
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 29 September 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.970242
This article is part of the Research TopicCirculating Tumor DNA in Cancer: A Role as a Response and Monitoring “Next-Generation” Biomarker in Cancer therapyView all 11 articles
 Rachel C. T. Lam1
Rachel C. T. Lam1 David Johnson2Gigi Lam1
David Johnson2Gigi Lam1 Michelle L. Y. Li1Joyce W. L. Wong1W. K. Jacky Lam3K. C. Allen Chan3
Michelle L. Y. Li1Joyce W. L. Wong1W. K. Jacky Lam3K. C. Allen Chan3 Brigette Ma2*
Brigette Ma2*Advances in Next Generation Sequencing (NGS) technologies have enabled the accurate detection and quantification of circulating tumor-derived (ct)DNA in most gastrointestinal (GI) cancers. The prognostic and predictive utility of ctDNA in patiets with different stages of colorectal (CRC), gastro-esophageal (GEC) and pancreaticobiliary cancers (PBC) are currently under active investigation. The most mature clinical data to date are derived from studies in the prognostic utility of personalized ctDNA-based NGS assays in the detection of minimal residual disease (MRD) and early recurrence after surgery in CRC and other GI cancers. These findings are being validated in several prospective studies which are designed to test if ctDNA could outperform conventional approaches in guiding adjuvant chemotherapy, and in post-operative surveillance in some GI cancers. Several adaptive studies using ctDNA as a screening platform are also being used to identify patients with actionable genomic alterations for clinical trials of targeted therapies. In the palliative setting, ctDNA monitoring during treatment has shown promise in the detection and tracking of clonal variants associated with acquired resistance to targeted therapies and immune-checkpoint inhibitors (ICI). Moreover, ctDNA may help to guide the therapeutic re-challenge of targeted therapies in patients who have prior exposure to such treatment. This review will examine the most updated research findings on ctDNA as a biomarker in CRC, GEC and PBCs. It aims to provide insights into how the unique strengths of this biomarker could be optimally leveraged in improving the management of these GI cancers.
According to GLOBOCAN 2020, several gastrointestinal (GI) cancers are amongst the top ten most prevalent and lethal cancers in certain parts of the world (1). Colorectal cancer (CRC) accounts for one in every 10 cancer-related deaths and is most prevalent in Western countries. Gastric cancer (GC) and esophageal squamous cancer (ESCC) are more common in East Asia and are responsible for one in every 13 and one in 18 cancer-related deaths in the world, respectively. The overall incidence rates of CRC, pancreatic cancer (PC) and biliary cancers (BLC) are stable or declining, but GC and esophageal adenocarcinoma (GEA) show rising trends in younger people from developed countries (1). Systemic therapy is integral to the management of some advanced GI cancers and the use of biomarkers in guiding treatment decisions may improve patient’s outcome (2–4).
Circulating tumor DNA (ctDNA) is a non-invasive and promising biomarker which is under active investigation in patients with GI cancers. The term ‘liquid biopsy’ refers to the process of sampling ctDNA, which is a component found in cell-free DNA (cfDNA) originating from the direct release, active secretion, necrosis or apoptosis of tumor cells into the circulation (5, 6). Each fragment of ctDNA usually has an average size of 166 base pairs, which resembles mononucleosomal units originating from cellular apoptosis (7). In recent years, research studies have evaluated the utility of ctDNA in the management of some GI cancers in these clinical settings: 1) the detection of minimal (or molecular) residual disease (MRD) following surgical resection of the primary tumor and in guiding adjuvant therapy; 2) assessment of clinical response to neoadjuvant chemotherapy and/or radiotherapy; 3) monitoring of response to palliative drug therapies; 4) tracking of clonal dynamics and evolution during targeted therapy, as well as in 5) the enrichment and selection of patients for clinical trials of novel anti-cancer therapies (Figure 1). The main objective of this article is to review the latest and most salient research studies on the clinical application of ctDNA in patients with advanced CRC, GEA, ESCC, PC and BLC. This review will also focus on how the strengths of ctDNA can be optimally leveraged in improving the treatment of these GI cancers.
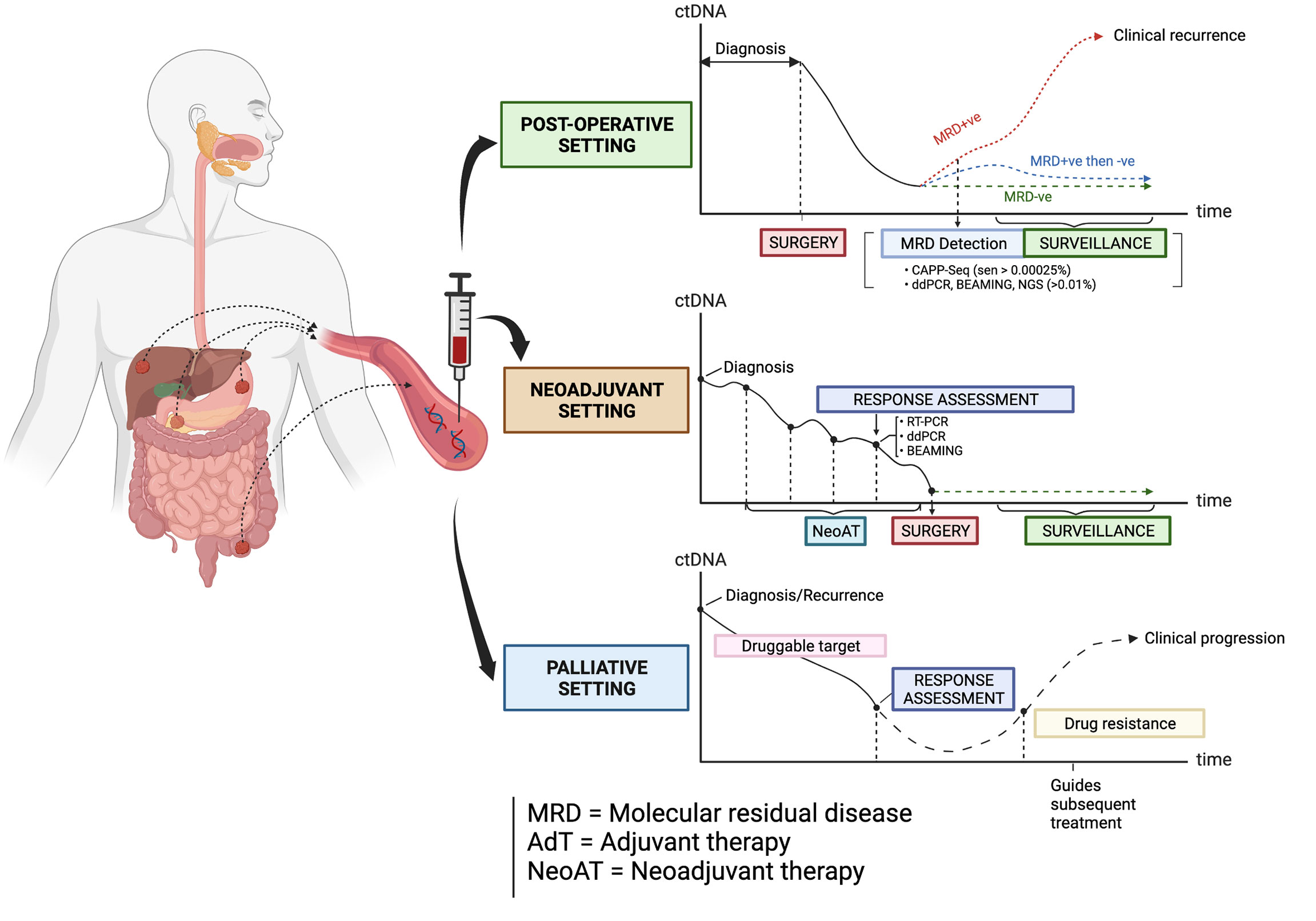
Figure 1 The types of clinical settings where ctDNA is being investigated as a biomarker in guiding the management of some gastrointestinal cancers. Created with BioRender.com.
The quantification of ctDNA in solid tumors generally involves two broad categories of assays: tumor-informed and tumor-agnostic assays (8, 9). Tumor-informed assays require a prior knowledge of tumor-specific genomic alterations. One of the commonest platforms used is the polymerase chain reaction (PCR)-based assays, which include droplet digital PCR (ddPCR), quantitative real-time (RT-qPCR) and ‘Beads, Emulsion, Amplification and Magnetics’ (BEAMing) PCR (8, 9). Another type of platform is to apply Next-Generation Sequencing (NGS) on a target panel of genomic alterations, examples of which include the Tagged-Amplicon deep sequencing (TAm-seq), Safe-sequencing System (Safe-SeqS) and CAncer Personalized Profiling by deep sequencing (CAPP-Seq) (8, 10). Such NGS-based assays are highly sensitive with a Limit of Detection (LOD) of variant allelic frequencies (VAF) as low as 0.01%, and specific in detecting various mutations including indels, rearrangements and copy number alterations (CNAs) in GI cancers. In contrast, tumor-agnostic assays are broad, panel-based sequencing assays that detect genomic alterations and methylation changes (9). They allow real-time tracking of novel mutational changes and cancer-specific variants simultaneously (8, 9).
In general, the detection rates of ctDNA can vary between different types of GI cancers. Bettegowda et al. found that ctDNA could be detected in around 73%, 57% and 48% of patients with CRC, GEC and PC, respectively (11). Strickler et al. reported a high correlation between the rates of ctDNA-derived and tumor-derived NGS-based detection of 20 most commonly mutated genes in CRC (12). However, the detection and interpretation of ctDNA are potentially limited by several patient-related and assay-related factors. Discordance between tumor and plasma samples may be influenced by intra-tumoral heterogeneity, tumor histology, anatomical location of metastases and the patient’s tumor burden. For instance, GC has a higher level of genomic heterogeneity than PC and CRCs, resulting in more variable interpatient rates of ctDNA detection. The level of tumor DNA shedding into plasma is lower with mucinous tumors and locoregional metastases, compared with liver metastases in CRC (11, 13, 14).
Limitations resulting from these pre-analytical and assay-related factors may undermine the accuracy of ctDNA results. False-negative results may be caused by the low VAF of specific variants or from inadequate volumes of plasma sampled (9). Since cfDNA is also released by blood cells, the expansion of blood cells in clonal hematopoiesis of indeterminate potential (CHIP) may increase the level of background noise signals and false-positive ctDNA measurements (15). According to a consensus statement by the National Cancer Institute (NCI) Colon-Rectal-Anal Taskforce, limitations related to ctDNA assays could be minimized by standardization of a common protocol for blood collection, sample processing, DNA extraction and analysis (16).
To date, the most mature clinical data on ctDNA are derived from patients with CRC. The detection rate of ctDNA in CRC is relatively high compared with other GI cancers that are discussed in this review - from an overall 73% in localized CRC (10), to 95.8% in patients with liver metastasis (16). Figure 2 is a chronological overview of some of the key studies on the clinical application of ctDNA in the management of early and advanced CRC. Details of these studies will be discussed in the following sections.
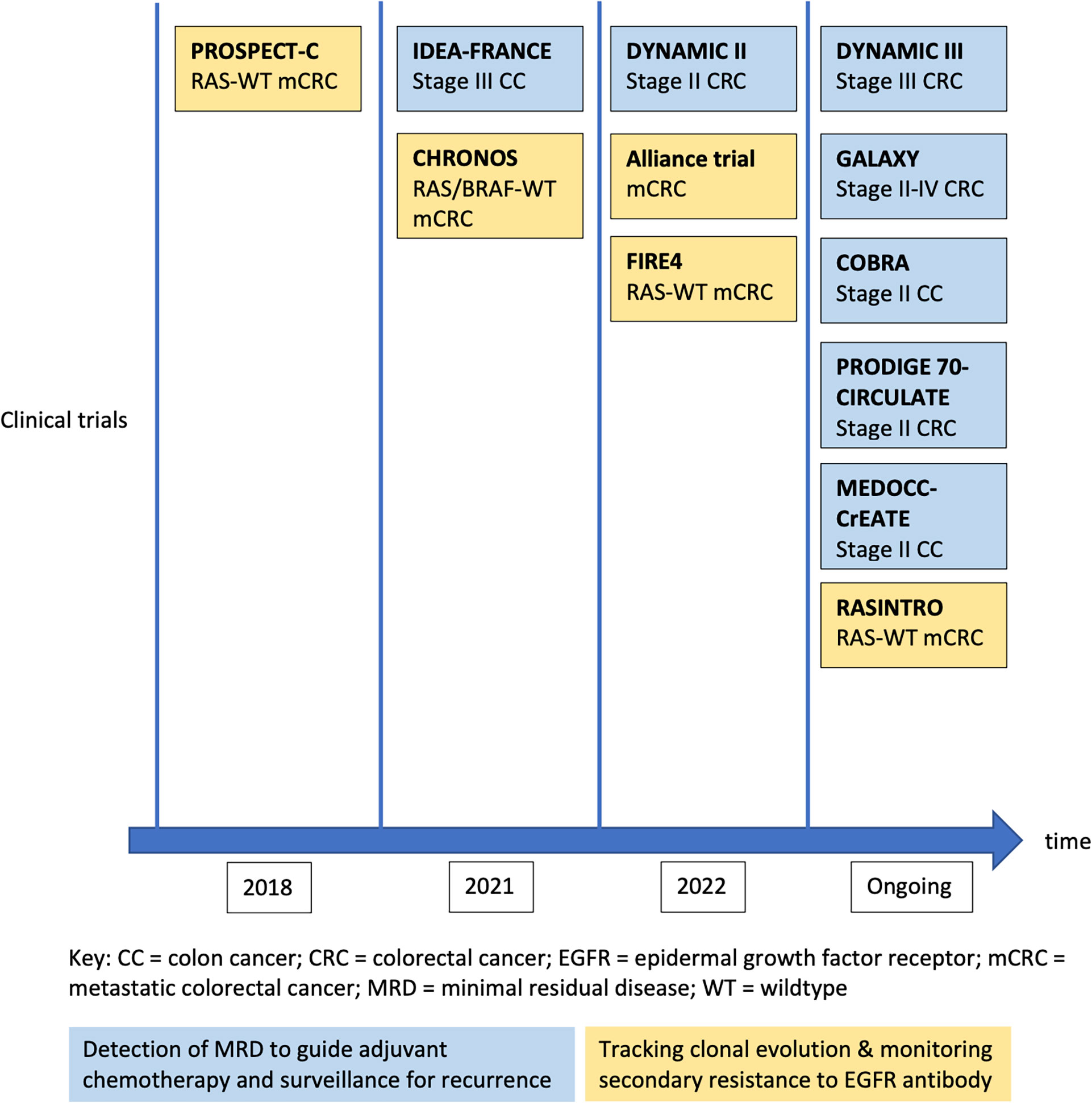
Figure 2 Development of major clinical trials for ctDNA application in colorectal cancers.
ctDNA allows the detection of MRD - residual cancer cells that are not detectable by conventional diagnostic tools (17). The current standard of care for stage III and some high-risk stage II colon cancers are surgery followed by adjuvant chemotherapy and then surveillance. The NCI Colon-Rectal-Anal Taskforce recommends the minimum time-points for perioperative sample collection to be 4-8 weeks post-resection, as cell damage during surgical resection and wound healing may lead to a surge in cfDNA (16).
Earlier trials have consistently shown that ctDNA is a powerful prognostic biomarker for the early detection of recurrence in resectable CRC, independent of clinico-pathological factors. Several Australian studies have reported that the ctDNA detection rate in patients with stage II colon cancer following surgery is 7.9%; while the ctDNA detection rate in patients with stage III colon cancer following surgery is 21% (18, 19). In a cohort of 486 patients with stage II-III colon cancers and locally advanced rectal cancer (LARC), patients with detectable ctDNA (MRD+ve) after surgery experienced lower 5-year recurrence-free survival (RFS) rates of 38.6% versus (vs) 85.5% (P < 0.001) and overall survival (OS) rates of 64.6% vs 89.4% (P < 0.001), when compared with those patients with undetectable ctDNA (MRD-ve) (20). In addition, the risk of recurrence is proportionately increased with higher levels of ctDNA VAF (hazard ratio [HR] = 1.2, 2.5 and 5.8 for VAFs of 0.1%, 0.5% and 1%, respectively) (20).
A Danish trial showed that the risk of post-operative recurrence increases if ctDNA becomes first detectable at the following time-points: 7 times higher risk if soon after surgery, 17.5 times if detected after adjuvant chemotherapy, and 43.5 times during surveillance (21). Interestingly, the duration of adjuvant chemotherapy may affect the prognostic significance of ctDNA (22). In a post-hoc analysis from the IDEA-FRANCE trial (which investigated the optimal duration of adjuvant chemotherapy by comparing 6 months vs 3 months of treatment in stage III colon cancer), ctDNA was prognostic in patients who received 3 months of adjuvant chemotherapy and with T4 and/or N2 tumors, but not in those treated for 6 months and with T1-3/N1 tumors (22).
Besides investigating the prognostic significance of postoperative ctDNA status, other studies compared the performance of ctDNA and radiological imaging in the detection of post-operative recurrence. However, the results are mixed because of the different imaging intervals used across different studies (21, 23, 24). A Danish study showed that checking ctDNA every 6 months could detect cancer recurrences up to 16.5 months earlier than radiologic imaging (21). This result is supported by a prospective study using the Signatera assay (23), where ctDNA could detect recurrences at a median of 9.08 months earlier than imaging in 193 patients with stage II-III CRC. In the TRACC study (25), post-operative MRD+ve status was the most powerful prognostic factor associated with increased RFS (HR = 28.8; 95% CI = 3.5 - 234.1; P < 0.001), compared with clinical factors, microsatellite (MSI) and tumor mutational burden (TMB) in 122 patients with stage II-III CRC. In contrast, a retrospective study (n = 48, Signatera assay) found no significant differences in the lead time or rate of detecting postoperative recurrence between ctDNA and imaging - the latter performed at intervals recommended by the United States (US) National Comprehensive Cancer Network (NCCN) guideline (24). The ongoing observational study (BESPOKE) will evaluate the impact of ctDNA testing (Signatera assay) on adjuvant treatment decisions and detection of recurrence in stage I-IV CRC across over 200 US sites (26). In conclusion, these observational studies have shown that ctDNA could accurately detect MRD status after surgery and predict disease recurrence, thus prospective randomized trials are warranted to determine if ctDNA will influence treatment decisions.
There are at least 7 ongoing phase III studies with an interventional, observational or adaptive-platform design. These studies investigate the utility of postoperative MRD detection using ctDNA in guiding de-escalating or escalating adjuvant approaches in the management of stage II-III and/or resectable stage IV CRC (Table 1).
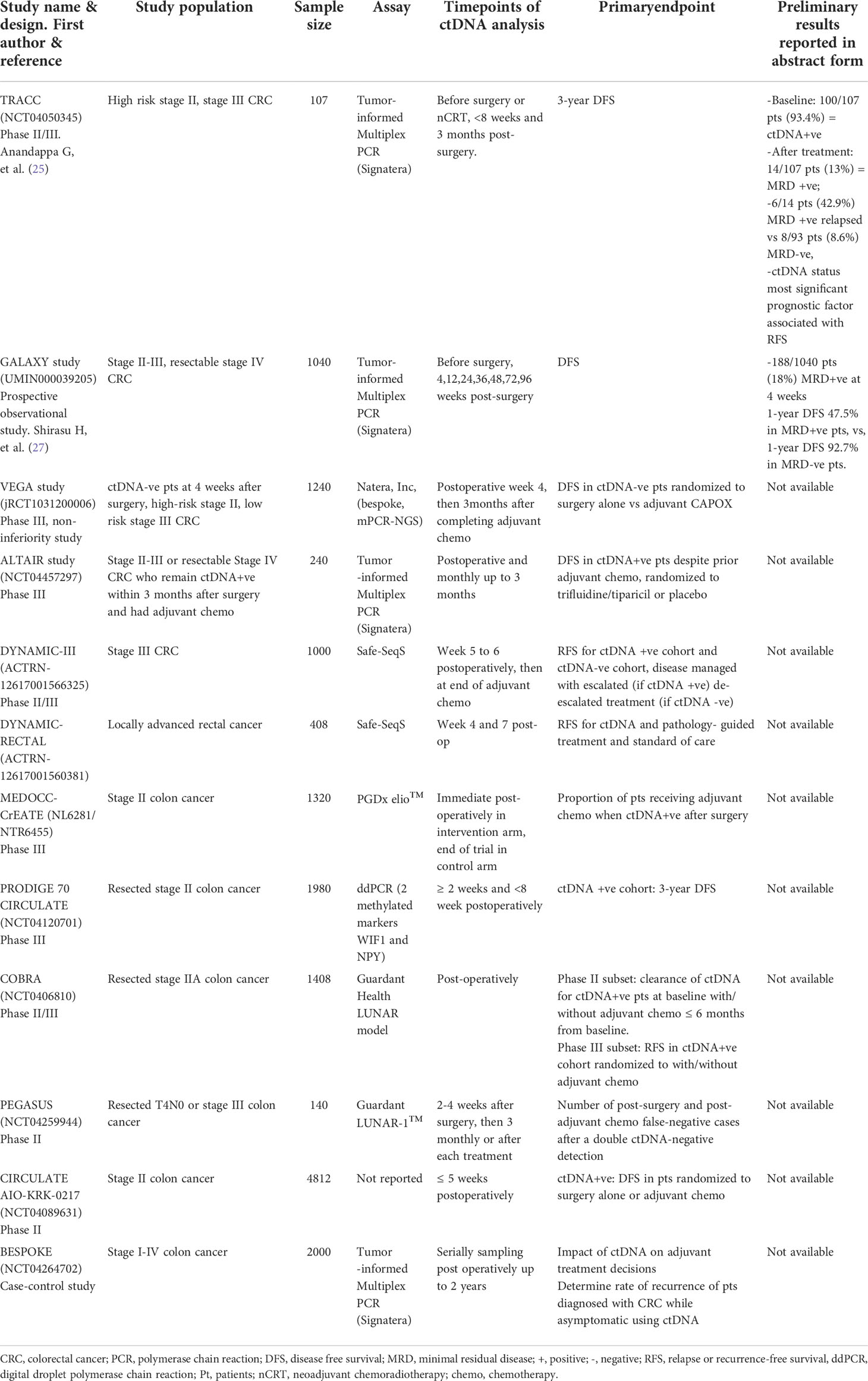
Table 1 Ongoing phase III or large observational studies or abstract-only reports on minimal residual disease.
Several trials are investigating whether ctDNA-detected MRD can outperform conventional methods of directing adjuvant chemotherapy (Table 1). The DYNAMIC II (28) and III (ACTRN-12617001566325) studies are interventional studies led by the Australasian Gastro-Intestinal Trials Group for patients with stage II to III CRC. The pivotal DYNAMIC II trial is the first of these studies to be published recently, where 455 patients with stage II colon cancer (T3 or T4, N0, M0) were randomized in a 2:1 ratio to have treatment decisions guided by either ctDNA results (using Safe-Seq assay in a central laboratory) or standard clinicopathological features (29). Patients with a positive ctDNA result at either 4-week or 7-week after surgery received adjuvant fluoropyrimidine or oxaliplatin-based chemotherapy. Designed to detect non-inferiority between the 2 arms, the study met its primary endpoint by showing that ctDNA-guided approach was non-inferior to standard management (93.5% and 92.4% respectively; 95% CI = 4.1 - 6.2 [non-inferiority margin, −8.5% points]) in terms of 2-year RFS. Moreover, fewer patients in the ctDNA-guided arm received adjuvant chemotherapy (15% vs 28%; relative risk = 1.82; 95% CI = 1.25 - 2.65). This study is the first to show that ctDNA-guided approach to the management of stage II colon cancer could reduce the use of adjuvant chemotherapy use without compromising RFS. There are several other ongoing trials which investigate the utility of ctDNA in guiding adjuvant decisions. These include the PEGASUS study (NCT04259944, uses the LUNAR1 assay, Guardant Health) which has a novel, real-time adaptive design, where patients with stage II-III CRC will switch chemotherapy regimens based on the MRD status monitored at 3-month intervals (30). The US NRG-led COBRA (NCT04068103) is an escalation trial for stage II colon cancer, where MRD+ve patients will receive adjuvant chemotherapy, while MRD-ve patients will undergo surveillance alone (31). The French PRODIGE 70-CIRCULATE (NCT04120701) study will screen over 2600 patients with stage II CRC and randomize 198 MRD+ve patients post-surgery to either adjuvant FOLFOX (infusional 5-fluorouracil, leucovorin and oxaliplatin) for 6 months or observation. Similar to the DYNAMIC studies, the MEDOCC-CrEATE study will enroll 1320 stage II colon cancer patients without indication for adjuvant chemotherapy based on current practice guidelines, and randomize them into two possible interventional arms: ctDNA-uninformed (standard observation without adjuvant chemotherapy) vs ctDNA-informed (adjuvant CAPOX or observation, depending on MRD status) (29).
The largest study to date is the colossal ‘CIRCULATE-Japan’, an adaptive platform study which investigates the utility of ctDNA in MRD detection for patients with resectable stage II to IV CRC via. Eligible patients are first enrolled into an observational screening study (called GALAXY) and undergo ctDNA testing (Signatera assay) before treatment and at defined intervals after surgery. Each participant’s ctDNA results are made available to the treating physicians to guide adjuvant treatment or enrollment into either one of two interventional phase III studies – the VEGA and ALTAIR (32). Preliminary result of the GALAXY study on the ctDNA dynamics of 1040 (out of a target of 5200) patients have been reported (27, 33). For all stages of CRC, patients who were MRD+ve at 4 weeks post-surgery (18%, 188/1040 patients) had 1-year disease-free survival (DFS) rate of 47.5%; while patients who were MRD-ve at 4 weeks post-surgery (82%, 852/1040 patients) had a 1-year DFS rate of 92.7% with a HR of 10.9 (95% CI = 7.8 - 15.4; P < 0.001). Adjuvant chemotherapy may be able to convert patients who were initially MRD+ve into MRD-ve. The use of adjuvant chemotherapy resulted in a higher proportion of patients (68%) being converted to MRD-ve at 12-weeks post-surgery, while only 10% of patients who did not receive chemotherapy were converted into MRD-ve. Patients who were successfully converted from MRD+ve to MRD-ve status had better DFS. In contrast, in patient subgroups who were MRD-ve at 4 weeks post-surgery, the use of adjuvant chemotherapy did not influence the DFS (HR = 1.3; 95% CI = 0.5 - 3.6%). In conclusion, this study supports the use of ctDNA at 4 weeks post-surgery to guide adjuvant therapy. The ongoing ALTAIR study will randomize patients who are ctDNA+ve at any time-point within 2 years after curative-intent surgery, to 6 months of oral trifluridine/tipiracil or placebo. The VEGA trial investigates a de-escalation strategy of randomizing patients who were MRD-ve at 4 weeks post-surgery to either surgery alone or 3 months of adjuvant CAPOX.
LARC is usually treated with multimodality treatment with either concurrent chemoradiotherapy (chemo-RT) or total neoadjuvant therapy (TNT) followed by total mesorectal excision surgery (TME). In general, ctDNA can be detected in approximately 57 - 77% of patients before surgery, 15.6 - 22.3% of patients after neoadjuvant therapy and 10.5 - 12% of patients after surgery - allowing for differences in the patients and assays across studies (34–36). Sampling of ctDNA at any of these perioperative time-points are also prognostic to a different extent. Appelt et al. (37) found that patients with baseline detectable hypermethylated ctDNA predicted improved OS (HR = 2.08; 95% CI = 1.23 - 1.51) and freedom from distant metastases (HR = 2.20; 95% CI = 1.19 - 4.07). Zhou et al. (34) showed that median VAF in baseline ctDNA was a strong independent predictor of metastasis-free survival (MFS) (HR = 1.27; P < 0.001). However, another study showed no association between ctDNA at baseline and MFS (38).
There is increasing interest in organ preservation strategies to spare patients the morbidity of a TME surgery. The recently reported phase II OPRA trial (NCT02008656) showed that up to half of patients may be able to achieve a clinical complete response (cCR) with a TNT approach without a detriment to DFS. ctDNA may complement conventional approach of predicting response to neoadjuvant therapy. Wang et al. (39) constructed a risk model unifying baseline ctDNA, ctDNA clearance, tumoral mutation status and magnetic resonance imaging (MRI)-based tumor regression grade in the prediction of pathological complete remission (pCR) after neoadjuvant therapy. This model was shown to be more accurate than models that were derived from only ctDNA or only MRI-based tumor regression grade.
The role of adjuvant chemotherapy after chemo-RT and surgery is controversial, with only one study showing a progression-free survival (PFS) benefit with FOLFOX in stage III LARC (40). Patients with detectable ctDNA after surgery are significantly associated with worse RFS if ctDNA is detected as early as 4-6 weeks after neoadjuvant chemo-RT (HR = 6.6; P < 0.001) or as late as 4-10 weeks after surgery (HR = 13.0; P < 0.001). This prognostic significance of MRD+ve status is independent of adjuvant chemotherapy and clinicopathological risk factors (36). This finding has formed the basis of the ongoing DYNAMIC RECTAL study (ACTRN-12617001560381) which will randomize patients to chemotherapy or surveillance after surgery, depending on the MRD status.
In conclusion, ctDNA has the potential of directing adjuvant therapy and improving the accuracy of assessing response to neoadjuvant therapy. These in turn may help to identify patients who might be candidates for de-escalated approaches, such as surveillance alone after TME, sphincter-preserving surgery or a wait-and-watch approach without surgery after chemo-RT. Validation in larger prospective trials using a risk-adapted approach in the management of LARC are ongoing.
In stage 4 CRC, ctDNA VAF is significantly associated with the number of metastatic sites (41) and is prognostic in resectable or unresectable metastatic CRC (42). Chemotherapy and targeted therapy are part of the standard treatments for stage 4 CRC, and various biomarker-guided therapies targeting BRAF mutation and EGFR-mediated signaling have improved patient survival (3). Many studies have reported a high concordance between tumor and plasma in the detection of KRAS and BRAF mutations using ddPCR (43, 44). For patients with resectable oligometastatic CRC, detectable levels of ctDNA after surgery and/or post-operative chemotherapy are associated with shorter RFS (45).
The dynamic changes of ctDNA during the first few cycles of chemotherapy may predict radiologic response. In a study by Tie et al., 74% of patients had a 10-fold decrease in ctDNA level before cycle 2, which correlated with radiologic responses at 8-10 weeks (odds ratio [OR] = 5.25; 95% CI = 1.38 – 19.93; P = 0.016) (46). Conversely, an increase in ctDNA during the first cycle of chemotherapy could predict inferior outcome (47). The findings by Tie et al. are supported by another prospective study by Garlan et al., where ‘ctDNA responders’ had superior radiologic response, PFS and OS than those who were ctDNA non-responders (48).
In clinical practice, RAS and BRAF mutations are routinely analyzed to guide anti-EGFR therapy in mCRC. Other molecular alterations also contribute to resistance to EGFR antibody therapy, such as PIK3CA mutation, HER2, MET and ERBB2 amplifications (49–51). Several landmark studies have suggested that when cancer cells are subjected to therapeutic pressure during anti-EGFR therapy, they acquire secondary genetic alterations in a process known as clonal evolution, which may contribute to drug resistance. Emergence of resistant clones can be tracked serially using ctDNA during anti-EGFR therapy, at as early as 10 months before the overt development of clinical resistance (52). Diaz et al. suggested that these drug-resistant KRAS mutant cancer cells are already present before a patients is started on EGFR antibody treatment (53). To confirm these findings, the PROSPECT-C phase II study was carried out to track the clonal evolution of resistant subclones using ctDNA during EGFR antibody therapy in patients with RAS-wildtype (WT) CRC. At baseline, 50% of patients already harbored aberrations in RAS pathway and BRAF V600E mutations in their ctDNA (54), and most patients (86.3%) would have detectable ctDNA levels of these resistant mutations at clinical progression (54).
ctDNA has also been used to track clonal evolution in treatment-naïve patients receiving EGFR antibodies together with chemotherapy. The CALGB/SWOG 80405 (Alliance) (55) is a first line study which randomizes patients to two different drug sequences of first-line therapy for stage IV CRC: chemotherapy plus EGFR antibody (cetuximab) or chemotherapy plus VEGF antibody (bevacizumab). In the post-hoc analysis of 133 patients with RAS/BRAF-WT CRC, ctDNA tracking showed a trend towards a higher prevalence of acquired mutations associated with resistance to EGFR antibody in patients (n = 11; 15.3%) randomized to the bevacizumab arm than patients in the cetuximab arm (n = 5; 8.2%) (OR = 2.0; P = 0.29). These provocative findings seem to suggest that exposure to bevacizumab in the first-line setting may increase the chance of patients acquiring EGFR antibody resistance-associated genomic alteration, thus further validation is warranted.
ctDNA is useful in selecting patients for EGFR antibody rechallenge. Clonal evolution is a dynamic process and therefore the optimal time-point and ctDNA VAF thresholds for determining whether a patient could be re-challenged with EGFR antibody therapy need to be defined. Siravegna et al. reported that the circulating level of mutant RAS clones increase initially during anti-EGFR therapy and gradually fall when therapy was withdrawn (49). Furthermore, circulating RAS and EGFR VAF undergo exponential decay at cessation of EGFR antibody therapy with a cumulative half-life of around 4.4 months (56). In a cohort of 80 patients who were re-treated with EGFR antibody, an overall response rate (ORR) of 23% was observed. A non-statistical trend towards higher ORR and PFS was noted if these patients were re-challenged after a longer drug holiday (in terms of <1 vs 2 half-lives) from the last EGFR antibody therapy (56). This knowledge may provide insight into the optimal timing of EGFR antibody re-challenge, however, the most appropriate VAF threshold that can guide treatment remains unclear (16). A meta-analysis showed that in patients without detectable RAS mutation in ctDNA, re-challenge with EGFR antibody therapy was associated with a larger benefit in PFS (HR = 0.40; 95% CI = 0.22 - 0.70; P = 0.001) and OS (HR = 0.37; 95% CI = 0.16 - 0.85; P = 0.02) than in patients with detectable RAS mutation (57). Several trials investigating the clinical impact of ctDNA-guided re-challenge of EGFR antibody therapy are ongoing, these include the RASINTRO (NCT03259009), FIRE4 trial (58) and the CHRONOS study (59) (Table 2). The CHRONOS is an interventional study which enrolls responders to EGFR antibody therapy who are ‘triple wild-type’ in RAS, BRAF and EGFR ectodomain in ctDNA. Patients will be re-challenged with panitumumab while ddPCR and NGS are used to track clonal evolution. Of the 52 patients screened in a preliminary report, 36 (69%) were triple wild-type, 27 received panitumumab with an ORR of 30% (59).
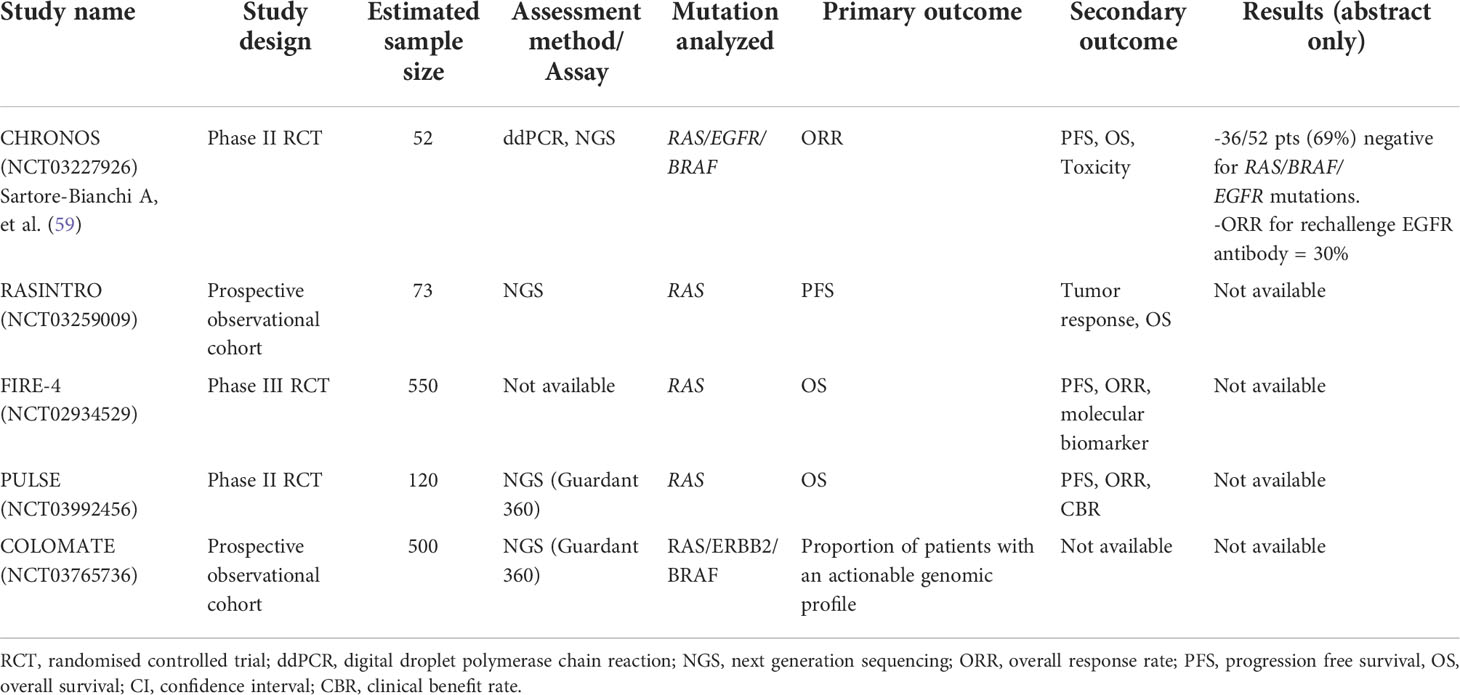
Table 2 Studies evaluating circulating tumor DNA as a screening tool to detect patients who could benefit from EGFR antibody re-challenge in metastatic colorectal cancer.
For other rarer molecular subgroups such as BRAF mutant, HER2 amplified and MSI-high (MSI-H) CRC, newer drug therapies are becoming available in the clinic (3). ctDNA monitoring has been used to track clonal evolutions in patient subgroups with BRAF V600E mutations and HER2 alterations in some clinical trials. In an exploratory analysis of the phase III BEACON trial (60) which demonstrated the superiority of targeting BRAF-EGFR-MEK inhibition with encorafenib-binimetinib-cetuximab over cetuximab-chemotherapy, over 90% of patients had detectable BRAF V600E mutations in ctDNA (GuardantOMNI assay). ctDNA VAF was found to be prognostic but not predictive of drug response (61). Patients with low ctDNA VAF (defined as lower than the median VAF) had longer median OS (14.8 months; 95% CI = 11.7 – 23.0) than in those with higher VAF (5.4 months; 95% CI = 4.4 – 6.1) when treated with encorafenib-cetuximab (61). In the phase II TRIUMPH trial (UMIN000027887) which tested the combination of trastuzumab plus pertuzumab in patients with HER2-amplified stage IV CRC, both tissue and ctDNA were used for determining HER2 status. The ORR were similar in patients who were tested HER2+ve with tissue compared with ctDNA (ORR = 30% vs 28% respectively) (62).The COLOMATE study (NCT03765736) is an ongoing, seamless adaptive protocol that primarily uses ctDNA (Guardant 360) to screen patients with secondary resistance to targeted therapies, for enrolment into 3 different clinical trials depending on their ctDNA genotype: panitumumab re-challenge (PULSE study NCT03992456); tucatinib, trastuzumab, and TAS-102 for patients if ctDNA show HER2-alteration; and re-challenge with encorafenib, cetuximab and binimetinib if patient is BRAF V600E-mutant (63).
The application of ctDNA in gastric cancers (GC) remains challenging due to the relatively low frequency of genomic alterations, the larger inter-patient and intra-patient temporo-spatial heterogeneity in tumors and plasma, as well as impaired tumor shredding from peritoneal metastases (64–66). Around 37% of GC tumors contain actionable somatic mutations (e.g. KRAS, TP53, PIK3CA) or gene amplifications (e.g. HER2, MET, EGFR, FGFR2, ERBB2) (64, 67–69). Ichikawa et al. found that 68.1% of cancer-related genes identified in ctDNA of patients with GC are actionable, with TP53 mutation and ERBB2 being the most common (70). Maron et al. reported that in a large cohort over 1600 patients with GEA, the presence of some actionable RTK amplifications (e.g. HER2, EGFR, MET, FGFR2) are of prognostic significance (64).
Several comparative analyses of genomic profiling using ctDNA and tumors have been carried out in GC. The Korean VIKTORY trial of stage IV GC reported a 89.5% concordance between liquid and tumor biopsy for MET amplification (71). Schrock et al. reported a 86% concordance in genomic alterations detected in tissue and plasma derived from 417 patients with GI cancers; however, only 63% of alterations found in ctDNA were detected in tumor, suggesting intra-tumoral heterogeneity (69). Moreover, the concordance rate was lower (50%) for gene amplifications such as HER2. Studies on the concordance between HER2 amplification in tumors using conventional methods (immunohistochemistry [IHC], or FISH) and ctDNA have shown mixed results. Some studies found high concordance with ddPCR (72, 73), but another showed that only 62% of patients with known HER2+ve tumors had detectable HER2 amplification in ctDNA (64). In conclusion, these studies suggest that genotype information from ctDNA is complementary but cannot replace tumor-based NGS in GC (64).
Similar to CRC, ctDNA has been investigated in the detection of MRD detection in resectable GC, gastroesophageal junction (GEJ) and esophageal adenocarcinoma (EAC). Data are limited by the relatively low level of ctDNA found before surgery (42 - 47%) in GC or EAC (74, 75). In one of the largest study in GEA, Maron et al. evaluated the utility of a commercial ctDNA-NGS assay (Guardant 360) 1630 patients with GC and EACs. MRD detection after curative surgery of EACs is strongly associated with an increased risk of recurrence (64). Kim et al. found that postoperative MRD+ve status in stage I-III GC precedes radiographic progression by 6 months (76), and is associated with shorter DFS (HR = 14.78; 95% CI = 7.991 – 61.29; P < 0.0001) and OS (HR = 7.664; 95% CI = 2.916 – 21.06; P = 0.002) (75). Similar findings are also reported by Openshaw et al. in GEJ cancers with shorter RFS (HR = 3.7; P = 0.028) (77).
In patients with advanced GC undergoing systemic therapy, Maron et al. showed that the maximal tumor VAF (maxVAF) in ctDNA could reflect tumor burden, such that in patients with a baseline maxVAF level of > 0.5%, who experienced a ≥ 50% fall in the maxVAF level during the first 5 months of systemic treatment, had superior median OS of 13.7 vs 8.6 months than those who had not (HR = 0.3; 95% CI = 0.1 – 0.8; P = 0.02) (64). The role of ctDNA in tracking clonal evolution in patients undergoing trastuzumab or lapatinib-based therapy has been evaluated in another study, where ctDNA monitoring has revealed multiple alterations that are purportedly associated with secondary resistance to anti-HER2 therapies, such as MYC, EGFR, FGFR2 and MET amplifications (78), as well as PIK3CA, ERBB2/4, NF1 and KRAS Q61R mutations (79, 80).
The PANGEA is the first reported prospective study using a biomarker-guided platform to individualize patients with stage IV GEA for systemic therapy (81). Pre-treatment tumor and ctDNA-based NGS target sequencing, IHC of programmed death receptor-1 ligand (PD-L1) expression, TMB and Epstein-Barr virus (EBV) status were used to stratify and assign patients to receive 1 out of 6 matched monoclonal antibody against PD1, EGFR, HER2, FGFR2 or VEGFR2 (81). The PANGEA met its primary endpoint with 45 of 68 (66%) patients alive at 12 months - exceeding the 50% historical control rate (81). The PLAGAST (NCT02674373) study is an ongoing non-interventional study which is aimed at evaluating the association of ctDNA dynamics with prognosis and response in patients with GC undergoing systemic therapy. The Oesophageal Cancer Clinical Molecular Stratification (OCCAMS) Consortium is leading an ongoing study of patients with resectable EAC where ctDNA will be performed (Signatera assay) during postoperative surveillance. A preliminary report on 12 patients showed that MRD+ve has a sensitivity and specificity of 100% in detecting early postoperative recurrence (82). Ococks et al. reported that ctDNA+ve patients have a longer median cancer-specific survival (10.0 months) than ctDNA negative patients (29.9 months) (HR = 5.55; 95% CI = 2.42 - 12.71; P = 0.0003) (83). Bonazzi et al. reported that detectable ctDNA variants in post-treatment plasma is associated with inferior disease-specific survival, and VAF increased with recurrence (84). In conclusion, these studies validate that ctDNA is prognostic for relapse and survival, and could be incorporated for risk stratification of patients for adjuvant chemotherapy escalation or de-escalation.
The mutational profile of ESCC is different from that of esophageal adenocarcinomas (EAC), but similar to that of other squamous cell cancers (85, 86). In a meta-analysis on sequencing methodologies including ctDNA analysis in ESCC, ctDNA assays have a relatively low sensitivity of 48.9% (29.4 - 68.8%), but high specificity of 95.5% (90.6 - 97.9%) for detecting recurrence post-surgery (87). The data on the utility of ctDNA in MRD detection in ESCC are mostly derived from small, retrospective studies. Two reports reported a decrease in ctDNA VAF in patients post-surgery (88, 89). In patients with localized ESCC undergoing neoadjuvant therapy, MRD+ve status post-treatment was associated with increased risk of tumor progression (HR = 18.7; P < 0.0001), distant metastases (HR = 32.1; P < 0.0001) and shorter disease-specific survival (HR = 23.1; P < 0. 0001) (14).
The genomic characterization of pancreatic cancer (PC) shows that somatic mutations of KRAS, TP53, CDKN2A are common (90). In a meta-analysis on 369 patients, KRAS mutation can be detected in ctDNA with a pooled sensitivity of 70% and specificity of 86% (91). However, one of the major limitations on the clinical applicability of ctDNA-NGS in PC is the low concordance rate of 31.9% in the tumor vs ctDNA-derived result (91). This may be due to the hepatic clearance of ctDNA released from the PC primary at the hepatic portal vein (92). Another limitation is the false-positive ddPCR results caused by benign conditions such as pancreatitis, therefore the additional use of methylation markers has been suggested to minimize this possibility (92, 93).
The prognostic value of ctDNA has been evaluated in a recent meta-analysis of 48 studies of over 3000 patients with different stages of PC. This study found that the detection of KRAS mutations via ctDNA has a negative impact on OS and PFS in PC (HR = 2.42; 95% CI = 1.95 - 2.99 and HR = 2.46; 95% CI = 2.01 - 3.00, respectively) (94). In localized PC, detection of ctDNA preoperatively is associated with poorer RFS (HR = 4.1; P = 0.002) and OS (HR = 4.0; P = 0.003) (95). This is consistent in another study, where ctDNA detection is associated with inferior RFS and PFS (HR = 2.27; 95% CI = 1.59 – 3.24; P < 0.001) and OS (HR = 2.04; 95% CI = 1.29 – 3.21; P = 0.002) (96). These studies suggest that MRD detection using ctDNA in the early postoperative period is prognostic in resectable PC (95, 97, 98), but may be affected by the use of neoadjuvant chemotherapy (99). Nevertheless, subsequent detection of ctDNA during surveillance strongly predicts recurrence with a 90% sensitivity and 88% specificity (99). The ongoing interventional phase III DYNAMIC-Pancreas study (ACTRN-12618000335291) in early-stage PC will evaluate the utility of ctDNA in guiding adjuvant therapy in resectable PC.
Most studies which investigated the potential of ctDNA in monitoring response to chemotherapy in advanced PC used KRAS genotyping, while a few targeted other clonal mutations. KRAS mutation can be detected in 36 out of 54 (67%) of patients with advanced PC (100). Collectively, several studies have shown that ctDNA increase tends to precede clinical progression as determined by imaging and serum Ca19.9 level by a few months (100–102). In a meta-analysis of studies on patients with detectable KRAS before treatment, conversion to undetectable KRAS after treatment is associated with better prognosis (94). ctDNA has also been used to track other cancer-specific mutations such as TP53, APC, ATM, FBXW7, SMAD4, CDKN2A and other variants (101, 103). BRCA1/2 mutations can be found between 1-10% of PC and may predict response to PARP inhibitors in the palliative setting (104). A study has found a high degree of concordance in BRCA mutation detected in tissue and plasma (103). Larger studies are needed to test the feasibility of using ctDNA to select and monitor patients for BRCA1/2 mutation and PARP inhibitor therapy. In conclusion, the development of ctDNA in monitoring response to systemic treatment is still at an early stage and requires further validation.
Most patients with cholangiocarcinomas (CC) - including IHCC, EHCC and gallbladder cancer, are usually diagnosed at an advanced stage where post-operative recurrence risk is high (105). FGFR1-3 fusions and IDH1/2 mutations can be found in 15 - 20% of IHCC, where the concordance of tumor and ctDNA-derived is higher for IHCC (92%) than that of EHCC (55%) (105). The detection of ctDNA using target-panel NGS has been used to track clonal evolution during chemotherapy, demonstrating that over 60% of patients may develop new driver genes at progression (105). There have been significant advances in the development of new targeted therapies for CC such as IDH1 inhibitor for IDH1 mutant tumors (106), and FGFR inhibitors for tumors harboring FGFR2 fusions (107, 108). ctDNA has been investigated in selecting patients for such therapies and in tracking emergence of secondary resistance to these agents. Goyal et al. were the first to describe the molecular basis of acquired resistance to a FGFR2 antibody (BGJ39) (109) by using serial cfDNA monitoring during treatment. An acquired V564F mutation was found in 3 out of 4 patients who progressed, while 2 progressors had multiple FGFR point mutations. There was a high concordance between tissue and ctDNA in detecting these resistant variants. This study may pave the way for larger studies on ctDNA in guiding anti-FGFR2 therapy for CC.
Immune-checkpoint inhibitors (ICI) such as PD1 and CTLA-4 therapy are now part of the standard therapeutic options for stage IV MSI-H CRC in the first and subsequent line settings. In addition, patients with other GI cancers that are MSI-H or TMB > 10 mut/Mb (110) may be suitable for anti-PD1 therapy in the palliative setting. Several studies have investigated the feasibility of ctDNA in assessing MSI, TMB status in GI cancers. Nakamura et al. compared ctDNA NGS (Guardant 360) and tissue based MSI assessments in a cohort of 658 patients with advanced GI cancer in the SCRUM-Japan GOZILA study - an observational ctDNA-based study which screens patients with GI cancers for enrollment into clinical trials within a nation-wide trial network (111). The concordance between tumor and ctDNA for detection of MSI is high with an overall percent agreement of 98.2% (95% CI = 96.8 - 99.1). In particular, ctDNA was able to identify patients with MSI-high tumors who might benefit from anti-PD1 therapy (111). Using the Guardant360 assay, Maron et al. reported a 100% concordance between tumor-derived MMR status (IHC) and plasma-derived MSI-status using ctDNA-NGS in 6 patients (64). In contrast, there is significant discordance between tumor and ctDNA-derived TMB assessment. In a ‘real-life’ retrospective study of 410 patients (82 had GI cancers) with matching TMB results from tumor and plasma-based commercial NGS assays in the community setting, the median TMB was higher in plasma (m = 10.5 mut/Mb) than in tumor (m = 6.0 mut/Mb; P < 0.001). This will have obvious implication on selecting patients with non-CRC GI cancers for PD1 inhibitors based on ctDNA TMB alone, since the drug label for the tissue-agnostic approval of pembrolizumab recommends that the TMB threshold should be ≥ 10 mut/mb. In conclusion, if ctDNA TMB is used to select patients with GI cancers for PD1 inhibitor, a much higher ctDNA TMB threshold (up to 12 to 40 mut/Mb depending on the assays used) should be used to guide treatment decisions (112).
The monitoring of ctDNA during ICI therapy has been investigated in stage IV MSI-H CRC in a number of small cases reports. In these studies, the following endpoints were analyzed: the quantitation of VAF, measurement of TMB and tracking of tumor-specific mutations such as TP53, RAS and BRAF (113). Some studies have suggested that MSI-H CRC are often poorly-differentiated and produce significantly lower levels of serum tumor markers such as CEA and CA 19.9 (114–116) than well-differentiated tumors. Therefore, ctDNA holds promise as a blood-based predictive biomarker of response to ICI for such patients.
In Zhang et al.’s study of 978 patients across 16 tumor types (48 had GEAs, 32 had PC and 58 had MSI-H solid tumors) who were undergoing ICI therapy, changes in VAF during treatment could predict drug response, such that patients who could completely clear ctDNA (VAF = 0) had longer PFS and OS (P < 0.0001) than those who could not (117). Similarly, Kim et al. also found that in a study of 61 patients with stage IV GC treated with a PD1 inhibitor, changes in the ctDNA levels at 6 weeks post-treatment correlated with PFS and ORR (118).
Apart from VAF clearance as an endpoint, another study by Jin et al. investigated other endpoints e.g. ‘decline in maxVAF’ and ‘ctDNA-positivity’ via a NGS ctDNA assay, in 46 patients treated with PD1 inhibitor alone or in combination with chemotherapy (119). The median PFS was significantly longer in patients who experienced > 25% decline in maxVAF (7.3 months vs 3.6 months, P = 0.0011; 53.3% vs 13.3%, P = 0.06), and in those who had undetectable ctDNA (7.4 months vs. 4.9 months, P = 0.025) after ICI-based therapy (119).
Advances in NGS and PCR technologies have enabled the accurate detection and quantification of ctDNA in patients with different stages of GI cancers. There is a practical need to identify an informative and less invasive biomarker to help guide adjuvant, neoadjuvant and palliative drug therapies. The strongest evidence available to date showed that ctDNA is a strong prognostic marker when used to detect MRD following curative intent surgery in resectable GI cancer. The DYNAMIC II study is the first to show that ctDNA can direct adjuvant chemotherapy in the management of stage II colon cancer without compromising RFS. Several interventional studies with adaptive design using ctDNA as a screening platform are ongoing in patients with resectable CRC, PC and LARC. These studies are designed to definitively address the questions of whether ctDNA is superior to conventional methods of guiding adjuvant chemotherapy on patient’s survival, and whether ctDNA guided escalation or de-escalation of adjuvant therapy may help to improve survival and minimize the risk of long-term treatment-related morbidities.
In the palliative setting, there are emerging data to suggest that ctDNA dynamics during the early treatment period are both prognostic and/or predictive of subsequent response to systemic treatments. Furthermore, serial measurement of ctDNA during targeted therapies has enabled tracking of clonal evolution and emergence of secondary resistance-related variants to some targeted therapies. Some evidence supports the use of ctDNA in guiding EGFR antibody rechallenge followed by ICI, while more evidence is needed for other targeted therapies e.g. HER2 or BRAF.
As NGS technologies and other pre-analyzed variables are refined continuously with time, the cost and accuracy of ctDNA are likely to improve with time. There remain challenges that need to be overcome. It is unclear whether it is more informative to do both blood and tumor NGS at baseline than other modality alone in guiding treatment decisions or in selecting patients for clinical trials, given the intratumoral heterogeneity in GI cancers. Furthermore, there is a lack in consensus on determining the most biologically meaningful thresholds of ctDNA-related metrics (e.g. maxVAF, percentage change in VAF) to guide oncologists in practice. In postoperative surveillance, it is unlikely that ctDNA will completely replace conventional diagnostic and staging tools e.g. imaging and protein-based serum cancer markers in the management of GI cancers. In clinical trials, ctDNA may potentially accelerate drug development by facilitating the molecular genotyping of patients for clinical trials of novel targeted therapies, in detecting early signals of drug response and in tracking emerging clonal resistance (120).
In conclusion, it is important to reach consensus on how ctDNA as a biomarker should be practically incorporated into current complex treatment algorithms to guide the treatment of GI cancers in potentially curative and palliative settings. One of the possible directions is to use the massive volume of genomic data derived from the systemic profiling of ctDNA for the development of artificial intelligence driven computational models and programs that can be applied in the routine oncological care of patients with GI cancers.
RL and BM are responsible for overall design, planning, writing and editing of manuscript and figure. DJ is responsible for writing up of specific sections and the table. WL, KC, GL, ML, and JW are responsible for planning and editing of specific sections. All authors contributed to the article and approved the submitted version.
This review is supported in part by the Kingboard Precision Oncology Program and the Charlie Lee Precision Immuno-oncology program, The Chinese University of Hong Kong, Hong Kong SAR.
We would like to thank Ms. Alice Kong for clerical assistance.
KC is a director of Take2, DRA and Novostics. KC holds equities in Take2, DRA, Grail/Illumina. KC and WL were previous consultants to Grail. WL holds equity in Grail/Illumina. KC holds patents portfolio in molecular diagnostics and receive royalties from Take2, DRA, Grail, Illumina, Sequenome, Xcelom.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Nakamura Y, Kawazoe A, Lordick F, Janjigian YY, Shitara K. Biomarker-targeted therapies for advanced-stage gastric and gastro-oesophageal junction cancers: an emerging paradigm. Nat Rev Clin Oncol (2021) 18(8):473–87. doi: 10.1038/s41571-021-00492-2
3. Di Nicolantonio F, Vitiello PP, Marsoni S, Siena S, Tabernero J, Trusolino L, et al. Precision oncology in metastatic colorectal cancer - from biology to medicine. Nat Rev Clin Oncol (2021) 18(8):506–25. doi: 10.1038/s41571-021-00495-z
4. Hosein AN, Dougan SK, Aguirre AJ, Maitra A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat Cancer (2022) 3(3):272–86. doi: 10.1038/s43018-022-00349-2
5. Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch R-D, et al. DNA Fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic Cells1. Cancer Res (2001) 61(4):1659–65.
6. Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res (2014) 24(6):766–9. doi: 10.1038/cr.2014.44
7. Jiang P, Chan CWM, Chan KCA, Cheng SH, Wong J, Wong VW-S, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci United States America (2015) 112(11):E1317–E25. doi: 10.1073/pnas.1500076112
8. Siravegna G, Mussolin B, Venesio T, Marsoni S, Seoane J, Dive C, et al. How liquid biopsies can change clinical practice in oncology. Ann Oncol (2019) 30(10):1580–90. doi: 10.1093/annonc/mdz227
9. Larribère L, Martens UM. Advantages and challenges of using ctDNA NGS to assess the presence of minimal residual disease (MRD) in solid tumors. Cancers (Basel) (2021) 13(22):5698. doi: 10.3390/cancers13225698
10. To YH, Lee B, Wong HL, Gibbs P, Tie J. Circulating tumour DNA to guide treatment of gastrointestinal malignancies. Visc Med (2020) 36(5):388–96. doi: 10.1159/000509657
11. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med (2014) 6(224):224ra24. doi: 10.1126/scitranslmed.3007094
12. Strickler JH, Loree JM, Ahronian LG, Parikh AR, Niedzwiecki D, Pereira AAL, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov (2018) 8(2):164–73. doi: 10.1158/2159-8290.CD-17-1009
13. Vidal J, Muinelo L, Dalmases A, Jones F, Edelstein D, Iglesias M, et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann Oncol (2017) 28(6):1325–32. doi: 10.1093/annonc/mdx125
14. Azad TD, Chaudhuri AA, Fang P, Qiao Y, Esfahani MS, Chabon JJ, et al. Circulating tumor DNA analysis for detection of minimal residual disease after chemoradiotherapy for localized esophageal cancer. Gastroenterology (2020) 158(3):494–505 e6. doi: 10.1053/j.gastro.2019.10.039
15. Abbosh C, Swanton C, Birkbak NJ. Clonal haematopoiesis: a source of biological noise in cell-free DNA analyses. Ann Oncol (2019) 30(3):358–9. doi: 10.1093/annonc/mdy552
16. Dasari A, Morris VK, Allegra CJ, Atreya C, Benson AB 3rd, Boland P, et al. ctDNA applications and integration in colorectal cancer: an NCI colon and rectal-anal task forces whitepaper. Nat Rev Clin Oncol (2020) 17(12):757–70. doi: 10.1038/s41571-020-0392-0
17. Cescon DW, Bratman SV, Chan SM, Siu LL. Circulating tumor DNA and liquid biopsy in oncology. Nat Cancer (2020) 1(3):276–90. doi: 10.1038/s43018-020-0043-5
18. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med (2016) 8(346):346ra92. doi: 10.1126/scitranslmed.aaf6219
19. Tie J, Cohen JD, Wang Y, Christie M, Simons K, Lee M, et al. Circulating tumor DNA analyses as markers of recurrence risk and benefit of adjuvant therapy for stage III colon cancer. JAMA Oncol (2019) 5(12):1710–7. doi: 10.1001/jamaoncol.2019.3616
20. Tie J, Cohen JD, Lo SN, Wang Y, Li L, Christie M, et al. Prognostic significance of postsurgery circulating tumor DNA in nonmetastatic colorectal cancer: Individual patient pooled analysis of three cohort studies. Int J Cancer (2021) 148(4):1014–26. doi: 10.1002/ijc.33312
21. Reinert T, Henriksen TV, Christensen E, Sharma S, Salari R, Sethi H, et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol (2019) 5(8):1124–31. doi: 10.1001/jamaoncol.2019.0528
22. Taieb J, Taly V, Henriques J, Bourreau C, Mineur L, Bennouna J, et al. Prognostic value and relation with adjuvant treatment duration of ctDNA in stage III colon cancer: a Post hoc analysis of the PRODIGE-GERCOR IDEA-France trial. Clin Cancer Res (2021) 27(20):5638–46. doi: 10.1158/1078-0432.CCR-21-0271
23. Tarazona N, Henriksen TV, Carbonell-Asins JA, Reinert T, Sharma S, Roda D, et al. Circulating tumor DNA to detect minimal residual disease, response to adjuvant therapy, and identify patients at high risk of recurrence in patients with stage I-III CRC. J Clin Oncol (2020) 38(15_suppl):4009. doi: 10.1200/JCO.2020.38.15_suppl.4009
24. Fakih M, Sandhu J, Wang C, Kim J, Chen YJ, Lai L, et al. Evaluation of comparative surveillance strategies of circulating tumor DNA, imaging, and carcinoembryonic antigen levels in patients with resected colorectal cancer. JAMA Netw Open (2022) 5(3):e221093. doi: 10.1001/jamanetworkopen.2022.1093
25. Anandappa G, Starling N, Begum R, Bryant A, Sharma S, Renner D, et al. Minimal residual disease (MRD) detection with circulating tumor DNA (ctDNA) from personalized assays in stage II-III colorectal cancer patients in a U.K. multicenter prospective study (TRACC). J Clin Oncol (2021) 39(3_suppl):102–. doi: 10.1200/JCO.2021.39.3_suppl.102
26. Kasi PM, Sawyer S, Guilford J, Munro M, Ellers S, Wulff J, et al. BESPOKE study protocol: a multicentre, prospective observational study to evaluate the impact of circulating tumour DNA guided therapy on patients with colorectal cancer. BMJ Open (2021) 11(9):e047831. doi: 10.1136/bmjopen-2020-047831
27. Shirasu H, Taniguchi H, Watanabe J, Kotaka M, Yamazaki K, Hirata K, et al. O-11 monitoring molecular residual disease by circulating tumor DNA in resectable colorectal cancer: Molecular subgroup analyses of a prospective observational study GALAXY in CIRCULATE-Japan. Ann Oncol (2021) 32:S222–S3. doi: 10.1016/j.annonc.2021.05.015
28. Tie J, Cohen JD, Lahouel K, Lo SN, Wang Y, Kosmider S, et al. Circulating tumor DNA analysis guiding adjuvant therapy in stage II colon cancer. N Engl J Med (2022) 386(24):2261–72. doi: 10.1056/NEJMoa2200075
29. Schraa SJ, van Rooijen KL, van der Kruijssen DEW, Rubio Alarcón C, Phallen J, Sausen M, et al. Circulating tumor DNA guided adjuvant chemotherapy in stage II colon cancer (MEDOCC-CrEATE): study protocol for a trial within a cohort study. BMC Cancer (2020) 20(1):790. doi: 10.1186/s12885-020-07252-y
30. Lonardi S, Montagut C, Pietrantonio F, Elez E, Sartore-Bianchi A, Tarazona N, et al. The PEGASUS trial: Post-surgical liquid biopsy-guided treatment of stage III and high-risk stage II colon cancer patients. J Clin Oncol (2020) 38(15_suppl):TPS4124–TPS. doi: 10.1200/jco.2020.38.15_suppl.tps4124
31. Morris VK, Yothers G, Kopetz S, Jacobs SA, Lucas PC, Iqbal A, et al. Phase II/III study of circulating tumOr DNA as a predictive BiomaRker in adjuvant chemotherapy in patients with stage II colon cancer: NRG-GI005 (COBRA). J Clin Oncol (2021) 39(15_suppl):TPS3622–TPS. doi: 10.1200/JCO.2021.39.15_suppl.TPS3622
32. Nakamura Y, Fujisawa T, Taniguchi H, Bando H, Okamoto W, Tsuchihara K, et al. SCRUM-Japan GI-SCREEN and MONSTAR-SCREEN: Path to the realization of biomarker-guided precision oncology in advanced solid tumors. Cancer Sci (2021) 112(11):4425–32. doi: 10.1111/cas.15132
33. Taniguchi H, Nakamura Y, Kotani D, Yukami H, Mishima S, Sawada K, et al. CIRCULATE-Japan: Circulating tumor DNA-guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci (2021) 112(7):2915–20. doi: 10.1111/cas.14926
34. Zhou J, Wang C, Lin G, Xiao Y, Jia W, Xiao G, et al. Serial circulating tumor DNA in predicting and monitoring the effect of neoadjuvant chemoradiotherapy in patients with rectal cancer: A prospective multicenter study. Clin Cancer Res (2021) 27(1):301–10. doi: 10.1158/1078-0432.CCR-20-2299
35. Murahashi S, Akiyoshi T, Sano T, Fukunaga Y, Noda T, Ueno M, et al. Serial circulating tumour DNA analysis for locally advanced rectal cancer treated with preoperative therapy: prediction of pathological response and postoperative recurrence. Br J Cancer (2020) 123(5):803–10. doi: 10.1038/s41416-020-0941-4
36. Tie J, Cohen JD, Wang Y, Li L, Christie M, Simons K, et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut (2019) 68(4):663–71. doi: 10.1136/gutjnl-2017-315852
37. Appelt AL, Andersen RF, Lindebjerg J, Jakobsen A. Prognostic value of serum NPY hypermethylation in neoadjuvant chemoradiotherapy for rectal cancer: Secondary analysis of a randomized trial. Am J Clin Oncol (2020) 43(1):9–13. doi: 10.1097/COC.0000000000000609
38. Khakoo S, Carter PD, Brown G, Valeri N, Picchia S, Bali MA, et al. MRI Tumor regression grade and circulating tumor DNA as complementary tools to assess response and guide therapy adaptation in rectal cancer. Clin Cancer Res (2020) 26(1):183–92. doi: 10.1158/1078-0432.CCR-19-1996
39. Wang Y, Yang L, Bao H, Fan X, Xia F, Wan J, et al. Utility of ctDNA in predicting response to neoadjuvant chemoradiotherapy and prognosis assessment in locally advanced rectal cancer: A prospective cohort study. PloS Med (2021) 18(8):e1003741. doi: 10.1371/journal.pmed.1003741
40. Hong YS, Kim SY, Lee JS, Nam B-H, K-p K, JE K, et al. Oxaliplatin-based adjuvant chemotherapy for rectal cancer after preoperative chemoradiotherapy (ADORE): Long-term results of a randomized controlled trial. J Clin Oncol (2019) 37(33):3111–23. doi: 10.1200/JCO.19.00016
41. Osumi H, Shinozaki E, Takeda Y, Wakatsuki T, Ichimura T, Saiura A, et al. Clinical relevance of circulating tumor DNA assessed through deep sequencing in patients with metastatic colorectal cancer. Cancer Med (2019) 8(1):408–17. doi: 10.1002/cam4.1913
42. Jones RP, Pugh SA, Graham J, Primrose JN, Barriuso J. Circulating tumour DNA as a biomarker in resectable and irresectable stage IV colorectal cancer; a systematic review and meta-analysis. Eur J Cancer (2021) 144:368–81. doi: 10.1016/j.ejca.2020.11.025
43. Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med (2014) 20(4):430–5. doi: 10.1038/nm.3511
44. Spindler KL, Pallisgaard N, Appelt AL, Andersen RF, Schou JV, Nielsen D, et al. Clinical utility of KRAS status in circulating plasma DNA compared to archival tumour tissue from patients with metastatic colorectal cancer treated with anti-epidermal growth factor receptor therapy. Eur J Cancer (2015) 51(17):2678–85. doi: 10.1016/j.ejca.2015.06.118
45. Wang DS, Yang H, Liu XY, Chen ZG, Wang Y, Fong WP, et al. Dynamic monitoring of circulating tumor DNA to predict prognosis and efficacy of adjuvant chemotherapy after resection of colorectal liver metastases. Theranostics. (2021) 11(14):7018–28. doi: 10.7150/thno.59644
46. Tie J, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol (2015) 26(8):1715–22. doi: 10.1093/annonc/mdv177
47. Lyskjaer I, Kronborg CS, Rasmussen MH, Sorensen BS, Demuth C, Rosenkilde M, et al. Correlation between early dynamics in circulating tumour DNA and outcome from FOLFIRI treatment in metastatic colorectal cancer. Sci Rep (2019) 9(1):11542. doi: 10.1038/s41598-019-47708-1
48. Garlan F, Laurent-Puig P, Sefrioui D, Siauve N, Didelot A, Sarafan-Vasseur N, et al. Early evaluation of circulating tumor DNA as marker of therapeutic efficacy in metastatic colorectal cancer patients (PLACOL study). Clin Cancer Res (2017) 23(18):5416–25. doi: 10.1158/1078-0432.CCR-16-3155
49. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med (2015) 21(7):795–801. doi: 10.1038/nm.3870
50. Mohan S, Heitzer E, Ulz P, Lafer I, Lax S, Auer M, et al. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PloS Genet (2014) 10(3):e1004271. doi: 10.1371/journal.pgen.1004271
51. Pietrantonio F, Vernieri C, Siravegna G, Mennitto A, Berenato R, Perrone F, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res (2017) 23(10):2414–22. doi: 10.1158/1078-0432.CCR-16-1863
52. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature (2012) 486(7404):532–6. doi: 10.1038/nature11156
53. Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature (2012) 486(7404):537–40. doi: 10.1038/nature11219
54. Khan KH, Cunningham D, Werner B, Vlachogiannis G, Spiteri I, Heide T, et al. Longitudinal liquid biopsy and mathematical modeling of clonal evolution forecast time to treatment failure in the PROSPECT-c phase II colorectal cancer clinical trial. Cancer Discov (2018) 8(10):1270–85. doi: 10.1158/2159-8290.CD-17-0891
55. Raghav KPS, Ou F-S, Venook AP, Innocenti F, Sun R, Lenz H-J, et al. Circulating tumor DNA dynamics on front-line chemotherapy with bevacizumab or cetuximab in metastatic colorectal cancer: A biomarker analysis for acquired genomic alterations in CALGB/SWOG 80405 (Alliance) randomized trial. J Clin Oncol (2022) 40(4_suppl):193–. doi: 10.1200/JCO.2022.40.4_suppl.193
56. Parseghian CM, Loree JM, Morris VK, Liu X, Clifton KK, Napolitano S, et al. Anti-EGFR-resistant clones decay exponentially after progression: implications for anti-EGFR re-challenge. Ann Oncol (2019) 30(2):243–9. doi: 10.1093/annonc/mdy509
57. Vlachou MS, Mauri D, Zarkavelis G, Ntellas P, Tagkas C, Gkoura S, et al. Plasma ctDNA RAS status selects patients for anti-EGFR treatment rechallenge in metastatic colorectal cancer: a meta-analysis. Exp Oncol (2021) 43(3):252–6. doi: 10.32471/exp-oncology.2312-8852.vol-43-no-3.16592
58. Stintzing S, Weikersthal Lv, Fuchs M, Kaiser F, Heinrich K, Modest DP, et al. Randomized study to investigate a switch maintenance concept with 5-FU plus bevacizumab after FOLFIRI plus cetuximab induction treatment versus continued treatment with FOLFIRI plus cetuximab: Report of a secondary endpoint of the phase-III FIRE-4 study (AIO KRK-0114). J Clin Oncol (2022) 40(16_suppl):3519–. doi: 10.1200/JCO.2022.40.16_suppl.3519
59. Sartore-Bianchi A, Pietrantonio F, Lonardi S, Mussolin B, Rua F, Fenocchio E, et al. Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: The CHRONOS trial. J Clin Oncol (2021) 39(15_suppl):3506–. doi: 10.1200/JCO.2021.39.15_suppl.3506
60. Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med (2019) 381(17):1632–43. doi: 10.1056/NEJMoa1908075
61. Kopetz S, Murphy DA, Pu J, Yaeger R, Ciardiello F, Desai J, et al. Evaluation of baseline BRAF V600E mutation in circulating tumor DNA and efficacy response from the BEACON study. J Clin Oncol (2022) 40(4_suppl):162–. doi: 10.1200/JCO.2022.40.4_suppl.162
62. Nakamura Y, Okamoto W, Kato T, Esaki T, Kato K, Komatsu Y, et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: a phase 2 trial. Nat Med (2021) 27(11):1899–903. doi: 10.1038/s41591-021-01553-w
63. Yoshino T. COLOMATE challenge to overcome resistance in metastatic colorectal cancer. Oncol (Williston Park) (2021) 35(10):656. doi: 10.46883/ONC.2021.3510.0656
64. Maron SB, Chase LM, Lomnicki S, Kochanny S, Moore KL, Joshi SS, et al. Circulating tumor DNA sequencing analysis of gastroesophageal adenocarcinoma. Clin Cancer Res (2019) 25(23):7098–112. doi: 10.1158/1078-0432.CCR-19-1704
65. Frankell AM, Jammula S, Li X, Contino G, Killcoyne S, Abbas S, et al. The landscape of selection in 551 esophageal adenocarcinomas defines genomic biomarkers for the clinic. Nat Genet (2019) 51(3):506–16. doi: 10.1038/s41588-018-0331-5
66. Pectasides E, Stachler MD, Derks S, Liu Y, Maron S, Islam M, et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discovery (2018) 8(1):37–48. doi: 10.1158/2159-8290.CD-17-0395
67. Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. (2012) 61(5):673–84. doi: 10.1136/gutjnl-2011-301839
68. Kato S, Okamura R, Baumgartner JM, Patel H, Leichman L, Kelly K, et al. Analysis of circulating tumor DNA and clinical correlates in patients with esophageal, gastroesophageal junction, and gastric adenocarcinoma. Clin Cancer Res (2018) 24(24):6248–56. doi: 10.1158/1078-0432.CCR-18-1128
69. Schrock AB, Pavlick D, Klempner SJ, Chung JH, Forcier B, Welsh A, et al. Hybrid capture-based genomic profiling of circulating tumor DNA from patients with advanced cancers of the gastrointestinal tract or anus. Clin Cancer Res (2018) 24(8):1881–90. doi: 10.1158/1078-0432.CCR-17-3103
70. Ichikawa H, Nagahashi M, Shimada Y, Hanyu T, Ishikawa T, Kameyama H, et al. Actionable gene-based classification toward precision medicine in gastric cancer. Genome Med (2017) 9(1):93. doi: 10.1186/s13073-017-0484-3
71. Lee J, Kim ST, Kim K, Lee H, Kozarewa I, Mortimer PGS, et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY umbrella trial. Cancer Discovery (2019) 9(10):1388–405. doi: 10.1158/2159-8290.CD-19-0442
72. Wang H, Li B, Liu Z, Gong J, Shao L, Ren J, et al. HER2 copy number of circulating tumour DNA functions as a biomarker to predict and monitor trastuzumab efficacy in advanced gastric cancer. Eur J Cancer (2018) 88:92–100. doi: 10.1016/j.ejca.2017.10.032
73. Shoda K, Ichikawa D, Fujita Y, Masuda K, Hiramoto H, Hamada J, et al. Monitoring the HER2 copy number status in circulating tumor DNA by droplet digital PCR in patients with gastric cancer. Gastric Cancer (2017) 20(1):126–35. doi: 10.1007/s10120-016-0599-z
74. Paschold L, Binder M. Circulating tumor DNA in gastric and gastroesophageal junction cancer. Curr Oncol (2022) 29(3):1430–41. doi: 10.3390/curroncol29030120
75. Yang J, Gong Y, Lam VK, Shi Y, Guan Y, Zhang Y, et al. Deep sequencing of circulating tumor DNA detects molecular residual disease and predicts recurrence in gastric cancer. Cell Death Dis (2020) 11(5):346. doi: 10.1038/s41419-020-2531-z
76. Kim YW, Kim YH, Song Y, Kim HS, Sim HW, Poojan S, et al. Monitoring circulating tumor DNA by analyzing personalized cancer-specific rearrangements to detect recurrence in gastric cancer. Exp Mol Med (2019) 51(8):1–10. doi: 10.1038/s12276-019-0292-5
77. Openshaw MR, Mohamed AA, Ottolini B, Fernandez-Garcia D, Richards CJ, Page K, et al. Longitudinal monitoring of circulating tumour DNA improves prognostication and relapse detection in gastroesophageal adenocarcinoma. Br J Cancer (2020) 123(8):1271–9. doi: 10.1038/s41416-020-1002-8
78. Kim ST, Banks KC, Pectasides E, Kim SY, Kim K, Lanman RB, et al. Impact of genomic alterations on lapatinib treatment outcome and cell-free genomic landscape during HER2 therapy in HER2+ gastric cancer patients. Ann Oncol (2018) 29(4):1037–48. doi: 10.1093/annonc/mdy034
79. Wang DS, Liu ZX, Lu YX, Bao H, Wu X, Zeng ZL, et al. Liquid biopsies to track trastuzumab resistance in metastatic HER2-positive gastric cancer. Gut. (2019) 68(7):1152–61. doi: 10.1136/gutjnl-2018-316522
80. Wang Y, Zhao C, Chang L, Jia R, Liu R, Zhang Y, et al. Circulating tumor DNA analyses predict progressive disease and indicate trastuzumab-resistant mechanism in advanced gastric cancer. EBioMedicine. (2019) 43:261–9. doi: 10.1016/j.ebiom.2019.04.003
81. Catenacci DVT, Moya S, Lomnicki S, Chase LM, Peterson BF, Reizine N, et al. Personalized antibodies for gastroesophageal adenocarcinoma (PANGEA): A phase II study evaluating an individualized treatment strategy for metastatic disease. Cancer Discovery (2021) 11(2):308–25. doi: 10.1158/2159-8290.CD-20-1408
82. Ococks E, Ng A, Devonshire G, Dashner S, Chan WC, Sharma S, et al. 1491P bespoke circulating tumor DNA assay for the detection of minimal residual disease in esophageal adenocarcinoma patients. Ann Oncol (2020) 31:S926. doi: 10.1016/annonc/annonc284
83. Ococks E, Frankell AM, Masque Soler N, Grehan N, Northrop A, Coles H, et al. Longitudinal tracking of 97 esophageal adenocarcinomas using liquid biopsy sampling. Ann Oncol (2021) 32(4):522–32. doi: 10.1016/j.annonc.2020.12.010
84. Bonazzi VF, Aoude LG, Brosda S, Lonie JM, Patel K, Bradford JJ, et al. ctDNA as a biomarker of progression in oesophageal adenocarcinoma. ESMO Open (2022) 7(3):100452. doi: 10.1016/j.esmoop.2022.100452
85. Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet (2014) 46(5):467–73. doi: 10.1038/ng.2935
86. Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet (2014) 46(10):1097–102. doi: 10.1038/ng.3076
87. Chidambaram S, Markar SR. Clinical utility and applicability of circulating tumor DNA testing in esophageal cancer: a systematic review and meta-analysis. Dis Esophagus (2022) 35(2):doab046. doi: 10.1093/dote/doab046
88. Luo H, Li H, Hu Z, Wu H, Liu C, Li Y, et al. Noninvasive diagnosis and monitoring of mutations by deep sequencing of circulating tumor DNA in esophageal squamous cell carcinoma. Biochem Biophys Res Commun (2016) 471(4):596–602. doi: 10.1016/j.bbrc.2016.02.011
89. Meng P, Wei J, Geng Y, Chen S, Terpstra MM, Huang Q, et al. Targeted sequencing of circulating cell-free DNA in stage II-III resectable oesophageal squamous cell carcinoma patients. BMC Cancer (2019) 19(1):818. doi: 10.1186/s12885-019-6025-2
90. Network CGAR. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell (2017) 32(2):185–203.e13. doi: 10.1016/j.ccell.2017.07.007
91. Luchini C, Veronese N, Nottegar A, Cappelletti V, Daidone MG, Smith L, et al. Liquid biopsy as surrogate for tissue for molecular profiling in pancreatic cancer: A meta-analysis towards precision medicine. Cancers (Basel) (2019) 11(8):1152. doi: 10.3390/cancers11081152
92. Rashid S, Singh N, Gupta S, Rashid S, Nalika N, Sachdev V, et al. Progression of chronic pancreatitis to pancreatic cancer: Is there a role of gene mutations as a screening tool? Pancreas (2018) 47(2):227–32. doi: 10.1097/MPA.0000000000000975
93. Kisiel JB, Raimondo M, Taylor WR, Yab TC, Mahoney DW, Sun Z, et al. New DNA methylation markers for pancreatic cancer: Discovery, tissue validation, and pilot testing in pancreatic juice. Clin Cancer Res (2015) 21(19):4473–81. doi: 10.1158/1078-0432.CCR-14-2469
94. Milin-Lazovic J, Madzarevic P, Rajovic N, Djordjevic V, Milic N, Pavlovic S, et al. Meta-analysis of circulating cell-free DNA's role in the prognosis of pancreatic cancer. Cancers (Basel) (2021) 13(14):3378. doi: 10.3390/cancers13143378
95. Lee B, Lipton L, Cohen J, Tie J, Javed AA, Li L, et al. Circulating tumor DNA as a potential marker of adjuvant chemotherapy benefit following surgery for localized pancreatic cancer. Ann Oncol (2019) 30(9):1472–8. doi: 10.1093/annonc/mdz200
96. Guven DC, Sahin TK, Yildirim HC, Aktepe OH, Dizdar O, Yalcin S. A systematic review and meta-analysis of the association between circulating tumor DNA (ctDNA) and prognosis in pancreatic cancer. Crit Rev Oncol Hematol (2021) 168:103528. doi: 10.1016/j.critrevonc.2021.103528
97. Bernard V, Kim DU, San Lucas FA, Castillo J, Allenson K, Mulu FC, et al. Circulating nucleic acids are associated with outcomes of patients with pancreatic cancer. Gastroenterology. (2019) 156(1):108–18.e4. doi: 10.1053/j.gastro.2018.09.022
98. Hadano N, Murakami Y, Uemura K, Hashimoto Y, Kondo N, Nakagawa N, et al. Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br J Cancer (2016) 115(1):59–65. doi: 10.1038/bjc.2016.175
99. Groot VP, Mosier S, Javed AA, Teinor JA, Gemenetzis G, Ding D, et al. Circulating tumor DNA as a clinical test in resected pancreatic cancer. Clin Cancer Res (2019) 25(16):4973–84. doi: 10.1158/1078-0432.CCR-19-0197
100. Kruger S, Heinemann V, Ross C, Diehl F, Nagel D, Ormanns S, et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann Oncol (2018) 29(12):2348–55. doi: 10.1093/annonc/mdy417
101. Park G, Park JK, Son DS, Shin SH, Kim YJ, Jeon HJ, et al. Utility of targeted deep sequencing for detecting circulating tumor DNA in pancreatic cancer patients. Sci Rep (2018) 8(1):11631. doi: 10.1038/s41598-018-30100-w
102. Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B, et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol (2016) 10(4):635–43. doi: 10.1016/j.molonc.2015.11.012
103. Berger AW, Schwerdel D, Ettrich TJ, Hann A, Schmidt SA, Kleger A, et al. Targeted deep sequencing of circulating tumor DNA in metastatic pancreatic cancer. Oncotarget. (2018) 9(2):2076–85. doi: 10.18632/oncotarget.23330
104. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med (2019) 381(4):317–27. doi: 10.1056/NEJMoa1903387
105. Ettrich TJ, Schwerdel D, Dolnik A, Beuter F, Blatte TJ, Schmidt SA, et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci Rep (2019) 9(1):13261. doi: 10.1038/s41598-019-49860-0
106. Abou-Alfa GK, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol (2020) 21(6):796–807. doi: 10.1016/S1470-2045(20)30157-1
107. Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol (2018) 36(3):276–82. doi: 10.1200/JCO.2017.75.5009
108. Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol (2020) 21(5):671–84. doi: 10.1016/S1470-2045(20)30109-1
109. Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov (2017) 7(3):252–63. doi: 10.1158/2159-8290.CD-16-1000
110. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite Instability/Mismatch repair-deficient cancer: Results from the phase II KEYNOTE-158 study. J Clin Oncol (2020) 38(1):1–10. doi: 10.1200/JCO.19.02105
111. Nakamura Y, Okamoto W, Denda T, Nishina T, Komatsu Y, Yuki S, et al. Clinical validity of plasma-based genotyping for microsatellite instability assessment in advanced GI cancers: SCRUM-Japan GOZILA substudy. JCO Precis Oncol (2022) 6:e2100383. doi: 10.1200/PO.21.00383
112. Schlauch D, Fu X, Jones SF, Burris HA, Spigel DR, Reeves J, et al. Tumor-specific and tumor-agnostic molecular signatures associated with response to immune checkpoint inhibitors. JCO Precis Oncol (2021) 5:1625–38. doi: 10.1200/PO.21.00008
113. Fatima S, Ma Y, Safrachi A, Haider S, Spring KJ, Vafaee F, et al. Harnessing liquid biopsies to guide immune checkpoint inhibitor therapy. Cancers (Basel) (2022) 14(7):1669. doi: 10.3390/cancers14071669
114. Xiao H, Yoon YS, Hong S-M, Roh SA, Cho D-H, Yu CS, et al. Poorly differentiated colorectal cancers: Correlation of microsatellite instability with clinicopathologic features and survival. Am J Clin Pathol (2013) 140(3):341–7. doi: 10.1309/AJCP8P2DYNKGRBVI
115. Siregar GA, Sibarani H. Comparison of carcinoembryonic antigen levels among degree of differentiation and colorectal cancer's location in medan. Open Access Maced J Med Sci (2019) 7(20):3447–50. doi: 10.3889/oamjms.2019.442
116. Kasi PM, Kamatham S, Shahjehan F, Li Z, Johnson PW, Merchea A, et al. BRAF-V600E and microsatellite instability prediction through CA-19-9/CEA ratio in patients with colorectal cancer. J Gastroint Oncol (2020) 11(2):236–41. doi: 10.21037/jgo.2019.12.08
117. Zhang Q, Luo J, Wu S, Si H, Gao C, Xu W, et al. Prognostic and predictive impact of circulating tumor DNA in patients with advanced cancers treated with immune checkpoint blockade. Cancer Discov (2020) 10(12):1842–53. doi: 10.1158/2159-8290.CD-20-0047
118. Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med (2018) 24(9):1449–58. doi: 10.1038/s41591-018-0101-z
119. Jin Y, Chen DL, Wang F, Yang CP, Chen XX, You JQ, et al. The predicting role of circulating tumor DNA landscape in gastric cancer patients treated with immune checkpoint inhibitors. Mol Cancer (2020) 19(1):154. doi: 10.1186/s12943-020-01274-7
Keywords: ctDNA, gastrointestinal cancer, minimal residual disease, prognostic and predictive biomarker, next generation sequencing
Citation: Lam RCT, Johnson D, Lam G, Li MLY, Wong JWL, Lam WKJ, Chan KCA and Ma B (2022) Clinical applications of circulating tumor-derived DNA in the management of gastrointestinal cancers – current evidence and future directions. Front. Oncol. 12:970242. doi: 10.3389/fonc.2022.970242
Received: 15 June 2022; Accepted: 15 September 2022;
Published: 29 September 2022.
Edited by:
Reza Safaralizadeh, University of Tabriz, IranReviewed by:
Sandra Brosda, The University of Queensland, AustraliaCopyright © 2022 Lam, Johnson, Lam, Li, Wong, Lam, Chan and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brigette Ma, YnJpZ2V0dGVAY2xvLmN1aGsuZWR1Lmhr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.