
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Oncol. , 16 September 2022
Sec. Pediatric Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.970076
This article is part of the Research Topic Molecular Characteristics and Personalized Treatment for Pediatric Brain Tumors View all 6 articles
 Haoxiang Jiang1†
Haoxiang Jiang1† Lu Qiu1†
Lu Qiu1† Juan Song1Dandan Xu1Lei Sun1Yinbo Feng1Jun Zhao1Jun Qian2Zhiwei Yu2*Jin Peng3*
Juan Song1Dandan Xu1Lei Sun1Yinbo Feng1Jun Zhao1Jun Qian2Zhiwei Yu2*Jin Peng3*Background: Diffuse leptomeningeal glioneuronal tumors are rare leptomeningeal neoplasms composed of oligodendrocyte-like cells characterized by neuronal differentiation and a lack of isocitrate dehydrogenase gene mutation.
Purpose: We aimed to analyze the clinical progression, pathological characteristics, and radiological findings of diffuse leptomeningeal glioneuronal tumors in children, as well as the relevance of clinico-radiological data.
Data Sources: We searched MEDLINE, PubMed, and Web of Science to identify case reports, original articles, and review articles discussing diffuse leptomeningeal glioneuronal tumors published between 2000 and 2021.
Study Selection: The analysis included 145 pediatric patients from 43 previous studies.
Data Analysis: Data regarding patient pathology, MRI manifestations, clinical symptoms, and progression were collected. The relationship between imaging classification and pathological findings was using chi-square tests. Overall survival was analyzed using Kaplan–Meier curves.
Data Synthesis: Parenchymal tumors were mainly located in the intramedullary areas of the cervical and thoracic spine, and patients which such tumors were prone to 1p-deletion (χ2 = 4.77, p=0.03) and KIAA1549-BRAF fusion (χ2 = 12.17, p<0.001). The median survival time was 173 months, and the survival curve fell significantly before 72 months. Parenchymal tumor location was associated with overall survival (p=0.03), patients with KIAA 1549-BRAF (+) and treated with chemotherapy exhibited a better clinical course (p<0.001).
Limitations: The analysis included case reports rather than consecutively treated patients due to the rarity of diffuse leptomeningeal glioneuronal tumors, which may have introduced a bias.
Conclusions: Early integration of clinical, pathological, and radiological findings is necessary for appropriate management of this tumor, as this may enable early treatment and improve prognosis.
The 2021 World Health Organization (WHO) classification of brain tumors lists diffuse leptomeningeal glioneuronal tumor (DLGNT) as a tumor type alone (1), which has not yet been assigned a WHO grade. DLGNTs are rare leptomeningeal neoplasms composed of oligodendrocyte-like cells characterized by neuronal differentiation and a lack of isocitrate dehydrogenase (IDH) gene mutation. Additionally, DLGNTs have been associated with KIAA1549-BRAF gene fusion and 1p deletion or 1p/19q co-deletion (2, 3). Given the rarity of this tumor type, its histological, radiological, and clinical features and pattern of progression remain to be fully elucidated (4).
Although they frequently occur as leptomeningeal tumors (5), some studies have reported DLGNTs without leptomeningeal dissemination, which manifests as intraspinal or intracerebral cystic or solid masses (6–8). Notably, clinical follow-up is often incomplete, and some patients present with indolent chronic disease while others experience an aggressive clinical course (4). Moreover, the differential diagnosis often includes tuberculous meningitis, cryptococcal neoformans meningitis, and meningeal metastatic tumor. Therefore, a systematic review may aid in clarifying the characteristics of DLGNT, which can in turn promote early treatment and improve prognosis/management.
DLGNT mainly occurs in children, and adult cases are rare. In this review, we focused on literature concerning pediatric DLGNT published from 2000 to 2021. To determine the relevance of clinico-radiological data for diagnosis and decision-making concerning DLGNT, we aimed to analyze the clinical progression, pathological characteristics, and MRI findings among these cases.
This study was conducted according to the Preferred Reporting Items for Systematic reviews and Meta-Analyses (9).
We searched for articles related to DLGNT, published between 2000 and 2021, using PubMed, MEDLINE, and Web of Science. The following keywords were used: diffuse leptomeningeal glioneuronal tumor, DLGNT, diffuse leptomeningeal oligodendroglioma, and diffuse leptomeningeal gliomatosis/neurocytomas. Inclusion criteria were as follows: (i) age ≤18 years; (ii) availability of MRI and clinical data; (iii) case reports, original articles, and review articles. Exclusion criteria were as follows: (i) incomplete MRI or clinical data; (ii) meeting abstracts, letters, or comments; (iii) lack of relevant data; (iv) radiology or pathology not conforming to the diagnostic criteria for DLGNT (10).
Risk of bias and adequacy of reporting were assessed by two investigators using the Quality Assessment Tool for case reports published by the Australia Joanna Briggs Institute (https://synthesismanual.jbi.global) (11). For each case, the first author, publication year, country, age, sex, cerebrospinal fluid analysis, progression, pathology, and MRI manifestations were documented. All imaging studies were independently reviewed by two trained radiologists (LQ, with 3 years of pediatric radiology experience and JS, with 8 years of pediatric radiology experience). If no agreement could be reached, a decision was made in consultation with a third author (HJ, with 17 years of pediatric radiology experience).
Clinical data were described using ranges, mean value, medians, and percentages as appropriate. The relationships between imaging classifications and pathological findings were analyzed using chi-square tests. The difference in prognosis between surgical resection and nonsurgical resection groups were analyzed using chi-square tests. Overall survival was analyzed using Kaplan–Meier curves and the log-rank test. P values <0.05 were considered significant.
Figure 1 displays the flow of the review process. The initial search of electronic databases identified 603 records. The titles and abstracts of these records were screened for their eligibility, resulting in a first selection of 70 papers. The full text was further assessed for each of these articles. Studies were excluded if they were irrelevant to our purpose (n = 21) or did not include complete MRI or clinical data (n = 1). Among the 48 studies remaining, 43 studies were finally included based on the diagnostic criteria for DLGNT. The analyses thus included 145 patients from the previous literature. Based on the risk of bias and adequate reporting assessment, studies were rated as good (Online Tables 1-2).
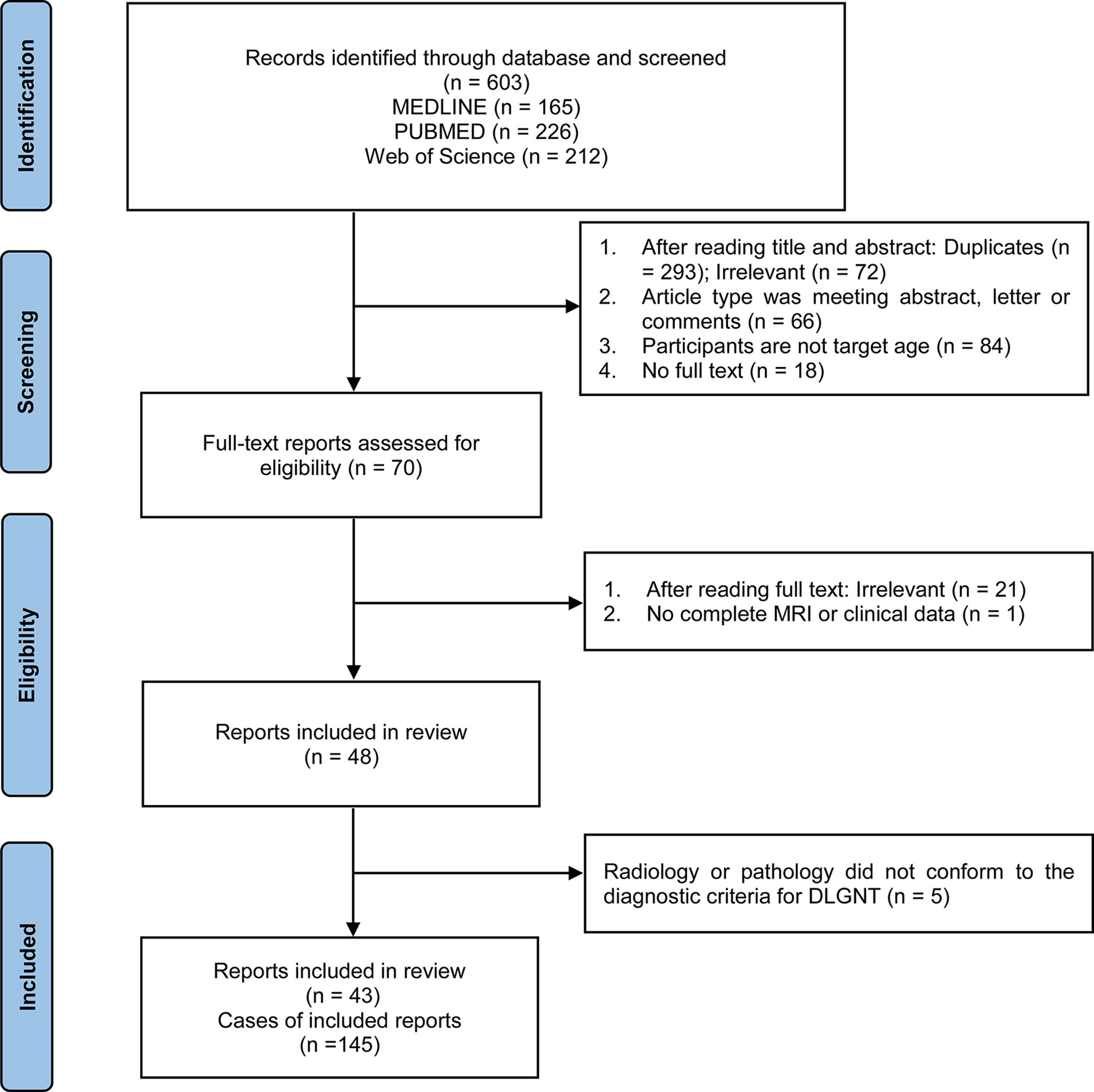
Figure 1 Flow chart showing the selection of reports.
In the combined analysis of the previous literature [145 pediatric patients with DLGNT (Table 1 and Figure 2); average age: 6.91 years; median age: 5 years], male predominance was observed in the overall sample (male/female: 1.7/1). The most reported symptoms were headache, nausea, and vomiting, which may have been related to hydrocephalus. Other symptoms included seizures, ataxia, speech problems, and sensorimotor impairments. Cerebrospinal fluid samples were positive for protein and negative for malignant cells except 3 cases, of which 2 cases showed malignant tumor cells with increased protein content and 1 case showed normal protein content without malignant tumor cells.
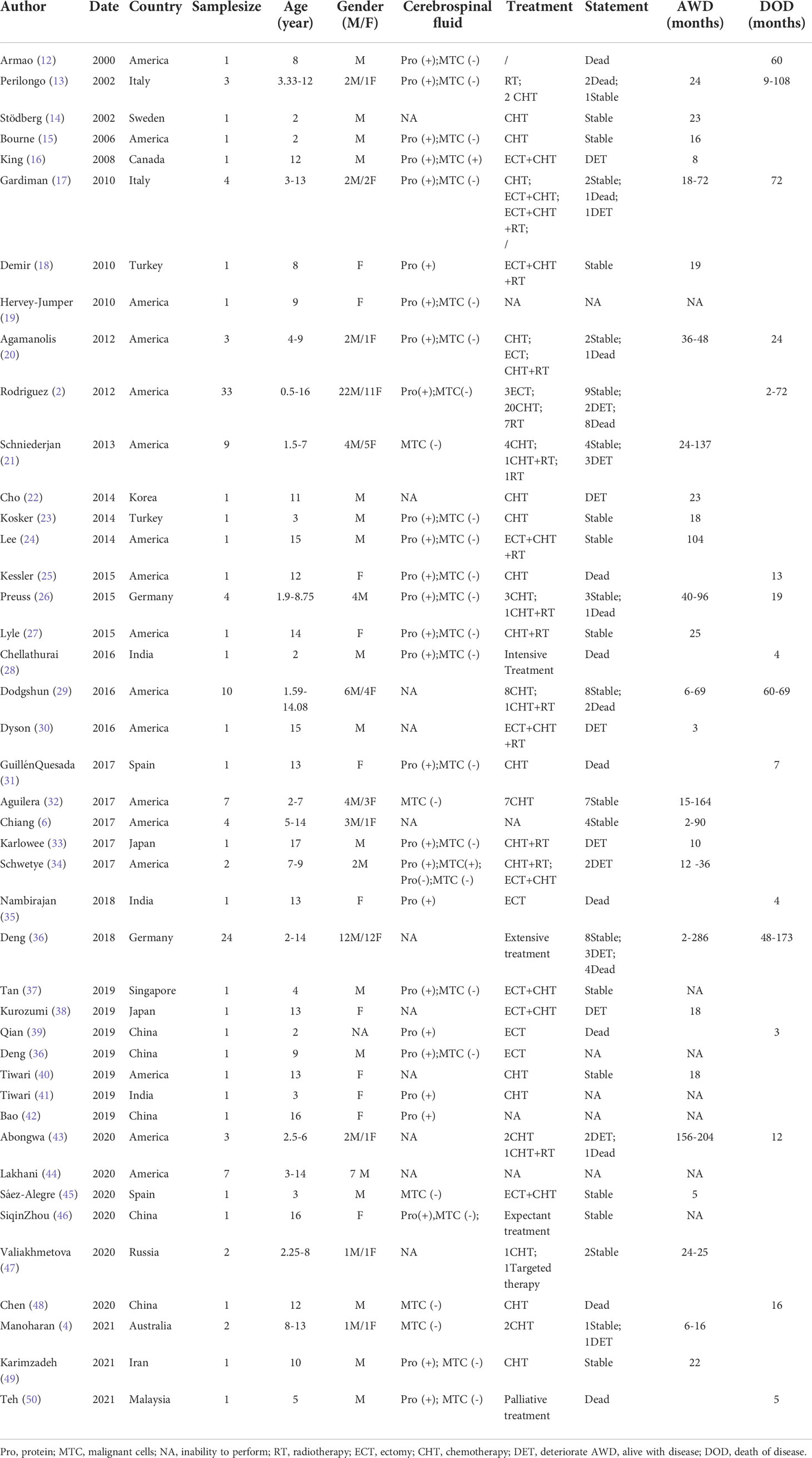
Table 1 Literature review of DLGNT.
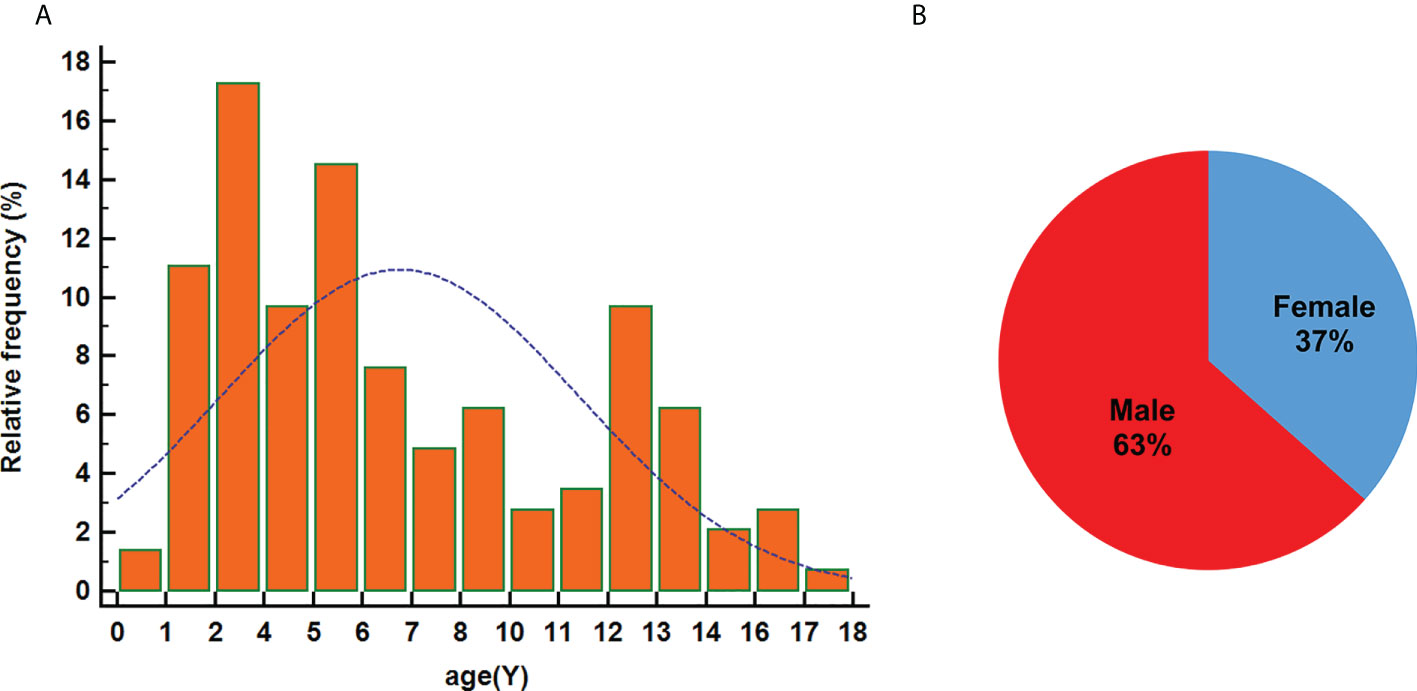
Figure 2 Age at diagnosis (A) and sex distribution (B).
The combined analysis of data from previously published cases revealed that the common histological findings were oligodendrocyte-like cells in the desmoplastic leptomeninges, which were composed of round to ovoid nuclei with mild atypia, clear cytoplasm, and perinuclear halos. Most DLGNTs were histologically low-grade, with a Ki67 index of <20%. Among patients with available data (Figure 3) (Online Table 3), S100 and oligodendrocyte transcription factor 2 positivity were observed in 96% (51 of 53) and 98% (39 of 40) of cases, respectively. All patients with available data were negative for IDH1 (0 of 53), while 60% (58 of 96) were positive for glial fibrillary acidic protein positive, and 81% (75 of 93) were positive for synaptophysin. In addition, neuronal nuclear antigen findings were positive in 24% (9/38) of patients, while epithelial membrane antigen findings were positive in only 4% (1/20) of patients. Moreover, 60% (30/50) of patients exhibited KIAA1549-BRAF fusion, 10% (3/30) exhibited BRAF V600E mutation, 75% (61/81) exhibited 1p-deletion, 28% (22/78) exhibited 19q-deletion, and 27% (21/78) exhibited co-deletion of 1p/19q.
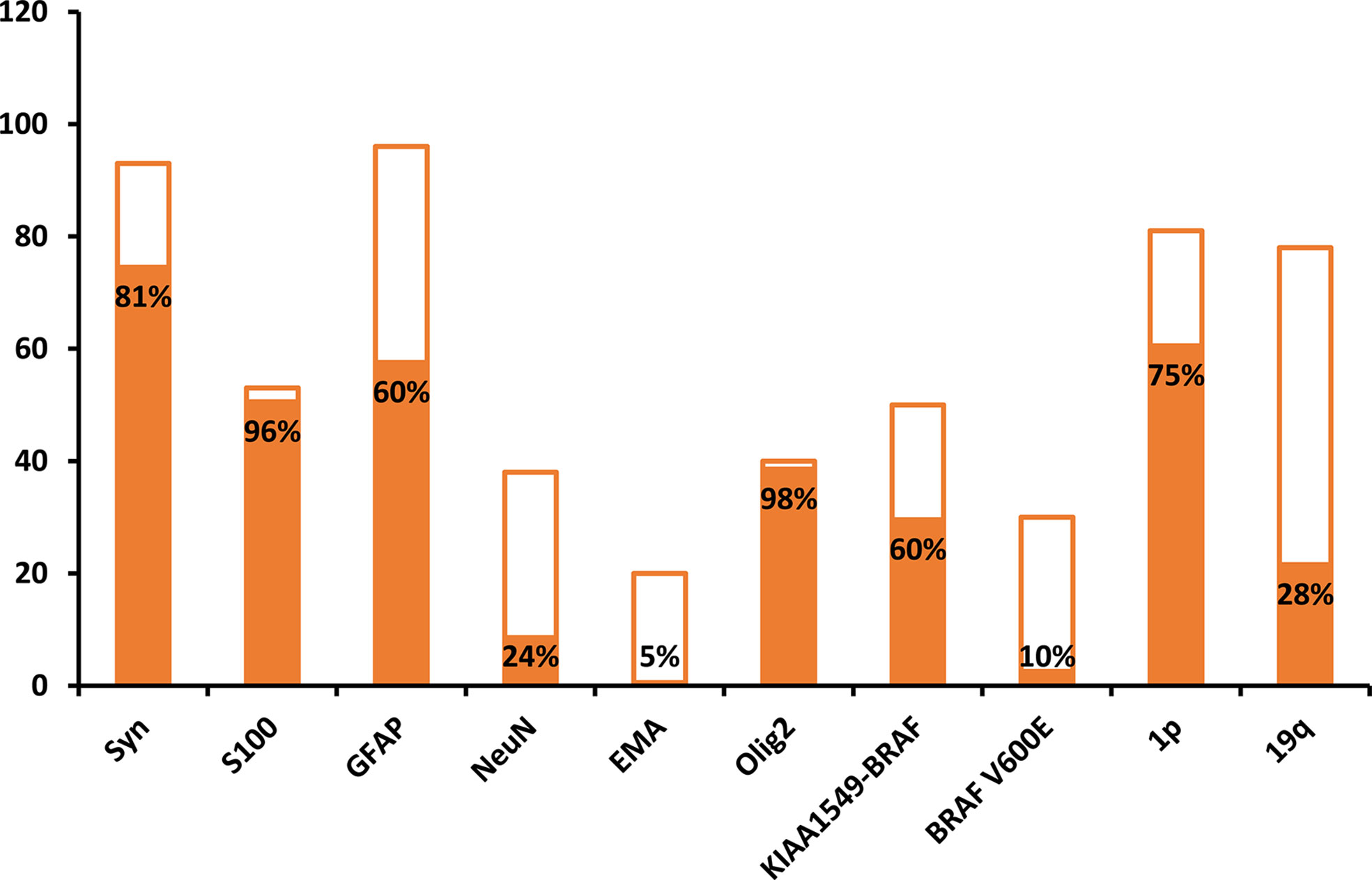
Figure 3 Analysis of immunohistochemical and molecular markers in patients with diffuse leptomeningeal glioneuronal tumors. Y-axis stands for the number of cases, and the solid bars represent the percentage of positive cases.
Sixty-nine percent (66/95) of patients with available data presented with hydrocephalus, which was the most common finding at the first hospital admission and was associated with headache and vomiting. In addition, 85% (111/131) of patients developed extensive contrast enhancement of the intracranial and spinal leptomeninges on MRI. Nodular and cystic changes of the leptomeninges were also detected in 50% (52/103) and 52% (57/109) of patients, respectively. Unusual unequivocal masses and parenchymal invasion were observed in 53% (72/137) of patients (Figure 4A). DLGNT lesions were mainly located in the subtentorial area (72%) (107/148), cervical spinal (72%) (107/148) and thoracic spine (71%) (105/148) (Figures 4B, 5A). In contrast, parenchymal tumors were mainly located in the intramedullary regions of the cervical (53%) (39/73) and thoracic spine (62%) (45/73) (Figure 5B).
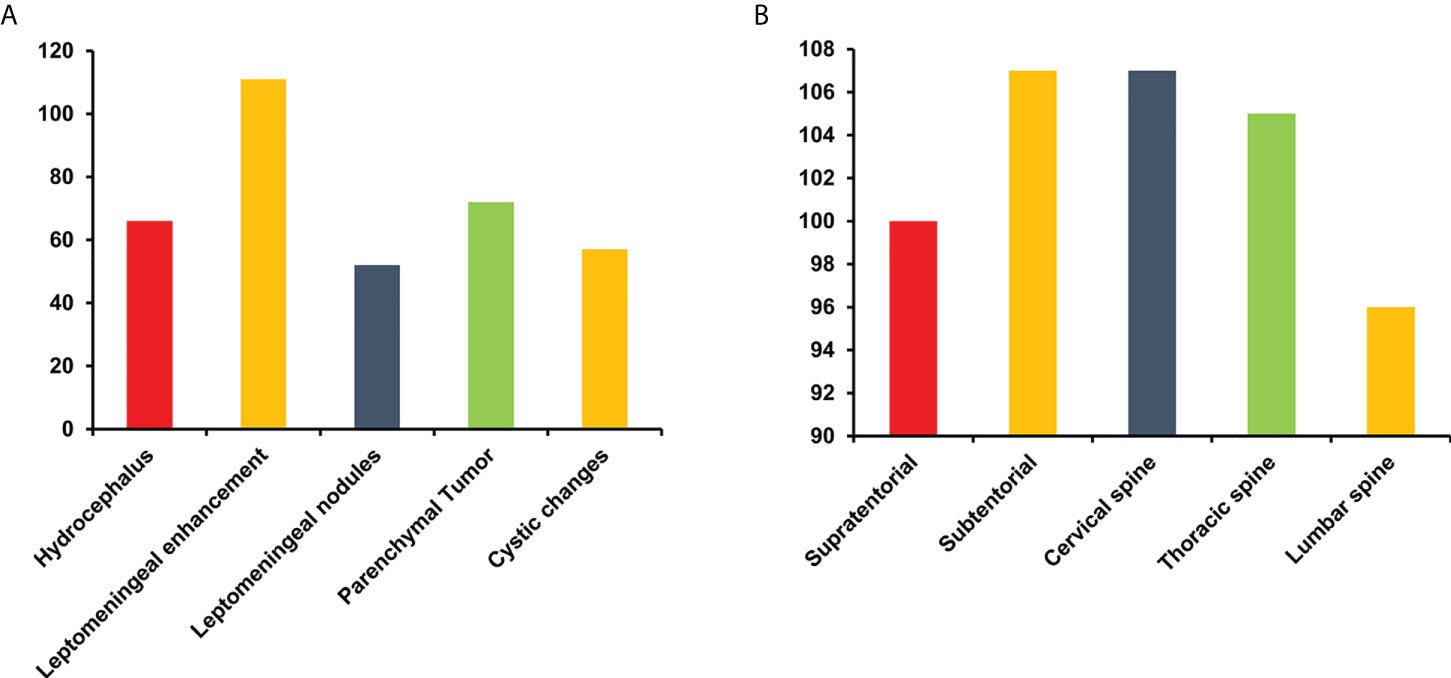
Figure 4 The finding (A) and distribution of lesions (B) on MRI in patients with diffuse leptomeningeal glioneuronal tumors. Y-axis represents the number of cases.
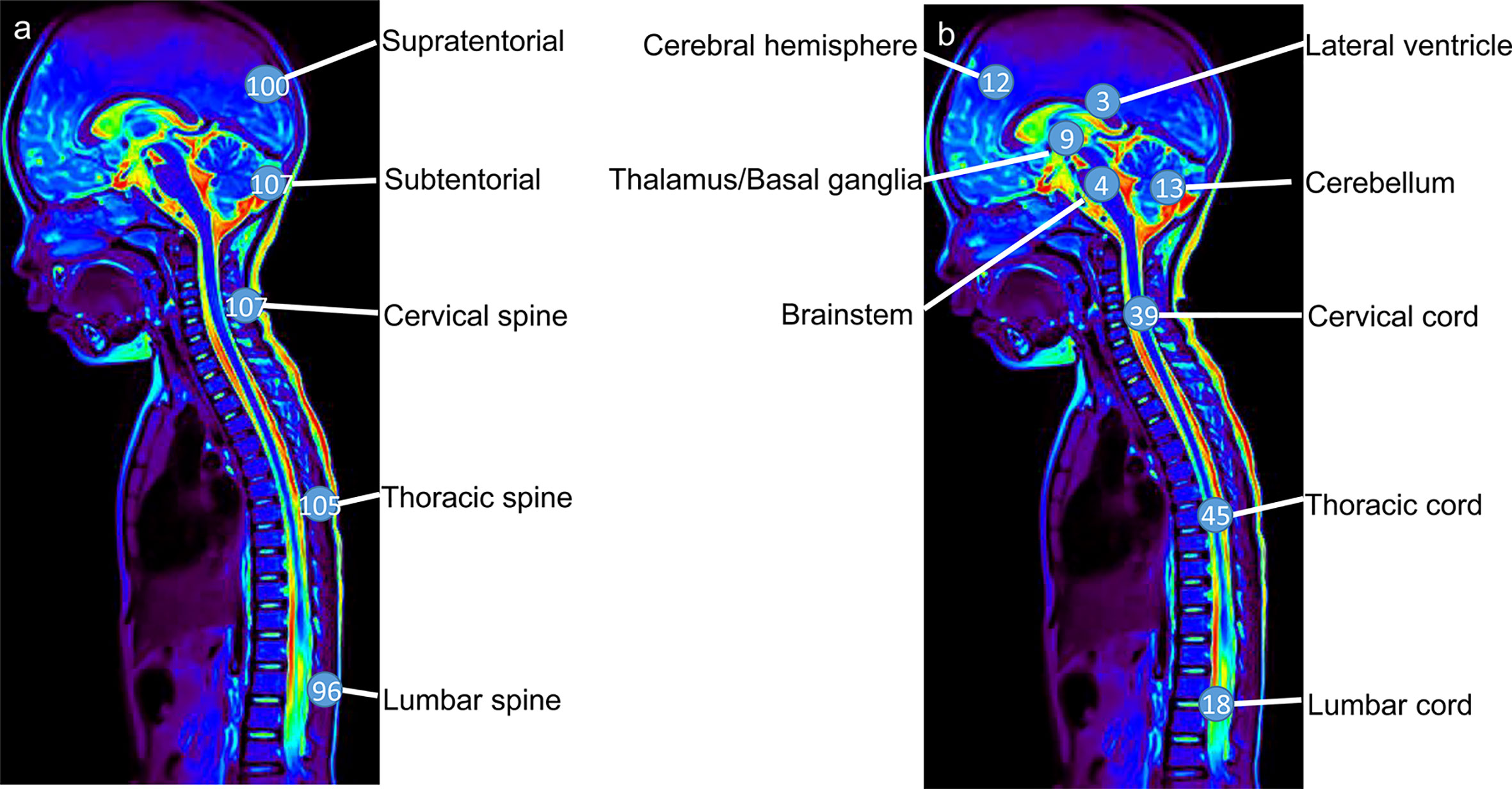
Figure 5 The lesions (A) and parenchymal tumor (B) location in patients with diffuse leptomeningeal glioneuronal tumors. Numbers in circles represent the numbers of lesions (A) and parenchymal tumors (B) in the specific region.
In addition, patients with parenchymal tumors were prone to 1p-deletion (χ2 = 4.77, p=0.03) and KIAA1549-BRAF fusion (χ2 = 12.17, p<0.001). Other imaging classifications were not significantly associated with pathological findings (p>0.05).
In total 145 cases, 94 patients were successfully followed up for 2–286 months (median follow-up: 25 months). 70% (66/94) of follow-up patients were still living. The total mortality rate was 19% (28/145). Among the 28 deceased patients, only 7 underwent postmortem examinations. One patient died of sepsis and multiple organ failure, and an autopsy revealed extensive necrotizing pneumonia due to Legionella pneumophila. More patients died after a devastating course of progressive neurological deterioration. Of the 20 patients who underwent surgical tumor resection, 18 were successfully followed up, of whom 5 died, 6 progressed and 7 were stable. Average survival time was 36 months with a 25% (5/20) overall mortality rate. Of the 74 patients who underwent biopsy due to the multifocality of lesions, 56 were successfully followed up, of whom 16 died, 10 progressed and 40 were stable. Average survival was 45 months with a 22% (16/74) overall mortality rate. There was no significant difference in prognosis between tumor resection and non-resection groups (χ2 = 5.00, P=0.08). Chemotherapy (85%) (79/93) was usually performed following confirmation of the diagnosis. Radiotherapy and ventricular-peritoneal shunting were performed in 23% (21/93) and 38% (36/94) of patients, respectively.
Furthermore, different molecular subtypes have different prognoses. 23 KIAA1549-BRAF positive patients were followed up, of which 5 died, 4 progressed and 14 stable. All 3 BRAF-V600E positive patients were stable. 42 1p deleted patients were followed up, of which 8 died, 9 progressed, and 25 stable. 14 19q deleted patients were followed up, of which 3 died, 3 progressed and 8 stable.
Overall survival time was analyzed after the first pathologic diagnosis (Figure 6). The median survival time was 173 months, and the survival curve fell significantly before 72 months. For patients with parenchymal tumors, location was associated with overall survival (60 months; 26% for those located in the cerebrum vs. 74% located in spine, p=0.03). Patients treated with chemotherapy exhibited a better clinical course (overall survival of 60 months, 0% vs. 72% with chemotherapy, p<0.001) (Figure 7). In immunohistochemical, KIAA-BRAF positive patients had a better clinical course than negative patients (overall survival of 60 months, 34% vs. 71%, p=0.01). There were no differences in GFAP, 1p, 19q (Figure 8), or treatment with surgical resection, radiotherapy, or ventricular-peritoneal shunting (p>0.05).
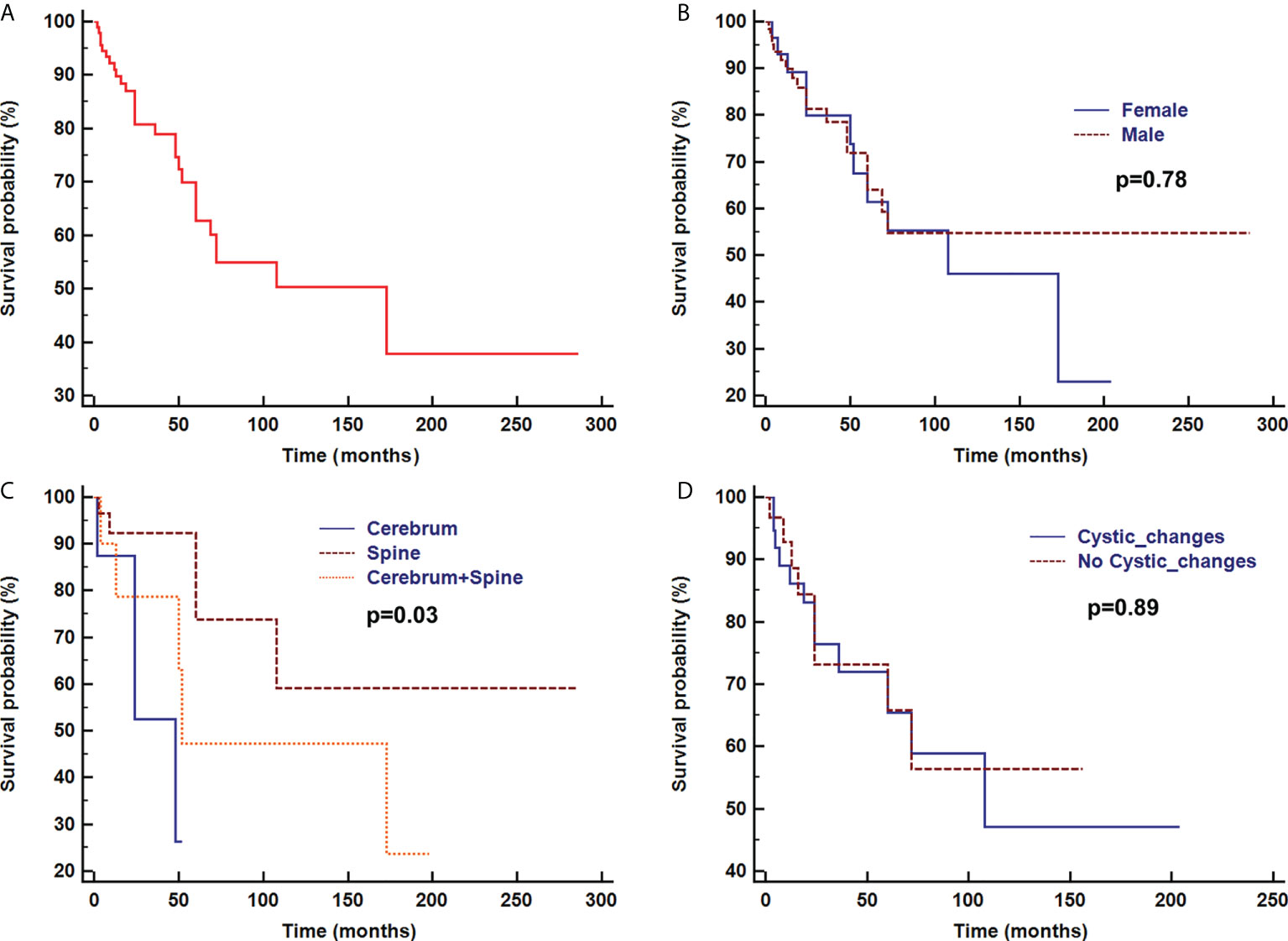
Figure 6 Survival curve analysis of overall survival (A), sex (B), and radiological findings of parenchymal tumor location (C), and with or without cystic changes (D) were analyzed using Kaplan–Meier curves and log-rank tests.
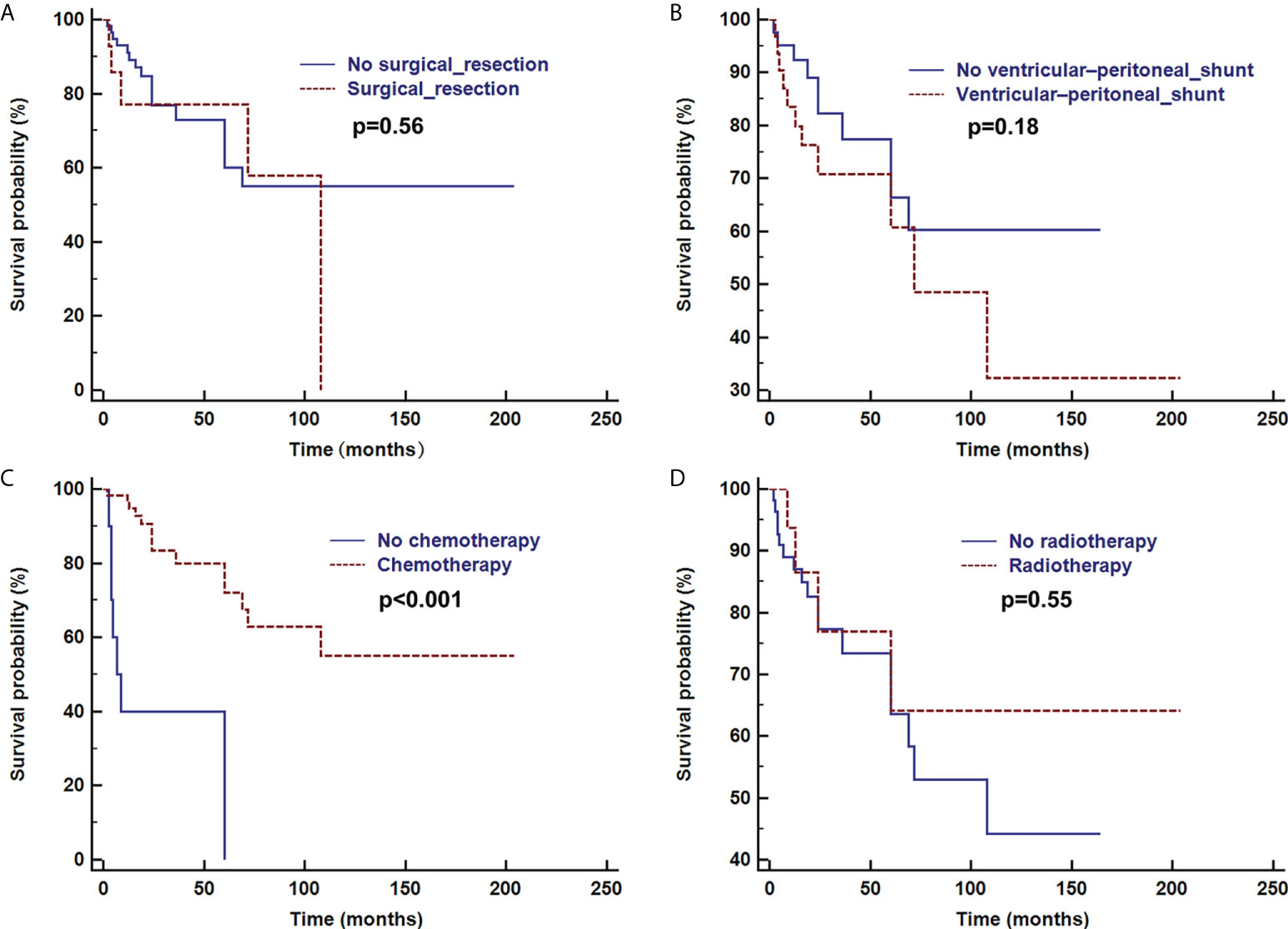
Figure 7 Survival curve analysis of treatment with or without surgical resection (A), ventricular-peritoneal shunt (B), chemotherapy (C), and radiotherapy (D) were analyzed using Kaplan–Meier curves and log-rank tests.
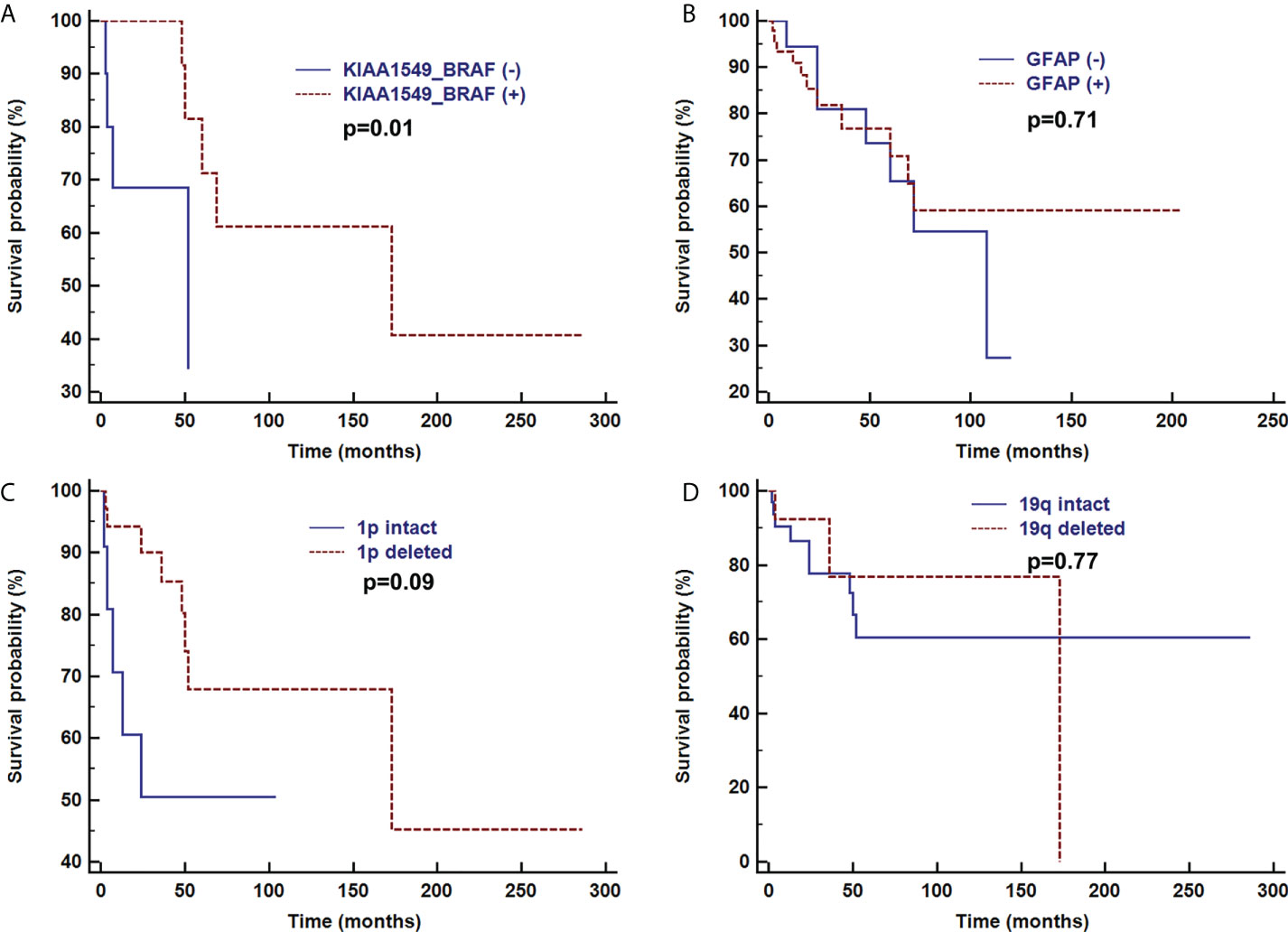
Figure 8 Survival curve analysis of pathological findings of KIAA1549-BRAF (A), GFAP (B), 1p intact (C), and 19q intact (D) were analyzed using Kaplan–Meier curves and log-rank tests.
In this study, we comprehensively reviewed DLGNT studies published from 2000 to 2021, following which we analyzed data for 145 pediatric patients included in 43 previous studies. To the best of our knowledge, this clinical imaging-focused series is the largest in the literature to date.
DLGNT is more common in children than in people aged >18 years and demonstrates a male predominance (2, 27). In this study, the peak age at onset ranged from 1–6 years old, with DLGNT affecting more boys than girls. In addition, 69% patients presented with hydrocephalus, which was associated with headache and vomiting caused by intracranial hypertension. It is a bite higher than previous literatures (2, 29). Some cases did not mention whether there was hydrocephalus. We did not take this part of cases into consideration, which may lead to literature bias.
The cerebrospinal fluid samples from most patients except 3 cases exhibit increased protein content without increases in cytological components, suggesting an increased likelihood of leptomeningeal proliferation of tumor cells (2). Two cases showed malignant tumor cells. For the first case, surgical resection and followed chemotherapy did not stop progression. The second patient died one year after diagnosis despite prompt chemotherapy. We cannot figure out whether there is a difference in prognosis between malignant tumor cells positive and negative patients due to the small sample size, further studies are needed in the future.
In total, 96% of patients tested were positive for S100 and negative for epithelial membrane antigen, indicating that the tumors originated from nerve cells rather than epithelial cells. Unlike gangliogliomas, DLGNTs are composed of a single-cell-type population of both glial and neuronal cell components. Consistently, 98% of patients tested positive for oligodendrocyte transcription factor 2, a transcription factor known as a common precursor of glial and neuronal cells (51). In our study, all patients with available data were negative for IDH1, consistent with previous findings (21).
In molecular testing, BRAF and 1p/19q gene alterations are important for differential diagnosis (3). Both pilomyxoid astrocytoma and DLGNT exhibit a tendency for leptomeningeal spread as well as similarities in histological features. However, pilomyxoid astrocytoma exhibits a mucinous background, in which bipolar tumor cells radiate around blood vessels along with rare gangliocytic tumor cells, and 1p/19q chromosome arm deletion is absent in such cases (2). A previous study reported 1p deletion and BRAF-KIAA1549 fusion in 59% and 75% of patients with DLGNT, respectively (3). Similarly, these mutations occurred in 75% and 60% of patients in this study. KIAA1549-BRAF fusion has also been associated with pilocytic astrocytoma (7). Thus, 1p deletion may represent a specific diagnostic index.
As noted in a previous MRI study (44), DLGNT presented with hydrocephalus, diffuse meningeal enhancement, nodules and diffuse cystic changes. The nodules and cystic changes mainly located the leptomeningeal surfaces of the brain and spine, as well as subependymal regions of the ventricle. A DLGNT autopsy study conducted by Louis (10) revealed extensive dilation and fibrosis of the subarachnoid space, accompanying intraventricular mass and cystic changes, as well as intraparenchymal extensions along the perivascular space. In this study, 53% of patients had parenchymal tumors in the cerebrum or spinal cord, which were mainly located in the intramedullary area of the cervical-thoracic cord. However, Chiang et al. (6) reported masses in the spinal cord but no diffuse lesions in their patients. This finding suggests the biological evolution of the tumor from the parenchyma to the cerebrospinal fluid and blood vessel space. Additionally, our findings indicated that patients with parenchymal tumors were prone to 1p-deletion and KIAA1549-BRAF fusion, highlighting the need for additional studies. Previous literature showed different molecular subtypes were associated with MRI findings that leptomeninges enhancement was more common in DLGNT-MC-2. (Online Figure 1) (7).
The biological behavior of DLGNT remains uncertain and has not been recommended by WHO for histological classification. Our study found most patients had stable prognosis. The 19% mortality rate supports the classification of DLGNT as an indolent disease in children. In this study, good clinical relief was observed after urgent ventriculoperitoneal shunting, although hyperproteinorrachia is often detected in these lesions, which can produce shunt obstruction (52). Surgical tumor resection and ventricular-peritoneal shunting alone are insufficient for improving overall survival, chemotherapy can significantly improve survival, as noted in previous studys (2, 53). Additionally, inhibition of the MAPK signaling pathway represents a potential therapeutic approach (29). In particular, patients with parenchymal tumors located in the cerebrum and those treated without chemotherapy experienced a worse clinical course, which suggests the importance of early diagnosis, repeated biopsy, localization, and standardized treatment.
In addition, previous research indicated histological and molecular features are associated with overall survival (2, 7). In our study, KIAA1549-BRAF positive patients had significantly higher overall survival than negative ones. However, there were no differences in GFAP, 1p, 19q.
Our study had some limitations, including its retrospective nature. In addition, the analysis included case reports rather than consecutively treated patients due to the rarity of DLGNT, which may have biased the results. Further, details regarding clinical progression and the extent of the correlation analysis between radiographic and histopathological features were limited to insufficient availability of follow-up data.
In conclusion, the current study suggests that DLGNT presents with specific, regular changes, including hydrocephalus, diffuse enhancement of the meninges, multiple nodules, and cystic changes on the brain surface. Notably, clinical progression, pathological characteristics, and radiological findings were associated with overall survival. Our findings highlight the need to integrate clinical and imaging features to improve the early and accurate diagnosis of DLGNT, simplify clinical decision-making, and improve prognosis.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
HJ and LQ drafted the initial manuscript, and summarized and analyzed the data. JS, DX, LS, YF, JZ, and JQ designed the data collection instruments, collected data, and carried out the initial analyses. JP and ZY conceptualized and designed the article, reviewed and revised the manuscript, and reviewed the manuscript for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Natural Science Foundation of Shaanxi Province (2021JM-558), Medical Innovation Team of Jiangsu Province (CXTDB2017016), and Wuxi Health Commission Precision Medicine Key Projects and Funding(j202107).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.970076/full#supplementary-material
Supplementary Figure 1 | Radiological features of 3 representative cases of DLGNTs. [DLGNT-MC-1_10 (A-C), DLGNT-MC-2_09 (D, E), DLGNT-MC-2_03 (F)]. Multicystic lesions with nodular enhancement in the cerebellum and cervical spinal cord spanning with leptomeningeal enhancement covering the cerebellum. Reproduced with permission from Springer Nature. Deng MY, Sill M, Chiang J, et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 2018;136(2):239-53.
DLGNT, diffuse leptomeningeal glioneuronal tumors; IDH, isocitrate dehydrogenase; WHO, World Health Organization.
1. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
2. Rodriguez FJ, Perry A, Rosenblum MK, Krawitz S, Cohen KJ, Lin D, et al. Disseminated oligodendroglial-like leptomeningeal tumor of childhood: A distinctive clinicopathologic entity. Acta Neuropathol (2012) 124(5):627–41. doi: 10.1007/s00401-012-1037-x
3. Rodriguez FJ, Schniederjan MJ, Nicolaides T, Tihan T, Burger PC, Perry A, et al. High rate of concurrent BRAF-KIAA1549 gene fusion and 1p deletion in disseminated oligodendroglioma-like leptomeningeal neoplasms (DOLN). Acta Neuropathol (2015) 129(4):609–10. doi: 10.1007/s00401-015-1400-9
4. Manoharan N, Ajuyah P, Senapati A, Wong M, Mullins A, Rodriguez M, et al. Diffuse leptomeningeal glioneuronal tumour (DLGNT) in children: The emerging role of genomic analysis. Acta Neuropathol Commun (2021) 9(1):147. doi: 10.1186/s40478-021-01248-w
5. Gardiman MP, Fassan M, Nozza P, Orvieto E, Garre ML, Milanaccio C, et al. Diffuse leptomeningeal glioneuronal tumours: Clinico-pathological follow-up. Pathologica (2012) 104(6):428–31.
6. Chiang J, Harreld JH, Orr BA, Sharma S, Ismail A, Segura AD, et al. Low-grade spinal glioneuronal tumors with BRAF gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol (2017) 134(1):159–62. doi: 10.1007/s00401-017-1728-4
7. Deng MY, Sill M, Chiang J, Schittenhelm J, Ebinger M, Schuhmann MU, et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol (2018) 136(2):239–53. doi: 10.1007/s00401-018-1865-4
8. Appay R, Pages M, Colin C, Jones D, Varlet P, Figarella-Branger D. Diffuse leptomeningeal glioneuronal tumor: A double misnomer? A report of two cases. Acta Neuropathol Commun (2020) 8(1):95. doi: 10.1186/s40478-020-00978-7
9. Shamseer L, Moher D, Clarke M, Ghersi D, Liberati A, Petticrew M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: Elaboration and explanation. BMJ (2015) 350:g7647. doi: 10.1136/bmj.g7647
10. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
11. Moola S, Munn Z, Tufanaru C, Aromataris E, Sears K, Sfetcu R, et al. Chapter 7: Systematic reviews of etiology and risk. In: Aromataris E, Munn Z, editors. JBI Manual for Evidence Synthesis. BI (2020). Available at: https://synthesismanual.jbi.global. doi: 10.46658/JBIMES-20-08
12. Armao DM, Stone J, Castillo M, Mitchell KM, Bouldin TW, Suzuki K. Diffuse leptomeningeal oligodendrogliomatosis: Radiologic/pathologic correlation. AJNR Am J Neuroradiol (2000) 21(6):1122–6.
13. Perilongo G, Gardiman M, Bisaglia L, Rigobello L, Calderone M, Battistella A, et al. Spinal low-grade neoplasms with extensive leptomeningeal dissemination in children. Childs Nerv Syst (2002) 18(9-10):505–12. doi: 10.1007/s00381-002-0626-8
14. Stödberg T, Deniz Y, Esteitie N, Jacobsson B, Mousavi-Jazi M, Dahl H, et al. A case of diffuse leptomeningeal oligodendrogliomatosis associated with HHV-6 variant A. Neuropediatrics (2002) 33(5):266–70. doi: 10.1055/s-2002-36739
15. Bourne TD, Mandell JW, Matsumoto JA, Jane JJ, Lopes MB. Primary disseminated leptomeningeal oligodendroglioma with 1p deletion. Case report. J Neurosurg (2006) 105(6 Suppl):465–9. doi: 10.3171/ped.2006.105.6.465
16. King JA, Halliday W, Drake JM. High-grade primary diffuse leptomeningeal gliomatosis in a child with neurofibromatosis Type 1. J Neurosurg Pediatr (2008) 2(6):402–5. doi: 10.3171/PED.2008.2.12.402
17. Gardiman MP, Fassan M, Orvieto E, D'Avella D, Denaro L, Calderone M, et al. Diffuse leptomeningeal glioneuronal tumors: A new entity? Brain Pathol (2010) 20(2):361–6. doi: 10.1111/j.1750-3639.2009.00285.x
18. Demir HA, Varan A, Akyüz C, Söylemezoğlu F, Cila A, Büyükpamukçu M. Spinal low-grade neoplasm with leptomeningeal dissemination mimicking tuberculous meningitis in a child. Childs Nerv Syst (2011) 27(1):187–92. doi: 10.1007/s00381-010-1218-7
19. Hervey-Jumper SL, Jumper M, Blaivas M, Parmar HA, Robertson PL, Maher CO. Primary diffuse leptomeningeal oligodendroglioma. Pediatr Neurosurg (2010) 46(4):326–8. doi: 10.1159/000317261
20. Agamanolis DP, Katsetos CD, Klonk CJ, Bartkowski HM, Ganapathy S, Staugaitis SM, et al. An unusual form of superficially disseminated glioma in children: report of 3 cases. J Child Neurol (2012) 27(6):727–33. doi: 10.1177/0883073811426500
21. Schniederjan MJ, Alghamdi S, Castellano-Sanchez A, Mazewski C, Brahma B, Brat DJ, et al. Diffuse leptomeningeal neuroepithelial tumor: 9 pediatric cases with chromosome 1p/19q deletion status and IDH1 (R132H) immunohistochemistry. Am J Surg Pathol (2013) 37(5):763–71. doi: 10.1097/PAS.0b013e31827bf4cc
22. Cho HJ, Myung JK, Kim H, Park CK, Kim SK, Chung CK, et al. Primary diffuse leptomeningeal glioneuronal tumors. Brain Tumor Pathol (2015) 32(1):49–55. doi: 10.1007/s10014-014-0187-z
23. Kosker M, Sener D, Kilic O, Hasiloglu ZI, Islak C, Kafadar A, et al. Primary diffuse leptomeningeal gliomatosis mimicking tuberculous meningitis. J Child Neurol (2014) 29(12):NP171–175. doi: 10.1177/0883073813509121
24. Lee SL, Wong A, Stadler JAI Jr., McClendon J, Smith TR, Wadhwani NR, et al. Cranial and spinal oligodendrogliomatosis: A case report and review of the literature. Childs Nerv Syst (2015) 31(1):147–53. doi: 10.1007/s00381-014-2506-4
25. Kessler BA, Bookhout C, Jaikumar S, Hipps J, Lee YZ. Disseminated oligodendroglial-like leptomeningeal tumor with anaplastic progression and presumed extraneural disease: Case report. Clin Imaging (2015) 39(2):300–4. doi: 10.1016/j.clinimag.2014.11.018
26. Preuss M, Christiansen H, Merkenschlager A, Hirsch FW, Kiess W, Mueller W, et al. Disseminated oligodendroglial-like leptomeningeal tumors: preliminary diagnostic and therapeutic results for a novel tumor entity [corrected]. J Neurooncol (2015) 124(1):65–74. doi: 10.1007/s11060-015-1735-z
27. Lyle MR, Dolia JN, Fratkin J, Nichols TA, Herrington BL. Newly identified characteristics and suggestions for diagnosis and treatment of diffuse leptomeningeal glioneuronal/neuroepithelial tumors: A case report and review of the literature. Child Neurol Open (2015) 2(1):2329048X14567531. doi: 10.1177/2329048X14567531
28. Chellathurai A, Vaidya JS, Kathirvelu G, Alagappan P. Primary diffuse leptomeningeal oligodendrogliomatosis: A case report and literature review. Indian J Radiol Imaging (2016) 26(3):337–41. doi: 10.4103/0971-3026.190424
29. Dodgshun AJ, SantaCruz N, Hwang J, Ramkissoon SH, Malkin H, Bergthold G, et al. Disseminated glioneuronal tumors occurring in childhood: treatment outcomes and BRAF alterations including V600E mutation. J Neurooncol (2016) 128(2):293–302. doi: 10.1007/s11060-016-2109-x
30. Dyson K, Rivera-Zengotita M, Kresak J, Weaver K, Stover B, Fort J, et al. FGFR1 N546K and H3F3A K27M mutations in a diffuse leptomeningeal tumour with glial and neuronal markers. Histopathology (2016) 69(4):704–7. doi: 10.1111/his.12983
31. Guillen Quesada A, Puerta Roldan P, Morales La Madrid A, Sunol M, Jou C, Muchart Lopez J, et al. Diffuse leptomeningeal Glioneural tumour simulating tuberculous meningitis in a 13-year-old girl. Pediatr Neurosurg (2018) 53(2):140–2. doi: 10.1159/000485249
32. Aguilera D, Castellino RC, Janss A, Schniederjan M, McNall R, MacDonald T, et al. Clinical responses of patients with diffuse leptomeningeal glioneuronal tumors to chemotherapy. Childs Nerv Syst (2018) 34(2):329–34. doi: 10.1007/s00381-017-3584-x
33. Karlowee V, Kolakshyapati M, Amatya VJ, Takayasu T, Nosaka R, Sugiyama K, et al. Diffuse leptomeningeal glioneuronal tumor (DLGNT) mimicking Whipple’s disease: A case report and literature review. Childs Nerv Syst (2017) 33(8):1411–4. doi: 10.1007/s00381-017-3405-2
34. Schwetye KE, Kansagra AP, McEachern J, Schmidt RE, Gauvain K, Dahiya S. Unusual high-grade features in pediatric diffuse leptomeningeal glioneuronal tumor: comparison with a typical low-grade example. Hum Pathol (2017) 70:105–12. doi: 10.1016/j.humpath.2017.06.004
35. Nambirajan A, Suri V, Kedia S, Goyal K, Malgulwar PB, Khanna G, et al. Paediatric diffuse leptomeningeal tumor with glial and neuronal differentiation harbouring chromosome 1p/19q co-deletion and H3.3 K27M mutation: Unusual molecular profile and its therapeutic implications. Brain Tumor Pathol (2018) 35(3):186–91. doi: 10.1007/s10014-018-0325-0
36. Deng D, Guo J, Li H, Xu S, Wang W. Diffuse leptomeningeal glioneuronal tumor: A case report. Chin J Rad (2019) 53:145–6.
37. Tan G, Merchant K, Tan E, Low D, Ng LP, Seow WT, et al. A germline variant of TP53 in paediatric diffuse leptomeningeal glioneuronal tumour. Childs Nerv Syst (2019) 35(6):1021–7. doi: 10.1007/s00381-019-04128-w
38. Kurozumi K, Nakano Y, Ishida J, Tanaka T, Doi M, Hirato J, et al. High-grade glioneuronal tumor with an ARHGEF2-NTRK1 fusion gene. Brain Tumor Pathol (2019) 36(3):121–8. doi: 10.1007/s10014-019-00345-y
39. Lu Q, Zou LP, Gui QP, Shi XY, Zhai X, Yang G, et al. Diffuse leptomeningeal glioneuronal tumor presented with language developmental delay. Neuro Endocrinol Lett (2019) 40(4):161–5.
40. Tiwari N, Tamrazi B, Robison N, Krieger M, Ji J, Tian D. Unusual radiological and histological presentation of a diffuse leptomeningeal glioneuronal tumor (DLGNT) in a 13-year-old girl. Childs Nerv Syst (2019) 35(9):1609–14. doi: 10.1007/s00381-019-04074-7
41. Tiwari S, Yadav T, Pamnani J, Mathew JM, Elhence P, Praneeth K, et al. Diffuse leptomeningeal glioneuronal tumor: A unique leptomeningeal tumor entity. World Neurosurg (2020) 135:297–300. doi: 10.1016/j.wneu.2019.12.119
42. Bao Y, Xiao J, Cheng Z, Qin Y, Xie D, Wu H, et al. Characteristics of diffuse leptomeningeal glioneuronal tumor with first-episode headache and rapid blindness misdiagnosed as viral meningoencephalitis. J Med cases (2019) 10(8):241–5. doi: 10.14740/jmc3347
43. Abongwa C, Cotter J, Tamrazi B, Dhall G, Davidson T, Margol A. Primary diffuse leptomeningeal glioneuronal tumors of the central nervous system: Report of three cases and review of literature. Pediatr Hematol Oncol (2020) 37(3):248–58. doi: 10.1080/08880018.2019.1711270
44. Lakhani DA, Mankad K, Chhabda S, Feizi P, Patel R, Sarma A, et al. Diffuse leptomeningeal glioneuronal tumor of childhood. AJNR Am J Neuroradiol (2020) 41(11):2155–9. doi: 10.3174/ajnr.A6737
45. Sáez-Alegre M, Saceda GJ, Utrilla CC, Aracil SF, García-Feijoo P, Carceller BF. Diffuse leptomeningeal glioneuronal tumour: Where to biopsy? Case report and literature review. Childs Nerv Syst (2021) 37(7):2405–8. doi: 10.1007/s00381-020-04955-2
46. Zhou S, Zhang M, Wu H, Liu G, Wang Y, Bao Y. Vision treatment strategy for acute blindness in adolescent female with diffuse leptomeningeal glioneuronal tumor. Int J Neurosci (2020) 130(11):1170–3. doi: 10.1080/00207454.2020.1730364
47. Valiakhmetova A, Papusha L, Druy A, Yasko L, Ektova A, Karachunsky A, et al. Pediatric diffuse leptomeningeal glioneuronal tumor: Two clinical cases of successful targeted therapy. Pediatr Blood Cancer (2020) 67(12):e28478. doi: 10.1002/pbc.28478
48. Chen W, Kong Z, Fu J, Zhao D, Wang R, Ma W, et al. Diffuse leptomeningeal glioneuronal tumour (DLGNT) with hydrocephalus as an initial symptom: A case-based update. Childs Nerv Syst (2020) 36(3):459–68. doi: 10.1007/s00381-019-04481-w
49. Karimzadeh P, Nilipour Y, Khalili M, Nikkhah A, Taghavijelodar M, Moradi E. A case of diffuse leptomeningeal glioneuronal tumor in a 10-year-old boy: First report from Iran. Clin Case Rep (2021) 9(12):e05199. doi: 10.1002/ccr3.5199
50. Teh YG, Azizan N, Mohd NN, Ng CY, Wong KJ, Mohd ZF. Case report: unusual high-grade diffuse leptomeningeal glioneuronal tumor mimicking tuberculous meningitis in a child from an endemic region. Front Pediatr (2021) 9:767614. doi: 10.3389/fped.2021.767614
51. Matsumura N, Yokoo H, Mao Y, Yin W, Nakazato Y. Olig2-positive cells in glioneuronal tumors show both glial and neuronal characters: The implication of a common progenitor cell? Neuropathology (2013) 33(3):246–55. doi: 10.1111/j.1440-1789.2012.01355.x
52. Garibotto F, Pavanello M, Milanaccio C, Gaggero G, Fiaschi P. Management of hydrocephalus related to diffuse leptomeningeal glioneuronal tumour: A multifaceted condition. Childs Nerv Syst (2021) 37(4):1039–40. doi: 10.1007/s00381-020-04867-1
Keywords: clinical progression, pathology, radiology, Diffuse Leptomeningeal Glioneuronal Tumor, pediatrics, systematic review
Citation: Jiang H, Qiu L, Song J, Xu D, Sun L, Feng Y, Zhao J, Qian J, Yu Z and Peng J (2022) Clinical progression, pathological characteristics, and radiological findings in children with diffuse leptomeningeal glioneuronal tumors: A systematic review. Front. Oncol. 12:970076. doi: 10.3389/fonc.2022.970076
Received: 15 June 2022; Accepted: 10 August 2022;
Published: 16 September 2022.
Edited by:
Weijun Feng, Fudan University, ChinaReviewed by:
Andrea Carai, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2022 Jiang, Qiu, Song, Xu, Sun, Feng, Zhao, Qian, Yu and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiwei Yu, eXU5ODIwNEBhbGl5dW4uY29t; Jin Peng, cGVuZ2ppbmxpdGFvQDE2My5jb20=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.