 Nathan Eaton1,2†‡
Nathan Eaton1,2†‡ Emily K. Boyd1,2‡Ratnashree Biswas1‡Melissa M. Lee-Sundlov1‡Theresa A. Dlugi1‡
Emily K. Boyd1,2‡Ratnashree Biswas1‡Melissa M. Lee-Sundlov1‡Theresa A. Dlugi1‡ Haley E. Ramsey3,4†Shikan Zheng1
Haley E. Ramsey3,4†Shikan Zheng1 Robert T. Burns1‡
Robert T. Burns1‡ Martha C. Sola-Visner3,4‡
Martha C. Sola-Visner3,4‡ Karin M. Hoffmeister1,5‡
Karin M. Hoffmeister1,5‡ Hervé Falet1,2*‡
Hervé Falet1,2*‡- 1Translational Glycomics Center, Versiti Blood Research Institute, Milwaukee, WI, United States
- 2Department of Cell Biology, Neurobiology, and Anatomy, Medical College of Wisconsin, Milwaukee, WI, United States
- 3Division of Newborn Medicine, Boston Children’s Hospital, Boston, MA, United States
- 4Department of Pediatrics, Harvard Medical School, Boston, MA, United States
- 5Departments of Medicine and Biochemistry, Medical College of Wisconsin, Milwaukee, WI, United States
Dnm2fl/fl Pf4-Cre (Dnm2Plt–/–) mice lacking the endocytic GTPase dynamin 2 (DNM2) in platelets and megakaryocytes (MKs) develop hallmarks of myelofibrosis. At the cellular level, the tyrosine kinase JAK2 is constitutively active but decreased in expression in Dnm2Plt–/– platelets. Additionally, Dnm2Plt–/– platelets cannot endocytose the thrombopoietin (TPO) receptor Mpl, leading to elevated circulating TPO levels. Here, we assessed whether the hyperproliferative phenotype of Dnm2Plt–/– mice was due to JAK2 constitutive activation or to elevated circulating TPO levels. In unstimulated Dnm2Plt–/– platelets, STAT3 and, to a lower extent, STAT5 were phosphorylated, but their phosphorylation was slowed and diminished upon TPO stimulation. We further crossed Dnm2Plt–/– mice in the Mpl–/– background to generate Mpl–/–Dnm2Plt–/– mice lacking Mpl ubiquitously and DNM2 in platelets and MKs. Mpl–/– Dnm2Plt–/– platelets had severely reduced JAK2 and STAT3 but normal STAT5 expression. Mpl–/– Dnm2Plt–/– mice had severely reduced bone marrow MK and hematopoietic stem and progenitor cell numbers. Additionally, Mpl–/– Dnm2Plt–/– mice had severe erythroblast (EB) maturation defects, decreased expression of hemoglobin and heme homeostasis genes and increased expression of ribosome biogenesis and protein translation genes in spleen EBs, and developed anemia with grossly elevated plasma erythropoietin (EPO) levels, leading to early fatality by postnatal day 25. Mpl–/– Dnm2Plt+/+ mice had impaired EB development at three weeks of age, which normalized with adulthood. Together, the data shows that DNM2-dependent Mpl-mediated endocytosis in platelets and MKs is required for steady-state hematopoiesis and provides novel insights into a developmentally controlled role for Mpl in normal erythropoiesis, regulating hemoglobin and heme production.
Key Points
● Dynamin 2 (DNM2)-dependent Mpl-mediated endocytosis in platelets and megakaryocytes is required for steady-state hematopoiesis.
● Mpl developmentally regulates mouse erythropoiesis.
Introduction
Blood platelets play an essential role in maintaining hemostasis and the integrity of the vasculature. High blood platelet count and platelet hyperactivation increase the risk of thrombosis and stroke, while low blood platelet count and platelet dysfunction predispose to hemorrhage. Platelets also participate in antimicrobial host defense and secrete cytokines that can induce inflammation and growth factors contributing to tissue repair. Thus, platelet homeostasis must be tightly regulated to avoid adverse effects of high or low platelet count.
Signaling of the hematopoietic cytokine thrombopoietin (TPO) through its receptor Mpl is essential for thrombopoiesis (1–3) and hematopoietic stem and progenitor cell (HSPC) maintenance (4–10). Patients with loss-of-function mutations in TPO or Mpl develop congenital amegakaryocytic thrombocytopenia (CAMT) and subsequent bone marrow failure (11–13). Mice lacking either TPO or Mpl have low megakaryocyte (MK) numbers and consequently develop severe thrombocytopenia (14–16). Hepatocytes are a major source of TPO, secreting the cytokine into the blood circulation (17). The mechanisms regulating circulating TPO levels are being debated. In one model, levels of circulating TPO are maintained solely by its uptake and metabolism by high-affinity Mpl receptors on platelets and MKs (18–23). In another model, circulating platelet levels regulate TPO mRNA expression in the liver by the proinflammatory cytokine interleukin 6 (IL-6), providing a regulated pathway to increase platelet production during acute inflammatory responses (24–26). More recent data suggest that the removal of aged, asialylated platelets stimulates hepatic TPO synthesis to maintain steady-state circulating TPO and platelet levels (27).
In platelets and MKs, the interaction between TPO and its receptor Mpl initiates an intracellular signaling cascade that involves the phosphorylation and activation of the tyrosine kinase JAK2 and the subsequent phosphorylation of signal transducer and activator of transcription (STAT) proteins (28). TPO binding to Mpl is also associated with cellular uptake of TPO and its subsequent degradation in a process regulated by receptor-mediated endocytosis (RME) (29, 30). RME plays an integral and physiologically relevant part in regulating plasma TPO levels. Cells expressing the JAK2V617F mutant commonly found in myeloproliferative neoplasm (MPN) patients display reduced recycling and increased degradation of Mpl, leading to elevated circulating TPO levels (31, 32). Mice specifically lacking Mpl or JAK2 in platelets and MKs, in which Mpl-mediated TPO endocytosis is blunted, display severe HSPC and MK hyperplasia and consequent thrombocytosis (33–35). However, the role of impaired TPO homeostasis in the HSPC and MK hyperplasia has not been conclusively demonstrated.
Dynamin 2 (DNM2) is a highly conserved GTPase essential for RME (36). DNM2 mutations in humans have been associated with Charcot-Marie-Tooth disease, centronuclear myopathy, and early T-cell precursor acute lymphoblastic leukemia (37–39). Ubiquitous Dnm2 deletion or loss of function in mice results in early embryonic lethality (40, 41). We have previously shown that Dnm2fl/fl Pf4-Cre (Dnm2Plt–/–) mice lacking DNM2 in platelets and MKs develop HSPC and MK hyperplasia, extramedullary hematopoiesis, and splenomegaly (42). Additionally, Dnm2Plt–/– mice develop severe macrothrombocytopenia and bleeding (43). At the cellular level, Dnm2Plt–/– platelets display constitutive activation but decreased expression of JAK2 and are unable to endocytose Mpl, leading to elevated circulating TPO levels.
Here, we assessed whether the hyperproliferative phenotype of Dnm2Plt–/– mice was due to JAK2 constitutive activation or elevated circulating TPO levels. STAT3 and to a lower extent STAT5 were phosphorylated in unstimulated Dnm2Plt–/– platelets. However, their phosphorylation was slowed and diminished when Dnm2Plt–/– platelets were stimulated with TPO. Additional Mpl deletion resulted in the loss of JAK2 and STAT3, but not STAT5 in Mpl–/– Dnm2Plt–/– platelets, linking JAK2 and STAT3 expression to Mpl and DNM2. At three weeks of age, Mpl–/– Dnm2Plt–/– mice displayed a near complete depletion of bone marrow MKs and significantly reduced HSPCs, indicating that Mpl is the primary receptor contributing to the hyperproliferative phenotype of Dnm2Plt–/– mice. However, Mpl–/– Dnm2Plt–/– mice showed severe anemia, erythroblast (EB) maturation defects, decreased expression of hemoglobin and heme homeostasis genes and increased expression of ribosome biogenesis and protein translation genes in spleen EBs, and grossly elevated plasma erythropoietin (EPO) levels, resulting in early fatality by postnatal day 25. Mpl–/– Dnm2Plt+/+ mice also displayed reduced EB development, which returned to normal with adulthood. Taken together, the data shows that DNM2-dependent Mpl-mediated endocytosis in platelets and MKs is required for steady-state hematopoiesis and provides novel insights into a developmentally controlled role for Mpl in normal erythropoiesis, regulating hemoglobin and heme production.
Methods
Mice
Dnm2Plt–/– mice were crossed with Mpl–/– mice to obtain mice lacking DNM2 in platelets and MKs and Mpl ubiquitously (14, 42). Mouse genotyping was confirmed by PCR of ear tissue DNA using primers: CCCTGCTAGTGACCTTTCTTGA (forward) and GCAGGAAGACACACAACTGAAC (reverse; Dnm2+ 172bp and Dnm2fl 271bp); CCTGTATTCCCAGAGTGTGCC (forward), GGAGCTTGAGCAGGTAGAGAG (reverse; Mpl+ 203bp), and CCAGCTCATTCCTCCCACTC (reverse; Mpl– 295bp); and AGATGCCAGGACATCAGGAACCTG (forward) and ATCAGCCACACCAGACACAGAGATC (reverse; Pf4-iCre 237bp). Mice were treated according to the National Institutes of Health and Medical College of Wisconsin Institutional Animal Care and Use Committee guidelines (Animal Use Application 5600).
Platelet preparation
Mouse blood was collected from the retroorbital plexus in anticoagulant citrate dextrose solution (43). Platelet-rich plasma was obtained by centrifugation of the blood at 100 g for 8 min, followed by centrifugation of the supernatant and buffy coat at 100 g for 6 min. After washing twice in washing buffer (140 mM NaCl, 5 mM KCl, 12 mM trisodium citrate, 10 mM glucose, and 12.5 mM sucrose, pH 6.0), platelets were resuspended at 4 x 10 (8) platelets/ml in resuspension buffer (140 mM NaCl, 3 mM KCl, 0.5 mM MgCl2, 5 mM NaHCO3, 10 mM glucose, 10 mM HEPES, pH 7.4) and were allowed to rest for 30 min before use.
Immunoblot analysis
Platelets were lysed in 1% Nonidet P-40, 150 mM NaCl, and 50 mM Tris/HCl, pH 7.4, containing 1 mM EGTA, 1 mM sodium orthovanadate, and cOmplete Protease Inhibitor Cocktail (Roche). SDS-PAGE buffer was added to lysates in the presence of 1% β-mercaptoethanol. Proteins were resolved by SDS-PAGE following quantification by Bradford protein assay and transferred onto PVDF membrane. After blocking overnight with 1% BSA in 0.2% Tween-20, 100 mM NaCl, and 20 mM Tris/HCl, pH 7.4, membranes were probed with rabbit antibodies directed against total or phosphorylated STAT3 (Tyr705) and STAT5 (Tyr694 in STAT5A; Tyr699 in STAT5B) (Cell Signaling), JAK2, β-actin, or GAPDH, followed by secondary horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (Thermo Fisher Scientific). Detection was performed by enhanced chemiluminescence.
Complete blood counts
Mouse blood was collected from retroorbital plexus and diluted in Cellpack (Sysmex) supplemented with EDTA and PGE1 (43, 44). Complete blood counts were measured on a Sysmex XT-2000i automatic hematology analyzer.
TPO and EPO levels
Plasma EPO and TPO levels were quantified using a Mouse Thrombopoietin and Erythropoietin Quantikine ELISA kit (R&D Systems), respectively, following manufacturer recommendations (42).
Cryosectioning and immunolabeling
Femurs of mice were fixed overnight in 1% paraformaldehyde/phosphate-lysine-sodium periodate and cryoprotected for at least 72 h in a 30% sucrose/phosphate buffer solution at 4°C before subsequent freezing in Sakura Tissue Tek O.C.T. compound (Andwin Scientific). Frozen cryosectioning on slides was performed at the Medical College of Wisconsin Histological Laboratory and Core Center. For MK counts, 7-µm femur sections were rehydrated and permeabilized in TBS-T for 15 min at room temperature (RT) then blocked overnight in a 5% BSA/PBS solution at 4°C. Sections were incubated for 2 h at RT with monoclonal rat-anti-GPIbα (Emfret Analytics) and polyclonal rabbit anti-laminin (Sigma-Aldrich) followed by a 1 h RT incubation with conjugated secondary antibodies (Molecular Probes). Sections were washed and mounted with Prolong Diamond Antifade Mountant with DAPI (Invitrogen) and imaged on a Nikon Eclipse Ti2-E platform equipped with a DS-Qi2 camera and Plan Apo 10x/0.45 (NIS-Elements AR 5.02.00 software). Data were image-processed using Imaris (Bitplane) and Matlab (Mathworks) softwares. Surfaces were created toward the greatest signal intensities GPIbα-positive cells and quantitatively analyzed using Imaris and Excel (Microsoft).
Bone marrow and spleen histology
Mouse femurs and spleens were fixed overnight in 4% paraformaldehyde/PBS. Bones were decalcified in 0.5 M EDTA, pH 8.0 (Boston BioProducts) for 7 days under rotation, exchanging EDTA twice daily. Tissues were paraffin embedded, and sections were stained with hematoxylin and eosin (H&E) at the Versiti Blood Research Institute and Medical College of Wisconsin Histology Core.
Blood smears
Blood smears were performed via Wright-Giemsa stain. Anticoagulated whole blood was thinly smeared across a glass slide and fixed for 3 min in methanol, stained 1 min in Wright-Giemsa, and washed for 5 min in PBS. Imaging was performed on a Nikon Eclipse E600 microscope equipped with a SPOT insight firewire color mosaic camera (SPOT imaging solutions) and Plan Apo 40x/0.75 objective, with SPOT imaging 5.1.3 software.
Flow cytometry analysis
Spleen and bone marrow cells were collected and homogenized through a 70-µm filter. For the EB analysis, spleen cells were stained with FITC-conjugated anti-CD71 and PE-conjugated anti-TER-119 (BD Biosciences) after homogenization and washing (45, 46). For the HSPC analysis, bone marrow cells were prepared for staining by erythrocyte lysis (BD Pharm Lyse; BD Biosciences). Cells were then stained in ice-cold PBS containing 2% FBS using the following antibodies: lineage cocktail containing TER-119, CD11b (Mac-1), Ly-6G/Ly-6C (Gr-1), CD3ϵ, and CD45R (B220); CD117 (Kit); Ly-6A/E (Sca-1); CD150 (Slamf1); CD48 (Slamf2); CD16/32; CD34; CD41 (Itga2b); and CD105 (endoglin) (BioLegend and eBiosciences). 4′,6 Diamidino-2-phenylindole (Invitrogen) was used for dead cell discrimination. SLAM and MKEP panels used were described previously (47–49). Samples were analyzed by flow cytometry using an LSR II (BD Biosciences). Post-acquisition analysis of data was performed with FlowJo software.
Erythroblast Dnm2 DNA analysis
Spleen cells were collected and homogenized through a 70-µm filter and leukocytes were depleted using anti-CD45 beads (Miltenyi Biotec). Remaining cells were stained with FITC-conjugated anti-CD71, PE-conjugated anti-TER-119, and APC-conjugated anti-CD45 as control. Immature CD71high EBs were collected on a FACSAria II cell sorter (BD Biosciences). EB genomic DNA was obtained using the QIAamp Mini DNA kit (Qiagen). Duplicates of real-time PCR experiments were performed on a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems) amplifying Dnm2 and Rn18s as reference (50). Primers used were: CCCTGCTAGTGACCTTTCTTGA (forward) and GCAGGAAGACACACAACTGAAC (reverse; Dnm2fl 271bp); and TTGACGGAAGGGCACCACCAG (forward) and GCACCACCACCCACGGAATCG (reverse; Rn18s 131bp). Ct numbers were extracted for both Dnm2 and Rn18s with auto baseline and manual threshold.
Erythroblast Bulk RNA sequencing analysis
EB RNA was isolated using Trizol reagent (Invitrogen) and an Autogen Prep-245 system and was assessed with the Bioanalyzer RNA Nano Assay (Agilent). All samples had observed RNA Integrity Number values >7.4 with DV200 over 81%. RNA libraries were prepared (Illumina TruSeq Stranded mRNA, single indexed) and run on the Illumina High Seq-2500 for 125bp paired end reads at the Medical College of Wisconsin Genomic Sciences and Precision Medicine Center. Samples were sequenced to an average depth of 40 million reads. All data was quality controlled using FastQC and RSeQC, followed by manual review and data visualization (51). Bulk RNA-seq data were aligned to the Mus musculus mm10 genome and quality control was performed using Nextflow pipeline (nf-core/rnaseq 1.4.2) (DOI:10.5281/zenodo.1400710) (52). Gene expression was quantified at the gene level using Salmon. RNA-seq libraries were then normalized and genes were tested for differential expression between Dnm2Plt+/+, Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/– samples with DESeq2 v1.24.0 (53). DESeq2 Wald tests were used to determine whether fold changes were significantly different from zero. For visualization, data were transformed using the regularized logarithm transformation (53). Pre-ranked gene set enrichment analyses were conducted using shrunken fold-changes and clusterProfiler v3.12.0 (54). Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, and GO databases were used for Gene Set Enrichment Analysis (GSEA) (55–57). The Benjamini-Hochberg method was used to adjust p-values for false-discovery in both differential expression and GSEA analyses (58). Genes were defined as differentially expressed if they were upregulated or downregulated 1.5-fold with an adjusted P-value <.05.
Statistical analysis
Results were compared statistically with the unpaired Student’s t-test (mean comparison between two groups), one- and two-way ANOVA (mean comparison between multiple groups), and the Log-rank test (survival distribution comparison) using Prism software (GraphPad). Differences were considered statistically significant when P <.05.
Results
Impaired STAT signaling in Dnm2Plt–/– platelets
The TPO-Mpl interaction initiates a signaling cascade that involves JAK2 activation and the subsequent phosphorylation of STAT proteins (28). Dnm2Plt–/– mice specifically lacking DNM2 in platelets and MKs develop HSPC and MK hyperplasia, extramedullary hematopoiesis, and splenomegaly (42). While JAK2 expression is decreased in Dnm2Plt–/– platelets, its TPO-independent phosphorylation at tyrosine residues 1007 and 1008 indicates constitutive activation (42). To understand the alterations in JAK2-STAT signaling, we evaluated the phosphorylation of STAT3 and STAT5 in Dnm2Plt–/– platelets (Figures 1A−C). As expected, STAT3 and STAT5 were not phosphorylated in unstimulated control Dnm2Plt+/+ platelets. Incubation of Dnm2Plt+/+ platelets with 50 ng/ml of TPO resulted in STAT3 and STAT5 phosphorylation on tyrosine residues 705 and 694/699, respectively, that began at 2 min and became maximal at 5 min. We observed STAT3 and to a lower extent STAT5 phosphorylation in unstimulated Dnm2Plt–/– platelets. However, compared to controls, STAT3 and STAT5 phosphorylation was slowed and diminished in Dnm2Plt–/– platelets following stimulation with TPO. The data is consistent with JAK2 constitutive activation but decreased expression in Dnm2Plt–/– platelets.
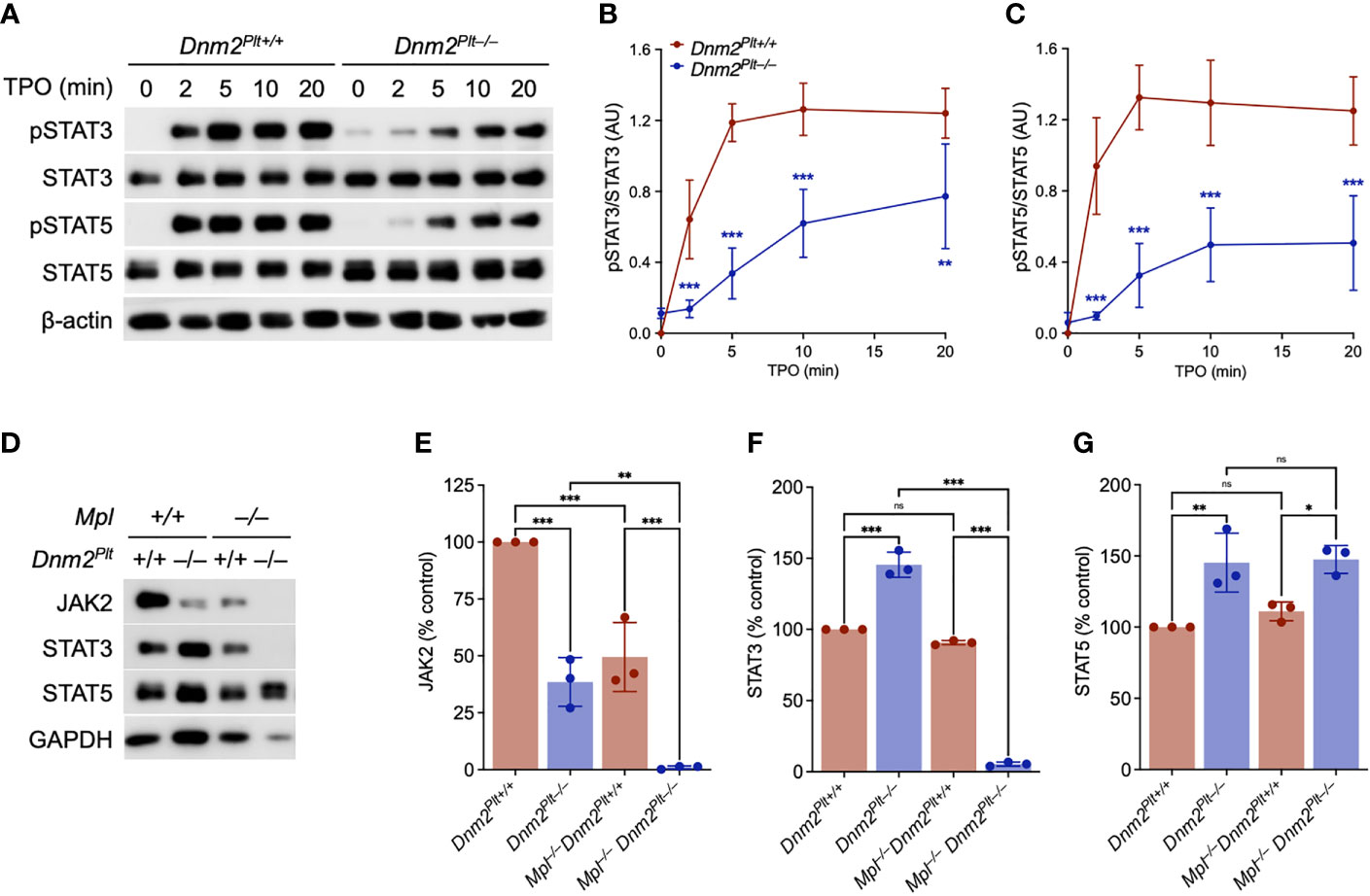
Figure 1 JAK2-STAT signaling defects in Dnm2Plt–/– platelets. (A) Dnm2Plt+/+ and Dnm2Plt–/– platelets were activated with 50 ng/ml TPO for 10 min at 37°C, lysed, subjected to SDS-PAGE, and probed for total and phosphorylated STAT3 (pSTAT3; Tyr705), total and phosphorylated STAT5 (pSTAT5; Tyr694 in STAT5A, Tyr699 in STAT5B), and β-actin as loading control, as indicated. Densitometry analysis of STAT3 (B) and STAT5 (C) phosphorylation. Results represent mean ± SD of 4 independent experiments and are compared statistically by two-way ANOVA (**,P <.01; ***, P <.001). (D) Platelet lysates of Dnm2Plt+/+, Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/– mice at P24 corresponding to 2 µg of protein were subjected to SDS-PAGE and probed for JAK2, STAT3, STAT5, and GAPDH as loading control, as indicated. Results are representative of 3 independent experiments. Densitometry analysis of JAK2 (E), STAT3 (F), and STAT5 (G) expression. Results represent mean ± SD of 3 independent experiments and are compared statistically by one-way ANOVA (ns, not significant, *P <.05; **P <.01; ***P <.001).
To ascertain the role of impaired Mpl-mediated endocytosis in the hyperproliferative phenotype, we crossed Dnm2Plt–/– mice in the Mpl–/– background to generate mice lacking DNM2 in platelets and MKs and Mpl ubiquitously. We measured a ~60% and ~50% reduction in JAK2 expression in platelets lacking DNM2 and Mpl, respectively (Figures 1D, E). By contrast, expression of STAT3 and STAT5 was increased by ~40% in platelets lacking DNM2, but was unaffected by Mpl deletion (Figures 1D, F, G). The combined deletion of DNM2 and Mpl resulted in the loss of JAK2 and STAT3, but not STAT5 in Mpl–/– Dnm2Plt–/– platelets (Figures 1D−G). Together, the data links JAK2 and STAT3, but not STAT5 homeostasis in platelets to Mpl and DNM2 expression. While proteins were loaded according to protein amount, no standard controls (β-actin, β-tubulin, GAPDH) gave a good signal for Mpl–/– Dnm2Plt–/– platelets, suggesting major protein up- and down-regulation.
Early lethality in Mpl–/– Dnm2Plt–/– mice
We crossed Mpl–/– Dnm2Plt+/+ and Mpl+/– Dnm2Plt–/– mice and obtained four offspring genotypes, i.e., Mpl+/– Dnm2Plt+/+, Mpl+/– Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/–, with a normal Mendelian inheritance ratio at birth (data not shown). Mpl+/– Dnm2Plt+/+, Mpl+/– Dnm2Plt–/–, and Mpl–/– Dnm2Plt+/+ mice reached adulthood (Figure 2A), as described previously for Dnm2Plt–/– and Mpl–/– mice (14, 42). By contrast, Mpl–/– Dnm2Plt–/– mice became pale and moribund (data not shown) and died at a median age of 25 days postnatally (Log-rank P <.001).
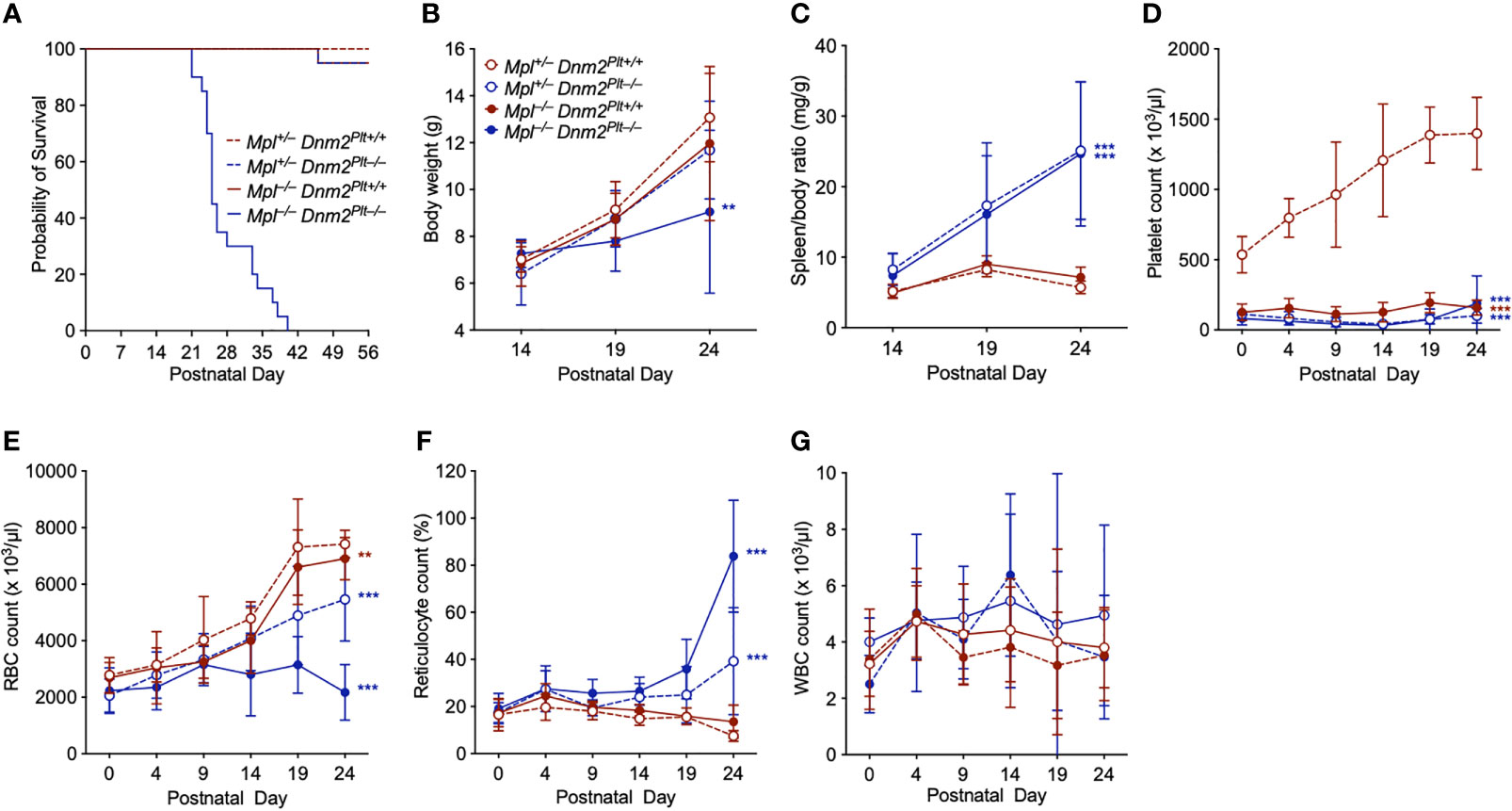
Figure 2 Mpl–/– Dnm2Plt–/– mice exhibit early lethality, failure to thrive, splenomegaly, and severe anemia. (A) Survival of Mpl+/– Dnm2Plt+/+, Mpl+/– Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/– littermates. Results are estimated using the Kaplan-Meier method and are compared statistically using the Log-rank test (n = 20 mice in each group: ***, Log-rank P <.001). Body weights (B) and spleen/body ratio (C) at P14, P19, and P24. Platelet (D), RBC (E), reticulocyte (F), and WBC (G) counts from birth (P0) to postnatal day 24 (P24). Results represent mean ± SD of 5-19 independent experiments and are compared statistically by two-way ANOVA (**P <.01; ***P <.001).
We measured body weights at postnatal day 14 (P14), P19, and P24 (Figure 2B). While Mpl+/– Dnm2Plt+/+, Mpl+/– Dnm2Plt–/–, and Mpl–/– Dnm2Plt+/+ mice gained weight over time, Mpl–/– Dnm2Plt–/– mice failed to thrive after P14 and showed diminutive growth at P24, with a body weight of 9.05 ± 3.48 g, compared to 13.07 ± 1.88 g in Mpl+/– Dnm2Plt+/+ littermates, a 31% decrease (P = .005). DNM2 deletion in platelets and MKs led to severe splenomegaly, independently of Mpl expression (Figure 2C), indicating that the extramedullary hematopoiesis of Dnm2Plt–/– mice was not related to impaired Mpl-mediated endocytosis in platelets and MKs.
Severe anemia in Mpl–/– Dnm2Plt–/– mice
To understand the cause of early mortality of Mpl–/– Dnm2Plt–/– mice, we measured hematological parameters between birth (P0) and P24. In Mpl+/– Dnm2Plt+/+ mice, the platelet count increased from 536 ± 130 x 103/µl at P0 to 1398 ± 257 x 103/µl at P24 (Figure 2D), as described for control Mpl+/+ mice (44). Consistent with previous observations (14, 42), mice lacking DNM2 in platelets and MKs and/or Mpl ubiquitously developed severe thrombocytopenia, with platelet counts constantly below 200 x 103/µl, which was observed at birth and throughout development.
The RBC count rose continuously in Mpl+/– Dnm2Plt+/+, Mpl+/– Dnm2Plt–/–, and Mpl–/– Dnm2Plt+/+ mice (Figure 2E). By contrast, the RBC count failed to increase after P14 in Mpl–/– Dnm2Plt–/– mice, which displayed severe anemia and an aberrant increase in reticulocyte count at P24 (Figure 2F). All four mouse genotypes had a normal white blood cell (WBC) count throughout development (Figure 2G). Together, the data suggested that DNM2 deletion in platelets and MKs combined with Mpl ubiquitous deletion induced an age-dependent lethal anemia in Mpl–/– Dnm2Plt–/– mice. We, therefore, investigated megakaryopoiesis and erythropoiesis in further detail.
The MK hyperplasia of Dnm2Plt–/– mice requires Mpl expression
We evaluated platelet counts and bone marrow megakaryopoiesis at P24 and P56 (Figure 3). At P24, DNM2 deletion in platelets and MKs and/or Mpl ubiquitous deletion resulted in comparable severe thrombocytopenia with platelet counts of 111 ± 43 x 103/µl in Dnm2Plt–/–, 158 ± 53 x 103/µl in Mpl–/– Dnm2Plt+/+, and 189 ± 196 x 103/µl in Mpl–/– Dnm2Plt–/–, compared to 1243 ± 300 x 103/µl in control Dnm2Plt+/+ mice (P <.0001) (Figure 3A). Similar low platelet counts were obtained at P56, with 152 ± 57 x 103/µl in Dnm2Plt–/– and 151 ± 79 103/µl in Mpl–/– Dnm2Plt+/+ mice, compared to 1299 ± 227 x 103/µl in control Dnm2Plt+/+ mice (P <.001) (Figure 3E). Due to their early lethality, hematological parameters could not be evaluated at P56 for Mpl–/– Dnm2Plt–/– mice.
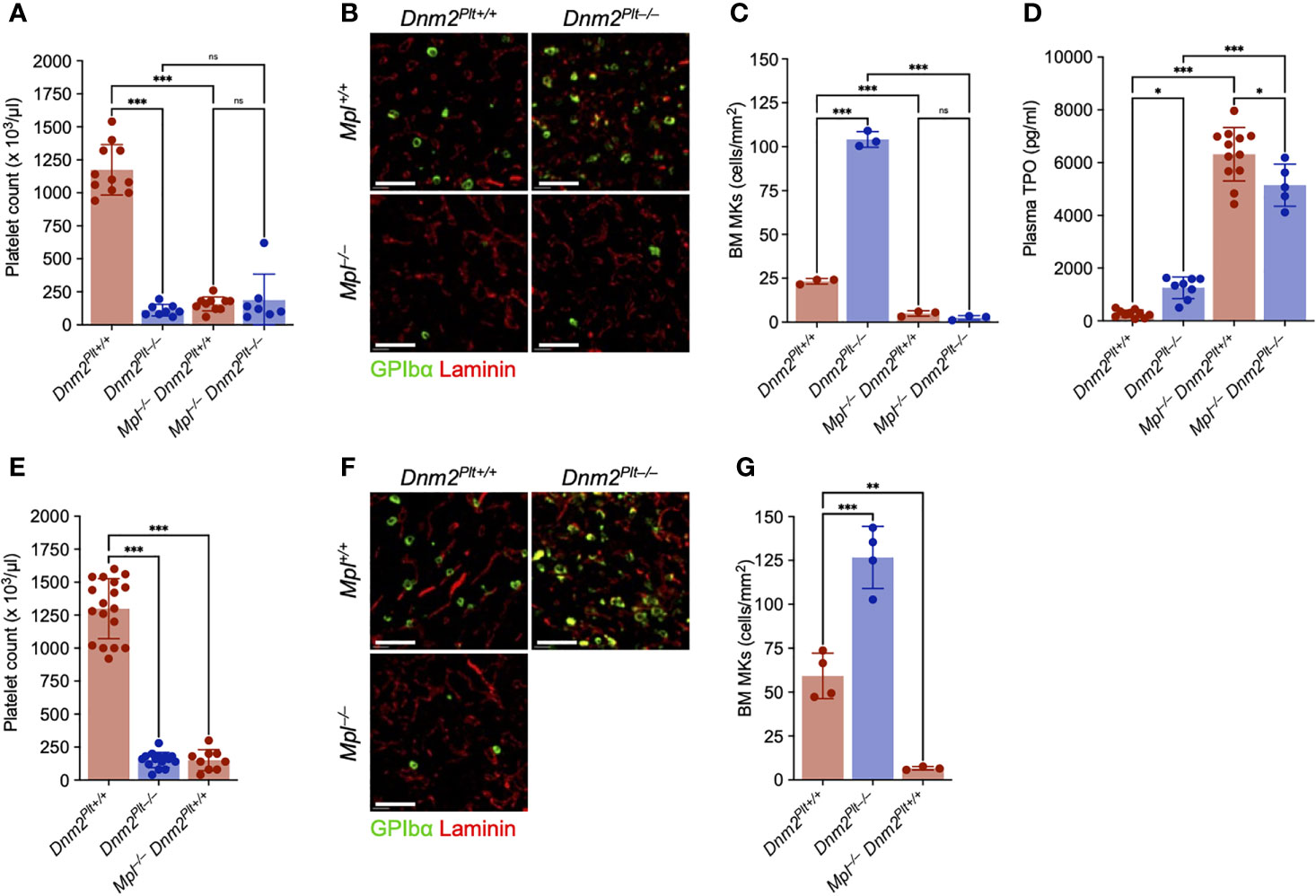
Figure 3 The MK hyperplasia of Dnm2Plt–/– mice requires Mpl expression. Platelet count at P24 (A) and P56 (E). Results represent mean ± SD of 7-13 independent experiments and are compared statistically by one-way ANOVA (ns, not significant, ***, P <.001). Seven-µm frozen bone marrow sections of Dnm2Plt+/+, Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/– mice at P24 (B) and P56 (F) were probed for resident MKs (GPIbα, green) and bone marrow vasculature (laminin, red). Sections shown are representative of 3-4 mice in each genotype. Scale bars represent 150 µm. Bone marrow MK numbers at P24 (C) and P56 (G). Results represent mean ± SD of 3-4 independent experiments and are compared statistically by one-way ANOVA (ns, not significant, **P <.01; ***P <.001). (D) Plasma TPO levels at P24. Results represent mean ± SD of 5-16 independent experiments and are compared statistically by one-way ANOVA (*P <.05; **P <.01; ***P <.001).
We evaluated bone marrow MKs by immunofluorescence microscopy using an antibody directed against GPIbα. At P24, Dnm2Plt–/– mice displayed a severe MK hyperplasia, with 104.1 ± 4.5 MKs/mm2, compared to 23.3 ± 1.6 MKs/mm2 in control Dnm2Plt+/+ mice (P <.001), a 4.5-fold increase (Figures 3B, C). Mpl deletion resulted in near-complete depletion of bone marrow MKs in Mpl–/– Dnm2Plt–/– mice (2.5 ± 1.3 MKs/mm2), like Mpl–/– Dnm2Plt+/+ mice (5.0 ± 1.6 MKs/mm2). Comparable results were obtained by H&E staining in femur bone marrow and spleen tissues (Supplementary Figures 1A, B), and the MK hypoplasia of Mpl–/– Dnm2Plt+/+ mice remained at P56 (Figures 3F, G). The data demonstrated that the MK hyperplasia of Dnm2Plt–/– mice required the expression of the TPO receptor Mpl.
Consistent with the loss of DNM2-dependent Mpl-mediated endocytosis, plasma TPO levels were elevated in mice lacking DNM2 in platelets and MKs and/or Mpl ubiquitously: 1253 ± 411 pg/ml in Dnm2Plt–/–, 6318 ± 1014 pg/ml in Mpl–/– Dnm2Plt+/+, and 5144 ± 800 pg/ml in Mpl–/– Dnm2Plt–/–, compared to 269 ± 139 pg/ml in control Dnm2Plt+/+ mice (P <.001) (Figure 3D). Together, the data showed that Mpl-mediated endocytosis in platelets and MKs required DNM2 expression to regulate plasma TPO levels.
Severe erythroid maturation defects in Mpl–/– Dnm2Plt–/– mice
Because of the escalating RBC deficit and death at P25, we evaluated RBC counts and erythroid maturation more in detail at P24 (Figures 4A−E). Dnm2Plt–/– and Mpl–/– Dnm2Plt+/+ mice had normal RBC counts with and 7247 ± 513 x 103/µl and 6900 ± 750 x 103/µl, respectively, compared to 7255 ± 727 x 103/µl in control Dnm2Plt+/+ mice (Figure 4A), indicating that individual loss of either DNM2 in platelets and MKs or Mpl ubiquitously does not affect RBC counts. By contrast, DNM2 deletion in platelets and MKs combined with ubiquitous Mpl deletion led to a significant decrease in RBC count to 2171 ± 983 x 103/µl in Mpl–/– Dnm2Plt–/– mice (P <.001). The severe anemia of Mpl–/– Dnm2Plt–/– mice was accompanied by a grossly elevated reticulocyte count of 83.8 ± 23.8%, compared to 7.3 ± 1.4%, 14.6 ± 7.1%, and 13.5 ± 7.0% in Dnm2Plt+/+, Dnm2Plt–/–, and Mpl–/– Dnm2Plt+/+ mice, respectively (P <.001) (Figure 4B). Analyzing thin blood smears showed that Mpl–/– Dnm2Plt–/– mice developed marked polychromasia (Supplementary Figure 1C), suggesting premature release during RBC formation.
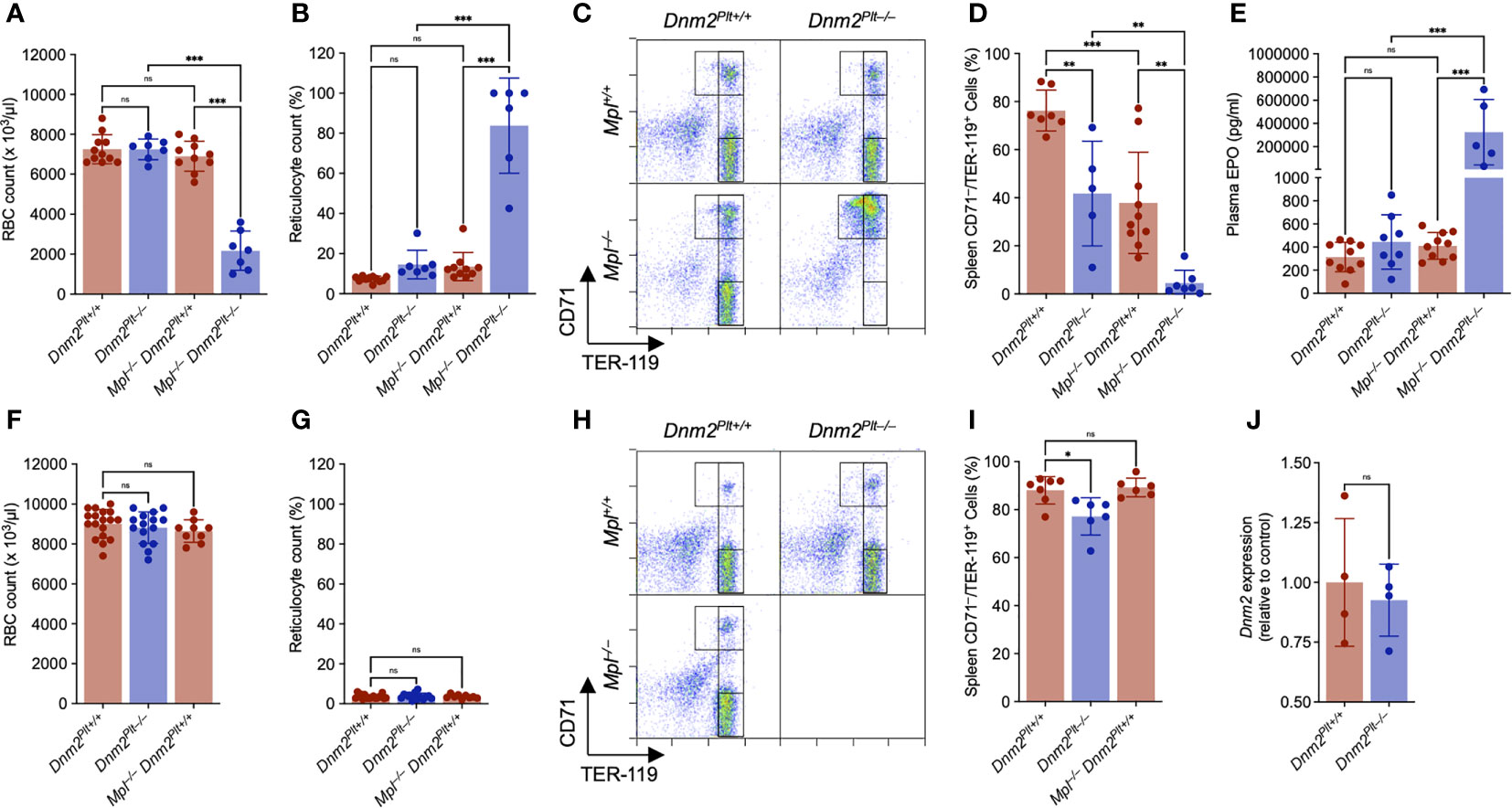
Figure 4 Mpl–/– Dnm2Plt–/– mice exhibit impaired erythroid development at P24. RBC count at P24 (A) and P56 (F). Reticulocyte count at P24 (B) and P56 (G). Results represent mean ± SD of 4-18 independent experiments and are compared statistically by one-way ANOVA (ns, not significant, ***P <.001). Flow cytometry profiles of spleen EBs at P24 (C) and P56 (H) using the erythroid markers CD71 and TER-119. Data shown are representative of 3-13 mice in each genotype. Percentage of mature spleen CD71low/TER-119high EBs at P24 (D) and P56 (I). Results represent mean ± SD of 5-10 independent experiments and are compared statistically by one-way ANOVA (ns, not significant, *P <.05; **P <.01; ***P <.001). Plasma EPO levels at P24 (E). Results represent mean ± SD of 5-15 independent experiments and are compared statistically by one-way ANOVA (ns, not significant, ***P <.001). (J) Genomic Dnm2 expression in isolated spleen CD71high EBs was evaluated by qPCR and normalized to Dnm2Plt+/+ cells. Results represent mean ± SD of 4 independent experiments and are compared statistically by Student’s t-test. (ns, not significant).
Erythroid maturation at P24 was evaluated by flow cytometry analysis using CD71 and TER-119 as surface erythroid markers (Figure 4C), where immature EBs are defined as CD71high/TER-119low and mature EBs as CD71low/TER-119high (45, 46). In the spleens of control Dnm2Plt+/+ mice, 76.3 ± 8.5% of erythroid cells were mature CD71low/TER-119high EBs (Figure 4D). The population decreased to 41.7 ± 21.7% in Dnm2Plt–/– mice (P = .006) and 37.9 ± 21.1% in Mpl–/– Dnm2Plt+/+ mice (P <.001). In the spleens of Mpl–/– Dnm2Plt–/– mice, only 4.5 ± 5.3% (P <.001) of erythroid cells were marked as mature EBs, as the majority failed to properly develop beyond an earlier CD71high stage.
While plasma EPO levels were in the normal range in Dnm2Plt–/– and Mpl–/– Dnm2Plt+/+ mice, they were grossly elevated in Mpl–/– Dnm2Plt–/– mice, with 325 ± 282 ng/ml, compared to 314 ± 128 pg/ml in control Dnm2Plt+/+ mice (P <.001), a ~1000-fold increase (Figure 4E). The data showed that the severe anemia of Mpl–/– Dnm2Plt–/– mice was due to a blockade of erythroid maturation at an early CD71high stage. Thus, the loss of DNM2 in platelets and MKs combined with Mpl ubiquitous deletion caused a severe defect in erythrocyte development and maturation.
Mpl contributes to erythroblast maturation during early mouse development
While Mpl deficiency has been associated with pancytopenia in humans (59), anemia has not been observed in mice lacking either Mpl or TPO, which mainly develop thrombocytopenia (14–16). To understand how our observations at P24 differ from these previous studies, we evaluated RBC counts and erythroid maturation in adult mice (Figures 4F−I). As observed at P24, the RBC and reticulocyte count at P56 was normal in Dnm2Plt–/– and Mpl–/– Dnm2Plt+/+ mice (Figures 4F, G). Evaluating erythroid maturation further, the mature CD71low/TER-119high EB population in Dnm2Plt–/– mice increased between P24 and P56 to 77.2 ± 7.8%, although it did not reach control levels (P = .01) (Figures 4H, I). Mpl–/– Dnm2Plt+/+ mice had normal erythropoiesis at P56, with 89.3 ± 3.9% mature EBs, compared to 88.1 ± 5.7% in control Dnm2Plt+/+ mice. Thus, the TPO-Mpl dependence of erythroid maturation was not evident when mice reached adulthood, demonstrating a developmental role for Mpl in erythropoiesis.
DNM2 is normally expressed in Dnm2Plt–/– erythroblasts
Because ubiquitous Dnm2 deletion or loss of function leads to microcytic anemia and embryonic lethality (40, 41), we investigated whether Dnm2 was erroneously excised in Dnm2Plt–/– EBs (Figure 4J). Genomic DNA was collected from Dnm2Plt+/+ and Dnm2Plt–/– CD71high spleen EBs and probed by quantitative PCR using primers flanking the 5’ Dnm2 loxP site, in conditions where the reverse Dnm2 primer would not have a template to anneal to and no PCR product would be generated if the region between the two Dnm2 loxP sites had been excised (40). Dnm2Plt+/+ and Dnm2Plt–/– EBs yielded comparable amount of Dnm2fl PCR product, compared to control 18S rRNA, demonstrating that Dnm2 was not excised and therefore DNM2 was normally expressed in Dnm2Plt–/– EBs.
Hematopoietic stem and progenitor cell (HSPC) dysregulation in Mpl–/– Dnm2Plt–/– mice
We performed a quantitative assessment of the bone marrow HSPC compartment at P24 using flow cytometry and well-established immunophenotypic markers (Figure 5) (47–49). Compared to control Dnm2Plt+/+ mice, the lineage–/Sca-1+/Kit+ (LSK) compartment of Dnm2Plt–/– mice was significantly expanded (Figure 5A), including long-term CD150+/CD48– (Figure 5B) and short-term CD150–/CD48– (Figure 5C) HSCs (LT- and ST-HSCs, respectively). The LSK compartment was severely and equally reduced in Mpl–/– Dnm2Plt+/+ and Mpl–/– Dnm2Plt–/– mice. The lack of LT- and ST-HSCs in the absence of Mpl is consistent with the reported requirement for TPO and Mpl in regulating HSCs (15, 60, 61).
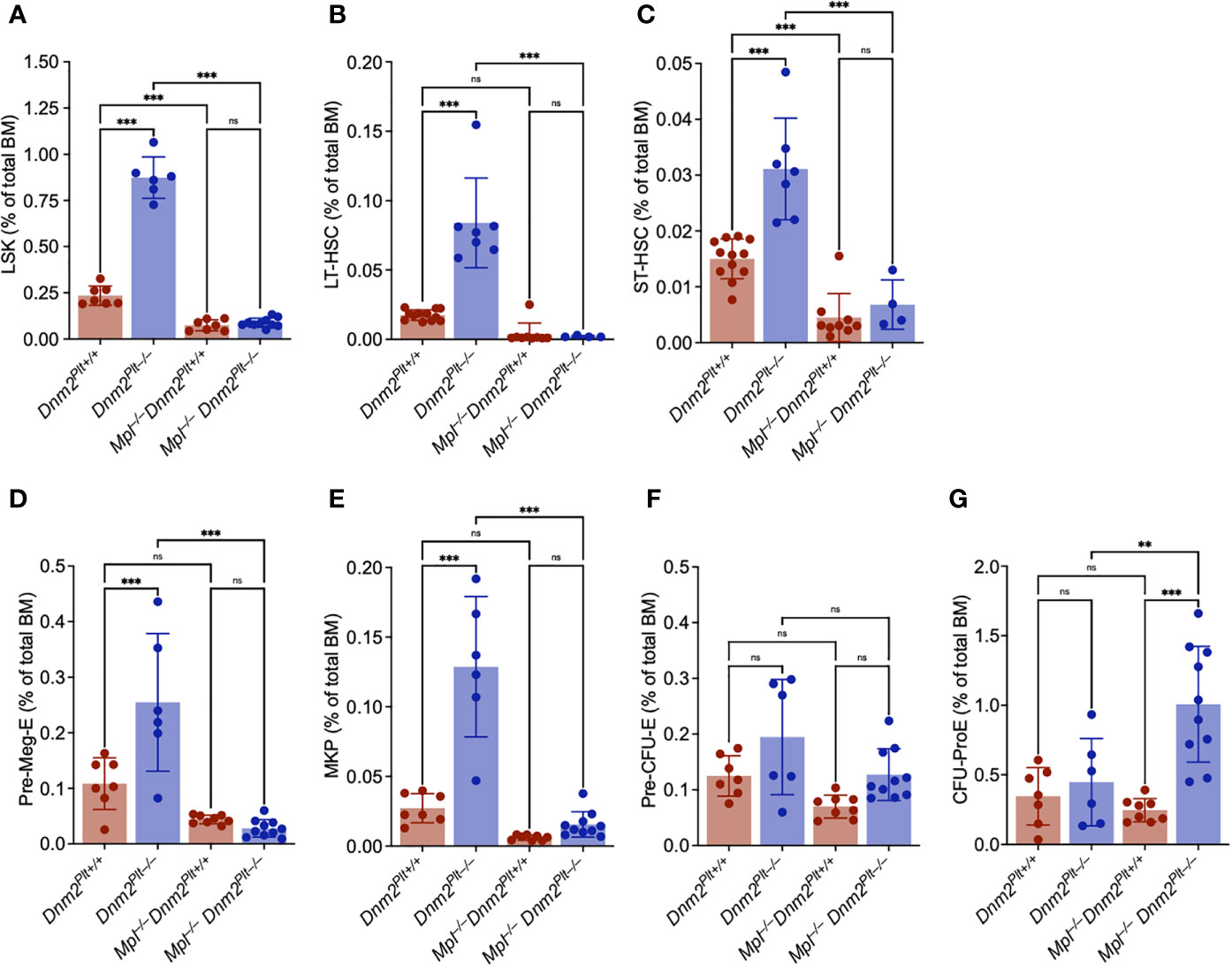
Figure 5 The increased HSPC expansion of Dnm2Plt–/– mice requires Mpl expression. Frequency of bone marrow Lin–/Sca1+/Kit+ (LSK) (A), long-term (LT)-HSC (B), short-term (ST)-HSC (C), Pre-Meg-E (D), MK progenitor (MKP) (E), Pre-CFU-E (F), and CFU-ProE (G) in Dnm2Plt+/+, Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/– mice at P24. Results represent mean ± SD of 4-12 independent experiments and are compared statistically by one-way ANOVA (**P <.01; ***P <.001). ns, not significant.
MK and erythroid progenitors were evaluated in further details. Consistent with the observed MK hyperplasia and studies in adult mice (42), Dnm2Plt–/– mice displayed an expansion of the Pre-Meg-E (Figure 5D) and MK progenitor (MKP) (Figure 5E) compartments. The Pre-Meg-E and MKP expansion was abrogated in Mpl–/– Dnm2Plt–/– mice, demonstrating that it was mediated by Mpl ubiquitous expression. While the Pre-CFU-E compartment was minimally affected by deletion of either DNM2 in platelets and MKs or Mpl ubiquitously (Figure 5F), the CFU-ProE compartment was elevated in Mpl–/– Dnm2Plt–/– mice (Figure 5G). Together, the data demonstrated that the loss of DNM2-dependent Mpl-mediated endocytosis in platelets and MKs was responsible for the expansion of the LSK, Pre-Meg-E, and MKP compartments in Dnm2Plt–/– mice. It also led to an expansion of the CFU-ProE compartment in Mpl–/– Dnm2Plt–/– mice, consistent with the observed erythroid maturation arrest at the stage of CD71high immature EBs.
Genome-wide transcriptome effects in erythroblasts
To evaluate the role of Mpl ubiquitous deletion and DNM2 deletion in platelets and MKs on erythropoiesis, spleen EBs isolated from Dnm2Plt+/+, Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/– mice at P24 were subjected to transcriptional profiling (n = 3 in each cohort). A total of 35,324 genes were retained, and two separate gene sets were generated to assess the effects of Mpl ubiquitous deletion or DNM2 deletion in platelets and MKs.
Following the analysis of Mpl–/– Dnm2Plt+/+ and Mpl–/– Dnm2Plt–/–, compared to Dnm2Plt+/+ and Dnm2Plt–/– mice, the Mpl effect gene set contained 45 upregulated and 401 downregulated genes (Figures 6A, B; Supplementary Table 1). Notably, hemoglobin (Hbb-bs, Hbb-bt, Hbq1a) and heme homeostasis genes (Alas2, Fech, Bpgm, Ftl1, Slc48a1) were downregulated. Delving further into the transcriptomic data, the heat map of average FPKM values from all four groups showed that hemoglobin and heme homeostasis genes were severely decreased in mice lacking Mpl ubiquitously, independently of DNM2 deletion in platelets and MKs (Figure 6E). Together, the data indicate that Mpl plays a critical role in EB maturation by regulating hemoglobin and heme production, both critical to erythropoiesis (62). Additional relevant downregulated genes are involved in ubiquitination (Ubb, Mkrn1, Marchf2, Usp15) and ribosome recruitment and translation initiation (Pabpc1). Alternatively, H2A cluster histone genes (H2ac7, H2ac10, H2ac12, H2ac13, H2ac14) were upregulated in Mpl–/– >Dnm2Plt+/+ and Mpl–/–Dnm2Plt–/– EBs, compared to Dnm2Plt+/+ and Dnm2Plt–/– EBs. Genes involved in inflammation (S100a8, S100a9, Cyba, Cybb, Ncf1) were decreased in all three thrombocytopenic genotypes, while platelet genes (Gp1bb, Pf4) were elevated in Dnm2Plt–/– mice presenting MK hyperplasia (Figure 6E).
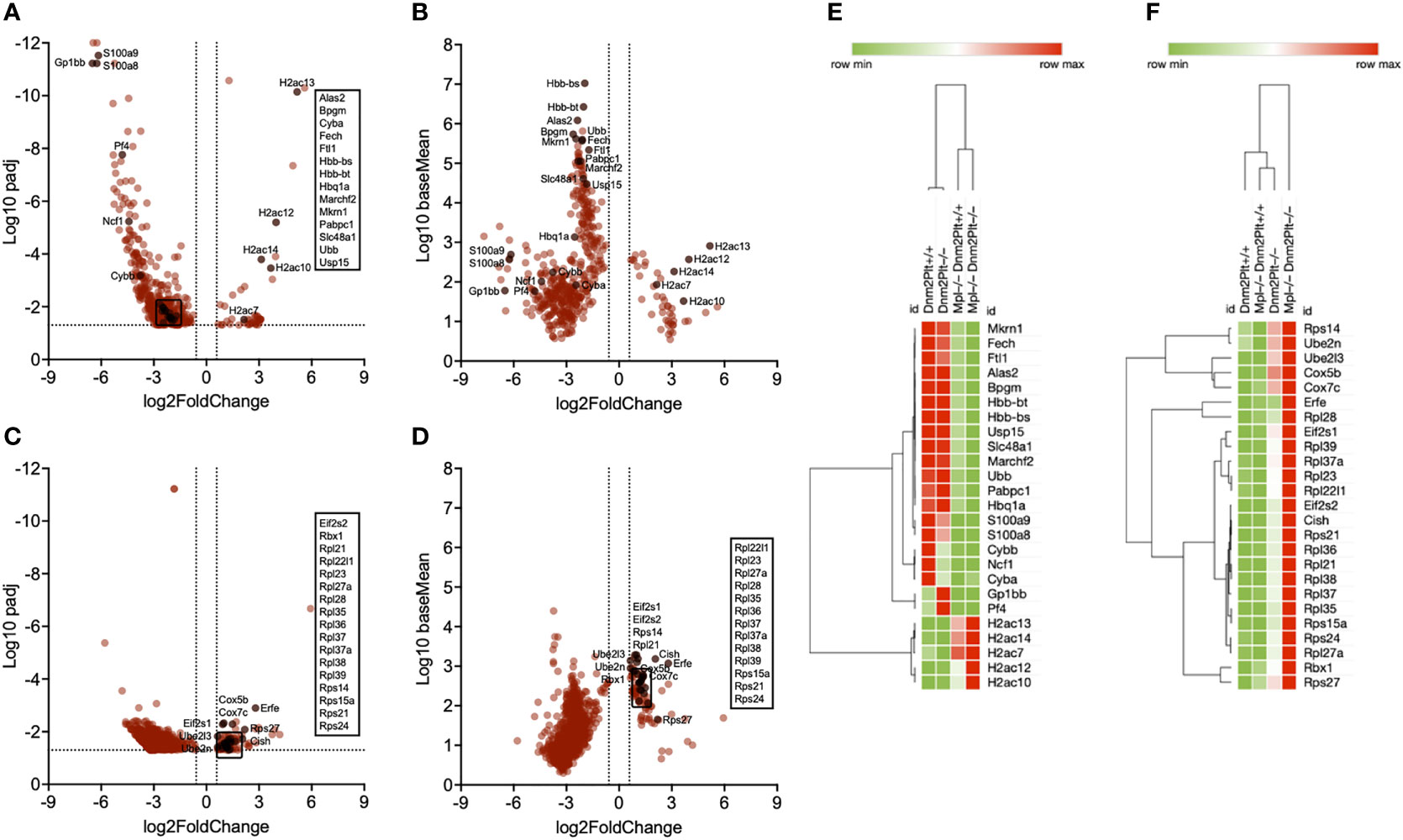
Figure 6 Transcriptional profiling of spleen EBs in Dnm2Plt+/+, Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/– mice. Volcano plots showing genes differentially expressed in EBs of Mpl–/– Dnm2Plt+/+ + Mpl–/– Dnm2Plt–/– versus Dnm2Plt+/+ + Dnm2Plt–/– mice (Mpl effect), displayed as adjusted P-value (A) and baseMean (B). Volcano plots showing genes differentially expressed in EBs of Dnm2Plt–/– + Mpl–/– Dnm2Plt–/– versus Dnm2Plt+/+ + Mpl–/– Dnm2Plt+/+ mice (DNM2 effect), displayed as adjusted P-value (C) and baseMean (D). The log2FoldChange indicates the mean expression level change for each gene. Dashed lines indicate fold changes of -1.5 and 1.5 (x-axis) and adjusted P-value of.05 (y-axis). Each dot denotes one gene. Heat maps showing relevant genes identified using the Mpl (E) and DNM2 (F) effects in all four groups.
Following the analysis of Dnm2Plt–/– and Mpl–/– Dnm2Plt–/–, compared to Dnm2Plt+/+ and Mpl–/– Dnm2Plt+/+ mice, the DNM2 effect gene set contained 89 upregulated and 803 downregulated genes (Figures 6C, D; Supplementary Table 2). E2 ubiquitin-conjugating enzymes (Ube2l3, Ube2n) and cytochrome c oxidase genes (Cox5b, Cox7c) were upregulated in mice lacking DNM2 in platelets and MKs, independently of Mpl ubiquitous deletion (Figure 6F). E3 ubiquitin ligase genes (Cish, Rbx1), genes involved in ribosome biogenesis (Rpl21, Rpl22l1, Rpl23, Rpl27a, Rpl28, Rpl35, Rpl36, Rpl37, Rpl37a, Rpl38, Rpl39, Rps14, Rps15a, Rps21, Rps24, Rps27), and translation initiation factor eIF2α genes (Eif2s1, Eif2s2) were upregulated in Mpl–/– Dnm2Plt–/– mice. The observed increase in erythroferrone (Erfe) is consistent with severe anemia and grossly elevated plasma EPO levels (63).
Discussion
Here, we assessed whether the hyperproliferative phenotype of Dnm2Plt–/– mice specifically lacking DNM2 in platelets and MKs was due to JAK2 constitutive activation or elevated circulating TPO levels (42). Our data shows that DNM2-dependent Mpl-mediated endocytosis in the MK/platelet lineage is required for steady-state hematopoiesis and provides novel insights into a developmentally controlled role for Mpl in normal erythropoiesis.
STAT3 and, to a lower extent, STAT5 were tyrosine phosphorylated in Dnm2Plt–/– platelets in the absence of TPO, consistent with JAK2 constitutive activation (42). However, STAT3 and STAT5 phosphorylation were slowed and diminished following stimulation with TPO. The slowing of STAT phosphorylation indicated that the MK hyperplasia of Dnm2Plt–/– mice was not due to constitutive Mpl signaling in MKs. Instead, the phenotype of Dnm2Plt–/– mice resembles that of mice lacking Mpl or JAK2 in platelets and MKs, in which Mpl-mediated JAK-STAT signaling and TPO endocytosis are blunted (33–35). To ascertain whether elevated plasma TPO levels stimulating Mpl-expressing HSCs led to the severe MK hyperplasia and HSPC expansion, we generated Mpl–/– Dnm2Plt–/– mice lacking DNM2 in platelets and MKs and Mpl ubiquitously. JAK2 expression was further diminished in Mpl–/– Dnm2Plt–/– platelets. STAT3, but not STAT5 expression was also decreased, suggesting that JAK2 and STAT3 expression in platelets and MKs are intimately regulated by DNM2 and Mpl. JAK2 expression was also decreased in Mpl–/– platelets, consistent with a scaffolding role for Mpl (64–66). Further proteomics and RNAseq analysis data may reveal how Mpl and DNM2 regulate the expression of these and other platelet and MK proteins.
Ubiquitous Mpl deletion in Mpl–/– Dnm2Plt–/– mice resulted in a severe deficiency of bone marrow MKs and HSCs, like in Mpl–/– mice, demonstrating that impaired Mpl-mediated endocytosis in platelets and MKs lacking DNM2 is responsible for the MK hyperplasia and HSC expansion of Dnm2Plt–/– mice. Ablation of the MK population and HSC niche in Mpl–/– mice is consistent with previous studies characterizing the mouse model (14, 15, 67), and others using a signaling deficient, cell surface truncated form of the receptor (68, 69). Mpl deficiency is associated with abnormal maturation of neonatal MKs and developmental stage-specific defects in platelet function (44). Hence the MK defect and thrombocytopenia result from defective and inefficient MKs. Comparing Mpl–/– and Thpo–/– mice revealed that Mpl expression, but not TPO, was critical for the hyperproliferative phenotype of a JAK2V617F+ MPN mouse model, including the splenomegaly (66). The authors hypothesized that expression of hyperactive JAK2V617F in HSCs was likely decreased in the absence of its chaperone, Mpl. In our experiments, Mpl deletion did not alleviate the severe splenomegaly of Mpl–/– Dnm2Plt–/– mice, indicating that other mechanisms are at play. The data suggests that extramedullary hematopoiesis, including the production of premature erythrocytes, occurs in the spleen despite the loss of DNM2-dependent endocytosis in platelets and MKs and Mpl deletion. However, the precise role of DNM2 and Mpl in regulating spleen homeostasis remains to be determined.
We did not anticipate the premature death of Mpl–/– Dnm2Plt–/– mice at P25, which was attributed to severe anemia and disrupted EB maturation in early development. Increased circulating reticulated RBCs and grossly elevated plasma EPO levels confirmed a rise in stress erythropoiesis. Dnm2 was not excised in EBs isolated from Dnm2Plt–/– mice, confirming that DNM2 was normally expressed in erythroid progenitors. The data, therefore, excludes defective DNM2-dependent CD71-mediated transferrin uptake in EBs as the cause of the severe anemia, as has been described in mice expressing the Dnm2 loss-of-function mutation V235G ubiquitously (41). This poses the question of how additional Mpl deletion in the platelet- and MK-specific Dnm2Plt–/– background aggravates the RBC phenotype.
Our RNA sequencing analysis supports the notion that Mpl plays a critical role in regulating hemoglobin and heme homeostasis in EBs, as associated genes (Hbb-bs, Hbb-bt, Hbq1a, Alas2, Fech, Bpgm, Ftl1, Slc48a1) were significantly decreased in EBs lacking Mpl, independently of DNM2 deletion in platelets and MKs. Additional downregulated genes are involved in ubiquitination (Ubb, Mkrn1, Marchf2, Usp15) and ribosome recruitment and translation initiation (Pabpc1). Hemoglobin production depends on the fine-tuned sequential process of joining two α-globin and two β-globin subunits with the addition of attached iron-binding heme groups. In β-thalassemia, overabundant α-globin is polyubiquitinated and targeted for protein degradation, thereby preventing proteotoxicity (70, 71). The data further suggests poor proteome integrity in EBs lacking Mpl, as the RNA-binding E3 ubiquitin ligase Makorin 1 (Mkrn1) interacts with poly(A)-binding protein 1 (Pabpc1) to maintain ribosome-associated quality control of poly(A) translation (72). Alternatively, H2A cluster histone genes (H2ac7, H2ac10, H2ac12, H2ac13, H2ac14) were upregulated in EBs lacking Mpl. Whether this increase contributes to chromatin condensation and enucleation required for RBC formation remains to be determined (73, 74).
Remarkably, DNM2 deletion in platelets and MKs resulted in upregulation of E2 ubiquitin-conjugating enzymes (Ube2l3, Ube2n) and cytochrome c oxidase (Cox5b, Cox7c) in EBs, independently of Mpl ubiquitous deletion. Ube2l3 and Ube2n have been implicated in autophagic clearance of depolarized mitochondria (75), suggesting increased mitophagy in EBs in mice lacking DNM2 in platelets and MKs. E3 ubiquitin ligase genes (Cish, Rbx1), genes involved in ribosome biogenesis (Rpl21, Rpl22l1, Rpl23, Rpl27a, Rpl28, Rpl35, Rpl36, Rpl37, Rpl37a, Rpl38, Rpl39, Rps14, Rps15a, Rps21, Rps24, Rps27), and translation initiation factor eIF2α genes (Eif2s1, Eif2s2) were upregulated in Mpl–/– Dnm2Plt–/– mice, consistent with increased protein translation (76, 77). Together, the data suggests that the severe anemia and early mortality of Mpl–/– Dnm2Plt–/– mice is due to the combined effects of decreased hemoglobin and heme production, mitochondrial dysfunction, and proteotoxicity, resulting from increased, but poorly quality-controlled protein translation in EBs.
How does specific DNM2 deletion in the MK/platelet lineage affect the expression of these genes and ultimately erythroid development? One possibility is that MKs and platelets internalize cytokines to contribute to regulating EB maturation. In the absence of DNM2-dependent endocytosis in the MK/platelet lineage, combined with the severe MK hypoplasia and thrombocytopenia in the Mpl–/– background, increased levels of these cytokines lead to EB maturation blockage. MKs are the primary source of transforming growth factor β1 (TGF-β1) and as such regulate steady-state erythropoiesis by restraining progenitor cell and EB production (78). MK TGF-β1 and platelet factor 4 (PF4) also maintains HSC quiescence during homeostasis and promotes HSC regeneration after chemotherapeutic stress (79, 80). A second hypothesis is that Mpl deficiency greatly limits the proliferation of the bone marrow HSPC pool, thereby reducing the availability of differentiating HSCs to develop along the erythroid lineage. The consequences of Mpl loss on erythroid development have been reported previously in studies using induced pluripotent stem cells (iPSCs) derived from CAMT patients, where deficient Mpl signaling results in a loss of MEP differentiation and is critical for successful erythropoiesis (81).
While their circulating RBC and reticulocyte counts were normal at P24 and P56, Mpl–/– Dnm2Plt+/+ mice developed a transient erythropoiesis defect, with a percentage of mature EBs about half that of Dnm2Plt+/+ mice, which was only apparent during early development (P24). By adulthood (P56), Mpl–/– Dnm2Plt+/+ mice were able to normalize the EB maturation defect. The data indicates that Mpl regulates erythropoiesis during early development in mice. TPO expands erythroid progenitors, increases RBC production, and enhances erythroid recovery following myelosuppressive therapy (82). Others have implicated an EPO-independent, macrophage-associated pathway supporting terminal erythropoiesis in this expansion system in humans (83). The fetal/neonatal hematopoietic system must generate enough blood cells to meet the demands of rapid growth. This unique challenge might underlie the high incidence of thrombocytopenia among preterm neonates (44). It is possible that under developmental stress and increased need for blood production, there is crosstalk between EPO and TPO signaling in regulating hematopoiesis to produce platelets and RBCs efficiently simultaneously. Therefore, Mpl-dependent erythropoiesis is likely more significant than expected under pathological and developmental pressure to produce platelets and RBCs rapidly. Conversely, EPO can also induce megakaryopoiesis, supporting the notion that the two pathways cooperate to ensure platelet and RBC numbers (84).
In conclusion, DNM2-dependent Mpl-mediated endocytosis in platelets and MKs is required for steady-state hematopoiesis. It provides novel insights into a developmentally controlled role for Mpl in normal erythropoiesis, regulating hemoglobin and heme production.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found in the NCBI GEO repository under accession number GSE206343.
Ethics statement
The animal study was reviewed and approved by Medical College of Wisconsin Institutional Animal Care and Use Committee guidelines (Animal Use Application 5600).
Author contributions
NE, EB, RB, ML-S, TD, and HR designed and performed experiments, collected, analyzed, and interpreted data, and revised the manuscript. SZ and RB analyzed the RNAseq data. MS-V and KH analyzed and interpreted data and revised the manuscript. HF conceived and designed the study, designed and performed experiments, collected, analyzed, and interpreted data, and wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the American Society of Hematology Foundation (H.F.) and National Institutes of Health, National Heart, Lung, and Blood Institute grants HL046925 (M.C.S.V.), HL089224, HL141954, HL151333 (K.M.H.), and HL126743 (H.F.).
Acknowledgments
We thank Jon Wieser for technical assistance and Drs Hartmut Weiler and Anthony Veltri for helpful discussion.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.959806/full#supplementary-material
Supplementary Figure 1 | The MK hyperplasia of Dnm2Plt–/– mice requires Mpl expression. H&E staining of femur bone marrow (A) and spleen (B) sections, and thin blood smears (C) of Dnm2Plt+/+, Dnm2Plt–/–, Mpl–/– Dnm2Plt+/+, and Mpl–/– Dnm2Plt–/– mice at P24. Data shown are representative of 3 mice in each genotype. Scale bars represent 40 μm.
References
1. Kaushansky K, Lok S, Holly RD, Broudy VC, Lin N, Bailey MC, et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-mpl ligand thrombopoietin. Nature (1994) 369(6481):568–71. doi: 10.1038/369568a0
2. Lok S, Kaushansky K, Holly RD, Kuijper JL, Lofton-Day CE, Oort PJ, et al. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production. Vivo Nature (1994) 369(6481):565–8. doi: 10.1038/369565a0
3. de Sauvage FJ, Hass PE, Spencer SD, Malloy BE, Gurney AL, Spencer SA, et al. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-mpl ligand. Nature (1994) 369(6481):533–8. doi: 10.1038/369533a0
4. Kimura S, Roberts AW, Metcalf D, Alexander WS. Hematopoietic stem cell deficiencies in mice lacking c-mpl, the receptor for thrombopoietin. Proc Natl Acad Sci U S A (1998) 95(3):1195–200. doi: 10.1073/pnas.95.3.1195
5. Qian H, Buza-Vidas N, Hyland CD, Jensen CT, Antonchuk J, Mansson R, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell (2007) 1(6):671–84. doi: 10.1016/j.stem.2007.10.008
6. Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell (2007) 1(6):685–97. doi: 10.1016/j.stem.2007.10.020
7. Plo I, Bellanne-Chantelot C, Mosca M, Mazzi S, Marty C, Vainchenker W. Genetic alterations of the Thrombopoietin/MPL/JAK2 axis impacting megakaryopoiesis. Front Endocrinol (Lausanne) (2017) 8:234. doi: 10.3389/fendo.2017.00234
8. Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest (2005) 115(12):3339–47. doi: 10.1172/JCI26674
9. Kaushansky K. Molecular mechanisms of thrombopoietin signaling. J Thromb Haemost (2009) 7 Suppl 1:235–8. doi: 10.1111/j.1538-7836.2009.03419.x
10. Hitchcock IS, Kaushansky K. Thrombopoietin from beginning to end. Br J Haematol (2014) 165(2):259–68. doi: 10.1111/bjh.12772
11. Ballmaier M, Germeshausen M, Krukemeier S, Welte K. Thrombopoietin is essential for the maintenance of normal hematopoiesis in humans: development of aplastic anemia in patients with congenital amegakaryocytic thrombocytopenia. Ann N Y Acad Sci (2003) 996:17–25. doi: 10.1111/j.1749-6632.2003.tb03228.x
12. Seo A, Ben-Harosh M, Sirin M, Stein J, Dgany O, Kaplelushnik J, et al. Bone marrow failure unresponsive to bone marrow transplant is caused by mutations in thrombopoietin. Blood (2017) 130(7):875–80. doi: 10.1182/blood-2017-02-768036
13. Dasouki MJ, Rafi SK, Olm-Shipman AJ, Wilson NR, Abhyankar S, Ganter B, et al. Exome sequencing reveals a thrombopoietin ligand mutation in a Micronesian family with autosomal recessive aplastic anemia. Blood (2013) 122(20):3440–9. doi: 10.1182/blood-2012-12-473538
14. Gurney AL, Carver-Moore K, de Sauvage FJ, Moore MW. Thrombocytopenia in c-mpl-deficient mice. Science (1994) 265(5177):1445–7. doi: 10.1126/science.8073287
15. Alexander WS, Roberts AW, Nicola NA, Li R, Metcalf D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-mpl. Blood (1996) 87(6):2162–70. doi: 10.1182/blood.V87.6.2162.bloodjournal8762162
16. de Sauvage FJ, Carver-Moore K, Luoh SM, Ryan A, Dowd M, Eaton DL, et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med (1996) 183(2):651–6. doi: 10.1084/jem.183.2.651
17. Hoffmeister KM, Falet H. Platelet clearance by the hepatic ashwell-morrell receptor: mechanisms and biological significance. Thromb Res (2016) 141 Suppl 2:S68–72. doi: 10.1016/S0049-3848(16)30370-X
18. Kuter DJ, Rosenberg RD. The reciprocal relationship of thrombopoietin (c-mpl ligand) to changes in the platelet mass during busulfan-induced thrombocytopenia in the rabbit. Blood (1995) 85(10):2720–30. doi: 10.1182/blood.V85.10.2720.bloodjournal85102720
19. Cohen-Solal K, Villeval JL, Titeux M, Lok S, Vainchenker W, Wendling F. Constitutive expression of mpl ligand transcripts during thrombocytopenia or thrombocytosis. Blood (1996) 88(7):2578–84. doi: 10.1182/blood.V88.7.2578.bloodjournal8872578
20. Fielder PJ, Gurney AL, Stefanich E, Marian M, Moore MW, Carver-Moore K, et al. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood (1996) 87(6):2154–61. doi: 10.1182/blood.V87.6.2154.bloodjournal8762154
21. Cerutti A, Custodi P, Duranti M, Noris P, Balduini CL. Thrombopoietin levels in patients with primary and reactive thrombocytosis. Br J Haematol (1997) 99(2):281–4. doi: 10.1046/j.1365-2141.1997.3823196.x
22. Shinjo K, Takeshita A, Nakamura S, Naitoh K, Yanagi M, Tobita T, et al. Serum thrombopoietin levels in patients correlate inversely with platelet counts during chemotherapy-induced thrombocytopenia. Leukemia (1998) 12(3):295–300. doi: 10.1038/sj.leu.2400946
23. Engel C, Loeffler M, Franke H, Schmitz S. Endogenous thrombopoietin serum levels during multicycle chemotherapy. Br J Haematol (1999) 105(3):832–8. doi: 10.1046/j.1365-2141.1999.01459.x
24. Wolber EM, Fandrey J, Frackowski U, Jelkmann W. Hepatic thrombopoietin mRNA is increased in acute inflammation. Thromb Haemost (2001) 86(6):1421–4. doi: 10.1055/s-0037-1616745
25. Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood (2001) 98(9):2720–5. doi: 10.1182/blood.V98.9.2720
26. McIntosh B, Kaushansky K. Transcriptional regulation of bone marrow thrombopoietin by platelet proteins. Exp Hematol (2008) 36(7):799–806. doi: 10.1016/j.exphem.2008.02.012
27. Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, et al. The ashwell-morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med (2015) 21(1):47–54. doi: 10.1038/nm.3770
28. Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene (2013) 32(21):2601–13. doi: 10.1038/onc.2012.347
29. Hitchcock IS, Chen MM, King JR, Kaushansky K. YRRL motifs in the cytoplasmic domain of the thrombopoietin receptor regulate receptor internalization and degradation. Blood (2008) 112(6):2222–31. doi: 10.1182/blood-2008-01-134049
30. Saur SJ, Sangkhae V, Geddis AE, Kaushansky K, Hitchcock IS. Ubiquitination and degradation of the thrombopoietin receptor c-mpl. Blood (2010) 115(6):1254–63. doi: 10.1182/blood-2009-06-227033
31. Pecquet C, Diaconu CC, Staerk J, Girardot M, Marty C, Royer Y, et al. Thrombopoietin receptor down-modulation by JAK2 V617F: Restoration of receptor levels by inhibitors of pathologic JAK2 signaling and of proteasomes. Blood (2012) 119(20):4625–35. doi: 10.1182/blood-2011-08-372524
32. Besancenot R, Roos-Weil D, Tonetti C, Abdelouahab H, Lacout C, Pasquier F, et al. JAK2 and MPL protein levels determine TPO-induced megakaryocyte proliferation versus differentiation. Blood (2014) 124(13):2104–15. doi: 10.1182/blood-2014-03-559815
33. Ng AP, Kauppi M, Metcalf D, Hyland CD, Josefsson EC, Lebois M, et al. Mpl expression on megakaryocytes and platelets is dispensable for thrombopoiesis but essential to prevent myeloproliferation. Proc Natl Acad Sci USA (2014) 111(16):5884–9. doi: 10.1073/pnas.1404354111
34. Meyer SC, Keller MD, Woods BA, LaFave LM, Bastian L, Kleppe M, et al. Genetic studies reveal an unexpected negative regulatory role for Jak2 in thrombopoiesis. Blood (2014) 124(14):2280–4. doi: 10.1182/blood-2014-03-560441
35. Alvarez MB, Xu L, Childress PJ, Maupin KA, Mohamad SF, Chitteti BR, et al. Megakaryocyte and osteoblast interactions modulate bone mass and hematopoiesis. Stem Cells Dev (2018) 27(10):671–82. doi: 10.1089/scd.2017.0178
36. Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol (2012) 13(2):75–88. doi: 10.1038/nrm3266
37. Züchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate charcot-Marie-Tooth disease. Nat Genet (2005) 37(3):289–94. doi: 10.1038/ng1514
38. Bitoun M, Maugenre S, Jeannet PY, Lacene E, Ferrer X, Laforet P, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet (2005) 37(11):1207–9. doi: 10.1038/ng1657
39. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature (2012) 481(7380):157–63. doi: 10.1038/nature10725
40. Ferguson SM, Raimondi A, Paradise S, Shen H, Mesaki K, Ferguson A, et al. Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell (2009) 17(6):811–22. doi: 10.1016/j.devcel.2009.11.005
41. Brown FC, Collett M, Tremblay CS, Rank G, De Camilli P, Booth CJ, et al. Loss of dynamin 2 GTPase function results in microcytic anaemia. Br J Haematol (2017) 178(4):616–28. doi: 10.1111/bjh.14709
42. Bender M, Giannini S, Grozovsky R, Jonsson T, Christensen H, Pluthero FG, et al. Dynamin 2-dependent endocytosis is required for normal megakaryocyte development in mice. Blood (2015) 125(6):1014–24. doi: 10.1182/blood-2014-07-587857
43. Eaton N, Drew C, Wieser J, Munday AD, Falet H. Dynamin 2 is required for GPVI signaling and platelet hemostatic function in mice. Haematologica (2020) 105(5):1414–23. doi: 10.3324/haematol.2019.218644
44. Lorenz V, Ramsey H, Liu ZJ, Italiano J Jr., Hoffmeister K, Bihorel S, et al. Developmental stage-specific manifestations of absent TPO/c-MPL signalling in newborn mice. Thromb Haemost (2017) 117(12):2322–33. doi: 10.1160/TH17-06-0433
45. Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood (2001) 98(12):3261–73. doi: 10.1182/blood.V98.12.3261
46. Ishikawa Y, Maeda M, Pasham M, Aguet F, Tacheva-Grigorova SK, Masuda T, et al. Role of the clathrin adaptor PICALM in normal hematopoiesis and polycythemia vera pathophysiology. Haematologica (2015) 100(4):439–51. doi: 10.3324/haematol.2014.119537
47. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell (2005) 121(7):1109–21. doi: 10.1016/j.cell.2005.05.026
48. Pronk CJ, Rossi DJ, Mansson R, Attema JL, Norddahl GL, Chan CK, et al. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell (2007) 1(4):428–42. doi: 10.1016/j.stem.2007.07.005
49. Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell (2013) 13(1):102–16. doi: 10.1016/j.stem.2013.05.014
50. Huang YS, Delgadillo LF, Cyr KH, Kingsley PD, An X, McGrath KE, et al. Circulating primitive erythroblasts establish a functional, protein 4.1R-dependent cytoskeletal network prior to enucleating. Sci Rep (2017) 7(1):5164. doi: 10.1038/s41598-017-05498-4
51. Subramaniam S, Ogoti Y, Hernandez I, Zogg M, Botros F, Burns R, et al. A thrombin-PAR1/2 feedback loop amplifies thromboinflammatory endothelial responses to the viral RNA analogue poly(I:C). Blood Adv (2021) 5(13):2760–74. doi: 10.1182/bloodadvances.2021004360
52. Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol (2020) 38(3):276–8. doi: 10.1038/s41587-020-0439-x
53. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
54. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an r package for comparing biological themes among gene clusters. OMICS (2012) 16(5):284–7. doi: 10.1089/omi.2011.0118
55. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res (2000) 28(1):27–30. doi: 10.1093/nar/28.1.27
56. Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, et al. The reactome pathway knowledgebase. Nucleic Acids Res (2018) 46(D1):D649–55. doi: 10.1093/nar/gkx1132
57. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. the gene ontology consortium. Nat Genet (2000) 25(1):25–9. doi: 10.1038/75556
58. Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol (1995) 57(1):289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
59. Germeshausen M, Ballmaier M, Welte K. MPL mutations in 23 patients suffering from congenital amegakaryocytic thrombocytopenia: the type of mutation predicts the course of the disease. Hum Mutat (2006) 27(3):296. doi: 10.1002/humu.9415
60. Ku H, Yonemura Y, Kaushansky K, Ogawa M. Thrombopoietin, the ligand for the mpl receptor, synergizes with steel factor and other early acting cytokines in supporting proliferation of primitive hematopoietic progenitors of mice. Blood (1996) 87(11):4544–51. doi: 10.1182/blood.V87.11.4544.bloodjournal87114544
61. Sitnicka E, Lin N, Priestley GV, Fox N, Broudy VC, Wolf NS, et al. The effect of thrombopoietin on the proliferation and differentiation of murine hematopoietic stem cells. Blood (1996) 87(12):4998–5005. doi: 10.1182/blood.V87.12.4998.bloodjournal87124998
62. Chiabrando D, Mercurio S, Tolosano E. Heme and erythropoieis: more than a structural role. Haematologica (2014) 99(6):973–83. doi: 10.3324/haematol.2013.091991
63. Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet (2014) 46(7):678–84. doi: 10.1038/ng.2996
64. Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem (2005) 280(29):27251–61. doi: 10.1074/jbc.M501376200
65. Lu X, Levine R, Tong W, Wernig G, Pikman Y, Zarnegar S, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci USA (2005) 102(52):18962–7. doi: 10.1073/pnas.0509714102
66. Sangkhae V, Etheridge SL, Kaushansky K, Hitchcock IS. The thrombopoietin receptor, MPL, is critical for development of a JAK2V617F-induced myeloproliferative neoplasm. Blood (2014) 124(26):3956–63. doi: 10.1182/blood-2014-07-587238
67. Alexander WS, Roberts AW, Maurer AB, Nicola NA, Dunn AR, Metcalf D. Studies of the c-mpl thrombopoietin receptor through gene disruption and activation. Stem Cells (1996) 14 Suppl 1:124–32. doi: 10.1002/stem.5530140716
68. Alexander WS, Maurer AB, Novak U, Harrison-Smith M. Tyrosine-599 of the c-mpl receptor is required for shc phosphorylation and the induction of cellular differentiation. EMBO J (1996) 15(23):6531–40. doi: 10.1002/j.1460-2075.1996.tb01044.x
69. Kohlscheen S, Wintterle S, Schwarzer A, Kamp C, Brugman MH, Breuer DC, et al. Inhibition of Thrombopoietin/Mpl signaling in adult hematopoiesis identifies new candidates for hematopoietic stem cell maintenance. PloS One (2015) 10(7):e0131866. doi: 10.1371/journal.pone.0131866
70. Khandros E, Thom CS, D'Souza J, Weiss MJ. Integrated protein quality-control pathways regulate free alpha-globin in murine beta-thalassemia. Blood (2012) 119(22):5265–75. doi: 10.1182/blood-2011-12-397729
71. Pla-Prats C, Thoma NH. Quality control of protein complex assembly by the ubiquitin-proteasome system. Trends Cell Biol (2022) 32(8):696–706. doi: 10.1016/j.tcb.2022.02.005
72. Hildebrandt A, Bruggemann M, Ruckle C, Boerner S, Heidelberger JB, Busch A, et al. The RNA-binding ubiquitin ligase MKRN1 functions in ribosome-associated quality control of poly(A) translation. Genome Biol (2019) 20(1):216. doi: 10.1186/s13059-019-1814-0
73. Ji P, Murata-Hori M, Lodish HF. Formation of mammalian erythrocytes: chromatin condensation and enucleation. Trends Cell Biol (2011) 21(7):409–15. doi: 10.1016/j.tcb.2011.04.003
74. Gautier EF, Leduc M, Ladli M, Schulz VP, Lefevre C, Boussaid I, et al. Comprehensive proteomic analysis of murine terminal erythroid differentiation. Blood Adv (2020) 4(7):1464–77. doi: 10.1182/bloodadvances.2020001652
75. Geisler S, Vollmer S, Golombek S, Kahle PJ. The ubiquitin-conjugating enzymes UBE2N, UBE2L3 and UBE2D2/3 are essential for parkin-dependent mitophagy. J Cell Sci (2014) 127(Pt 15):3280–93. doi: 10.1242/jcs.146035
76. Chen JJ, Zhang S. Heme-regulated eIF2alpha kinase in erythropoiesis and hemoglobinopathies. Blood (2019) 134(20):1697–707. doi: 10.1182/blood.2019001915
77. Testa U. Apoptotic mechanisms in the control of erythropoiesis. Leukemia (2004) 18(7):1176–99. doi: 10.1038/sj.leu.2403383
78. Di Giandomenico S, Kermani P, Molle N, Yabut MM, Abu-Zeinah G, Stephens T, et al. Megakaryocyte TGFbeta1 partitions erythropoiesis into immature progenitor/stem cells and maturing precursors. Blood (2020) 136(9):1044–54. doi: 10.1182/blood.2019003276
79. Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med (2014) 20(11):1315–20. doi: 10.1038/nm.3707
80. Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med (2014) 20(11):1321–6. doi: 10.1038/nm.3706
81. Hirata S, Takayama N, Jono-Ohnishi R, Endo H, Nakamura S, Dohda T, et al. Congenital amegakaryocytic thrombocytopenia iPS cells exhibit defective MPL-mediated signaling. J Clin Invest (2013) 123(9):3802–14. doi: 10.1172/JCI64721
82. Kaushansky K, Broudy VC, Grossmann A, Humes J, Lin N, Ren HP, et al. Thrombopoietin expands erythroid progenitors, increases red cell production, and enhances erythroid recovery after myelosuppressive therapy. J Clin Invest (1995) 96(3):1683–7. doi: 10.1172/JCI118210
83. Belay E, Miller CP, Kortum AN, Torok-Storb B, Blau CA, Emery DW. A hyperactive mpl-based cell growth switch drives macrophage-associated erythropoiesis through an erythroid-megakaryocytic precursor. Blood (2015) 125(6):1025–33. doi: 10.1182/blood-2014-02-555318
Keywords: mpl, dnm2, megakaryopoeiesis, erythropoiesis, hematopoiesis
Citation: Eaton N, Boyd EK, Biswas R, Lee-Sundlov MM, Dlugi TA, Ramsey HE, Zheng S, Burns RT, Sola-Visner MC, Hoffmeister KM and Falet H (2022) Endocytosis of the thrombopoietin receptor Mpl regulates megakaryocyte and erythroid maturation in mice. Front. Oncol. 12:959806. doi: 10.3389/fonc.2022.959806
Received: 02 June 2022; Accepted: 29 July 2022;
Published: 30 August 2022.
Edited by:
Anna Rita Migliaccio, Campus Bio-Medico University, ItalyReviewed by:
Marco Vitale, University of Parma, ItalyKenneth Kaushansky, Stony Brook University, United States
Copyright © 2022 Eaton, Boyd, Biswas, Lee-Sundlov, Dlugi, Ramsey, Zheng, Burns, Sola-Visner, Hoffmeister and Falet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hervé Falet, aGZhbGV0QHZlcnNpdGkub3Jn
†Present address: Nathan Eaton, Division of Hemostasis and Thrombosis, Beth Israel Deaconess Medical Center and Department of Medicine, Harvard Medical School, Boston, MA, United States
Haley E. Ramsey, Division of Hematology and Oncology, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, United States
‡ORCID: Nathan Eaton, orcid.org/0000-0001-9479-1365
Emily K. Boyd, orcid.org/0000-0001-9925-0824
Ratnashree Biswas, orcid.org/0000-0002-8307-2880
Melissa M. Lee-Sundlov, orcid.org/0000-0002-8290-8586
Theresa A. Dlugi, orcid.org/0000-0002-4716-784X
Robert T. Burns, orcid.org/0000-0001-5334-3486
Martha C. Sola-Visner, orcid.org/0000-0002-0013-598X
Karin M. Hoffmeister, orcid.org/0000-0001-6526-7919
Hervé Falet, orcid.org/0000-0003-0788-9204