 Ninon Very
Ninon Very Ikram El Yazidi-Belkoura
Ikram El Yazidi-Belkoura
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 17 August 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.960312
This article is part of the Research TopicDrug-Resistance in Cancer Cells: A New Wine in an old BottleView all 15 articles
In cancer cells, metabolic reprogramming is associated with an alteration of the O-GlcNAcylation homeostasis. This post-translational modification (PTM) that attaches O-GlcNAc moiety to intracellular proteins is dynamically and finely regulated by the O-GlcNAc Transferase (OGT) and the O-GlcNAcase (OGA). It is now established that O-GlcNAcylation participates in many features of cancer cells including a high rate of cell growth, invasion, and metastasis but little is known about its impact on the response to therapies. The purpose of this review is to highlight the role of O-GlcNAc protein modification in cancer resistance to therapies. We summarize the current knowledge about the crosstalk between O-GlcNAcylation and molecular mechanisms underlying tumor sensitivity/resistance to targeted therapies, chemotherapies, immunotherapy, and radiotherapy. We also discuss potential benefits and strategies of targeting O-GlcNAcylation to overcome cancer resistance.
One of the main hallmarks of cancer cells is energy metabolism reprogramming to support continuous proliferation (1). This mechanism, known as the Warburg effect, shifts energy production from the oxidative phosphorylation that operates in normal cells to the faster aerobic glycolysis. To compensate for the low energy yield of aerobic glycolysis, tumor cells overexpress Glucose Transporters (GLUTs) (2) and increase their glucose uptake by a factor of 10. In addition to glucose, cancer cells excessively consume glutamine as another source of carbon and nitrogen to produce nucleic acids, lipids, and proteins (3). The conversion of glucose into fructose-6-phosphate is a common step in both glycolysis and the hexosamine biosynthetic pathways (HBP), which metabolizes 2 to 3% of total glucose entering the cell (4).
The final product of the HBP is the nucleotide-sugar uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) that requires building blocks produced by glucose, amino acids (mainly glutamine but also glucogenic and ketogenic amino acids), fatty acids (acetyl-coenzyme A, acetyl-CoA) and nucleotides (uridine triphosphate, UTP) metabolisms. UDP-GlcNAc is therefore considered a cellular nutritional sensor. UDP-GlcNAc serves as a substrate of OGT to O-GlcNAcylate serine (Ser) and threonine (Thr) residues of cytoplasmic, nuclear, and mitochondrial proteins; the OGA removes it. The activity of both OGT and OGA makes this ubiquitous intracellular post-translational modification highly dynamic as has been widely described (5–7). A fine “Yin-Yang” occupancy competition mechanism between O-GlcNAcylation and phosphorylation on the same or adjacent Ser/Thr residues regulates protein’s interaction, stability, subcellular localization, and enzymatic activity of their common target proteins (8). Elevated nutrients uptake and metabolism are correlated with increased HBP flow, UDP-GlcNAc level, and global protein O-GlcNAcylation in a wide variety of cancers including chronic lymphocytic leukemia (CLL), breast, lung, liver, prostate, endometrium, pancreas, colon, larynx and bladder (9–17). This aberrant hyper-O-GlcNAcylation is also the result of an alteration in the expression and activity of HBP enzymes and OGT (18–21). Interestingly, decreased OGA and/or increased OGT and O-GlcNAcylation levels are associated with poor cancer grade progression (10, 14, 16, 20, 22, 23).
For the past two decades, a growing body of evidence has demonstrated the crucial role of abnormal O-GlcNAcylation of many oncogenes, tumor suppressors, and signaling actors on the growth, adhesion, migration, and invasion of cancer cells. However, while the role of O-GlcNAcylation in carcinogenesis and tumor progression remains of high interest in cancer research (24, 25), the impact of this glycosylation in the response of cancer to therapies is poorly investigated. The donor substrate for O-GlcNAcylation UDP-GlcNAc can also be epimerized to uridine diphosphate N-acetylgalactosamine (UDP-GalNAc) or modified into cytidine monophosphate-N-acetylneuraminic acid (CMP-Neu5Ac). UDP-GlcNAc, UDP-GalNAc, and CMP-Neu5Ac are nucleotide-sugar substrates for the complex glycosylation of membrane or secretory proteins. In addition to their role in the development and progression of cancer, complex glycosylation alterations have also been correlated with resistance to anti-cancer therapies by interfering with metabolism and modulating tumor cell aggressiveness (26, 27).
In this review, we summarize recent evidence highlighting that cancer therapeutics affect cellular O-GlcNAcylation homeostasis and, reciprocally, that O-GlcNAcylation modulates the response of cancer cells to therapies. Finally, we discuss the benefits of targeting O-GlcNAcylation as a novel promising therapeutic strategy for cancer.
Multiple lines of proof demonstrate that cellular stress (including glucose deprivation, chemotherapeutic and DNA-damaging agents) dramatically increases global O-GlcNAcylation of protein and that the sugar might be protective (28–30).
Notably, chemotherapeutic drugs (i.e. doxorubicin (DOX), 5-fluorouracil (5-FU), camptothecin (CPT), and cisplatin induce an accumulation of intracellular UDP-GlcNAc levels and global protein O-GlcNAcylation in different cancer cell lines (30–33). The increased flux through the HBP is at least partially mediated by the induction of Glutamine : Fructose-6-phosphate Amidotransferase (GFAT) through the AKT/X-Box Binding Protein 1 (XBP1) transcription factor pathway in an unfolded protein response (UPR)-independent manner (30) and by activation of the direct transcriptional activator Forkhead box A2 (FOXA2) (31). Interestingly, cellular UDP-GlcNAc levels are increased in cisplatin-sensitive brain tumor cells (32). Wang et al. (2021) demonstrated that cisplatin enhances UDP-GlcNAc production and global O-GlcNAcylation levels in vitro and in vivo by OGT, GFAT1 activation, and OGA inhibition (33). Conversely, we recently demonstrated that 5-FU decreases intracellular O-GlcNAcylation in vitro by reducing OGT at both protein and transcriptional levels but also in vivo most likely by reducing OGT activity (34). Owing to the fact that OGT enzymatic activity is inhibited by elevated UDP, UTP, and UDP-GlcNAc (35), we suggest that 5-FU metabolites may inhibit OGT by producing fluorinated derivates or uridine compounds (36). Overall, it seems well defined that cellular O-GlcNAcylation is increased in response to anti-cancer therapies-induced stress but the molecular structure of some therapeutic agents could also interfere with UDP-GlcNAc metabolism and OGT activity. UDP-GlcNAc could thus be a potential candidate for monitoring patient response to some anti-cancer therapies.
O-GlcNAcylation is considered a DNA damage-induced PTM since OGT relocates to the sites of damaged DNA caused by several agents (i.e. ionizing radiation, etoposide, methyl methanesulfonate (MMS), cisplatin, mitomycin C (MMC)) and catalyzes O-GlcNAcylation of several proteins of the repair machinery (37). By interfering with the other post-translational modifications of histones defining the “histone code”, it is suggested that O-GlcNAcylation regulates chromatin compaction and gene transcription in response to stress (38). Histone O-GlcNAcylation and DNA condensation are concomitantly increased under heat shock-induced stress (39). Treatment with AUY922 and ganetespib HSP90 inhibitors induces O-GlcNAcylation of core histones (Ser122 of H2A, Thr45 of H3, and Thr30 of H4) in bladder carcinoma cells (40). This study outlines the association between histone PTMs and proteomic changes in response to HSP90 inhibitor treatment in bladder carcinoma cells. Bibliographic analysis of therapies’ impact on O-GlcNAcylation shows a steady increase in the number of papers demonstrating that global O-GlcNAcylation is moderately increased by chemotherapeutic agents in sensitive cells but more significantly in resistant ones. To have a more precise vision, we will detail below the link between O-GlcNAcylation and anticancer response by type of therapy.
Breast cancer is the most common cancer in women and the leading cause of cancer-related death. Among different subtypes, luminal breast cancers expressing the Estrogen Receptor alpha (ERα) represent approximately 80% of cases (41). ERα plays a crucial role in cancer initiation and progression by binding to estrogen response elements sequence in promotor of target genes upon association with estradiol, its natural ligand, and other transcription factors and activation of downstream signaling pathways such as Phosphatidylinositol 3-Kinase (PI3K)/AKT and Mitogen-Activated Protein Kinase (MAPK) (42). Hormone therapies are the mainstay of the treatment of hormone receptor positive breast tumors. Since its approval by the U.S. Food and Drug Administration (FDA) in 1977, tamoxifen is one of the most commonly used hormone therapy and acts as a partial antagonist of ERα. However, many breast tumors exhibit de novo or acquired resistance to hormone therapies and some potential mechanisms include deregulation of endoplasmic reticulum (ER) pathway components, modification of cell cycle, and survival regulators or activation of escape signaling pathways (43). Thus, low expression of ERα is generally associated with resistance and poor prognosis (44).
Since cancer initiation and progression are fueled by metabolic reprogramming, the detection of metabolic biomarkers is an emerging approach for the prediction of cancer recurrence (45). In a retrospective study, Kuo et al. (2021) recently revealed that O-GlcNAcylation and Pyruvate Kinase M2 (PKM2) served as potentially independent prognostic markers in luminal breast cancers treated with endocrine treatment including tamoxifen. High levels of O-GlcNAcylation or PKM2 are positively associated with a high risk of cancer recurrence and poor long-term disease-free survival (46). PKM2 is a PK isoform preferentially expressed in cancer (47) for which hyper-O-GlcNAcylation is observed in breast tumors. O-GlcNAcylation of Ser362, Thr365, Thr405, and Ser406 causes nuclear translocation of PKM2 leading to up-regulation of GLUT1 and Lactate Dehydrogenase A (LDHA) glycolysis components. Thus, the glycosylation by targeting PKM2 would promote the Warburg effect and breast tumor growth (21, 48) (Figure 1).
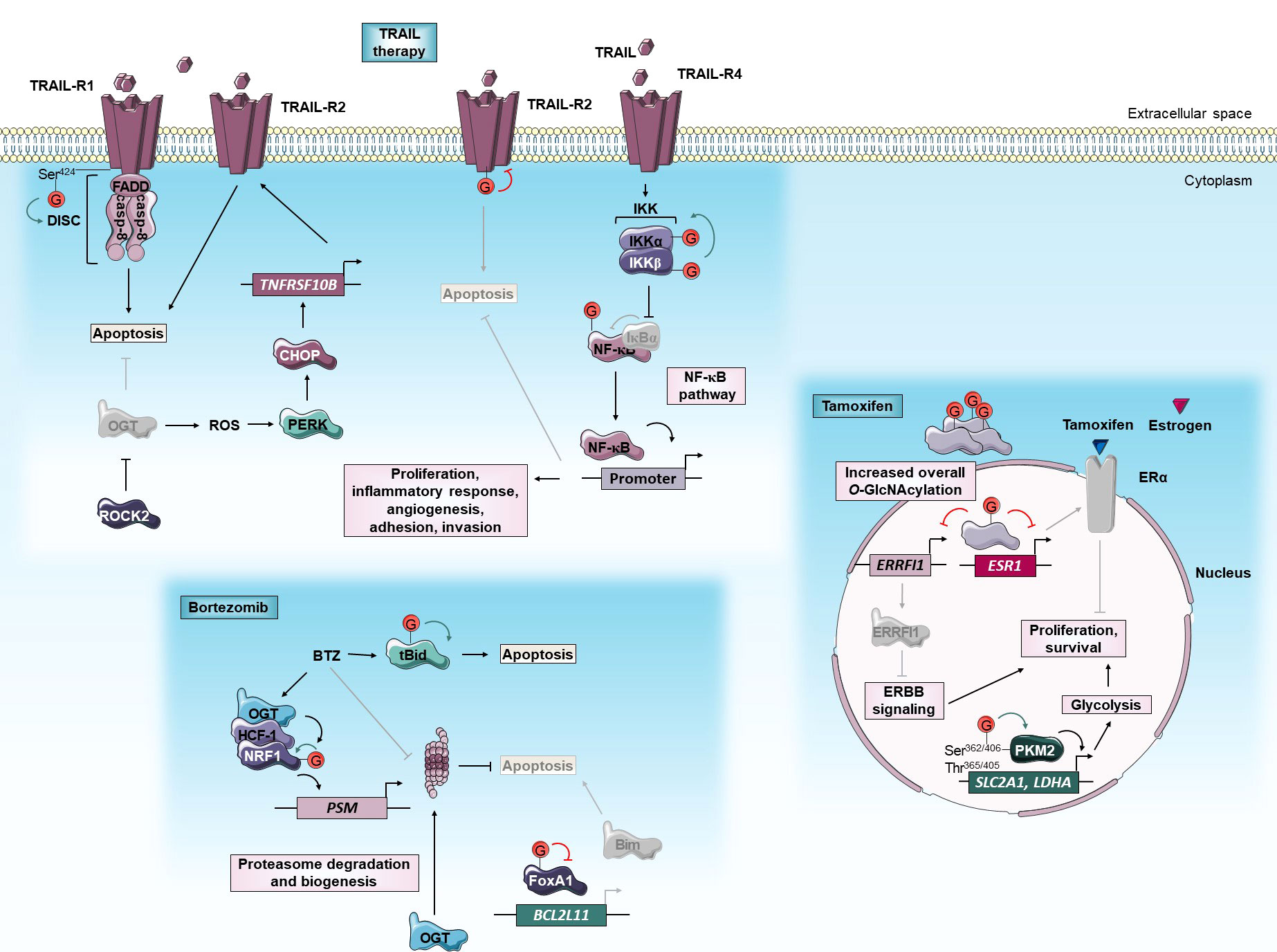
Figure 1 O-GlcNAcylation and anti-cancer targeted therapies. O-GlcNAcylation modulates sensitivity of cancer cells to TRAIL, bortezomib and tamoxifen targeted therapies. Green and red arrows indicate respectively activation and inhibition of protein targeted by O-GlcNAcylation. The gray color indicates that the protein is inactivated/absent or that the cellular mechanism is not taking place.BCL2L11, Bcl-2-Like protein 11; BTZ, bortezomib; Casp, caspase; CHOP, C/EBP Homologous Protein; DISC, death-inducing signaling complex; ERα, Estrogen receptor α; ERRFI1, ERBB Receptor Feedback Inhibitor 1; ESR1, Estrogen Receptor 1; FADD, Fas-Associated protein with Death Domain; FoxA1, Forkhead Box A1; HCF-1, Host Cell Factor-1; IKK, Inhibitory κB Kinase; LDHA, Lactate Dehydrogenase A; NF-κB, Nuclear Factor-Kappa B; NRF1, Nuclear Respiratory Factor 1; OGT, O-GlcNAc Transferase; PERK, Protein kinase RNA-like Kinase; PKM2, Pyruvate Kinase M2; PSM, Proteasome subunit; ROCK2, Rho-associated Coiled-coil forming protein Kinase 2; ROS, reactive oxygen species; Ser, serine; SLC2A1, Solute Carrier family 2 member 1; tBid, truncated BH3 interacting domain death agonist; TNFRSF10B, Tumor Necrosis Factor (TNF) Receptor Superfamily member 10B; Thr, threonine; TRAIL, TNF-Related Apoptosis Inducing Ligand; TRAIL-R, TRAIL Receptor.
Beyond being a recurrence biomarker of endocrine-treated breast tumors, O-GlcNAcylation could also be an interesting therapeutic target to sensitize anti-estrogen-resistant breast tumors. First, O-GlcNAcylation promotes resistance of MCF-7 HR+ breast cancer cells to tamoxifen by reducing ERα mRNA levels. In these cells, inhibition of OGT by siRNA or OSMI-1 specific inhibitor potentiates the cytotoxic effect of the drug (49, 50). Interestingly, tamoxifen-resistant cells are also dependent on high OGT activity. The OGT inhibition by OSMI-1 treatment sensitizes tamoxifen-resistant MCF-7 more than parental ones by inducing ERBB Receptor Feedback Inhibitor 1 (ERRFI1) expression. ERRFI1 is a tumor-suppressor that inhibits ErbB receptor tyrosine kinase-signaling which is a known driver of tamoxifen resistance (51) (Figure 1). Thus, high expression of ERRFI1 is associated with extended survival in patients with tamoxifen-treated ERα-positive breast tumors (52). Altogether, therapeutic approach leading to O-GlcNAcylation inhibition could improve the sensitivity of ERα+ breast cancer to tamoxifen.
The proteasome is a large intracellular protease complex composed of a 20S proteolytic core and two 19S regulatory particles. Proteasome activity is essential for the control of various cellular processes such as cell cycle, DNA repair, signal transduction, and protein quality control. Bortezomib (BTZ) is a peptide boronic acid that reversibly acts on the chymotrypsin-like activity of the 20S particle. BTZ has become a target of choice for the treatment of cancers that present high proteasome activity (53). Since its FDA authorization in 2003, it is used for the treatment of relapsed/refractory mantle cell lymphoma (MCL) and multiple myeloma and is further undergoing clinical evaluation in solid tumors including breast, colorectal, ovarian, pancreatic, prostate, and squamous cell carcinomas. However, innate and acquired resistance to BTZ are frequently observed. Some BTZ resistance mechanisms include mutations or up-regulation of proteasome subunits, alteration of stress response, and cell survival pathways, or multi-drug resistance (54).
Interestingly, OGT is included in the list of BTZ sensitizers in myeloma cells (55). Several pieces of evidence demonstrate that O-GlcNAcylation up-regulates the bounce-back response that restores proteasome activity by transcriptional activation of proteasome subunit genes. There is a positive correlation between OGT and proteasome subunits expression in clinical cancer samples including breast invasive carcinoma and colorectal adenocarcinoma. Sekine et al. (2018) firstly demonstrated that O-GlcNAcylation is critical for the maintenance of proteasome activity by regulating Nuclear Respiratory Factor 1 (NRF1) transcription factor through its interaction with Host Cell Factor-1 (HCF-1). In response to BTZ, OGT targets and stabilizes NRF1. In turn, HCF-1 promotes the binding of NRF1 at promoter regions of proteasome subunit genes. OGT knock-down sensitizes MDA-MB-231 breast and NCI-460 pancreatic cancer cells to BTZ in vitro and in xenograft in a mouse model by blocking NRF1-dependent proteasome bounce-back response (56). It was also demonstrated that O-GlcNAcylation promotes the bounce-back response in an NRF1-independent-manner by promoting the turnover of the proteasome. Under BTZ-induced proteasome inhibition, O-GlcNAcylation enhances both degradation and biogenesis of proteasome allowing the recovery of its activity (Figure 1). The underlying mechanism is not yet clarified but the translation or the stability of proteasome subunits may be improved by O-GlcNAcylation since neither the subunit mRNA levels of the proteasome nor its assembly pathway is affected by OGT inhibition (57).
Other studies reveal that O-GlcNAcylation could regulate the BTZ-mediated activation of extrinsic and intrinsic apoptosis triggered by the accumulation of pro-apoptotic proteins including BH3 interacting domain death agonist (Bid) and Bim proteins (58). Increased O-GlcNAcylation of FOXA1, a forkhead transcription factor that activates Bim expression, is involved in breast cancer resistance to BTZ. There is an association between the elevation of O-GlcNAcylation content and resistance to BTZ in mammary cancer samples. BTZ dynamically induces an increase of global O-GlcNAcylation levels in intrinsic and extrinsic BTZ resistant breast cell lines but not in sensitive ones. In BTZ resistant cells, O-GlcNAcylation targets and reduces the stability of FOXA1 allowing Bim attenuation. OGT silencing or inhibition with L01 small molecule sensitizes resistant cells to BTZ by increasing FOXA1 and Bim protein levels (59). In contrast, Luanpitpong et al. (2019) demonstrated that O-GlcNAcylation of truncated Bid (tBid) pro-apoptotic protein could sensitize MCL to BTZ. The use of pharmacological inhibitors of OGT (alloxan) and OGA (PUGNAc, Thiamet-G, ketoconazole (KCZ)) respectively abrogate and sensitize to BTZ-induced apoptosis in MCL cell lines. O-GlcNAcylation targets tBid and interferes with its ubiquitination and its subsequent degradation by the 26S proteasome (60). Glycosylation-mediated stabilization and accumulation of tBid intensify the apoptosis signal induced by BTZ (Figure 1). Thus, OGA inhibition by KCZ treatment increased the sensitivity of patient-derived primary cells and BTZ-resistant MCL cells (61).
Together, these data indicate that, in a cancer type-dependent manner, activation or inhibition of O-GlcNAcylation in combination with BTZ proteasome inhibitor is a promising clinical strategy against resistance to therapy.
Upon Tumor necrosis factor (TNF)-Related Apoptosis Inducing Ligand (TRAIL) cytokine binding, TRAIL-Receptor 1 and 2 (TRAIL-R1 and TRAIL-R2 also known as respectively Death Receptor 4 and 5 (DR4 and DR5)) trigger the assembly of death-inducing signaling complex (DISC). The latter leads to apoptosis by activation of initiator caspases-8/10 and downstream effector caspases such as caspase-3. Unlike TNF-α and FAS extrinsic apoptosis-inducing ligands, TRAIL has an attractive ability to selectively induce apoptosis in tumor cells while sparing normal cells (62). Therefore, several clinical trials are currently underway to assess the efficacy of TRAIL therapy in cancer. Circularly permuted TRAIL (CPT) is tested in myeloma and some antibodies have also been developed: an anti-TRAIL-R (dulanermin) against lymphoma, colorectal and lung cancers, an anti-TRAIL-R1 (mapatumumab) or an anti-TRAIL-R2 (anti-DR5) (tigatuzumab) against several solid tumors (63). However, such clinical trials have failed to achieve a beneficial anticancer activity because a large number of cancers develop intrinsic and acquired resistance to TRAIL. Inhibition of apoptosis can be due to impaired TRAIL-R signaling, reduced caspases function, or disrupted balance between pro-apoptotic and anti-apoptotic proteins (64).
The death domain (DD) of TRAIL-R1 regulates both apoptosis and necrosis upon TRAIL ligand binding. In response to TRAIL, O-GlcNAcylation at the Ser424 DD residue of TRAIL-R1 facilitates receptors clustering within lipid rafts, DISC assembly, and induction of cell death (Figure 1). Several TRAIL-resistant cancer cell lines display a TRAIL-R1-Ser424 mutation and therefore defect of O-GlcNAcylation. The mutation at Ser424 residue could then be a potential genetic diagnostic marker of patients with cancer to predict TRAIL therapy response (65). O-GlcNAcylation of TRAIL-R2 also plays an important role in pancreatic cancer TRAIL resistance (66). Gain- and loss-of-function of OGT respectively render TRAIL-sensitive pancreatic cells more resistant to tigatuzumab TRAIL-R2 agonist and promote tigatuzumab-induced apoptosis in TRAIL-resistant cells in vitro and in a mouse model. Since O-GlcNAcylation of TRAIL-R2 regulates its trimerization and the activation of apoptotic signals, inhibiting O-GlcNAcylation could increase the effectiveness of TRAIL therapy (Figure 1). In addition, the overall level of global O-GlcNAcylation or O-GlcNAcylated TRAIL-R2 could be a biomarker of pancreatic cancer sensitivity to TRAIL therapy. In addition to activating apoptosis, TRAIL has also been described to induce the Nuclear Factor-Kappa (NF-κB) survival pathway that may promote drug resistance (67). Several studies have already revealed that O-GlcNAcylation regulates this pathway (68–72). Recently, Lee et al. (2021) demonstrate that a combination of TRAIL and OSMI-1 (OGT inhibitor) enhances TRAIL-induced apoptosis in colon cancer cells and xenograft tumors by lowering the activity of Inhibitory κB Kinase β (IKKβ) and inhibition of downstream NF-κB pro-survival signaling (73) (Figure 1). Additionally, OGT inhibition by OSMI-1 improves TRAIL-induced apoptosis by a parallel mechanism involving ER stress response. Hypo-O-GlcNAcylation induces the accumulation of reactive oxygen species (ROS) that leads to ER stress and activation of UPR signaling pathways including Protein Kinase RNA (PKR)-like kinase (PERK). Activation of PERK enhances expression of the pro-apoptotic protein C/EBP Homologous Protein (CHOP) that, in turn, up-regulates expression of TRAIL-R2 leading to TRAIL sensitization (73) (Figure 1). Concordantly, O-GlcNAcylation prevents activation of CHOP (74). Together, these studies suggest that a combination of TRAIL and OGT inhibition may induce synergistic effects and provide a promising therapeutic strategy for the treatment of various cancers.
Finally, a recent study identifies the GTPase RhoA effector Rho-associated Coiled-coil forming protein Kinase 2 (ROCK2) as a key regulator of O-GlcNAcylation and TRAIL sensitivity in osteosarcoma (OS). ROCK2 inhibits TRAIL-mediated apoptosis in OS cells by affecting OGT ubiquitination and degradation, and subsequently increasing protein O-GlcNAcylation levels (Figure 1). ROCK2 overexpression is an independent predictor of poor prognosis in OS patients and knock-down of ROCK2 causes a reduction in O-GlcNAcylation level and increases the OS cell sensitivity to TRAIL. Based on these data, the authors suggest ROCK2 as a potential biomarker for OS diagnostic and therapeutic tools (75).
5-FU is a fluorinated uracil analog that acts as an antimetabolite to disrupt nucleic acid synthesis and repair in highly proliferating cancer cells. It was approved in 1962 by FDA and is widely used in the treatment of solid tumors including colorectal, breast, anus, esophagus, pancreas, stomach, head, neck, and ovary cancers. The major cytotoxic activity of 5-FU consists of its conversion by Thymidine Kinase (TK) and Thymidine Phosphorylase (TP) into the active metabolite 5-fluorodeoxyuridine monophosphate (FdUMP) which sequesters and inhibits Thymidylate Synthase (TS), a key enzyme involved in the de novo synthesis of deoxythymidine monophosphate (dTMP). Cancer resistance to 5-FU can be caused by an alteration in metabolism, TS target level, recognition or repair of DNA damages, and/or inhibition of apoptosis (76).
Several studies have already correlated OGT expression with 5-FU sensitivity. Notably, Temmink et al. (2010) showed that H630 colon cancer cells resistant to trifluorothymidine (TFT), a fluorinated thymidine analog that is part of TAS-102 chemotherapy and shares the anabolic pathway of TS inhibition with 5-FU, underexpress OGT, Solute Carrier family 29A (SLC29A), and TK (77). On the contrary, a transcriptomic study on human NCI-60 tumors showed that OGT expression is negatively correlated with FdUMP sensitivity, no correlation with 5-FU sensitivity was established (78). OGT was also identified in a cluster of co-expressed genes associated with 5-FU resistance in colorectal cancer (CRC) (79). Moreover, the 5-FU pathway actors SLC29A1 (80), TP (10), TK (81, 82), and TS (34) are direct targets for O-GlcNAcylation (Figure 2). Nevertheless, modified sites and roles of this PTM on these proteins as well as their impacts on 5-FU sensitivity are largely not elucidated yet. Very recently, we demonstrated that TS is stabilized by O-GlcNAcylation at Thr251 and Thr306 residues. OGT knock-down decreases 5-FU-induced apoptosis by enhancing TS proteasomal degradation and, by reducing TS level and enzyme activity in HT-29 cell line but not in 5-FU resistant HT-29 counterpart (34) (Figure 2). The latter exhibits TS gene amplification and TS overexpression which is a current biomarker of 5-FU resistance in CRC (83). Our data highlight the importance to distinguish TS gene overexpression and the corresponding enzyme post-translational stabilization. We have shown that the regulation of 5-FU response by O-GlcNAcylation is finely tuned and depends on a proper quantity of TS proteins. In a CRC mouse model, the combination of 5-FU with the OGA inhibitor Thiamet-G had a synergistic inhibitory effect on tumor grade and progression (34). As the O-GlcNAcylation homeostasis-TS axis mediates the response to 5-FU, we propose to combine an OGA inhibitor with 5-FU-based therapies to enhance CRC patient response to 5-FU.
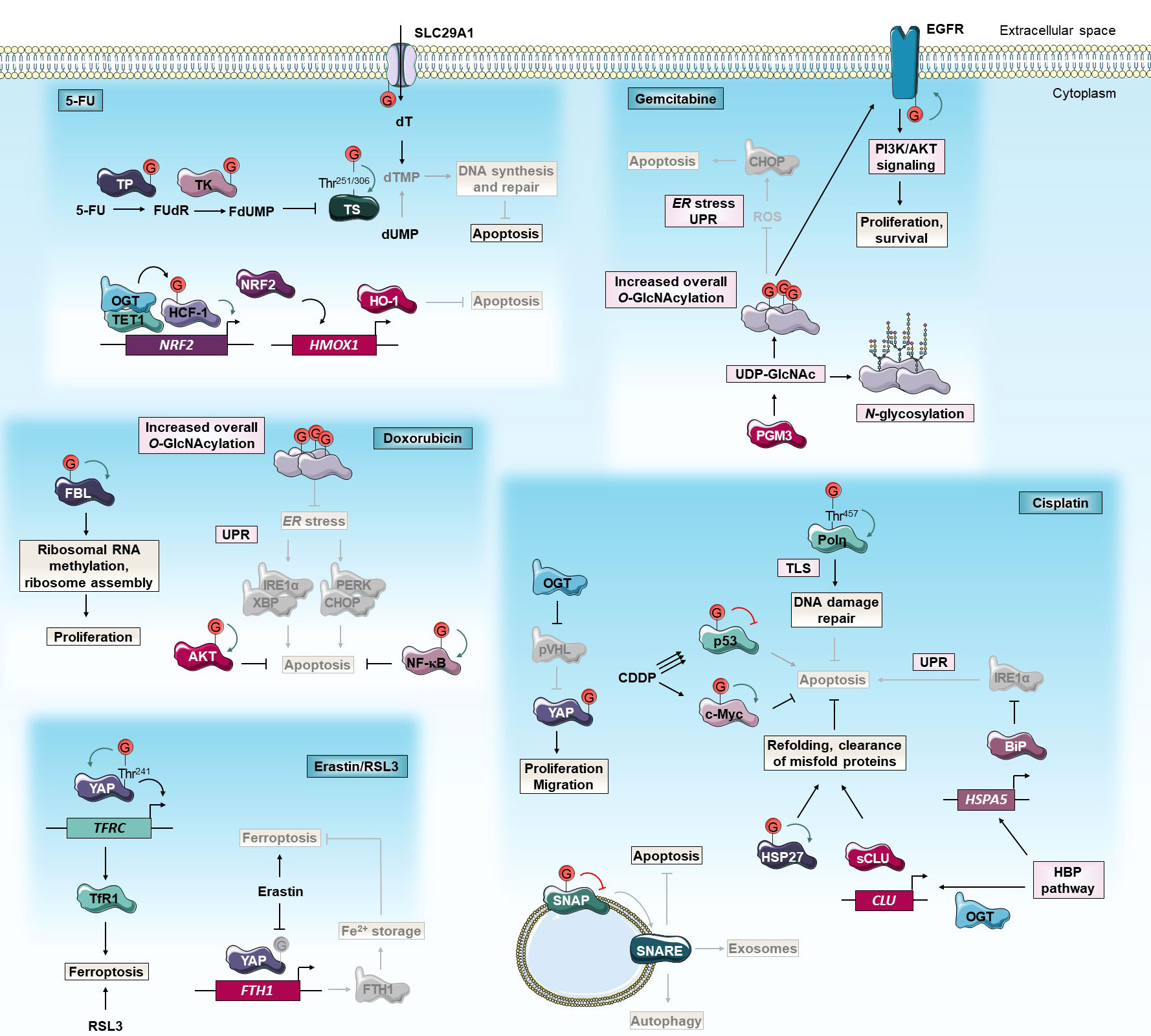
Figure 2 O-GlcNAcylation and anti-cancer chemotherapies. O-GlcNAcylation modulates cancer cell response to 5-FU, cisplatin, gemcitabine, doxorubicin and erastin/RSL3 chemotherapies. Green and red arrows indicate respectively activation and inhibition of protein targeted by O-GlcNAcylation. The gray color indicates that the protein is inactivated/absent or that the cellular mechanism is not taking place.5-FU, 5-fluorouracil; BiP, Binding immunoglobulin Protein; CDDP, cis-diaminedichloroplatinium(II); CHOP, C/EBP Homologous Protein; CLU, Clusterin; dUMP, deoxyuridine monophosphate; dT, deoxythymidine; dTMP, dT monophosphate; EGFR, Epidermal Growth Factor Receptor; ER, endoplasmic reticulum; FBL, Fibrillarin; FTH1, Ferritin Heavy chain 1; FdUMP, fluorodUMP; FUdR, fluorodeoxyuridine; HBP, Hexosamine Biosynthetic Pathway; HCF-1, Host Cell Factor-1; HMOX1, Heme Oxygenase-1; HO-1, Heme Oxygenase-1; HSP27, Heat Shock Protein 27 kD; IRE1α, Inositol-Requiring Enzyme 1α; NF-κB, Nuclear Factor-Kappa B; NRF2, Nuclear factor erythroid-2-Related Factor 2; OGT, O-GlcNAc Transferase; PERK, Protein kinase RNA-like Kinase; PGM3, Phosphoacetylglucosamine Mutase 3; PI3K, Phosphatidylinositol 3-Kinase; Polη, DNA Polymerase η; pVHL, von Hippel-Lindau protein; ROS, reactive oxygen species; RSL3, Ras-selective lethal small molecule 3; sCLU, secretory CLU; SLC29A1, Solute Carrier family 29 member 1; SNAP, Synaptosomal Associated Protein; SNARE, Soluble N-ethylmaleimide-sensitive-factor Attachment protein Receptor; TET1, Tet methylcytosine dioxygenase 1; TfR1, Transferrin Receptor 1; TFRC, Transferrin Receptor; Thr, threonine; TK, Thymidine Kinase; TLS, translesion DNA synthesis; TP, Thymidine Phosphorylase; TS, Thymidylate Synthase; UDP-GlcNAc, uridine disphosphate N-acetylglucosamine; UPR, unfolded protein response; XBP, AKT/X-Box Binding Protein; YAP, Yes-Associated Protein.
Another study showed that, in comparison to parental cells, 5-FU resistant SNUC5 colon cancer cells undergo oxidative stress due to 5-FU-induced accumulation of ROS. These cells overexpressed Tet methylcytosine dioxygenase 1 (TET1) and OGT. Both enzymes interact together at the promoter of Nuclear factor erythroid-2-Related Factor 2 (NRF2), a transcription factor that regulates the expression of antioxidant enzymes such as Heme Oxygenase-1 (HO-1). OGT indirectly activates the transcription of NRF2 (Figure 2). Since oxaliplatin-resistant SNUC5 cells and cisplatin-resistant ovarian cancer cells express a high level of NRF2, inhibition of O-GlcNAcylation may potentiate the 5-FU-induced oxidative stress in these cells by decreasing NRF2 and HO-1 expression (84, 85). Chen et al. (2017) revealed an opposite functional connection between O-GlcNAcylation and NRF2 antioxidant pathway. In fact, Kelch-like ECH-Associated Protein 1 (KEAP1), the primary negative regulator of NRF2, O-GlcNAcylation at Ser104 is required for proteasomal degradation of NRF2. Gene expression signatures of low OGT activity and high NRF2 activation are strongly correlated in several breast tumor datasets and OGT inhibition induces NRF2 and subsequently reduces cysteine-deprivation induced-oxidative stress in breast MDA-MB-231 cells (86). Additionally, the inhibitory effect of O-GlcNAcylation on oxidative stress was confirmed as GFAT inhibition with 6-diazo-5-oxo-norleucine (DON) sensitizes cancer cells to oxidative stress-induced apoptosis (83). Of note, oxidative stress-resistant Phosphatase and tensin homolog (Pten) KO-mice tumors show increased GFAT2 and O-GlcNAcylation levels compared to wild-type (87).
Cisplatin is a platinum-based alkylating agent that, mechanistically, reacts with DNA bases to cross-link adjacent purines, blocks DNA replication and induce apoptosis in rapidly dividing cells. It was firstly approved by FDA in 1978 for the treatment of testicular, ovarian, and bladder cancers. Nowadays, it is an important component of combination therapy for a wide range of solid tumors. The resistance to cisplatin can be caused by decreased drug accumulation, increased drug detoxification, increased DNA repair or DNA damage tolerance, and cell death escape (68). We describe below how O-GlcNAcylation interferes with the three last resistance mechanisms.
Autophagy is a self-protection mechanism that occurs as an emergency response and consists of phagocytosis of cytoplasmic proteins or damaged organelles into autophagosomes to meet the basic metabolic needs of cells. This cellular defensive pathway has a pro-survival role but can induce cell death under certain conditions. Autophagy plays a crucial role in the response of cancer cells to chemotherapeutic drugs including cisplatin (69, 70). Zhou et al. (2018) showed that OGT expression is significantly lower in cisplatin chemoresistant ovarian cancerous tissues compared to sensitive tissues. Additionally, OGT knock-down increases resistance to cisplatin of ovarian cancer cells and xenografted in mice (71). Reduced OGT expression regulates Synaptosomal Associated Protein 29 (SNAP 29) component of the Soluble N-ethylmaleimide-sensitive-factor Attachment protein Receptor (SNARE) vesicle formation machinery (72) which increases pro-survival autophagic flux that correlates with decreased apoptosis and increased cisplatin resistance (71) (Figure 2). Conversely, Wang et al. (2018) showed that siRNA invalidation of OGT increases autophagic flux and cisplatin-induced apoptosis in T24 and UMUC-3 bladder cancer cells (17, 88) (Figure 2). In addition to the degradation of long-lived macromolecules, the autophagy-lysosome system has broader functions and is involved in the selective degradation of other intracellular components including some oncogenes. The transcriptional co-activator Yes-Associated Protein (YAP) is a major effector of the Hippo pathway and its overactivation promotes cancer cell growth and chemoresistance. O-GlcNAcylation can block the von Hippel-Lindau protein (pVHL)-mediated lysosomal degradation of YAP and thus enhance cellular proliferation, migration, and cisplatin resistance in lung adenocarcinoma cells (89) (Figure 2). SNARE proteins also regulate the fusion of exosome-containing multivesicular bodies (MVB) to the plasma membrane and their secretion into the extracellular environment. Compared to normal cells, cancer cells secrete an increasing number of exosomes that can facilitate the efflux of intracellular chemotherapeutic drugs, hence promoting chemoresistance (90). In A2780 and SKOV3 ovarian cancer cells, down-regulation of OGT reduces O-GlcNAcylation of SNAP-23 promoting the formation of SNARE complex and exosomes. Thus, this O-GlcNAcylation-mediated mechanism causes chemoresistance by increasing the exosome-mediated efflux of intracellular cisplatin (91) (Figure 2).
The tumor suppressor p53 plays a crucial role in cell biology and is considered the “guardian of the genome”. In response to DNA damages, caused by chemotherapeutic agents such as cisplatin, p53 is stabilized and activates downstream pathways to arrest the cell cycle, repair the damaged DNA or induce apoptosis. Loss of function of p53 is a common feature in more than 50% of human cancers (92) and drives tumor growth and chemotherapy resistance. Several studies reveal that O-GlcNAcylation could regulate cisplatin resistance in a p53-dependent manner. Luanpitpong et al. (2017) showed that, depending on the cellular context and the level of cisplatin-activation of p53, increased O-GlcNAcylation could lead to cisplatin resistance in lung cancer cells by distinct molecular mechanisms. Indeed, in NCI-H292 cells, cisplatin weakly induces p53 activation and O-GlcNAcylation stabilizes the c-Myc oncoprotein to promote cell survival. In parallel, in NCI-H460 cells in which cisplatin induces strong activation of p53, O-GlcNAcylation of p53 increases its ubiquitination and its proteasomal degradation resulting in an enhanced oncogenic and anti-apoptotic phenotype (19) (Figure 2). Of note, literature shows some discrepancy since previous studies showed that O-GlcNAcylation stabilizes p53 (93) as in ovarian cancer cells (94). In these cells, the combination of OGA inhibitor Thiamet-G with cisplatin increases the drug sensitivity by inducing cell cycle arrest in wild-type p53 and to a lesser extent in silenced p53 cells. These data suggest that the Thiamet-G and cisplatin synergistic anti-tumoral effect is partially dependent on wild-type p53 pathway activation but also on other unknown growth-related pathways (94). Altogether, these results indicate that the effect of O-GlcNAcylation on cisplatin chemotherapy response could depend on the type of cancer and/or induction level, mutation status, and site-specific O-GlcNAcylation of p53.
After cisplatin exposure, DNA damage responses are initiated to maintain genome integrity. DNA cross-links are removed by the Nucleotide Excision Repair (NER) pathway or bypassed during replication through translesion DNA synthesis (TLS). TLS is mediated by specialized DNA polymerases (Pol) including Polη characterized by a low fidelity and the ability to replicate damaged DNA on stalled replication forks. TLS process is believed to contribute to the development of cisplatin resistance (95). In response to DNA damage, OGT relocates to DNA lesions and catalyzed O-GlcNAcylation of several substrates (37). Notably, Polη undergoes O-GlcNAcylation at Thr457 which allows its polyubiquitination at Lys462 and its subsequent dissociation from replication forks. Thus, cells expressing the O-GlcNAc-deficient T457A mutant Polη exhibit a higher sensitivity to cisplatin (96) (Figure 2). Given the key role of Polη in TLS and cisplatin chemoresistance of ovarian cancer stem cells (95), targeting Polη O-GlcNAcylation could be a chemotherapy-enhancing strategy in ovarian cancer treatment.
Stress conditions can destabilize the folding of cytoplasmic proteins leading to exposure of hydrophobic regions that interact with each other to form deleterious proteolysis resistant aggregates. In these conditions, molecular chaperones can form multimeric complexes to refold denatured or aggregated proteins. Elevated activity of chaperones allows cancer cells to grow and resist stress conditions including chemotherapy (97). Several studies showed that O-GlcNAcylation modulates cisplatin response by targeting directly or indirectly molecular chaperons. Based on the initial observation that higher O-GlcNAcylation levels in hepatoblastoma tissues might be associated with the pathogenesis, Song and collaborators (2019) realized a global proteomic analysis using O-GlcNAc antibody beads and immobilized metal affinity chromatography (IMAC) enrichments followed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) to identify potential O-GlcNAcylated and phosphorylated therapeutic targets. Among them, the anti-apoptotic chaperone Heat Shock Protein 27 kD (HSP27, also known as Heat Shock Protein family Beta-1 (HSPB1)) was identified, and its O-GlcNAcylation was further shown to promote cell proliferation and cisplatin resistance in hepatic cancer cell lines (98) (Figure 2). Overexpression of HSP27 is related to tumorigenesis, metastasis, and therapy resistance by acting as an upstream regulator of oncogenic (i.e. Hippo pathway, AKT, Glycogen Synthase Kinase 3 Beta (GSK3β), or β-catenin) and anti-apoptotic (i.e. NF-κB, Mothers Against Decapentaplegic homolog 3 (SMAD3), p38/MAPK) pathways. Targeting HSPs to enhance the effects of anti-cancer drugs is a promising approach and several molecules that modulate HSP protein functions are currently investigated in preclinical and clinical trials. For instance, HSP70 client protein inhibitor sorafenib has been approved to treat hepatocellular, renal, and thyroid carcinoma (97, 99). Studies revealed that O-GlcNAcylation stimulates aggressiveness and resistance of several cancers to chemotherapies and targeted therapies by modulating levels, activity, or subcellular localization of HSP proteins (100) including HSP27 (101–103). In this way, decreasing O-GlcNAcylation of HSPs could be a new anti-cancer therapeutic strategy. Secretory clusterin (sCLU) is a chaperon that facilitates the extracellular clearance of misfolded proteins. Like HSP proteins, this pro-survival factor is involved in cancer cell proliferation and drug resistance. OGT and sCLU expression are elevated in cervical cancer cell lines, and O-GlcNAc-induced up-regulation of sCLU leads to cisplatin resistance (Figure 2). sCLU silencing antisense oligonucleotides have been developed and approved for anti-cancer clinical trials. However, there is no drug currently available that completely blocks sCLU expression. Then, inhibiting OGT in combination with the knock-down of sCLU could be a new therapeutic strategy to overcome cisplatin resistance (104). Binding immunoglobulin Protein (BiP) is a major ER HSP70 family chaperone that binds to misfolded or unfolded proteins and releases UPR sensors (Inositol-Requiring Enzyme 1α (IRE1α), PERK, and Activating Transcription Factor 6 (ATF6)) to initiate the UPR pathway. Inhibition of the HBP pathway with the glutamine analog DON enhances cisplatin-induced apoptosis of lung cancer cells by repressing BiP expression and activating IRE1α (Figure 2). Up-regulation of BiP expression by HBP flux is supported by a previous study carried out in hepatoblastoma cells (105). Together, these data suggest that the combination of cisplatin with HBP inhibitors could improve lung cancer platinum-based chemotherapies (106).
Gemcitabine (GEM) acts as an antimetabolite and is a deoxycytidine nucleoside analog. It was approved by FDA in 1996 as chemotherapy for pancreatic cancer and later on for non-small cell lung, ovarian and breast cancers. GEM is metabolized to the active metabolite, 2’,2’-difluoro-2’-deoxycytidine triphosphate (dFdCTP) that can incorporate into DNA and RNA leading to the termination of DNA synthesis and faulty translation respectively. However, many cancers resist to GEM and the main involved mechanism consists of an up-regulation of the catabolic enzyme Cytidine Deaminase (CDA), deficiency in the anabolism enzyme Deoxycytidine Kinase (DCK), and alterations in nucleoside influx transporters (107).
Interestingly, increased metabolism of glucose and glutamine, two substrates for the HBP, fuel GEM resistance of pancreatic cancer (108, 109). Moreover, the HBP enzyme Phosphoacetylglucosamine Mutase 3 (PGM3) was shown to be overexpressed in GEM resistant human pancreatic tumors. Upon increasing GEM doses, PGM3 and global protein O-GlcNAcylation levels are decreased in sensitive BxPC-3 pancreatic cancer cells but increased in PANC-1 and MIA PaCa-2 resistant ones (110). Based on these observations, Ricchiardiello et al. (2020) recently investigated the effect of HBP flux inhibition in avoiding GEM resistance. Treatment with PGM3 inhibitor FR054 decreases tri-/tetra-antennary complex N-glycosylation of membrane proteins, O-GlcNAcylation of intracellular proteins, and enhances GEM efficiency in vitro and in vivo. Mechanistically, chemical inhibition of PGM3 causes a sustained activation of the pro-apoptotic UPR protein CHOP associated with a reduction in membrane localization of pro-tumorigenic Epidermal Growth Factor Receptor (EGFR) and a strong attenuation of the EGFR-Akt axis (110) (Figure 2). It was recently demonstrated that EGFR is directly O-GlcNAcylated and that OGT knock-down downregulates EGFR and its downstream PI3K/AKT pathway, and promotes cell cycle arrest and apoptosis in 786-O renal cell carcinoma cells (111). Pending further investigations to determine whether hypo-O-GlcNAcylation induced by PGM3 inhibition is directly involved in GEM sensitization mechanism, we speculate that hypo-O-GlcNAcylation could activate CHOP by inducing ROS accumulation, ER stress, and UPR activation (73, 74) (Figure 2). Therapeutic approaches involving PGM3 inhibition, and possibly inhibition of protein O-GlcNAcylation, in combination with GEM could be promising to bypass the drug resistance in pancreatic cancer.
Doxorubicin (DOX) is an anthracycline class medication approved by FDA and routinely applied to the treatment of several cancers such as breast, lung, gastric, ovarian, thyroid, non-Hodgkin’s, and Hodgkin’s lymphoma, sarcoma and pediatric tumors. This chemotherapeutic agent acts by intercalation with DNA, disruption of Topoisomerase II (Top-II), and production of quinone-type free radicals triggering cell death. Chemoresistance involves alteration of efflux ATP-Binding Cassette (ABC) transporter proteins, epigenetics, and signaling pathways (e.g. MAPK, PI3K/AKT/mammalian Target Of Rapamycin (mTOR), Wnt/β-catenin, Notch and Transforming Growth Factor Beta (TGF-β)) (112).
As previously mentioned, DOX treatment stimulates HBP flux and protein O-GlcNAcylation through the XBP1/AKT axis and FOXA2 leading to activation of pro-survival pathways (30, 31). Interestingly, recent studies showed that DOX treatment in combination with OSMI-1 has a synergistic apoptotic effect not only in several cancer types including sensitive and resistant liver (30, 113), prostate (114), and breast cancer cell lines but also in primary cells from newly diagnosed, refractory and relapsed acute myeloid leukemia (AML) patients (30). Hypo-O-GlcNAcylation enhances the sensitivity toward DOX by preventing pro-survival NF-κB and AKT activation (30, 113) and promoting ER stress response which leads to increased IRE1α-XBP and PERK-CHOP signaling (113) (Figure 2). Conversely, O-GlcNAcylation could protect MCF-7 breast cancer cells against DOX by stabilizing box C/D small nucleolar ribonucleoprotein complexes (snoRNPs) core component fibrillarin (FBL) on Ser142 and maintaining ribosomal RNA methylation and ribosome assembly (110) (Figure 2). Dysregulation of ribosome biogenesis promotes cancer cell proliferation (115). To note that treatment of HepG2 hepatocarcinoma cells with a low dose of DOX (1 μM) or a suboptimal dose of OSMI-1 (20 μM) alone did not induce apoptosis while the combined treatment did (113). In addition to improve therapeutic efficacy and alleviate DOX resistance in different cancer types, therapy combination with OGT inhibition could allow the use of smaller doses of DOX, hence reducing the associated side effects.
Ferroptosis is a newly identified form of programmed cell death that is characterized by an iron-dependent accumulation of lipid peroxides. Induction of ferroptosis emerged as a new strategy to trigger cancer cell death and ferroptosis-inducing compounds have been categorized into two classes based on their inhibition mode of Glutathione Peroxidase 4 (GPX4). The latter detoxifies lipid hyperoxides within biological membranes. The first class (e.g., erastin, Sorafenib) indirectly inhibits GPX4 by blocking the cysteine-glutamate transporter (system XC-) and depleting the GPX4 cofactor glutathione (GSH). The second class (e.g., Ras-selective lethal small molecule 3 (RSL3)) directly inhibits GPX4 leading to lipid-ROS accumulation (116). YAP has been reported to play a pivotal role in ferroptosis by upregulating critical modulators including Acyl-CoA Synthetase Long-chain family member 4 (ACSL4), Transferrin Receptor 1 (TfR1) (117), and Ferritin heavy chain 1 (FTH1) (118). O-GlcNAcylation of YAP on Thr241 residue prevents its degradation and enhances its transcriptional activity (12, 119). Under ferroptotic stress with erastin, system XC- inhibition leads indirectly to Thr241 hypo-O-GlcNAcylation of YAP, FTH1 down-regulation, free iron release, and increased ferroptosis in lung adenocarcinoma cells (118) (Figure 2). In contrast, Zhu et al. (2021) demonstrate that, under high glucose levels, O-GlcNAcylation enhances RSL3-mediated ferroptosis by targeting YAP on Thr241 and increasing its transcriptional activity on TfR1 in HCC cell lines (120) (Figure 2). Further study of YAP O-GlcNAcylation’s role in ferroptosis, which seems to depend on the ferroptosis-inducing compound and the type of cancer tissue, would be useful to clarify these discrepancies.
Programmed cell Death protein 1 (PD-1) is a suppressive receptor expressed in immune T cells upon T cell activation. It is involved in T cell activation, proliferation, survival, and cytotoxic activity. PD-1 is highly selective for Programmed Death-Ligand 1 (PD-L1), an immune-inhibitory ligand overexpressed by cancer cells as an “adaptive immune mechanism” to escape immune responses. A range of monoclonal antibodies specific to the PD-1 and PD-L1 immune checkpoints have been approved for the treatment of a wide variety of human cancers. However, compensatory up-regulation of PD-L1 gradually causes drug resistance in some cancer patients (121).
Shang et al. (2021) recently showed that the embryo- and tumor-specific folate cycle enzyme Methylenetetrahydrofolate Dehydrogenase 2 (MTHFD2) can rescue cancer cells from T cell cytotoxicity by driving the folate cycle and by sustaining sufficient uridine-related metabolites including UDP-GlcNAc substrate donor. Thus, MTHFD2 is highly positively correlated to global O-GlcNAcylation levels which, in turn, targets c-Myc at Thr58 and promotes its stability and its direct activation of PD-L1 transcription (122). The c-Myc O-GlcNAcylation at Thr58 prevents its interaction with F-box/wD repeat-containing protein 7 (Fbw7) ubiquitin E3-ligase, increases its stability, and its transcriptional activity on target genes involved in cell proliferation, metabolism, and apoptosis (123). More particularly, O-GlcNAcylation mediated-stabilization of c-Myc is involved in renewal, clonal expansion and malignant transformation of T cell progenitors (88). MTHFD2, which is rarely expressed in normal adult tissues, emerges as a potential safe target. Thus, inhibiting the O-GlcNAcylation-c-Myc-PD-L1 signaling axis could be a promising therapeutic strategy to stimulate anti-cancer T cell cytotoxicity, hence improving immunotherapy.
Radiation therapy is given to about 50% of all cancer patients and contributes to 40% of curative anti-cancer treatments. The mode of action is to prevent cancer cells from proliferation by inducing extensive DNA damage. Exposure to radiation leads to DNA double-strand breaks (DSB) and triggers the DNA damage response (DDR), a complex signal transduction pathway that regulates DNA repair, cell cycle checkpoints activation, chromatin remodeling, cell senescence, and apoptosis. One of the key DNA damage-induced epigenetic modifications is the phosphorylation of H2A histone family member X (H2AX, also known as γH2AX) by a group of PI3-like kinases including Ataxia Telangiectasia Mutated (ATM), ATM and Rad3 related protein (ATR) and DNA-dependent Protein Kinase (DNA-PK). Aberrant DDR pathway activation directly confers tumor radioresistance (124).
In non-irradiated cells, OGT functions to suppress genomic instability and reduce cellular stress probably by protecting cells from oxidative stress and/or cell-cycle defects. Thus, under oxidative stress conditions, increased OGT and O-GlcNAc levels were required to induce DDR and proliferation of Drosophila intestinal stem/progenitor cells. In an autoregulatory feedback loop, Checkpoint Kinase 1 (CHK1)/CHK2 DDR effectors stabilize OGT (125). Moreover, OGT inhibition can induce cellular stress resulting in constitutive activation of DDR (126). O-GlcNAcylation can directly target many proteins of the DDR in response to heat-stress-induced DNA damage (i.e. DNA-PK, Coactivator-associated Arginine Methyltransferase 1 (CARM1), Ubiquitin-Associated Protein 2-Like (UBAP2L), Nuclear Factor 45 (NF45), NF90, RuvB-Like 1 (RUVBL1)) (127). A quantitative phosphoproteomic study showed that loss of O-GlcNAcylation in OGT null cells affects phosphorylation of cell cycle and DDR proteins including ATM and Checkpoint Kinase 1 (CHK1). Notably, hypo-O-GlcNAcylation increases activating phosphorylation events on ATM at Ser1981 and on its downstream substrates p53, H2AX, and CHK2 (128). In irradiated cells, OGT is essential for DNA damage repair, recovery from checkpoint arrest, blockage onset of cellular senescence, and cell survival (126, 129, 130). Quantitative proteomic profiling of irradiated MCF-7 breast cancer cells revealed a strong regulation of chromatin modification and organization and DDR-associated factors upon alteration of O-GlcNAcylation (126). The H3K27 methyltransferase (HMT) Enhancer of Zeste 2 (EZH2) of Polycomb Related Complex 2 (PRC2) is considered a potential link between O-GlcNAcylation levels and irradiation induced DDR. EZH2-mediated histone methylation is found to promote DSB repair (131) and its Ser76 and Ser84 O-GlcNAcylation increases its stability and its HMT activity (132, 133). Irradiated tumor xenografts treated with shOGT or alloxan OGT inhibitor displayed a decreased EZH2 level, persistent DNA damages, a reduced proliferation, and an increased senescence. PUGNAc-induced elevation of O-GlcNAcylation or feeding animals with GlcNAc protects tumors against irradiation (126). Moreover, upon irradiation, O-GlcNAcylated H2B at Ser122 interacts with Nijmegen Breakage Sydrome 1 protein a member of MRE11-RAD50-NBS1 (MRN) complex, and promotes its accumulation at DSB where it normally acts as a bridge spanning the broken ends (129).
Other study showed contradictory results and point out OGT as a negative regulator to limit the expansion of DDR signaling in response to irradiation. Notably, in irradiated HeLa cervical cancer cells, OGT O-GlcNAcylates H2AX at Ser139 and Mediator of DNA Damage Checkpoint 1 (MDC1), and restrains the expansion of the DSBs-induced phosphorylation from the DNA damage sites. Thus, depletion of OGT in these cells does not affect DSB repair but prolongs the G2/M checkpoint, delays cell cycle recovery, and reduces cell viability following DNA damages (37).
Since its discovery in the 1980s (134, 135), the community have extensively proven the implication of O-GlcNAcylation in the etiology of chronic human diseases such as metabolic, neurodegenerative, cardiovascular diseases and cancer (6, 136, 137) but the role of the sugar in the resistance to various anti-cancer therapies has arisen more recently. Although many molecular mechanisms remain to be elucidated, targeting O-GlcNAcylation emerges as a promising strategy to impede cancer resistance. The current development of metabolic inhibitors, inhibitors of HBP and O-GlcNAcylation enzymes, RNA aptamers and nanobodies appear as promising tools and must be investigated in the light of cancer therapies to prevent or obliviate chemoresistance. The reprogramming of glucose and amino acid metabolisms provides energy and metabolites to cancer cells, including substrates for O-GlcNAcylation, to support growth, progression, metastasis, and resistance to anti-cancer therapies. Thus, a first level of action is to use metabolic inhibitors to modulate enzymes and transporters involved in nutrient metabolisms. The 2-deoxy-D-glucose (2DG) and dapagliflozin to target glucose metabolism, and GRASPA and ERY001 to target amino acid metabolism are currently under investigation in clinical trials (138). Direct targeting of HBP enzymes could be an alternative strategy. Thus, some inhibitors more or less specific for the rate-limiting GFAT (i.e. DON, azaserine and RO0509347), PGM3 (i.e. FR054) or UDP-N-Acetylglucosamine Pyrophosphorylase 1 (UAP1) (i.e. UTP a,b-methylenebisphosphonate analogue (meUTP) and GAL-012) are currently used (5). Notably, FR054 PGM3 specific inhibitor appears to have an in vivo antitumor efficacy by inhibiting the HBP flux (139). A third considered strategy is to use OGT or OGA inhibitors to directly restore the homeostasis of O-GlcNAcylation (5). The small molecule OSMI-1 is a highly specific OGT inhibitor of the quinolinone-6-sulfonamide (Q6S) class. Despite being membrane permeable and relatively large, having low aqueous solubility and inducing toxicity limiting its activity in cellulo (140), OSMI-1 could still have therapeutic potential since it synergistically enhances some cancer therapies (i.e. TRAIL and DOX)-induced apoptosis in vivo in tumor xenograft models (73, 113). The aminothiazoline derivative Thiamet-G is the most widely used OGA inhibitor in vitro and in vivo due to its stability, selectivity, oral bioavailability, and ability to cross the blood-brain barrier (141). Chronic elevation of O-GlcNAcylation by prolonged treatment with Thiamet-G (more than 5 months) was shown nontoxic in vivo in mice (142). We recently shown the efficiency of Thiamet-G to improve global O-GlcNAcylation levels and 5-FU response in murine carcinogen-induced CRC tumors (34). In addition, other selective OGA inhibitors, MK-8719 (143) and ASN120290 (previously known as ASN-561) (144) have obtained the orphan drug designation by the FDA for the treatment of the progressive supranuclear palsy (PSP) in 2016 and 2018 respectively. However, modulating global cellular O-GlcNAcylation levels have limits in term of therapeutic strategy. More than 5000 proteins have been identified as O-GlcNAcylated to date. Analysis of this O-GlcNAcome revealed the diversity of cellular mechanisms in which it is involved (145). In this way, global regulation of O-GlcNAcylation levels could affect proteins essential to cell/tissue physiology and induces severe side effects or induces other pathologies (6). To overcome this limitation, tools to implement a fourth strategy are under development which consists on targeting the O-GlcNAcylation of specific protein(s) of interest. Several writing and erasing O-GlcNAcylation engineering have been developed and some can allow the glycosylation/deglycosylation of a protein without the need for prior identification of glyco-sites (146). Notably and very recently, a series of modulated RNA aptamers (147) and nanobody-OGT/OGA (148–150) able to direct these enzymes to a specific target protein of interest have been designed and delivered to living cells. These new approaches seem promising but require further studies to prove their efficiency and explore their possible in vivo applications.
NV and IEB co-conceived, co-organized and co-wrote the manuscript. NV prepared the figures. All authors have read and approved the final manuscript.
This work was supported by the “Ligue Contre le Cancer/Comité du Nord/Comité de la Somme”, the University of Lille and the “Center National de la Recherche Scientifique”.
We thank Dr Stéphan Hardivillé and Dr Isabel Gonzalez Mariscal for proofreading the review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Carvalho KC, Cunha IW, Rocha RM, Ayala FR, Cajaíba MM, Begnami MD, et al. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics (Sao Paulo) (2011) 66:965–72. doi: 10.1590/s1807-59322011000600008
3. Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev (2008) 18:54–61. doi: 10.1016/j.gde.2008.02.003
4. Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem (1991) 266:4706–12.
5. Ma J, Wu C, Hart GW. Analytical and biochemical perspectives of protein O-GlcNAcylation. Chem Rev (2021) 121:1513–81. doi: 10.1021/acs.chemrev.0c00884
6. Chatham JC, Zhang J, Wende AR. Role of O-linked n-acetylglucosamine protein modification in cellular (patho)physiology. Physiol Rev (2021) 101:427–93. doi: 10.1152/physrev.00043.2019
7. Ma J, Hou C, Wu C. Demystifying the O-GlcNAc code: A systems view. Chem Rev (2022). doi: 10.1021/acs.chemrev.1c01006
8. van der Laarse SAM, Leney AC, Heck AJR. Crosstalk between phosphorylation and O-GlcNAcylation: friend or foe. FEBS J (2018) 285:3152–67. doi: 10.1111/febs.14491
9. Shi Y, Tomic J, Wen F, Shaha S, Bahlo A, Harrison R, et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia (2010) 24:1588–98. doi: 10.1038/leu.2010.152
10. Champattanachai V, Netsirisawan P, Chaiyawat P, Phueaouan T, Charoenwattanasatien R, Chokchaichamnankit D, et al. Proteomic analysis and abrogated expression of O-GlcNAcylated proteins associated with primary breast cancer. Proteomics (2013) 13:2088–99. doi: 10.1002/pmic.201200126
11. Mi W, Gu Y, Han C, Liu H, Fan Q, Zhang X, et al. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta (2011) 1812:514–9. doi: 10.1016/j.bbadis.2011.01.009
12. Zhang X, Qiao Y, Wu Q, Chen Y, Zou S, Liu X, et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat Commun (2017) 8:15280. doi: 10.1038/ncomms15280
13. Olivier-Van Stichelen S, Dehennaut V, Buzy A, Zachayus J-L, Guinez C, Mir A-M, et al. O-GlcNAcylation stabilizes β-catenin through direct competition with phosphorylation at threonine 41. FASEB J (2014) 28:3325–38. doi: 10.1096/fj.13-243535
14. Krześlak A, Wójcik-Krowiranda K, Forma E, Bieńkiewicz A, Bryś M. Expression of genes encoding for enzymes associated with O-GlcNAcylation in endometrial carcinomas: clinicopathologic correlations. Ginekol Pol (2012) 83:22–6.
15. Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-linked β-n-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem (2012) 287:11070–81. doi: 10.1074/jbc.M111.302547
16. Starska K, Forma E, Brzezińska-Błaszczyk E, Lewy-Trenda I, Bryś M, Jóźwiak P, et al. Gene and protein expression of O-GlcNAc-cycling enzymes in human laryngeal cancer. Clin Exp Med (2015) 15:455–68. doi: 10.1007/s10238-014-0318-1
17. Wang L, Chen S, Zhang Z, Zhang J, Mao S, Zheng J, et al. Suppressed OGT expression inhibits cell proliferation while inducing cell apoptosis in bladder cancer. BMC Cancer (2018) 18:1141. doi: 10.1186/s12885-018-5033-y
18. Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res (2013) 73:5277–87. doi: 10.1158/0008-5472.CAN-13-0549
19. Luanpitpong S, Angsutararux P, Samart P, Chanthra N, Chanvorachote P, Issaragrisil S. Hyper-O-GlcNAcylation induces cisplatin resistance via regulation of p53 and c-myc in human lung carcinoma. Sci Rep (2017) 7:10607. doi: 10.1038/s41598-017-10886-x
20. Xu D, Wang W, Bian T, Yang W, Shao M, Yang H. Increased expression of O-GlcNAc transferase (OGT) is a biomarker for poor prognosis and allows tumorigenesis and invasion in colon cancer. Int J Clin Exp Pathol (2019) 12:1305–14.
21. Singh JP, Qian K, Lee J-S, Zhou J, Han X, Zhang B, et al. O-GlcNAcase targets pyruvate kinase M2 to regulate tumor growth. Oncogene (2020) 39:560–73. doi: 10.1038/s41388-019-0975-3
22. Trinca GM, Goodman ML, Papachristou EK, D’Santos CS, Chalise P, Madan R, et al. O-GlcNAc-Dependent regulation of progesterone receptor function in breast cancer. Horm Cancer (2018) 9:12–21. doi: 10.1007/s12672-017-0310-9
23. Gu Y, Mi W, Ge Y, Liu H, Fan Q, Han C, et al. GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res (2010) 70:6344–51. doi: 10.1158/0008-5472.CAN-09-1887
24. Lee JB, Pyo K-H, Kim HR. Role and function of O-GlcNAcylation in cancer. Cancers (Basel) (2021) 13:5365. doi: 10.3390/cancers13215365
25. Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: A new cancer hallmark? Front Endocrinol (Lausanne) (2013) 4:99. doi: 10.3389/fendo.2013.00099
26. Very N, Lefebvre T, El Yazidi-Belkoura I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget (2017) 9:1380–402. doi: 10.18632/oncotarget.22377
27. Rodrigues JG, Duarte HO, Reis CA, Gomes J. Aberrant protein glycosylation in cancer: implications in targeted therapy. Biochem Soc Trans (2021) 49:843–54. doi: 10.1042/BST20200763
28. Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. a survival response of mammalian cells. J Biol Chem (2004) 279:30133–42. doi: 10.1074/jbc.M403773200
29. Cheung WD, Hart GW. AMP-activated protein kinase and p38 MAPK activate O-GlcNAcylation of neuronal proteins during glucose deprivation. J Biol Chem (2008) 283:13009–20. doi: 10.1074/jbc.M801222200
30. Liu Y, Cao Y, Pan X, Shi M, Wu Q, Huang T, et al. O-GlcNAc elevation through activation of the hexosamine biosynthetic pathway enhances cancer cell chemoresistance. Cell Death Dis (2018) 9:485. doi: 10.1038/s41419-018-0522-0
31. Huang H, Wang Y, Huang T, Wang L, Liu Y, Wu Q, et al. FOXA2 inhibits doxorubicin-induced apoptosis via transcriptionally activating HBP rate-limiting enzyme GFPT1 in HCC cells. J Physiol Biochem (2021) 77:625–38. doi: 10.1007/s13105-021-00829-6
32. Pan X, Wilson M, Mirbahai L, McConville C, Arvanitis TN, Griffin JL, et al. In vitro metabonomic study detects increases in UDP-GlcNAc and UDP-GalNAc, as early phase markers of cisplatin treatment response in brain tumor cells. J Proteome Res (2011) 10:3493–500. doi: 10.1021/pr200114v
33. Wang D, Wu J, Wang D, Huang X, Zhang N, Shi Y. Cisplatin enhances protein O−GlcNAcylation by altering the activity of OGT, OGA and AMPK in human non−small cell lung cancer cells. Int J Oncol (2021) 58:27. doi: 10.3892/ijo.2021.5207
34. Very N, Hardivillé S, Decourcelle A, Thévenet J, Djouina M, Page A, et al. Thymidylate synthase O-GlcNAcylation: a molecular mechanism of 5-FU sensitization in colorectal cancer. Oncogene (2022) 41:745–56. doi: 10.1038/s41388-021-02121-9
35. Haltiwanger RS, Blomberg MA, Hart GW. Glycosylation of nuclear and cytoplasmic proteins. purification and characterization of a uridine diphospho-n-acetylglucosamine:polypeptide beta-n-acetylglucosaminyltransferase. J Biol Chem (1992) 267:9005–13.
36. Pederson NV, Zanghi JA, Miller WM, Knop RH. Discrimination of fluorinated uridine metabolites in n-417 small cell lung cancer cell extracts via 19F- and 31P-NMR. Magn Reson Med (1994) 31:224–8. doi: 10.1002/mrm.1910310217
37. Chen Q, Yu X. OGT restrains the expansion of DNA damage signaling. Nucleic Acids Res (2016) 44:9266–78. doi: 10.1093/nar/gkw663
38. Dehennaut V, Leprince D, Lefebvre T. O-GlcNAcylation, an epigenetic mark. focus on the histone code, TET family proteins, and polycomb group proteins. Front Endocrinol (Lausanne) (2014) 5:155. doi: 10.3389/fendo.2014.00155
39. Sakabe K, Wang Z, Hart GW. β-n-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U.S.A. (2010) 107:19915–20. doi: 10.1073/pnas.1009023107
40. Li QQ, Hao J-J, Zhang Z, Krane LS, Hammerich KH, Sanford T, et al. Proteomic analysis of proteome and histone post-translational modifications in heat shock protein 90 inhibition-mediated bladder cancer therapeutics. Sci Rep (2017) 7:201. doi: 10.1038/s41598-017-00143-6
41. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers (2019) 5:66. doi: 10.1038/s41572-019-0111-2
42. Tokunaga E, Hisamatsu Y, Tanaka K, Yamashita N, Saeki H, Oki E, et al. Molecular mechanisms regulating the hormone sensitivity of breast cancer. Cancer Sci (2014) 105:1377–83. doi: 10.1111/cas.12521
43. Hartkopf AD, Grischke E-M, Brucker SY. Endocrine-resistant breast cancer: Mechanisms and treatment. BRC (2020) 15:347–54. doi: 10.1159/000508675
44. Kim C, Tang G, Pogue-Geile KL, Costantino JP, Baehner FL, Baker J, et al. Estrogen receptor (ESR1) mRNA expression and benefit from tamoxifen in the treatment and prevention of estrogen receptor-positive breast cancer. J Clin Oncol (2011) 29:4160–7. doi: 10.1200/JCO.2010.32.9615
45. Gandhi N, Das GM. Metabolic reprogramming in breast cancer and its therapeutic implications. Cells (2019) 8:E89. doi: 10.3390/cells8020089
46. Kuo W-L, Tseng L-L, Chang C-C, Chen C-J, Cheng M-L, Cheng H-H, et al. Prognostic significance of O-GlcNAc and PKM2 in hormone receptor-positive and HER2-nonenriched breast cancer. Diagnostics (Basel) (2021) 11:1460. doi: 10.3390/diagnostics11081460
47. Yang Y, Wu K, Liu Y, Shi L, Tao K, Wang G. Prognostic significance of metabolic enzyme pyruvate kinase M2 in breast cancer: A meta-analysis. Med (Baltimore) (2017) 96:e8690. doi: 10.1097/MD.0000000000008690
48. Wang Y, Liu J, Jin X, Zhang D, Li D, Hao F, et al. O-GlcNAcylation destabilizes the active tetrameric PKM2 to promote the warburg effect. Proc Natl Acad Sci U.S.A. (2017) 114:13732–7. doi: 10.1073/pnas.1704145115
49. Kanwal S, Fardini Y, Pagesy P, N’tumba-Byn T, Pierre-Eugène C, Masson E, et al. O-GlcNAcylation-inducing treatments inhibit estrogen receptor α expression and confer resistance to 4-OH-tamoxifen in human breast cancer-derived MCF-7 cells. PloS One (2013) 8:e69150. doi: 10.1371/journal.pone.0069150
50. Barkovskaya A, Seip K, Hilmarsdottir B, Maelandsmo GM, Moestue SA, Itkonen HM. O-GlcNAc transferase inhibition differentially affects breast cancer subtypes. Sci Rep (2019) 9:5670. doi: 10.1038/s41598-019-42153-6
51. Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res (2008) 68:826–33. doi: 10.1158/0008-5472.CAN-07-2707
52. Barkovskaya A, Seip K, Prasmickaite L, Mills IG, Moestue SA, Itkonen HM. Inhibition of O-GlcNAc transferase activates tumor-suppressor gene expression in tamoxifen-resistant breast cancer cells. Sci Rep (2020) 10:16992. doi: 10.1038/s41598-020-74083-z
53. D’Arcy P, Brnjic S, Olofsson MH, Fryknäs M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med (2011) 17:1636–40. doi: 10.1038/nm.2536
54. Lü S, Wang J. The resistance mechanisms of proteasome inhibitor bortezomib. biomark Res (2013) 1:13. doi: 10.1186/2050-7771-1-13
55. Zhu YX, Tiedemann R, Shi C-X, Yin H, Schmidt JE, Bruins LA, et al. RNAi screen of the druggable genome identifies modulators of proteasome inhibitor sensitivity in myeloma including CDK5. Blood (2011) 117:3847–57. doi: 10.1182/blood-2010-08-304022
56. Sekine H, Okazaki K, Kato K, Alam MM, Shima H, Katsuoka F, et al. O-GlcNAcylation signal mediates proteasome inhibitor resistance in cancer cells by stabilizing NRF1. Mol Cell Biol (2018) 38:e00252–18. doi: 10.1128/MCB.00252-18
57. Hashimoto E, Okuno S, Hirayama S, Arata Y, Goto T, Kosako H, et al. Enhanced O-GlcNAcylation mediates cytoprotection under proteasome impairment by promoting proteasome turnover in cancer cells. iScience (2020) 23:101299. doi: 10.1016/j.isci.2020.101299
58. Chen S, Zhang Y, Zhou L, Leng Y, Lin H, Kmieciak M, et al. A bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood (2014) 124:2687–97. doi: 10.1182/blood-2014-03-564534
59. Liu Y, Wang X, Zhu T, Zhang N, Wang L, Huang T, et al. Resistance to bortezomib in breast cancer cells that downregulate bim through FOXA1 O-GlcNAcylation. J Cell Physiol (2019) 234:17527–37. doi: 10.1002/jcp.28376
60. Azakir BA, Desrochers G, Angers A. The ubiquitin ligase itch mediates the antiapoptotic activity of epidermal growth factor by promoting the ubiquitylation and degradation of the truncated c-terminal portion of bid. FEBS J (2010) 277:1319–30. doi: 10.1111/j.1742-4658.2010.07562.x
61. Luanpitpong S, Chanthra N, Janan M, Poohadsuan J, Samart P, U-Pratya Y, et al. Inhibition of O-GlcNAcase sensitizes apoptosis and reverses bortezomib resistance in mantle cell lymphoma through modification of truncated bid. Mol Cancer Ther (2018) 17:484–96. doi: 10.1158/1535-7163.MCT-17-0390
62. Holoch PA, Griffith TS. TNF-related apoptosis-inducing ligand (TRAIL): a new path to anti-cancer therapies. Eur J Pharmacol (2009) 625:63–72. doi: 10.1016/j.ejphar.2009.06.066
63. Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev (2018) 37:733–48. doi: 10.1007/s10555-018-9728-y
64. Ching NS, Sonia WHM. Trailing TRAIL resistance for targeted cancer therapy. Biomed J Sci Tech Res (2018) 4:3668–71.
65. Lee H, Oh Y, Jeon Y-J, Lee S-Y, Kim H, Lee H-J, et al. DR4-Ser424 O-GlcNAcylation promotes sensitization of TRAIL-tolerant persisters and TRAIL-resistant cancer cells to death. Cancer Res (2019) 79:2839–52. doi: 10.1158/0008-5472.CAN-18-1991
66. Yang S, Xu F, Yuan K, Sun Y, Zhou T, Zhao X, et al. Regulation of pancreatic cancer TRAIL resistance by protein O-GlcNAcylation. Lab Invest (2020) 100:777–85. doi: 10.1038/s41374-019-0365-z
67. Yang J, LeBlanc FR, Dighe SA, Hamele CE, Olson TL, Feith DJ, et al. TRAIL mediates and sustains constitutive NF-κB activation in LGL leukemia. Blood (2018) 131:2803–15. doi: 10.1182/blood-2017-09-808816
68. Chen S-H, Chang J-Y. New insights into mechanisms of cisplatin resistance: From tumor cell to microenvironment. Int J Mol Sci (2019) 20:E4136. doi: 10.3390/ijms20174136
69. He J, Yu J-J, Xu Q, Wang L, Zheng JZ, Liu L-Z, et al. Downregulation of ATG14 by EGR1-MIR152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophagy. Autophagy (2015) 11:373–84. doi: 10.1080/15548627.2015.1009781
70. Kim M, Jung J-Y, Choi S, Lee H, Morales LD, Koh J-T, et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy (2016) 13:149–68. doi: 10.1080/15548627.2016.1239676
71. Zhou F, Yang X, Zhao H, Liu Y, Feng Y, An R, et al. Down-regulation of OGT promotes cisplatin resistance by inducing autophagy in ovarian cancer. Theranostics (2018) 8:5200–12. doi: 10.7150/thno.27806
72. Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol (2014) 16:1215–26. doi: 10.1038/ncb3066
73. Lee S-J, Lee D-E, Choi S-Y, Kwon O-S. OSMI-1 enhances TRAIL-induced apoptosis through ER stress and NF-κB signaling in colon cancer cells. Int J Mol Sci (2021) 22:11073. doi: 10.3390/ijms222011073
74. Jang I, Kim HB, Seo H, Kim JY, Choi H, Yoo JS, et al. O-GlcNAcylation of eIF2α regulates the phospho-eIF2α-mediated ER stress response. Biochim Biophys Acta (2015) 1853:1860–9. doi: 10.1016/j.bbamcr.2015.04.017
75. Deng X, Yi X, Huang D, Liu P, Chen L, Du Y, et al. ROCK2 mediates osteosarcoma progression and TRAIL resistance by modulating O-GlcNAc transferase degradation. Am J Cancer Res (2020) 10:781–98.
76. Ghafouri-Fard S, Abak A, Tondro Anamag F, Shoorei H, Fattahi F, Javadinia SA, et al. 5-fluorouracil: A narrative review on the role of regulatory mechanisms in driving resistance to this chemotherapeutic agent. Front Oncol (2021) 11:658636. doi: 10.3389/fonc.2021.658636
77. Temmink OH, Bijnsdorp IV, Prins H-J, Losekoot N, Adema AD, Smid K, et al. Trifluorothymidine resistance is associated with decreased thymidine kinase and equilibrative nucleoside transporter expression or increased secretory phospholipase A2. Mol Cancer Ther (2010) 9:1047–57. doi: 10.1158/1535-7163.MCT-09-0932
78. Gmeiner WH, Reinhold WC, Pommier Y. Genome-wide mRNA and microRNA profiling of the NCI 60 cell-line screen and comparison of FdUMP[10] with fluorouracil, floxuridine, and topoisomerase 1 poisons. Mol Cancer Ther (2010) 9:3105–14. doi: 10.1158/1535-7163.MCT-10-0674
79. Cao S, Chang W, Wan C, Zang Y, Zhao J, Chen J, et al. Bi-clustering based biological and clinical characterization of colorectal cancer in complementary to CMS classification. bioRxiv (2018) 508275. doi: 10.1101/508275
80. Wang Z, Park K, Comer F, Hsieh-Wilson LC, Saudek CD, Hart GW. Site-specific GlcNAcylation of human erythrocyte proteins. Diabetes (2009) 58:309–17. doi: 10.2337/db08-0994
81. Jochmann R, Pfannstiel J, Chudasama P, Kuhn E, Konrad A, Stürzl M. O-GlcNAc transferase inhibits KSHV propagation and modifies replication relevant viral proteins as detected by systematic O-GlcNAcylation analysis. Glycobiology (2013) 23:1114–30. doi: 10.1093/glycob/cwt028
82. Hahne H, Sobotzki N, Nyberg T, Helm D, Borodkin VS, van Aalten DM, et al. Proteome wide purification and identification of O-GlcNAc modified proteins using click chemistry and mass spectrometry. J Proteome Res (2013) 12:927–36. doi: 10.1021/pr300967y
83. Palmirotta R, Carella C, Silvestris E, Cives M, Stucci SL, Tucci M, et al. SNPs in predicting clinical efficacy and toxicity of chemotherapy: walking through the quicksand. Oncotarget (2018) 9:25355–82. doi: 10.18632/oncotarget.25256
84. Kang KA, Piao MJ, Kim KC, Kang HK, Chang WY, Park IC, et al. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: involvement of TET-dependent DNA demethylation. Cell Death Dis (2014) 5:e1183. doi: 10.1038/cddis.2014.149
85. Kang KA, Piao MJ, Ryu YS, Kang HK, Chang WY, Keum YS, et al. Interaction of DNA demethylase and histone methyltransferase upregulates Nrf2 in 5-fluorouracil-resistant colon cancer cells. Oncotarget (2016) 7:40594–620. doi: 10.18632/oncotarget.9745
86. Chen P, Smith TJ, Wu J, Siesser PF, Bisnett BJ, Khan F, et al. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J (2017) 36:2233–50. doi: 10.15252/embj.201696113
87. Walter LA, Lin YH, Halbrook CJ, Chuh KN, He L, Pedowitz NJ, et al. Inhibiting the hexosamine biosynthetic pathway lowers O-GlcNAcylation levels and sensitizes cancer to environmental stress. Biochemistry (2020) 59:3169–79. doi: 10.1021/acs.biochem.9b00560
88. Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DMF, et al. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol (2016) 17:712–20. doi: 10.1038/ni.3439
89. Hu L, Wu H, Jiang T, Kuang M, Liu B, Guo X, et al. pVHL promotes lysosomal degradation of YAP in lung adenocarcinoma. Cell Signal (2021) 83:110002. doi: 10.1016/j.cellsig.2021.110002
90. Dorayappan KDP, Wanner R, Wallbillich JJ, Saini U, Zingarelli R, Suarez AA, et al. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: a novel mechanism linking STAT3/Rab proteins. Oncogene (2018) 37:3806–21. doi: 10.1038/s41388-018-0189-0
91. Qian L, Yang X, Li S, Zhao H, Gao Y, Zhao S, et al. Reduced O-GlcNAcylation of SNAP-23 promotes cisplatin resistance by inducing exosome secretion in ovarian cancer. Cell Death Discovery (2021) 7:112. doi: 10.1038/s41420-021-00489-x
92. Wang X, Simpson ER, Brown KA. p53: Protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res (2015) 75:5001–7. doi: 10.1158/0008-5472.CAN-15-0563
93. Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, et al. Modification of p53 with O-linked n-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol (2006) 8:1074–83. doi: 10.1038/ncb1470
94. de Queiroz RM, Madan R, Chien J, Dias WB, Slawson C. Changes in O-linked n-acetylglucosamine (O-GlcNAc) homeostasis activate the p53 pathway in ovarian cancer cells. J Biol Chem (2016) 291:18897–914. doi: 10.1074/jbc.M116.734533
95. Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, et al. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc Natl Acad Sci U.S.A. (2015) 112:4411–6. doi: 10.1073/pnas.1421365112
96. Ma X, Liu H, Li J, Wang Y, Ding Y-H, Shen H, et al. Polη O-GlcNAcylation governs genome integrity during translesion DNA synthesis. Nat Commun (2017) 8:1941. doi: 10.1038/s41467-017-02164-1
97. Yun CW, Kim HJ, Lim JH, Lee SH. Heat shock proteins: Agents of cancer development and therapeutic targets in anti-cancer therapy. Cells (2019) 9:E60. doi: 10.3390/cells9010060
98. Song H, Ma J, Bian Z, Chen S, Zhu J, Wang J, et al. Global profiling of O-GlcNAcylated and/or phosphorylated proteins in hepatoblastoma. Sig Transduct Target Ther (2019) 4:1–9. doi: 10.1038/s41392-019-0067-4
99. Shevtsov M, Multhoff G, Mikhaylova E, Shibata A, Guzhova I, Margulis B. Combination of anti-cancer drugs with molecular chaperone inhibitors. Int J Mol Sci (2019) 20:5284. doi: 10.3390/ijms20215284
100. Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science (2001) 291:2376–8. doi: 10.1126/science.1058714
101. Guo K, Gan L, Zhang S, Cui FJ, Cun W, Li Y, et al. Translocation of HSP27 into liver cancer cell nucleus may be associated with phosphorylation and O-GlcNAc glycosylation. Oncol Rep (2012) 28:494–500. doi: 10.3892/or.2012.1844
102. Netsirisawan P, Chaiyawat P, Chokchaichamnankit D, Lirdprapamongkol K, Srisomsap C, Svasti J, et al. Decreasing O-GlcNAcylation affects the malignant transformation of MCF-7 cells via Hsp27 expression and its O-GlcNAc modification. Oncol Rep (2018) 40:2193–205. doi: 10.3892/or.2018.6617
103. Balana AT, Levine PM, Craven TW, Mukherjee S, Pedowitz NJ, Moon SP, et al. O-GlcNAc modification of small heat shock proteins enhances their anti-amyloid chaperone activity. Nat Chem (2021) 13:441–50. doi: 10.1038/s41557-021-00648-8
104. Kim MJ, Choi MY, Lee DH, Roh GS, Kim HJ, Kang SS, et al. O-Linked n-acetylglucosamine transferase enhances secretory clusterin expression via liver X receptors and sterol response element binding protein regulation in cervical cancer. Oncotarget (2018) 9:4625–36. doi: 10.18632/oncotarget.23588
105. Sage AT, Walter LA, Shi Y, Khan MI, Kaneto H, Capretta A, et al. Hexosamine biosynthesis pathway flux promotes endoplasmic reticulum stress, lipid accumulation, and inflammatory gene expression in hepatic cells. Am J Physiology-Endocrinol Metab (2010) 298:E499–511. doi: 10.1152/ajpendo.00507.2009
106. Chen W, Do KC, Saxton B, Leng S, Filipczak P, Tessema M, et al. Inhibition of the hexosamine biosynthesis pathway potentiates cisplatin cytotoxicity by decreasing BiP expression in non-small-cell lung cancer cells. Mol Carcinog (2019) 58:1046–55. doi: 10.1002/mc.22992
107. Han H, Li S, Zhong Y, Huang Y, Wang K, Jin Q, et al. Emerging pro-drug and nano-drug strategies for gemcitabine-based cancer therapy. Asian J Pharm Sci (2022) 17:35–52. doi: 10.1016/j.ajps.2021.06.001
108. Chen R, Lai LA, Sullivan Y, Wong M, Wang L, Riddell J, et al. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci Rep (2017) 7:7950. doi: 10.1038/s41598-017-08436-6
109. Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell (2017) 32:71–87.e7. doi: 10.1016/j.ccell.2017.06.004
110. Ricciardiello F, Gang Y, Palorini R, Li Q, Giampà M, Zhao F, et al. Hexosamine pathway inhibition overcomes pancreatic cancer resistance to gemcitabine through unfolded protein response and EGFR-akt pathway modulation. Oncogene (2020) 39:4103–17. doi: 10.1038/s41388-020-1260-1
111. Wang L, Chen S, Zhang J, Mao S, Mao W, Zhang W, et al. Suppressed OGT expression inhibits cell proliferation and modulates EGFR expression in renal cell carcinoma. Cancer Manag Res (2019) 11:2215–23. doi: 10.2147/CMAR.S190642
112. Sritharan S, Sivalingam N. A comprehensive review on time-tested anticancer drug doxorubicin. Life Sci (2021) 278:119527. doi: 10.1016/j.lfs.2021.119527
113. Lee SJ, Kwon O-S. O-GlcNAc transferase inhibitor synergistically enhances doxorubicin-induced apoptosis in HepG2 cells. Cancers (Basel) (2020) 12:E3154. doi: 10.3390/cancers12113154
114. Makwana V, Dukie AS-A, Rudrawar S. Investigating the impact of OGT inhibition on doxorubicin- and docetaxel-induced cytotoxicity in PC-3 and WPMY-1 cells. Int J Toxicol (2020) 39:586–93. doi: 10.1177/1091581820948433
115. Qin W, Lv P, Fan X, Quan B, Zhu Y, Qin K, et al. Quantitative time-resolved chemoproteomics reveals that stable O-GlcNAc regulates box C/D snoRNP biogenesis. Proc Natl Acad Sci U.S.A. (2017) 114:E6749–58. doi: 10.1073/pnas.1702688114
116. Wang H, Lin D, Yu Q, Li Z, Lenahan C, Dong Y, et al. A promising future of ferroptosis in tumor therapy. Front Cell Dev Biol (2021) 9:629150. doi: 10.3389/fcell.2021.629150
117. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature (2019) 572:402–6. doi: 10.1038/s41586-019-1426-6
118. Zhang X, Yu K, Ma L, Qian Z, Tian X, Miao Y, et al. Endogenous glutamate determines ferroptosis sensitivity via ADCY10-dependent YAP suppression in lung adenocarcinoma. Theranostics (2021) 11:5650–74. doi: 10.7150/thno.55482
119. Peng C, Zhu Y, Zhang W, Liao Q, Chen Y, Zhao X, et al. Regulation of the hippo-YAP pathway by glucose sensor O-GlcNAcylation. Mol Cell (2017) 68:591–604.e5. doi: 10.1016/j.molcel.2017.10.010
120. Zhu G, Murshed A, Li H, Ma J, Zhen N, Ding M, et al. O-GlcNAcylation enhances sensitivity to RSL3-induced ferroptosis via the YAP/TFRC pathway in liver cancer. Cell Death Discovery (2021) 7:1–12. doi: 10.1038/s41420-021-00468-2
121. Chocarro de Erauso L, Zuazo M, Arasanz H, Bocanegra A, Hernandez C, Fernandez G, et al. Resistance to PD-L1/PD-1 blockade immunotherapy. a tumor-intrinsic or tumor-extrinsic phenomenon? Front Pharmacol (2020) 11:441. doi: 10.3389/fphar.2020.00441
122. Shang M, Yang H, Yang R, Chen T, Fu Y, Li Y, et al. The folate cycle enzyme MTHFD2 induces cancer immune evasion through PD-L1 up-regulation. Nat Commun (2021) 12:1940. doi: 10.1038/s41467-021-22173-5
123. Kamemura K, Hayes BK, Comer FI, Hart GW. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: alternative glycosylation/phosphorylation of THR-58, a known mutational hot spot of c-myc in lymphomas, is regulated by mitogens. J Biol Chem (2002) 277:19229–35. doi: 10.1074/jbc.M201729200
124. Huang R-X, Zhou P-K. And targets for radiotherapy sensitization in cancer. Sig Transduct Target Ther (2020) 5:1–27. doi: 10.1038/s41392-020-0150-x
125. Na H, Akan I, Abramowitz LK, Hanover JA. Nutrient-driven O-GlcNAcylation controls DNA damage repair signaling and Stem/Progenitor cell homeostasis. Cell Rep (2020) 31(6):107632. doi: 10.1016/j.celrep.2020.107632
126. Efimova EV, Appelbe OK, Ricco N, Lee SS-Y, Liu Y, Wolfgeher DJ, et al. O-GlcNAcylation enhances double-strand break repair, promotes cancer cell proliferation, and prevents therapy-induced senescence in irradiated tumors. Mol Cancer Res (2019) 17:1338–50. doi: 10.1158/1541-7786.MCR-18-1025
127. Zachara NE, Molina H, Wong KY, Pandey A, Hart GW. The dynamic stress-induced “O-GlcNAc-ome” highlights functions for O-GlcNAc in regulating DNA damage/repair and other cellular pathways. Amino Acids (2011) 40:793–808. doi: 10.1007/s00726-010-0695-z
128. Zhong J, Martinez M, Sengupta S, Lee A, Wu X, Chaerkady R, et al. Quantitative phosphoproteomics reveals crosstalk between phosphorylation and O-GlcNAc in the DNA damage response pathway. Proteomics (2015) 15:591–607. doi: 10.1002/pmic.201400339
129. Wang P, Peng C, Liu X, Liu H, Chen Y, Zheng L, et al. OGT mediated histone H2B S112 GlcNAcylation regulates DNA damage response. J Genet Genomics (2015) 42:467–75. doi: 10.1016/j.jgg.2015.07.002
130. Efimova EV, Takahashi S, Shamsi NA, Wu D, Labay E, Ulanovskaya OA, et al. Linking cancer metabolism to DNA repair and accelerated senescence. Mol Cancer Res (2016) 14:173–84. doi: 10.1158/1541-7786.MCR-15-0263
131. Campbell S, Ismail IH, Young LC, Poirier GG, Hendzel MJ. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle (2013) 12:2675–83. doi: 10.4161/cc.25795
132. Lo P-W, Shie J-J, Chen C-H, Wu C-Y, Hsu T-L, Wong C-H. O-GlcNAcylation regulates the stability and enzymatic activity of the histone methyltransferase EZH2. Proc Natl Acad Sci (2018) 115:7302–7. doi: 10.1073/pnas.1801850115
133. Chu C-S, Lo P-W, Yeh Y-H, Hsu P-H, Peng S-H, Teng Y-C, et al. O-GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci (2014) 111:1355–60. doi: 10.1073/pnas.1323226111
134. Torres CR, Hart GW. Topography and polypeptide distribution of terminal n-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence O-linked GlcNAc J Biol Chem (1984) 259:3308–17.
135. Holt GD, Hart GW. The subcellular distribution of terminal n-acetylglucosamine moieties. localization of a novel protein-saccharide linkage, O-linked GlcNAc. J Biol Chem (1986) 261:8049–57.
136. Very N, Vercoutter-Edouart A-S, Lefebvre T, Hardivillé S, El Yazidi-Belkoura I. Cross-dysregulation of O-GlcNAcylation and PI3K/AKT/mTOR axis in human chronic diseases. Front Endocrinol (Lausanne) (2018) 9:602. doi: 10.3389/fendo.2018.00602
137. Nie H, Yi W. O-GlcNAcylation, a sweet link to the pathology of diseases. J Zhejiang Univ Sci B (2019) 20:437–48. doi: 10.1631/jzus.B1900150
138. Tuerhong A, Xu J, Shi S, Tan Z, Meng Q, Hua J, et al. Overcoming chemoresistance by targeting reprogrammed metabolism: the achilles’ heel of pancreatic ductal adenocarcinoma. Cell Mol Life Sci (2021) 78:5505–26. doi: 10.1007/s00018-021-03866-y
139. Ricciardiello F, Votta G, Palorini R, Raccagni I, Brunelli L, Paiotta A, et al. Inhibition of the hexosamine biosynthetic pathway by targeting PGM3 causes breast cancer growth arrest and apoptosis. Cell Death Dis (2018) 9:1–17. doi: 10.1038/s41419-018-0405-4
140. Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, et al. A small molecule that inhibits OGT activity in cells. ACS Chem Biol (2015) 10:1392–7. doi: 10.1021/acschembio.5b00004
141. Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau. vivo Nat Chem Biol (2008) 4:483–90. doi: 10.1038/nchembio.96
142. Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, et al. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol (2012) 8:393–9. doi: 10.1038/nchembio.797
143. Selnick HG, Hess JF, Tang C, Liu K, Schachter JB, Ballard JE, et al. Discovery of MK-8719, a potent O-GlcNAcase inhibitor as a potential treatment for tauopathies. J Med Chem (2019) 62:10062–97. doi: 10.1021/acs.jmedchem.9b01090
144. Permanne B, Quattropani A, Hantson J, Neny M, Ousson S, Sand A, et al. O3-04-04: Pharmacological intervention with the novel o-glcnacase inhibitor ASN-561 reduces pathological tau in transgenic mice. Alzheimer’s Dementia (2015) 11:P227–7. doi: 10.1016/j.jalz.2015.07.257
145. Love DC, Hanover JA The hexosamine signaling pathway: deciphering the “O-GlcNAc code.” Sci STKE (2005) 2005:re13. doi: 10.1126/stke.3122005re13
146. Ge Y, Woo CM. Writing and erasing O-GlcNAc from target proteins in cells. Biochem Soc Trans (2021) 49:2891–901. doi: 10.1042/BST20210865
147. Zhu Y, Hart G. Targeting the O-GlcNAc transferase to specific proteins using RNA aptamers. FASEB J (2019) 33:799.5–5. doi: 10.1096/fasebj.2019.33.1_supplement.799.5
148. Ramirez DH, Ge Y, Woo CM. O-GlcNAc engineering on a target protein in cells with nanobody-OGT and nanobody-splitOGA. Curr Protoc (2021) 1:e117. doi: 10.1002/cpz1.117
149. Ramirez DH, Aonbangkhen C, Wu H-Y, Naftaly JA, Tang S, O’Meara TR, et al. Engineering a proximity-directed O-GlcNAc transferase for selective protein O-GlcNAcylation in cells. ACS Chem Biol (2020) 15:1059–66. doi: 10.1021/acschembio.0c00074
Keywords: 6845/12000 O-GlcNAcylation, cancer, therapy resistance, therapeutic strategy, post-translational modifications (PTMs)
Citation: Very N and El Yazidi-Belkoura I (2022) Targeting O-GlcNAcylation to overcome resistance to anti-cancer therapies. Front. Oncol. 12:960312. doi: 10.3389/fonc.2022.960312
Received: 02 June 2022; Accepted: 19 July 2022;
Published: 17 August 2022.
Edited by:
Takeo Tatsuta, Tohoku Medical and Pharmaceutical University, JapanReviewed by:
Junfeng Ma, Georgetown University, United StatesCopyright © 2022 Very and El Yazidi-Belkoura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ikram El Yazidi-Belkoura, aWtyYW0uZWwteWF6aWRpQHVuaXYtbGlsbGUuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.