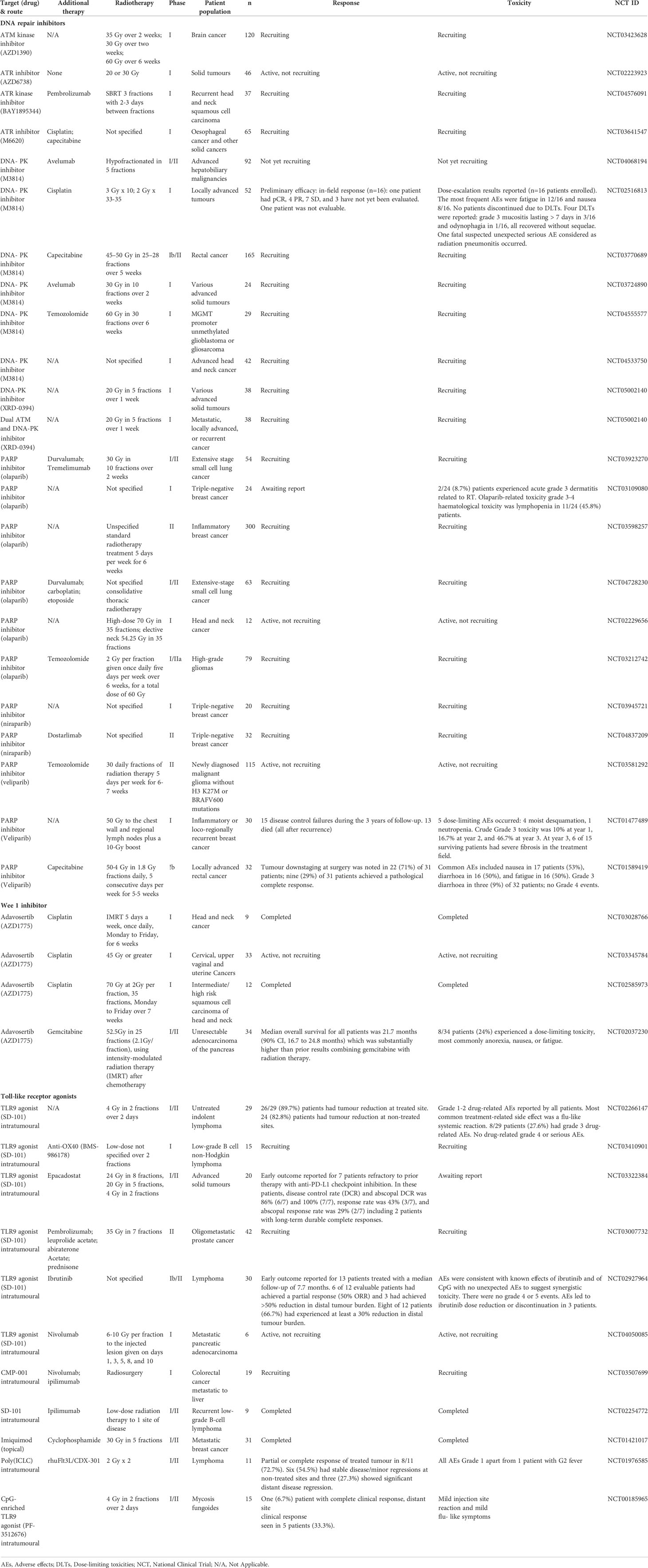
Table 2 Selected clinical trials investigating radiotherapy in combination with DDR inhibitor and/or other agents.
ATR is activated by single-stranded DNA (ssDNA) structures that may arise at resected DNA DSBs or stalled replication forks. ATR is recruited via interaction of ATR-interacting protein (ATRIP) with ssDNA-bound replication protein A (RPA) (105). RPA-ssDNA complexes stimulate loading of the RAD9–HUS1–RAD1 (9–1–1) heterotrimer, that recruits DNA topoisomerase II binding protein 1 (TopBP1) which activates ATR (106). Once ATR is activated, downstream targets, including checkpoint kinase 1 (Chk1), promote DNA repair (107, 108), restart of stalled replication forks (109) and intra-S and G2/M cell cycle arrest (110, 111). In response to DNA damage, activation of the intra-S-phase cell cycle checkpoint slows progression of DNA replication to allow time for resolution (110, 111). In addition, the ATR-dependent G2/M cell cycle checkpoint is activated through degradation of cell division cycle 25A (Cdc25A) (111), and phosphorylation of Cdc25C phosphatase inhibits its ability to activate nuclear cell division cycle 2 (Cdc2) and, hence, mitosis entry (112). Most cancer cells are defective in DNA damage-induced checkpoints through e.g. p53 pathway mutations, which leads to dependence on the intra-S-phase and G2/M checkpoints for cell survival (69). Therefore, ATR inhibition will lead to accumulation of DNA damage, premature entry into mitosis, mitotic catastrophe and cell death (69).
ATR inhibitors include schisandrin B (113), NU6027 (114), NVP-BEZ235 (115), VE-821 (116), VE-822 (117), AZ20 (118) and ceralasertib (AZD6738) (119, 120). NVP-BEZ235 has been reported to induce marked radiosensitivity in Ras-overexpressing cancers (121), and NU6027 has been shown to increase sensitivity to DNA-damaging agents in breast and ovarian cell lines (114). VE-822 results in selective sensitisation of pancreatic tumours to radiation in vivo by increasing persistent DNA damage, decreasing cell cycle checkpoint maintenance and reducing homologous recombination repair (117). In vitro, ATR inhibition downregulates radiotherapy-induced programmed death-ligand 1/2 (PD-L1/2) expression to sensitise cancer cells to T-cell killing, in addition to potentiating DNA damage (122). Promising preclinical in vivo studies (Table 1) of the ATR inhibitor ceralasertib (AZD6738) in combination with radiotherapy have shown an enhanced type I/II interferon response and increased immune cell infiltrate (88), increased RT-stimulated CD8+ T cell infiltration (87, 89), NK-mediated anti-tumour immunity (90), as well as reversal of the Treg immunosuppressive effect (87, 89). In addition, further addition of ICI (i.e. anti-PD-1, anti-PD-L1, anti-TIGIT (T-cell immunoglobulin and ITIM domain)) to the ceralasertib (AZD6738) and radiotherapy combination further improved response and long-lasting immunity in a CD8+ (87, 89) and NK-dependent manner (90).
There are, to date, three early phase clinical studies investigating ATR inhibition and radiotherapy. PATRIOT, a phase I study of ceralasertib (AZD6738) in combination with palliative radiotherapy, has completed recruitment and is awaiting report (NCT02223923). BAY1895344 in combination with radiotherapy and pembrolizumab in recurrent head and neck squamous cell carcinoma (HNSCC) (NCT04576091) and M6620 with radiotherapy and chemotherapy in solid cancers (NCT03641547) are ongoing studies (Table 2).
A downstream target of ATR, Chk1, has also been investigated as a potential therapeutic target, due to its ability to activate intra-S and G2/M cell cycle checkpoints and modulate the replication stress response (123), particularly as a sensitiser to radiotherapy (124). Chk1 inhibitors, to date, include UCN-01 (125), LY2606368 (126), PF-00477736 (127), MK8776 (128) and CCT244747 (129), AZD7762 (130) and LY2603618 (131). Although there have been promising results in refractory acute myeloid leukaemia and advanced cancer with MK-8776 (132, 133) and LY2606368 (134), unfortunately severe adverse effects such as drug-related cardiac toxicity have also been reported during the clinical development of these drugs, e.g. AZD7762 (135). Thus far, no clinical trials are investigating the combination of Chk1 inhibition and radiotherapy.
DNA-PK is pivotal for the initiation of DNA repair following DSBs, which ultimately results in recruitment of proteins involved in DNA damage repair progressing and ligating the broken DNA ends most recognised via the NHEJ pathway (136). Various cancer cell lines with reduced levels of DNA-PKcs show increased radiosensitivity compared to unirradiated controls (137–139) due to defective DNA DSB repair, inhibition of phosphorylated protein kinase B (Akt) on Ser473 and reduction of radiotherapy-induced transcription factor hypoxia-inducible factor-1 α levels (HIF-1 α) (138).
Given that DNA-PKcs is critical in radiotherapy-induced DDR, DNA-PKcs inhibition is an emerging therapeutic target for potentiating radiotherapy responses (140, 141), and many agents have already been tested in clinical trials. Non-selective DNA-PKcs inhibitors include wortmannin, which also inhibits ATM (142), and LY294002, which has a similar structure (143, 144). More selective DNA-PKcs inhibitors include NU7026 (145), NU7441 (146), IC86621, IC87102, IC87361 (147), vanillin (148), OK-1035 (149), SU11752 (150), BVAN08 (151), IC486241 (152) and NK314 (153). More recently, novel inhibitors have been discovered including M3814 (154), AZD7648 (155) and VX-984 (156). Doxycycline was first approved by US Food and Drug Administration (FDA) in 1967 as a broad-spectrum antibiotic and has recently been recognised to function also as an DNA-PK inhibitor (157). Mechanisms by which DNA-PKcs helps to sensitise to radiotherapy include prolongation of radiotherapy-induced G2/M phase arrest (158) and reduced repair of radiotherapy-induced DSB (147, 150, 159) leading to the induction of autophagic cell death and mitotic catastrophe (66).
In terms of DNA-PKcs inhibition leading to stimulation of the innate immune system, a recent study showed that combining radiation with M3814-induced DNA-PK inhibition increased cytosolic dsDNA and tumour type I interferon signalling in a cGAS-STING-independent, but RNA Polymerase III-, RIG-I- and MAVS-dependent manner, in pancreatic cancer models (91). Furthermore, radiotherapy and M3814 increased PD-L1 expression and sensitised to anti-PD-L1 treatment in poorly immunogenic pancreatic cancers (91). DNA-PKcs itself also functions as a DNA sensor that activates innate immunity. It has been reported to function as a PRR by binding to cytoplasmic DNA and can trigger a type I IFN response in a STING/IRF-3/TBK1-dependent manner (160) as well as a STING-independent manner via phosphorylation of heat shock protein HSPA8/heat shock cognate HSC70 (161). It is still unclear whether pharmacological inhibition of DNA-PKcs kinase activity may dampen anti-tumour immunity in contrast to inhibition of other DDR kinases described such as ATM or ATR.
Clinical studies of DNA repair inhibitors, M3814 (NCT04533750) and XRD-0394 (NCT05002140), in combination with radiotherapy are recruiting. In addition, triple combination of M3814 with radiotherapy and chemotherapy (NCT02516813, NCT03770689, NCT04555577) or anti-PD-L1 (NCT04068194, NCT03724890) are also awaiting report (Table 2).
PARP-1 has been the most extensively studied of the PARP superfamily and is a key regulator of DNA damage repair (162, 163). In response to DNA damage, such as that induced by radiotherapy, an initial response is poly(ADP-ribosyl)ation (PARylation) of proteins including nuclear DDR proteins, such as DNA-PKcs, to provide a local signal of DNA damage (163–165). Inhibitors of PARP generally function by inhibiting PARylation or suppressing PARP-1 release by ‘trapping’. PARP-1 inhibition has been reported to sensitise cancer cells to various forms of ionising radiation including conventional gamma irradiation (166, 167), proton-beam irradiation (167) and radionuclide therapy (168, 169) (Table 2). Although SSBs are primary repaired by PARP-1, its inhibition may not be lethal due to other available repair pathways, such as homologous recombination. However, deficiency in BRCA1/2 functionality, which are key components in the HR pathway of DSB repair, leads to synthetic lethality and selective sensitivity to PARP inhibition (170).
Beyond DNA repair, PARP-1 also plays an immunomodulatory role by regulating gene transcription of several immune cell types, modulating the stimulatory ability of DCs, and by directly affecting the differentiation and function of T and B cells (171, 172). PARP-1 knockout mice show reduced T helper type 2 (Th2) differentiation responses (172). PARP-1 is also involved in the differentiation of Foxp3+ regulatory T cells (Treg) and promotion of Treg cell apoptosis during inflammatory responses (172). PARP inhibitors generate cytoplasmic chromatin fragments with micronuclei characteristics which activate cGAS-STING, downstream type I interferon signalling and chemokine ligand 5 (CCL5) secretion in excision repair cross-complementation group 1 (ERCC1)-defective non-small cell lung cancer (NSCLC) cells (173). The capacity of PARP1 inhibitors to upregulate innate immune and inflammasome-like signalling events, such as cGAS-STING signalling, closely depends on their PARP1-trapping abilities (174, 175). In the context of viral infection, activated DNA-PK has been reported to phosphorylate PARP1 leading to its cytoplasmic translocation (176). Cytoplasmic PARP1 can then interact with and directly PARylate cGAS to inhibit its DNA-binding ability (176). This has implications to how PARP inhibition, in the context of cancer-induced genome instability, can positively modulate the host anti-tumour immune response.
Early PARP-1 inhibitors were non-specific and non-selective, such as nicotinamide (177), AG14361 (178) and 4-amino-1,8-naphthalimide (179). Newer PARP-1 inhibitors, such as olaparib and niraparib, are now used in routine clinical practice following approval by the FDA and European Union (180, 181). They are licensed for use in patients with advanced BRCA-mutated ovarian cancer, metastatic-castration-resistant prostate cancer with BRCA1/2 or ATM mutation (182), suspected germline HR repair gene mutated mCRPC who have progressed on enzalutamide or abiraterone (183) and, most recently, recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer which has responded to first-line platinum chemotherapy (184, 185).
Combining PARP-1 inhibition and radiotherapy has been supported by preclinical studies. Particularly in BRCA1-mutant cancers, PARP inhibition showed radiation hypersensitivity in lymphoblastoid cells (186). In various models, PARP-1 inhibitors KJ-28d (187), ABT-888 (188) and the PARP-1/2 inhibitor MK-4827 (189) increased cancer cell radiation sensitivity.
Many clinical trials are underway investigating the combination of PARP inhibitors and radiotherapy, with addition of chemotherapy and/or immunotherapy agents (Table 1). The mechanisms underlying radiosensitisation by PARP inhibitors are still not completely clear and, indeed, recent studies have revealed a wider immunological role for PARP-1 that could potentially be exploited through new therapeutic approaches (190). For example, one study showed through multiomics profiling that macrophage-mediated immune suppression is a liability of PARP inhibition (191). Following this evidence, the rationale for combining CSF-1R blocking antibodies with PARP inhibitors led to reprogramming of the TME and significantly enhanced innate and adaptive anti-tumour immunity, which was CD8+-mediated in BRCA-deficient tumours in vivo (191).
Wee1 is a cell cycle checkpoint negative regulator at the G2/M transition. The process by which Wee1 activation leads to phosphorylation and inactivation of the cyclin B1/CDK1 complex blocking entry into mitosis is well described (192).
Emerging studies have highlighted the role of Wee1 directly and indirectly in immune signalling (193). For example, ineffective CDK-1-dependent nuclear laminin degradation abrogates apoptosis induction, leading to immune resistance in tumour cells (194). Accordingly, Wee1 inhibition reconstitutes CDK1 activity to reverse resistance of these cancer cells to immune attack (194). In various cancer models, Wee1 inhibition promotes accumulation of cytosolic dsDNA, leading to activation of the cGAS-STING pathway (Figure 1), increased type I interferon target gene expression when delivered alone (195), as well as in combination with ATR inhibitors (196) or immune checkpoint blockade (197). A STING-independent pathway by which Wee1 inhibition induces the interferon response has also been reported. In cGAS-STING-defective tumour models, Wee1 inhibition can upregulate immune signalling through the dsRNA anti-viral defence pathway by promoting expression of endogenous retroviral element (ERV) (198). ERVs trigger dsRNA stress and the interferon response, resulting in the recruitment of anti-tumour T-cells, and increased expression of PD-L1 with sensitisation to anti-PD-L1 blockade in multiple cancer models (198).
Wee1 inhibitors, some of which are concomitant CDK1 inhibitors, are promising as a combination partner with radiotherapy (199). This combination has shown synergistic effects in various cancer models (200–202). Wee1 inhibitors such as 681641 (203), PD0166285 (204) and adavosertib (MK1775/AZD1775) (92, 202, 205) have been reported to increase the radiosensitivity of cancer cells. Cancer cells very frequently harbour G1 checkpoint deficiencies and Wee1 inhibitor-mediated prevention of DNA repair following radiotherapy may lead to premature entry into mitosis and, ultimately, cell death via mitotic catastrophe (206). Other mechanisms include blocking radiotherapy-induced DNA damage repair (204) by impairing DNA repair protein RAD51 homolog 1 (RAD51) focus formation (202) and suppression of Sirt1 (silent mating type information regulation 2 homolog 1). Sirt1 interacts with and deacetylates HR-repair machinery proteins including Nibrin (NBS1) and RAD51, thus, Wee1-induced Sirt1 suppression impairs HR-repair activity (207).
Several clinical trials are exploring the combination of Wee1 inhibition by adavosertib (MK1775/AZD1775) with radiotherapy and chemotherapy (NCT03028766, NCT03345784, NCT02585973, NCT02037230) (Table 2). The emerging immune-mediating effects of Wee1 inhibition provide a strong rationale for its combination with immune checkpoint inhibitors (198).
The ability to detect cytosolic nucleic acids by PRRs, arising from pathogens or disruption of cellular functions from genotoxic stress such as DNA damage, is part of the protective cellular response against infection or injury. These mechanisms are an evolutionary product of anti-microbial responses and can trigger an inflammatory signalling cascade and subsequent activation of the innate immune system. Targeting these nucleic acid sensing mechanisms has the potential to further amplify the DDR-induced anti-tumour innate immunity in conjunction with radiotherapy.
Stimulator of interferon genes (STING) is an endoplasmic reticulum adaptor that senses self and foreign cytoplasmic DNA, via cyclic GMP–AMP synthase (cGAS), and is crucial for effective innate immune signalling (208). Cytosolic DNA induces synthesis of the cyclic dinucleotide (CDN) cyclic GMP–AMP (cGAMP) from ATP and GTP by a cyclase enzyme called cGAS. cGAMP directly binds to STING to cause its dimerization and activation (209, 210), leading to activation of both NF-κB and IRF3 transcription pathways to induce expression of type I interferon, recruitment of immune cells, promotion of DC maturation and antigen-specific immune priming (211).
The cGAS-STING pathway is essential for anti-tumour T cell responses (212). One proposed mechanism is that CD8α+ DCs engulf apoptotic or necrotic tumour cells, and tumour cell-derived DNA triggers STING signalling in DCs (212–214). The subsequent type I IFN production by these DCs facilitates antigen cross-presentation and T-cell priming independent of the TLR or RIG-I/MAVS pathways (212). Recent studies have also suggested that STING signalling in the TME can suppress the immunosuppressive activity of MDSCs (215, 216). STING signalling is critical for radiation-induced anti-tumour responses (214) and, thus, it is an attractive potential treatment combination with radiotherapy. Preclinical data have shown that consideration needs to be given to radiotherapy dose per fraction as doses above 12-18 Gy induce the DNA exonuclease Trex1, which degrades the cytosolic DNA required to stimulate an effective STING-dependent type I IFN response (217).
The first generation STING agonist, 5,6-Dimethylxanthenone-4-acetic Acid (DMXAA), was originally developed as a vascular-disrupting agent (218, 219) and its anti-tumour effect is based on vascular necrosis leading to tumour starvation and haemorrhagic necrosis (218, 220). DMXAA has previously been shown to synergise with radiotherapy in mouse models in a hypoxia-preferential manner (221). However, the TME was found to remain immunologically sterile and tumours eventually progressed with time without durable protective anti-tumour immunity (222, 223). High local STING concentrations can lead to rapid T-cell apoptosis (224) whereas low-dose administration can lead to ‘vascular normalisation’ and favourably transform the TME to allow use of effective combinatorial anti-tumour immunotherapy (225–227).
There are two categories of STING agonists in clinical development: synthetic cyclic dinucleotides (CDNs) or non-CDN small molecules (228). These drugs are generally administered intratumourally due to their poor stability and bioavailability. This caveat limits their use to accessible tumours and recent efforts have been focused on development of STING agonists for systemic delivery (intravenously (228), orally (229, 230) and even as an inhalable nanoparticulate (231)). In addition, novel STING antibody-drug conjugates show promising preclinical results (232). There have only been a handful of preclinical studies investigating novel STING agonists with radiotherapy in vivo (Table 1). In mouse models, STING agonists synergise with radiotherapy to control local and distant disease and mediate rejection of tumour rechallenge (93, 231) via early T-cell-independent and TNF-α-dependent haemorrhagic necrosis, followed by a later stage of CD8 T-cell-dependent control (93). A number of clinical trials have looked into combining STING agonists with ICI or conventional chemotherapy (233); however, at the time of this review no radiotherapy and STING agonist combination clinical trials are in progress.
Toll-like receptors (TLRs) are a form of PRR expressed on sentinel immune cells which activate innate defence systems by detecting PAMPs. Genotoxic stress and DNA damage are increasingly recognised to signal through TLRs and cause the upregulation of TLR expression (234) via p53 (235). TLR signalling leads to maturation of APCs such as DCs, which are key mediators of T-cell activation and subsequent adaptive immunity. There is growing preclinical evidence that TLR agonists in combination with radiotherapy may lead to enhanced anti-tumour immunity, particularly through the mechanism of enhanced DC-mediated T-cell priming following radiotherapy (236). This occurs at various stages of this pathway; for example, TLR activation enhances type I IFN-signalling in many immune cells, modulates chemokine expression to enhance DC migration to lymphoid tissues (237–239) and upregulates CD80 and CD86 co-stimulatory molecules on DCs, which bind to CD28 on naïve T-cells for antigen/MHC-complex mediated TCR stimulation (240). TLRs can also stimulate DC-mediated release of IL-6 to dampen Treg suppressive signalling (241).
Given these observations, TLR agonists are seen as an attractive combination partner with radiotherapy. There have been numerous preclinical studies (Table 1) and early phase clinical trials (Table 2) of different TLR agonists, particularly of TLR3, TLR7/8 and TLR9, in combination with radiotherapy.
TLR3 senses dsRNA as a PAMP and polyinosinic-polycytidylic acid or poly (I:C) is a synthetic mimic of dsRNA which can stimulate TLR3-signalling pathways and lead to type I-IFN-dependent (242, 243) DC antigen cross-priming in vivo (244, 245). Poly(I:C) also has several immunostimulatory effects, including maturation and activation of DCs (246–248), T-cell stimulation (249, 250), enhanced cytotoxicity of Natural Killer (NK) cells (251–253), reprogramming of MDSCs (254) and repolarisation of macrophage populations from an M2 (classically activated macrophages) to M1 (alternatively activated macrophages) phenotype (255) (Figure 1). Pre-clinical studies exploring TLR3 agonists with radiotherapy in a radioresistant mouse model of lung cancer showed that poly(I:C) enhanced radiotherapy anti-tumour effects (256). The results from initial clinical trials have been disappointing, likely due to the short half-life of poly(I:C) (257). To address this, a degradation-resistant derivative polyinosinic-polycytidylic acid, and poly-L-lysine or poly(ICLC) was developed that has shown efficacy in clinical trials, although toxicity remains an issue (257). Preclinical studies in a murine lymphoma model have investigated the Fms-like tyrosine kinase 3 (Flt3)-ligand with radiotherapy and poly(ICLC) (258). Flt3-ligand is a cytokine which increases migration of DCs into the tumour and radiotherapy then stimulates maturation of DCs via ICD and HMGB-1 signalling for antigen uptake and processing (259). This combination with the addition of poly(ICLC) further maximises DC maturation and activation (246–248). There is a clinical study investigating intratumoral delivery of poly(ICLC) in combination with an in-situ vaccine rhuFlt3L/CDX-301 and radiotherapy which was well-tolerated and showed promising results (258) (NCT01976585) (Table 2). Two phase 2 studies in glioblastoma patients are also investigating the efficacy of poly(ICLC) in combination with radiotherapy (260, 261).
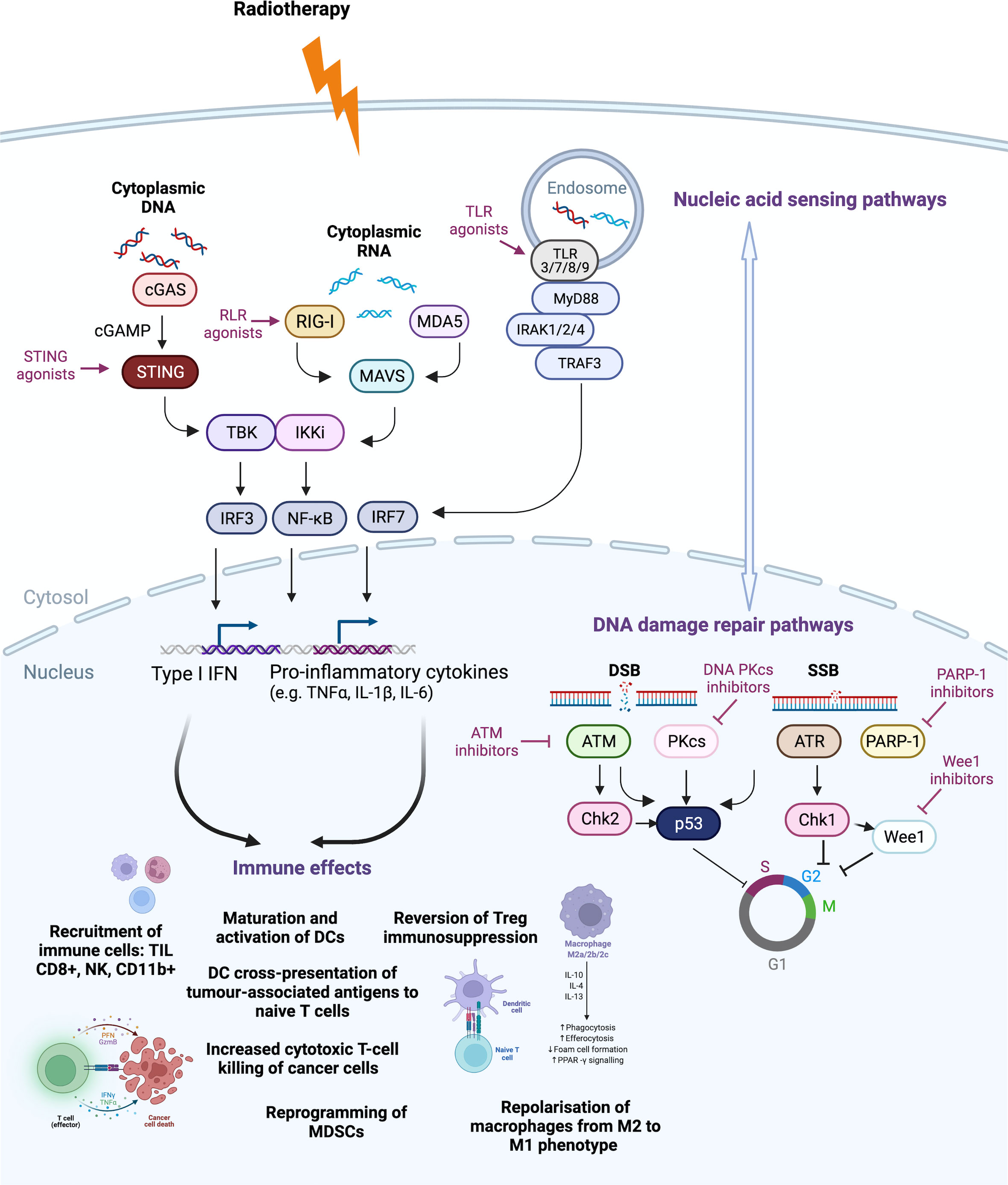
Figure 1 Druggable targets of the DNA damage response (DDR) pathway currently tested in clinical trials. Radiotherapy induces DNA damage and cell death. Nucleic acid sensing pathways detect cytoplasmic DNA and RNA to stimulate downstream pathways. Cytoplasmic DNA activates the Cyclic GMP–AMP synthase (cGAS) to produce cyclic GMP–AMP (cGAMP) that activates the stimulator of interferon genes (STING) pathway, leading to type I interferon (IFN) production. Radiotherapy-induced type I interferon (IFN) can induce RNA sensor activation through RNA polymerase III conversion of DNA to double-stranded RNA (dsRNA), radiotherapy-induced small non-coding RNA (sncRNA) or STAT1-induced dsRNA synthesis from endogenous retroviral elements (ERVs). These activate (RIG-I)-like receptors (RLRs), melanoma differentiation-associated protein 5 (MDA5) and retinoic acid-inducible gene-I (RIG-I), which also drives pro-inflammatory signalling through type I IFN and pro-inflammatory cytokine production. Toll-like receptors (TLRs) can recognise damage-associated molecular patterns (DAMPs) of single-stranded RNA (ssRNA), dsRNA or unmethylated CpG DNA in intracellular compartments such as endosomes, to lead to activation of nuclear factor-κB (NF-κB), mitogen-activated protein kinase (MAPKs) and interferon regulatory factors (IRFs). DNA damage repair mechanisms of single- (SSB) and double-strand breaks (DSB) are often upregulated by cancer cells to avoid cell cycle arrest or death. Inhibitors of DNA damage repair components, such as ataxia telangiectasia- mutated (ATM), ataxia telangiectasia and Rad3-related protein (ATR), DNA-dependent protein kinase, catalytic subunit (DNA-PKcs), poly(ADP- ribose) polymerase 1 (PARP-1) and Wee1 (Wee1-like protein kinase) function to propel the cell through the cell cycle, despite the presence of unrepaired damage, leading to accumulation of cytosolic DNA. This leads to cross-talk with the nucleic acid sensing pathway via activation of the cGAS-STING pathway and dsRNA stress pathway via promotion of ERV expression. These two pathways, through positive and negative cross-talk, shape the radiotherapy-induced DDR response that feeds into anti-tumour immune effects, including recruitment of tumour-infiltrating CD8+ T-cells, natural killer (NK) cells and CD11b+ innate immune cells, such as macrophages and neutrophils. Maturation and activation of dendritic cells (DCs) is increased, including DC cross-presentation of tumour-associated antigens to naive T-cells, which can become activated leading to T-cell-mediated cytotoxic-killing of cancer cells. Furthermore, the immunosuppressive effects of myeloid-derived suppressor cells (MDSCs) and regulatory T-cells (Tregs) can be reversed and macrophages can be repolarised from M2 to an M1 pro-inflammatory phenotype. Chk, checkpoint kinase; IKKi, inducible IκB kinase; IL, interleukin; IRAK, Interleukin 1 Receptor-Associated Kinase; MAVS, mitochondrial anti-viral-signalling protein; MyD88, Myeloid differentiation primary response 88; TBK, TANK-binding kinase 1; TNFα, tumour necrosis factor alpha; TRAF3, TNF Receptor-Associated Factor 3. Created with BioRender.com.
TLR7 and TLR8 detect guanosine or uridine-rich single-stranded RNA and their activation can directly induce MDSCs to lose their immunosuppressive function and acquire an APC-like phenotype that can induce tumour-specific T-cell responses (262), convert MDSCs to M1-like macrophages (263), activate NK cells (264–267) and revert Treg immunosuppressive effects (268). The imidazoquinolines are synthetic agonists for TLR7/8 of which topical imiquimod is the most extensively studied as well as being currently licensed for the treatment of superficial basal cell carcinoma (269). A preclinical study in breast cancer has investigated topical imiquimod in combination with radiotherapy and low-dose cyclophosphamide (94), and found that this triple combination had synergistic anti-cancer effects at both irradiated and unirradiated (abscopal) sites. Long-term surviving mice were able to reject tumour rechallenge, likely due to the establishment of anti-tumour immunological memory (94) (Table 1). A phase 2 clinical trial in metastatic breast cancer testing the efficacy of this triple therapy has finished recruiting (NCT01421017) (Table 2). Synergistic effects of subcutaneous TLR7 agonist and radiotherapy have also been observed in a preclinical model of melanoma (95) (Table 1). The efficacy of systemic delivery of the TLR7 agonists R848 (96), DSR-6434 (97), DSR-29133 (98) and 3M-011 (99), in combination with radiotherapy, has been explored in the treatment of several preclinical models of solid cancers. Dual therapy works synergistically to enhance tumour control, generate tumour-antigen-specific T-cells, suppress tumour growth (96–99) after rechallenge in long-term surviving mice (97) (98) and reduce the formation of distant metastases (99). Systemically-administered TLR7/8 agonists are not currently being investigated in a clinical setting; notably a phase I clinical trial investigating systemic TLR7 agonist ANA975 in chronic hepatitis C virus (270) had to be withdrawn due to excessive toxicity in extended preclinical studies (271), highlighting the need for caution when delivering systemic TLR7/8 agonists, especially in combination with radiotherapy (236).
Finally, TLR9 is expressed on APCs and B-cells and senses unmethylated CpG oligonucleotides present in bacterial and viral DNA (272–274). Again, TLR9 agonism can lead to activation and maturation of DCs, cytokine release from T helper type 1 (Th1) cells, differentiation of MDSC towards an M1 phenotype (275–279) and inhibition of Treg immunosuppressive effects (280). Several preclinical studies (281–284) have shown that TLR9 agonists can lead to anti-tumour effects in an NK- and CD8 T-cell-dependent manner (285). Preclinical studies showed enhanced tumour control in combination with radiotherapy in a model of murine fibrosarcoma and lung cancer (100–103), and induction of immunological memory by mice rejecting tumour rechallenge (102). The synergistic effects of radiotherapy and TLR9 agonists are dependent on a competent host immune system (102). Early clinical studies, although in small patient numbers, have tested TRL9 agonists in combination with radiotherapy. CpG-enriched oligodeoxynucleotide delivered intratumorally in combination with radiotherapy, 4 Gy in two fractions, led to overall objective response rates of 27% in the non-treated lesions of patients with relapsed low-grade B cell lymphoma (286).
RIG-I and melanoma differentiation-associated gene 5 (MDA5) are collectively (RIG-I)-like receptors (RLR) which detect cytosolic RNA and are a key PRR in anti-viral responses (287). RIG-I preferentially binds to short (>10 bp) dsRNAs whereas MDA5 detects long accessible dsRNAs (>2 kbp) (288, 289), and downstream signalling of either activates IRF3 and NF-κB pathways to induce type I IFN and other inflammatory cytokines. In the context of DNA damage, RIG-I interacts with X-ray repair cross complementing 4 (XRCC4) to impede formation of the XRCC4/LIG4 (DNA ligase 4)/XLF (XRCC4-like factor) at DSBs. High expression of RIG-I compromises DNA repair and sensitises cancer cells to irradiation treatment. In contrast, depletion of RIG-I renders cells resistant to irradiation in vitro and in vivo (290).
In the anti-tumour response, there is increasing evidence that RLR activation in various cancer models by RNA ligands can induce cancer cell apoptosis in a type I IFN-dependent (291), or -independent manner (292, 293). RIG-I signalling can induce ICD of ovarian and pancreatic cancer cells in vivo by systemic activation of DCs, NK cells and CD8+ T cells (294, 295). In a pancreatic cancer model, tumour-derived type I IFN activates DCs and CD8α+ DCs engulf apoptotic tumour material and cross-present tumour-associated antigen to naïve CD8+ T cells (296). RIG-I may also inhibit tumour growth indirectly through regulation of tumour hypoxia (297) and the gut microbiota (298). The efficacy of anti-cancer treatments such as radiotherapy and many chemotherapy agents has also been shown to depend on the RLR pathway through endogenous non-coding RNAs, and depletion of RIG-I in human tumours confers treatment resistance (299).
Harnessing the RLR-pathway through RLR agonists is an attractive therapeutic target and several RLR mimetics or agonists have been developed which have shown promise in preclinical studies. For example, a unique RIG-I agonist in the form of RNA stem-loop of 14 bp (SLR14), when delivered intratumorally, significantly inhibited B16 tumour growth locally and systemically in bilateral and tumour metastasis models, with cured mice developing immunological memory (300). SLR14 was mainly taken up by CD11b+ myeloid cells in the TME leading to subsequent increase in the number of CD8+ T lymphocytes, NK cells, and CD11b+ cells in SLR14-treated tumours (300). MK4621 (or RGT100), a synthetic RNA oligonucleotide RIG-I activator is currently in phase 1 clinical trials for the treatment of advanced/metastatic solid tumours (NCT03739138).
Combining RLR agonists and radiotherapy is an attractive strategy to activate multiple DDR pathways via cytosolic RNA sensing and radiotherapy-induced cytosolic DNA/DNA damage detection. In vitro, an RLR agonist Poly(I:C)-HMW (High Molecular Weight)/LyoVec™ [Poly(I:C)-HMW] sensitised in vitro human lung cancer cells to Fas ligand (FasL)-induced apoptosis by radiotherapy (301). In vivo intratumoral cytoplasmic delivery of the dsRNA mimic poly(I:C) by polyethylenimine (PEI), prior to diffusing alpha-emitting radiation therapy (DaRT), resulted in synergistic tumour and metastatic disease control. Furthermore, immunological memory was demonstrated, whereby splenocytes from treated mice adoptively transferred to naïve tumour-bearing mice, resulted in delayed tumour development and protection from rechallenge (104). Combining RLR-agonists and radiotherapy has not yet been translated into clinical practice and to the best of our knowledge there are no clinical trials investigating this combination.
We have discussed in detail the various druggable targets related to the DDR pathway, in particular agonists of the nucleic acid sensing pathways and inhibitors of DNA damage repair mechanisms. Next, this review will explore the clinical challenges and implications of combining radiotherapy with DDR-targeted agents.
Conventional chemotherapy has historically been used in the backbone of radical chemoradiation (CRT) in many locally advanced tumours such as rectal, cervical and head and neck cancers. Chemotherapy agents traditionally used as radiosensitisers include platin salts (e.g. Cisplatin, Carboplatin) or fluoropyrimidines (e.g. 5-fluorouracil or its prodrug Capecitabine), which trigger cell death by instigating DNA damage (302). Chemotherapy-induced cell death can lead to DNA leakage into the cytosol and trigger intrinsic STING pathway stimulation and activation of the immune system (303). Some may argue that investigating novel DDR-pathway specific agents is redundant given that chemotherapy may exert its anti-cancer effects partly by stimulating the innate immune system (303). However, it is recognised that chemotherapy (304), radiotherapy (305) or concomitant CRT (306) in various cancers can result in lymphocyte depletion which can potentially negate a sustained effective anti-tumour response. Lymphocyte depletion post-treatment is a poor prognostic factor in patients who have undergone radiotherapy for Stage III lung cancer (305) or CRT for newly diagnosed glioblastoma (306). Furthermore, defects in DDR signalling may contribute to chemoresistance in some cancer types (303) and, as such, development of specific DDR-targeting agents remains an important avenue for research.
The anti-tumour innate immunity initiated by radiotherapy and DDR inhibitors is likely to be complementary to the effect of immune checkpoint inhibitors (ICIs), which can sustain and maintain the adaptive arm of the anti-tumour immune response. For example, preclinical studies in lymphoma have shown that treatment with Flt3L, radiation and poly(ICLC) led to PD-L1 upregulation in both tumour cells and intratumoural DCs, and that the further addition of anti-PD-1 antibody led to improved local and systemic tumour control (258). There is an increasing number of early phase clinical studies investigating the addition of ICI with radiotherapy and DDR-targeted agents, such as TLR agonists (NCT03007732, NCT04050085, NCT03507699, NCT02254772) and DNA-PK inhibitors (NCT04068194, NCT03724890, NCT04576091, NCT03923270).
Clinical response to ICIs is typically predicted by tumour mutational burden and neoantigen load (307, 308). Preclinical data suggests that radiotherapy and DDR inhibitors may replicate the phenotype of high mutational and neoantigen burden and rationally direct therapeutic combinations with ICIs. However, the caveat is that radiotherapy-induced subclonal neoantigens may translate into poorer responses to ICI in some tumour types (307). The combination of radiotherapy and anti-CTLA-4 increases the diversity of TIL TCR repertoire, leading to increased tumour control in vivo; however, these tumours remain dominated by a small number of high-frequency T-cell clones (30, 32). It is still unknown whether it is more important to have an immune response against pre-existing tumour antigens or new radiotherapy-generated tumour antigens. As we await the results of the ongoing triple combination treatments (RT + DDR agents + ICI) in early phase clinical trials, further work is needed to investigate such combinations in the context of creation of subclonal neoantigens.
A key principle of radiation oncology is that the dose delivered to the tumour is limited by the surrounding normal tissue organs-at-risk (OARs). Hence, strategies in designing clinical trials arguably should have some basis for a selective effect of any combination drug on the tumour (309). Preclinical studies in mouse models, for example, show that M3814, a DNA-PK inhibitor given with radiotherapy, shows marked improvement in tumour control (310). However, when translated into clinical practice, a clinical trial of M3814 with radiation (NCT02516813) reported enhanced normal tissue reactions including dysphagia, prolonged stomatitis and radiation dermatitis (311). Pre-clinical models are also severely limited in predicting long-term treatment toxicity in humans.
A further therapeutic challenge of using DDR pathway agents with radiotherapy is that there may be high variability in drug pharmacokinetics leading to varying degrees of radiosensitisation between tumour versus normal tissues, which makes it difficult to predict the therapeutic index for each individual patient (309). Therefore, unless there is a clear mechanism for tumour-specific radiosensitisation, clinical trials combining DNA repair inhibitors and radiotherapy may be severely compromised by unacceptable toxicity. Potential solutions may be an intratumoural route of drug delivery, as taken by certain trials of TLR9 agonists and STING agonists (Table 2), or conditional drug activation, such as with a hypoxia-activated DNA-PK inhibitor (312, 313). Increased knowledge of biomarkers and access to routine tumour profiling may guide the best selection of which DDR agent to use in a particular cancer subtype, for example PARP-inhibitors in BRCA-mutant or ATM/ATR inhibitors in p53-mutant tumours. Advances in radiotherapy delivery techniques using stereotactic techniques to irradiate tumour volumes highly selectively is a further way to reduce off-target combination effects of DDR-targeting agents. For example, a Phase I trial in recurrent head and neck squamous cell carcinoma investigating combining an ATR kinase inhibitor BAY1895344 with pembrolizumab and stereotactic body radiotherapy (SBRT) (NCT04576091) represents one such promising approach.
In some occasions, radiotherapy can result in the regression of disease outside of the irradiated field in the so-called abscopal effect, which is thought to be immune-mediated (314). Inducing such systemic anti-tumour immune responses is likely highly dependent on radiotherapy dose and fractionation and these factors, therefore, need to be an important consideration in combination treatments with DDR agents and/or ICI (315).
Irradiation of regional lymph nodes in cancer treatment is common practice either with high doses in macroscopic disease or prophylactic lower doses, if lymph nodes are deemed to be at risk of harbouring micrometastatic disease. This approach has recently become more controversial given that we know these lymphoid organs have an important role in DC-mediated T-cell priming, activation and subsequent tumour infiltration following radiotherapy (31). Routine irradiation of regional lymph nodes may potentially deplete important immune cells and have a detrimental effect on the anti-tumour immune response (316).
The biological effects of radiotherapy, such as DNA damage complexity, depend on radiation quality and degree of linear energy transfer (LET). High LET radiation (e.g. protons, carbon ions, α-particle-emitting radionuclides) can differentially affect cell fate (317). For example, protons mainly induce apoptosis not necrosis which may reduce the leakage of nucleic acids into the cytoplasm to serve as danger signals, hence impacting on the innate immune response (317). The effects of radiotherapy were previously thought to be mainly due to nuclear DNA damage and their repair mechanisms. However, the outcome of irradiation depends also on the activation and regulation of other organelles that determine cellular metabolism, survival and immunological responses such as the mitochondria (318). Recent studies have shown that mitochondrial DNA DSBs activate a type I IFN response and mitochondrial RNA release into the cytoplasm triggers a RIG-I-MAVS-dependent immune response (319, 320). Low-dose versus high-dose radiation, as well as radiation quality, can also have different effects on mitochondria-mediated innate and adaptive immune responses (318). Interestingly, high LET particle radiotherapy which are more efficient in ROS production is reportedly more likely to lead to mitochondria-mediated apoptosis and anti-tumour immune responses (318, 321).
The most appropriate scheduling of DDR agents with respect to radiotherapy also needs to be investigated further. For example, a study investigating a novel TLR7/8 agonist in combination with radiotherapy showed that the optimal combination efficacy required the drug to be administered concurrently at the start rather than end of radiotherapy (98). However, another investigation of a TLR9 agonist showed maximum synergy was observed when mice received the agent three days after radiotherapy in the adjuvant setting (102). Clinical trials investigating TLR3 agonists used in the concurrent or adjuvant setting with respect to radiotherapy both showed activity (258, 260, 261, 322). More preclinical studies investigating the biological basis of optimal scheduling are required, although it may be that optimal scheduling may ultimately be both treatment- and tumour-specific.
Our increasing knowledge of the mechanisms of how radiotherapy-induced DDR interacts intimately with the host immune response is critical to the discovery of novel therapeutic targets and effective strategies against cancer. DDR-targeted agents are an exciting avenue for overcoming radioresistance and improving patient outcomes through enhancement of anti-tumour immunity. Understanding the molecular mechanisms and immunological effects of these DDR agents, through rigorous preclinical testing and translational analyses, is key to guiding rational clinical trial design in terms of drug route of delivery, schedules and choice of additional combination treatments, such as chemotherapy or immunotherapy.
CC conceptualised this review and drafted the manuscript. AR, EP, MP, AM and KH contributed to the writing and critical revision of this article. All authors contributed to the article and approved the submitted version.
This work was supported by the Wellcome Trust, ICR/RM NIHR Biomedical Research Centre, The Institute of Cancer Research/Royal Marsden Hospital Centre for Translational Immunotherapy, CRUK Head and Neck Programme Grant (C7224/A23275) and ICR/RM CRUK RadNet Centre of Excellence (C7224/A28724).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer (2005) 104(6):1129–37. doi: 10.1002/cncr.21324
2. Verheij M. Clinical biomarkers and imaging for radiotherapy-induced cell death. Cancer Metastasis Rev (2008) 27(3):471–80. doi: 10.1007/s10555-008-9131-1
3. Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res (2008) 68(5):1485–94. doi: 10.1158/0008-5472.CAN-07-0562
4. Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci (2016) 17(1). doi: 10.3390/ijms17010102
5. Withers HR. The four r’s of radiotherapy. In: Lett JTAH, editor. Advances in radiation biology. New York: Academic Press (1975).
6. Steel GG, McMillan TJ, Peacock JH. The 5Rs of radiobiology. Int J Radiat Biol (1989) 56(6):1045–8. doi: 10.1080/09553008914552491
7. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology (2014) 3:e28518. doi: 10.4161/onci.28518
8. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer (2020) 20(4):203–17. doi: 10.1038/s41568-020-0246-1
9. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
10. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
11. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature (2009) 461(7267):1071–8. doi: 10.1038/nature08467
12. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
13. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12(12):860–75. doi: 10.1038/nrc3380
14. Schaue D, McBride WH. Links between innate immunity and normal tissue radiobiology. Radiat Res (2010) 173(4):406–17. doi: 10.1667/RR1931.1
15. Zhou J, Wang G, Chen Y, Wang H, Hua Y, Cai Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J Cell Mol Med (2019) 23(8):4854–65. doi: 10.1111/jcmm.14356
16. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38(4):729–41. doi: 10.1016/j.immuni.2013.03.003
17. Ma Y, Adjemian S, Yang H, Catani JP, Hannani D, Martins I, et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology (2013) 2(6):e24568. doi: 10.4161/onci.24568
18. Kono K, Mimura K. Immunogenic tumor cell death induced by chemoradiotherapy in a clinical setting. Oncoimmunology (2013) 2(1):e22197. doi: 10.4161/onci.22197
19. Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget (2014) 5(2):403–16. doi: 10.18632/oncotarget.1719
20. Lhuillier C, Rudqvist NP, Elemento O, Formenti SC, Demaria S. Radiation therapy and anti-tumor immunity: exposing immunogenic mutations to the immune system. Genome Med (2019) 11(1):40. doi: 10.1186/s13073-019-0653-7
21. Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol (2015) 6:588. doi: 10.3389/fimmu.2015.00588
22. Sprung CN, Forrester HB, Siva S, Martin OA. Immunological markers that predict radiation toxicity. Cancer Lett (2015) 368(2):191–7. doi: 10.1016/j.canlet.2015.01.045
23. Ozsoy HZ, Sivasubramanian N, Wieder ED, Pedersen S, Mann DL. Oxidative stress promotes ligand-independent and enhanced ligand-dependent tumor necrosis factor receptor signaling. J Biol Chem (2008) 283(34):23419–28. doi: 10.1074/jbc.M802967200
24. Schaue D, Xie MW, Ratikan JA, McBride WH. Regulatory T cells in radiotherapeutic responses. Front Oncol (2012) 2:90. doi: 10.3389/fonc.2012.00090
25. Jia S, Ge S, Fan X, Leong KW, Ruan J. Promoting reactive oxygen species generation: a key strategy in nanosensitizer-mediated radiotherapy. Nanomed (Lond) (2021) 16(9):759–78. doi: 10.2217/nnm-2020-0448
26. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer (2015) 15(7):409–25. doi: 10.1038/nrc3958
27. Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol (2008) 181(5):3099–107. doi: 10.4049/jimmunol.181.5.3099
28. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi: 10.1038/32588
29. Wilkins AC, Patin EC, Harrington KJ, Melcher AA. The immunological consequences of radiation-induced DNA damage. J Pathol (2019) 247(5):606–14. doi: 10.1002/path.5232
30. Rudqvist NP, Pilones KA, Lhuillier C, Wennerberg E, Sidhom JW, Emerson RO, et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T cells. Cancer Immunol Res (2018) 6(2):139–50. doi: 10.1158/2326-6066.CIR-17-0134
31. Dovedi SJ, Cheadle EJ, Popple AL, Poon E, Morrow M, Stewart R, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin Cancer Res (2017) 23(18):5514–26. doi: 10.1158/1078-0432.CCR-16-1673
32. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520(7547):373–7. doi: 10.1038/nature14292
33. Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol (2004) 14(6):433–9. doi: 10.1016/j.semcancer.2004.06.006
34. Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest (2007) 117(5):1175–83. doi: 10.1172/JCI31537
35. Li Q, Withoff S, Verma IM. Inflammation-associated cancer: NF-kappaB is the lynchpin. Trends Immunol (2005) 26(6):318–25. doi: 10.1016/j.it.2005.04.003
36. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
37. Hiniker SM, Chen DS, Reddy S, Chang DT, Jones JC, Mollick JA, et al. A systemic complete response of metastatic melanoma to local radiation and immunotherapy. Transl Oncol (2012) 5(6):404–7. doi: 10.1593/tlo.12280
38. Harris TJ, Hipkiss EL, Borzillary S, Wada S, Grosso JF, Yen HR, et al. Radiotherapy augments the immune response to prostate cancer in a time-dependent manner. Prostate (2008) 68(12):1319–29. doi: 10.1002/pros.20794
39. Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol (2014) 5:489. doi: 10.3389/fimmu.2014.00489
40. Kachikwu EL, Iwamoto KS, Liao YP, DeMarco JJ, Agazaryan N, Economou JS, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys (2011) 81(4):1128–35. doi: 10.1016/j.ijrobp.2010.09.034
41. Qu Y, Jin S, Zhang A, Zhang B, Shi X, Wang J, et al. Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat Res (2010) 173(2):148–57. doi: 10.1667/RR0978.1
42. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res (2014) 74(19):5458–68. doi: 10.1158/0008-5472.CAN-14-1258
43. Rodriguez-Ruiz ME, Rodriguez I, Garasa S, Barbes B, Solorzano JL, Perez-Gracia JL, et al. Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mAbs and are dependent on CD8 T cells and crosspriming. Cancer Res (2016) 76(20):5994–6005. doi: 10.1158/0008-5472.CAN-16-0549
44. Hsieh RC, Krishnan S, Wu RC, Boda AR, Liu A, Winkler M, et al. ATR-mediated CD47 and PD-L1 up-regulation restricts radiotherapy-induced immune priming and abscopal responses in colorectal cancer. Sci Immunol (2022) 7(72):eabl9330. doi: 10.1126/sciimmunol.abl9330
45. Mantovani A, Romero P, Palucka AK, Marincola FM. Tumour immunity: effector response to tumour and role of the microenvironment. Lancet (2008) 371(9614):771–83. doi: 10.1016/S0140-6736(08)60241-X
46. Finkelstein SE, Iclozan C, Bui MM, Cotter MJ, Ramakrishnan R, Ahmed J, et al. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo-adjuvant treatment of high-risk soft tissue sarcoma patients. Int J Radiat Oncol Biol Phys (2012) 82(2):924–32. doi: 10.1016/j.ijrobp.2010.12.068
47. Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, et al. Stereotactic radiotherapy increases functionally suppressive regulatory T cells in the tumor microenvironment. Cancer Immunol Res (2017) 5(11):992–1004. doi: 10.1158/2326-6066.CIR-17-0040
48. Hellevik T, Pettersen I, Berg V, Winberg JO, Moe BT, Bartnes K, et al. Cancer-associated fibroblasts from human NSCLC survive ablative doses of radiation but their invasive capacity is reduced. Radiat Oncol (2012) 7:59. doi: 10.1186/1748-717X-7-59
49. Mantoni TS, Lunardi S, Al-Assar O, Masamune A, Brunner TB. Pancreatic stellate cells radioprotect pancreatic cancer cells through β1-integrin signaling. Cancer Res (2011) 71(10):3453–8. doi: 10.1158/0008-5472.CAN-10-1633
50. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-Dependent manner. Cancer Cell (2010) 17(2):135–47. doi: 10.1016/j.ccr.2009.12.041
51. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science (2010) 330(6005):827–30. doi: 10.1126/science.1195300
52. Kugeratski FG, Atkinson SJ, Neilson LJ, Lilla S, Knight JRP, Serneels J, et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci Signal (2019) 12(567):1–33. doi: 10.1126/scisignal.aan8247
53. Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PloS One (2009) 4(11):e7965. doi: 10.1371/journal.pone.0007965
54. Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol (2019) 10:925. doi: 10.3389/fimmu.2019.00925
55. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene (2010) 29(39):5346–58. doi: 10.1038/onc.2010.292
56. Montalbán Del Barrio I, Penski C, Schlahsa L, Stein RG, Diessner J, Wöckel A, et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages - a self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J Immunother Cancer (2016) 4:49. doi: 10.1186/s40425-016-0154-9
57. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology (2017) 6(6):e1320011. doi: 10.1080/2162402X.2017.1320011
58. d'Almeida SM, Kauffenstein G, Roy C, Basset L, Papargyris L, Henrion D, et al. The ecto-ATPDase CD39 is involved in the acquisition of the immunoregulatory phenotype by m-CSF-macrophages and ovarian cancer tumor-associated macrophages: Regulatory role of IL-27. Oncoimmunology (2016) 5(7):e1178025. doi: 10.1080/2162402X.2016.1178025
59. Hilchey SP, Kobie JJ, Cochran MR, Secor-Socha S, Wang JC, Hyrien O, et al. Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. J Immunol (2009) 183(10):6157–66. doi: 10.4049/jimmunol.0900475
60. Gourdin N, Bossennec M, Rodriguez C, Vigano S, Machon C, Jandus C, et al. Autocrine adenosine regulates tumor polyfunctional CD73(+)CD4(+) effector T cells devoid of immune checkpoints. Cancer Res (2018) 78(13):3604–18. doi: 10.1158/0008-5472.CAN-17-2405
61. Canale FP, Ramello MC, Núñez N, Araujo Furlan CL, Bossio SN, Gorosito Serrán M, et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8(+) T cells. Cancer Res (2018) 78(1):115–28. doi: 10.1158/0008-5472.CAN-16-2684
62. Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer (2005) 5(7):516–25. doi: 10.1038/nrc1650
63. Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med (2011) 17(7):860–6. doi: 10.1038/nm.2385
64. Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol (2015) 31:28–35. doi: 10.1016/j.semcancer.2014.07.001
65. Lagadec C, Vlashi E, Della Donna L, Meng Y, Dekmezian C, Kim K, et al. Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res (2010) 12(1):R13. doi: 10.1186/bcr2479
66. Huang RX, Zhou PK. DNA Damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther (2020) 5(1):60. doi: 10.1038/s41392-020-0150-x
67. Mladenov E, Magin S, Soni A, Iliakis G. DNA Double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol (2013) 3:113. doi: 10.3389/fonc.2013.00113
68. Iliakis G, Mladenov E, Mladenova V. Necessities in the processing of DNA double strand breaks and their effects on genomic instability and cancer. Cancers (Basel) (2019) 11(11). doi: 10.3390/cancers11111671
69. Weber AM, Ryan AJ. ATM And ATR as therapeutic targets in cancer. Pharmacol Ther (2015) 149:124–38. doi: 10.1016/j.pharmthera.2014.12.001
70. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature (2005) 436(7054):1186–90. doi: 10.1038/nature03884
71. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science (2003) 300(5625):1542–8. doi: 10.1126/science.1083430
72. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, et al. ATM And ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science (2007) 316(5828):1160–6. doi: 10.1126/science.1140321
73. Wang L, Yang L, Wang C, Zhao W, Ju Z, Zhang W, et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J Clin Invest (2020) 130(11):5951–66. doi: 10.1172/JCI130445
74. Savitsky K, Platzer M, Uziel T, Gilad S, Sartiel A, Rosenthal A, et al. Ataxia-telangiectasia: structural diversity of untranslated sequences suggests complex post-transcriptional regulation of ATM gene expression. Nucleic Acids Res (1997) 25(9):1678–84. doi: 10.1093/nar/25.9.1678
75. Taylor AM, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, et al. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature (1975) 258(5534):427–9. doi: 10.1038/258427a0
76. Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, Wakeham A, et al. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol (2002) 22(18):6521–32. doi: 10.1128/MCB.22.18.6521-6532.2002
77. Powell SN, DeFrank JS, Connell P, Eogan M, Preffer F, Dombkowski D, et al. Differential sensitivity of p53(-) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Res (1995) 55(8):1643–8. doi: 10.1016/0360-3016(95)97825-L
78. Price BD, Youmell MB. The phosphatidylinositol 3-kinase inhibitor wortmannin sensitizes murine fibroblasts and human tumor cells to radiation and blocks induction of p53 following DNA damage. Cancer Res (1996) 56(2):246–50.
79. Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res (2008) 68(18):7466–74. doi: 10.1158/0008-5472.CAN-08-0763
80. Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res (2004) 64(24):9152–9. doi: 10.1158/0008-5472.CAN-04-2727
81. Golding SE, Rosenberg E, Adams BR, Wignarajah S, Beckta JM, O'Connor MJ., et al. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle (2012) 11(6):1167–73. doi: 10.4161/cc.11.6.19576
82. Batey MA, Zhao Y, Kyle S, Richardson C, Slade A, Martin NM, et al. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol Cancer Ther (2013) 12(6):959–67. doi: 10.1158/1535-7163.MCT-12-0707
83. Vecchio D, Daga A, Carra E, Marubbi D, Raso A, Mascelli S, et al. Pharmacokinetics, pharmacodynamics and efficacy on pediatric tumors of the glioma radiosensitizer KU60019. Int J Cancer (2015) 136(6):1445–57. doi: 10.1002/ijc.29121
84. Zhang T, Shen Y, Chen Y, Hsieh JT, Kong Z. The ATM inhibitor KU55933 sensitizes radioresistant bladder cancer cells with DAB2IP gene defect. Int J Radiat Biol (2015) 91(4):368–78. doi: 10.3109/09553002.2015.1001531
85. Vecchio D, Daga A, Carra E, Marubbi D, Baio G, Neumaier CE, et al. Predictability, efficacy and safety of radiosensitization of glioblastoma-initiating cells by the ATM inhibitor KU-60019. Int J Cancer (2014) 135(2):479–91. doi: 10.1002/ijc.28680
86. Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res (2019) 79(15):3940–51. doi: 10.1158/0008-5472.CAN-19-0761
87. Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J Clin Invest (2018) 128(9):3926–40. doi: 10.1172/JCI96519
88. Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, et al. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res (2019) 25(11):3392–403. doi: 10.1158/1078-0432.CCR-18-1821
89. Sheng H, Huang Y, Xiao Y, Zhu Z, Shen M, Zhou P, et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J Immunother Cancer (2020) 8(1). doi: 10.1136/jitc-2019-000340
90. Patin EC, Dillon MT, Nenclares P, Grove L, Soliman H, Leslie I, et al. Harnessing radiotherapy-induced NK-cell activity by combining DNA damage-response inhibition and immune checkpoint blockade. J Immunother Cancer (2022) 10(3). doi: 10.1136/jitc-2021-004306
91. Wang W, McMillan MT, Zhao X, Wang Z, Jiang L, Karnak D, et al. DNA-PK inhibition and radiation promote anti-tumoral immunity through RNA polymerase III in pancreatic cancer. Mol Cancer Res (2022) 20(7):1137–1150. doi: 10.1158/1541-7786.MCR-21-0725
92. Patel P, Sun L, Robbins Y, Clavijo PE, Friedman J, Silvin C, et al. Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. Oncoimmunology (2019) 8(11):e1638207. doi: 10.1080/2162402X.2019.1638207
93. Baird JR, Friedman D, Cottam B, Dubensky TW Jr, Kanne DB, Bambina S, et al. Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res (2016) 76(1):50–61. doi: 10.1158/0008-5472.CAN-14-3619
94. Dewan MZ, Vanpouille-Box C, Kawashima N, DiNapoli S, Babb JS, Formenti SC, et al. Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clin Cancer Res (2012) 18(24):6668–78. doi: 10.1158/1078-0432.CCR-12-0984
95. Cho JH, Lee HJ, Ko HJ, Yoon BI, Choe J, Kim KC, et al. The TLR7 agonist imiquimod induces anti-cancer effects via autophagic cell death and enhances anti-tumoral and systemic immunity during radiotherapy for melanoma. Oncotarget (2017) 8(15):24932–48. doi: 10.18632/oncotarget.15326
96. Dovedi SJ, Melis MH, Wilkinson RW, Adlard AL, Stratford IJ, Honeychurch J, et al. Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood (2013) 121(2):251–9. doi: 10.1182/blood-2012-05-432393
97. Adlard AL, Dovedi SJ, Telfer BA, Koga-Yamakawa E, Pollard C, Honeychurch J, et al. A novel systemically administered toll-like receptor 7 agonist potentiates the effect of ionizing radiation in murine solid tumor models. Int J Cancer (2014) 135(4):820–9. doi: 10.1002/ijc.28711
98. Dovedi SJ, Adlard AL, Ota Y, Murata M, Sugaru E, Koga-Yamakawa E, et al. Intravenous administration of the selective toll-like receptor 7 agonist DSR-29133 leads to anti-tumor efficacy in murine solid tumor models which can be potentiated by combination with fractionated radiotherapy. Oncotarget (2016) 7(13):17035–46. doi: 10.18632/oncotarget.7928
99. Schölch S, Rauber C, Tietz A, Rahbari NN, Bork U, Schmidt T, et al. Radiotherapy combined with TLR7/8 activation induces strong immune responses against gastrointestinal tumors. Oncotarget (2015) 6(7):4663–76. doi: 10.18632/oncotarget.3081
100. Mason KA, Ariga H, Neal R, Valdecanas D, Hunter N, Krieg AM, et al. Targeting toll-like receptor 9 with CpG oligodeoxynucleotides enhances tumor response to fractionated radiotherapy. Clin Cancer Res (2005) 11(1):361–9. doi: 10.1158/1078-0432.361.11.1
101. Milas L, Mason KA, Ariga H, Hunter N, Neal R, Valdecanas D, et al. CpG oligodeoxynucleotide enhances tumor response to radiation. Cancer Res (2004) 64(15):5074–7. doi: 10.1158/0008-5472.CAN-04-0926
102. Meng Y, Carpentier AF, Chen L, Boisserie G, Simon JM, Mazeron JJ, et al. Successful combination of local CpG-ODN and radiotherapy in malignant glioma. Int J Cancer (2005) 116(6):992–7. doi: 10.1002/ijc.21131
103. Zhang H, Liu L, Yu D, Kandimalla ER, Sun HB, Agrawal S, et al. An in situ autologous tumor vaccination with combined radiation therapy and TLR9 agonist therapy. PloS One (2012) 7(5):e38111. doi: 10.1371/journal.pone.0038111
104. Domankevich V, Efrati M, Schmidt M, Glikson E, Mansour F, Shai A, et al. RIG-1-Like receptor activation synergizes with intratumoral alpha radiation to induce pancreatic tumor rejection, triple-negative breast metastases clearance, and antitumor immune memory in mice. Front Oncol (2020) 10:990. doi: 10.3389/fonc.2020.00990
105. Wold MS. Replication protein a: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem (1997) 66:61–92. doi: 10.1146/annurev.biochem.66.1.61
106. Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell (2006) 124(5):943–55. doi: 10.1016/j.cell.2005.12.041
107. Chen J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res (2000) 60(18):5037–9.
108. Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, et al. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev (2000) 14(23):2989–3002. doi: 10.1101/gad.851000
109. Errico A, Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol (2012) 47(3):222–35. doi: 10.3109/10409238.2012.655374
110. Sørensen CS, Syljuåsen RG, Falck J, Schroeder T, Rönnstrand L, Khanna KK, et al. Chk1 regulates the s phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell (2003) 3(3):247–58. doi: 10.1016/S1535-6108(03)00048-5
111. Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, et al. Chk1 mediates s and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem (2003) 278(24):21767–73. doi: 10.1074/jbc.M300229200
112. Graves PR, Lovly CM, Uy GL, Piwnica-Worms H. Localization of human Cdc25C is regulated both by nuclear export and 14-3-3 protein binding. Oncogene (2001) 20(15):1839–51. doi: 10.1038/sj.onc.1204259
113. Nishida H, Tatewaki N, Nakajima Y, Magara T, Ko KM, Hamamori Y, et al. Inhibition of ATR protein kinase activity by schisandrin b in DNA damage response. Nucleic Acids Res (2009) 37(17):5678–89. doi: 10.1093/nar/gkp593
114. Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer (2011) 105(3):372–81. doi: 10.1038/bjc.2011.243
115. Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther (2008) 7(7):1851–63. doi: 10.1158/1535-7163.MCT-08-0017
116. Charrier JD, Durrant SJ, Golec JM, Kay DP, Knegtel RM, MacCormick S, et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J Med Chem (2011) 54(7):2320–30. doi: 10.1021/jm101488z
117. Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis (2012) 3(12):e441. doi: 10.1038/cddis.2012.181
118. Foote KM, Blades K, Cronin A, Fillery S, Guichard SS, Hassall L, et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J Med Chem (2013) 56(5):2125–38. doi: 10.1021/jm301859s
119. Foote KM, Nissink JWM, McGuire T, Turner P, Guichard S, Yates JWT, et al. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. J Med Chem (2018) 61(22):9889–907. doi: 10.1021/acs.jmedchem.8b01187
120. Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, et al. Radiosensitization by the ATR inhibitor AZD6738 through generation of acentric micronuclei. Mol Cancer Ther (2017) 16(1):25–34. doi: 10.1158/1535-7163.MCT-16-0239
121. Konstantinidou G, Bey EA, Rabellino A, Schuster K, Maira MS, Gazdar AF, et al. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res (2009) 69(19):7644–52. doi: 10.1158/0008-5472.CAN-09-0823
122. Sun LL, Yang RY, Li CW, Chen MK, Shao B, Hsu JM, et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res (2018) 8(7):1307–16.
123. González Besteiro MA, Gottifredi V. The fork and the kinase: a DNA replication tale from a CHK1 perspective. Mutat Res Rev Mutat Res (2015) 763:168–80. doi: 10.1016/j.mrrev.2014.10.003
124. Chen Z, Xiao Z, Gu WZ, Xue J, Bui MH, Kovar P, et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int J Cancer (2006) 119(12):2784–94. doi: 10.1002/ijc.22198
125. Bunch RT, Eastman A. Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin Cancer Res (1996) 2(5):791–7.
126. King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, et al. LY2606368 causes replication catastrophe and antitumor effects through CHK1-dependent mechanisms. Mol Cancer Ther (2015) 14(9):2004–13. doi: 10.1158/1535-7163.MCT-14-1037
127. Güster JD, Weissleder SV, Busch CJ, Kriegs M, Petersen C, Knecht R, et al. The inhibition of PARP but not EGFR results in the radiosensitization of HPV/p16-positive HNSCC cell lines. Radiother Oncol (2014) 113(3):345–51. doi: 10.1016/j.radonc.2014.10.011
128. Suzuki M, Yamamori T, Bo T, Sakai Y, Inanami O. MK-8776, a novel Chk1 inhibitor, exhibits an improved radiosensitizing effect compared to UCN-01 by exacerbating radiation-induced aberrant mitosis. Transl Oncol (2017) 10(4):491–500. doi: 10.1016/j.tranon.2017.04.002
129. Barker HE, Patel R, McLaughlin M, Schick U, Zaidi S, Nutting CM, et al. CHK1 inhibition radiosensitizes head and neck cancers to paclitaxel-based chemoradiotherapy. Mol Cancer Ther (2016) 15(9):2042–54. doi: 10.1158/1535-7163.MCT-15-0998
130. Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther (2008) 7(9):2955–66. doi: 10.1158/1535-7163.MCT-08-0492
131. Weiss GJ, Donehower RC, Iyengar T, Ramanathan RK, Lewandowski K, Westin E, et al. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m(2) every 21 days in patients with cancer. Invest N Drugs (2013) 31(1):136–44. doi: 10.1007/s10637-012-9815-9
132. Karp JE, Thomas BM, Greer JM, Sorge C, Gore SD, Pratz KW, et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor sch 900776 in refractory acute leukemias. Clin Cancer Res (2012) 18(24):6723–31. doi: 10.1158/1078-0432.CCR-12-2442
133. Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol (2015) 33(9):1060–6. doi: 10.1200/JCO.2014.57.5027
134. Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, et al. Phase I study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J Clin Oncol (2016) 34(15):1764–71. doi: 10.1200/JCO.2015.64.5788
135. Seto T, Esaki T, Hirai F, Arita S, Nosaki K, Makiyama A, et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother Pharmacol (2013) 72(3):619–27. doi: 10.1007/s00280-013-2234-6
136. Lu H, Saha J, Beckmann PJ, Hendrickson EA, Davis AJ. DNA-PKcs promotes chromatin decondensation to facilitate initiation of the DNA damage response. Nucleic Acids Res (2019) 47(18):9467–79. doi: 10.1093/nar/gkz694
137. Douglas P, Gupta S, Morrice N, Meek K, Lees-Miller SP. DNA-PK-dependent phosphorylation of Ku70/80 is not required for non-homologous end joining. DNA Repair (Amst) (2005) 4(9):1006–18. doi: 10.1016/j.dnarep.2005.05.003
138. Liu Y, Zhang L, Liu Y, Sun C, Zhang H, Miao G, et al. DNA-PKcs deficiency inhibits glioblastoma cell-derived angiogenesis after ionizing radiation. J Cell Physiol (2015) 230(5):1094–103. doi: 10.1002/jcp.24841
139. Mamo T, Mladek AC, Shogren KL, Gustafson C, Gupta SK, Riester SM, et al. Inhibiting DNA-PK(CS) radiosensitizes human osteosarcoma cells. Biochem Biophys Res Commun (2017) 486(2):307–13. doi: 10.1016/j.bbrc.2017.03.033
140. Mohiuddin IS, Kang MH. DNA-PK as an emerging therapeutic target in cancer. Front Oncol (2019) 9:635. doi: 10.3389/fonc.2019.00635
141. Willoughby CE, Jiang Y, Thomas HD, Willmore E, Kyle S, Wittner A, et al. Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J Clin Invest (2020) 130(1):258–71. doi: 10.1172/JCI127483
142. Critchlow SE, Bowater RP, Jackson SP. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr Biol (1997) 7(8):588–98. doi: 10.1016/S0960-9822(06)00258-2
143. Davidson D, Amrein L, Panasci L, Aloyz R. Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front Pharmacol (2013) 4:5. doi: 10.3389/fphar.2013.00005
144. Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene (2005) 24(6):949–61. doi: 10.1038/sj.onc.1208332
145. Gurung RL, Lim HK, Venkatesan S, Lee PS, Hande MP. Targeting DNA-PKcs and telomerase in brain tumour cells. Mol Cancer (2014) 13:232. doi: 10.1186/1476-4598-13-232
146. Yang C, Wang Q, Liu X, Cheng X, Jiang X, Zhang Y, et al. NU7441 enhances the radiosensitivity of liver cancer cells. Cell Physiol Biochem (2016) 38(5):1897–905. doi: 10.1159/000445551
147. Shinohara ET, Geng L, Tan J, Chen H, Shir Y, Edwards E, et al. DNA-Dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res (2005) 65(12):4987–92. doi: 10.1158/0008-5472.CAN-04-4250
148. Durant S, Karran P. Vanillins–a novel family of DNA-PK inhibitors. Nucleic Acids Res (2003) 31(19):5501–12. doi: 10.1093/nar/gkg753
149. Take Y, Kumano M, Hamano Y, Fukatsu H, Teraoka H, Nishimura S, et al. OK-1035, a selective inhibitor of DNA-dependent protein kinase. Biochem Biophys Res Commun (1995) 215(1):41–7. doi: 10.1006/bbrc.1995.2431
150. Ismail IH, Mårtensson S, Moshinsky D, Rice A, Tang C, Howlett A, et al. SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene (2004) 23(4):873–82. doi: 10.1038/sj.onc.1207303
151. Zhang B, Huang B, Guan H, Zhang SM, Xu QZ, He XP, et al. Proteomic profiling revealed the functional networks associated with mitotic catastrophe of HepG2 hepatoma cells induced by 6-bromine-5-hydroxy-4-methoxybenzaldehyde. Toxicol Appl Pharmacol (2011) 252(3):307–17. doi: 10.1016/j.taap.2011.03.003
152. Davidson D, Grenier J, Martinez-Marignac V, Amrein L, Shawi M, Tokars M, et al. Effects of the novel DNA dependent protein kinase inhibitor, IC486241, on the DNA damage response to doxorubicin and cisplatin in breast cancer cells. Invest N Drugs (2012) 30(4):1736–42. doi: 10.1007/s10637-011-9678-5
153. Guo L, Liu X, Nishikawa K, Plunkett W. Inhibition of topoisomerase IIalpha and G2 cell cycle arrest by NK314, a novel benzo[c]phenanthridine currently in clinical trials. Mol Cancer Ther (2007) 6(5):1501–8. doi: 10.1158/1535-7163.MCT-06-0780
154. Wise HC, Iyer GV, Moore K, Temkin SM, Gordon S, Aghajanian C, et al. Activity of M3814, an oral DNA-PK inhibitor, in combination with topoisomerase II inhibitors in ovarian cancer models. Sci Rep (2019) 9(1):18882. doi: 10.1038/s41598-019-54796-6
155. Fok JHL, Ramos-Montoya A, Vazquez-Chantada M, Wijnhoven PWG, Follia V, James N, et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat Commun (2019) 10(1):5065. doi: 10.1038/s41467-019-12836-9
156. Khan AJ, Misenko SM, Thandoni A, Schiff D, Jhawar SR, Bunting SF, et al. VX-984 is a selective inhibitor of non-homologous end joining, with possible preferential activity in transformed cells. Oncotarget (2018) 9(40):25833–41. doi: 10.18632/oncotarget.25383
157. Lamb R, Fiorillo M, Chadwick A, Ozsvari B, Reeves KJ, Smith DL, et al. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: Implications for more effective radiation therapy. Oncotarget (2015) 6(16):14005–25. doi: 10.18632/oncotarget.4159
158. Dong J, Zhang T, Ren Y, Wang Z, Ling CC, He F, et al. Inhibiting DNA-PKcs in a non-homologous end-joining pathway in response to DNA double-strand breaks. Oncotarget (2017) 8(14):22662–73. doi: 10.18632/oncotarget.15153
159. Sun Q, Guo Y, Liu X, Czauderna F, Carr MI, Zenke FT, et al. Therapeutic implications of p53 status on cancer cell fate following exposure to ionizing radiation and the DNA-PK inhibitor M3814. Mol Cancer Res (2019) 17(12):2457–68. doi: 10.1158/1541-7786.MCR-19-0362
160. Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife (2012) 1:e00047. doi: 10.7554/eLife.00047
161. Burleigh K, Maltbaek JH, Cambier S, Green R, Gale M Jr, James RC, et al. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci Immunol (2020) 5(43). doi: 10.1126/sciimmunol.aba4219
162. Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, Kraus WL. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol Cell (2019) 75(6):1270–1285.e14. doi: 10.1016/j.molcel.2019.06.020
163. Maltseva EA, Rechkunova NI, Sukhanova MV, Lavrik OI. Poly(ADP-ribose) polymerase 1 modulates interaction of the nucleotide excision repair factor XPC-RAD23B with DNA via Poly(ADP-ribosyl)ation. J Biol Chem (2015) 290(36):21811–20. doi: 10.1074/jbc.M115.646638
164. Sodhi RK, Singh N, Jaggi AS. Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul Pharmacol (2010) 53(3-4):77–87. doi: 10.1016/j.vph.2010.06.003
165. Han Y, Jin F, Xie Y, Liu Y, Hu S, Liu XD, et al. DNA−PKcs PARylation regulates DNA−PK kinase activity in the DNA damage response. Mol Med Rep (2019) 20(4):3609–16. doi: 10.3892/mmr.2019.10640
166. Vormoor B, Schlosser YT, Blair H, Sharma A, Wilkinson S, Newell DR, et al. Sensitizing Ewing sarcoma to chemo- and radiotherapy by inhibition of the DNA-repair enzymes DNA protein kinase (DNA-PK) and poly-ADP-ribose polymerase (PARP) 1/2. Oncotarget (2017) 8(69):113418–30. doi: 10.18632/oncotarget.21300
167. Hirai T, Saito S, Fujimori H, Matsushita K, Nishio T, Okayasu R, et al. Radiosensitization by PARP inhibition to proton beam irradiation in cancer cells. Biochem Biophys Res Commun (2016) 478(1):234–40. doi: 10.1016/j.bbrc.2016.07.062
168. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer (2011) 11(4):239–53. doi: 10.1038/nrc3007
169. Jannetti SA, Carlucci G, Carney B, Kossatz S, Shenker L, Carter LM, et al. PARP-1-Targeted radiotherapy in mouse models of glioblastoma. J Nucl Med (2018) 59(8):1225–33. doi: 10.2967/jnumed.117.205054
170. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434(7035):917–21. doi: 10.1038/nature03445
171. Laudisi F, Sambucci M, Pioli C. Poly (ADP-ribose) polymerase-1 (PARP-1) as immune regulator. Endocr Metab Immune Disord Drug Targets (2011) 11(4):326–33. doi: 10.2174/187153011797881184
172. Rosado MM, Bennici E, Novelli F, Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology (2013) 139(4):428–37. doi: 10.1111/imm.12099
173. Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest (2019) 129(3):1211–28. doi: 10.1172/JCI123319
174. Kim C, Wang XD, Yu Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife (2020) 9. doi: 10.7554/eLife.60637
175. McLaughlin LJ, Stojanovic L, Kogan AA, Rutherford JL, Choi EY, Yen RC, et al. Pharmacologic induction of innate immune signaling directly drives homologous recombination deficiency. Proc Natl Acad Sci U.S.A. (2020) 117(30):17785–95. doi: 10.1073/pnas.2003499117
176. Wang F, Zhao M, Chang B, Zhou Y, Wu X, Ma M, et al. Cytoplasmic PARP1 links the genome instability to the inhibition of antiviral immunity through PARylating cGAS. Mol Cell (2022) 82(11):2032–2049.e7. doi: 10.1016/j.molcel.2022.03.034
177. Ieraci A, Herrera DG. Nicotinamide inhibits ethanol-induced caspase-3 and PARP-1 over-activation and subsequent neurodegeneration in the developing mouse cerebellum. Cerebellum (2018) 17(3):326–35. doi: 10.1007/s12311-017-0916-z
178. Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst (2004) 96(1):56–67. doi: 10.1093/jnci/djh005
179. Noël G, Godon C, Fernet M, Giocanti N, Mégnin-Chanet F, Favaudon V. Radiosensitization by the poly(ADP-ribose) polymerase inhibitor 4-amino-1,8-naphthalimide is specific of the s phase of the cell cycle and involves arrest of DNA synthesis. Mol Cancer Ther (2006) 5(3):564–74. doi: 10.1158/1535-7163.MCT-05-0418
180. Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, et al. FDA Approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res (2015) 21(19):4257–61. doi: 10.1158/1078-0432.CCR-15-0887
181. Mullard A. European Regulators approve first PARP inhibitor. Nat Rev Drug Discov (2014) 13(12):877–7. doi: 10.1038/nrd4508
182. L, a. FDA grants breakthrough designation to niraparib for metastatic CRPC (2019). Available at: https://www.targetedonc.com/view/fda-grants-breakthrough-designation-to-niraparib-for-metastatic-crpc.
183. FDA Approves olaparib for HRR gene-mutated metastatic castration-resistant prostate cancer. In: Drug approvals and databases 2020. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer#:~:text=On%20May%2019%2C%202020%2C%20the,(mCRPC)%2C%20who%20have%20progressed. (Silver Spring, MD: U.S. Food and Drug Administration)
184. FDA Approves olaparib tablets for maintenance treatment in ovarian cancer. In: Drug approvals and databases 2017. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-tablets-maintenance-treatment-ovarian-cancer. (Silver Spring, MD: U.S. Food and Drug Administration)
185. Ison G, Howie LJ, Amiri-Kordestani L, Zhang L, Tang S, Sridhara R, et al. FDA Approval summary: Niraparib for the maintenance treatment of patients with recurrent ovarian cancer in response to platinum-based chemotherapy. Clin Cancer Res (2018) 24(17):4066–71. doi: 10.1158/1078-0432.CCR-18-0042
186. Bourton EC, Ahorner PA, Plowman PN, Zahir SA, Al-Ali H, Parris CN. The PARP-1 inhibitor olaparib suppresses BRCA1 protein levels, increases apoptosis and causes radiation hypersensitivity in BRCA1(+/-) lymphoblastoid cells. J Cancer (2017) 8(19):4048–56. doi: 10.7150/jca.21338
187. Ryu H, Kim HJ, Song JY, Hwang SG, Kim JS, Kim J, et al. A small compound KJ-28d enhances the sensitivity of non-small cell lung cancer to radio- and chemotherapy. Int J Mol Sci (2019) 20(23):6026. doi: 10.3390/ijms20236026
188. Guillot C, Favaudon V, Herceg Z, Sagne C, Sauvaigo S, Merle P, et al. PARP inhibition and the radiosensitizing effects of the PARP inhibitor ABT-888 in in vitro hepatocellular carcinoma models. BMC Cancer (2014) 14:603. doi: 10.1186/1471-2407-14-603
189. Wang L, Mason KA, Ang KK, Buchholz T, Valdecanas D, Mathur A, et al. MK-4827, a PARP-1/-2 inhibitor, strongly enhances response of human lung and breast cancer xenografts to radiation. Invest N Drugs (2012) 30(6):2113–20. doi: 10.1007/s10637-011-9770-x
190. Pernin V, Mégnin-Chanet F, Pennaneach V, Fourquet A, Kirova Y, Hall J. [PARP inhibitors and radiotherapy: rational and prospects for a clinical use]. Cancer Radiother (2014) 18(8):790–8; quiz 799-802. doi: 10.1016/j.canrad.2014.05.012
191. Mehta AK, Cheney EM, Hartl CA, Pantelidou C, Oliwa M, Castrillon JA, et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat Cancer (2021) 2(1):66–82. doi: 10.1038/s43018-020-00148-7
192. Qiu L, Wang JJ, Ying SH, Feng MG. Wee1 and Cdc25 control morphogenesis, virulence and multistress tolerance of beauveria bassiana by balancing cell cycle-required cyclin-dependent kinase 1 activity. Environ Microbiol (2015) 17(4):1119–33. doi: 10.1111/1462-2920.12530
193. Koh SB. The expanding role of WEE1. Cell Signal (2022) 94:110310. doi: 10.1016/j.cellsig.2022.110310
194. Hamilton DH, Huang B, Fernando RI, Tsang KY, Palena C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial-mesenchymal transition. Cancer Res (2014) 74(9):2510–9. doi: 10.1158/0008-5472.CAN-13-1894
195. Li C, Shen Q, Zhang P, Wang T, Liu W, Li R, et al. Targeting MUS81 promotes the anticancer effect of WEE1 inhibitor and immune checkpoint blocking combination therapy via activating cGAS/STING signaling in gastric cancer cells. J Exp Clin Cancer Res (2021) 40(1):315. doi: 10.1186/s13046-021-02120-4
196. Wu X, Kang X, Zhang X, Xie W, Su Y, Liu X, et al. WEE1 inhibitor and ataxia telangiectasia and RAD3-related inhibitor trigger stimulator of interferon gene-dependent immune response and enhance tumor treatment efficacy through programmed death-ligand 1 blockade. Cancer Sci (2021) 112(11):4444–56. doi: 10.1111/cas.15108
197. Hai J, Zhang H, Zhou J, Wu Z, Chen T, Papadopoulos E, et al. Generation of genetically engineered mouse lung organoid models for squamous cell lung cancers allows for the study of combinatorial immunotherapy. Clin Cancer Res (2020) 26(13):3431–42. doi: 10.1158/1078-0432.CCR-19-1627
198. Guo E, Xiao R, Wu Y, Lu F, Liu C, Yang B, et al. WEE1 inhibition induces anti-tumor immunity by activating ERV and the dsRNA pathway. J Exp Med (2022) 219(1):e20210789. doi: 10.1084/jem.20210789
199. Lee YY, Cho YJ, Shin SW, Choi C, Ryu JY, Jeon HK, et al. Anti-tumor effects of Wee1 kinase inhibitor with radiotherapy in human cervical cancer. Sci Rep (2019) 9(1):15394. doi: 10.1038/s41598-019-51959-3
200. Lescarbeau RS, Lei L, Bakken KK, Sims PA, Sarkaria JN, Canoll P, et al. Quantitative phosphoproteomics reveals Wee1 kinase as a therapeutic target in a model of proneural glioblastoma. Mol Cancer Ther (2016) 15(6):1332–43. doi: 10.1158/1535-7163.MCT-15-0692
201. Matheson CJ, Venkataraman S, Amani V, Harris PS, Backos DS, Donson AM, et al. A WEE1 inhibitor analog of AZD1775 maintains synergy with cisplatin and demonstrates reduced single-agent cytotoxicity in medulloblastoma cells. ACS Chem Biol (2016) 11(4):921–30. doi: 10.1021/acschembio.5b00725
202. Kausar T, Schreiber JS, Karnak D, Parsels LA, Parsels JD, Davis MA, et al. Sensitization of pancreatic cancers to gemcitabine chemoradiation by WEE1 kinase inhibition depends on homologous recombination repair. Neoplasia (2015) 17(10):757–66. doi: 10.1016/j.neo.2015.09.006
203. Havelek R, Cmielova J, Kralovec K, Bruckova L, Bilkova Z, Fousova I, et al. Specific inhibition of Wee1 kinase and Rad51 recombinase: a strategy to enhance the sensitivity of leukemic T-cells to ionizing radiation-induced DNA double-strand breaks. Biochem Biophys Res Commun (2014) 453(3):569–75. doi: 10.1016/j.bbrc.2014.09.123
204. PosthumaDeBoer J, Würdinger T, Graat HC, van Beusechem VW, Helder MN, van Royen BJ, et al. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer (2011) 11:156. doi: 10.1186/1471-2407-11-156
205. Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, et al. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res (2011) 17(17):5638–48. doi: 10.1158/1078-0432.CCR-11-0650
206. Fu S, Wang Y, Keyomarsi K, Meric-Bernstam F, Meric-Bernstein F. Strategic development of AZD1775, a Wee1 kinase inhibitor, for cancer therapy. Expert Opin Investig Drugs (2018) 27(9):741–51. doi: 10.1080/13543784.2018.1511700
207. Chen G, Zhang B, Xu H, Sun Y, Shi Y, Luo Y, et al. Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene (2017) 36(50):6863–72. doi: 10.1038/onc.2017.297
208. Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A, et al. STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell (2013) 50(1):5–15. doi: 10.1016/j.molcel.2013.01.039
209. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339(6121):786–91. doi: 10.1126/science.1232458
210. Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U.S.A. (2009) 106(21):8653–8. doi: 10.1073/pnas.0900850106
211. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
212. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity (2014) 41(5):830–42. doi: 10.1016/j.immuni.2014.10.017
213. Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U.S.A. (2012) 109(47):19386–91. doi: 10.1073/pnas.1215006109
214. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity (2014) 41(5):843–52. doi: 10.1016/j.immuni.2014.10.019
215. Cheng H, Xu Q, Lu X, Yuan H, Li T, Zhang Y, et al. Activation of STING by cGAMP regulates MDSCs to suppress tumor metastasis via reversing epithelial-mesenchymal transition. Front Oncol (2020) 10:896. doi: 10.3389/fonc.2020.00896
216. Zhang CX, Ye SB, Ni JJ, Cai TT, Liu YN, Huang DJ, et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ (2019) 26(11):2314–28. doi: 10.1038/s41418-019-0302-0
217. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA Exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun (2017) 8:15618. doi: 10.1038/ncomms15618
218. Baguley BC. Antivascular therapy of cancer: DMXAA. Lancet Oncol (2003) 4(3):141–8. doi: 10.1016/S1470-2045(03)01018-0
219. Baguley BC, Siemann DW. Temporal aspects of the action of ASA404 (vadimezan; DMXAA). Expert Opin Investig Drugs (2010) 19(11):1413–25. doi: 10.1517/13543784.2010.529128
220. Ching LM, Cao Z, Kieda C, Zwain S, Jameson MB, Baguley BC. Induction of endothelial cell apoptosis by the antivascular agent 5,6-Dimethylxanthenone-4-acetic acid. Br J Cancer (2002) 86(12):1937–42. doi: 10.1038/sj.bjc.6600368
221. Wilson WR, Li AE, Cowan DS, Siim BG. Enhancement of tumor radiation response by the antivascular agent 5,6-dimethylxanthenone-4-acetic acid. Int J Radiat Oncol Biol Phys (1998) 42(4):905–8. doi: 10.1016/S0360-3016(98)00358-7
222. Matthews KE, Hermans IF, Roberts JM, Ching LM, Ronchese F. 5,6-Dimethylxanthenone-4-acetic acid treatment of a non-immunogenic tumour does not synergize with active or passive CD8+ T-cell immunotherapy. Immunol Cell Biol (2006) 84(4):383–9. doi: 10.1111/j.1440-1711.2006.01448.x
223. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res (2016) 76(8):2076–81. doi: 10.1158/0008-5472.CAN-15-1456
224. Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun (2017) 8(1):427. doi: 10.1038/s41467-017-00573-w
225. Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res (2013) 73(10):2943–8. doi: 10.1158/0008-5472.CAN-12-4354
226. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest (2019) 129(10):4350–64. doi: 10.1172/JCI125413
227. Chelvanambi M, Fecek RJ, Taylor JL, Storkus WJ. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J Immunother Cancer (2021) 9(2):e001906. doi: 10.1136/jitc-2020-001906
228. Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, Luke JJ. STING agonists as cancer therapeutics. Cancers (Basel) (2021) 13(11):2695. doi: 10.3390/cancers13112695
229. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature (2018) 564(7736):439–43. doi: 10.1038/s41586-018-0705-y
230. Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An orally available non-nucleotide STING agonist with antitumor activity. Science (2020) 369(6506):eaba6098. doi: 10.1126/science.aba6098
231. Liu Y, Crowe WN, Wang L, Lu Y, Petty WJ, Habib AA, et al. An inhalable nanoparticulate STING agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nat Commun (2019) 10(1):5108. doi: 10.1038/s41467-019-13094-5
232. Raghida A, Bukhalid JRD, Naniye Malli C, Catcott KC, Avocetien K, Bentley KW, et al. Abstract 6706: Systemic administration of STING agonist antibody-drug conjugates elicit potent anti-tumor immune responses with minimal induction of circulating cytokines. Cancer Res (2020) . 80(16_Supplement):6706. doi: 10.1158/1538-7445.AM2020-6706
233. Le Naour J, Zitvogel L, Galluzzi L, Vacchelli E, Kroemer G. Trial watch: STING agonists in cancer therapy. Oncoimmunology (2020) 9(1):1777624. doi: 10.1080/2162402X.2020.1777624
234. Harberts E, Gaspari AA. TLR signaling and DNA repair: are they associated? J Invest Dermatol (2013) 133(2):296–302. doi: 10.1038/jid.2012.288
235. Menendez D, Shatz M, Azzam K, Garantziotis S, Fessler MB, Resnick MA. The toll-like receptor gene family is integrated into human DNA damage and p53 networks. PloS Genet (2011) 7(3):e1001360. doi: 10.1371/journal.pgen.1001360
236. Walshaw RC, Honeychurch J, Choudhury A, Illidge TM. Toll-like receptor agonists and radiation therapy combinations: An untapped opportunity to induce anticancer immunity and improve tumor control. Int J Radiat Oncol Biol Phys (2020) 108(1):27–37. doi: 10.1016/j.ijrobp.2020.04.020
237. Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Aït-Yahia S, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med (1998) 188(2):373–86. doi: 10.1084/jem.188.2.373
238. Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell (1999) 99(1):23–33. doi: 10.1016/S0092-8674(00)80059-8
239. Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, Mackay CR, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol (1998) 28(9):2760–9. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N
240. Janeway CA Jr, Travers P, Walport M, Shlomchik MJ. Immunobiology: The immune system in health and disease. 5th ed. New York: Garland Science (2001).
241. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science (2003) 299(5609):1033–6. doi: 10.1126/science.1078231
242. Durand V, Wong SY, Tough DF, Le Bon A. IFN-alpha/beta-dependent cross-priming induced by specific toll-like receptor agonists. Vaccine (2006) 24 Suppl 2:S2–22-3. doi: 10.1016/j.vaccine.2005.01.115
243. McBride S, Hoebe K, Georgel P, Janssen E. Cell-associated double-stranded RNA enhances antitumor activity through the production of type I IFN. J Immunol (2006) 177(9):6122–8. doi: 10.4049/jimmunol.177.9.6122
244. Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, et al. A subset of toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol (2003) 170(8):4102–10. doi: 10.4049/jimmunol.170.8.4102
245. Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature (2005) 433(7028):887–92. doi: 10.1038/nature03326
246. Benwell RK, Hruska JE, Fritsche KL, Lee DR. Double stranded RNA- relative to other TLR ligand-activated dendritic cells induce extremely polarized human Th1 responses. Cell Immunol (2010) 264(2):119–26. doi: 10.1016/j.cellimm.2010.05.008
247. Smits EL, Ponsaerts P, Van de Velde AL, Van Driessche A, Cools N, Lenjou M, et al. Proinflammatory response of human leukemic cells to dsRNA transfection linked to activation of dendritic cells. Leukemia (2007) 21(8):1691–9. doi: 10.1038/sj.leu.2404763
248. Verdijk RM, Mutis T, Esendam B, Kamp J, Melief CJ, Brand A, et al. Polyriboinosinic polyribocytidylic acid (poly(I:C)) induces stable maturation of functionally active human dendritic cells. J Immunol (1999) 163(1):57–61.
249. Salem ML, Diaz-Montero CM, El-Naggar SA, Chen Y, Moussa O, Cole DJ. The TLR3 agonist poly(I:C) targets CD8+ T cells and augments their antigen-specific responses upon their adoptive transfer into naïve recipient mice. Vaccine (2009) 27(4):549–57. doi: 10.1016/j.vaccine.2008.11.013
250. Currie AJ, van der Most RG, Broomfield SA, Prosser AC, Tovey MG, Robinson BW. Targeting the effector site with IFN-alphabeta-inducing TLR ligands reactivates tumor-resident CD8 T cell responses to eradicate established solid tumors. J Immunol (2008) 180(3):1535–44. doi: 10.4049/jimmunol.180.3.1535
251. Lauzon NM, Mian F, MacKenzie R, Ashkar AA. The direct effects of toll-like receptor ligands on human NK cell cytokine production and cytotoxicity. Cell Immunol (2006) 241(2):102–12. doi: 10.1016/j.cellimm.2006.08.004
252. Schmidt KN, Leung B, Kwong M, Zarember KA, Satyal S, Navas TA, et al. APC-independent activation of NK cells by the toll-like receptor 3 agonist double-stranded RNA. J Immunol (2004) 172(1):138–43. doi: 10.4049/jimmunol.172.1.138
253. Miyake T, Kumagai Y, Kato H, Guo Z, Matsushita K, Satoh T, et al. Poly I:C-induced activation of NK cells by CD8 alpha+ dendritic cells via the IPS-1 and TRIF-dependent pathways. J Immunol (2009) 183(4):2522–8. doi: 10.4049/jimmunol.0901500
254. Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci U.S.A. (2012) 109(6):2066–71. doi: 10.1073/pnas.1113099109
255. Vidyarthi A, Khan N, Agnihotri T, Negi S, Das DK, Aqdas M, et al. TLR-3 stimulation skews M2 macrophages to M1 through IFN-αβ signaling and restricts tumor progression. Front Immunol (2018) 9:1650. doi: 10.3389/fimmu.2018.01650
256. Yoshida S, Shime H, Takeda Y, Nam JM, Takashima K, Matsumoto M, et al. Toll-like receptor 3 signal augments radiation-induced tumor growth retardation in a murine model. Cancer Sci (2018) 109(4):956–65. doi: 10.1111/cas.13543
257. Levine AS, Sivulich M, Wiernik PH, Levy HB. Initial clinical trials in cancer patients of polyriboinosinic-polyribocytidylic acid stabilized with poly-l-lysine, in carboxymethylcellulose [poly(ICLC)], a highly effective interferon inducer. Cancer Res (1979) 39(5):1645–50.
258. Hammerich L, Marron TU, Upadhyay R, Svensson-Arvelund J, Dhainaut M, Hussein S, et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med (2019) 25(5):814–24. doi: 10.1038/s41591-019-0410-x
259. Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature (2009) 462(7269):99–103. doi: 10.1038/nature08512
260. Rosenfeld MR, Chamberlain MC, Grossman SA, Peereboom DM, Lesser GJ, Batchelor TT, et al. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol (2010) 12(10):1071–7. doi: 10.1093/neuonc/noq071
261. Butowski N, Chang SM, Junck L, DeAngelis LM, Abrey L, Fink K, et al. A phase II clinical trial of poly-ICLC with radiation for adult patients with newly diagnosed supratentorial glioblastoma: a north American brain tumor consortium (NABTC01-05). J Neurooncol (2009) 91(2):175–82. doi: 10.1007/s11060-008-9693-3
262. Spinetti T, Spagnuolo L, Mottas I, Secondini C, Treinies M, Rüegg C, et al. TLR7-based cancer immunotherapy decreases intratumoral myeloid-derived suppressor cells and blocks their immunosuppressive function. Oncoimmunology (2016) 5(11):e1230578. doi: 10.1080/2162402X.2016.1230578
263. Wang J, Shirota Y, Bayik D, Shirota H, Tross D, Gulley JL, et al. Effect of TLR agonists on the differentiation and function of human monocytic myeloid-derived suppressor cells. J Immunol (2015) 194(9):4215–21. doi: 10.4049/jimmunol.1402004
264. Rechtsteiner G, Warger T, Osterloh P, Schild H, Radsak MP. Cutting edge: priming of CTL by transcutaneous peptide immunization with imiquimod. J Immunol (2005) 174(5):2476–80. doi: 10.4049/jimmunol.174.5.2476
265. Shackleton M, Davis ID, Hopkins W, Jackson H, Dimopoulos N, Tai T, et al. The impact of imiquimod, a toll-like receptor-7 ligand (TLR7L), on the immunogenicity of melanoma peptide vaccination with adjuvant Flt3 ligand. Cancer Immun (2004) 4:9.
266. Gorski KS, Waller EL, Bjornton-Severson J, Hanten JA, Riter CL, Kieper WC, et al. Distinct indirect pathways govern human NK-cell activation by TLR-7 and TLR-8 agonists. Int Immunol (2006) 18(7):1115–26. doi: 10.1093/intimm/dxl046
267. Navi D, Huntley A. Imiquimod 5 percent cream and the treatment of cutaneous malignancy. Dermatol Online J (2004) 10(1):4. doi: 10.5070/D34VW339W4
268. Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science (2005) 309(5739):1380–4. doi: 10.1126/science.1113401
269. Panelli MC, Stashower ME, Slade HB, Smith K, Norwood C, Abati A, et al. Sequential gene profiling of basal cell carcinomas treated with imiquimod in a placebo-controlled study defines the requirements for tissue rejection. Genome Biol (2007) 8(1):R8. doi: 10.1186/gb-2007-8-1-r8
270. Xiang AX, Webber SE, Kerr BM, Rueden EJ, Lennox JR, Haley GJ, et al. Discovery of ANA975: an oral prodrug of the TLR-7 agonist isatoribine. Nucleosides Nucleotides Nucleic Acids (2007) 26(6-7):635–40. doi: 10.1080/15257770701490472
271. PRESS RELEASE. Anadys Pharmaceuticals Discontinues Development of Hepatitis C Drug [press release]. San Diego, CA: Fierce Biotech, (2007).
272. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature (2000) 408(6813):740–5. doi: 10.1038/35047123
273. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
274. Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, et al. CpG motifs in bacterial DNA trigger direct b-cell activation. Nature (1995) 374(6522):546–9. doi: 10.1038/374546a0
275. Häcker H, Mischak H, Häcker G, Eser S, Prenzel N, Ullrich A, et al. Cell type-specific activation of mitogen-activated protein kinases by CpG-DNA controls interleukin-12 release from antigen-presenting cells. EMBO J (1999) 18(24):6973–82. doi: 10.1093/emboj/18.24.6973
276. Hartmann G, Weiner GJ, Krieg AM. CpG DNA: a potent signal for growth, activation, and maturation of human dendritic cells. Proc Natl Acad Sci U.S.A. (1999) 96(16):9305–10. doi: 10.1073/pnas.96.16.9305
277. Jakob T, Walker PS, Krieg AM, Udey MC, Vogel JC. Activation of cutaneous dendritic cells by CpG-containing oligodeoxynucleotides: a role for dendritic cells in the augmentation of Th1 responses by immunostimulatory DNA. J Immunol (1998) 161(6):3042–9.
278. Sparwasser T, Koch ES, Vabulas RM, Heeg K, Lipford GB, Ellwart JW, et al. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol (1998) 28(6):2045–54. doi: 10.1002/(SICI)1521-4141(199806)28:06<2045::AID-IMMU2045>3.0.CO;2-8
279. Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol (2012) 188(4):1592–9. doi: 10.4049/jimmunol.1101304
280. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
281. Aurisicchio L, Peruzzi D, Conforti A, Dharmapuri S, Biondo A, Giampaoli S, et al. Treatment of mammary carcinomas in HER-2 transgenic mice through combination of genetic vaccine and an agonist of toll-like receptor 9. Clin Cancer Res (2009) 15(5):1575–84. doi: 10.1158/1078-0432.CCR-08-2628
282. Baines J, Celis E. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clin Cancer Res (2003) 9(7):2693–700.
283. Blazar BR, Krieg AM, Taylor PA. Synthetic unmethylated cytosine-phosphate-guanosine oligodeoxynucleotides are potent stimulators of antileukemia responses in naive and bone marrow transplant recipients. Blood (2001) 98(4):1217–25. doi: 10.1182/blood.V98.4.1217
284. Lonsdorf AS, Kuekrek H, Stern BV, Boehm BO, Lehmann PV, Tary-Lehmann M. Intratumor CpG-oligodeoxynucleotide injection induces protective antitumor T cell immunity. J Immunol (2003) 171(8):3941–6. doi: 10.4049/jimmunol.171.8.3941
285. Kawarada Y, Ganss R, Garbi N, Sacher T, Arnold B, Hämmerling GJ. NK- and CD8(+) T cell-mediated eradication of established tumors by peritumoral injection of CpG-containing oligodeoxynucleotides. J Immunol (2001) 167(9):5247–53. doi: 10.4049/jimmunol.167.9.5247
286. Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol (2010) 28(28):4324–32. doi: 10.1200/JCO.2010.28.9793
287. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5(7):730–7. doi: 10.1038/ni1087
288. Fitzgerald ME, Rawling DC, Vela A, Pyle AM. An evolving arsenal: viral RNA detection by RIG-i-like receptors. Curr Opin Microbiol (2014) 20:76–81. doi: 10.1016/j.mib.2014.05.004
289. Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, et al. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep (2012) 2(5):1187–96. doi: 10.1016/j.celrep.2012.10.005
290. Guo G, Gao M, Gao X, Zhu B, Huang J, Tu X, et al. Reciprocal regulation of RIG-I and XRCC4 connects DNA repair with RIG-I immune signaling. Nat Commun (2021) 12(1):2187. doi: 10.1038/s41467-021-22484-7
291. Poeck H, Wintges A, Dahl S, Bassermann F, Haas T, Heidegger S. Tumor cell-intrinsic RIG-I signaling governs synergistic effects of immunogenic cancer therapies and checkpoint inhibitors in mice. Eur J Immunol (2021) 51(6):1531–4. doi: 10.1002/eji.202049158
292. Besch R, Poeck H, Hohenauer T, Senft D, Häcker G, Berking C, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest (2009) 119(8):2399–411. doi: 10.1172/JCI37155
293. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell (2015) 162(5):961–73. doi: 10.1016/j.cell.2015.07.056
294. Ellermeier J, Wei J, Duewell P, Hoves S, Stieg MR, Adunka T, et al. Therapeutic efficacy of bifunctional siRNA combining TGF-β1 silencing with RIG-I activation in pancreatic cancer. Cancer Res (2013) 73(6):1709–20. doi: 10.1158/0008-5472.CAN-11-3850
295. Kübler K, Gehrke N, Riemann S, Böhnert V, Zillinger T, Hartmann E, et al. Targeted activation of RNA helicase retinoic acid-inducible gene-I induces proimmunogenic apoptosis of human ovarian cancer cells. Cancer Res (2010) 70(13):5293–304. doi: 10.1158/0008-5472.CAN-10-0825
296. Duewell P, Steger A, Lohr H, Bourhis H, Hoelz H, Kirchleitner SV, et al. RIG-i-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8(+) T cells. Cell Death Differ (2014) 21(12):1825–37. doi: 10.1038/cdd.2014.96
297. Engel C, Brügmann G, Lambing S, Mühlenbeck LH, Marx S, Hagen C, et al. RIG-I resists hypoxia-induced immunosuppression and dedifferentiation. Cancer Immunol Res (2017) 5(6):455–67. doi: 10.1158/2326-6066.CIR-16-0129-T
298. Zhu H, Xu WY, Hu Z, Zhang H, Shen Y, Lu S, et al. RNA Virus receptor rig-I monitors gut microbiota and inhibits colitis-associated colorectal cancer. J Exp Clin Cancer Res (2017) 36(1):2. doi: 10.1186/s13046-016-0471-3
299. Ranoa DR, Parekh AD, Pitroda SP, Huang X, Darga T, Wong AC, et al. Cancer therapies activate RIG-i-like receptor pathway through endogenous non-coding RNAs. Oncotarget (2016) 7(18):26496–515. doi: 10.18632/oncotarget.8420
300. Jiang X, Muthusamy V, Fedorova O, Kong Y, Kim DJ, Bosenberg M, et al. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J Exp Med (2019) 216(12):2854–68. doi: 10.1084/jem.20190801
301. Sato Y, Yoshino H, Tsuruga E, Kashiwakura I. Fas ligand enhances apoptosis of human lung cancer cells cotreated with RIG-i-like receptor agonist and radiation. Curr Cancer Drug Targets (2020) 20(5):372–81. doi: 10.2174/1568009620666200115161717
302. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity (2013) 39(1):74–88. doi: 10.1016/j.immuni.2013.06.014
303. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol (2015) 15(12):760–70. doi: 10.1038/nri3921
304. Verma R, Foster RE, Horgan K, Mounsey K, Nixon H, Smalle N, et al. Lymphocyte depletion and repopulation after chemotherapy for primary breast cancer. Breast Cancer Res (2016) 18(1):10. doi: 10.1186/s13058-015-0669-x
305. Matiello J, Dal Pra A, Zardo L, Silva R, Berton DC. Impacts of post-radiotherapy lymphocyte count on progression-free and overall survival in patients with stage III lung cancer. Thorac Cancer (2020) 11(11):3139–44. doi: 10.1111/1759-7714.13621
306. Ahn S, Park JS, Jang J, Ahn KJ, Hong YK, Yang SH, et al. The association between total lymphocyte count after concomitant chemoradiation and overall survival in patients with newly diagnosed glioblastoma. J Clin Neurosci (2020) 71:21–5. doi: 10.1016/j.jocn.2019.11.017
307. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
308. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (2015) 350(6257):207–11. doi: 10.1126/science.aad0095
309. Brown JM. Beware of clinical trials of DNA repair inhibitors. Int J Radiat Oncol Biol Phys (2019) 103(5):1182–3. doi: 10.1016/j.ijrobp.2018.11.063
310. Zenke FT, Zimmermann A, Sirrenberg C, Dahmen H, Kirkin V, Pehl U, et al. Pharmacologic inhibitor of DNA-PK, M3814, potentiates radiotherapy and regresses human tumors in mouse models. Mol Cancer Ther (2020) 19(5):1091–101. doi: 10.1158/1535-7163.MCT-19-0734
311. Mau-Sorensen M, Kuipers M, Nielsen DL, Verheul HM, Aftimos P, de Jonge MJA, et al. Safety, clinical activity and pharmacological biomarker evaluation of the DNA-dependent protein kinase (DNA-PK) inhibitor M3814: results from two phase I trials, in ESMO 2017 congress. 2018. Annals of Oncology (2018) 29(suppl_8):viii649–viii669. doi: 10.1093/annonc/mdy303.015
312. Wong WW, Jackson RK, Liew LP, Dickson BD, Cheng GJ, Lipert B, et al. Hypoxia-selective radiosensitisation by SN38023, a bioreductive prodrug of DNA-dependent protein kinase inhibitor IC87361. Biochem Pharmacol (2019) 169:113641. doi: 10.1016/j.bcp.2019.113641
313. Alastair HK, Baker JHE, Fryer KH, Banaáth J, Wang T, Porres SS, et al. Hypoxia-selective DNA-PK inhibitor [abstract]. In: AACR-NCI-EORTC international conference. Philadelphia, PA: AACR; Mol Cancer Ther (2017).
314. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys (2004) 58(3):862–70. doi: 10.1016/j.ijrobp.2003.09.012
315. Ko EC, Benjamin KT, Formenti SC. Generating antitumor immunity by targeted radiation therapy: Role of dose and fractionation. Adv Radiat Oncol (2018) 3(4):486–93. doi: 10.1016/j.adro.2018.08.021
316. Marciscano AE, Ghasemzadeh A, Nirschl TR, Theodros D, Kochel CM, Francica BJ, et al. Elective nodal irradiation attenuates the combinatorial efficacy of stereotactic radiation therapy and immunotherapy. Clin Cancer Res (2018) 24(20):5058–71. doi: 10.1158/1078-0432.CCR-17-3427
317. Nikitaki Z, Velalopoulou A, Zanni V, Tremi I, Havaki S, Kokkoris M, et al. Key biological mechanisms involved in high-LET radiation therapies with a focus on DNA damage and repair. Expert Rev Mol Med (2022) 24:e15. doi: 10.1017/erm.2022.6
318. Averbeck D, Rodriguez-Lafrasse C. Role of mitochondria in radiation responses: Epigenetic, metabolic, and signaling impacts. Int J Mol Sci (2021) 22(20):11047. doi: 10.3390/ijms222011047
319. Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, Sfeir A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature (2021) 591(7850):477–81. doi: 10.1038/s41586-021-03269-w
320. Yamazaki T, Kirchmair A, Sato A, Buqué A, Rybstein M, Petroni G, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol (2020) 21(10):1160–71. doi: 10.1038/s41590-020-0751-0
321. Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res (2018) 202:52–68. doi: 10.1016/j.trsl.2018.07.014
Keywords: DNA damage, innate immunity, radiotherapy, immunotherapy, combination therapy, cancer therapy
Citation: Chan Wah Hak CML, Rullan A, Patin EC, Pedersen M, Melcher AA and Harrington KJ (2022) Enhancing anti-tumour innate immunity by targeting the DNA damage response and pattern recognition receptors in combination with radiotherapy. Front. Oncol. 12:971959. doi: 10.3389/fonc.2022.971959
Received: 17 June 2022; Accepted: 01 August 2022;
Published: 29 August 2022.
Edited by:
Qiang Zhang, University of Michigan, United StatesReviewed by:
Michael Orth, LMU Munich University Hospital, GermanyCopyright © 2022 Chan Wah Hak, Rullan, Patin, Pedersen, Melcher and Harrington. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charleen M. L. Chan Wah Hak, Y2hhcmxlZW4uY2hhbkBpY3IuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.