
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 17 August 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.968351
This article is part of the Research TopicThe Impact of Alkalizing the Acidic Tumor Microenvironment to Improve Efficacy of Cancer TreatmentView all 13 articles
 Thomas N. Seyfried1*
Thomas N. Seyfried1* Gabriel Arismendi-Morillo2
Gabriel Arismendi-Morillo2 Giulio Zuccoli3
Giulio Zuccoli3 Derek C. Lee1
Derek C. Lee1 Tomas Duraj4
Tomas Duraj4 Ahmed M. Elsakka5
Ahmed M. Elsakka5 Joseph C. Maroon6
Joseph C. Maroon6 Purna Mukherjee1Linh Ta1Laura Shelton7Dominic D'Agostino8
Purna Mukherjee1Linh Ta1Laura Shelton7Dominic D'Agostino8 Michael Kiebish9
Michael Kiebish9 Christos Chinopoulos10
Christos Chinopoulos10Glioblastoma (GBM), similar to most cancers, is dependent on fermentation metabolism for the synthesis of biomass and energy (ATP) regardless of the cellular or genetic heterogeneity seen within the tumor. The transition from respiration to fermentation arises from the documented defects in the number, the structure, and the function of mitochondria and mitochondrial-associated membranes in GBM tissue. Glucose and glutamine are the major fermentable fuels that drive GBM growth. The major waste products of GBM cell fermentation (lactic acid, glutamic acid, and succinic acid) will acidify the microenvironment and are largely responsible for drug resistance, enhanced invasion, immunosuppression, and metastasis. Besides surgical debulking, therapies used for GBM management (radiation, chemotherapy, and steroids) enhance microenvironment acidification and, although often providing a time-limited disease control, will thus favor tumor recurrence and complications. The simultaneous restriction of glucose and glutamine, while elevating non-fermentable, anti-inflammatory ketone bodies, can help restore the pH balance of the microenvironment while, at the same time, providing a non-toxic therapeutic strategy for killing most of the neoplastic cells.
Glioblastoma (GBM) has among the highest mortality rates for primary brain tumors and remains largely unmanageable. Despite the hype surrounding newer therapies, median life expectancy following GBM diagnosis is only about 11-15 months with some large patient data bases reporting few survivors beyond 30 months (1–7). The poor overall GBM patient survival is also astonishingly consistent across many surgical institutions Figure 1. Although remarkable advances in science and technology have occurred over the last 100 years in Western societies, no significant advances have been made over this same period in improving survival for GBM patients (2, 7, 8). This abysmal lack of therapeutic progress can be due in large part to the inability to recognize GBM as a metabolic disorder (7, 12). Acidification of the GBM microenvironment arises as a consequence of the fermentation metabolism within the neoplastic tumor cells and is largely responsible for therapy failure. This review provides the evidence supporting this statement.
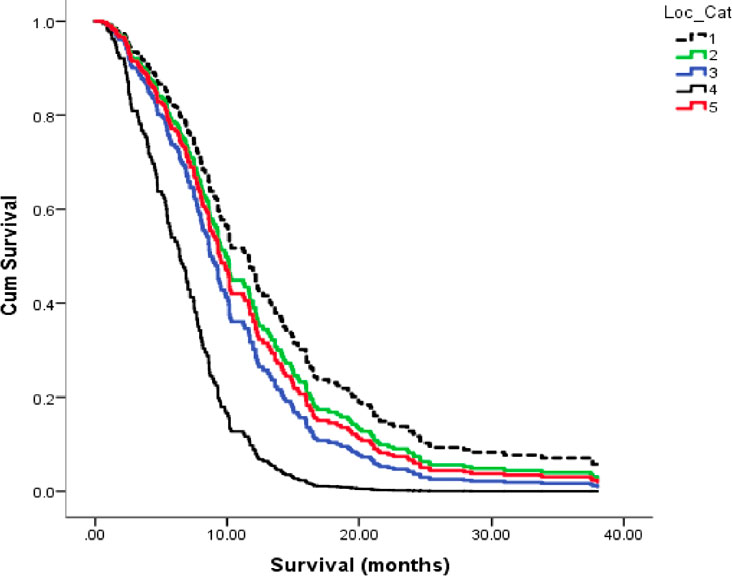
Figure 1 Kaplan-Meier plots for overall survival of GBM patients across five (1–5), Canadian surgical institutions. Each line represents patient survival for a particular institution as described (8). The GBM survival statistics recorded for these Canadian institutions are similar to those recorded in surgical institutions of other countries (2, 9, 10). These findings support the view of no major improvement of GBM patient survival in almost 100 years (8, 11). Image reproduced under a Creative Commons license from (8).
GBM, like most major cancers, is dependent on fermentation metabolism for the synthesis of biomass and energy (ATP) regardless of the cellular or the genetic heterogeneity observed within the tumor (13, 14). A dependency on fermentation metabolism is the consequence of the well-documented abnormalities in the number, the structure, and the function of GBM mitochondria and mitochondrial associated membranes (MAM) and shown in Figure 2, and as described previously in detail (7, 14, 15, 17–25). In light of these structural and functional abnormalities, it would not be possible for GBM mitochondria to synthesize much if any ATP through OxPhos based on the foundational principle in evolutionary biology that structure determines function (14, 26, 27). The numerous reports suggesting that OxPhos is either normal or not seriously impaired in GBM cells is inconsistent with this foundational principle (28–40). It is important to recognize that oxygen consumption is not a reliable marker for OxPhos function in cancer cells (see below). It is unlikely that ATP synthesis through OxPhos could be normal in GBM cells that have documented abnormalities in mitochondria ultrastructure and function. Moreover, the large numbers of somatic mutations seen in GBM and in many other cancers, for that matter, arise as down-stream effects of OxPhos dysfunction with consequent ROS production (12). The somatic mutations in tumor cells will prevent adaptive versatility according to the evolutionary concepts of Darwin and Potts, thus locking in a dependency on fermentation metabolism for growth (41–44). It should be known, especially in the oncology field, that nothing in either general biology or in cancer biology can make sense except in the light of evolution (12, 45).
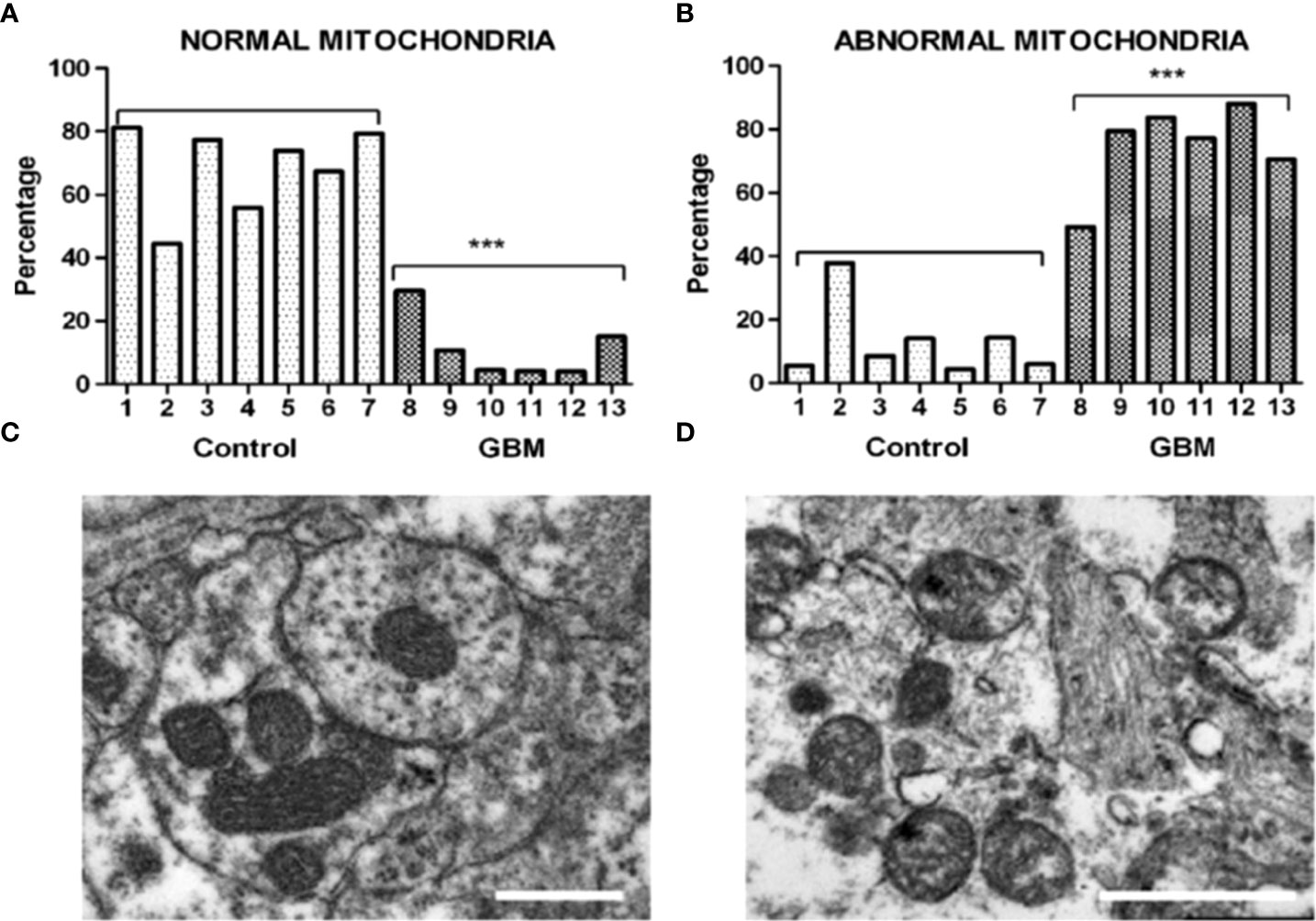
Figure 2 Morphological abnormalities seen in GBM mitochondria from the work of Deighton et al. (15). The morphology of 150 mitochondria was assessed in six GBM samples and in seven peri-tumoral control samples using Electron Microscopy (EM). (A) Percentage of normal mitochondria where cristae were visible throughout the mitochondria in peri-tumoral control and GBM samples (each bar represents one sample; *** p-value = 0.0001); (B) Percentage of abnormal mitochondria where cristae were sparse and abnormal in peri-tumoral control and GBM samples; *** p-value = 0.0001). (C, D) Representative EM images of normal and abnormal mitochondria, respectively. Cristolysis was significantly greater in mitochondria from GBM tissue than in mitochondria from normal surrounding brain tissue. The scale bars represent 0.5 um. The authors reported 117 mitochondrial proteins altered in GBM in association with ultrastructural mitochondrial abnormalities, similar to those described previously by Arismendi-Morillo et al. (16). ATP synthesis through OxPhos cannot be normal in tumor cells with these abnormalities. Image reproduced under a Creative Commons license from Deighton et al. (15).
It is important to emphasize that a reduction in OxPhos of ~50% would dissipate the protonmotive force causing a reversal of the Fo-F1 ATP synthase (7). The Fo-F1 ATP synthase generally operates in forward mode (i.e., synthesizing ATP) only when the mitochondria are sufficiently polarized. The Fo-F1 ATP synthase would be unable to generate ATP under a loss of electron transport chain operation on the order of 45-50% (7). This degree of loss would cause ATP hydrolysis, thus pumping protons out of the matrix. Reversal of the ATP synthase is what affords glutamine-driven mitochondrial substrate phosphorylation (mSLP) the critical role of providing ATP directly within the matrix when OxPhos becomes inhibited or impaired (7, 12). An inverse relationship between OxPhos efficiency and tumor aggression has been reported (46). A similar phenomenon has also been described with respect to the degree of fermentation and tumor growth, i.e., the greater is the fermentation, the more aggressive is the cancer (14, 47–49). GBM cells, regardless of their cellular origin or genetic heterogeneity, are dependent on fermentation for survival due to abnormalities in mitochondrial structure and function.
A large part of the confusion on mitochondrial dysfunction in cancer comes from the incorrect assumption that oxygen consumption observed in cancer cells is linked to ATP synthesis through OxPhos (14, 28, 29, 40, 50–53). Many cancers, including GBM, can survive in hypoxia (0.1% oxygen) or in a solution of potassium cyanide, a Complex IV inhibitor, findings that would exclude normal OxPhos as a source of ATP synthesis (54–57). Cells with normal OxPhos function cannot survive for very long in cyanide or in hypoxia. While oxygen is necessary for cholesterol synthesis, GBM cells can obtain cholesterol from the microenvironment under hypoxic conditions (54, 58). Many normal cells and tumor cells will consume oxygen and ferment lactic acid when grown in vitro, but only tumor cells continue to ferment when grown in vivo (14, 47, 48).
The oxygen consumption in tumor cells is uncoupled and is used more for ROS production than for ATP synthesis through OxPhos (14, 23, 59–61). High-resolution oxygen consumption measurements and extracellular flux analysis, such as produced by Seahorse XF technology, cannot accurately measure OxPhos-driven ATP synthesis (53). Moreover, these measurements are highly variable in inter-laboratory settings (cell lines with exactly the same genetic background can display opposite metabolic profiles), are extrapolated using general, non-cancer specific ATP/O stoichiometries, and are limited by non-physiological and artefactual cell culture conditions (53, 62). It is not clear if most investigators using general purpose respirometry are aware of these facts.
Also contributing to misinformation on oxygen consumption and ATP synthesis is the failure to recognize glutamine-driven mSLP as a major source of energy for GBM cells (7, 14). Warburg was also unaware of this linkage, as he assumed that oxygen consumption was linked to OxPhos in his cancer cell preparations (7, 47, 48). Viewed collectively, these findings indicate that oxygen consumption alone cannot be used as a measure of OxPhos-derived ATP synthesis in most tumor cells including GBM.
Recent studies have described how mSLP at the succinyl CoA synthetase reaction in the glutaminolysis pathway can provision ATP synthesis in GBM (7, 14, 53, 63, 64). The glutamine nitrogen produced from glutaminolysis is essential for the synthesis of nucleotides and amino acids. The waste products of glutaminolysis (primarily glutamic acid and succinic acid) would also contribute to acidification of the GBM microenvironment (7, 14, 65, 66). The catabolism of glutamine towards succinate will generate CO2 from the oxidative decarboxylation of the alpha-ketoglutarate dehydrogenation complex thus further acidifying the microenvironment. Additionally, succinate can stimulate NF-κB-driven inflammation and facilitate Hif-1α-driven glycolysis (7, 65, 67, 68). While the energetic competence of mitochondria in GBM and most other cancers is compromised in producing ATP through OxPhos, these mitochondria remain functional for other biosynthetic roles and in producing sufficient ATP through mSLP. Unlike OxPhos, however, that produces water and CO2 as waste products, mSLP produces glutamic acid and succinate acid as waste products that contribute to microenvironment acidification.
The metabolic waste products of glucose and glutamine fermentation (lactic acid, glutamic acid, and succinic acid) will together acidify the GBM microenvironment. This acidification is ultimately responsible for drug resistance, enhanced invasion, immunosuppression, and metastasis (7, 14, 53, 69). Glucose carbons are essential for biomass synthesis through the glycolysis and the pentose phosphate pathways, with lactic acid and nucleic acid precursors produced as major end products (14, 70, 71). The pyruvate kinase M2 (PKM2) isoform, which is abundantly expressed in most malignant cancers, produces pyruvate-derived lactic acid with minimal ATP synthesis (14, 72–75). In other words, most of the glucose-derived lactic acid coming from the tumor cells is produced with little ATP synthesis through glycolysis. Some of the lactate acid produced in cancer cells can be returned to the tumor as glucose through the Cori cycle thus maintaining a constant supply of glucose to the tumor (76).
Calorie restriction, which lowers blood glucose and elevates blood D-β-OHB, reduces nuclear expression of phosphorylated NF-κB (p65), cytosolic expression of phosphorylated IκB, total IκB, and DNA promoter binding activity of activated NF-κB in the CT-2A astrocytoma (77). NF-κB is a major driver of inflammation in the GBM microenvironment. Figure 3A shows how the waste products of glucose and glutamine fermentation are largely responsible for the inflammation and acidification in the GBM microenvironment. Hence, therapies that can lower blood glucose while elevating D-β-OHB will mitigate microenvironment inflammation and acidification through multiple mechanisms.
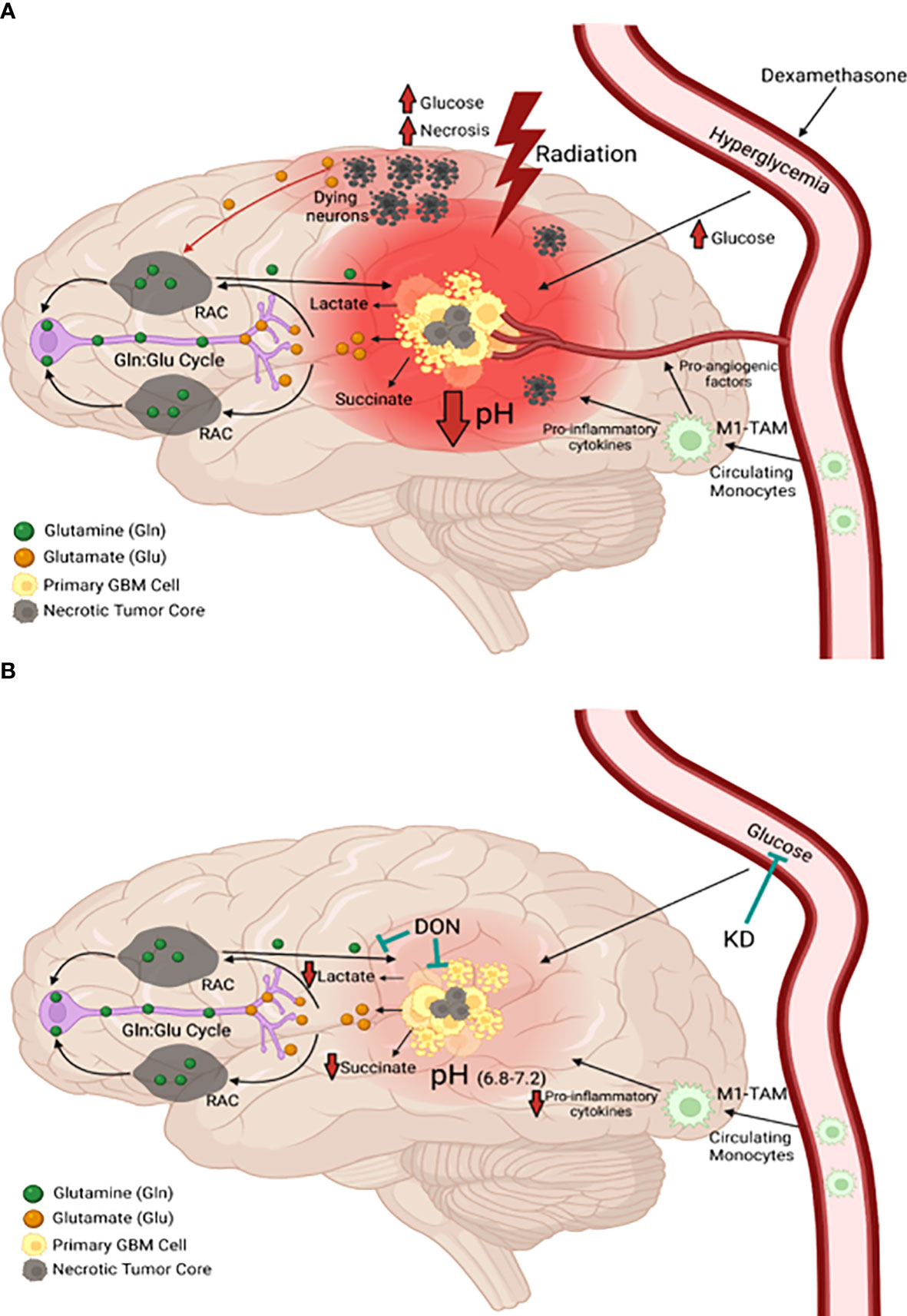
Figure 3 Origin and management of microenvironment acidity in GBM. (A) Glucose and glutamine are the primary energy metabolites necessary for driving rapid GBM growth. Glucose is the metabolic fuel necessary for nearly all brain functions under normal physiological conditions and is the major source of carbons for biomass synthesis through the glycolytic and pentose phosphate pathways in tumor cells. Tumor cells metabolize glutamine to glutamate, which is then metabolized to alpha-ketoglutarate. Significant energy is generated from the succinyl CoA ligase reaction (substrate level phosphorylation) in the glutaminolysis pathway using alpha-ketoglutarate-derived succinyl CoA as substrate (see Figure 7). In contrast to extracranial tissues, where glutamine is the most available amino acid, glutamine is tightly regulated in the brain through its involvement in the glutamate-glutamine cycle of neurotransmission (1, 78, 79). Glutamate is a major excitatory neurotransmitter that must be cleared rapidly following synaptic release in order to prevent excitotoxic damage to neurons (1, 79–81). Glial cells possess transporters for the clearance of extracellular glutamate, which is then metabolized to glutamine for delivery back to neurons. Neurons metabolize the glutamine to glutamate, which is then repackaged into synaptic vesicles for synaptic release (1). This cycle maintains low extracellular levels of both glutamate and glutamine in the normal neural parenchyma. Disruption of the cycle can provide neoplastic GBM cells access to glutamine. Besides serving as a metabolic fuel for the neoplastic tumor cells, glutamine is also an important fuel for cells of myeloid linage, which include macrophages, monocytes, microglia, and especially the invasive mesenchymal cells in GBM (1, 13, 82–84). In contrast to the proliferative GBM stem cells, the neoplastic GBM mesenchymal cells are thought to be derived from microglia or from microglia-stem cell fusion hybrids, which would have immuno-suppressive properties (82, 85). As long as GBM cells have access to glucose and glutamine, the tumor will grow and acidify the microenvironment making long-term management difficult. The current treatments for GBM (radiation and TMZ chemotherapy) will further increase glucose and glutamine availability, creating an unnecessary metabolic storm that will enhance microenvironment acidification and rapid tumor recurrence. The red hue is indicative of the inflammation and acidification of the tumor microenvironment (see text for further details). (B) The simultaneous restriction of glucose and glutamine, while elevating non-fermentable, anti-inflammatory ketone bodies, will reduce acidification, restore the pH balance of the microenvironment, and growth arrest or kill most of the neoplastic cells (11–13). RAC, reactive astrocytes; TAM, tumor-associated macrophages; Gln, glutamine; Glu, glutamate. These images were modified from that in (86).
The current treatment for GBM management involves debulking surgery, radiotherapy, and temozolomide chemotherapy (TMZ) (1, 8, 9, 87). While the waste products of glucose and glutamine fermentation will contribute to microenvironment acidification and the rapid growth of untreated GBM, the current treatments used for GBM management could also accelerate these processes after a growth delay following surgical debulking (1, 88, 89). It is documented that radiotherapy produces significant necrosis and hypoxia in the tumor microenvironment (1, 90–92). Radiotherapy disrupts the tightly regulated glutamine-glutamate cycle in the neural parenchyma thus increasing the levels of glutamine and glutamic acid as described further in Figure 3A.
Glutamic acid is an excitotoxic amino acid that enhances GBM invasion (1, 80, 81, 86, 93–96). Radiotherapy also damages the brain microenvironment, which increases glucose and glutamine availability to the tumor cells thus driving tumor growth through hyperglycolysis, necrosis, and acidification. While chemo-radiotherapy might have a role in the treatment of low-grade non-neural tumors, these confounding variables are ultimately responsible for GBM therapy resistance (1, 90, 97–99).
Linear regression analysis showed that blood glucose could predict the growth rate of the CT-2A malignant mouse astrocytoma, a stem-cell tumor (100, 101) (Figures 4A–C). Evidence also shows that survival is lower in GBM patients with higher blood glucose levels than in GBM patients with lower glucose levels (1, 103–111). Although the dexamethasone steroid is often prescribed along with standard treatments to reduce vasogenic edema, steroids will elevate blood glucose levels thus contributing indirectly to tumor growth (1, 112–114). Alternatives to dexamethasone for reducing vasogenic edema should receive consideration (115). Radiotherapy also increases blood glucose levels and facilitates hybridizations between tumor cells and macrophage/microglia thus producing highly invasive metastatic cells (1, 82, 108, 116–118). As glucose-derived lactic acid is the end product of glycolysis, GBM treatments that would elevate blood glucose levels will contribute to elevated lactic acid, microenvironment acidification, and tumor recurrence. Conversely, therapeutic strategies that would reduce glucose levels will lower lactic acid production, microenvironment acidification, and tumor recurrence (Figure 3B). It is clear from Figure 5 that calorie restriction, which lowers blood glucose while elevating ketone bodies, reduces microvessel density (angiogenesis) and increases tumor cell apoptosis in the CT-2A malignant astrocytoma. Hence, the dietary restriction of blood glucose can reduce microenvironment acidification through therapeutic effects on inflammation, angiogenesis and apoptosis.
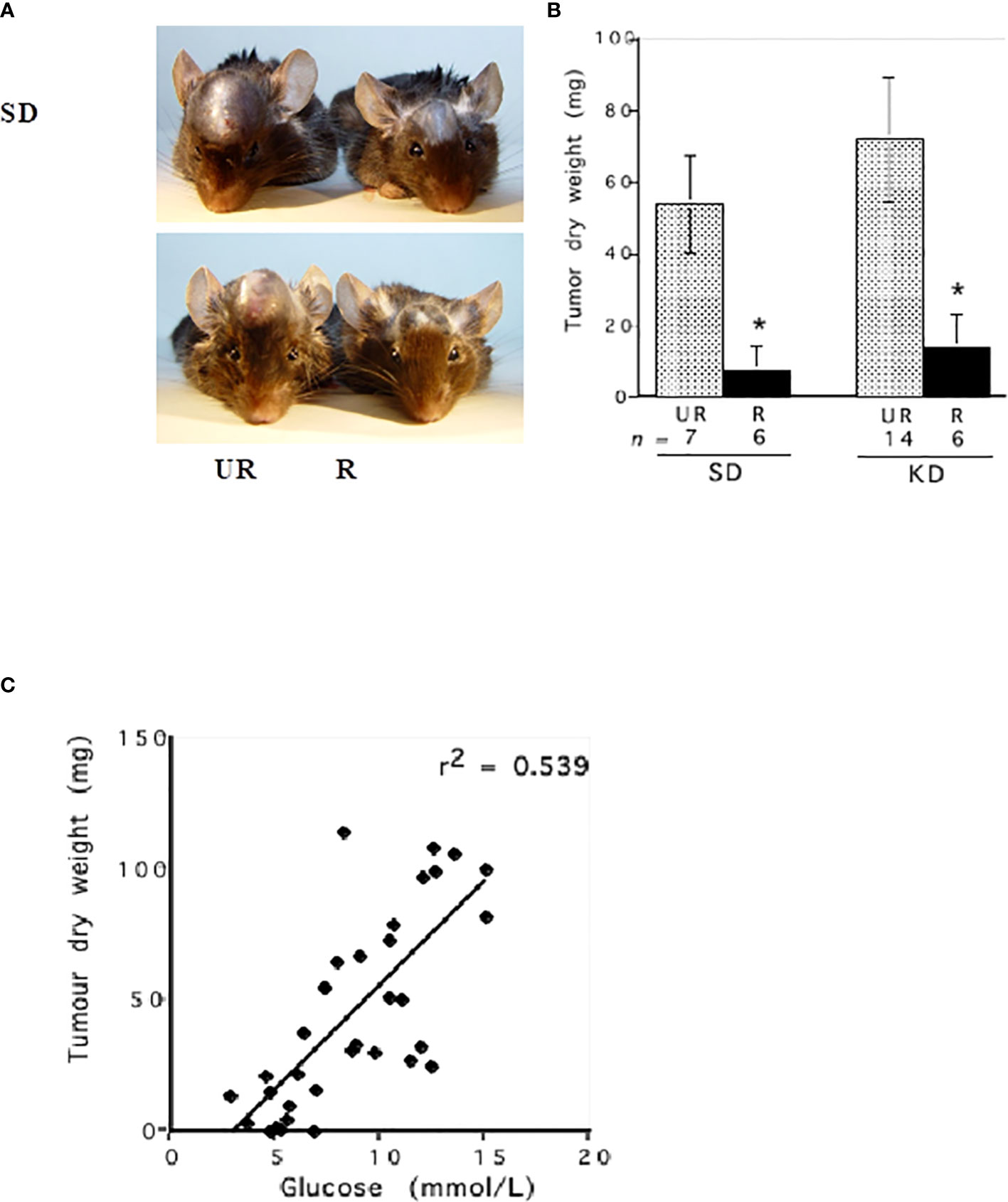
Figure 4 Influence of diet on the intracerebral growth of the CT-2A malignant astrocytoma. Dietary treatment was initiated 1 day after tumor implantation and was continued for 13 days. The visual representation (A) and quantitative assessment (B) of tumor growth in C57BL/6J mice receiving either the standard diet (SD) or ketogenic diet (KD) under either unrestricted (UR) or restricted (R) feeding. The asterisk indicates that the dry weights of the tumours in R groups were significantly lower than those in the UR groups at P < 0.01. (C) Linear regression analysis of plasma glucose and CT-2A-tumor growth in mice from both the SD and KD dietary groups combined (n = 34). These analyses included the values for plasma glucose and tumor growth of individual mice from both the UR and R-fed groups. The linear regression was highly significant at P < 0.001, indicating that blood glucose levels predict CT-2A tumor growth rate (100). The failure of the KD-UR to reduce blood glucose levels and tumor growth could be due to insulin insensitivity in this mouse strain (102). Images reproduced under a Creative Commons license.
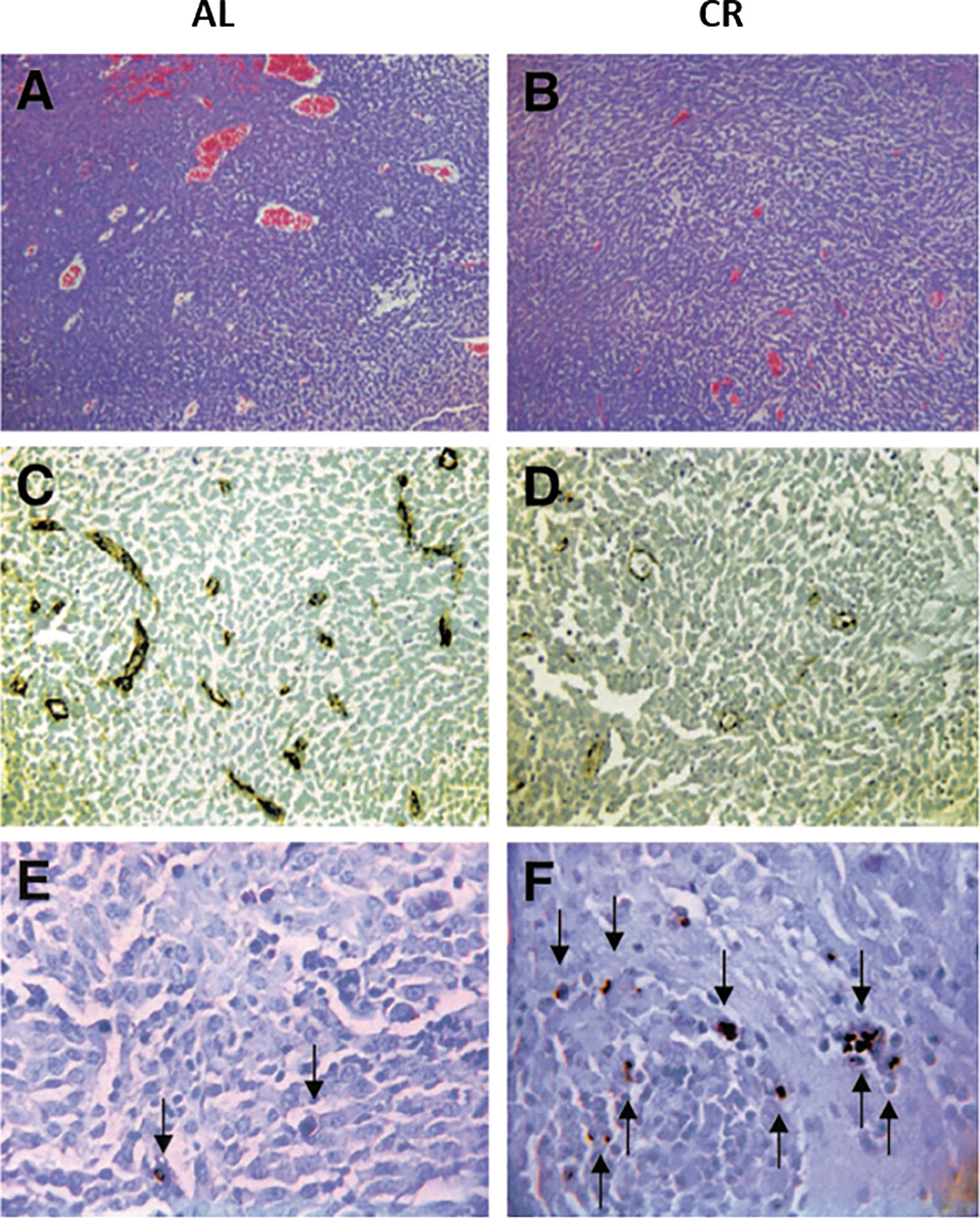
Figure 5 Influence of calorie restriction (CR) on microvessel density and apoptosis in the CT-2A malignant astrocytoma. CR was initiated 7 days before intracerebral tumor implantation and was continued for 11 days. H & E stained tumor sections in an ad libitum (AL) mouse (A) and in a CR mouse (B) (100X). Factor VIII immunostaining from the tumor grown in an AL mouse (C) and in a CR mouse (D) (200X). TUNEL positive apoptotic cells (arrows) from the tumor grown in an AL mouse (E) and in a CR mouse (F) (400X). Each stained section was representative of the entire tumor. All images were produced from digital photography. Image reproduced under a Creative Commons license from (119).
Accumulating evidence shows that the highly invasive mesenchymal cells seen in GBM are derived from neoplastic microglia or from microglia/macrophages that hybridize with non-invasive cancer stem cells, similar to that reported for other highly invasive metastatic cancers (82, 118, 120–124). Indeed, up to 60% of the cells in some GBM contain macrophage characteristics (125–128). We described how the neoplastic GBM cells with mesenchymal characteristics can be derived from transformed macrophages/microglia (13, 82, 129–131). As activated macrophages are immunosuppressive and acidify the microenvironment, it should be no surprise why immunotherapies have been largely ineffective in managing GBM (1, 132–134). The mesenchymal cells seen in GBM, whether part of the neoplastic cell population or part of the infiltrating cell population, will acidify the microenvironment through a variety of inflammation-linked mechanisms (135, 136). Some researchers also consider tumor cell-derived lactate as a checkpoint due to its ability to block immunotherapies (69). As lactate is derived from glucose, glucose restriction should reduce this “so called” checkpoint inhibitor. Hence, the mesenchymal cell populations in GBM will not only contribute to microenvironment acidification, but will also contribute to their own survival using glutamine as a metabolic fuel (13, 137, 138).
Immunotherapies have not yet been effective GBM management, but could be effective if there is evidence showing that they will not increase availability of glucose and glutamine in the tumor microenvironment, enhance inflammation, or cause hyper-progressive disease, as was documented in non-small cell lung cancer (139). Inflammatory oncotaxis, arising from surgical resection or from biopsy of lower-grade brain tumors, could also contribute to the transformation to high-grade secondary GBM (140–143). As the neoplastic macrophage/mesenchymal cells seen in GBM are dependent to a large degree on glutamine (13, 144), glutamine restriction will be essential for targeting these cells as we recently demonstrated (13). Recent studies show that a ketogenic diet can enhance the efficacy of immunotherapy (145). Most importantly, the simultaneous restriction of glucose and glutamine could improve the therapeutic efficacy of immunotherapies.
TMZ chemotherapy can contribute to microenvironment acidification through adverse effects on mitochondrial OxPhos function and increased production of GBM driver mutations (1, 9, 146, 147). In addition to increasing blood glucose levels, dexamethasone also increases glutamine levels through its induction of glutamine synthetase activity (7, 11, 86, 113, 148, 149). Bevacizumab (Avastin) is also widely prescribed to GBM patients to reduce angiogenesis (150–152). Bevacizumab, however, increases tumor necrosis while selecting for the most invasive and therapy-resistant tumor cells (153, 154). As both bevacizumab and TMZ damage mitochondria (155), these drugs will contribute further to tumor cell reliance on fermentation metabolism thus increasing microenvironment acidification (7, 11). Considered together, the current GBM chemotherapies inflict damage to the microenvironment and facilitate availability of glucose and glutamine to the neoplastic tumor cells, all of which will contribute to tumor recurrence, further acidification, and rapid progression (Figure 3A). It is not likely that overall patient survival could be improved when using therapies that increase distal tumor cell invasion and microenvironment acidification.
It should also be recognized that human cytomegalovirus (HCMV) infects many GBM that would further facilitate tumor cell use of glutamine and glucose (1, 156, 157). Recent studies show that vaccine-targeting of the HCMV pp65 protein could increase progression free and overall survival of some GBM patients (158). It would be interesting to determine if this therapeutic effect resulted in part from inhibition of the glycolysis or the glutaminolysis pathways in GBM cells (159, 160). Glucose and glutamine are required for synthesis of glutathione while glutamine is essential for the action of manganese superoxide dismutase (161–163). Consequently, the elevated use of glucose and glutamine, which increases anti-oxidant potential, will contribute to the resistance of GBM cells to chemotherapy and radiotherapy.
It is known that elevated aerobic fermentation (Warburg effect) also drives the multidrug resistant (MDR) phenotype, which protects GBM cells from toxic chemotherapy (1, 5, 41, 164). Hence, the treatment-linked increases in fermentable energy metabolites and disruption of the tumor microenvironment can explain in large part how overall survival remains so poor for most GBM patients treated with current standard of care (7, 97). The information presented in Figure 3A describes how current therapies can facilitate rapid GBM recurrence. It is our view that these therapies can account in large part the remarkable reproducibility of poor patient survival across multiple surgical institutions as seen in Figure 1. It is unlikely that GBM patient survival will improve significantly if therapies that increase microenvironment acidification and are inherently ineffective are continuously used (165).
As fermentation metabolism is ultimately responsible for rapid GBM growth and the acidification of the microenvironment, then therapies that target fermentation metabolism should reduce acidification and GBM growth. Metabolic therapy involves diet/drug combinations that target the availability of glucose and glutamine while also elevating non-fermentable, anti-inflammatory ketone bodies (13, 41, 166–170). Most importantly, GBM and other tumors cannot use ketone bodies for energy due to deficiencies in SCOT; the key mitochondrial enzyme needed for ketone body metabolism (171–173). No evidence has been presented, to our knowledge, showing that ketone bodies can replace glucose or glutamine in serum free media for the survival of any tumor cell. Ketogenic diets and water-only therapeutic fasting will lower circulating glucose levels while elevating circulating D-β-hydroxybutyrate (D-β-OHB) levels (41, 174–176). Water-only fasting in humans is comparable to a 40% calorie restriction in mice due to differences in basal metabolic rate that about six times faster in mice than in humans (177). Therapeutic strategies that lower blood glucose while elevating blood ketone bodies are anti-angiogenic, anti-edematous, anti-inflammatory, and pro-apoptotic. Evidence supporting this statement was described previously (13, 178). Diets that lower glucose and elevate D-β-OHB can also reduce circulating levels of insulin-like growth factor 1 (IGF-1), a known driver of tumor growth (Table 1). There is no known drug that can produce the broad range of therapeutic effects as can diets that reduce glucose while elevating D-β-OHB.
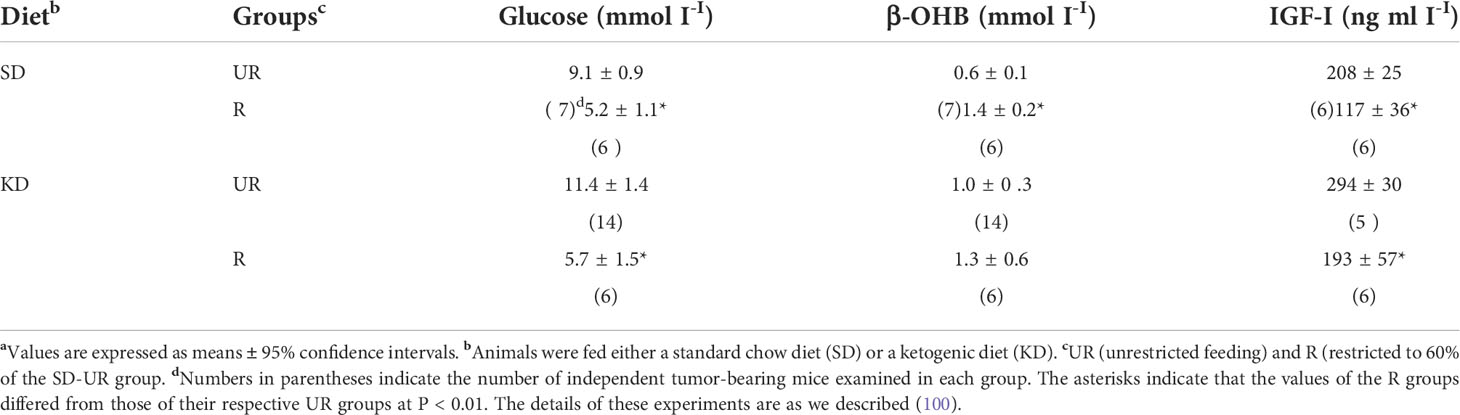
Table 1 Influence of diet on plasma glucose, β-OHB, and IGF-I levels in mice bearing the CT-2A intracerebral brain tumoura.
It is important to mention that blood glucose can be reduced to very low levels (less than 1.0 mM) in humans that are in therapeutic ketosis (6-8 mM, D-β-OHB) without producing hypoglycemic reactions (174, 179, 180). A whole-body transition from glucose-driven metabolism to D-β-OHB-driven metabolism will reduce circulating glucose levels thus reducing extracellular acidification from lactic acid production. At the same time, this transition will also produce metabolic stress on all neoplastic GBM cells that are dependent on glycolysis for growth (41, 172, 174, 181). Moreover, D-β-OHB metabolism enhances the ΔG’ATP hydrolysis in normal cells from -56 kJ/mole to -59 kJ/mole, thus providing normal cells with an energetic advantage over the fermentation-dependent tumor cells (41). We are not familiar with any therapies, besides ketogenic metabolic therapy (KMT), that can enhance the energetic advantage of normal cells over that of tumor cells (11, 41).
The energetic advantage of D−β-OHB metabolism in normal cells is seen predominantly with D-β-OHB, and is not seen with either the D/L- β-OHB racemic mixture or with fatty acids (174, 182, 183). On the other hand, racemic D/L-β-OHB tends to reduce blood glucose more through shifting redox state in the liver and can potentially increase ROS production in tumor cells through β-oxidation of the L-form (41). The L-β-OHB interconverts back to D-β-OHB (in tissues) through a racemase enzyme or gets converted to acetyl-CoA. The L-β-OHB also has greater potential as a signaling molecule since it remains in circulation longer and has similar effects at suppressing the NLRP3 inflammasome and epigenetic effects (184–186). Hence, D-β-OHB and D/L-β-OHB can stress tumor cell metabolism while enhancing the metabolism of normal cells through a variety of mechanisms.
The therapeutic effects seen with ketone bodies are generally best when blood glucose levels are low (generally below 3.6 mM), as little or no therapeutic benefit is seen in either preclinical GBM models or in human patients when glucose levels remain elevated (100, 171, 187). These therapeutic glucose levels could be difficult to achieve for many GBM patients, however, due to the glucose-elevating effects of the current standard treatments used to manage GBM. We also did not find any therapeutic benefit of sodium bicarbonate on the growth of the VM-M3 mouse glioblastoma suggesting that alkalinization using sodium bicarbonate was ineffective in managing this GBM model (L. Shelton, unpublished). It is the synergistic action of low blood glucose with elevated ketone bodies that provides the best therapeutic strategy for slowing growth and reducing microenvironment inflammation and acidification.
In addition to glucose, glutamine is the other major fuel that drives GBM growth especially the neoplastic mesenchymal cells (14, 137, 144). We showed that the glutamine-targeting analogue, 6-diazo-5-oxo-L-norleucine (DON), used with a calorie restricted ketogenic diet could significantly reduce growth and improve overall survival in preclinical models of GBM (Figure 6). Moreover, we found that ketogenic diets facilitated delivery of DON and other small molecules through the blood brain barrier (13, 188). This delivery may be due in part to the action of the content of caprylic acid in the diet (189). Hyperbaric oxygen therapy can also reduce angiogenesis and microenvironment inflammation especially in combination with therapeutic ketosis (41, 190–193). In addition to findings in preclinical models, we also described how the IDH1 mutation could act as a therapeutic drug that simultaneous targets the glycolysis and glutaminolysis pathways to improve survival in a GBM patient (11) (Figure 7). The long term survival of this patient, now at eight years, was attributed to a combination of his younger age, his low-carbohydrate ketogenic diet, his acquired IDH1 mutation, and finally to his avoidance of radiation, TMZ, and steroids (11). Ketogenic metabolic therapy involves the synergistic therapeutic action of the KD used with drugs and procedures that restrict availability of glucose and glutamine while providing normal cells with an energetic advantage over tumor cells that are limited to energy generation through fermentation (12, 41). More recent studies also support some of these observations in younger GBM patients (199). Persistent statements suggesting that tumor cells have a growth advantage over normal cells make no sense in the light of evolutionary theory (12). The metabolic pathways contributing to GBM microenvironment acidification and their management by KMT and the IDH1 mutation are described in Figure 7.
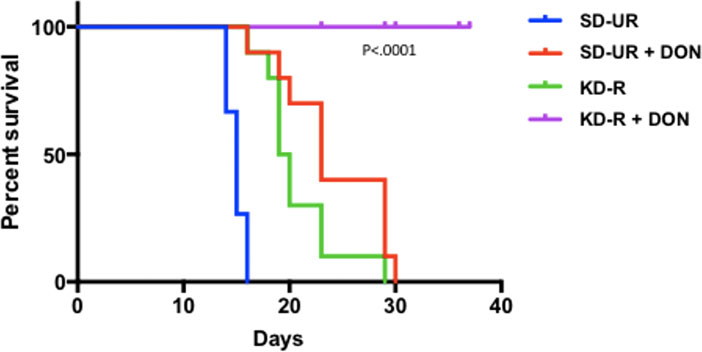
Figure 6 Influence of diet/drug therapy on overall survival of VM/Dk mice with the VM-M3 invasive GBM. A calorie restricted ketogenic diet (KD-R) was administered together with the glutaminase inhibitor, 6-diazo-5-oxo-L-norleucine (DON) as we described (13). Overall survival was significantly longer in the tumor bearing mice receiving the diet/drug combination (KD-R + DON) than in the mice receiving the standard high-carbohydrate diet (SD-UR), the KD-R alone, or DON alone. It is important to mention that 2-3x more DON was delivered to the tumor of the mice fed the KD-R than to the mice fed the SD-UR indicating that the KD facilitates a non-toxic delivery of small drug molecules through the blood brain barrier (13, 188). Image reproduced under a Creative Commons license from (13).
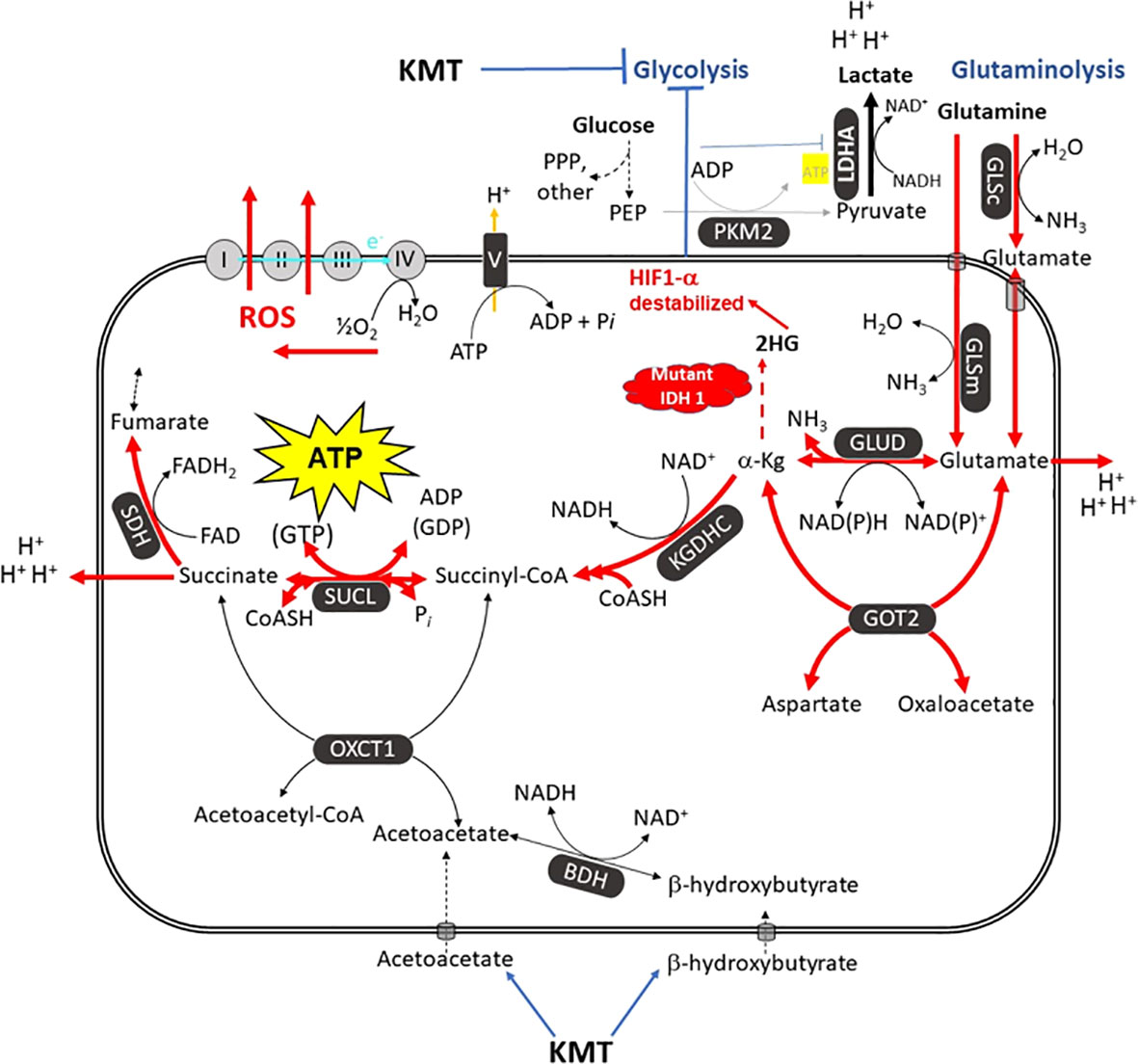
Figure 7 The metabolic pathways responsible for the acidification of the GBM microenvironment. GBM growth is dependent on glucose carbons for biomass synthesis through glycolysis and glutamine carbons for ATP synthesis through glutaminolysis. The glutamine nitrogen is necessary for protein and nucleic acid synthesis. The waste products of the glycolytic and the glutaminolysis pathways (lactic acid, glutamic acid, and succinic acid) will acidify the GBM microenvironment. The oxygen consumption is linked to ROS production, not to ATP synthesis. Excessive ROS produce somatic mutations and further increase inflammation and acidification of the microenvironment (7, 12, 14, 194). A calorie restricted KD will reduce glucose availability for glycolysis while also interfering with the glutaminolysis pathway (11). Glutamine-driven mSLP in the glutaminolysis pathway is a major source of ATP synthesis for GBM cells (7, 14). The glutaminolysis pathway (red) becomes dominant in tumor cells with inefficient OxPhos and that express the dimeric PKM2 isoform. PKM2 is expressed in GBM and produces less ATP through glycolysis than does the PKM1 isoform (73, 75, 195, 196). The elevation of ketone bodies (D-β-hydroxybutyrate and acetoacetate) through KD will indirectly reduce ATP synthesis through the succinate CoA ligase (SUCL) reaction by diverting CoA from succinate to acetoacetate. The IDH1 mutation could reduce ATP synthesis through mSLP by increasing synthesis of 2-hydroxyglutarate that is derived from α-ketoglutarate and thus reduce the succinyl CoA substrate for the SUCL reaction (11, 14, 197). Besides its potential effect in reducing glutaminolysis, 2-hydroxyglutarate can also target multiple HIF1α-responsive genes and enzymes in the glycolysis pathway thus limiting synthesis of metabolites and one-carbon metabolism needed for rapid tumor growth (7, 14, 68, 198). The down regulation of Hif1-α-regulated lactate dehydrogenase A (LDHA), through the action of both the KD and the IDH1 mutation, will reduce extracellular lactate levels thus further reducing microenvironment inflammation, acidification, and tumor cell invasion. Hence, the simultaneous inhibition of glycolysis and glutaminolysis through the synergistic effects KMT and the IDH1 mutation will reduce the majority of signaling pathways necessary for rapid GBM growth and acidification of the microenvironment. BDH, β-hydroxybutyrate dehydrogenase; FAD, flavin adenine dinucleotide; GLSc, glutaminase cytosolic; GLSm, glutaminase mitochondrial; GLUD, glutamate dehydrogenase; GOT2, aspartate aminotransferase; KGDHC, α-ketoglutarate dehydrogenase complex; LDHA, lactate dehydrogenase A; NME, nucleoside diphosphate kinase; OXCT1, succinyl-CoA:3-ketoacid coenzyme A transferase 1; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PEP, phosphoenolpyruvate; PKM2, pyruvate kinase M2; SDH, succinate dehydrogenase; SUCL, succinate-CoA ligase. KMT, Ketogenic metabolic therapy. Reprinted with modifications from (14).
There are several limitations that currently prevent the application of metabolic therapy for reducing microenvironment acidification and the growth of GBM. First, the dosage, timing, and scheduling of the diet/drug combinations that can best target glucose and glutamine availability have yet to be optimized for most GBM patients (1, 11, 41, 87). Second, the findings that GBM is largely dependent on glucose and glutamine fermentation for growth due to OxPhos deficiency is inconsistent with the current dogmatic view that GBM and most other cancers are exceedingly complex genetic diseases requiring complicated Rube Goldberg-type solutions (12). Finally, the most important limitation for adapting metabolic therapy in the clinic is the absence of a business model that can generate sufficient replacement revenue using cost-effective, non-toxic metabolic therapies (200–203). We predict that major advances in overall GBM patient survival will be realized once GBM becomes recognized as a mitochondrial metabolic disease and when non-toxic metabolic therapies become the standard of care for management.
Microenvironment acidification is largely responsible for drug resistance, enhanced invasion, immunosuppression, and metastasis. The acidic waste products of glucose and glutamine fermentation metabolism (lactic acid, glutamic acid, and succinic acid), generated within the neoplastic tumor cells, are responsible for the acidification of the GBM microenvironment. Stated simply: The greater is the availability of fermentable fuels, the greater is the resistance to therapy. The cancer microenvironment will heal itself if the origin of the acidification can be removed. Therapeutic strategies that restrict the availability of fermentable fuels, while increasing levels of non-fermentable ketone bodies, will reduce acidification, eliminate the majority of neoplastic tumor cells, and thus improve GBM management.
All authors read and approved the information presented in the manuscript. ST wrote the manuscript. A-MG, ZG, LD, DT, EA, MJ, MP, TL, D’AD, KM, and CC made material contributions to content and revisions.
We thank the Foundation for Metabolic Cancer Therapies, CrossFit Inc., The Nelson and Claudia Peltz Family Foundation, Lewis Topper, The John and Kathy Garcia Foundation, Mr. Edward Miller, Kenneth Rainin Foundation, the Corkin Family Foundation, Children with Cancer UK, and the Boston College Research Expense Fund for their support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author KM was employed by the company BERG LLC. Author SL was employed by the company Matterworks.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as the potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
PPP, pentose phosphate pathway; OxPhos, oxidative phosphorylation; mSLP, mitochondrial substrate level phosphorylation; TMZ, temozolomide; D-β-OHB, D-β-hydroxybutyrate; KD, ketogenic diet; KMT, Ketogenic metabolic therapy; ROS, reactive oxygen species.
1. Seyfried TN, Shelton L, Arismendi-Morillo G, Kalamian M, Elsakka A, Maroon J, et al. Provocative question: Should ketogenic metabolic therapy become the standard of care for glioblastoma? Neurochem Res (2019) 44(10):2392–404. doi: 10.1007/s11064-019-02795-4
2. Efremov L, Abera SF, Bedir A, Vordermark D, Medenwald D. Patterns of glioblastoma treatment and survival over a 16-years period: pooled data from the german cancer registries. J Cancer Res Clin Oncol (2021) 147(11):3381–90. doi: 10.1007/s00432-021-03596-5
3. Polivka J Jr., Polivka J, Holubec L, Kubikova T, Priban V, Hes O, et al. Advances in experimental targeted therapy and immunotherapy for patients with glioblastoma multiforme. Anticancer Res (2017) 37(1):21–33. doi: 10.21873/anticanres.11285
4. Fabbro-Peray P, Zouaoui S, Darlix A, Fabbro M, Pallud J, Rigau V, et al. Association of patterns of care, prognostic factors, and use of radiotherapy-temozolomide therapy with survival in patients with newly diagnosed glioblastoma: a French national population-based study. J Neurooncol (2018) 142(1):91–101. doi: 10.1007/s11060-018-03065-z
5. Geraldo LHM, Garcia C, da Fonseca ACC, Dubois LGF, de Sampaio ESTCL, Matias D, et al. Glioblastoma therapy in the age of molecular medicine. Trends Cancer (2019) 5(1):46–65. doi: 10.1016/j.trecan.2018.11.002
6. Wegman-Ostrosky T, Reynoso-Noveron N, Mejia-Perez SI, Sanchez-Correa TE, Alvarez-Gomez RM, Vidal-Millan S, et al. Clinical prognostic factors in adults with astrocytoma: Historic cohort. Clin Neurol Neurosurg (2016) 146:116–22. doi: 10.1016/j.clineuro.2016.05.002
7. Chinopoulos C, Seyfried TN. Mitochondrial substrate-level phosphorylation as energy source for glioblastoma: Review and hypothesis. ASN Neuro (2018) 10:1759091418818261. doi: 10.1177/1759091418818261
8. Fatehi M, Hunt C, Ma R, Toyota BD. Persistent disparities in survival for patients with glioblastoma. World Neurosurg (2018) 120:e511–e6. doi: 10.1016/j.wneu.2018.08.114
9. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7
10. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
11. Seyfried TN, Shivane AG, Kalamian M, Maroon JC, Mukherjee P, Zuccoli G. Ketogenic metabolic therapy, without chemo or radiation, for the long-term management of idh1-mutant glioblastoma: An 80-month follow-up case report. Front Nutr (2021) 8:682243. doi: 10.3389/fnut.2021.682243
12. Seyfried TN, Chinopoulos C. Can the mitochondrial metabolic theory explain better the origin and management of cancer than can the somatic mutation theory? Metabolites (2021) 11(9):572. doi: 10.3390/metabo11090572
13. Mukherjee P, Augur ZM, Li M, Hill C, Greenwood B, Domin MA, et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun Biol (2019) 2:200. doi: 10.1038/s42003-019-0455-x
14. Seyfried TN, Arismendi-Morillo G, Mukherjee P, Chinopoulos C. On the origin of atp synthesis in cancer. iScience (2020) 23(11):101761. doi: 10.1016/j.isci.2020.101761
15. Deighton RF, Le Bihan T, Martin SF, Gerth AM, McCulloch M, Edgar JM, et al. Interactions among mitochondrial proteins altered in glioblastoma. J neuro-oncol (2014) 118(2):247–56. doi: 10.1007/s11060-014-1430-5
16. Arismendi-Morillo GJ, Castellano-Ramirez AV. Ultrastructural mitochondrial pathology in human astrocytic tumors: potentials implications pro-therapeutics strategies. J Electron Micros (Tokyo) (2008) 57(1):33–9. doi: 10.1093/jmicro/dfm038
17. Feichtinger RG, Weis S, Mayr JA, Zimmermann F, Geilberger R, Sperl W, et al. Alterations of oxidative phosphorylation complexes in astrocytomas. Glia (2014) 62(4):514–25. doi: 10.1002/glia.22621
18. Katsetos CD, Anni H, Draber P. Mitochondrial dysfunction in gliomas. Semin Pediatr neurol (2013) 20(3):216–27. doi: 10.1016/j.spen.2013.09.003
19. Deighton RF, Le Bihan T, Martin SF, Barrios-Llerena ME, Gerth AM, Kerr LE, et al. The proteomic response in glioblastoma in young patients. J neuro-oncol (2014) 8(27):44141x–58. doi: 10.1007/s11060-014-1474-6
20. Oudard S, Boitier E, Miccoli L, Rousset S, Dutrillaux B, Poupon MF. Gliomas are driven by glycolysis: Putative roles of hexokinase, oxidative phosphorylation and mitochondrial ultrastructure. Anticancer Res (1997) 17(3C):1903–11.
21. Scheithauer BW, Bruner JM. The ultrastructural spectrum of astrocytic neoplasms. Ultrastructural Pathol (1987) 11(5-6):535–81. doi: 10.3109/01913128709048447
22. Sipe JC, Herman MM, Rubinstein LJ. Electron microscopic observations on human glioblastomas and astrocytomas maintained in organ culture systems. Am J pathol (1973) 73(3):589–606.
23. Rhodes CG, Wise RJ, Gibbs JM, Frackowiak RS, Hatazawa J, Palmer AJ, et al. In vivo disturbance of the oxidative metabolism of glucose in human cerebral gliomas. Ann Neurol (1983) 14(6):614–26. doi: 10.1002/ana.410140604
24. Meixensberger J, Herting B, Roggendorf W, Reichmann H. Metabolic patterns in malignant gliomas. J Neurooncol (1995) 24(2):153–61. doi: 10.1007/BF01078485
25. Arismendi-Morillo G, Castellano-Ramirez A, Seyfried TN. Ultrastructural characterization of the mitochondria-associated membranes abnormalities in human astrocytomas: Functional and therapeutics implications. Ultrastruct Pathol (2017) 41(3):234–44. doi: 10.1080/01913123.2017.1300618
26. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J (2011) 435(2):297–312. doi: 10.1042/BJ20110162
27. Lehninger AL. The mitochondrion: Molecular basis of structure and function. New York: W.A. Benjamin, INC (1964). 263 p.
28. Michl J, Wang Y, Monterisi S, Blaszczak W, Beveridge R, Bridges EM, et al. CRISPR-Cas9 screen identifies oxidative phosphorylation as essential for cancer cell survival at low extracellular pH. Cell Rep (2022) 38(10):110493. doi: 10.1016/j.celrep.2022.110493
29. Orang AV, Petersen J, McKinnon RA, Michael MZ. Micromanaging aerobic respiration and glycolysis in cancer cells. Mol Metab (2019) 23:98–126. doi: 10.1016/j.molmet.2019.01.014
30. Strickland M, Stoll EA. Metabolic reprogramming in glioma. Front Cell Dev Biol (2017) 5:43. doi: 10.3389/fcell.2017.00043
31. Vaupel P, Multhoff G. Revisiting the warburg effect: historical dogma versus current understanding. J Physiol (2021) 599(6):1745–57. doi: 10.1113/JP278810
32. Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res (2018) 28(3):265–80. doi: 10.1038/cr.2017.155
33. Moreno-Sanchez R, Marin-Hernandez A, Saavedra E, Pardo JP, Ralph SJ, Rodriguez-Enriquez S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol (2014) 50:10–23. doi: 10.1016/j.biocel.2014.01.025
34. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med (2018) 24(7):1036–46. doi: 10.1038/s41591-018-0052-4
35. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11(5):325–37. doi: 10.1038/nrc3038
36. Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev (2012) 26(17):1926–44. doi: 10.1101/gad.188292.112
37. Garofano L, Migliozzi S, Oh YT, D’Angelo F, Najac RD, Ko A, et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat Cancer (2021) 2(2):141–56. doi: 10.1038/s43018-020-00159-4
38. Cairns RA. Drivers of the warburg phenotype. Cancer J (2015) 21(2):56–61. doi: 10.1097/PPO.0000000000000106
39. Biswas S, Lunec J, Bartlett K. Non-glucose metabolism in cancer cells–is it all in the fat? Cancer Metastasis Rev (2012) 31(3-4):689–98. doi: 10.1007/s10555-012-9384-6
40. Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun (2004) 313(3):459–65. doi: 10.1016/j.bbrc.2003.11.136
41. Seyfried TN, Yu G, Maroon JC, D’Agostino DP. Press-pulse: a novel therapeutic strategy for the metabolic management of cancer. Nutr Metab (Lond) (2017) 14:19. doi: 10.1186/s12986-017-0178-2
42. Seyfried TN. Nothing in cancer biology makes sense except in the light of evolution. In: Cancer as a metabolic disease: On the origin, management, and prevention of cancer. Hoboken, NJ: John Wiley & Sons (2012). p. 261–75.
43. Potts R. Environmental hypotheses of hominin evolution. Am J Phys Anthropol (1998) Suppl 27:93–136. doi: 10.1002/(SICI)1096-8644(1998)107:27+<93::AID-AJPA5>3.0.CO;2-X
44. Potts R. Complexity of adaptibility in human evolution. In: Goodman M, Moffat AS, editors. Probing human origins. Cambridge, MA: American Academy of Arts & Sciences (2002). p. 33–57.
45. Dobzhansky T. Nothing in biology makes sense except in the light of evolution. Amer Biol Teacher (1973) 35(March):125–9. doi: 10.2307/4444260
46. Solaini G, Sgarbi G, Baracca A. Oxidative phosphorylation in cancer cells. Biochim Biophys Acta (2011) 1807(6):534–42. doi: 10.1016/j.bbabio.2010.09.003
47. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
48. Warburg O. On the respiratory impairment in cancer cells. Science (1956) 124:269–70. doi: 10.1126/science.124.3215.269
49. Burk D, Woods M, Hunter J. On the significance of glucolysis for cancer growth, with special reference to morris rat hepatomas. J Natl Cancer Inst (1967) 38(6):839–63.
50. Weinhouse S. On respiratory impairment in cancer cells. Science (1956) 124(3215):267–9. doi: 10.1126/science.124.3215.267
51. Weinhouse S. The warburg hypothesis fifty years later. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol (1976) 87(2):115–26. doi: 10.1007/BF00284370
52. Viale A, Corti D, Draetta GF. Tumors and mitochondrial respiration: A neglected connection. Cancer Res (2015) 75(18):3685–6. doi: 10.1158/0008-5472.CAN-15-0491
53. Duraj T, Carrion-Navarro J, Seyfried TN, Garcia-Romero N, Ayuso-Sacido A. Metabolic therapy and bioenergetic analysis: The missing piece of the puzzle. Mol Metab (2021) 54:101389. doi: 10.1016/j.molmet.2021.101389
54. Ta NL, Seyfried TN. Influence of serum and hypoxia on incorporation of [(14)C]-D-Glucose or [(14)C]-L-Glutamine into lipids and lactate in murine glioblastoma cells. Lipids (2015) 50(12):1167–84. doi: 10.1007/s11745-015-4075-z
55. Ceruti S, Mazzola A, Abbracchio MP. Resistance of human astrocytoma cells to apoptosis induced by mitochondria-damaging agents: Possible implications for anticancer therapy. J Pharmacol Exp Ther (2005) 314(2):825–37. doi: 10.1124/jpet.105.085340
56. Barron ES. The catalytic effect of methylene blue on the oxygen consumption of tumors and normal tissues. J Exp Med (1930) 52(3):447–56. doi: 10.1084/jem.52.3.447
57. Renner C, Asperger A, Seyffarth A, Meixensberger J, Gebhardt R, Gaunitz F. Carnosine inhibits ATP production in cells from malignant glioma. Neurol Res (2010) 32(1):101–5. doi: 10.1179/016164109X12518779082237
58. Ahmad F, Sun Q, Patel D, Stommel JM. Cholesterol metabolism: A potential therapeutic target in glioblastoma. Cancers (Basel) (2019) 11(2):146. doi: 10.3390/cancers11020146
59. Warburg O. The metabolism of carcinoma cells. J Cancer Res (1925) 9(1):148–63. doi: 10.1158/jcr.1925.148
60. Velez J, Hail N Jr., Konopleva M, Zeng Z, Kojima K, Samudio I, et al. Mitochondrial uncoupling and the reprograming of intermediary metabolism in leukemia cells. Front Oncol (2013) 3:67. doi: 10.3389/fonc.2013.00067
61. Zhu Y, Dean AE, Horikoshi N, Heer C, Spitz DR, Gius D. Emerging evidence for targeting mitochondrial metabolic dysfunction in cancer therapy. J Clin Invest (2018) 128(9):3682–91. doi: 10.1172/JCI120844
62. Fisher-Wellman KH, Hagen JT, Kassai M, Kao LP, Nelson MAM, McLaughlin KL, et al. Alterations in sphingolipid composition and mitochondrial bioenergetics represent synergistic therapeutic vulnerabilities linked to multidrug resistance in leukemia. FASEB J (2022) 36(1):e22094. doi: 10.1096/fj.202101194RRR
63. Gao C, Shen Y, Jin F, Miao Y, Qiu X. Cancer stem cells in small cell lung cancer cell line h446: Higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS One (2016) 11(5):e0154576. doi: 10.1371/journal.pone.0154576
64. Flores RE, Brown AK, Taus L, Khoury J, Glover F, Kami K, et al. Mycoplasma infection and hypoxia initiate succinate accumulation and release in the VM-M3 cancer cells. Biochim Biophys Acta (2018) 1859(9):975–83. doi: 10.1016/j.bbabio.2018.03.012
65. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
66. Slaughter AL, D’Alessandro A, Moore EE, Banerjee A, Silliman CC, Hansen KC, et al. Glutamine metabolism drives succinate accumulation in plasma and the lung during hemorrhagic shock. J Trauma Acute Care Surg (2016) 81(6):1012–9. doi: 10.1097/TA.0000000000001256
67. Zhang S, Liang Y, Li L, Chen Y, Wu P, Wei D. Succinate: a novel mediator to promote atherosclerotic lesion progression. DNA Cell Biol (2022) 41(3):285–91. doi: 10.1089/dna.2021.0345
68. Chesnelong C, Chaumeil MM, Blough MD, Al-Najjar M, Stechishin OD, Chan JA, et al. Lactate dehydrogenase a silencing in IDH mutant gliomas. Neuro Oncol (2014) 16(5):686–95. doi: 10.1093/neuonc/not243
69. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell (2022) 40(2):201–18 e9. doi: 10.1016/j.ccell.2022.01.001
70. Ge T, Yang J, Zhou S, Wang Y, Li Y, Tong X. The role of the pentose phosphate pathway in diabetes and cancer. Front Endocrinol (Lausanne) (2020) 11:365. doi: 10.3389/fendo.2020.00365
71. Ahmad F, Dixit D, Sharma V, Kumar A, Joshi SD, Sarkar C, et al. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis (2016) 7:e2213. doi: 10.1038/cddis.2016.117
72. Stanke KM, Wilson C, Kidambi S. High expression of glycolytic genes in clinical glioblastoma patients correlates with lower survival. Front Mol Biosci (2021) 8:752404. doi: 10.3389/fmolb.2021.752404
73. Mukherjee J, Phillips JJ, Zheng S, Wiencke J, Ronen SM, Pieper RO. Pyruvate kinase M2 expression, but not pyruvate kinase activity, is up-regulated in a grade-specific manner in human glioma. PLoS One (2013) 8(2):e57610. doi: 10.1371/journal.pone.0057610
74. Chinopoulos C. From glucose to lactate and transiting intermediates through mitochondria, bypassing pyruvate kinase: Considerations for cells exhibiting dimeric pkm2 or otherwise inhibited kinase activity. Front Physiol (2020) 11:543564. doi: 10.3389/fphys.2020.543564
75. Liu YR, Song DD, Liang DM, Li YJ, Yan YF, Sun HF, et al. Oncogenic TRIB2 interacts with and regulates PKM2 to promote aerobic glycolysis and lung cancer cell procession. Cell Death Discov (2022) 8(1):306. doi: 10.1038/s41420-022-01095-1
76. Pouliquen DL. Hepatic mitochondrial function and brain tumours. Curr Opin Clin Nutr Metab Care (2007) 10(4):475–9. doi: 10.1097/MCO.0b013e328108f452
77. Mulrooney TJ, Marsh J, Urits I, Seyfried TN, Mukherjee P. Influence of caloric restriction on constitutive expression of nf-kappab in an experimental mouse astrocytoma. PLoS One (2011) 6(3):e18085. doi: 10.1371/journal.pone.0018085
78. McKenna MC, Gruetter R, Sonnewald U, Waagepetersen HS, Schousboe A. Energy metabolism of the brain. In: Siegel GJ, Albers RW, Bradey ST, Price DP, editors. Basic neurochemistry: Molecular, cellular, and medical aspects. New York: Elsevier Academic Press (2006). p. 531–57.
79. Sonnewald U, Schousboe A. Introduction to the glutamate-glutamine cycle. Adv Neurobiol (2016) 13:1–7. doi: 10.1007/978-3-319-45096-4_1
80. Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res (1999) 59(17):4383–91.
81. Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med (2001) 7(9):1010–5. doi: 10.1038/nm0901-1010
82. Huysentruyt LC, Akgoc Z, Seyfried TN. Hypothesis: are neoplastic macrophages/microglia present in glioblastoma multiforme? ASN neuro (2011) 3(4):e00064. doi: 10.1042/AN20110011
83. Newsholme P. Why is l-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J Nutr (2001) 131(9 Suppl):2515S–22S. doi: 10.1093/jn/131.9.2515S
84. Lewis C, Murdoch C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am J Pathol (2005) 167(3):627–35. doi: 10.1016/S0002-9440(10)62038-X
85. Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol (1999) 100(1-2):216–32. doi: 10.1016/S0165-5728(99)00203-9
86. Seyfried TN, Shelton LM, Mukherjee P. Does the existing standard of care increase glioblastoma energy metabolism? Lancet Oncol (2010) 11(9):811–3. doi: 10.1016/S1470-2045(10)70166-2
87. Elsakka AMA, Bary MA, Abdelzaher E, Elnaggar M, Kalamian M, Mukherjee P, et al. Management of glioblastoma multiforme in a patient treated with ketogenic metabolic therapy and modified standard of care: a 24-month follow-up. Front Nutr (2018) 5:20. doi: 10.3389/fnut.2018.00020
88. Ahmadloo N, Kani AA, Mohammadianpanah M, Nasrolahi H, Omidvari S, Mosalaei A, et al. Treatment outcome and prognostic factors of adult glioblastoma multiforme. J Egypt Natl Canc Inst (2013) 25(1):21–30. doi: 10.1016/j.jnci.2012.11.001
89. Stensjoen AL, Solheim O, Kvistad KA, Haberg AK, Salvesen O, Berntsen EM. Growth dynamics of untreated glioblastomas in vivo. Neuro Oncol (2015) 17(10):1402–11. doi: 10.1093/neuonc/nov029
90. Winter SF, Loebel F, Loeffler J, Batchelor TT, Martinez-Lage M, Vajkoczy P, et al. Treatment-induced brain tissue necrosis: A clinical challenge in neuro-oncology. Neuro Oncol (2019) 21(9):1118–30. doi: 10.1093/neuonc/noz048
91. Lawrence YR, Blumenthal DT, Matceyevsky D, Kanner AA, Bokstein F, Corn BW. Delayed initiation of radiotherapy for glioblastoma: How important is it to push to the front (or the back) of the line? J Neurooncol (2011) 105(1):1–7. doi: 10.1007/s11060-011-0589-2
92. Lawrence YR, Wang M, Dicker AP, Andrews D, Curran WJ Jr., Michalski JM, et al. Early toxicity predicts long-term survival in high-grade glioma. Br J cancer (2011) 104(9):1365–71. doi: 10.1038/bjc.2011.123
93. Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J neurochem (2008) 105(2):287–95. doi: 10.1111/j.1471-4159.2008.05301.x
94. Dahlberg D, Struys EA, Jansen EE, Morkrid L, Midttun O, Hassel B. Cyst fluid from cystic, malignant brain tumors: A reservoir of nutrients, including growth factor-like nutrients, for tumor cells. Neurosurgery (2017) 80(6):917–24. doi: 10.1093/neuros/nyw101
95. Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol (2015) 17(12):1556–68. doi: 10.1038/ncb3272
96. Seyfried TN, Flores R, Poff AM, D’Agostino DP, Mukherjee P. Metabolic therapy: a new paradigm for managing malignant brain cancer. Cancer Lett (2015) 356(2 Pt A):289–300. doi: 10.1016/j.canlet.2014.07.015
97. Duan C, Yang R, Yuan L, Engelbach JA, Tsien CI, Rich KM, et al. Late effects of radiation prime the brain microenvironment for accelerated tumor growth. Int J Radiat Oncol Biol Phys (2019) 103(1):190–4. doi: 10.1016/j.ijrobp.2018.08.033
98. Rovlias A, Kotsou S. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery (2000) 46(2):335–42. doi: 10.1097/00006123-200002000-00015
99. Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg (1997) 86(2):241–51. doi: 10.3171/jns.1997.86.2.0241
100. Seyfried TN, Sanderson TM, El-Abbadi MM, McGowan R, Mukherjee P. Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br J Cancer (2003) 89(7):1375–82. doi: 10.1038/sj.bjc.6601269
101. Binello E, Qadeer ZA, Kothari HP, Emdad L, Germano IM. Stemness of the ct-2a immunocompetent mouse brain tumor model: characterization in vitro. J Cancer (2012) 3:166–74. doi: 10.7150/jca.4149
102. Meidenbauer JJ, Ta N, Seyfried TN. Influence of a ketogenic diet, fish-oil, and calorie restriction on plasma metabolites and lipids in C57BL/6J mice. Nutr Metab (2014) 11:23. doi: 10.1186/1743-7075-11-23
103. Derr RL, Ye X, Islas MU, Desideri S, Saudek CD, Grossman SA. Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. J Clin Oncol (2009) 27(7):1082–6. doi: 10.1200/JCO.2008.19.1098
104. Mayer A, Vaupel P, Struss HG, Giese A, Stockinger M, Schmidberger H. Strong adverse prognostic impact of hyperglycemic episodes during adjuvant chemoradiotherapy of glioblastoma multiforme. Strahlenther Onkol. (2014) 190(10):933–8. doi: 10.1007/s00066-014-0696-z
105. McGirt MJ, Chaichana KL, Gathinji M, Attenello F, Than K, Ruiz AJ, et al. Persistent outpatient hyperglycemia is independently associated with decreased survival after primary resection of malignant brain astrocytomas. Neurosurgery (2008) 63(2):286–91. doi: 10.1227/01.NEU.0000315282.61035.48
106. Schwartzbaum J, Edlinger M, Zigmont V, Stattin P, Rempala GA, Nagel G, et al. Associations between prediagnostic blood glucose levels, diabetes, and glioma. Sci Rep (2017) 7(1):1436. doi: 10.1038/s41598-017-01553-2
107. Strowd RE 3rd, Grossman SA. The role of glucose modulation and dietary supplementation in patients with central nervous system tumors. Curr Treat opt Oncol (2015) 16(8):356. doi: 10.1007/s11864-015-0356-2
108. Tieu MT, Lovblom LE, McNamara MG, Mason W, Laperriere N, Millar BA, et al. Impact of glycemia on survival of glioblastoma patients treated with radiation and temozolomide. J Neurooncol (2015) 124(1):119–26. doi: 10.1007/s11060-015-1815-0
109. Zhao S, Cai J, Li J, Bao G, Li D, Li Y, et al. Bioinformatic profiling identifies a glucose-related risk signature for the malignancy of glioma and the survival of patients. Mol Neurobiol (2016) 54(10):8203–10. doi: 10.1007/s12035-016-0314-4
110. Decker M, Sacks P, Abbatematteo J, De Leo E, Brennan M, Rahman M. The effects of hyperglycemia on outcomes in surgical high-grade glioma patients. Clin Neurol Neurosurg (2019) 179:9–13. doi: 10.1016/j.clineuro.2019.02.011
111. Link TW, Woodworth GF, Chaichana KL, Grossman SA, Mayer RS, Brem H, et al. Hyperglycemia is independently associated with post-operative function loss in patients with primary eloquent glioblastoma. J Clin Neurosci (2012) 19(7):996–1000. doi: 10.1016/j.jocn.2011.09.031
112. Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, et al. Corticosteroids compromise survival in glioblastoma. Brain (2016) 139(Pt 5):1458–71. doi: 10.1093/brain/aww046
113. Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer (2015) 113(2):232–41. doi: 10.1038/bjc.2015.238
114. Chang SM, Parney IF, Huang W, Anderson FA Jr., Asher AL, Bernstein M, et al. Patterns of care for adults with newly diagnosed malignant glioma. JAMA (2005) 293(5):557–64. doi: 10.1001/jama.293.5.557
115. Stokum JA, Gerzanich V, Sheth KN, Kimberly WT, Simard JM. Emerging pharmacological treatments for cerebral edema: Evidence from clinical studies. Annu Rev Pharmacol Toxicol (2020) 60:291–309. doi: 10.1146/annurev-pharmtox-010919-023429
116. Champ CE, Klement RJ. Commentary on “Strong adverse prognostic impact of hyperglycemic episodes during adjuvant chemoradiotherapy of glioblastoma multiforme”. Strahlenther Onkol. (2015) 191(3):281–2. doi: 10.1007/s00066-014-0788-9
117. Davies PS, Powell AE, Swain JR, Wong MH. Inflammation and proliferation act together to mediate intestinal cell fusion. PLoS One (2009) 4(8):e6530. doi: 10.1371/journal.pone.0006530
118. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev oncogenesis (2013) 18(1-2):43–73. doi: 10.1615/CritRevOncog.v18.i1-2.40
119. Mukherjee P, El-Abbadi MM, Kasperzyk JL, Ranes MK, Seyfried TN. Dietary restriction reduces angiogenesis and growth in an orthotopic mouse brain tumour model. Br J Cancer (2002) 86(10):1615–21. doi: 10.1038/sj.bjc.6600298
120. Shabo I, Midtbo K, Andersson H, Akerlund E, Olsson H, Wegman P, et al. Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer (2015) 15:922. doi: 10.1186/s12885-015-1935-0
121. Shabo I, Olsson H, Sun XF, Svanvik J. Expression of the macrophage antigen CD163 in rectal cancer cells is associated with early local recurrence and reduced survival time. Int J Cancer (2009) 125(8):1826–31. doi: 10.1002/ijc.24506
122. Munzarova M, Kovarik J. Is cancer a macrophage-mediated autoaggressive disease? Lancet (1987) 1(8539):952–4. doi: 10.1016/S0140-6736(87)90295-9
123. Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer (2008) 8(5):377–86. doi: 10.1038/nrc2371
124. Lindstrom A, Midtbo K, Arnesson LG, Garvin S, Shabo I. Fusion between M2-macrophages and cancer cells results in a subpopulation of radioresistant cells with enhanced DNA-repair capacity. Oncotarget (2017) 8(31):51370–86. doi: 10.18632/oncotarget.17986
125. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res (1996) 56(20):4625–9.
126. Roggendorf W, Strupp S, Paulus W. Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol (1996) 92:288–93. doi: 10.1007/s004010050520
127. Wood GW, Morantz RA. Immunohistologic evaluation of the lymphoreticular infiltrate of human central nervous system tumors. J Natl Cancer Inst (1979) 62(3):485–91. doi: 10.1093/jnci/62.3.485
128. Morantz RA, Wood GW, Foster M, Clark M, Gollahon K. Macrophages in experimental and human brain tumors. part 2: Studies of the macrophage content of human brain tumors. J Neurosurg (1979) 50(3):305–11. doi: 10.3171/jns.1979.50.3.0305
129. Huysentruyt LC, Mukherjee P, Banerjee D, Shelton LM, Seyfried TN. Metastatic cancer cells with macrophage properties: Evidence from a new murine tumor model. Int J Cancer (2008) 123(1):73–84. doi: 10.1002/ijc.23492
130. Shelton LM, Mukherjee P, Huysentruyt LC, Urits I, Rosenberg JA, Seyfried TN. A novel pre-clinical in vivo mouse model for malignant brain tumor growth and invasion. J Neurooncol (2010) 99(2):165–76. doi: 10.1007/s11060-010-0115-y
131. Seyfried TN, Shelton LM, Huysentruyt LC. The VM mouse model of glioblastoma multiforme. In: Martinez-Murillo RA, editor. Neuromethods. Springer Science+Business Media, LLC New York : Humana Press : Springer (2012).
132. Kurz SC, Cabrera LP, Hastie D, Huang R, Unadkat P, Rinne M, et al. PD-1 inhibition has only limited clinical benefit in patients with recurrent high-grade glioma. Neurology (2018) 91(14):e1355–e9. doi: 10.1212/WNL.0000000000006283
133. Weller M, Le Rhun E. Immunotherapy for glioblastoma: quo vadis? Nat Rev Clin Oncol (2019) 16(7):405–6. doi: 10.1038/s41571-019-0195-3
134. Ratnam NM, Gilbert MR, Giles AJ. Immunotherapy in CNS cancers: the role of immune cell trafficking. Neuro Oncol (2019) 21(1):37–46. doi: 10.1093/neuonc/noy084
135. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis (2009) 30(7):1073–81. doi: 10.1093/carcin/bgp127
136. Gast CE, Silk AD, Zarour L, Riegler L, Burkhart JG, Gustafson KT, et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci Adv (2018) 4(9):eaat7828. doi: 10.1126/sciadv.aat7828
137. Oizel K, Yang C, Renoult O, Gautier F, Do QN, Joalland N, et al. Glutamine uptake and utilization of human mesenchymal glioblastoma in orthotopic mouse model. Cancer Metab (2020) 8:9. doi: 10.1186/s40170-020-00215-8
138. Pecqueur C, Oliver L, Oizel K, Lalier L, Vallette FM. Targeting metabolism to induce cell death in cancer cells and cancer stem cells. Int J Cell Biol (2013) 2013:805975. doi: 10.1155/2013/805975
139. Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, et al. Hyperprogressive disease in patients with advanced non-small cell lung cancer treated with pd-1/pd-l1 inhibitors or with single-agent chemotherapy. JAMA Oncol (2018) 4(11):1543–52. doi: 10.1001/jamaoncol.2018.3676
140. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol (2005) 109(1):93–108. doi: 10.1007/s00401-005-0991-y
141. Trojanowski P, Jarosz B, Szczepanek D. The diagnostic quality of needle brain biopsy specimens obtained with different sampling methods - experimental study. Sci Rep (2019) 9(1):8077. doi: 10.1038/s41598-019-44622-4
142. Alieva M, Margarido AS, Wieles T, Abels ER, Colak B, Boquetale C, et al. Preventing inflammation inhibits biopsy-mediated changes in tumor cell behavior. Sci Rep (2017) 7(1):7529. doi: 10.1038/s41598-017-07660-4
143. Walter ND, Rice PL, Redente EF, Kauvar EF, Lemond L, Aly T, et al. Wound healing after trauma may predispose to lung cancer metastasis: Review of potential mechanisms. Am J Respir Cell Mol Biol (2011) 44(5):591–6. doi: 10.1165/rcmb.2010-0187RT
144. Oizel K, Chauvin C, Oliver L, Gratas C, Geraldo F, Jarry U, et al. Efficient mitochondrial glutamine targeting prevails over glioblastoma metabolic plasticity. Clin Cancer Res (2017) 23(20):6292–304. doi: 10.1158/1078-0432.CCR-16-3102
145. Ferrere G, Tidjani Alou M, Liu P, Goubet AG, Fidelle M, Kepp O, et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight (2021) 6(2):e145207. doi: 10.1172/jci.insight.145207
146. Oliva CR, Nozell SE, Diers A, McClugage SG 3rd, Sarkaria JN, Markert JM, et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J Biol Chem (2010) 285(51):39759–67. doi: 10.1074/jbc.M110.147504
147. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science (2014) 343(6167):189–93. doi: 10.1126/science.1239947
148. Klement RJ, Champ CE. Corticosteroids compromise survival in glioblastoma in part through their elevation of blood glucose levels. Brain (2017) 140(3):e16. doi: 10.1093/brain/aww324
149. Arcuri C, Tardy M, Rolland B, Armellini R, Menghini AR, Bocchini V. Glutamine synthetase gene expression in a glioblastoma cell-line of clonal origin: Regulation by dexamethasone and dibutyryl cyclic AMP. Neurochem Res (1995) 20(10):1133–9. doi: 10.1007/BF00995375
150. Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med (2017) 377(20):1954–63. doi: 10.1056/NEJMoa1707358
151. Seyfried TN. Strategies. cancer as a metabolic disease: On the origin, management, and prevention of cancer. Hoboken, NJ: John Wiley & Sons (2012). p. 227–89.
152. Iwamoto FM, Abrey LE, Beal K, Gutin PH, Rosenblum MK, Reuter VE, et al. Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology (2009) 73(15):1200–6. doi: 10.1212/WNL.0b013e3181bc0184
153. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell (2009) 15(3):220–31. doi: 10.1016/j.ccr.2009.01.027
154. de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol (2010) 12(3):233–42. doi: 10.1093/neuonc/nop027
155. Nanegrungsunk D, Apaijai N, Yarana C, Sripetchwandee J, Limpastan K, Watcharasaksilp W, et al. Bevacizumab is superior to temozolomide in causing mitochondrial dysfunction in human brain tumors. Neurol Res (2016) 38(4):285–93. doi: 10.1080/01616412.2015.1114233
156. Rahbar A, Orrego A, Peredo I, Dzabic M, Wolmer-Solberg N, Straat K, et al. Human cytomegalovirus infection levels in glioblastoma multiforme are of prognostic value for survival. J Clin Virol (2013) 57(1):36–42. doi: 10.1016/j.jcv.2012.12.018
157. Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J virol (2011) 85(4):1573–80. doi: 10.1128/JVI.01967-10
158. Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res (2017) 23(8):1898–909. doi: 10.1158/1078-0432.CCR-16-2057
159. Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K, Michikawa M. Altered cholesterol metabolism in niemann-pick type C1 mouse brains affects mitochondrial function. J Biol Chem (2005) 280(12):11731–9. doi: 10.1074/jbc.M412898200
160. Yu Y, Clippinger AJ, Alwine JC. Viral effects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol (2011) 19(7):360–7. doi: 10.1016/j.tim.2011.04.002
161. Kathagen A, Schulte A, Balcke G, Phillips HS, Martens T, Matschke J, et al. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol (2013) 126(5):763–80. doi: 10.1007/s00401-013-1173-y
162. Ria F, Landriscina M, Remiddi F, Rosselli R, Iacoangeli M, Scerrati M, et al. The level of manganese superoxide dismutase content is an independent prognostic factor for glioblastoma. Biol Mech Clin Implications Br J Cancer (2001) 84(4):529–34. doi: 10.1054/bjoc.2000.1594
163. Aiken KJ, Bickford JS, Kilberg MS, Nick HS. Metabolic regulation of manganese superoxide dismutase expression via essential amino acid deprivation. J Biol Chem (2008) 283(16):10252–63. doi: 10.1074/jbc.M709944200
164. Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res (2005) 65(2):613–21. doi: 10.1158/0008-5472.613.65.2
166. VanItallie TB, Nufert TH. Ketones: Metabolism’s ugly duckling. Nutr Rev (2003) 61(10):327–41. doi: 10.1301/nr.2003.oct.327-341
167. Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids (2004) 70(3):309–19. doi: 10.1016/j.plefa.2003.09.007
168. Klement RJ. The emerging role of ketogenic diets in cancer treatment. Curr Opin Clin Nutr Metab Care (2019) 22(2):129–34. doi: 10.1097/MCO.0000000000000540
169. Fine EJ, Miller A, Quadros EV, Sequeira JM, Feinman RD. Acetoacetate reduces growth and ATP concentration in cancer cell lines which over-express uncoupling protein 2. Cancer Cell Int (2009) 9:14. doi: 10.1186/1475-2867-9-14
170. Winter SF, Loebel F, Dietrich J. Role of ketogenic metabolic therapy in malignant glioma: A systematic review. Crit Rev Oncol Hematol (2017) 112:41–58. doi: 10.1016/j.critrevonc.2017.02.016
171. Zhou W, Mukherjee P, Kiebish MA, Markis WT, Mantis JG, Seyfried TN. The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr Metab (Lond) (2007) 4:5. doi: 10.1186/1743-7075-4-5
172. Tisdale MJ, Brennan RA. Loss of acetoacetate coenzyme a transferase activity in tumours of peripheral tissues. Br J Cancer (1983) 47(2):293–7. doi: 10.1038/bjc.1983.38
173. Fredericks M, Ramsey RB. 3-oxo acid coenzyme a transferase activity in brain and tumors of the nervous system. J neurochem (1978) 31(6):1529–31. doi: 10.1111/j.1471-4159.1978.tb06581.x
174. Cuenoud B, Hartweg M, Godin JP, Croteau E, Maltais M, Castellano CA, et al. Metabolism of exogenous d-beta-hydroxybutyrate, an energy substrate avidly consumed by the heart and kidney. Front Nutr (2020) 7:13. doi: 10.3389/fnut.2020.00013
175. Klein P, Tyrlikova I, Zuccoli G, Tyrlik A, Maroon JC. Treatment of glioblastoma multiforme with “classic” 4:1 ketogenic diet total meal replacement. Cancer Metab (2020) 8(1):24. doi: 10.1186/s40170-020-00230-9
176. Voss M, Wenger KJ, von Mettenheim N, Bojunga J, Vetter M, Diehl B, et al. Short-term fasting in glioma patients: Analysis of diet diaries and metabolic parameters of the ERGO2 trial. Eur J Nutr (2022) 61(1):477–87. doi: 10.1007/s00394-021-02666-1
177. Mahoney LB, Denny CA, Seyfried TN. Caloric restriction in C57BL/6J mice mimics therapeutic fasting in humans. Lipids Health Dis (2006) 5(1):13. doi: 10.1186/1476-511X-5-13
178. Jiang YS, Wang FR. Caloric restriction reduces edema and prolongs survival in a mouse glioma model. J neuro-oncol (2013) 114(1):25–32. doi: 10.1007/s11060-013-1154-y
179. Drenick EJ, Alvarez LC, Tamasi GC, Brickman AS. Resistance to symptomatic insulin reactions after fasting. J Clin Invest (1972) 51(10):2757–62. doi: 10.1172/JCI107095
180. Cahill GF Jr., Veech RL. Ketoacids? good medicine? Trans Am Clin Climatol Assoc (2003) 114:149–61.
181. LaManna JC, Salem N, Puchowicz M, Erokwu B, Koppaka S, Flask C, et al. Ketones suppress brain glucose consumption. Adv Exp Med Biol (2009) 645:301–6. doi: 10.1007/978-0-387-85998-9_45
182. Veech RL, Todd King M, Pawlosky R, Kashiwaya Y, Bradshaw PC, Curtis W. The “great” controlling nucleotide coenzymes. IUBMB Life (2019) 71(5):565–79. doi: 10.1002/iub.1997
183. Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF Jr. Ketone bodies, potential therapeutic uses. IUBMB Life (2001) 51(4):241–7. doi: 10.1080/152165401753311780
184. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med (2015) 21(3):263–9. doi: 10.1038/nm.3804
185. Shang S, Wang L, Zhang Y, Lu H, Lu X. The beta-hydroxybutyrate suppresses the migration of glioma cells by inhibition of nlrp3 inflammasome. Cell Mol Neurobiol (2018) 38(8):1479–89. doi: 10.1007/s10571-018-0617-2
186. Poff AM, Ari C, Arnold P, Seyfried TN, D’Agostino DP. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int J Cancer J Int Cancer (2014) 135(7):1711–20. doi: 10.1002/ijc.28809
187. Rieger J, Bahr O, Maurer GD, Hattingen E, Franz K, Brucker D, et al. ERGO: a pilot study of ketogenic diet in recurrent glioblastoma. Int J Oncol (2014) 44(6):1843–52. doi: 10.3892/ijo.2014.2382
188. Denny CA, Heinecke KA, Kim YP, Baek RC, Loh KS, Butters TD, et al. Restricted ketogenic diet enhances the therapeutic action of n-butyldeoxynojirimycin towards brain GM2 accumulation in adult sandhoff disease mice. J Neurochem (2010) 113(6):1525–35. doi: 10.1111/j.1471-4159.2010.06733.x
189. Altinoz MA, Ozpinar A, Seyfried TN. Caprylic (Octanoic) acid as a potential fatty acid chemotherapeutic for glioblastoma. Prostaglandins Leukot Essent Fatty Acids (2020) 159:102142. doi: 10.1016/j.plefa.2020.102142
190. Iyikesici MS, Slocum AK, Slocum A, Berkarda FB, Kalamian M, Seyfried TN. Efficacy of metabolically supported chemotherapy combined with ketogenic diet, hyperthermia, and hyperbaric oxygen therapy for stage iv triple-negative breast cancer. Cureus (2017) 9(7):e1445. doi: 10.7759/cureus.1445
191. Iyikesici MS, Slocum AK, Winters N, Kalamian M, Seyfried TN. Metabolically supported chemotherapy for managing end-stage breast cancer: A complete and durable response. Cureus (2021) 13(4):e14686. doi: 10.7759/cureus.14686
192. Poff AM, Ari C, Seyfried TN, D’Agostino DP. The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS One (2013) 8(6):e65522. doi: 10.1371/journal.pone.0065522
193. Stuhr LE, Raa A, Oyan AM, Kalland KH, Sakariassen PO, Petersen K, et al. Hyperoxia retards growth and induces apoptosis, changes in vascular density and gene expression in transplanted gliomas in nude rats. J Neuro-oncol (2007) 85(2):191–202. doi: 10.1007/s11060-007-9407-2
194. Bartesaghi S, Graziano V, Galavotti S, Henriquez NV, Betts J, Saxena J, et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc Natl Acad Sci USA (2015) 112(4):1059–64. doi: 10.1073/pnas.1413165112
195. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-myc deregulate pyruvate kinase mRNA splicing in cancer. Nature (2010) 463(7279):364–8. doi: 10.1038/nature08697
196. Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell (2014) 53(1):75–87. doi: 10.1016/j.molcel.2013.11.001
197. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19(1):17–30. doi: 10.1016/j.ccr.2010.12.014
198. Zhang K, Xu P, Sowers JL, Machuca DF, Mirfattah B, Herring J, et al. Proteome analysis of hypoxic glioblastoma cells reveals sequential metabolic adaptation of one-carbon metabolic pathways. Mol Cell Proteomics (2017) 16(11):1906–21. doi: 10.1074/mcp.RA117.000154
199. Schwartz KA, Noel M, Nikolai M, Olson LK, Hord NG, Zakem M, et al. Long term survivals in aggressive primary brain malignancies treated with an adjuvant ketogenic diet. Front Nutr (2022) 9:770796. doi: 10.3389/fnut.2022.770796
200. Runyan A, Banks J, Bruni DS. Current and future oncology management in the United States. J Manag Care Spec Pharm (2019) 25(2):272–81. doi: 10.18553/jmcp.2019.25.2.272
201. Bird N, Knox L, Palmer A, Heenen D, Blanc P, Scobie N, et al. When innovation and commercialization collide: A patient advocate view in neuroblastoma. J Clin Oncol (2022) 40(2):120–6. doi: 10.1200/JCO.21.01916
202. Song Z, Rose S, Safran DG, Landon BE, Day MP, Chernew ME. Changes in health care spending and quality 4 years into global payment. N Engl J Med (2014) 371(18):1704–14. doi: 10.1056/NEJMsa1404026
203. McGinnis AD. On the origin of financial toxicity for cancer patients. eScholarship@BC (2019), 1–26. Available to: http://hdl.handle.net/2345/bc-ir:108586.
Keywords: glutaminolysis, glycolysis, fermentation, succinate, lactate, glutamate, ketogenic diet, ketogenic metabolic therapy
Citation: Seyfried TN, Arismendi-Morillo G, Zuccoli G, Lee DC, Duraj T, Elsakka AM, Maroon JC, Mukherjee P, Ta L, Shelton L, D'Agostino D, Kiebish M and Chinopoulos C (2022) Metabolic management of microenvironment acidity in glioblastoma. Front. Oncol. 12:968351. doi: 10.3389/fonc.2022.968351
Received: 13 June 2022; Accepted: 15 July 2022;
Published: 17 August 2022.
Edited by:
Reo Hamaguchi, Juntendo University, JapanReviewed by:
Fahim Ahmad, National Cancer Institute at Frederick (NIH), United StatesCopyright © 2022 Seyfried, Arismendi-Morillo, Zuccoli, Lee, Duraj, Elsakka, Maroon, Mukherjee, Ta, Shelton, D’Agostino, Kiebish and Chinopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas N. Seyfried, VGhvbWFzLnNleWZyaWVkQGJjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.