
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 23 September 2022
Sec. Gastrointestinal Cancers: Colorectal Cancer
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.955313
This article is part of the Research Topic Gut Microbiota and Chemotherapy Resistance of Colorectal Cancer View all 6 articles
 Yen-Cheng Chen1,2
Yen-Cheng Chen1,2 Chia-Hsien Chuang3
Chia-Hsien Chuang3 Zhi-Feng Miao1Kwan-Ling Yip1Chung-Jung Liu4
Zhi-Feng Miao1Kwan-Ling Yip1Chung-Jung Liu4 Ling-Hui Li5Deng-Chyang Wu6,7Tian−Lu Cheng6
Ling-Hui Li5Deng-Chyang Wu6,7Tian−Lu Cheng6 Chung-Yen Lin3*†
Chung-Yen Lin3*† Jaw-Yuan Wang1,2,8,9,10,11*†
Jaw-Yuan Wang1,2,8,9,10,11*†Studies have reported the effects of the gut microbiota on colorectal cancer (CRC) chemotherapy, but few studies have investigated the association between gut microbiota and targeted therapy. This study investigated the role of the gut microbiota in the treatment outcomes of patients with metastatic CRC (mCRC). We enrolled 110 patients with mCRC and treated them with standard cancer therapy. Stool samples were collected before administering a combination of chemotherapy and targeted therapy. Patients who had a progressive disease (PD) or partial response (PR) for at least 12 cycles of therapy were included in the study. We further divided these patients into anti-epidermal growth factor receptor (cetuximab) and anti-vascular endothelial growth factor (bevacizumab) subgroups. The gut microbiota of the PR group and bevacizumab-PR subgroup exhibited significantly higher α-diversity. The β-diversity of bacterial species significantly differed between the bevacizumab-PR and bevacizumab-PD groups (P = 0.029). Klebsiella quasipneumoniae exhibited the greatest fold change in abundance in the PD group than in the PR group. Lactobacillus and Bifidobacterium species exhibited higher abundance in the PD group. The abundance of Fusobacterium nucleatum was approximately 32 times higher in the PD group than in the PR group. A higher gut microbiota diversity was associated with more favorable treatment outcomes in the patients with mCRC. Bacterial species analysis of stool samples yielded heterogenous results. K. quasipneumoniae exhibited the greatest fold change in abundance among all bacterial species in the PD group. This result warrants further investigation especially in a Taiwanese population.
Colorectal cancer (CRC), a highly prevalent malignant disease globally, is the third most common cancer and the fourth leading cause of cancer-related deaths (1). In Asia, the incidence and mortality rates of CRC are the highest among all cancers, and the prevalence of CRC has been increasing in various countries including Japan, Korea, China, and Taiwan (2). In Taiwan, CRC has been the most common cancer since 2006 (3). Approximately 20%–25% of patients with CRC are initially diagnosed as having stage IV CRC with distant metastasis (4, 5). Although the overall survival (OS) of patients with CRC has increased, the treatment of metastatic CRC (mCRC) remains a clinical challenge. For example, patients with mCRC and BRAF mutation exhibited a poor response to systemic treatment and had an unfavorable prognosis (6, 7). The RAS gene mutation is a crucial factor for CRC tumorigenesis, invasion, and metastasis and can thus serve as a therapeutic agent (8). For mCRC treatment, doublet or triplet chemotherapy with fluoropyridine, oxaliplatin, or irinotecan is commonly used as a neoadjuvant therapy (9). Targeted therapy, including the use of an antivascular endothelial growth factor (VEGF) agent (e.g., bevacizumab ramucirumab, and aflibercept) and anti-epidermal growth factor receptor (EGFR) agents (e.g., cetuximab and panitumumab), has been suggested for mCRC treatment in combination with chemotherapy (5, 10–12). However, treatment outcomes have been unfavorable. The 3-year OS rate is approximately 50% (9), and less than 20% of patients survive beyond 5 years from the time of mCRC diagnosis (4).
State-of-the-art therapies for mCRC have been widely researched to improve treatment outcomes (13). The human gut microbiota plays a crucial role in human health and CRC treatment (14–16). The balance of the gut microbiota is essential for human health, and it affects the immune system, bowel health, and protection against pathogens. The dysregulation of the gut microbiota, or dysbiosis, is harmful and can result in cancer formation (15, 17). The toxicity of chemotherapeutic agents can alter the balance of the gut microbiota and break down the mucosal barrier of the bowel (18, 19). This mechanism induces gastrointestinal mucositis, thus affecting the quality of life of patients and leading to lower treatment compliance (19).
In CRC treatment, the gut microbiota is strongly associated with chemotherapy-related side effects (19, 20). For example, diarrhea is an adverse event (AE) that is commonly associated with irinotecan use, and SN-38 (the active metabolite of irinotecan) is the primary toxic agent. Some bacteria in the human bowel can secrete β-glucuronidase that converts SN-38G (the inactive metabolite) to SN-38, which may cause diarrhea. Silymarin, a bioflavonoid complex, can inhibit β-glucuronidase activity to alleviate diarrhea (21). Apart from side effects, the gut microbiota can also affect treatment outcomes. For instance, Fusobacterium nucleatum was demonstrated to promote the resistance of CRC cells to oxaliplatin through the activation of the autophagy pathway (22).
Some studies have reported an association between the gut microbiota and CRC chemotherapy (19, 23, 24); however, data regarding the association between the gut microbiota and targeted therapies for mCRC are lacking. In this study, we analyzed fecal samples from patients with mCRC to identify potential candidate bacteria related to treatment outcomes and to AEs that are associated with combination chemotherapy and targeted therapy. The findings of this study can help elucidate the role of the gut microbiota in targeted therapies for CRC. The present study was approved by the Institutional Ethics Committee of our hospital (KMUHIRB-E(II)-20180012).
We enrolled 110 patients with mCRC between July 2017 and June 2018. Abdominal computed tomography (CT), colonoscopy, and histopathology were used to establish the definite diagnosis of mCRC. A multidisciplinary panel consisting of colorectal surgeons, gastroenterologists, medical oncologists, radiologists, radiation oncologists, and pathologists discussed the treatment program. We first collected stool samples before the initiation of chemotherapy or targeted therapy. Chemotherapy with FOLFIRI (folinic acid, fluorouracil [5-FU], and irinotecan) was administered to all the patients. According to the patients’ general condition and nutritional status as well as the presence of metastasis, comorbidities, and RAS gene variations, we selected targeted therapy involving the use of either an anti-VEGF agent (bevacizumab [Avastin, Roche, Basel, Switzerland]) or an anti-EGFR agent (cetuximab [Erbitux, Merck, Darmstadt, Germany]) as the first-line treatment. Bevacizumab can be used for patients with either the wild-type or RAS gene variants, whereas cetuximab can only be used for patients with the wild-type RAS gene because of the drug’s mechanism of action (25, 26). The treatment regimen comprised a 120-min intravenous (IV) infusion of cetuximab (500 mg/m2) or bevacizumab (5 mg/kg) on day 1, followed by a 4-h IV infusion of irinotecan (180 mg/m2) plus normal saline (500 mL) and then a 42–46-h IV infusion of leucovorin (200 mg/m2), 5-FU (2800 mg/m2), and normal saline (500 mL). This regimen was repeated once every 2 weeks. During this period, we measured the serum carcinoembryonic antigen (CEA) level throughout each chemotherapy cycle.
We performed enhanced abdominal CT or magnetic resonance imaging (MRI) after every six cycles of therapy or if the patients were determined to have abnormal serum CEA levels during two consecutive tests. Response measurements were based on the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1 (27, 28); partial response (PR), stable disease (SD), and progressive disease (PD) were evaluated on the basis of CT or MRI findings. The patients who exhibited a PR or PD during at least 12 cycles of therapy were included in the study. Those who exhibited a SD during 12 or more cycles of therapy were excluded. We divided the patients into the PD and PR groups according to their clinical response. For further stool analysis, the patients were divided into four subgroups according to the first-line targeted therapy they received: cetuximab-PD, bevacizumab-PD, cetuximab-PR, and bevacizumab-PR. The patient selection and classification processes are presented in Figure 1.
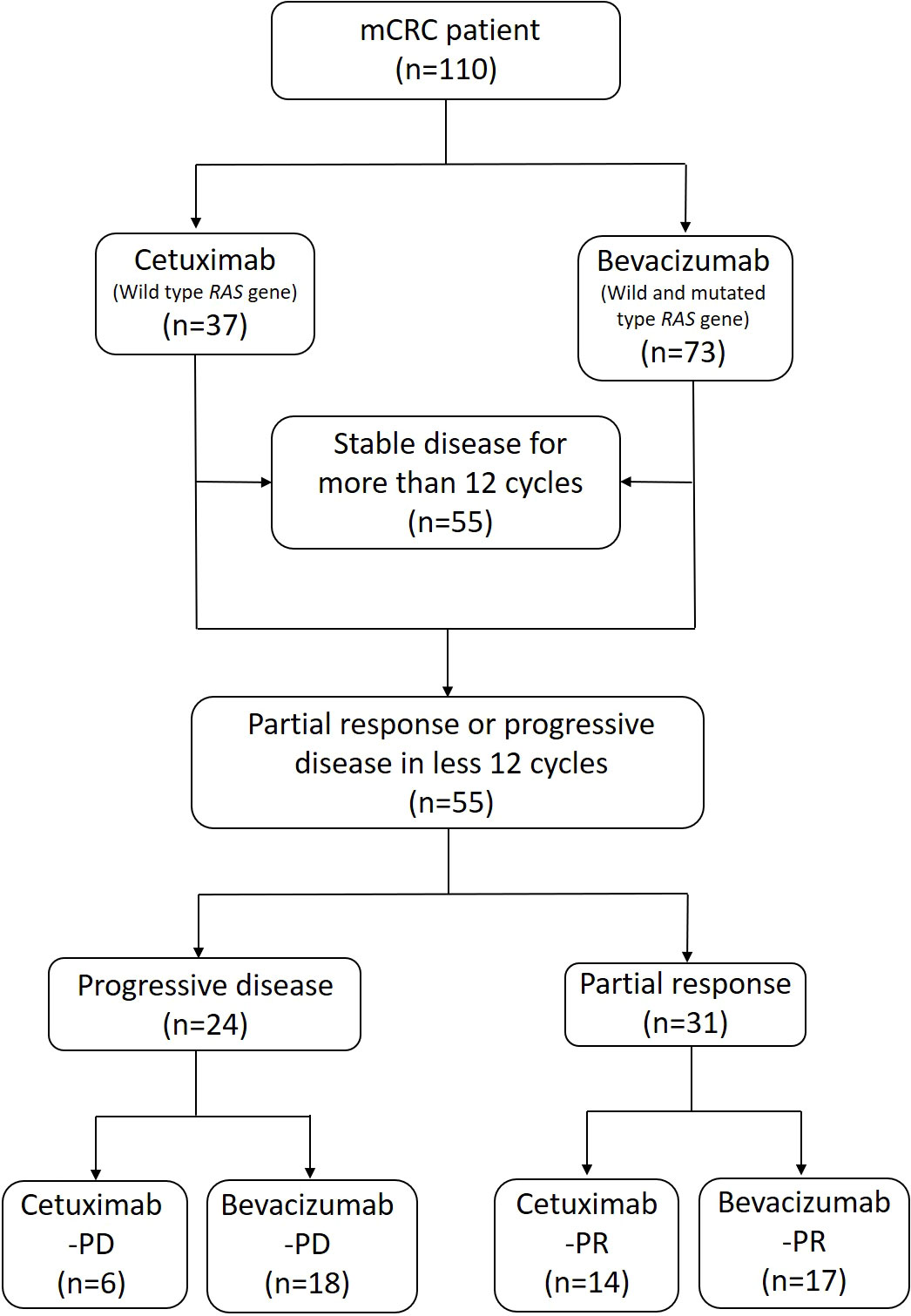
Figure 1 Flowchart of patient selection and classification. Anti-EGFR (cetuximab) and anti-VEGF (bevacizumab) agents were administered in accordance with the patients’ general condition and with results on RAS gene expression. The patients received standard treatment for mCRC in combination with a FOLFIRI (folinic acid, 5-FU, and irinotecan) chemotherapy regimen. According to the patients’ clinical response, we classified them into “progressive disease” and “partial response” groups. The patients were further classified into four subgroups with cetuximab-PD, bevacizumab-PD, cetuximab-PR, and bevacizumab-PR for the subsequent stool analysis.
During each cycle of neoadjuvant and adjuvant chemotherapy, AEs were examined using the Common Terminology Criteria for Adverse Events, version 4.0 (29). In the substantial diarrhea group, we included patients with grade 3 or grade 2 diarrhea and those who had grade 1 diarrhea at least five times during the treatment course. The median number of treatment cycles was 10 (range: 6 to 20). In the minor or no diarrhea group, we included the patients who did not experience the aforementioned diarrhea episodes.
All stool samples were collected in our ward during hospitalization with the assistance of a nurse. We used an Fe-Col fecal collection device (Alpha Laboratories, Eastleigh, United Kingdom) to collect the stool sample and prevent contamination. The samples were temporarily stored in a −4°C freezer and then transferred into a −80°C freezer within 6 h. We used the QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) to extract high-quality genomic DNA (gDNA) from frozen stool samples. DNA extraction was performed in accordance with a published protocol (30). The quantity and quality of the extracted gDNA were measured using an ND-1000 spectrophotometer (Nanodrop Technology, Wilmington, USA), Qubit 4 Fluorometer (Thermo Fisher, Waltham, USA), and Agilent 4200 TapeStation System (Agilent Technologies, Santa Clara, USA); these measurements included the concentration, purity, and integrity of gDNA. The protocol that yielded a higher quantity and quality of gDNA was then used to extract the gDNA of the remaining samples. The gDNA samples were stored at −80°C until library preparation and sequencing.
Sequencing libraries were prepared using the Nextera DNA Flex kit (Illumina) in accordance with the manufacturer’s instructions. One paired-end library with an insert size of approximately 320 bp was constructed for each sample. Libraries were normalized by performing the Qubit assay; the pooled library was then sequenced on one lane on a NovaSeq6000 platform (NovaSeq Control Software 1.6.0/RTA v3.4.4) with a 2 × 150 setup by using the NovaSeqXp workflow in the S1 mode flow cell. The Bcl to FastQ conversion was performed using bcl2fastq_v2.20.0.422 in CASAVA genetic analysis software. The quality scale used was Sanger/phred33/Illumina 1.8 +.
For library construction, we extracted DNA from stool samples. After performing quality control, we used the qualified samples for library construction. The sequencing library was constructed through the random fragmentation of the DNA sample, followed by 5′ and 3′ adapter ligation. Moreover, we used tagmentation to combine fragmentation and ligation reactions into a single step, thus considerably increasing the efficiency of the library construction process. Adapter-ligated fragments were amplified by performing the polymerase chain reaction and then purified on a gel.
For cluster generation, the library was loaded into a flow cell where fragments were captured on a lawn of surface-bound oligos that were complementary to library adapters. Each fragment was amplified into distinct clonal clusters through bridge amplification. When cluster generation was completed, the templates were ready for sequencing.
The quality control of sequencing data performed using KneadData v.0.7.10 (https://huttenhower.sph.harvard.edu/kneaddata/) included read quality checks, base quality trimming, and human genome decontamination (hg37dec_v0.1). The taxonomic assignment was conducted using Kraken2 v.2.1.2 (31) on the basis of the alignment of minimizers from k-mers against the complete genome database in RefSeq for archaea, bacteria, fungi, and viruses. In addition, we added the unexplored human microbiome genome from another study (32) to enhance the sensitivity for taxonomic classification. After classification, the abundance levels of taxa were re-estimated using Bracken v.2.2 (https://ccb.jhu.edu/software/bracken/) at the species level (33).
We examined the β-diversity of the samples in different subgroups by using QIIME2 v.2020.2.0 (https://qiime2.org) (34). The classification report from Bracken was converted using QIIME2, and the α-diversity (Shannon index) and β-diversity (Bray–Curtis) were calculated. The diversity results were visualized through boxplots and principal co-ordinate analysis plots in R software. We performed the Mann–Whitney U test to analyze differences in the α-diversity index and performed a permutational multivariate analysis of variance by using 999 iterations to examine differences in Bray–Curtis matrices between the groups. Statistical test results with P < 0.05 were considered significant.
The differential abundance analysis was performed using DESeq2 package v.1.30.1 (35). Our hypothesis was tested using the Wald test, and P values were adjusted using the Benjamini–Hochberg method. Taxa that met our statistical criteria (adjusted P < 0.05, >2 shrunken log2 fold changes, and >1000 base means) were considered differentially abundant between the two patient groups.
All statistical analyses were performed using SPSS version 20 (IBM, Armonk, NY, USA). The chi-square test was performed to compare categorical data. P < 0.05 indicated statistical significance. Descriptive statistics are presented as proportions and means.
The characteristics of the patients with mCRC at diagnosis and their gene variation profiles are presented in Table 1. Of the 110 patients with mCRC, 55 had a SD after more than 12 cycles of chemotherapy and were subsequently excluded from this study. The other 55 patients met the inclusion criteria of having a clear PD or PR. The PD and PR groups included 24 and 31 patients, respectively. Six patients exhibited a PD after receiving cetuximab and were included into the cetuximab-PD subgroup. Similarly, 18 patients were included into the bevacizumab-PD subgroup. The cetuximab-PR and bevacizumab-PR subgroups consisted of 14 and 17 patients, respectively. No patient presented a complete response after treatment.

Table 1 Patient Characteristics at Diagnosis and Gene Variation Profiles (n = 55).
Fourteen patients experienced substantial diarrhea during the treatment course. The number of patients with substantial diarrhea was equal in both the cetuximab (7 patients, 35%) and bevacizumab (7 patients, 20%) groups. We observed no significant difference in the clinical response or diarrhea frequency between the cetuximab and bevacizumab groups (Table 2).

Table 2 Results for patients by clinical response and diarrhea type.
α-Diversity was calculated to compare the diversity and richness of bacterial species among the groups and subgroups (36). The gut microbiota of the PR group exhibited significantly higher α-diversity than that of the PD group (P < 0.01, Figure 2A). The gut microbiota of the bevacizumab-PR subgroup demonstrated significantly higher α-diversity than that of the bevacizumab-PD subgroup (P < 0.01, Figure 2B). The α-diversity of the gut microbiota did not significantly differ between the cetuximab-PD and cetuximab-PD subgroups (P = 0.35, Figure 2C).
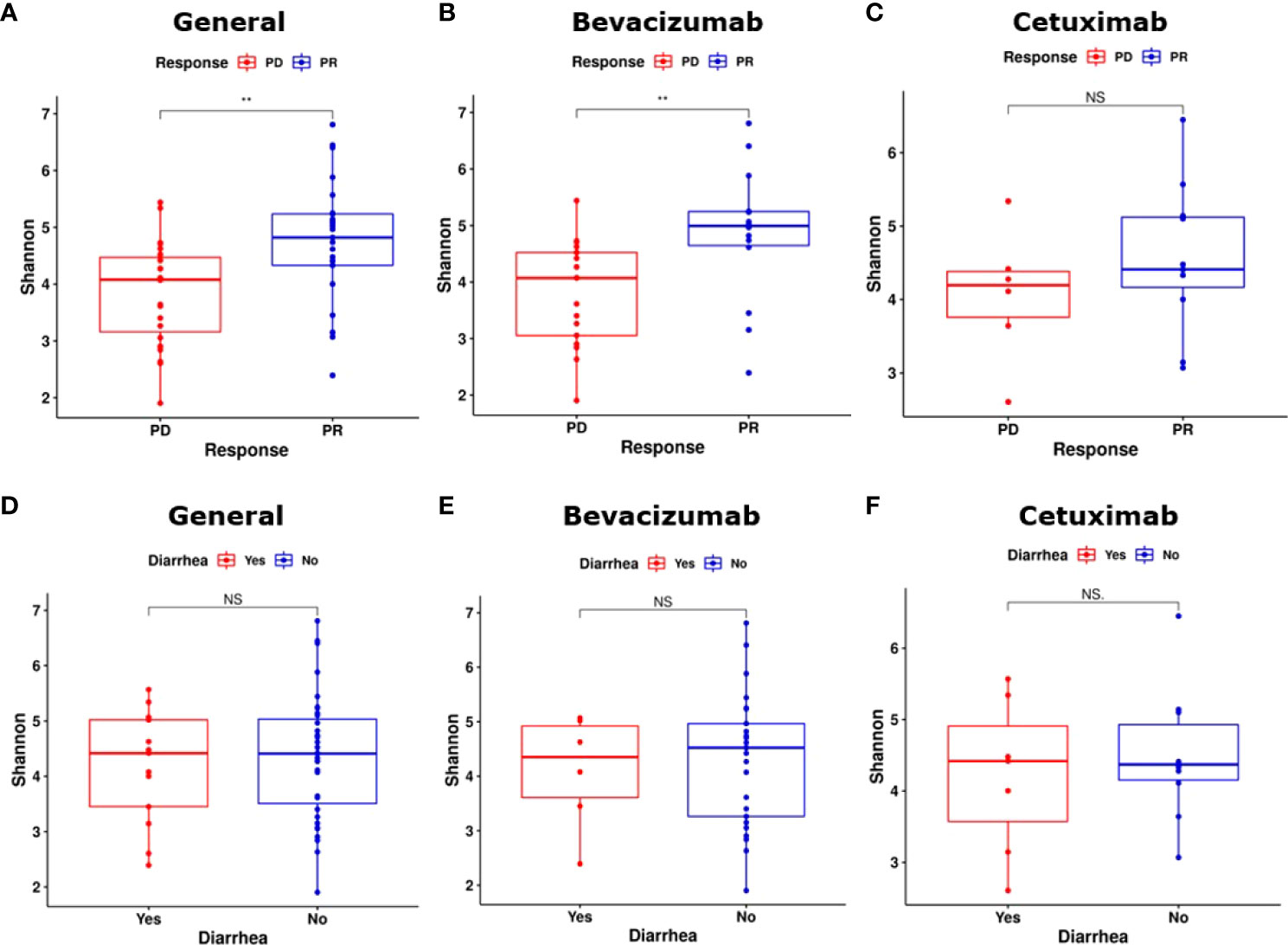
Figure 2 Results on α-diversity of groups and subgroups. (A) The PR group had a significantly higher α-diversity than did the PD group (P < 0.01). (B) α-diversity significantly differed between the bevacizumab-PD and PR subgroups (P < 0.01). (C) Bacterial species richness did not significantly differ between the cetuximab-PR and cetuximab-PD subgroups (P = 0.35). (D) α-diversity did not significantly differ between the “substantial diarrhea” and “minor or no diarrhea” groups (P = 0.57). (E, F) α-diversity did not significantly differ between the bevacizumab and cetuximab diarrhea subgroups (bevacizumab-PR vs. bevacizumab-PD and cetuximab-PR vs. cetuximab-PD, P = 0.72 and P = 1.00, respectively). NS, Not significant; **: P < 0.01.
We compared the α-diversity of the substantial diarrhea and minor or no diarrhea groups. The gut microbiota of the minor or no diarrhea group exhibited nonsignificant α-diversity (P = 0.57, Figure 2D). No significant differences in the α-diversity of the gut microbiota were noted between the patients who developed substantial diarrhea in the bevacizumab subgroups and those in the cetuximab subgroups (bevacizumab-PR vs. bevacizumab-PD and cetuximab-PR vs. cetuximab-PD, P = 0.72 and P = 1.00, Figures 2E, F, respectively).
β-Diversity was calculated to compare the compositional differences of bacterial species among the groups and subgroups (37). The β-diversity of the gut microbiota did not significantly differ between the PD and PR groups (P = 0.16, Figure 3A). However, the β-diversity of bacterial species in the bevacizumab-PR subgroup significantly differed from that of the bevacizumab-PD subgroup (P = 0.029, Figure 3B). The β-diversity of the gut microbiota did not significantly differ between the cetuximab-PD and cetuximab-PR subgroups (P = 0.784, Figure 3C).
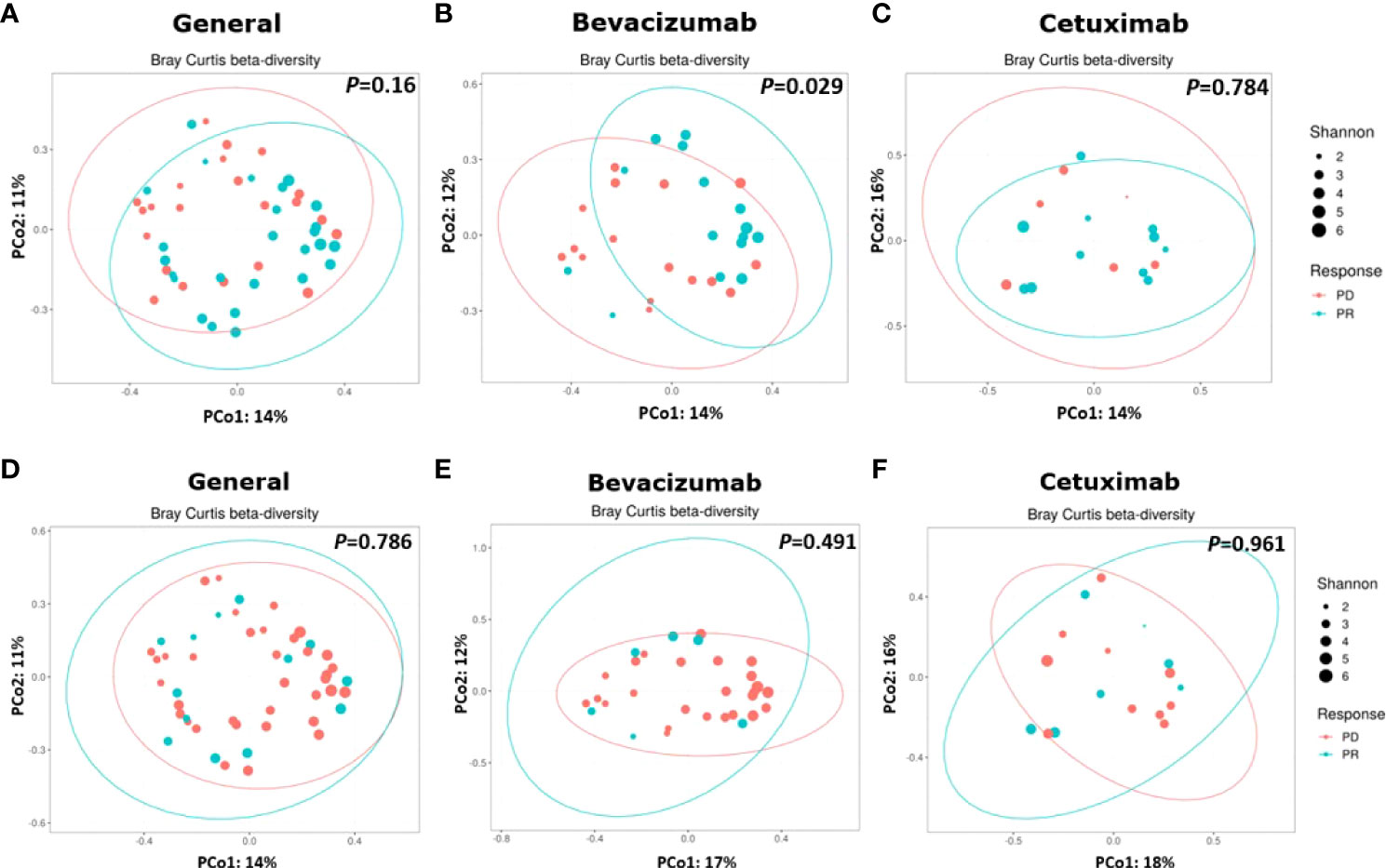
Figure 3 Results on β-diversity of treatment groups and subgroups. (A) β-diversity did not significantly differ between the PD and PR groups (P = 0.16); (B) Bacterial species significantly different between the bevacizumab-PD and PR subgroups (P = 0.029). (C) β-Diversity did not significantly differ between the cetuximab-PD and PR subgroups (P = 0.784). (D) β-diversity did not significantly differ between the substantial diarrhea and minor or no diarrhea groups (P = 0.786). (E, F) β-diversity did not significantly differ between the bevacizumab (P = 0.491) and cetuximab (P = 0.961) subgroups.
No significant difference in the β-diversity of the gut microbiota (P = 0.786, Figure 3D) was noted between the substantial diarrhea and minor or no diarrhea groups. Moreover, no significant difference in the β-diversity of the gut microbiota was noted in the patients with substantial diarrhea and those with minor or no diarrhea between the bevacizumab (P = 0.491, Figure 3E) and cetuximab (P = 0.961, Figure 3F) subgroups.
To analyze bacterial taxa, we compared bacterial species expression among the groups and subgroups. Bacterial species that were not significantly differentially abundant after treatment were screened out, and differences in abundance are expressed as log2 fold changes. Figure 4A illustrates the differential taxon abundance of the PD and PR groups. K. quasipneumoniae in the PD group had a greater fold change in abundance than did K. quasipneumoniae in the PR group, and the difference between them in the fold change in abundance was the greatest among all bacterial species in the PD group. Limosilactobacillus mucosae exhibited the second greatest difference in the fold change in abundance between the PD (the greater of the two) and PR groups. Veillonella atypica, Veillonella dispar, Veillonella nakazawae, and Veillonella S12025-13 in the PD group demonstrated greater fold changes in abundance than in the PR group. The Lactobacillus species had a higher abundance in the PD group than in the PR group. Bifidobacterium dentium, Bifidobacterium breve, and Bifidobacterium bifidum had higher abundance in the PR group than in the PD group. The abundance of F. nucleatum in the PD group was approximately 32 (25) times higher than that in the PR group.

Figure 4 Results of the log2 fold change in the disease progression and partial response groups and subgroups of bevacizumab and cetuximab. Each dot indicates a specific bacteria species with a significant difference (P < 0.05) indicated on the y-axis. Positive values on the x-axis indicate that the bacterial species was significantly more abundant in the PR than in the PD group and subgroups. Negative values indicate the opposite. The zero point indicates the equal fold of abundance in both the groups (20). Only log2 fold change values >|2| are shown. (A) Bacterial expression was compared between the PD and PR groups. Fusobacterium nucleatum in the PD group was located at approximately the −5 point (log2 fold change = −5.13, Padj = 3.37e-6), indicating that the expression of F. nucleatum was higher in the PD group than in the PR group by approximately 32 (25) times. Prevotella copri in the PR group was located at approximately 3 point (log2 fold change = 2.90, Padj = 0.043), indicating that the expression of P. copri was higher in the PR group than in the PD group by approximately 8 (23) times. (B) In the bevacizumab-PD subgroup, Lacticaseibacillus paracasei exhibited the highest fold change in abundance by more than 1000 (210) times compared with the bevacizumab-PR subgroup (log2 fold change = −10.23, Padj = 8.28e-8). Other Lactobacillus species, including Limosilactobacillus vaginalis, Limosilactobacillus fermentum, and Lactobacillus delbrueckii, exhibited a higher fold change in abundance in the PD group than in the PR subgroup (log2 fold change = −9.06, Padj = 4.15e-10; log2 fold change = −6.22, Padj = 8.90e-6; and log2 fold change = −2.32, Padj = 6.22e-6, respectively). Bifidobacterium dentium and Bifidobacterium breve presented a higher fold of abundance (log2 fold change = −5.85, Padj = 8.90e-6 and log2 fold change = −4.34, Padj = 4.66e-5, respectively). F. nucleatum exhibited a higher fold change in abundance by approximately 64 (26) times (log2 fold change = −5.77, Padj = 2.84e-4). In the bevacizumab-PR subgroup, P. dentalis and P. copri revealed a higher fold change in abundance than did the bevacizumab-PD subgroup by approximately 8 (23) times (log2 fold change = 2.77, Padj = 4.41e-3 and log2 fold change = 2.90, Padj = 7.58e-3, respectively). (C) In the cetuximab-PD subgroup, no bacterial species presented a significantly different fold change in abundance. Anaerostipes caccae (log2 fold change = 4.94; Padj = 2.47e-3) and Adlercrentzia equolifaciens (log2 fold change = 4.70; Padj = 6.92e-3) revealed a higher fold change in abundance in the cetuximab-PR subgroup than in the cetuximab-PD subgroup.
Phascolarctobacterium sp. Marseille-Q4147 in the PR group exhibited a greater fold change in abundance than in the PD group, and the difference between them in the fold change in abundance was the greatest among all bacterial species in the PR group. Similarly, Prevotella dentalis and Prevotella copri in the PR group exhibited greater fold changes in abundance than in the PD group.
In the bevacizumab-PD subgroup, Lacticaseibacillus paracasei, Li. Vaginalis, Limosilactobacillus fermentum, and Lactobacillus delbrueckii exhibited greater fold changes in abundance than in the bevacizumab-PR subgroup. Lacti paracasei demonstrated the largest fold change in abundance (>1000 [210] times) in the bevacizumab-PD subgroup. B. dentium and B. breve in the bevacizumab-PD subgroup presented greater fold changes in abundance than in the bevacizumab-PR subgroup. The fold change in the abundance of F. nucleatum in the bevacizumab-PD subgroup was nearly 64 (26) times greater than that of F. nucleatum in the bevacizumab-PR subgroup. The fold changes in the abundance of P. dentalis and P. copri in the bevacizumab-PR subgroup were approximately 8 (23) times greater than those of their counterparts in the bevacizumab-PD subgroup (Figure 4B).
In the cetuximab-PD subgroup, no bacterial species were significantly differentially abundant. Anaerostipes caccae and Adlercrentzia equolifaciens demonstrated greater fold changes in abundance in the cetuximab-PR subgroup than in the cetuximab-PD subgroup (Figure 4C).
The human gut microbiota is closely associated with CRC development (15, 18, 38, 39). In healthy individuals, the diversity and balance of the intestinal microbiota are essential to maintain the mucosal barrier and prevent pathogen invasion (15, 18, 38, 39). A balanced gut microbiota combined with favorable genetic or immune conditions, lifestyle, diet, and environmental factors contribute to intestinal homeostasis. The dysregulation of these factors can alter the gut microbiota and lead to gut dysbiosis (18, 38, 39). In the present study, the stool samples of the PR group and the bevacizumab-PR subgroup demonstrated significantly higher α-diversity of bacterial species than those of the PD group and the bevacizumab-PD subgroup, respectively. These results indicate that higher intestinal bacterial diversity may be helpful in the treatment of mCRC.
The α-diversity of bacterial species in the stool samples of the patients who received cetuximab did not significantly differ between the PR and PD groups. This finding may be attributable to 1) the limited number of the patients included in this study or 2) selection bias caused by the administration of cetuximab to only patients with the wild-type RAS gene. The KRAS gene, a member of the RAS gene family, is associated with many signaling pathways, including those related to inflammation, the immune system, and metabolism. The gut microbiota is a key factor because it maintains intestinal homeostasis through these pathways (40). Variations in the KRAS gene can disrupt normal signaling functions and hamper intestinal homeostasis. The accumulative effect can result in CRC tumorigenesis and affect CRC treatment outcomes (40). Microbiota can significantly differ between CRC tissues with and without KRAS gene variations (41). In clinical practice, anti-EGFR agents (e.g., cetuximab) are unsuitable for treating patients with KRAS gene variations (25, 26). In our study, those who received cetuximab were all wild-type KRAS gene carriers, whereas the patients who received bevacizumab comprised both wild-type and KRAS gene variation carriers (Figure 1). This selection bias may have affected the α-diversity results. However, more evidence is necessary to determine the relationship between intestinal bacterial diversity and KRAS gene variations.
We observed no significant difference in the β-diversity of bacterial species between the PD and PR groups. This result indicates the presence of similar intestinal bacterial composition in the PR and PD groups. The human gut microbiota can be individualized under the effect of both genetic and environmental factors (42–44). Dietary habit is an especially crucial environmental factor that can modulate microbiota composition (42, 44). Most of our included patients lived in southern Taiwan and shared similar food, culture, and climate. This may partially explain our β-diversity results, which indicated that the treatment outcome did not depend only on the gut microbiota. Age, sex, genetic factors, and nutritional status also affect CRC treatment outcomes. β-diversity was significantly different between the bevacizumab-PD and bevacizumab-PR subgroups. This result suggests that the composition of the gut microbiota is associated with the treatment response in specific subgroups. During our treatment course, the bevacizumab group comprised both the wild-type and RAS gene variation carriers (Figure 1). The KRAS gene is associated with various signaling pathways and can alter treatment outcomes (26, 40, 41). The relationship between the KRAS gene and human gut microbiota remains unclear. More studies, such as those investigating orthotopic rectal cancer by using animal models, should be conducted to elucidate this relationship in the future (16). Additional stool samples from the patients in the cetuximab-PD and cetuximab-PR subgroups may be necessary to obtain more compelling results.
In our experience, patients with mCRC typically require longer courses of systemic therapy. In this situation, oxaliplatin-based chemotherapy, such as FOLFOX (folinic acid, 5-FU, and oxaliplatin) may cause severe or irreversible peripheral neuropathy. Therefore, we used a FOLFIRI regimen as the initial therapy for mCRC. In this study, all our patients received chemotherapy with the FOLFIRI regimen, and the most frequent AE of the FOLFIRI regimen is diarrhea. The human gut microbiota plays a role in mitigating diarrhea caused by chemotherapy (45–47). Antibiotic-associated diarrhea caused by Clostridium difficile is a well-known condition that is the result of gut dysbiosis (45, 48). Normal intestinal commensal bacteria can inhibit pathogen growth and prevent pathogen-induced diarrhea (45). Many studies have reported that probiotics, including Bifidobacterium, Lactobacillus, and yeast, can improve gut microbiota balance and inhibit pathogen colonization. Through this mechanism, probiotics can prevent or treat pathogen-induced diarrhea (45, 47). Bacteria that produce β-glucuronidase can aggravate diarrhea (21, 23, 24), and β-glucuronidase inhibitors can be used for the symptomatic relief of diarrhea (21, 49). In theory, the elimination of bacteria that produce β-glucuronidase can also alleviate diarrhea. However, we failed to identify relevant bacterial species in our stool sample analysis.
DESeq2 analysis is a powerful parametric approach to compare targeted metagenomics (50, 51).We observed heterogeneous results when comparing the PD and PR groups. The Bifidobacterium and Lactobacillus species all exhibited significantly greater fold changes in abundance in the PD group than in the PR group. Moreover, the Bifidobacterium and Lactobacillus species demonstrated greater fold changes in abundance in the bevacizumab-PD subgroup than in the bevacizumab-PR subgroup. Bifidobacterium and Lactobacillus species are widely used in commercial probiotic supplements (52–54). Some studies have reported an association between Bifidobacterium species and CRC tumorigenesis (47). The effects of Bifidobacterium and Lactobacillus species on CRC chemotherapy and immune therapy outcomes have been studied (47, 55). However, how Bifidobacterium and Lactobacillus species affect the outcomes of targeted therapies for mCRC is not yet clearly understood.
Bifidobacterium species can metabolize carbohydrates to produce mainly lactate and acetate. These two acids maintain gut microbiota homeostasis and inhibit pathogen overgrowth (53, 56). Intestinal homeostasis reduces the risk of bowel inflammation and improves the human immune system, thus inhibiting tumorigenesis (15, 57). In cell line studies, Bifidobacterium species have been observed to inhibit colon cancer cell growth and proliferation (56, 58). However, in an animal study, Bifidobacterium species was observed both in normal and colon cancer mice (59). Lactobacillus species are common microorganisms that provide various health benefits, including exerting immunomodulatory, antidiabetic, and tumor suppressive effects (54). Some Lactobacillus species inhibit CRC tumorigenesis or enhance CRC treatment outcomes (55, 60). However, Lactobacillus is a complicated genus containing more than 170 species that can cause opportunistic infections under specific conditions (61, 62).
In the present study, F. nucleatum exhibited significantly greater fold changes in abundance in both the PD group and bevacizumab-PD subgroup than in the opposing group and subgroup. F. nucleatum is a gram-negative anaerobic bacterium that is strongly associated with CRC initiation and progression (63, 64); mechanisms underlying this association include metabolism alteration, immune modulation, and virulence factor expression (63). F. nucleatum is more abundant in colorectal adenomas and adenocarcinomas than in the normal colon tissue, and it can promote CRC tumorigenesis (63, 64). Moreover, animal studies have demonstrated that F. nucleatum can facilitate CRC distant metastasis and intensify chemoresistance (22, 65). Thus, the greater fold change in the abundance of F. nucleatum in the PD group and the bevacizumab-PD subgroup was expected.
The heterogeneity of our results indicates that interactions among the mCRC, host gene type, chemotherapy and targeted therapy, and gut microbiota involve a complex network instead of a simple pathway. Other studies examining the association between the gut microbiota and CRC tumorigenesis have reported heterogenous results. Liu et al. revealed that the abundance of some CRC-associated bacterial species, such as those of the Fusobacterium, Bacteroides, and Prevotella genera, varied among different biopsy specimens and that the compositions of the gut microbiota significantly differed between the adenoma and adenocarcinoma tissue (41). The composition of the gut microbiota also differed between the proximal and distal CRC tissues. However, the finding of the bacterial analysis performed using stool samples is not equivalent to that conducted using CRC tissues (66). We provide preliminary data regarding the association of the gut microbiota and combined chemotherapy and targeted therapy in mCRC.
P. dentalis and P. copri exhibited significantly greater fold changes in abundance in both the PR and bevacizumab-PR subgroups than in the PD group and bevacizumab-PD subgroup. Prevotella species are associated with the development of CRC because they were discovered to be more abundant in patients with CRC and in the adenoma tissue than in healthy individuals (41, 66, 67). The heterogeneity of Prevotella species in adenoma–adenocarcinoma sequences was also noted. Liu et al. reported that the abundance of Prevotella species was high in the adenoma tissue but low in the CRC tissue (41). Niccolai et al. demonstrated that Prevotella species are positively correlated with interleukin-9. Although interleukin-9 is associated with CRC tumorigenesis, the authors could not confirm the role of Prevotella species in CRC development (68). Because all the stool samples in our study were collected from the patients with CRC, a higher abundance of Prevotella species in the PR group is expected. However, the effects of Prevotella species on CRC treatment, especially with targeted therapy, are uncertain and require further investigation.
The fold change in the abundance of K. quasipneumoniae in the PR group was nearly 1000 (210) times greater than that of K. quasipneumoniae in the PD group. K. quasipneumoniae is closely related to Klebsiella pneumoniae (69), and interspecies and intraspecies gene transmission is possible between the two species (70). K. pneumoniae is a gram-negative pathogen that cause various infectious diseases (71) and is also associated with CRC development because it produces colibactin (72, 73). Colibactin, which was initially reported in Escherichia coli, is a bacterial toxin that contributes to CRC tumorigenesis (72–74). E. coli carries the polyketide synthase (pks) gene that can produce colibactin and induce CRC development (72, 74). K. pneumoniae also carry the pks gene and produce colibactin, causing DNA damage and subsequent CRC carcinogenesis (72, 73). Lai et al. reported that the incidence rate of pks-positive K. pneumoniae in Taiwan is approximately 25.6%, which is much higher than the 3.5% rate in Europe (73). Therefore, studying the role of pks-positive K. pneumoniae in Taiwanese patients undergoing treatment for mCRC is crucial to determine the role of K. pneumoniae as a potential target bacterium for future treatments.
Several blind spots were present in our data. For example, Veillonella species (V. atypica, V. dispar, and V. nakazawae) were highly abundant in the PR group, but little information is available regarding their clinical effects on CRC. Some bacteria that have been demonstrated to contribute to CRC development, such as Enterococcus faecalis, Bacteroides fragilis, and E. coli (15, 18, 39), were not found to be highly abundant in our results. Lactobacillus and Bifidobacterium species were highly abundant in the nonfavorable outcome group. Detailed dietary habit history, especially that related to probiotic intake, may be necessary to yield more robust results.
The major limitation of this study is the insufficiently small sample sizes of patients and stool samples. According to the RECIST criteria, the patients with a SD were considered to not exhibit the best response; thus, we excluded these patients from our study (28). Although some studies have classified patients without a PD as responders and compared outcomes between PD and PR + SD groups (75, 76), we identified the bacterial species that have the highest potential to affect mCRC treatment outcomes. Thus, we compared the gut microbiota between the PD and PR groups. This study design resulted in the small size of our patient sample. For instance, anti-EGFR agents are more effective in inducing CRC cytoreduction than do anti-VEGF agents in patients with the wild-type KRAS gene and mCRC, which means fewer patients exhibit a PD after using anti-EGFR agents as the first-line treatment (77, 78). In our study, only six patients met the criteria for inclusion in the cetuximab-PD subgroup, which greatly hampered the interpretation of our results. Nevertheless, this pilot study represents the first step to discovering the interaction between the human gut microbiota and targeted therapies for CRC. A larger sample size and more stool samples are required to clarify the role of the gut microbiota in mCRC treatment, especially its interaction with combined chemotherapy and targeted therapy.
Our results indicate that a higher gut microbiota diversity is associated with a more favorable treatment outcome in patients with mCRC. Considerable heterogeneity was observed in the bacterial species in the stool samples of the patients with mCRC. Lactobacillus and Bifidobacterium species exhibited significantly greater fold changes in abundance in the PD group than in the PR group. F. nucleatum exhibited significantly greater fold changes in abundance in the PD group and the bevacizumab-PD subgroup. K. quasipneumoniae exhibited the greatest fold change in abundance among all bacterial species in the PD group. This result warrants further investigation especially in a Taiwanese population. A prospective, randomized study with a larger sample size and greater number of stool samples is necessary to validate the correlation between microbiota and targeted therapy in mCRC.
The data presented in the study is deposited in the European Bioinformatics Institute (EMBL-EBI) repository. Accession number is PRJEB55366. The direct link is https://www.ebi.ac.uk/ena/browser/view/PRJEB55366.
The studies involving human participants were reviewed and approved by Institutional Ethics Committee of Kaohsiung Medical University Chung-Ho Memorial Hospital. The patients/participants provided their written informed consent to participate in this study.
Conceptualization, L-HL, D-CW, T-LC, C-YL, and J-YW; Methodology, C-HC, Z-FM, K-LY, C-JL, and C-YL; Software, C-HC and C-YL; Validation, C-YL and J-YW; Visualization, Y-CC and C-HC; Writing—original draft preparation, Y-CC; Writing—review and editing, Y-CC, C-HC, C-YL, and J-YW; All authors contributed to the article and approved the submitted version.
This work was supported by grants through funding from the Ministry of Science and Technology (MOST 109-2314-B-037-035, MOST 109-2314-B-037-040, MOST 109-2314-B-037-046-MY3, MOST110-2314-B-037-097) and the Ministry of Health and Welfare (MOHW109-TDU-B-212-134026, MOHW109-TDU-B-212-114006, MOHW110-TDU-B-212-1140026) and funded by the health and welfare surcharge of on tobacco products, and the Kaohsiung Medical University Hospital (KMUH110-0R37, KMUH110-0R38, KMUH110-0M34, KMUH110-0M35, KMUH110-0M36, KMUHSA11013, KMUH-DK(C)110010, KMUH-DK(B)110004-3) and KMU Center for Cancer Research (KMU-TC111A04-1) and KMU Center for Liquid Biopsy and Cohort Research Center Grant (KMU-TC109B05) and KMU Office for Industry-Academic Collaboration (S109036),Kaohsiung Medical University. In addition, this study was supported by the Grant of Taiwan Precision Medicine Initiative and Taiwan Biobank, Academia Sinica, Taiwan, R.O.C.
We acknowledge the support of Kaohsiung Medical University Hospital, Kaohsiung Medical University and Taiwan Academia Sinica.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: Cancer J Clin (2019) 69(1):7–34. doi: 10.3322/caac.21551
2. Onyoh EF, Hsu WF, Chang LC, Lee YC, Wu MS, Chiu HM. The rise of colorectal cancer in Asia: Epidemiology, screening, and management. Curr Gastroenterol Rep (2019) 21(8):36. doi: 10.1007/s11894-019-0703-8
3. Taiwan Health Promotion Administration. Staying away from the threat of colorectal cancer amounts to fecal occult blood test (FOBT) and early intervention to maintain intestinal health. Ministry of Health and Welfare, Taiwan. (2018). Available at: https://www.hpa.gov.tw/EngPages/Detail.aspx?nodeid=1038&pid=7615.
4. Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA (2021) 325(7):669–85. doi: 10.1001/jama.2021.0106
5. Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med (2009) 360(14):1408–17. doi: 10.1056/NEJMoa0805019
6. Hsieh YC, Chang TK, Su WC, Huang CW, Tsai HL, Chen YC, et al. UGT1A1 polymorphism for irinotecan dose escalation in patients with BRAF-mutated metastatic colorectal cancer treated with first-line bevacizumab and FOLFIRI. J Oncol (2021) 2021:6686517. doi: 10.1155/2021/6686517
7. Yeh JH, Tsai HL, Chen YC, Li CC, Huang CW, Chang TK, et al. BRAF, MEK, and EGFR triplet inhibitors as salvage therapy in BRAF-mutated metastatic colorectal cancer-a case series study target therapy of BRAF-mutated mCRC. Medicina (Kaunas Lithuania) (2021) 57(12):1339. doi: 10.3390/medicina57121339
8. Huang CW, Tsai HL, Chen YT, Huang CM, Ma CJ, Lu CY, et al. The prognostic values of EGFR expression and KRAS mutation in patients with synchronous or metachronous metastatic colorectal cancer. BMC Cancer (2013) 13:599. doi: 10.1186/1471-2407-13-599
9. Tsai HL, Huang CW, Lin YW, Wang JH, Wu CC, Sung YC, et al. Determination of the UGT1A1 polymorphism as guidance for irinotecan dose escalation in metastatic colorectal cancer treated with first-line bevacizumab and FOLFIRI (Pure fist). Eur J Cancer (Oxford England: 1990) (2020) 138:19–29. doi: 10.1016/j.ejca.2020.05.031
10. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med (2004) 350(23):2335–42. doi: 10.1056/NEJMoa032691
11. Peeters M, Price T. Biologic therapies in the metastatic colorectal cancer treatment continuum–applying current evidence to clinical practice. Cancer Treat Rev (2012) 38(5):397–406. doi: 10.1016/j.ctrv.2011.08.002
12. Modest DP, Pant S, Sartore-Bianchi A. Treatment sequencing in metastatic colorectal cancer. Eur J Cancer (Oxford England: 1990) (2019) 109:70–83. doi: 10.1016/j.ejca.2018.12.019
13. Tsai HL, Chen YC, Yin TC, Su WC, Chen PJ, Chang TK, et al. Comparison of UGT1A1 polymorphism as guidance of irinotecan dose escalation in RAS wild type metastatic colorectal cancer patients treated with cetuximab or bevacizumab plus FOLFIRI as the first-line therapy. Oncol Res (2022) 29(1):47–61. doi: 10.3727/096504022x16451187313084
14. Kim M, Vogtmann E, Ahlquist DA, Devens ME, Kisiel JB, Taylor WR, et al. Fecal metabolomic signatures in colorectal adenoma patients are associated with gut microbiota and early events of colorectal cancer pathogenesis. mBio (2020) 11(1):e03186–19. doi: 10.1128/mBio.03186-19
15. Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol (2016) 22(2):501–18. doi: 10.3748/wjg.v22.i2.501
16. Chen YC, Miao ZF, Yip KL, Cheng YA, Liu CJ, Li LH, et al. Gut fecal microbiota transplant in a mouse model of orthotopic rectal cancer. Front Oncol (2020) 10:568012. doi: 10.3389/fonc.2020.568012
17. Fiorentini C, Carlini F, Germinario EAP, Maroccia Z, Travaglione S, Fabbri A. Gut microbiota and colon cancer: A role for bacterial protein toxins? Int J Mol Sci (2020) 21(17):6201. doi: 10.3390/ijms21176201
18. Kang M, Martin A. Microbiome and colorectal cancer: Unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin Immunol (2017) 32:3–13. doi: 10.1016/j.smim.2017.04.003
19. Yixia Y, Sripetchwandee J, Chattipakorn N, Chattipakorn SC. The alterations of microbiota and pathological conditions in the gut of patients with colorectal cancer undergoing chemotherapy. Anaerobe (2021) 68:102361. doi: 10.1016/j.anaerobe.2021.102361
20. Wu X, Zhang T, Chen X, Ji G, Zhang F. Microbiota transplantation: Targeting cancer treatment. Cancer Lett (2019) 452:144–51. doi: 10.1016/j.canlet.2019.03.010
21. Chang TK, Yin TC, Su WC, Tsai HL, Huang CW, Chen YC, et al. A pilot study of silymarin as supplementation to reduce toxicities in metastatic colorectal cancer patients treated with first-line FOLFIRI plus bevacizumab. Oncol Res (2021) 28(7):801–9. doi: 10.3727/096504021x16218531628569
22. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell (2017) 170(3):548–63.e16. doi: 10.1016/j.cell.2017.07.008
23. Gori S, Inno A, Belluomini L, Bocus P, Bisoffi Z, Russo A, et al. Gut microbiota and cancer: How gut microbiota modulates activity, efficacy and toxicity of antitumoral therapy. Crit Rev oncology/hematology (2019) 143:139–47. doi: 10.1016/j.critrevonc.2019.09.003
24. Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol (2017) 14(6):356–65. doi: 10.1038/nrgastro.2017.20
25. Fornasier G, Francescon S, Baldo P. An update of efficacy and safety of cetuximab in metastatic colorectal cancer: A narrative review. Adv Ther (2018) 35(10):1497–509. doi: 10.1007/s12325-018-0791-0
26. Afrăsânie VA, Marinca MV, Alexa-Stratulat T, Gafton B, Păduraru M, Adavidoaiei AM, et al. KRAS, NRAS, BRAF, HER2 and Microsatellite Instability in Metastatic Colorectal Cancer - Practical Implications for the Clinician. Radiol Oncol (2019) 53(3):265–74. doi: 10.2478/raon-2019-0033
27. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, national cancer institute of the united states, national cancer institute of Canada. J Natl Cancer Institute (2000) 92(3):205–16. doi: 10.1093/jnci/92.3.205
28. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised recist guideline (Version 1.1). Eur J Cancer (Oxford England: 1990) (2009) 45(2):228–47. doi: 10.1016/j.ejca.2008.10.026
29. U.S. Department of Health and Human Services NIoH, National Cancer Institute, USA.gov. Common terminology criteria for adverse events (Ctcae) V4.0 (June 14, 2010). Available at: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_40.
30. Qiagen H. Germany. Qiaamp Fast DNA Stool Mini Kit. (2022). Available at: https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/dna-purification/genomic-dna/qiaamp-fast-dna-stool-mini-kit.
31. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with kraken 2. Genome Biol (2019) 20(1):257. doi: 10.1186/s13059-019-1891-0
32. Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell (2019) 176(3):649–62.e20. doi: 10.1016/j.cell.2019.01.001
33. Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput Sci (2017) 3:e104. doi: 10.7717/peerj-cs.104
34. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using qiime 2. Nat Biotechnol (2019) 37(8):852–7. doi: 10.1038/s41587-019-0209-9
35. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
36. Xun W, Li W, Xiong W, Ren Y, Liu Y, Miao Y, et al. Diversity-triggered deterministic bacterial assembly constrains community functions. Nat Commun (2019) 10(1):3833. doi: 10.1038/s41467-019-11787-5
37. Socolar JB, Gilroy JJ, Kunin WE, Edwards DP. How should beta-diversity inform biodiversity conservation? Trends Ecol Evol (2016) 31(1):67–80. doi: 10.1016/j.tree.2015.11.005
38. Saus E, Iraola-Guzmán S, Willis JR, Brunet-Vega A, Gabaldón T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol aspects Med (2019) 69:93–106. doi: 10.1016/j.mam.2019.05.001
39. Dai Z, Zhang J, Wu Q, Chen J, Liu J, Wang L, et al. The role of microbiota in the development of colorectal cancer. Int J Cancer (2019) 145(8):2032–41. doi: 10.1002/ijc.32017
40. Ternet C, Kiel C. Signaling pathways in intestinal homeostasis and colorectal cancer: KRAS at centre stage. Cell communication signaling: CCS (2021) 19(1):31. doi: 10.1186/s12964-021-00712-3
41. Liu W, Zhang X, Xu H, Li S, Lau HC, Chen Q, et al. Microbial community heterogeneity within colorectal neoplasia and its correlation with colorectal carcinogenesis. Gastroenterology (2021) 160(7):2395–408. doi: 10.1053/j.gastro.2021.02.020
42. Bibbò S, Ianiro G, Giorgio V, Scaldaferri F, Masucci L, Gasbarrini A, et al. The role of diet on gut microbiota composition. Eur Rev Med Pharmacol Sci (2016) 20(22):4742–9.
43. O’Keefe SJ. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol (2016) 13(12):691–706. doi: 10.1038/nrgastro.2016.165
44. Zmora N, Suez J, Elinav E. You are what you eat: Diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol (2019) 16(1):35–56. doi: 10.1038/s41575-018-0061-2
45. Li Y, Xia S, Jiang X, Feng C, Gong S, Ma J, et al. Gut microbiota and diarrhea: An updated review. Front Cell Infect Microbiol (2021) 11:625210. doi: 10.3389/fcimb.2021.625210
46. Cheng WY, Wu CY, Yu J. The role of gut microbiota in cancer treatment: Friend or foe? Gut (2020) 69(10):1867–76. doi: 10.1136/gutjnl-2020-321153
47. Wieczorska K, Stolarek M, Stec R. The role of the gut microbiome in colorectal cancer: Where are we? where are we going? Clin colorectal Cancer (2020) 19(1):5–12. doi: 10.1016/j.clcc.2019.07.006
48. Ramirez J, Guarner F, Bustos Fernandez L, Maruy A, Sdepanian VL, Cohen H. Antibiotics as major disruptors of gut microbiota. Front Cell infection Microbiol (2020) 10:572912. doi: 10.3389/fcimb.2020.572912
49. Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Sci (New York NY) (2010) 330(6005):831–5. doi: 10.1126/science.1191175
50. Calle ML. Statistical analysis of metagenomics data. Genomics Inf (2019) 17(1):e6. doi: 10.5808/GI.2019.17.1.e6
51. Goolam Mahomed T, Peters R, Pretorius G, Goolam Mahomed A, Ueckermann V, Kock MM, et al. Comparison of targeted metagenomics and is-pro methods for analysing the lung microbiome. BMC Microbiol (2021) 21(1):228. doi: 10.1186/s12866-021-02288-x
52. Kim MJ, Ku S, Kim SY, Lee HH, Jin H, Kang S, et al. Safety evaluations of bifidobacterium bifidum BGN4 and bifidobacterium longum bori. Int J Mol Sci (2018) 19(5):1422. doi: 10.3390/ijms19051422
53. Eslami M, Yousefi B, Kokhaei P, Hemati M, Nejad ZR, Arabkari V, et al. Importance of probiotics in the prevention and treatment of colorectal cancer. J Cell Physiol (2019) 234(10):17127–43. doi: 10.1002/jcp.28473
54. Slattery C, Cotter PD, O’Toole PW. Analysis of health benefits conferred by lactobacillus species from kefir. Nutrients (2019) 11(6):1252. doi: 10.3390/nu11061252
55. Zhuo Q, Yu B, Zhou J, Zhang J, Zhang R, Xie J, et al. Lysates of lactobacillus acidophilus combined with CTLA-4-Blocking antibodies enhance antitumor immunity in a mouse colon cancer model. Sci Rep (2019) 9(1):20128. doi: 10.1038/s41598-019-56661-y
56. Bahmani S, Azarpira N, Moazamian E. Anti-colon cancer activity of bifidobacterium metabolites on colon cancer cell line Sw742. Turkish J gastroenterology: Off J Turkish Soc Gastroenterol (2019) 30(9):835–42. doi: 10.5152/tjg.2019.18451
57. Lucas C, Barnich N, Nguyen HTT. Microbiota, inflammation and colorectal cancer. Int J Mol Sci (2017) 18(6):1310. doi: 10.3390/ijms18061310
58. Kim Y, Lee D, Kim D, Cho J, Yang J, Chung M, et al. Inhibition of proliferation in colon cancer cell lines and harmful enzyme activity of colon bacteria by bifidobacterium adolescentis Spm0212. Arch pharmacal Res (2008) 31(4):468–73. doi: 10.1007/s12272-001-1180-y
59. Luo J, Li Y, Xie J, Gao L, Liu L, Ou S, et al. The primary biological network of bifidobacterium in the gut. FEMS Microbiol Lett (2018) 365(8):fny057. doi: 10.1093/femsle/fny057
60. Sugimura N, Li Q, Chu ESH, Lau HCH, Fong W, Liu W, et al. Lactobacillus gallinarum modulates the gut microbiota and produces anti-cancer metabolites to protect against colorectal tumourigenesis. Gut (2021) gutjnl-2020-323951. doi: 10.1136/gutjnl-2020-323951
61. O’Callaghan J, O’Toole PW. Lactobacillus: Host-microbe relationships. Curr topics Microbiol Immunol (2013) 358:119–54. doi: 10.1007/82_2011_187
62. Goldstein EJ, Tyrrell KL, Citron DM. Lactobacillus species: Taxonomic complexity and controversial susceptibilities. Clin Infect diseases: an Off Publ Infect Dis Soc America (2015) 60 Suppl 2:S98–107. doi: 10.1093/cid/civ072
63. Hashemi Goradel N, Heidarzadeh S, Jahangiri S, Farhood B, Mortezaee K, Khanlarkhani N, et al. Fusobacterium nucleatum and colorectal cancer: A mechanistic overview. J Cell Physiol (2019) 234(3):2337–44. doi: 10.1002/jcp.27250
64. Shang FM, Liu HL. Fusobacterium nucleatum and colorectal cancer: A review. World J Gastrointestinal Oncol (2018) 10(3):71–81. doi: 10.4251/wjgo.v10.i3.71
65. Guo S, Chen J, Chen F, Zeng Q, Liu WL, Zhang G. Exosomes derived from fusobacterium nucleatum-infected colorectal cancer cells facilitate tumour metastasis by selectively carrying mir-1246/92b-3p/27a-3p and Cxcl16. Gut (2020) gutjnl-2020-321187. doi: 10.1136/gutjnl-2020-321187
66. Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, et al. Tumour-associated and non-Tumour-Associated microbiota in colorectal cancer. Gut (2017) 66(4):633–43. doi: 10.1136/gutjnl-2015-309595
67. Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of fusobacterium persistence and antibiotic response in colorectal cancer. Sci (New York NY) (2017) 358(6369):1443–8. doi: 10.1126/science.aal5240
68. Niccolai E, Russo E, Baldi S, Ricci F, Nannini G, Pedone M, et al. Significant and conflicting correlation of il-9 with prevotella and bacteroides in human colorectal cancer. Front Immunol (2020) 11:573158. doi: 10.3389/fimmu.2020.573158
69. Martin RM, Bachman MA. Colonization, infection, and the accessory genome of klebsiella pneumoniae. Front Cell infection Microbiol (2018) 8:4. doi: 10.3389/fcimb.2018.00004
70. Mathers AJ, Crook D, Vaughan A, Barry KE, Vegesana K, Stoesser N, et al. Klebsiella quasipneumoniae provides a window into carbapenemase gene transfer, plasmid rearrangements, and patient interactions with the hospital environment. Antimicrobial Agents Chemotherapy (2019) 63(6):e02513–18. doi: 10.1128/aac.02513-18
71. Wang G, Zhao G, Chao X, Xie L, Wang H. The characteristic of virulence, biofilm and antibiotic resistance of klebsiella pneumoniae. Int J Environ Res Public Health (2020) 17(17):6278. doi: 10.3390/ijerph17176278
72. Strakova N, Korena K, Karpiskova R. Klebsiella pneumoniae producing bacterial toxin colibactin as a risk of colorectal cancer development - a systematic review. Toxicon: Off J Int Soc Toxinology (2021) 197:126–35. doi: 10.1016/j.toxicon.2021.04.007
73. Lai YC, Lin AC, Chiang MK, Dai YH, Hsu CC, Lu MC, et al. Genotoxic klebsiella pneumoniae in Taiwan. PloS One (2014) 9(5):e96292. doi: 10.1371/journal.pone.0096292
74. Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes (2014) 5(5):675–80. doi: 10.4161/19490976.2014.969989
75. Bang JI, Lim Y, Paeng JC, Han SW, Park S, Lee JM, et al. Comparison of quantitative methods on FDG PET/CT for treatment response evaluation of metastatic colorectal cancer. Nucl Med Mol Imaging (2017) 51(2):147–53. doi: 10.1007/s13139-016-0449-2
76. Li L, Xu Y, Yang Y, Ye F, Zhang H, Zhou A, et al. Recist 1.1, Choi and mChoi criteria in the evaluation of tumor response in patients with metastatic colorectal cancer treated with Regorafenib and anti-PD-1 antibody. Eur J Radiol (2021) 141:109823. doi: 10.1016/j.ejrad.2021.109823
77. Arnold D, Lueza B, Douillard JY, Peeters M, Lenz HJ, Venook A, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann oncology: Off J Eur Soc Med Oncol (2017) 28(8):1713–29. doi: 10.1093/annonc/mdx175
78. Yoshino T, Arnold D, Taniguchi H, Pentheroudakis G, Yamazaki K, Xu RH, et al. Pan-Asian Adapted ESMO Consensus Guidelines for the Management of Patients with Metastatic Colorectal Cancer: A JSMO-ESMO Initiative Endorsed by CSCO, KACO, MOS, SSO and TOS. Ann Oncology: Off J Eur Soc Med Oncol (2018) 29(1):44–70. doi: 10.1093/annonc/mdx738
Keywords: metastatic colorectal cancer, targeted therapy, Lactobacillus species, Bifidobacterium species, Fusobacterium nucleatum, Klebsiella quasipneumoniae
Citation: Chen Y-C, Chuang C-H, Miao Z-F, Yip K-L, Liu C-J, Li L-H, Wu D-C, Cheng T−L, Lin C-Y and Wang J-Y (2022) Gut microbiota composition in chemotherapy and targeted therapy of patients with metastatic colorectal cancer. Front. Oncol. 12:955313. doi: 10.3389/fonc.2022.955313
Received: 28 May 2022; Accepted: 19 August 2022;
Published: 23 September 2022.
Edited by:
Yijia Wang, Nankai University, ChinaReviewed by:
Howard Chi Ho Yim, University of New South Wales, AustraliaCopyright © 2022 Chen, Chuang, Miao, Yip, Liu, Li, Wu, Cheng, Lin and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaw-Yuan Wang, amF3eXVhbndhbmdAZ21haWwuY29t; Y3k2MTQxMTJAbXMxNC5oaW5ldC5uZXQ=; Chung-Yen Lin, Y3lsaW5AaWlzLnNpbmljYS5lZHUudHc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.