 Yuping Li1,2†Yupeng Xie2†Di Wang3Hanyan Xu1,2Junru Ye1,2Jiani C. Yin3Junjie Chen1,2Junrong Yan3Bin Ye3Chengshui Chen1,2*
Yuping Li1,2†Yupeng Xie2†Di Wang3Hanyan Xu1,2Junru Ye1,2Jiani C. Yin3Junjie Chen1,2Junrong Yan3Bin Ye3Chengshui Chen1,2*- 1Key Laboratory of Interventional Pulmonology of Zhejiang Province, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
- 2Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
- 3Medical Department, Nanjing Geneseeq Technology Inc., Nanjing, Jiangsu, China
Background: Li-Fraumeni syndrome (LFS) and Li-Fraumeni-like (LFL) syndrome are rare hereditary diseases characterized by predisposition to a diverse spectrum of cancer types, primarily sarcoma. The pathogenic variants underlying the majority of LFL cases remain to be explored.
Methods: We performed whole-exome sequencing (WES) on 13 core members of a large LFL family with highly aggregated incidences of cancers, including cases with sarcoma, non-small cell lung cancer and cardiac angiosarcoma, and conducted a comprehensive literature review of candidate gene associations in LFS/LFL syndromes or sarcoma to identify potential pathogenic germline variants.
Results: No germline variants in the best-known LFL/LFS-associated gene TP53 were detected. Of all the genes associated with LFS/LFL or sarcoma that we have surveyed, we identified a novel p.P35L germline variant in POT1 (protection of telomeres 1). Germline and somatic alterations in POT1 have been implicated in a series of familial cancers, including angiosarcoma, glioma, melanoma and colorectal cancer. This particular variant is located in the telomere-binding OB1 domain, which is important in maintaining the proper telomere length, and showed high conservation across different POT1 orthologues. No record of the variant was found in any of the 1000 genomes, ExAC, gnomAD, dpSNP and COSMIC databases. Prediction algorithms and in silico structural analysis suggested completely disrupted protein structure and function of POT1 in the presence of this mutation.
Conclusions: Leveraging WES, we identified a novel germline risk allele, p.P35L in POT1, that likely predisposes to LFL syndrome. Our results support the routine testing of POT1 and other LFL/LFS-associated genes in the risk populations to enable early cancer diagnosis, prevention and intervention.
Introduction
Li-Fraumeni syndrome (LFS) and Li-Fraumeni-like (LFL) syndrome are clinically heterogeneous, autosomal dominant disorders (1), which are characterized by increased risks of early-onset cancers that may arise from multiple organ systems. The common cancer types associated with LFS/LFL are breast cancer, soft-tissue sarcoma, brain tumors, osteosarcoma and hematological malignancies (2). In addition, a wide range of cancer types, including lung, gastric, colorectal, liver, and pancreas cancers, among others, have been reported at a relatively low incidence in families with LFS/LFL (3). The classical criteria for LFS are a proband with sarcoma diagnosed before the age of 45 and a first-degree relative diagnosed with cancer before the age of 45, plus a first- or second-degree relative with cancer before the age of 45 or sarcoma at any age (3). The classification criteria for LFL syndrome are less stringent.
The best-characterized LFS/LFL gene is TP53, of which germline mutations can be found in approximately 70% of LFS and 20-40% of LFL families (3, 4). Carriers of germline TP53 mutations have a much higher risk for cancers at a young age (5), with an estimated lifetime risk of 70% in males and nearly 100% in females due to the high incidence of breast cancer (3, 6). The remaining TP53-negative LFS/LFL families indicate the presence of additional susceptibility genes. CHEK2 encodes a cell cycle checkpoint regulator that participates in the processes of DNA repair, cell death, and cell cycle control through stabilization of p53 (7). While loss of function CHEK2 mutations might contribute to increased risks of individual cancer development in LFS/LFL families, several studies have ruled out CHEK2 as a major LFS/LFL susceptibility gene (8, 9). However, due to the low incidence of LFS/LFL and limited tumor testing in the affected families, the causative alleles of the remaining LFS/LFL families are yet to be identified.
In this study, we report a LFL family with early-onset sarcoma and lung cancers. Whole exome sequencing (WES) was performed on core family members to identify candidate risk variants. Associations with cancer development and functional predictions of pathogenicity were performed to uncover potential pathogenic variants.
Method
Study subjects and sample collection
A Chinese LFL family of multiple affected individuals with sarcoma and NSCLCs were studied (Figure 1). WES was performed on 13 core members of the second and third generations. A comprehensive search of the Pubmed database was performed by using terms related to LFS, LFL, sarcoma, susceptibility variants, and familial cancer syndromes. Manual review of the relevant articles was conducted. Written informed consents were obtained from all family members. The study was approved by the Ethics Committee of the First Affiliated Hospital of WenZhou Medical University and was conducted in accordance with the Declaration of Helsinki.
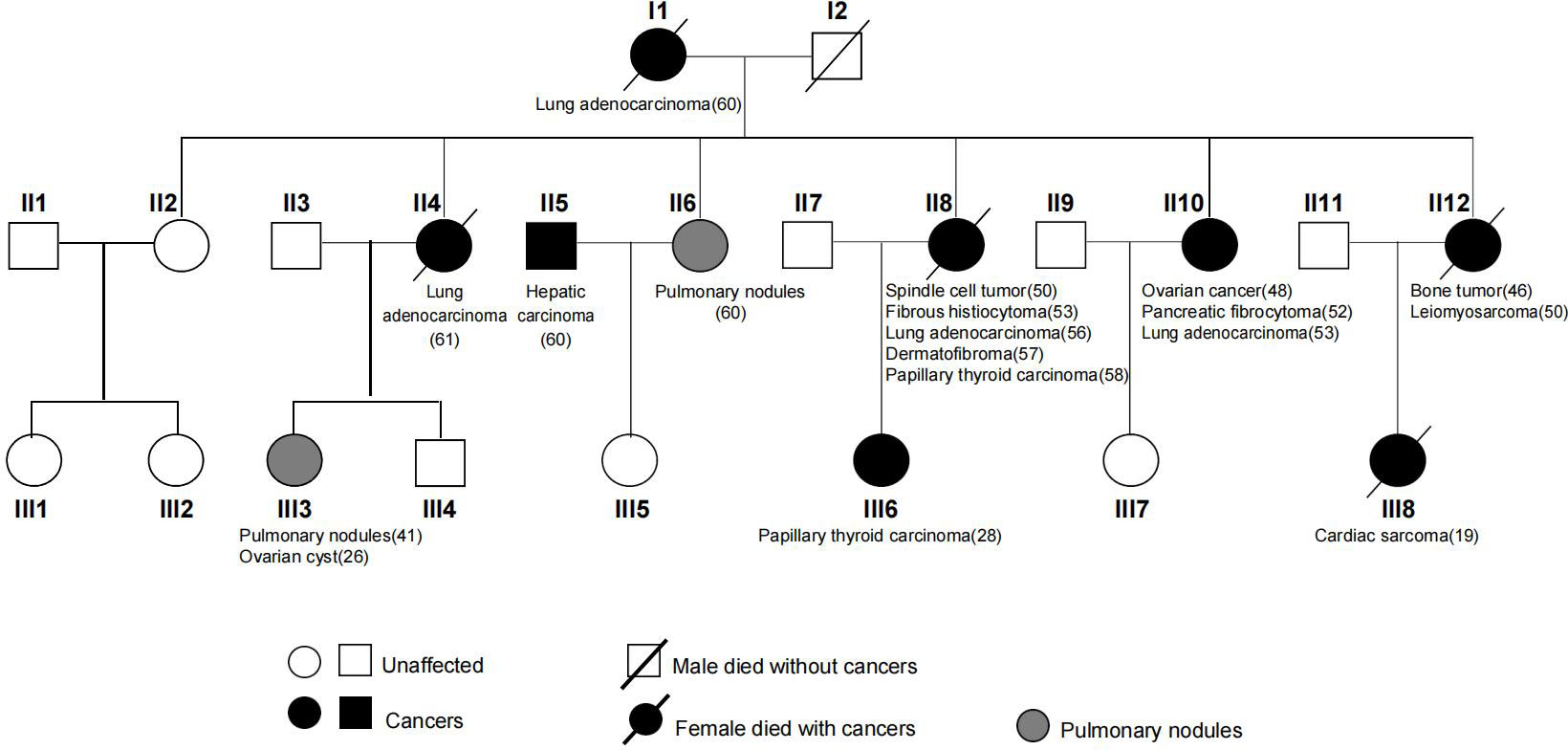
Figure 1 Pedigree of the LFL family across three generations in this study.
Exome sequencing
Sequencing tests were performed in a CLIA-certified and CAP-accredited NGS testing center (Nanjing Geneseeq Technology Inc., China). In brief, genomic DNA from whole blood and tumor tissues were extracted by using the DNeasy Blood and Tissue kit (Qiagen). Purified genomic DNA was qualified by Nanodrop2000 for A260/280 and A260/A230 ratios (Thermo Fisher Scientific). Fragmented DNA was subjected to library preparations using KAPA Hyper Prep Kit (KAPA Biosystems). Exome capture was performed using the xGen Exome Research Panel v2 (Integrated DNA Technologies) and Hybridization and Wash Reagents Kit according to manufacturer’s protocol. Sequencing was performed with enriched libraries on an Illumina HiSeq 4000 platform. The mean coverage depths were ~68X for the white cell controls and ~137X for the tumor tissue.
Mutation calling
Trimmomatic was used for FASTQ file quality control. Sequencing reads with low quality (quality reading below 20) or N bases were removed. Paired-end reads were then aligned to the reference human genome (build hg19), using the Burrows-Wheeler Aligner (BWA, https://github.com/lh3/bwa/tree/master/bwakit) with default parameters. PCR deduplication was performed using Picard (https://broadinstitute.github.io/picard/) and local realignment around indels and base quality score recalibration were performed using GATK3 (https://software.broadinstitute.org/gatk/). The GATK Haplotype caller was used for germline variant calling. Single-nucleotide variations (SNVs) and insertion/deletion mutations were detected using VarScan2 and Mutect2, with a minimum variant allele frequency threshold set at 0.01 and p-value threshold for calling variants set at 0.05 to generate Variant Call Format files. All SNVs/indels were annotated with ANNOVAR, and each SNV/indel was manually checked on the Integrative Genomics Viewer. Common SNPs with more than 1% of population frequency in the 1000 Genomes Project or the Exome Aggregation Consortium (ExAC) exome database were excluded. Copy number ratios were calculated by CNVKit (https://cnvkit.readthedocs.io). In silico structural modeling was performed by using PyMOL (pymol.org).
Result
History and clinical investigation of the LFL family
A 61-year-old female patient (II4) was admitted to our hospital and diagnosed with lung adenocarcinoma. Detailed clinical consultation revealed a strong family history of cancer diagnosed at relatively young ages, suggesting that cancer development might be hereditary. A complete family history across three generations was obtained, with the pedigree shown in Figure 1. Clinical characteristics of the patients are summarized in Table 1. The consultand’s mother was diagnosed with lung adenocarcinoma at the age of 60. In addition, three sisters (II8, II10, and II12) were also diagnosed with cancers at young ages. Of the three, two sisters (II8 and II10) were first diagnosed with spindle cell tumor and fibrous histiocytomas of the pancreas at 50 and 52 years of age, respectively. Both of them subsequently developed lung adenocarcinoma at ages 56 and 53, respectively. Moreover, II8 developed with bilateral papillary thyroid carcinoma at the age of 58. Another sister (II12) was diagnosed with bone cancer at age 46 and later with leiomyosarcoma at age 50. Two other sisters were otherwise unaffected, although one of them (II6) was detected with multiple pulmonary nodules at age 60. The consultand’s daughter (III3) was detected with pulmonary nodules and ovarian cysts at age 41 and 26, respectively. Extended family history was taken and we found two third-generation females (III6 and III8) with cancer histories. Notably, III8, the daughter of II12, had been diagnosed with rare early-onset cardiac angiosarcoma (CAS) and died from the disease at age 19. III6, the daughter of II8, were diagnosed with papillary thyroid carcinoma at the age of 28. All of the above tumors diagnosed in this kindred have been confirmed as primary lesions. At the time of manuscript preparation, II4, II8 and II12 died at ages of 65, 59 and 52 respectively. Taken into account the family histories, cancer types and age of onset, we considered the kindred to fit the LFL classification criteria.
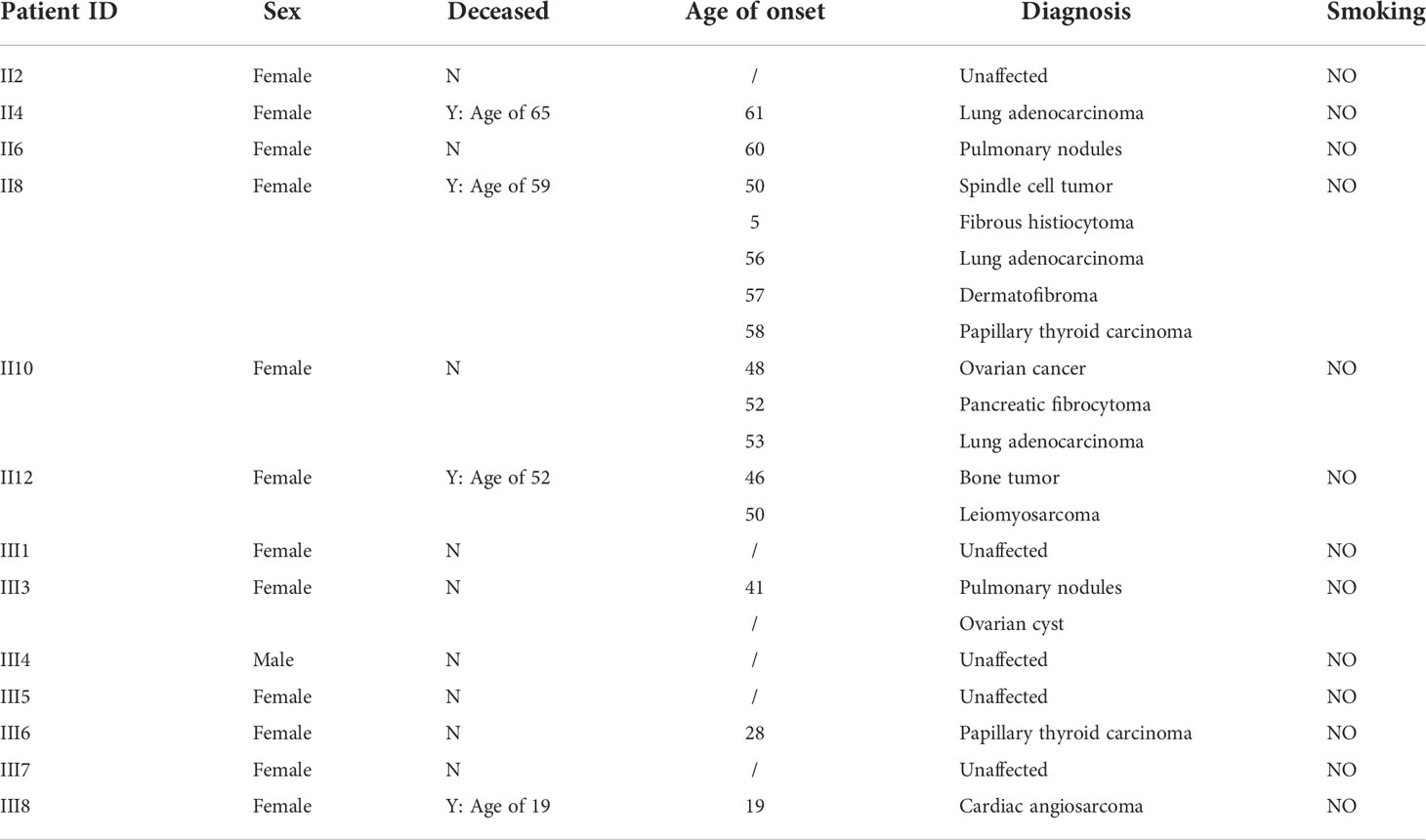
Table 1 Clinical characteristics of individuals in the LFL family.
Identification of candidate LFL variants
To identify candidate pathogenic germline variants in this LFL family, WES was performed on core family members, including six cancer patients (II4/8/10/12 and III6/8), as well as seven unaffected individuals (II2/6 and III1/3/4/5/7). To search for potential causative alleles, we assessed germline variants with frequency of less than 1% in the general or the East Asian populations based on the gnomAD_exome database. As LFS and LFL syndromes can be ascribed mostly to TP53 mutations, we first checked for the presence of TP53 variants in association with cancer development. However, no germline variants in TP53 were detected in any of the individuals. Next, we performed literature review on LFS, LFL or sarcoma-relevant susceptibility genes and narrowed down our search to 98 genes that might explain the increased cancer risks in our LFL family (Table 2).
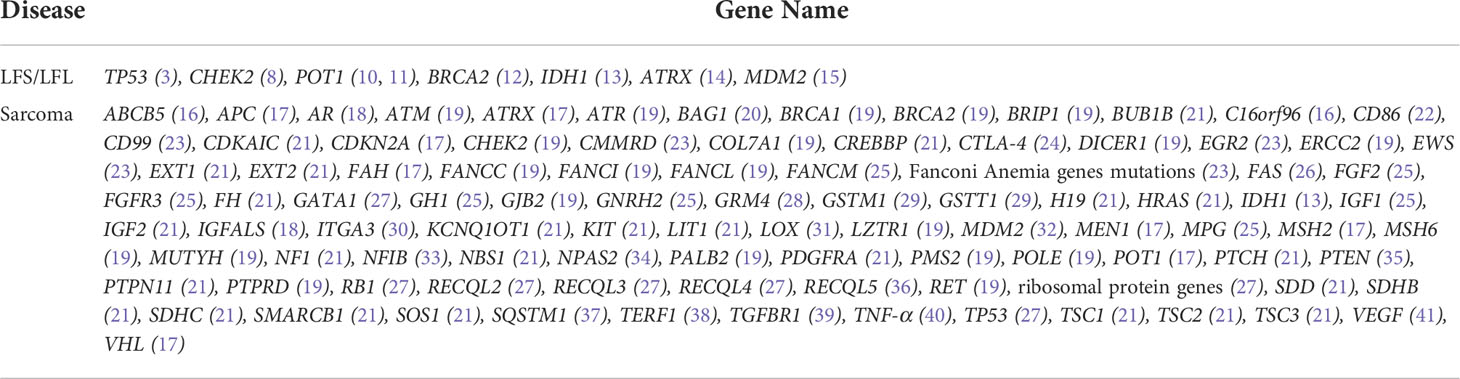
Table 2 List of candidate genes associated with LFS/LFL and sarcoma.
Interestingly, we detected a p.P35L variant in protection of telomeres (POT1). The POT1 p.P35L variant was detected in all of the affected individuals with cancers, including those with NSCLC, sarcoma, thyroid cancers, as well as the individual with childhood-onset cardiac angiosarcoma (III8) (Table 3). Germline and somatic mutations of POT1 have been reported to underlie a series of familial cancers, including angiosarcoma, glioma, melanoma and colorectal cancer (10, 42). POT1 is also frequently mutated in chronic lymphocytic leukemia (43) (Figure 2A). Notably, a p.R117C variant in POT1 has been reported in TP53-negative LFL families with cardiac and breast angiosarcoma (11). On the other hand, the p.P35L variant in our reported family has not been described in the literature or any of the existing databases, including the 1000 Genomes, ExAC, gnomAD, dpSNP and COSMIC databases, which is in accordance with the American College of Medical Genetics (ACMG) guidelines with moderate evidence for pathogenicity (PM2 evidence).
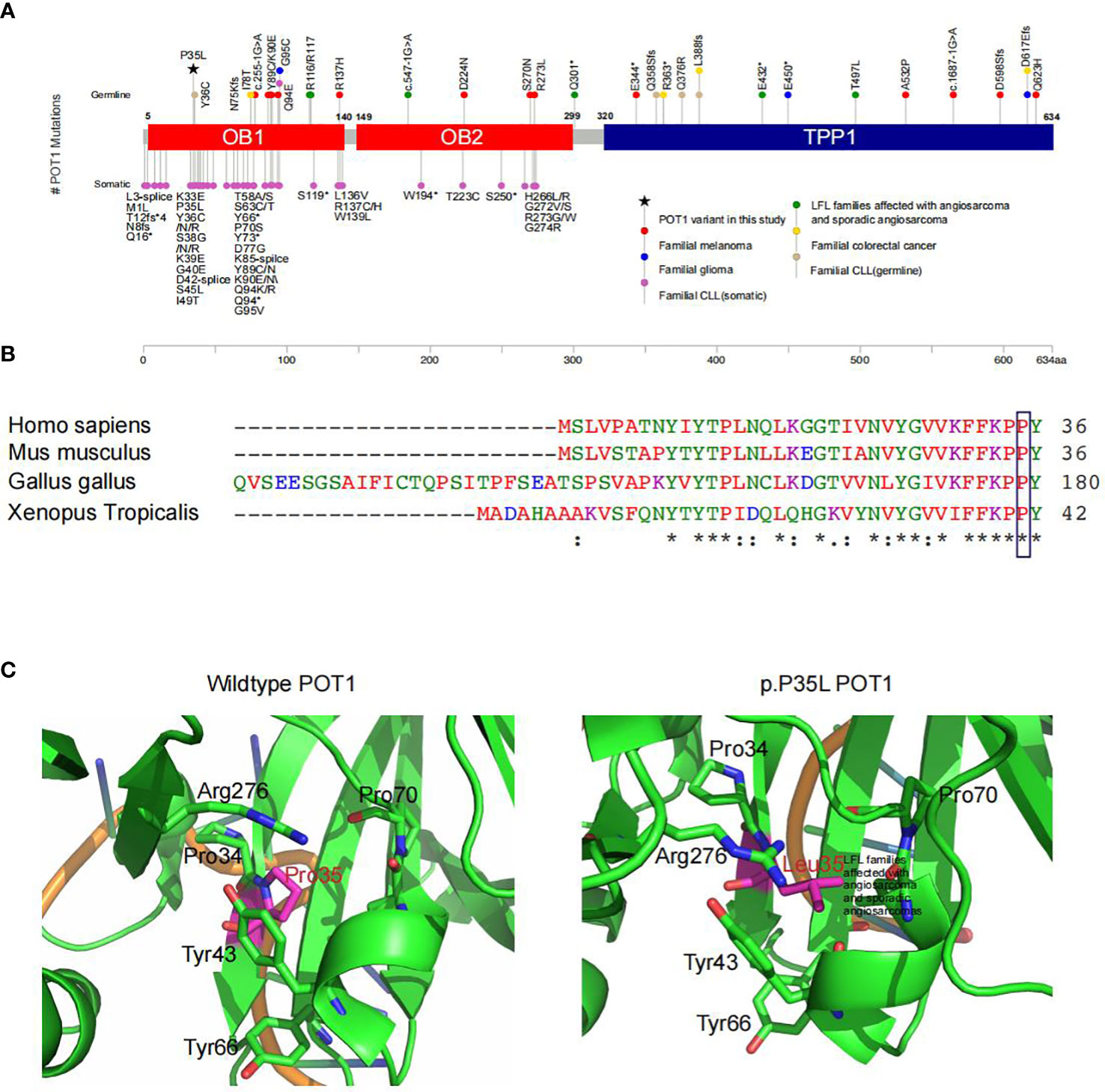
Figure 2 Functional prediction of the POT1 variant in association with LFL. (A) Summary of reported POT1 variants. Germline and somatic variants in POT1 underlie a series of familial cancers, including angiosarcoma (green), glioma (blue), melanoma (red), colorectal cancer (yellow) and chronic lymphocytic leukemia (pink and light brown) as indicated. The POT1 p.P35L variant detected in this pedigree is marked with a black asterisk, which is located in one of the OB fold domains. (B) Sequence alignment showing strong conservation of the POT1 p.P35 residue across different orthologues. An “*” (asterisk) indicates positions which have a single, fully conserved residue. A “:” (colon) indicates conservation between groups of strongly similar properties. A “.” (period) indicates conservation between groups of weakly similar properties. (C) In silico structural modeling of the POT1 protein indicated change of the proline residue at position 35 to leucine led to significant conformational change of the protein, which would affect binding of the telomeres.

Table 3 List of POT1 associated with cancer development in the LFL family.
POT1 encodes a core component of the shelterin complex, which functions to bind and protect the telomeres (44). The p.P35 residue is located in the one of the oligonucleotide/oligosaccharide binding (OB) fold domains that directly bind the telomeres (42) (Figure 2A). Comparisons of the POT1 amino acid sequences across different orthologues showed strong conservation of the p.P35 residue (Figure 2B). Indeed, alteration of the p.P35 residue was predicted to be deleterious to protein function by all existing algorithms such as SIFT (score=0.01), Polyphen (score=1) and GERP (score=5.71) (Table 3), which is consistent with PP3 evidence of the ACMG guidelines. Importantly, change of the proline residue at position 35 to leucine led to significant conformational change of the protein, and consequently would affect binding of the telomeres (Figure 2C).
Discussion
Given their low incidence and highly heterogeneous nature, the genetic mechanisms underlying TP53-negative LFS and LFL syndromes have not been extensively studied. In this study, we report a Chinese family with LFL syndrome, consisting of individuals with lung cancers, thyroid cancers and a case with highly rare cardiac angiosarcoma, all occurred at relatively young ages. WES profiling of core family members and comprehensive literature review revealed a potentially causative missense variant (p.P35L) in POT1. The p.P35 residue lies in the telomere binding domain and is highly conserved across different orthologues. Pathogenicity of the variant is further supported by functional prediction algorithms and in silico structural modeling analysis.
Previous studies have demonstrated that individuals with germline TP53 variants have significantly increased risks of a broad range of neoplasms, involving the lung, stomach, skin and gonadal germ cells (45). In over 70% LFS cases, cancer is driven by mutations in the TP53 gene. On the other hand, only 20-40% of LFL syndrome families can be accounted for by the presence of TP53 germline mutation (3, 4). Thus, additional susceptibility genes remain to be identified in approximately 30-60% of LFS/LFL syndromes. It has been reported that alterations in the cell cycle checkpoint kinase, CHEK2, might be associated with increased cancer risks in LFS/LFL families (8). However, CHEK2 has been excluded as a major LFS gene, but rather represents a moderate risk gene for individual cancer susceptibility within LFS clusters (46, 47). In this kindred, no germline variants in TP53 and CHEK2 were detected (Supplementary Table 1). It is worth noting that exon 1 of TP53 were not fully covered by the WES panel used in this study, so we cannot exclude the possibility that this family might also carry loss-of-function mutations in TP53 in addition to POT1 mutations. On the other hand, we performed additional genetic analysis on the available tumors to check for the presence of TP53, CHEK2 and POT1. WES was performed on tumor tissue samples from four patients (II4/8/10/12), consisting of lung tumor samples from II4/8/10 and a leiomyosarcoma sample from II12. Two somatic variants in TP53, p.D259Y and p.S215R, were detected in II10 and II12, respectively. The TP53 p.D259Y is well-defined pathogenic that can reduce the tumor suppressor function of p53, while the p.S215R alteration is of unknown significance. In addition, driven gene mutations were found in two of the patients, including an EGFR p.L858R in II10 and a KRAS p.G12D in II12. However, no additional POT1 variants were found in any of the tumor samples (Supplementary Table 2).
One previous study has described a p.R117C variant in POT1, which results in unstable telomeres, as a causative allele of cardiac angiosarcoma in LFL families (11). Further study by the same group has uncovered a wide spectrum of POT1 variants in patients with familial angiosarcoma, as well as sporadic angiosarcoma and sarcomas (10). A considerable proportion of reported POT1 mutations in familial cancer syndromes occur in the telomere-binding OB1 domain (Figure 2A). The novel p.P35L variant identified in our kindred is also located in the OB1 domain with potential deleterious effects to protein structure and function. Subsequent characterization of the spectrum of POT1 mutations in 62,368 solid tumors has revealed a strong association between the presence of POT1 mutations and angiosarcoma development (10). In line with this study, one of the POT1 variant carriers, the III8 patient in our LFL family, presented with cardiac angiosarcoma and died at an early age of 19. Angiosarcomas, which commonly occur in the skin, breast, liver and deep tissues, are rare malignancies with highly aggressive nature (48). Angiosarcomas of the heart are extremely rare and associated with high recurrence and metastasis rates, with an estimated frequency of 0.001% to 0.003% in the general population (49). Given the rarity of the disease and unspecific symptoms, such as shortness of breath, weight loss and fatigue, early diagnosis can be very challenging (48). Screening for POT1 and other LFS/LFL or angiosarcoma causative genes might be of great clinical relevance. It is worth noting that no POT1 mutations were detected in the two individuals (II6 and III3) with pulmonary nodules and/or ovarian cysts. As smoking status and environmental exposure might be related to the development of pulmonary nodules, it had been first confirmed that both II6 and III3 are non-smokers with no obvious environmental exposures. II6’s husband was diagnosed with hepatocellular carcinoma at the age of 60, although it is unclear whether this has anything to do with the family’s living habits. Moreover, the two patients and their family members have no history of tuberculosis infection, as tuberculosis infection may be one of the factors causing pulmonary nodules. On the other hand, it should be noted that the incidence of pulmonary nodules is extremely high (~ 50%) in China. Given the large number of individuals with pulmonary nodules and the absence of clear environmental causes, the two cases with pulmonary nodules may be accidental events and are likely unrelated to the hereditary condition in this LFL family. Meanwhile, long-term monitoring of all the unaffected individuals in this family might be necessary given the age-dependent penetrance of hereditary cancer syndromes. In addition, we noticed that three NSCLC patients whom were detected POT1 p.P35L variant are all non-smokers. Of the 13 individuals included in our study, 12 were females, including the three POT1-positive non-smoker patients with NSCLC. This is not surprising as a very low percentage of Chinese females are smokers. The fact that these patients had developed lung cancer is consistent with our hypothesis that genetic alterations, rather than environmental exposures, underlie pathogenesis in our patients. We think that the presence of this variant in non-smokers with lung cancer is interesting and further supports our finding that POT1 alteration is the driving event in our patients.
The molecular basis of most familial hereditary cancer syndromes remains elusive. Leveraging high throughput next-generation sequencing, our study, together with other published work (3, 50), has identified a novel causative variant in a LFL family that provide insight into the pathogenesis of hereditary cancer syndromes and might facilitate early prevention of the diseases. While further validation studies might be necessary to firmly establish the causal relationship between the POT1 p.P35L variant and multi-cancer phenotype, given the general relevance of POT1 in LFS/LFL and angiosarcoma patients, our study support the incorporation of POT1 testing into cancer gene panels for routine diagnostic purposes to enable early diagnosis, prevention and therapeutic intervention of individuals at risk.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: The Genome Sequence Archive for Human (GSA-Human), The National Genomics Data Center, HRA001931 at https://ngdc.cncb.ac.cn/search/?dbId=hra&q=HRA001931&page=1.
Ethics statement
The study was approved by the Ethics Committee (protocol number is KY2021-156d) of the First Affiliated Hospital of WenZhou Medical University and was conducted in accordance with the Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YL and CC involved in conception and design. HX, JC and JuY carried out provision of study material or patients. DW, JCY, JRY and BY interpreted the data. DW and JRY provided NGS technical support. YL and YX wrote the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We gratefully thank the patients and their families who gave consent on presenting the data in this study.
Conflict of interest
DW, JCY, JRY and BY are employees of Nanjing Geneseeq Technology Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.963364/full#supplementary-material
Supplementary Table 1 | All germline variants detected in the family members.
Supplementary Table 2 | All somatic variants detected in four second-generation patients diagnosed with cancer.
References
1. Gargallo P, Yáñez Y, Segura V, Juan A, Torres B, Balaguer J, et al. Li-Fraumeni syndrome heterogeneity. Clin Transl Oncol (2020) 22:978–88. doi: 10.1007/s12094-019-02236-2
2. Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science (1990) 250:1233–8. doi: 10.1126/science.1978757
3. McBride KA, Ballinger ML, Killick E, Kirk J, Tattersall MH, Eeles RA, et al. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol (2014) 11:260–71. doi: 10.1038/nrclinonc.2014.41
4. Varley JM, McGown G, Thorncroft M, Santibanez-Koref MF, Kelsey AM, Tricker KJ, et al. Germ-line mutations of TP53 in Li-fraumeni families: an extended study of 39 families. Cancer Res (1997) 57:3245–52.
5. Hwang SJ, Lozano G, Amos CI, Strong LC. Germline p53 mutations in a cohort with childhood sarcoma: Sex differences in cancer risk. Am J Hum Genet (2003) 72:975–83. doi: 10.1086/374567
6. Chompret A, Brugières L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer (2000) 82:1932–7. doi: 10.1054/bjoc.2000.1167
7. Bartek J, Falck J, Lukas J. CHK2 kinase–a busy messenger. Nat Rev Mol Cell Biol (2001) 2:877–86. doi: 10.1038/35103059
8. Ruijs MW, Broeks A, Menko FH, Ausems MG, Wagner A, Oldenburg R, et al. The contribution of CHEK2 to the TP53-negative Li-fraumeni phenotype. Hered Cancer Clin Pract (2009) 7:4. doi: 10.1186/1897-4287-7-4
9. Zhuang X, Li Y, Cao H, Wang T, Chen J, Liu J, et al. Case report of a Li-fraumeni syndrome-like phenotype with a de novo mutation in CHEK2. Med (Baltimore) (2016) 95:e4251. doi: 10.1097/md.0000000000004251
10. Calvete O, Garcia-Pavia P, Domínguez F, Bougeard G, Kunze K, Braeuninger A, et al. The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur J Hum Genet (2017) 25:1278–81. doi: 10.1038/ejhg.2017.134
11. Calvete O, Martinez P, Garcia-Pavia P, Benitez-Buelga C, Paumard-Hernández B, Fernandez V, et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li-fraumeni-like families. Nat Commun (2015) 6:8383. doi: 10.1038/ncomms9383
12. Evans DG, Wu CL, Birch JM. BRCA2: A cause of Li-fraumeni-like syndrome. J Med Genet (2008) 45:62–3. doi: 10.1136/jmg.2007.054494
13. Watanabe T, Vital A, Nobusawa S, Kleihues P, Ohgaki H. Selective acquisition of IDH1 R132C mutations in astrocytomas associated with Li-fraumeni syndrome. Acta Neuropathol (2009) 117:653–6. doi: 10.1007/s00401-009-0528-x
14. Nordfors K, Haapasalo J, Afyounian E, Tuominen J, Annala M, Häyrynen S, et al. Whole-exome sequencing identifies germline mutation in TP53 and ATRX in a child with genomically aberrant AT/RT and her mother with anaplastic astrocytoma. Cold Spring Harb Mol Case Stud 4 (2018) 4(3). doi: 10.1101/mcs.a002246
15. Ponti F, Corsini S, Gnoli M, Pedrini E, Mordenti M, Sangiorgi L. Evaluation of TP53 Pro72Arg and MDM2 SNP285-SNP309 polymorphisms in an Italian cohort of LFS suggestive patients lacking identifiable TP53 germline mutations. Fam Cancer (2016) 15:635–43. doi: 10.1007/s10689-016-9895-3
16. Jones RM, Melton PE, Pinese M, Rea AJ, Ingley E, Ballinger ML, et al. Identification of novel sarcoma risk genes using a two-stage genome wide DNA sequencing strategy in cancer cluster families and population case and control cohorts. BMC Med Genet (2019) 20:69. doi: 10.1186/s12881-019-0808-9
17. Mirabello L, Zhu B, Koster R, Karlins E, Dean M, Yeager M, et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol (2020) 6:724–34. doi: 10.1001/jamaoncol.2020.0197
18. Musselman JR, Bergemann TL, Ross JA, Sklar C, Silverstein KA, Langer EK, et al. Case-parent analysis of variation in pubertal hormone genes and pediatric osteosarcoma: a children’s oncology group (COG) study. Int J Mol Epidemiol Genet (2012) 3:286–93.
19. Capasso M, Montella A, Tirelli M, Maiorino T, Cantalupo S, Iolascon A., et al. Genetic predisposition to solid pediatric cancers. Front Oncol (2020) 10:590033. doi: 10.3389/fonc.2020.590033
20. Hu F, Yu Y, Chen JS, Hu H, Scheet P, Huff CD. Integrated case-control and somatic-germline interaction analyses of soft-tissue sarcoma. J Med Genet (2021) 58:145–53. doi: 10.1136/jmedgenet-2019-106814
21. Farid M, Ngeow J. Sarcomas associated with genetic cancer predisposition syndromes: A review. Oncologist (2016) 21:1002–13. doi: 10.1634/theoncologist.2016-0079
22. Wang W, Song H, Liu J, Song B, Cao X. CD86 + 1057G/A polymorphism and susceptibility to osteosarcoma. DNA Cell Biol (2011) 30:925–9. doi: 10.1089/dna.2011.1211
23. Gargallo P, Yáñez Y, Juan A, Segura V, Balaguer J, Torres B, et al. Review: Ewing sarcoma predisposition. Pathol Oncol Res (2020) 26:2057–66. doi: 10.1007/s12253-019-00765-3
24. He J, Wang J, Wang D, Dai S, Yv T, Chen P, et al. Association between CTLA-4 genetic polymorphisms and susceptibility to osteosarcoma in Chinese han population. Endocrine (2014) 45:325–30. doi: 10.1007/s12020-013-0050-8
25. Mirabello L, Yu K, Berndt SI, Burdett L, Wang Z, Chowdhury S, et al. A comprehensive candidate gene approach identifies genetic variation associated with osteosarcoma. BMC Cancer (2011) 11:209. doi: 10.1186/1471-2407-11-209
26. Koshkina NV, Kleinerman ES, Li G, Zhao CC, Wei Q, Sturgis EM, et al. Exploratory analysis of fas gene polymorphisms in pediatric osteosarcoma patients. J Pediatr Hematol Oncol (2007) 29:815–21. doi: 10.1097/MPH.0b013e3181581506
27. Gianferante DM, Mirabello L, Savage SA. Germline and somatic genetics of osteosarcoma - connecting aetiology, biology and therapy. Nat Rev Endocrinol (2017) 13:480–91. doi: 10.1038/nrendo.2017.16
28. Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, et al. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat Genet (2013) 45:799–803. doi: 10.1038/ng.2645
29. Lu XF, Yang WL, Wan ZH, Li J, Bi ZG. Glutathione s-transferase polymorphisms and bone tumor risk in China. Asian Pac J Cancer Prev (2011) 12:3357–60.
30. Yang W, He M, Zhao J, Wang Z. Association of ITGA3 gene polymorphisms with susceptibility and clinicopathological characteristics of osteosarcoma. Med Oncol (2014) 31:826. doi: 10.1007/s12032-013-0826-y
31. Liu Y, Lv B, He Z, Zhou Y, Han C, Shi G, et al. Lysyl oxidase polymorphisms and susceptibility to osteosarcoma. PloS One (2012) 7:e41610. doi: 10.1371/journal.pone.0041610
32. Naumov VA, Generozov EV, Solovyov YN, Aliev MD, Kushlinsky NE. Association of FGFR3 and MDM2 gene nucleotide polymorphisms with bone tumors. Bull Exp Biol Med (2012) 153:869–73. doi: 10.1007/s10517-012-1847-9
33. Mirabello L, Koster R, Moriarity BS, Spector LG, Meltzer PS, Gary J, et al. A genome-wide scan identifies variants in NFIB associated with metastasis in patients with osteosarcoma. Cancer Discov (2015) 5:920–31. doi: 10.1158/2159-8290.Cd-15-0125
34. Benna C, Lv B, He Z, Zhou Y, Han C, Shi G, et al. Associations of clock genes polymorphisms with soft tissue sarcoma susceptibility and prognosis. J Transl Med (2018) 16:338. doi: 10.1186/s12967-018-1715-0
35. Zhang P, Liu J, Li X, Gao M, Feng F, Wang W, et al. NF1 and PTEN gene polymorphisms and the susceptibility to soft tissue sarcomas in a Chinese population: A case-control study. Exp Mol Pathol (2021) 118:104603. doi: 10.1016/j.yexmp.2021.104603
36. Zhi LQ, Ma W, Zhang H, Zeng SX, Chen B. Association of RECQL5 gene polymorphisms and osteosarcoma in a Chinese han population. Tumour Biol (2014) 35:3255–9. doi: 10.1007/s13277-013-1425-4
37. Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U.S.A. (2014) 111:E5564–5573. doi: 10.1073/pnas.1419260111
38. Mirabello L, Richards EG, Duong LM, Yu K, Wang Z, Cawthon R, et al. Telomere length and variation in telomere biology genes in individuals with osteosarcoma. Int J Mol Epidemiol Genet (2011) 2:19–29.
39. Hu YS, Pan Y, Li WH, Zhang Y, Li J, Ma BA, et al. Association between TGFBR1*6A and osteosarcoma: a Chinese case-control study. BMC Cancer (2010) 10:169. doi: 10.1186/1471-2407-10-169
40. Patio-Garcia A, Sotillo-Pieiro E, Modesto C, Sierrases-Maga L. Analysis of the human tumour necrosis factor-alpha (TNFalpha) gene promoter polymorphisms in children with bone cancer. J Med Genet (2000) 37:789–92. doi: 10.1136/jmg.37.10.789
41. Wang Z, Wen P, Luo X, Fang X, Wang Q, Ma F, et al. Association of the vascular endothelial growth factor (VEGF) gene single-nucleotide polymorphisms with osteosarcoma susceptibility in a Chinese population. Tumour Biol (2014) 35:3605–10. doi: 10.1007/s13277-013-1475-7
42. Wu Y, Poulos RC, Reddel RR. Role of POT1 in human cancer. Cancers (Basel) (2020) 12(10):2739. doi: 10.3390/cancers12102739
43. Speedy HE, Kinnersley B, Chubb D, Broderick P, Law PJ, Litchfield K, et al. Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood (2016) 128:2319–26. doi: 10.1182/blood-2016-01-695692
44. Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science (2001) 292:1171–5. doi: 10.1126/science.1060036
45. Masciari S, Dewanwala A, Stoffel EM, Lauwers GY, Zheng H, Achatz MI, et al. Gastric cancer in individuals with Li-fraumeni syndrome. Genet Med (2011) 13:651–7. doi: 10.1097/GIM.0b013e31821628b6
46. Sodha N, Houlston RS, Bullock S, Yuille MA, Chu C, Turner G, et al. Increasing evidence that germline mutations in CHEK2 do not cause Li-fraumeni syndrome. Hum Mutat (2002) 20:460–2. doi: 10.1002/humu.10136
47. Evans DG, Birch JM, Narod SA. Is CHEK2 a cause of the Li-fraumeni syndrome? J Med Genet (2008) 45:63–4. doi: 10.1136/jmg.2007.054700
48. Patel SD, Peterson A, Bartczak A, Lee S, Chojnowski S, Gajewski Pc, et al. Primary cardiac angiosarcoma - a review. Med Sci Monit (2014) 20:103–9. doi: 10.12659/msm.889875
49. Look Hong NJ, Pandalai PK, Hornick JL, Shekar PS, Harmon DC, Chen YL, et al. Cardiac angiosarcoma management and outcomes: 20-year single-institution experience. Ann Surg Oncol (2012) 19:2707–15. doi: 10.1245/s10434-012-2334-2
Keywords: Li-Fraumeni-like syndrome, whole exome sequencing (WES), co-segregation, genetic risk alleles, POT1
Citation: Li Y, Xie Y, Wang D, Xu H, Ye J, Yin JC, Chen J, Yan J, Ye B and Chen C (2022) Whole exome sequencing identified a novel POT1 variant as a candidate pathogenic allele underlying a Li–Fraumeni-like family. Front. Oncol. 12:963364. doi: 10.3389/fonc.2022.963364
Received: 07 June 2022; Accepted: 17 October 2022;
Published: 01 November 2022.
Edited by:
Pavel Vodicka, Institute of Experimental Medicine (ASCR), CzechiaReviewed by:
Tania Lee Slatter, University of Otago, New ZealandSilvia R. Rogatto, University of Southern Denmark, Denmark
Copyright © 2022 Li, Xie, Wang, Xu, Ye, Yin, Chen, Yan, Ye and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengshui Chen, wzchencs@163.com
†These authors have contributed equally to this work