 Tucker Lemos
Tucker Lemos Akil Merchant
Akil Merchant- Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, CA, United States
The Hedgehog (HH) pathway is a promising therapeutic target in hematological malignancies. Activation of the pathway has been tied to greater chances of relapse and poorer outcomes in several hematological malignancies and inhibiting the pathway has improved outcomes in several clinical trials. One inhibitor targeting the pathway via the protein Smoothened (SMO), glasdegib, has been approved by the FDA for use with a low dose cytarabine regiment in some high-risk acute myeloid leukemia patients (AML). If further clinical trials in glasdegib produce positive results, there may soon be more general use of HH inhibitors in the treatment of hematological malignancies.While there is clinical evidence that HH inhibitors may improve outcomes and help prevent relapse, a full understanding of any mechanism of action remains elusive. The bulk of AML cells exhibit primary resistance to SMO inhibition (SMOi), leading some to hypothesize that that clinical activity of SMOi is mediated through modulation of self-renewal and chemoresistance in rare cancer stem cells (CSC). Direct evidence that CSC are being targeted in patients by SMOi has proven difficult to produce, and here we present data to support the alternative hypothesis that suggests the clinical benefit observed with SMOi is being mediated through stromal cells in the tumor microenvironment.This paper’s aims are to review the history of the HH pathway in hematopoiesis and hematological malignancy, to highlight the pre-clinical and clinical evidence for its use a therapeutic target, and to explore the evidence for stromal activation of the pathway acting to protect CSCs and enable self-renewal of AML and other diseases. Finally, we highlight gaps in the current data and present hypotheses for new research directions.
Introduction
The Hedgehog pathway was originally described by Nüsslein-Volhard and Wieschaus in reference to a mutant drosophila phenotype that produced a spiky embryo (1). It is one of several critical embryonic body patterning pathways and its role in development has been extensively studied and characterized since its original discovery. Its expression is limited in most healthy adult tissues.
The oncological relevance of the pathway was independently realized by Kinzler et al. when they discovered a gene that had a fifty-fold amplification in some human gliomas (2). It was later discovered that this Glioma-associated oncogene family (GLI1, GLI2, and GLI3) was the mammalian homolog for the HH-responsive transcription factor cubitus interuptus (Ci) (3, 4).
The link between aberrant activation of the pathway and tumorigenesis is not limited to gliomas; it has been implicated in many cancers including basal cell carcinoma, breast cancer, gastric cancer, pancreatic cancer, and various hematological malignancies (5–7). This paper will be limited to the last of these, but for a review of the HH pathway in cancer generally see Skoda et al., 2018 and Scales and de Sauvage 2009 (8, 9).
Canonical hedgehog signaling
A simplified schema of the activation of the Hedgehog signaling pathway (see Figure 1) principally focuses on four families of proteins: the Hedgehog ligands, the transmembrane protein Patched (PTCH), the transmembrane protein Smoothened (SMO), and the Gli family of transcription factors (Gli1, Gli2, Gli3) (8).
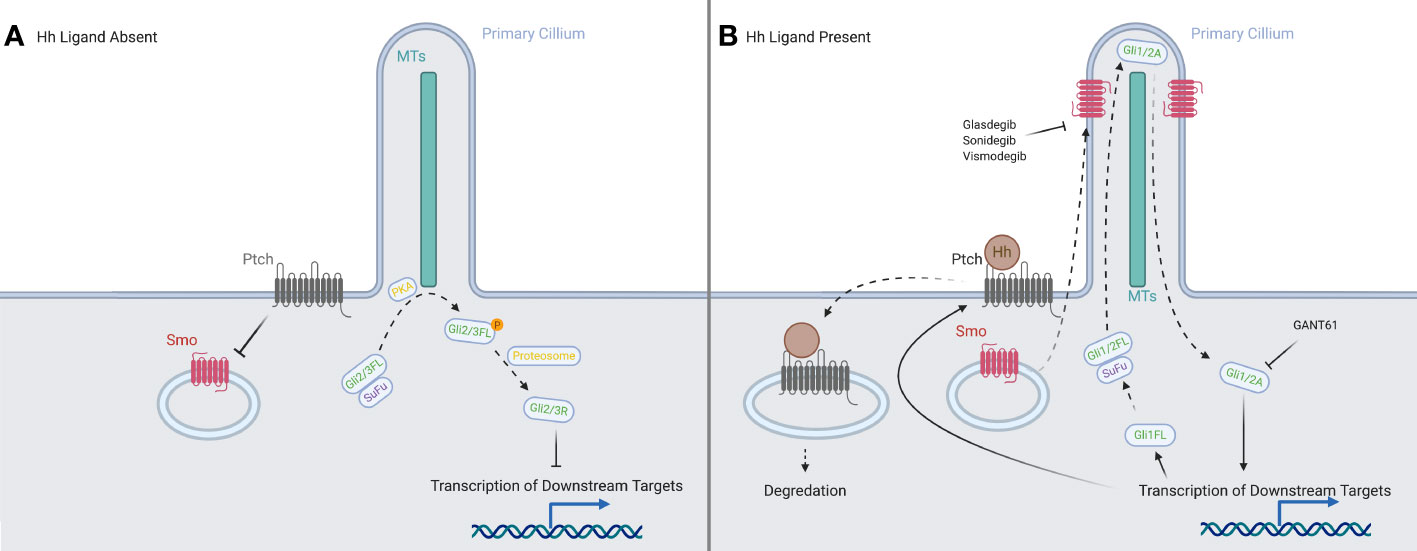
Figure 1 A representation of the canonical HH signaling in the presence and absence of a HH ligand. (A) With the ligands absent, PTCH prevents SMO from translocating to the primary cilium. In the absence of SMO, the full-length forms of GLI2 and GLI3 are phosphorylated by PKA, and then processed into their repressor forms, which inhibit downstream transcription. (B) When ligands are present, they are bound by PTCH and both are internalized and degraded. This allows the translocation of SMO to the tip of the primary cilium, where it processes the full-length forms of the GLI proteins into their activator forms, which then promote transcription of downstream targets in the nucleus. Created in BioRender.
There are three different HH ligands, Indian hedgehog (IHH) which is expressed in early hematopoietic tissues (10) and has a role in modulating chondrocyte development (11), Desert hedgehog (DHH) which regulates development of the peripheral nerves (12) and is essential to spermatogenesis (13), and Sonic hedgehog (SHH). SHH is the most widely expressed and best studied of the HH ligands, being involved in embryonic body patterning.
In the absence of the ligand, PTCH is localized to the base of the primary cilium (PC), where it inhibits SMO activity, likely by preventing SMO modification via cholesterol (14, 15). We have shown that human blood and bone marrow cells have primary cilia and mediate hedgehog signaling (16). The canonical activation of the signaling cascade is caused by the binding of a HH ligand to PTCH (17, 18) upon which both are internalized, and SMO moves from vesicles in the cytosol to the PC (19–21), joining the other members of the pathway: the GLI family and the machinery responsible for processing it (22). The translocation of SMO modulates the post-translational processing of GLI2 and GLI3 in the PC: allowing GLI2 to act as an activator and preventing GLI3 from being processed into its strong repressor form (23, 24). Another protein, Suppressor of Fused (SUFU), which normally acts to inhibit GLI activators and sequester them in the cytosol (25–27), allows them to travel to the nucleus, where they upregulate PTCH and GLI1 (28), creating both positive and negative feedback loops to tightly control the downstream signal. The activator forms of GLI1 and GLI2 set off a signaling cascade, reaching downstream targets such as MYCN (29), BCL-2 (30) and VEGF-A (31), many of which are associated with oncogenesis.
Further complexity in transducing this signal comes from its significant overlap and crosstalk with other developmental pathways, especially WNT and NOTCH (32).
Modes of HH signaling
The HH pathway has been shown to regulate proliferation, apoptosis, and angiogenesis, therefore it is not surprising that activation of HH signaling is a common factor in many cancers. Signaling in this context can be broken up into four types, based on the activity of the Hh ligand (see Figure 2), though these types are not necessarily mutually exclusive.
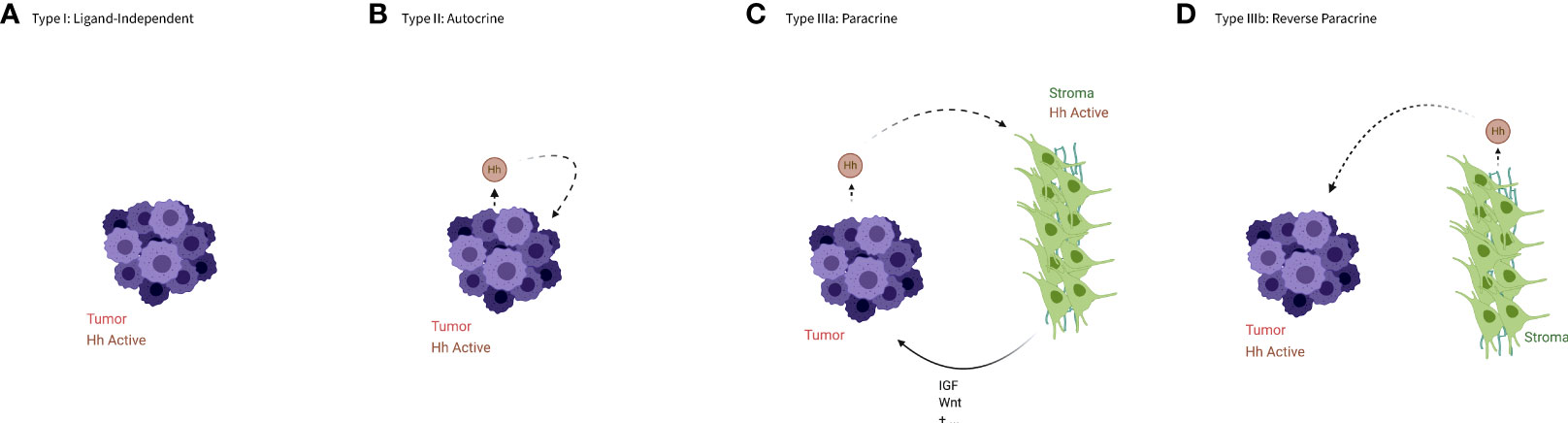
Figure 2 An illustration of the types of aberrant HH signaling. (A) In type I signaling, the Hh pathway is active despite the absence of the Hh ligand. (B) In type II or autocrine signaling tumor cells both produce and bind the ligands. (C) In type IIIa or paracrine signaling tumor cells produce ligands that activate HH signaling in the stroma, which in turn produces a more favorable niche for the tumor cells or cancer stem cells. (D) In type IIIb or reverse paracrine signaling the stroma produce ligands to activate HH signaling in the tumor cells. Created in BioRender.
Type I –ligand independent
Type I signaling occurs when the pathway is constitutively active without regard to the presence or absence of a HH ligand. This is often caused by loss of function mutations in the PTCH gene that produce a protein unable to prevent SMO from activating the pathway, as first demonstrated by patients with nevoid basal cell carcinoma syndrome, or Gorlin syndrome (33). These individuals carry an ineffective copy of the PTCH gene and are at an elevated risk of developing various tumors, especially basal cell carcinomas (BCCs) (34, 35). Alternatively, activating mutations in SMO are often found in spontaneous BCC, and rarely in other tumor types (36–38).
Constitutional activation can also be effected by mutations to other elements of the signaling pathway, such as gain of function mutations in SMO that counteract inhibition by PTCH demonstrated in sporadic BCCs (36), loss of the GLI3 suppressor via methylation in AML (39) and inactivation of SUFU in medulloblastoma (40).
Type II – autocrine/juxtacrine
Type II signaling occurs when the pathway is activated by ligands that are produced by the tumor cell itself, or by other nearby tumor cells. This self-targeted increase in HH expression has been observed in a variety of tumors, including but not limited to those of the digestive tract, brain, and lung (41–45).
Type IIIa – paracrine
Type III signaling occurs when the pathway is activated by ligands produced by other, functionally distinct cells. Type IIIa refers specifically to tumor cells producing ligands to induce HH signaling in non-malignant cells. This was first demonstrated in epithelial tumor cells that produced HH ligands but relied on the pathway being activated in nearby stromal cells rather than binding the ligands themselves (46–48). The stromal cells, upon binding HH, upregulate other signaling factors such as insulin-like growth factor (IGF) and Wnt (46), which can stimulate tumor growth both directly and by creating a tumor promoting microenvironment.
Type IIIa signaling has also been demonstrated to act in a tumor-suppressing manner in some solid cancers, further complicating our understanding of this pathway (49).
Type IIIb – reverse paracrine
Type IIIb signaling refers to paracrine signaling that occurs in a reverse manner, with stromal cells stimulating tumor growth through secretion of HH ligands, which activate the pathway in tumor cells. This signaling pattern has been demonstrated most clearly in B-cell malignancies (50, 51) and multiple myeloma (52). Though this mode of signaling has been almost exclusively tied to hematological malignancies, there is evidence of stroma-produced HH ligands in some gliomas (53).
Signaling in cancer stem cells
Hedgehog signaling in cancer stem cells is of particular interest and may employ any of the modes discussed above. CSCs have been implicated as an important part of understanding the response of various cancers to therapy. CSCs are a subset of slowly dividing cells that exist within a tumor that can proliferate, differentiate and reconstitute a heterogenous tumor (54). They are resistant to conventional chemotherapy due to their relatively slow growth and have been consistently implicated as a cause of relapse in many difficult to treat tumors. CSCs have been shown to be supported and maintained by HH signaling in a variety of tumors, such as CML (55), MM (52) and AML (56, 57). In some cases autocrine signaling between differentiated tumor cells and cancer stem cells has been demonstrated while in other cases paracrine signaling between cancer stem cells and the tumor microenvironment has been implicated. For a thorough review of the role of HH signaling in CSCs, see Cochrane et al., 2015 (54).
A possible requirement for stromal HH signaling in normal and malignant hematopoiesis
Conflicting data for the role of HH in normal hematopoiesis
Before exploring the HH pathway in hematologic malignancies it is helpful to understand what is known about the HH signaling in normal hematopoiesis.
Early studies showed increased hematopoietic stem cell (HSC) proliferation when SHH was added in vitro (58). A role for HH signaling in HSC differentiation was further supported by studies in zebrafish models (59).
The strongest evidence of the HH pathway’s role in HSC renewal comes from mouse models. Trowbridge et. al., found that mice hemizygous for Ptch (increasing HH pathway activity) had significantly increased numbers of HSC progenitor cells and, further, the prolonged HH signaling could exhaust the ability of HSCs to self-renew (60). Our group has shown that loss of Gli1 transcription factor is associated with decreased cell cycling of HSC and delayed recovery of myelopoiesis after radiation or chemotherapy. These mice had no loss of steady state bone marrow chimerism in transplantation experiments, demonstrating that Gli1 is likely only required for the burst of proliferation required during stress hematopoiesis (61).
While studies of Ptch and Gli1 suggest an important role of HH signaling in hematopoiesis, gene deletion studies of Smo, the central hub of hedgehog signaling, have been less clear. Dierks et al. reported that mice fetal liver cells lacking one or both copies of Smo were unable to retain their in vitro colony-forming potential in replating experiments, while cells hemizygous for Ptch were far more successful than the wildtype in forming colonies after being replated (62). In transplantation experiments, they showed that loss of Smo delayed engraftment kinetics, however, long-term, steady-state hematopoiesis does not require Smo, as no difference was observed between the number of HSCs, B-cells, or other cell types in mice transplanted with Smo knockout fetal liver HSCs (62). In contrast, Zhao et al. used conditional Smo knockout mice crossed with Vav-Cre mice, which results in conditional knockout of a Smo in the HSC compartment, to show that mice with Smo deficient HSCs had significantly lower rates of blood reconstitution after transplantation, demonstrating a requirement of Smo for HSC renewal (55). Hofmann et al. and Gao et al. both independently found that, using the same conditional Smo allele, this time crossed with an inducible Mx-1-Cre strain, Smo was dispensable for normal hematopoietic function (63, 64). Studies with human HSCs found no reduction in differentiation capacity with inhibiting Smo, both in vitro with human HSCs and in vivo with the hematopoietic compartments of mice transplanted with HSCs from human cord blood (65).
We have hypothesized that these seemingly contradictory results may be due to different patterns of Cre expression in Vav-Cre and Mx-1-Cre models (66). While both systems are active in the primitive stem cell compartment, Vav-cre shows additional activity in bone marrow stroma and endothelial precursors. An in-depth review of the various HSC-targeting Cre systems can be found in Joseph et al., 2016 (67). The differences in the two models are demonstrated in the similarly difficult issue of determining the role of Wnt/β-catenin signaling in HSCs, where the Vav-Cre model also indicated a requirement for β-catenin for normal HSC function and the Mx-1-Cre model revealed that it was dispensable (68). Therefore, a HSC extrinsic requirement for Hedgehog and Wnt signaling pathways in bone marrow stroma, but not intrinsic to HSCs themselves could explain this discrepancy.
Evidence for stromal signaling
We propose that, rather than targeting the bulk tumor, glasdegib and other Smo inhibitors are instead acting on stromal cells that promote tumor growth and reconstitution through maintenance of a hospitable niche.
In preclinical models, Smo inhibitors are not directly cytotoxic, but seem to reduce proliferation and self-renewal (55, 60, 69). This is borne out in clinical trials, where the addition of glasdegib to chemotherapy did not improve CR rates. Rather, the effects of glasdegib appear to increase the overall survival (OS) of patients by reducing relapse. One plausible mechanism for this observation is that glasdegib is targeting the leukemic stem cells that mediate relapse. However, direct evidence that SMO inhibitors act on CSC in a cell autonomous fashion are lacking. Meanwhile, evidence from animal vav-Cre models discussed above suggest that loss of Smo in the stroma leads to a loss of HSC self-renewal. This suggests the possibility that Smo inhibitors could act on the bone marrow stroma and thus modulate CSC self-renewal.
Indeed, evidence for such a mechanism can be found in studies on the interaction of HH and retinoid signaling in multiple myeloma. In an elegant series of studies Ghiaur et al. were able to show that stromal production of the enzyme CYP26 can inactivate retinoic acid signaling, preventing differentiation and functionally expanding HSC self-renewal capacity (70). They further demonstrated that stromal production of CYP26 dependent on Smo activity. In a follow up study using a mouse model there were able to show that stroma-specific knockout of Smo sensitizes otherwise refractory multiple myeloma to bortezomib (71).
Preclinical data supporting the HH pathway as a therapeutic target in hematologic malignancies
As HH is a common driver of tumorigenesis and chemo-resistance, inhibition of the pathway presents a clear target for therapy. HH components have been found to be aberrantly activated in a variety of hematological malignancies, and further, can often be implicated as markers of a poor prognosis in patients, in AML (57, 69) and in CML (65). In this section we summarize the preclinical studies in several tumor types that have demonstrated potential clinical applications of HH inhibitors.
Chronic myeloid leukemia
Chronic myeloid leukemia (CML) is a hematological malignancy that results when HSCs acquire the fusion oncogene BCR-ABL, leading to constitutive activation of the ABL tyrosine kinase. The advent of tyrosine kinase inhibitor (TKI) therapy has dramatically improved the prognosis of this disease; the treatment is able to consistently effect cytogenic and molecular level responses in early chronic-phase patients (72). CML is often used as an exemplar of a CSC-driven cancer and until recently TKI therapy was considered a lifelong requirement due to CSC-driven reconstitution of the disease upon the end of the treatment (73, 74). Although, recent evidence and practice has moved towards ending TKI therapy in select patients who have achieved deep-molecular responses, only half of these patients maintain their response after discontinuing TKI therapy and many never achieve the deep responses needed to attempt discontinuation (75). Therefore, for these patients or those who develop TKI resistant mutations, further treatment options are needed. One of these candidates has been HH inhibitors, as the pathway has been shown to be aberrantly activated in CML patients. There is increased expression of the pathway in progenitor cells, which becomes more pronounced as the disease progresses (55, 62, 65).
In vitro and in vivo experiments have returned promising results with HH inhibitors, with treatments reducing the proliferative and self-renewal capacity of CML cells.
Dierks et al. used a viral transgenic model of BCR-ABL in murine bone marrow to show that treatment with cyclopamine, a SMO inhibitor, caused a significant decrease in colony-forming potential and a reduction of the percentage of BCL-ABL+ myeloid progenitors (62). Using the same model, Zhao et al. demonstrate that cells with elevated levels of Numb, a cell determinant that is upregulated in SMO knockout cells, have reduced colony-forming potential. They show that treatment with a SMO inhibitor can reduce the colony number in murine models, in primary human CML, and in the imatinib-resistant T315I mutant of the disease (55).
Glasdegib, vismodegib and sonidegib, three SMO inhibitors, have all been found to significantly decreased progenitors in samples from blast crisis patients and chronic phase patients, while having no significant effect on the colony-forming potential of normal cord blood samples (65, 69). SMOi effect against CML is thought to be mediated, it part, through preventing hedgehog mediated quiescence of cancer stem cells. Transduction of progenitors with an inactive version of GLI2 led to abrogated cell-cycle dormancy, indicating that the pathway may be integral to maintaining this mechanism of chemoresistance (65).
In mice models, downregulation of the pathway through either inhibition or knockout of Smo increases survival, even in some TKI-resistant forms of the disease. Meanwhile, constitutive activation of Smo via the SmoM2 mutant allele, survival time significantly decreased when compared to control mice (55, 76). Transplant experiments with BCR-ABL+ Smo -/- fetal liver cells have shown that Smo was required for reconstitution of CML CSCs and thereby of the disease (62). Smo inhibition via either glasdegib or sonidegib, combined with TKI therapy drastically reduces engraftment and preventing serial transplantation, implying that Smo inhibition could potentially be combined with TKI to enhance their therapeutic activity (65, 69).
Taken together, these studies have formed a rationale for trials combining SMO inhibitors with TKI therapy, both as a treatment option in rare forms of TKI-resistant forms of the disease and to increase rates of complete eradication of the disease.
Acute myeloid leukemia
Acute myeloid leukemia (AML) has been one of the most promising targets of HH inhibition therapy, either with Smo inhibitors or with further downstream inhibitors of GLI1 and GLI2 activators.
High level activation of HH signaling is found in a subset of AML patients, is associated with progression of myelodysplastic syndrome (MDS) to AML, and with reduced rates of overall survival of AML patients (57, 77–79). The SMOi glasdegib achieved FDA approval based on a Phase 2 clinical trial and a review of pre-clinical data can help illuminate potential mechanisms of action for this observed benefit. Potential therapeutic mechanisms of HH inhibitors include down-regulation of pro-survival or apoptotic pathways, loss of CSC dormancy, or modulation of chemo-resistance.
The most straightforward mechanism is the pathway’s ability to regulate cell survival and apoptosis through downstream targets such as AKT. HL-60/RX, a radiation-resistant form of the AML model HL-60 cell-line, had elevated expression of GLI-1 and SMO compared to normal HL-60 cells (80). Inhibition of the pathway using a Smo inhibitor (sonidegib), sensitized the HL-60/RX to radiation. In addition, inhibiting Smo reduced expression of elements in the PI-3K/AKT pathway, downregulating apoptosis (81) and has been linked to aberrant activation of HH signaling in other cancers (82). These results are supported by RNA-seq data, which shows that relapsed/refractory AML patients are associated with higher expression of GLI1 and PI3K (56).
Another area where malignant HH signaling could enhance tumor chemoresistance is through the maintenance of malignant progenitor populations in dormancy. As chemotherapy is cell cycle selective, the slowed division of CSCs allows them to survive treatment, after which they can reconstitute the disease (83). Direct evidence connecting this effect to HH signaling is evident in AML, where cells treated with SMO inhibition (glasdegib) showed significantly fewer dormant CD45+ cells and were sensitized to Ara-C treatment (84).
HH signaling can also effect drug resistance to both cytotoxic and targeted chemotherapy via glucuronidation of the therapeutic molecules. Zahreddine et al., 2014, observed increased GLI1 levels upon relapse of patients (85). Other studies have found that patients with increased evidence of HH activity showed greater resistance to Ara-C and to the anti-viral drug ribavirin, which is being clinically evaluated as a therapy targeting the oncogenic eukaryotic translation initiation factor eIF4E (86). In GLI1-elevated patients, there was a decrease of ribvarin-bound eIF4E that could be reversed upon Smo inhibition. Further, GLI1 knockdown reduced levels of UGT1A, a drug- glucuronidating enzyme, and mass-spec analysis revealed that both Ara-C and ribavirin were glucuronidated in drug-resistant cells. Both the resistance and glucuronidation were reversed upon SMOi (85). In stabilizing UGT1A, GLI1 activation can be directly tied to drug-resistance via glucuronidation.
The FLT3 internal tandem duplication mutation (FLT3-ITD) appears in 25% of cases of AML and has a significant negative prognostic impact (87, 88). FLT3-ITD patient samples and cell lines have increased expression of GLI2, and FLT3-ITD transgenic mice show rapid progression from myeloproliferation to acute leukemia when crossed with SmoM2 transgenic mice that have constitutive HH activation (89). These data suggest that FLT3 and HH signaling can cooperate to drive leukemic progression, although the precise mechanisms of interaction between these pathways have not been worked out (89).
Complicating matters further, several recent papers have shown varied mechanisms of SMO-independent upregulation of GLI activators, all of which are inherently resistant to SMO inhibition. GLI1 upregulation has been shown to be integrated with PI3K independent of SMO (90). We have reported on SMO-independent activation of the pathway caused by loss of GLI3 expression associated with hypermethylation of the GLI3 locus. This leads to loss of GLI3R transcriptional repression and unopposed HH target activation through GLI1 and GLI2 (39). Further work has found that primary AML cells from some patients with high GLI1 expression will have reduced proliferation and self-renewal capacity when treated with the GLI1 and GLI2 inhibitor GANT61 but not when treated with SMOi (57). These data suggest that SMOi alone may not be adequate to target HH activation in AML and that direct GLI inhibitors may be required.
Chronic lymphocytic leukemia and acute lymphoblastic leukemia
While myeloid malignancies have been the main target of investigation for HH inhibition therapies, there is also preclinical evidence that patients diagnosed with lymphoid leukemias may also find clinical benefit in inhibition of this pathway.
As in AML and CML, both acute and chronic lymphoid leukemias seem to respond to HH inhibitors. In B-cell ALL, cancer stem cells that were treated with a Smo inhibitor showed decreased self-renewal potential (91), a result mirrored by the subset of GLI1 rich T-cells when they were treated with either a GLI or SMO inhibitor (92). In T-ALL specifically, there is evidence that inactivating mutations of PTCH speed progression of NOTCH-induced disease, and restoration of WT PTCH activity can induce apoptosis in PTCH mutant T-ALL cells in vitro.
In B-cell chronic lymphoblastic leukemia (CLL), HH activation increases proliferation and resistance of CLL cells and is associated with progression of the disease (93, 94). As in other malignancies, GLI1 upregulation is tied to a worse prognosis and predicts response of cells to HH inhibition (95, 96). Inhibition of HH signaling via SMOi can sensitize GLI1 positive B-CLL cells to chemotherapy and induce higher rates of apoptosis (95). However, many cases show primary resistance to SMOi (96), in which case a response requires alternative therapies, such as targeting downstream of SMO with GANT61 (93, 94, 97).
As in AML, finding different ways to target the HH pathway should be considered to properly account for tumors resistant to SMO inhibition. This could be in the form of new Smo inhibitors that can be effective against resistant mutants of the protein or in pursuing direct inhibitors of GLI activators such as GANT61.
Clinical data
The wealth of preclinical data on normal and aberrant HH signaling has guided our interpretation of clinical trials of HH inhibitors. The first FDA-approved HH inhibitor was the SMO inhibitor GDC-0449 (vismodegib, Genentech) for use in relapsed or advanced basal cell carcinomas (BCCs) in 2012 (109). Approval of vismodegib has since been followed by FDA approval of LDE-225 (sonidegib, Novartis) for advanced BCCs in 2015 (110).
Within the scope of hematological malignancies, PF-04449913 (glasdegib, Pfizer) is currently the only FDA-approved HH inhibitor. In 2018, glasdegib was approved for use in newly diagnosed cases of acute myeloid leukemia in combination with low dose cytarabine (LDAC) for patients that are not candidates for intensive induction chemotherapy (111).
Several SMO inhibitors have been tested in clinical trials for hematological malignancies, as summarized in Table 1. Vismodegib clinical trials have been run in myelofibrosis, multiple myeloma, and select lymphatic malignancies, but none of them demonstrated the efficacy required to encourage further study. An early phase 1 trial of Sonidegib combined with AZA in various myeloid malignancies, (NCT02129101) did not see decreased remission rates (108). However, especially in AML patients, the trial found increases in both OS and rates of SD. A later phase II trial was stopped early due to lack of efficacy (NCT01826214). Two other SMO inhibitors IPI-926 (Sardegib) and BMS-833923 were studied in myelofibrosis and CML, respectively. Sardegib treatment of myelofibrosis patients did show modest clinical activity, however it failed to reach pre-specified endpoints for clinical efficacy. Specifically, several patients had reductions in spleen size and/or GLI1 levels in the BM. (NCT01371617) (106). BMS-833923 was added to dasatinib in CML with the intention of reducing self-renewal capacity of CML stem cells. The study showed some efficacy, with 3 out of 10 dasatinib-resistant chronic phase CML patients demonstrating clinical benefit with one patient achieving a complete cytogenetic response. However, the addition of BMS-833923 did not seem to affect the potential for self-renewal as measured by colony forming culture assays, in contrast to in vitro study (70) and there was no observed clinical response in advanced CML patients or ALL patients. (NCT01218477) (104).
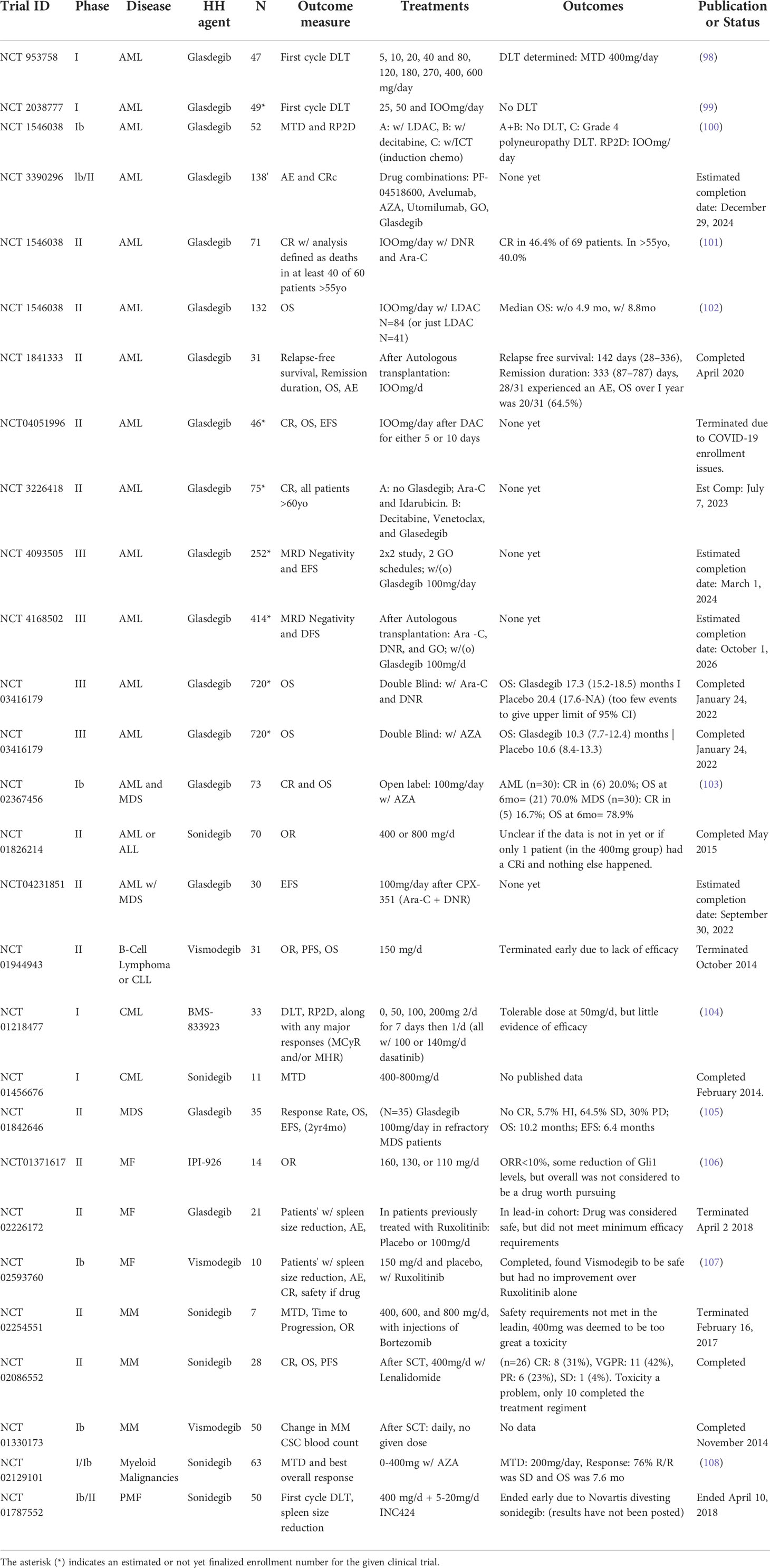
Table 1 List of clinical trials of SMO inhibitors in hematological malignancies (from clinical trials.gov).
Glasdegib phase I trials
Glasdegib is an oral, selective HH inhibitor that binds SMO (112) Clinical trials have been limited to the study of its effect of myeloid malignancies: Japanese phase I NCT 02038777 (99), US and Europe phase I NCT 00953758 (98) and a study in myelofibrosis (NCT 02226172)
The Japanese trial tested doses of 25, 50, and 100mg. In the AML group (n=7), there was 1 complete remission and four patients who achieved stable disease; in the MDS group there was one marrow complete remission and two patients who achieved stable disease. Further, GLI1 was found to be significantly downregulated in patients who were in the 50 or 100mg treatment group (99).
These results are mirrored in the American and Italian trial, which tested for doses ranging from 5 to 600mg. In the AML group (n=28), 16 showed some possible biological activity: 1 patient had complete remission, four had partial remission, four had a minor response, and seven achieved stable disease. 3 of 6 MDS patients achieved stable disease, with two of those showing hematological improvement, 2 of 7 myelofibrosis patients demonstrated some clinical improvement and the one enrolled CMML patient also achieved stable disease. One patient out of five with CML achieved a partial cytogenetic response (98). Martinelli et al. found a maximum tolerated dose of 400mg daily, however 100mg was suggested as the phase 2 dose based on tolerability and target inhibition (98).
Bright Aml 1003
BRIGHT AML 1003 (NCT 01546038) examined the use of Glasdegib in various combinations for AML or high-risk MDS. The Phase Ib/II study, tested glasdegib 100mg in combination with standard “7+3” induction chemotherapy (ICT) for fit patients and glasdegib in combination with LDAC for unfit patients unable to tolerate high dose chemotherapy.
For fit patients, the 1003 study was a single arm, phase II of glasdegib 100mg daily plus ICT with CR rate as the primary endpoint (102). Although the trial failed to meet the prespecified CR rate, relapse rates and overall survival (OS) was significantly improved in risk groups when compared to expected outcomes by European LeukemiaNet category (113). This pattern of decreased relapse rate without an effect on CR is characteristic of a CSC targeting agent, as one would expect a drug targeting CSCs to not affect the bulk tumor, but rather to inhibit the surviving CSCs ability to self-renew and reconstitute the disease.
Patients ineligible for intensive induction therapy on the 1003 study were randomized to LDAC alone or LDAC in combination with glasdegib in an open label design. CR rates and survival at one year was significantly increased in the glasdegib combination, with acceptably low toxicity (102). FDA approval was made largely based upon these data (111). A major weakness of this trial, however, was the open label design which may have contributed to the significantly shorter treatment duration for LDAC alone patients and could confound the results. More importantly, extremely favorable response rates to venetoclax combinations with hypomethylating agents in similar, unfit AML patient populations have limited clinical enthusiasm for the LDAC+glasdegib combination.
Glasdegib phase III and in-progress trials
There are currently three large phase III trials recently reported or in progress. BRIGHT AML 1019 (NCT 03416179) is a global randomized double-blinded placebo control trial of glasdegib in combination with standard therapy for front line AML. Fit patients eligible for induction chemotherapy are offered cytarabine and daunorubicin (7 + 3) plus glasdegib or placebo. Unfit patients are treated with azacitidine and glasdegib or placebo. Neither arm hit their primary overall survival endpoint. However, detailed subgroup analysis from this large phase III study has not yet been presented.
Two trials examine glasdegib in combination with gemtuzumab ozogamicin (GO), an antibody-drug conjugate that targets cells expressing CD33, a marker that is expressed in tumor cells in almost all AML patients (114). A German trial, GnG (NCT 04093505) is using a two-by-two factorial design with two different GO schedules followed by the use of 100mg per day glasdegib or a placebo. It aims to find the effect of the treatments on measurable residual disease (MRD) and event free survival (EFS) at 2 years. An Italian trial (NCT 04168502) is looking at younger patients and considers the use of glasdegib as maintenance after a patient receives an autologous or allogenic stem cell transplant (SCT). Patients receive 7 + 3 ICT (Ara-C and DNR), GO and a stem cell transplant, followed by 100mg glasdegib per day. The study will consider MRD following the SCT and GO treatments, and then measure disease free survival at 2 and 5 years.
Discussion
While SMOi has seen some success in clinical study for AML, the processes of interpreting the results and charting future directions for the treatment are hindered by current gaps in our mechanistic understanding of the role of HH signaling in hematological malignancies. It remains unclear whether the tumor or the microenvironment are the primary targets for SMOi. Demonstrating modulation of HH signaling in tumor samples has been challenging and has prevented the development of useful pharmacodynamic biomarkers.
Based on data from both clinical and preclinical study, we suggest that signaling is active in stromal cells that support the hematopoietic and leukemic stem cell microenvironments. This hypothesis would explain the apparently contradictory results from earlier examinations of HH signaling in hematopoiesis, as well as the pattern of SMOi preventing relapse rather improvement of initial response in clinical trials. However, this hypothesis needs to be specifically tested by specifically knocking out Smo in the stroma without affecting its function in HSCs/CSCs. Establishing a greater understanding of the mechanisms of HH signaling in hematological malignancy will allow for better interpretation of its clinical benefits and improved implementation as a targeted therapy.
Author contributions
Conception of idea was done by AM. Manuscript writing and figure preparation was done by TL. Manuscript editing was done by both authors. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from National Heart, Lung and Blood Institute (NHLBI R01 R01HL138414) to AM.
Acknowledgments
We would like to thank our colleagues in the Merchant Lab at the Cedars-Sinai Medical Center.
Conflict of interest
AM has received research funding from Pfizer and served on an advisory board for Novartis.
TL declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nüsslein-volhard C, Wieschaus E. Mutations affecting segment number and polarity in drosophila. Nature (1980) 287:795–801. doi: 10.1038/287795a0
2. Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O’Brien SJ, et al. Identification of an amplified, highly expressed gene in a human glioma. Science (1987) 236(4797):70–3. doi: 10.1126/science.3563490
3. Walterhouse D, Ahmed M, Slusarski D, Kalamaras J, Boucher D, Holmgren R, et al. Gli, a zinc finger transcription factor and oncogene, is expressed during normal mouse development. Dev Dyn (1993) 196(2):91–102. doi: 10.1002/aja.1001960203
4. Hui CC, Slusarski D, Platt KA, Holmgren R, Joyner AL. Expression of three mouse homologs of the drosophila segment polarity gene cubitus interruptus, gli, gli-2, and gli-3, in ectoderm-and mesoderm-derived tissues suggests multiple roles during postimplantation development. Dev Biol (1994) 162(2):402–13. doi: 10.1006/dbio.1994.1097
5. Wicking C, Smyth I, Bale A. The hedgehog signalling pathway in tumorigenesis and development. Oncogene (1999) 18:7844–51. doi: 10.1038/sj.onc.1203282
6. Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res (2004) 64(17):6071–4. doi: 10.1158/0008-5472.CAN-04-0416
7. Thayer SP, Di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature (2003) 425:851–6. doi: 10.1038/nature02009
8. Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L. The role of the hedgehog signaling pathway in cancer: A comprehensive review. Bosnian J Basic Med Sci (2018) 18(1):8–20. doi: 10.17305/bjbms.2018.2756
9. Scales SJ, de Sauvage FJ. Mechanisms of hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci (2009) 30(6):303–12. doi: 10.1016/j.tips.2009.03.007
10. Dyer MA, Farrington SM, Mohn D, Munday JR, Baron MH. Indian Hedgehog activates hematopoiesis and vasculogenesis and can respecifyprospective neurectodermal cell fate in the mouse embryo. Development (2001) 128(10):1717–30. doi: 10.1242/dev.128.10.1717
11. Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP, et al. BMP and lhh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development (2001) 128(22):4523–34. doi: 10.1242/dev.128.22.4523
12. Parmantier E, Lynn B, Lawson D, Turmaine M, Namini SS, Chakrabarti L, et al. Schwann cell–derived desert hedgehog controls the development of peripheral nerve sheaths. Neuron (1999) 23(4):713–24. doi: 10.1016/S0896-6273(01)80030-1
13. Bitgood MJ, Shen L, McMahon AP. Sertoli cell signaling by desert hedgehog regulates the male germline. Curr Biol (1996) 6(3):298–304. doi: 10.1016/S0960-9822(02)00480-3
14. Taipale J, Cooper M, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of smoothened. Nature (2002) 418:892–6. doi: 10.1038/nature00989
15. Xiao X, Tang JJ, Peng C, Wang Y, Fu L, Qiu ZP, et al. Cholesterol modification of smoothened is required for hedgehog signaling. Mol Cell (2017) 66(1):154–62. doi: 10.1016/j.molcel.2017.02.015
16. Singh M, Chaudhry P, Merchant AA. Primary cilia are present on human blood and bone marrow cells and mediate hedgehog signaling. Exp Hematol (2016) 44(12):1181–1187.e2. doi: 10.1016/j.exphem.2016.08.009
17. Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, et al. The tumour-suppressor gene patched encodes a candidate receptor for sonic hedgehog. Nature (1996) 384:129–34. doi: 10.1038/384129a0
18. Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the hedgehog receptor. Nature (1996) 384:176–79. doi: 10.1038/384176a0
19. Denef N, Neubüser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell (2000) 102(4):521–31. doi: 10.1016/S0092-8674(00)00056-8
20. Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science (2007) 317(5836):372–6. doi: 10.1126/science.1139740
21. Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF. Vertebrate smoothened functions at the primary cilium. Nature (2005) 437:1018–02. doi: 10.1038/nature04117
22. Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet (2005) 1(4):e53. doi: 10.1371/journal.pgen.0010053.eor
23. Sasaki H, Nishizaki Y, Hui CC, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of shh signaling. Development (1999) 126(17):3915–24. doi: 10.1242/dev.126.17.3915
24. Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell (2000) 100(4):423–34. doi: 10.1016/S0092-8674(00)80678-9
25. Merchant M, Vajdos FF, Ultsch M, Maun HR, Wendt U, Cannon J, et al. Suppressor of fused regulates gli activity through a dual binding mechanism. Mol Cell Biol (2004) 24(19):8627–41. doi: 10.1128/mcb.24.19.8627-8641.2004
26. Wang C, Pan Y, Wang B. Suppressor of fused and spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development (2010) 137(12). doi: 10.1242/dev.052126
27. Barnfield PC, Zhang X, Thanabalasingham V, Yoshida M, Hui CC. Negative regulation of Gli1 and Gli2 activator function by suppressor of fused through multiple mechanisms. Differentiation (2005) 73(8):397–405. doi: 10.1111/j.14320436.2005.00042.x
28. Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial shh signaling and ectopic activation of the shh pathway. Development (2002) 129(20):4753–61. doi: 10.1242/dev.129.20.4753
29. Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, et al. Transcriptional profiling of the sonic hedgehog response: A critical role for n-myc in proliferation of neuronal precursors. Proc Natl Acad Sci United States America (2003) 100(12):7331–36. doi: 10.1073/pnas.0832317100
30. Bigelow RLH, Chari NS, Undén AB, Spurgers KB, Lee S, Roop DR, et al. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J Biol Chem (2004) 279(2):1197–205. doi: 10.1074/jbc.M310589200
31. Morrow D, Cullen JP, Liu W, Guha S, Sweeney C, Birney YA, et al. Sonic hedgehog induces notch target gene expression in vascular smooth muscle cells via VEGF-a. Arterioscler Thromb Vasc Biol (2009) 29(7):1112–18. doi: 10.1161/ATVBAHA.109.186890
32. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting wnt, notch, and hedgehog pathways. Nat Rev Clin Oncol (2011) 8:97–106. doi: 10.1038/nrclinonc.2010.196
33. Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science (1996) 272(5268):1668–71. doi: 10.1126/science.272.5268.1668
34. Hahn H, Christiansen J, Wicking C, Zaphiropoulos PG, Chidambaram A, Gerrard B, et al. A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J Biol Chem (1996) 271(21):12125–128. doi: 10.1074/jbc.271.21.12125
35. Cohen MM. Nevoid basal cell carcinoma syndrome: Molecular biology and new hypotheses. Int J Oral Maxillofac Surg (1999) 28(3):216–23. doi: 10.1034/j.1399-0020.1999.283280314.x
36. Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature (1998) 391:90–2. doi: 10.1038/34201
37. Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell (2015) 27(3):327–41. doi: 10.1016/J.CCELL.2015.02.001
38. Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell (2015) 27(3):342–53. doi: 10.1016/j.ccell.2015.02.002
39. Chaudhry P, Singh M, Triche TJ, Guzman M, Merchant AA. GLI3 repressor determines hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood (2017) 129(26):3465–75. doi: 10.1182/blood-2016-05-718585
40. Taylor MD, Liu L, Raffel C, Hui C, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet (2002) 31:306–10. doi: 10.1038/ng916
41. Kawahira H, Scheel DW, Smith SB, German MS, Hebrok M. Hedgehog signaling regulates expansion of pancreatic epithelial cells. Dev Biol (2005) 280(1):111–21. doi: 10.1016/j.ydbio.2005.01.008
42. Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, et al. Effects of oncogenic mutations in smoothened and patched can be reversed by cyclopamine. Nature (2000) 406:1005–09. doi: 10.1038/35023008
43. Berman DM, Karhadkar SS, Maitra A, De Oca RM, Gerstenblith MR, Briggs K, et al. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature (2003) 425:846–51. doi: 10.1038/nature01972
44. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol (2007) 17(2):165–72. doi: 10.1016/j.cub.2006.11.033
45. Abe Y, Tanaka N. The hedgehog signaling networks in lung cancer: The mechanisms and roles in tumor progression and implications for cancer therapy. BioMed Res Int (2016) 2016:7969286. doi: 10.1155/2016/7969286
46. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature (2008) 455:406–10. doi: 10.1038/nature07275
47. Tian H, Callahan CA, Dupree KJ, Darbonne WC, Ahn CP, Scales SJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci United States America (2009) 106(11):4254–59. doi: 10.1073/pnas.0813203106
48. Theunissen JW, De Sauvage FJ. Paracrine hedgehog signaling in cancer. Cancer Res (2009) 69(15):6007–10. doi: 10.1158/0008-5472.CAN-09-0756
49. Roberts KJ, Kershner AM, Beachy PA. The stromal niche for epithelial stem cells: A template for regeneration and a brake on malignancy. Cancer Cell (2017) 32(4):404–10. doi: 10.1016/j.ccell.2017.08.007
50. Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, et al. Essential role of stromally induced hedgehog signaling in b-cell malignancies. Nat Med (2007) 13:944–51. doi: 10.1038/nm1614
51. Lindemann RK. Stroma-initiated hedgehog signaling takes center stage in b-cell lymphoma. Cancer Res (2008) 68(4):961–4. doi: 10.1158/0008-5472.CAN-07-5500
52. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci United States America (2007) 104(10):4048–53. doi: 10.1073/pnas.0611682104
53. Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau A-M, et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res (2008) 68(7):2241–50. doi: 10.1158/0008-5472.CAN-07-6350
54. Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog signaling in the maintenance of cancer stem cells. Cancers (2015) 7(3):1554–85. doi: 10.3390/cancers7030851
55. Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature (2009) 458:776–9. doi: 10.1038/nature07737
56. Liang H, Zheng QL, Fang P, Zhang J, Zhang T, Liu W, et al. Targeting the PI3K/AKT pathway via GLI1 inhibition enhanced the drug sensitivity of acute myeloid leukemia cells. Sci Rep (2017) 7:40361. doi: 10.1038/srep40361
57. Wellbrock J, Latuske E, Kohler J, Wagner K, Stamm H, Vettorazzi E, et al. Expression of hedgehog pathway mediator GLI represents a negative prognostic marker in human acute myeloid leukemia and its inhibition exerts antileukemic effects. Clin Cancer Res (2015) 21(10):2388–98. doi: 10.1158/10780432.CCR-14-1059
58. Bhardwaj G, Murdoch B, Wu D, Baker DP, Williams KP, Chadwick K, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol (2001) 2:172–80. doi: 10.1038/84282
59. Gering M, Patient R. Hedgehog signaling is required for adult blood stem cell formation in zebrafish embryos. Dev Cell (2005) 8(3):389–400. doi: 10.1016/j.devcel.2005.01.010
60. Trowbridge JJ, Scott MP, Bhatia M. Hedgehog modulates cell cycle regulators in stem cells to control hematopoietic regeneration. Proc Natl Acad Sci United States America (2006) 103(38):14134–39. doi: 10.1073/pnas.0604568103
61. Merchant A, Joseph G, Wang Q, Brennan S, Matsui W. Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood (2010) 115(12):2391–6. doi: 10.1182/blood-2009-09-241703
62. Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al. Expansion of bcr-Abl-Positive leukemic stem cells is dependent on hedgehog pathway activation. Cancer Cell (2008) 14(3):238–49. doi: 10.1016/j.ccr.2008.08.003
63. Hofmann I, Stover EH, Cullen DE, Mao J, Morgan KJ, Lee BH, et al. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell (2009) 4(6):559–67. doi: 10.1016/j.stem.2009.03.016
64. Gao J, Graves S, Koch U, Liu S, Jankovic V, Buonamici S, et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell (2009) 4(6):548–58. doi: 10.1016/j.stem.2009.03.015
65. Sadarangani A, Pineda G, Lennon KM, Chun HJ, Shih A, Schairer AE, et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J Trans Med (2015) 21:98. doi: 10.1186/s12967-0150453-9
66. Merchant AA, Matsui W. Smoothening the controversial role of hedgehog in hematopoiesis. Cell Stem Cell (2009) 4(6):470–1. doi: 10.1016/j.stem.2009.05.006
67. Joseph C, Quach JM, Walkley CR, Lane SW, Lo Celso C, Purton LE. Deciphering hematopoietic stem cells in their niches: a critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem Cell (2013) 13(5):520–33. doi: 10.1016/j.stem.2013.10.010
68. Malhotra S, Kincade PW. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell (2009) 4(1):27–36. doi: 10.1016/j.stem.2008.12.004
69. Irvine DA, Zhang B, Kinstrie R, Tarafdar A, Morrison H, Campbell VL, et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci Rep (2016) 6:25476. doi: 10.1038/srep25476
70. Ghiaur G, Yegnasubramanian S, Perkins B, Gucwa JL, Gerber JM, Jones RJ. Regulation of human hematopoietic stem cell self-renewal by the microenvironment’s control of retinoic acid signaling. Proc Natl Acad Sci United States America (2013) 6(40):16121–126. doi: 10.1073/pnas.1305937110
71. Alonso S, Su M, Jones JW, Ganguly S, Kane MA, Jones RJ, et al. Human bone marrow niche chemoprotection mediated by cytochrome p450 enzymes. Oncotarget (2015) 6:14905–12. doi: 10.18632/oncotarget.3614
72. Rosti G, Castagnetti F, Gugliotta G, Baccarani M. Tyrosine kinase inhibitors in chronic myeloid leukaemia: which, when, for whom? nature reviews. Clin Oncol (2017) 14(3):141–54. doi: 10.1038/nrclinonc.2016.139
73. Mahon FX, Réa D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre stop imatinib (STIM) trial. Lancet Oncol (2010) 11(11):1029–35. doi: 10.1016/S1470-2045(10)70233-3
74. Goldman JM. How I treat chronic myeloid leukemia in the imatinib era. Blood (2007) 110(8):2828–37. doi: 10.1182/blood-2007-04-038943
75. Hughes TP, Ross DM. Moving treatment-free remission into mainstream clinical practice in CML. Blood (2016) 128(1):17–23. doi: 10.1182/blood-2016-01-694265
76. Zeng X, Zhao H, Li Y, Fan J, Sun Y, Wang S, et al. Targeting hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABLpositive chronic myeloid leukemia. Autophagy (2015) 11(2):355–72. doi: 10.4161/15548627.2014.994368
77. Fiedler W, Vohwinkel G, Krauter J, Vettorazzi E, Loges S, Fuhrmann L, et al. Expression of Gli2, a downstream mediator of the activated hedgehog pathway, represents a negative prognostic marker in AML. Blood (2011) 118(21):3535. doi: 10.1182/blood.v118.21.3535.3535
78. Lau BW, Huh K, Madero-Marroquin R, De Marchi F, Lim Y, Wang Q, et al. Hedgehog/GLI1 activation leads to leukemic transformation of myelodysplastic syndrome in vivo and GLI1 inhibition results in antitumor activity. Oncogene (2019) 38:687–98. doi: 10.1038/s41388-018-0431-9
79. Kobune M, Iyama S, Kikuchi S, Horiguchi H, Sato T, Murase K, et al. Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms. Blood Cancer J (2012) 2:e87. doi: 10.1038/bcj.2012.36
80. Li X, Chen F, Zhu Q, Ding B, Zhong Q, Huang K, et al. Gli-1/PI3K/AKT/NF-kB pathway mediates resistance to radiation and is a target for reversion of responses in refractory acute myeloid leukemia cells. Oncotarget (2016) 7(22):33004–15. doi: 10.18632/oncotarget.8844
81. Osaki M, Oshimura M, Ito H. PI3K-akt pathway: Its functions and alterations in human cancer. Apoptosis 9(6):667–76. doi: 10.1023/B:APPT.0000045801.15585.dd
82. Zhou J, Zhu G, Huang J, Li L, Du Y, Gao Y, et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: A novel potential therapeutic target. Cancer Lett (2016) 370(2):313–23. doi: 10.1016/J.CANLET.2015.11.006
83. Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol (2010) 28:275–80. doi: 10.1038/nbt.1607
84. Fukushima N, Minami Y, Hayakawa F, Kiyoi H, Sadarangani A, Jamieson CH, et al. Treatment with hedgehog inhibitor, PF-04449913, attenuates leukemia-initiation potential in acute myeloid leukemia cells. Blood (2013) 122(21):1649. doi: 10.1182/blood.v122.21.1649.1649
85. Zahreddine HA, Culjkovic-Kraljacic B, Assouline S, Gendron P, Romeo AA, Morris SJ, et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature (2014) 511:90–3. doi: 10.1038/nature13283
86. Casaos J, Gorelick NL, Huq S, Choi J, Xia Y, Serra R, et al. The use of ribavirin as an anticancer therapeutic: Will it go viral? Mol Cancer Ther (2019) 18(7):1185–94. doi: 10.1158/1535-7163.MCT-18-0666
87. Fathi AT. Glasdegib with low-dose cytarabine: A new upfront option for the vulnerable AML patient. Clin Cancer Res (2019) 25(20):6015–17. doi: 10.1158/1078-0432.CCR-19-1986
88. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129(4):424–47. doi: 10.1182/blood-2016-08-733196
89. Lim Y, Gondek L, Li L, Wang Q, Ma H, Chang E, et al. Integration of hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci Trans Med (2015) 7(291):291ra96. doi: 10.1126/scitranslmed.aaa5731
90. Latuske EM, Stamm H, Klokow M, Vohwinkel G, Muschhammer J, Bokemeyer C, et al. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget (2017) 8:29187–201. doi: 10.18632/oncotarget.16304
91. Lin TL, Wang QH, Brown P, Peacock C, Merchant AA, et al. Self-renewal of acute lymphocytic leukemia cells is limited by the hedgehog pathway inhibitors cyclopamine and IPI-926. PLoS One (2010) 5(12):e15262. doi: 10.1371/journal.pone.0015262
92. Dagklis A, Demeyer S, DeBie J, Radaelli E, Pauwels D, Degryse S, et al. Hedgehog pathway activation in T-cell acute lymphoblastic leukemia predicts response to SMO and GLI1 inhibitors. Blood (2016) 128(23):2642–54. doi: 10.1182/blood-2016-03-703454
93. Ghia EM, Rassenti LZ, Neuberg DS, Blanco A, Yousif F, Smith EN, et al. Activation of hedgehog signaling associates with early disease progression in chronic lymphocytic leukemia. Blood (2019) 133(25):2651–63. doi: 10.1182/blood-2018-09-873695
94. Kern D, Regl G, Hofbauer SW, Altenhofer P, Achatz G, Dlugosz A, et al. Hedgehog/GLI and PI3K signaling in the initiation and maintenance of chronic lymphocytic leukemia. Oncogene (2015) 34:5341–51. doi: 10.1038/onc.2014.450
95. Hegde GV, Peterson KJ, Emanuel K, Mittal AK, Joshi AD, Dickinson JD, et al. Hedgehog-induced survival of b-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: a potential new therapeutic target. Mol Cancer Res MCR (2008) 6(12):1928–36. doi: 10.1158/1541-7786.MCR-08-0142
96. Decker S, Zirlik K, Djebatchie L, Hartmann D, Ihorst G, Schmitt-Graeff A, et al. Trisomy 12 and elevated GLI1 and PTCH1 transcript levels are biomarkers for hedgehog-inhibitor responsiveness in CLL. Blood (2012) 119(4):997–1007. doi: 10.1182/blood-2011-06-359075
97. Desch P, Asslaber D, Kern D, Schnidar H, Mangelberger D, Alinger B, et al. Inhibition of GLI, but not smoothened, induces apoptosis in chronic lymphocytic leukemia cells. Oncogene (2010) 29:4885–95. doi: 10.1038/onc.2010.243
98. Martinelli G, Oehler VG, Papayannidis C, Courtney R, Shaik MN, Zhang X, et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet (2015) 2(8):e339–e346. doi: 10.1016/S2352-3026(15)00096-4
99. Minami Y, Minami H, Miyamoto T, Yoshimoto G, Kobayashi Y, Munakata W, et al. Phase I study of glasdegib (PF-04449913), an oral smoothened inhibitor, in Japanese patients with select hematologic malignancies. Cancer Sci (2017) 108:1628–33. doi: 10.1111/cas.13285
100. Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, et al. Phase ib study of glasdegib, a hedgehog pathway inhibitor, in combination with standard chemotherapy in patients with AML or high-risk MDS. Clin Cancer Res (2018) 24(10):2294–303. doi: 10.1158/1078-0432.CCR-17-2824
101. Cortes JE, Douglas Smith B, Wang ES, Merchant A, Oehler VG, Arellano M, et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am J Hematol (2018) 93(11):1301–10. doi: 10.1002/ajh.25238
102. Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia (2019) 33(2):379–89. doi: 10.1038/s41375-018-0312-9
103. Sekeres MA, Schuster MW, Joris M, Krauter J, Maertens JA, Gyan E, et al. A phase 1b study of glasdegib in combination with azacitidine in patients with untreated higher-risk myelodysplastic syndromes, acute myeloid leukemia, and chronic myelomonocytic leukemia. Blood (2019) 134:117. doi: 10.1182/blood-2019-124050
104. Shah NP, Cortes JE, Martinelli G, Smith BD, Clarke E, Copland M, et al. Dasatinib plus smoothened (SMO) inhibitor BMS-833923 in chronic myeloid leukemia (CML) with resistance or suboptimal response to a prior tyrosine kinase inhibitor (TKI): Phase I study CA180323. Blood (2014) 124(21):4539. doi: 10.1182/blood.V124.21.4539.4539
105. Lancet JE, Komrokji RS, Sweet KL, Duong VH, McGraw KL, Zhang L, et al. Phase 2 trial of smoothened (SMO) inhibitor PF-04449913 (PF-04) in refractory myelodysplastic syndromes (MDS). Blood (2016) 128(22):3174. doi: 10.1182/blood.v128.22.3174.3174
106. Sasaki K, Gotlib JR, Mesa RA, Ravandi F, Cortes JE, Kelly PF, et al. A phase 2 study of IPI-926, an oral hedgehog inhibitor, in patients with myelofibrosis. J Clin Oncol (2014) 32(15_suppl):7111. doi: 10.1200/jco.2014.32.15_suppl.7111
107. Couban S, Benevolo G, Donnellan W, Cultrera J, Koschmieder S, Verstovsek S, et al. A phase ib study to assess the efficacy and safety of vismodegib in combination with ruxolitinib in patients with intermediate- or high-risk myelofibrosis. J Hematol Oncol (2018) 11(1):122. doi: 10.1186/s13045-018-0661-x
108. Tibes R, Kosiorek HE, Dueck AC, Sproat L, Palmer J, Slack JL, et al. Phase I/IB study of azacitidine and hedgehog pathway inhibition in myeloid malignancies. Blood (2015) 126(23):1347. doi: 10.1182/blood.V126.23.1347.1347
109. Axelson M, Liu K, Jiang X, He K, Wang J, Zhao H, et al. U.S. food and drug administration approval: Vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin Cancer Res (2013) 19(9):2289–93. doi: 10.1158/1078-0432.CCR-12-1956
110. Casey D, Demko S, Shord S, Zhao H, Chen H, He K, et al. FDA Approval summary: Sonidegib for locally advanced basal cell carcinoma. Clin Cancer Res (2017) 23(10):2377–81. doi: 10.1158/1078-0432.CCR-16-2051
111. Norsworthy KJ, By K, Subramaniam S, Zhuang L, Del Valle PL, Przepiorka D, et al. FDA Approval summary: Glasdegib for newly diagnosed acute myeloid leukemia. Clin Cancer Res (2019) 25(20):6021–5. doi: 10.1158/1078-0432.CCR-19-0365
112. Munchhof MJ, Li Q, Shavnya A, Borzillo GV, Boyden TL, Jones CS, et al. Discovery of PF-04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med Chem Lett (2012) 3(2):106–11. doi: 10.1021/ml2002423
113. Röllig C, Bornhäuser M, Thiede C, Taube F, Kramer M, Mohr B, et al. Long-term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: evaluation of the proposed reporting system. J Clin Oncol (2011) 29(20):2758–65. doi: 10.1200/JCO.2010.32.8500
Keywords: hematological malignancy, glasdegib, smoothened inhibition, Gli3, GLI1, acute myeloid leukemia, hedgehog (Hh), hematopoiesis
Citation: Lemos T and Merchant A (2022) The hedgehog pathway in hematopoiesis and hematological malignancy. Front. Oncol. 12:960943. doi: 10.3389/fonc.2022.960943
Received: 03 June 2022; Accepted: 02 August 2022;
Published: 25 August 2022.
Edited by:
Swami P. Iyer, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Patrick Kevin Reville, University of Texas MD Anderson Cancer Center, United StatesXin Wang, Shandong Provincial Hospital, China
Copyright © 2022 Lemos and Merchant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akil Merchant, YWtpbC5tZXJjaGFudEBjc2hzLm9yZw==