 Stefan Forster1,2
Stefan Forster1,2 Ramin Radpour1,2*
Ramin Radpour1,2*- 1Tumor Immunology, Department for BioMedical Research (DBMR), University of Bern, Bern, Switzerland
- 2Department of Medical Oncology, Inselspital, Bern University Hospital, University of Bern, Bern, Switzerland
Multiple myeloma (MM) is the most common malignant monoclonal disease of plasma cells. Aside from classical chemotherapy and glucocorticoids, proteasome inhibitors, immunomodulatory agents and monoclonal antibodies are used in the current treatment scheme of MM. The tumor microenvironment (TME) plays a fundamental role in the development and progression of numerous solid and non-solid cancer entities. In MM, the survival and expansion of malignant plasma cell clones heavily depends on various direct and indirect signaling pathways provided by the surrounding bone marrow (BM) niche. In a number of MM patients, single plasma cell clones lose their BM dependency and are capable to engraft at distant body sites or organs. The resulting condition is defined as an extramedullary myeloma (EMM). EMMs are highly aggressive disease stages linked to a dismal prognosis. Emerging literature demonstrates that the dynamic interactions between the TME and malignant plasma cells affect myeloma dissemination. In this review, we aim to summarize how the cellular and non-cellular BM compartments can promote plasma cells to exit their BM niche and metastasize to distant intra-or extramedullary locations. In addition, we list selected therapy concepts that directly target the TME with the potential to prevent myeloma spread.
Introduction
Multiple Myeloma (MM) is a hematologic malignancy defined by the expansion of a clonal plasma cell population within the bone marrow (BM) that results in the manifestation of clinical symptoms such as bone pain, pathologic fractures and anemia (1). In almost all MM patients, disease development is characterized by a distinct sequence of primary molecular alterations leading to the early transformation of a plasma cell into its malignant counterpart. The acquisition of additional secondary mutations results in further disease progression. In addition, the tumor microenvironment (TME) and treatment-related selection mechanisms promote the expansion of distinct plasma cell clones with the most aggressive phenotype (2, 3). According to the international myeloma working group, the diagnosis of MM requires the presence of ≥ 10% clonal BM plasma cells or a biopsy-proven extramedullary plasmacytoma combined with at least one myeloma defining event (e.g., evidence of end-organ damage) and positive detection of defined biomarkers (4).
In the last decade, new MM therapy options have substantially progressed. Combination of standard chemotherapy and glucocorticoids with proteasome inhibitors (PI), immunomodulatory drugs and monoclonal antibodies (mAb) as well as other substances with innovative mechanisms of action have evidently improved the outcome of many MM patients (5). However, despite the fundamental achievements in the understanding of MM pathogenesis and the continuous supply of novel therapies, almost all MM patients eventually suffer from disease relapse (6, 7).
Refractory or relapsed disease stages bear a fundamental risk for further disease progression into the highly aggressive MM end-stages extramedullary myeloma (EMM) and plasma cell leukemia (PCL) (8–11). Here, the malignant plasma cell clones are no longer dependent on the BM microenvironment (as their extrinsic support). They are able to survive and engraft at distant body sites or organs including soft tissue, skin, liver, kidney and the central nervous system (CNS) (Figure 1) (8, 12, 13). In around two-thirds of patients suffering from EMM, clonal plasma cells break through the cortical bone and infiltrate the adjacent soft tissue. In the remaining one-third, hematogeneous dissemination to distant sites or organs can be observed (14). Patients with EMM and PCL have a significantly shorter overall survival and sufficient therapy options are often limited (8, 14, 15).
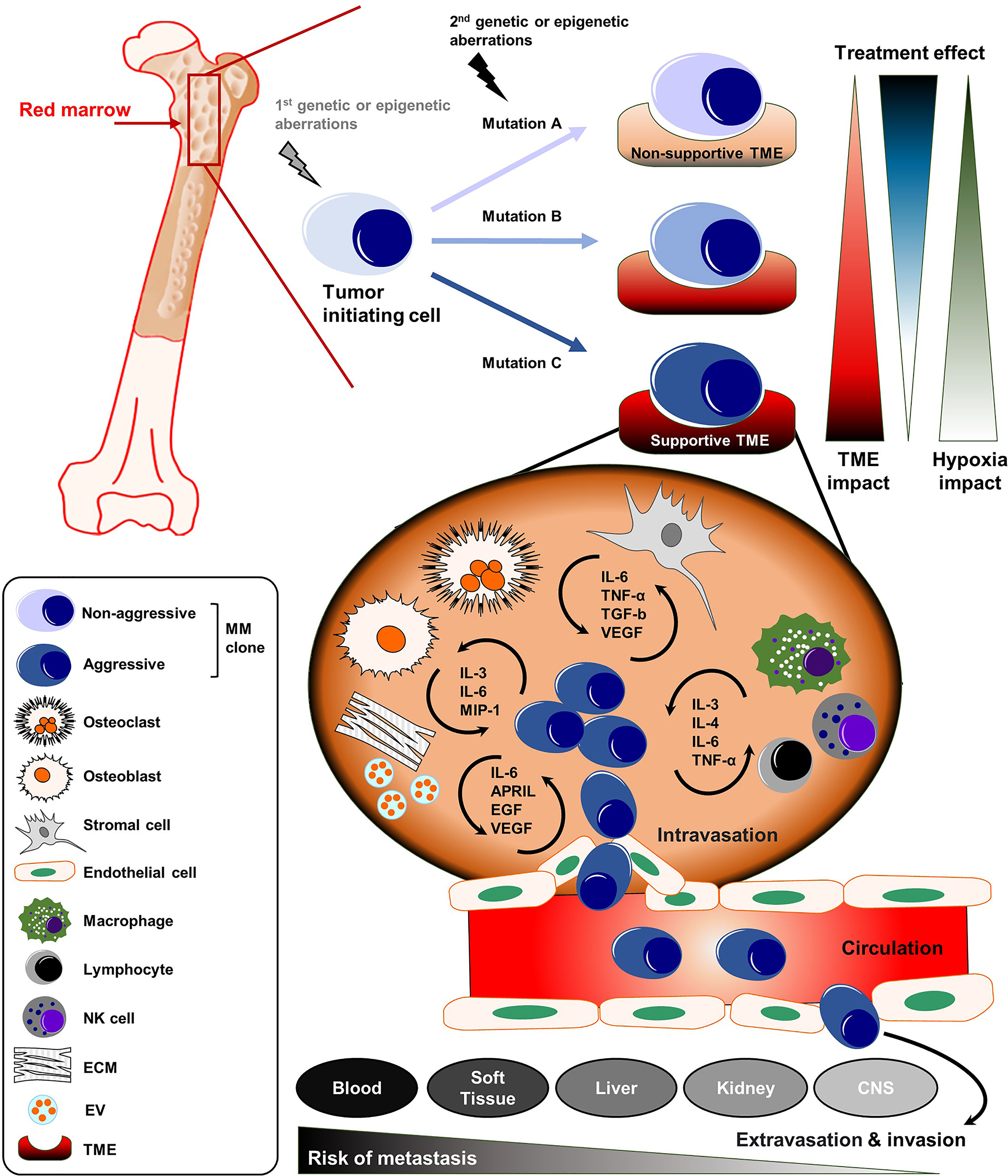
Figure 1 The complex organization of cancer initiation, progress, and distant metastasis of MM. MM is defined by the clonal expansion of a malignant plasma cell population upon genetic/epigenetic aberrations within the bone marrow (BM). The acquisition of secondary mutations in MM leads to further generation of sub-clones with a more aggressive phenotype. The dynamic influence of the TME and treatment-related mechanisms drive the progression of MM into even more aggressive disease stages. The malignant plasma cell clones can disseminate into the blood or to distant body sites or organs including soft tissue, liver, kidney and CNS. APRIL, a proliferation-inducing ligand; CNS, central nervous system; ECM, extracellular matrix; EGF, epidermal growth factor; EV, extracellular vesicle; IL, Interleukin; MIP-1, macrophage inflammatory protein 1; MM, multiple myeloma; NK, natural killer; TGF-b, transforming growth factor beta; TME, tumor microenvironment; TNF-α, Tumor necrosis factor alpha.
The tumor microenvironment (TME) is composed of different cellular compartments (e.g., endothelial cells, stromal cells, osteoclasts, osteoblasts and immune cells) and non-cellular fractions including the liquid milieu (e.g., growth factors, chemokines and cytokines) or the extracellular matrix (16). Emerging literature shows that the TME plays a key role in promoting myeloma survival, drug resistance and disease dissemination (Figure 1) (17). Preventing the infiltration and spread of myeloma cells to sites where they are capable to turn into quiescent states or colonize niches that are less accessible for standard therapies might play a significant role in overcoming EMM. In addition, therapies that can target or modulate the dynamic interactions of tumor cells with the TME, using different approaches such as immunotherapy or specific tyrosine kinase inhibitors, could overcome resistance and prevent the spread/metastasis of different solid and non-solid cancer entities including MM (Table 1) (18–20). In this review, we address the role of the TME on myeloma progression and dissemination and discuss potential therapies that could specifically target the TME to prevent manifestation of extramedullary disease (EMD).
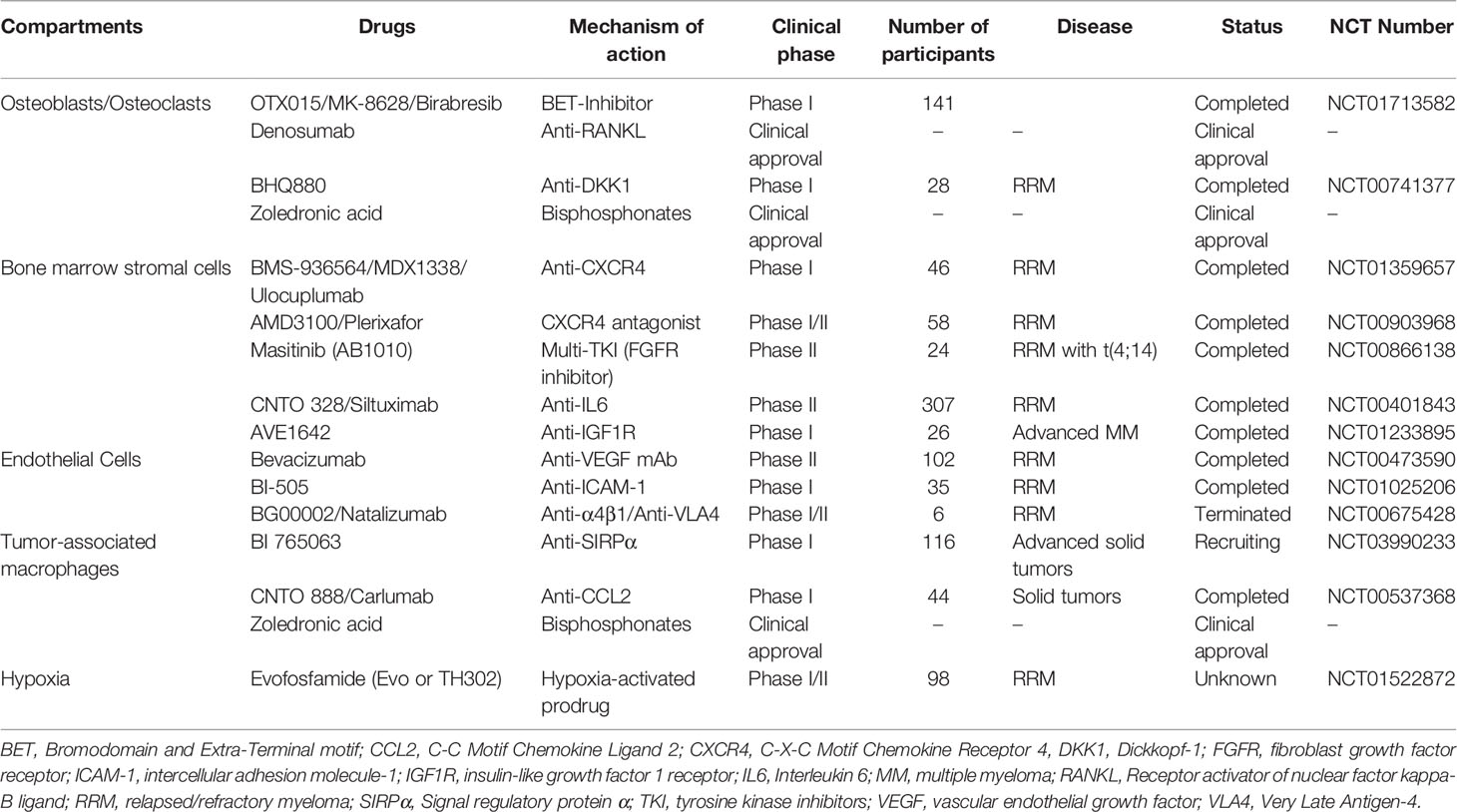
Table 1 Overview of therapy approaches targeting TME compartments in MM and related clinical trials.
Cellular Compartments of the TME
Bone Marrow Stromal and Vascular Endothelial Cells
The C-X-C motif chemokine ligand 12-C-X-C motif chemokine receptor 4 (CXCL12-CXCR4) interaction is one of the earliest steps regulating plasma cell homing to the BM. Bone marrow stromal cells (BMSCs) and vascular endothelial cells produce the chemoattractant stromal cell derived factor 1 alpha (SDF-1α/CXCL12) that binds to CXCR4 resulting in homing and/or retention of hematopoietic stem and progenitor cells to the BM (21, 22). The CXCL12/CXCR4 axis has been indicated to regulate cancer cell invasion and metastasis in different solid cancer entities such as breast cancer (23, 24). In MM, high CXCR4 expression levels are associated with a worse prognosis and enhanced risks for EMD development (25). To unravel the effect of CXCR4 on myeloma cell dissemination, Roccaro et al. intravenously injected the human myeloma cell line MM.1S into immunodeficient NOD/SCID gamma (NSG) mice to generate clones that primarily engrafted in the BM (BM-prone clones) and clones that survived outside the BM (EMD-prone clones). Comparative analysis between both populations showed higher CXCR4 expression levels, enhanced epithelial-to mesenchymal transition and higher properties for extramedullary dissemination in EMD-prone clones (26). Treatment with the anti-CXCR4 monoclonal antibody ulocuplumab (MDX1338) resulted in reduced homing and engraftment of injected MM.1S cells (22, 26). In contrast, treatment with the PI bortezomib and the immunomodulatory agent thalidomide have been reported to induce the downregulation of both molecules, CXCR4 and CXCL12, which could provoke the dissemination of plasma cell clones (27–29).
Additionally, other surface molecules are associated with myeloma dissemination. Vascular cell adhesion molecule 1 (VCAM-1) is expressed on both BMSCs and endothelial cells. The VCAM-1 ligand, very-late antigen 4 (VLA-4), is frequently overexpressed on malignant plasma cells in the BM and plays an important role in plasma cell homing and cell adhesion (30, 31). In this regard, Hathi et al. showed that CRISPR/Cas9 induced depletion of VLA-4 in murine 5TGM1 myeloma cells resulted in increased extramedullary spread to soft tissue sites, liver and spleen and to reduced intramedullary tumor burden compared to mice injected with 5TGM1-wildtype cells where no manifestation of EMD occurred (32). In line with these findings, heparanase-induced shedding of CD138 (Syndecan-1), expressed on normal and malignant plasma cells, promotes myeloma cell migration and invasion by coupling VLA-4 to the vascular endothelial cell growth factor receptor-2 (VEGFR2) (33). Loss of CD56 or downregulation of CD44, two molecules involved in the adhesion of myeloma cells to BMSCs and endothelial cells, are frequently observed in advanced and disseminated disease states (34–38). Platelets, produced by megakaryocytes in the BM, have been found to protect disseminated cancer cells from immune attack and shear stress in the blood stream (39, 40). Platelet-cancer cell interactions further facilitate the extravasation and engraftment of disseminated cancer cells at distant body sites (39, 41, 42). Co-incubation of the myeloma cell line OPM-2 with platelets promoted proliferation and resulted in increased rates of BM engraftment compared to untreated controls (42). Roundabout Guidance Receptor 1 (ROBO1) is another molecule involved in the adhesion of myeloma cells to BMSCs. ROBO1 knock-out in myeloma cell lines resulted in impaired engraftment and extramedullary dissemination in immunodeficient mice (43).
Besides direct cell-cell interactions, BMSCs and endothelial cells produce and secrete a variety of soluble factors such as interleukin 6 (IL-6), insulin-like growth factor-1 (IGF-1), vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) that are all reportedly linked to myeloma progression and cancer cell dissemination. Those soluble factors are promising targets for antibody-based therapy approaches tested in phase I and II clinical trials (Table 1) (44, 45). Therapy-and/or cytokine mediated disruption of ligand-receptor interactions that are involved in plasma cell homing or adhesion might increase the number of cancer cells invading the blood circulation (intravasation). Especially at late disease stages, where single cell clones might harbor the capacity to survive outside the BM microenvironment, the retention of cancer cells in the blood circulation might result in a leukemic spread or enable cells to engraft at distant body sites to form extramedullary tumor foci.
Tumor Infiltrating Lymphocytes
Recognition and elimination of dysfunctional cells is a pivotal task of the adaptive immune system, especially of CD8+ cytotoxic T lymphocytes (CTLs) (46). Upregulation of programmed cell death-ligand 1 (PD-L1) on cancer cells inhibits immune attack via binding to its receptor, programmed cell death protein 1 (PD1), expressed on activated T cells (47). High PD-L1 expression levels have been observed on malignant plasma cells making it a promising target for immune checkpoint inhibitors (ICIs) (18, 48, 49). In addition, enhanced numbers of PD1 positive and T cell immunoglobulin and mucin domain-containing protein 3 (TIM3) positive T cells were detected in the BM of MM patients suggesting reduced T cell activation (T cell exhaustion) and an immunosuppressive environment (50). However, up to now, ICI monotherapies have failed to show clinical benefits in myeloma patients (51). The combination of ICIs with the immunomodulatory agent pomalidomide even resulted in a higher risk for adverse or toxic side effects in phase III clinical trials (52).
Dendritic cells (DCs) in the TME might additionally protect myeloma cells from CTL mediated cell killing. Studies by Leone et al. demonstrated that myeloid DCs can downregulate the expression levels of proteasome subunits in myeloma cells thus preventing CTL induced elimination (53). Recent studies have shown promising concepts overcoming the limitations of ICI based therapies in MM. Exemplary, the combination of ICIs with anti-CD38 monoclonal antibodies has been tested in pre-clinical studies showing encouraging results (54). Though, further research is needed to elucidate the mechanisms of ICI failure in MM and to provide novel therapies that increase the efficacy of immune checkpoint blockade in myeloma patients.
Tumor-Associated Macrophages
Tumor-associated macrophages (TAMs) are known to promote disease progression in numerous solid malignancies including glioblastoma, melanoma, colorectal cancer, lung cancer or ovarian cancer (55–59). Emerging evidence highlights their contribution in driving dissemination of myeloma cells within the skeletal system and to extra-medullary sites. A variety of pre-clinical models has been used to investigate how TAMs affect myeloma cell survival and dissemination. Diphtheria-toxin mediated abrogation of tissue-resident CD169+ macrophages in CD169-DTR mice, intratibially injected with myeloma cells from a syngeneic Vk*MYC mouse model, resulted in reduced dissemination of myeloma cells into the blood circulation and to the contralateral tibial bone. Mechanistically, the pro-inflammatory cytokines IL-6 and tumor necrosis factor alpha (TNFα) secreted by CD169+TAMs, increase vascular leakage and downregulate CD138-mediated cell adhesion; thus, driving myeloma intravasation (60). Concomitantly, M2 polarized macrophages are known to promote angiogenesis (61). Reduced blood vessel density and VEGF expression levels were observed in myeloma xenografts injected with M2-polarized macrophages and treated with the macrophage-depleting agent clodronate. In contrast, xenografts in untreated mice showed enhanced tumor growth and increased VEGF blood concentrations (62). In line with these findings, transcriptomic profiling of the immune cell compartment in patients suffering from refractory or relapsed myeloma, revealed enrichment of a distinct subset of macrophages expressing genes linked to angiogenesis such as VEGF-A or diphtheria toxin receptor (Heparin-Binding EGF-Like Growth Factor) (63). BMI1, a member of a polycomb group multiprotein complex, is overexpressed in macrophages of 5T-myeloma mice compared to macrophages derived from healthy mice. Macrophages, isolated from myeloma bearing 5T BMI1-knock out mice showed reduced production of the pro-angiogenic factors VEGF and nitric oxide (NO). Besides their impact on angiogenesis, TAMs exert direct and indirect effects on myeloma cells preventing detection and elimination by the immune system or enhancing myeloma cell survival and drug resistance.
In vitro co-culture experiments using myeloma cells and macrophages revealed that the presence of M2 macrophages can prevent myeloma cells from undergoing apoptosis induced by bortezomib or melphalan treatment; hence, increasing resistance and promoting progression (64, 65). In addition, Wang et al. reported that high numbers of CD163+ TAMs within the tumor infiltrating immune cell population of myeloma foci are associated with a dismal outcome in myeloma patients receiving bortezomib-based treatment regimens (66). Reduced CXCR4, as a homing receptor for normal and malignant plasma cells in the BM, was detected in myeloma patients undergoing bortezomib-treatment. CXCR4 downregulation is reportedly linked to impaired adhesion and high levels of the macrophage migration inhibitory factor (MIF) (27, 67). Upregulation of CD47 expression can be observed in around 70% of MM patients. Binding of CD47 to signal regulatory protein α (SIRPα) on macrophages acts as a “don’t eat me signal” and protects myeloma cells from phagocytosis and apoptosis (68). Antibodies targeting CD47 or SIRPα are currently tested in clinical trials (Table 1).
In summary, TAMs might drive the extra-medullary expansion of myeloma cells from the BM into the blood circulation via stimulation of blood vessel formation and a cytokine-mediated increase of the vessel-wall permeability. TAMs additionally exert direct and indirect effects on neighboring myeloma cells driving the downregulation of important adhesion molecules such as CD138 and CXCR4. Pro-inflammatory cytokines such as IL-6 and TNFα that are released by TAMs, finally enhance cell migration and tissue invasion favoring cancer cell dissemination (60). Lastly, drug-treatment based selection mechanisms might generate myeloma cell clones that can survive without the essential factors provided by the BM microenvironment and gain the ability to engraft at different body sites and organs.
Osteoblasts and Osteoclasts
In contrast to TAMs or BMSCs, the impacts of osteoblasts and osteoclasts on myeloma cell dissemination are less understood. In MM patients, over-activation of osteoclasts and inhibition of osteoblastic differentiation often result in a dis-equilibrium that promotes resorption of healthy bone material and impairs osteoblastogenesis (69–71). As a consequence, myeloma patients frequently experience bone pain and spontaneous fractures that significantly affect morbidity and reduce quality of life (72, 73). Osteoclasts have been found to influence myeloma progression via direct crosstalk or release of distinct cytokines including IL-6, IL-3, macrophage inflammatory protein 1 alpha (MIP1α) or expression of receptor activator of NF-κB ligand (RANKL) (74). Expression of RANKL and secretion of IL-6, MIP1α or IL-3 by myeloma cells activates osteoclasts and thereby increases resorption of bone matrix (74–78). Enhanced resorption of bone material results in the release of different soluble factors such as IL-6, BAFF or APRIL that stimulate myeloma growth in a feed-forward mechanism also known as the “vicious cycle” (78, 79). IL-6 promotes myeloma cell dissemination via downregulation of CD138 expression and increases the vessel wall permeability; therefore, facilitating intravasation of cancer cells (60). In addition, osteoclasts can activate dormant myeloma cells in the BM, thereby, initiating micrometastases and provoking disease relapse (80). While osteoclasts have a pro-tumorigenic role, osteoblasts counteract osteoclast activity by keeping myeloma cells in quiescent cell states and via induction of apoptosis (80). For this reason, myeloma cells inhibit osteoblasts through the release of distinct secretory factors such as dickkopf-related protein 1 (DKK1), secreted frizzled-related protein 2 (sFRP2) and transforming growth factor beta (TGFβ) to overcome the tumor-suppressive effects of osteoblasts (76, 81, 82). In Phase I clinical trials, anti-DKK1 antibodies (BHQ880) were tested for their potential to overcome the myeloma-mediated inhibition of osteoblasts (83). In addition, preclinical in vivo studies using genetically modified C57BL6/KaLwRij mice with RUNX family transcription factor 2 (RUNX2) deficient osteoblasts indicate that RUNX2 deficiency in MM osteoblasts, triggers myeloma cells and enhances myeloma dissemination and growth (84).
Non-Cellular Compartments of the TME
Direct cell-cell interactions and soluble factors that are released by the TME are important stimuli that influence engraftment and dissemination of myeloma cells. Aside, additional TME related mediators have been found that can promote myeloma dissemination. Lack of nutrients with changes in the cell metabolism and/or increased hypoxic tension, influence the surface expression of distinct cell surface molecules on plasma cells (PCs) thereby favoring a pro-migratory phenotype with enhanced dissemination properties. In this regard, CD138 negative malignant PCs that had been isolated from tumor bearing Vk*MYC mice showed higher rates of migration, cancer cell dissemination and intravasation when injected into syngeneic recipients. In contrast, a high CD138 expression was associated with enhanced rates of tumor growth. Starvation experiments showed that CD138 is dynamically controlled by the availability of serum-components provided by the BM environment and that loss of CD138 upon starvation could be partially restored after addition of serum (85). In this regard, a high tumor burden with low nutrient levels might induce CD138 downregulation thus promoting single cell intravasation. In the blood stream with enhanced nutrient availability, PC might regain CD138 expression and exit the circulation to start another round of engraftment and growth. Besides, CD138 and CXCR4 expression levels are also dependent on the oxygen levels present in the BM. Especially at advanced and high-risk MM stages high rates of cancer cell proliferation might increase the consumption of nutrients and oxygen within the BM. In this regard, hypoxia has been shown to reduce CD138 and CXCR4 expression driving myeloma cells into an immature/stem-cell like state with enhanced properties for dissemination (86, 87).
Extracellular vesicles (EVs) are cellular particles released by all different types of cells and have been proposed to play an important role in the pathogenesis of EMD (88, 89). Different mechanisms have been described of how EVs can promote MM progression and myeloma cell dissemination. EVs can modulate angiogenesis and increase vessel wall permeability allowing myeloma cells to enter the blood circulation (90, 91). In addition, EVs can protect disseminated cells in the blood circulation from immune attack by providing an immunosuppressive “cover” (92). The presence of EVs also generates a cytokine-enriched niche for myeloma cell survival at extramedullary localizations (40).
The extracellular matrix (ECM) of the BM acts as a scaffold enabling cancer cells to adhere and to interact with its components or with other cells. Treatment with bortezomib and melphalan induces secretion of exosomes containing heparanase that results in degradation of ECM; thus, facilitating myeloma invasion (93). The TME provides a complex network of molecules that can affect the phenotype of PCs. Depending on the stimulus, PCs can acquire a pro-migratory profile that favors cell dissemination or stay in an adherent state allowing engraftment and tumor growth.
Conclusion
Even though the overall survival and response rate of MM patients have significantly improved due to the introduction of novel therapeutic agents, MM is still an incurable disease. Different studies have shown that the pathophysiology of MM is dynamically supported by strong and dynamic interactions with the surrounding microenvironment. Direct and indirect signaling pathways between malignant plasma cells and the TME can regulate plasma cell adhesion, cellular motility and the generation of new blood vessels. Additional factors such as hypoxia or nutrient-deprivation can further support the invasiveness of single plasma cell clones that finally enter the blood circulation. Cellular and non-cellular contents of the blood can improve the survival of disseminated plasma cells and facilitate the engraftment at distant body sites. Aside, acquisition of secondary mutations selects clones that are no longer dependent on the extrinsic support of the BM. A better understanding of the complex interactions between the TME and MM cells might potentially lead to new therapeutic approaches and result in an improved progression-free survival with reduced risks for disease relapse or extramedullary dissemination. For this reason, the TME is a promising therapeutic target, especially for aggressive, recurrent and/or advanced disease stages. In order to specify the relevance of those therapeutic approaches, future studies must be carried out in a larger and multicenter setting.
Author Contributions
SF and RR wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Open access funding provided by University Of Bern.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ramsenthaler C, Kane P, Gao W, Siegert RJ, Edmonds PM, Schey SA, et al. Prevalence of Symptoms in Patients With Multiple Myeloma: A Systematic Review and Meta-Analysis. Eur J Haematol (2016) 97(5):416–29. doi: 10.1111/ejh.12790
2. Röllig C, Knop S, Bornhäuser M. Multiple Myeloma. Lancet (2015) 385(9983):2197–208. doi: 10.1016/S0140-6736(14)60493-1
3. Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos M-V, et al. Multiple Myeloma. Nat Rev Dis Prim (2017) 3:17046. doi: 10.1038/nrdp.2017.46
4. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos M-V, et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol (2014) 15(12):e538–48. doi: 10.1016/S1470-2045(14)70442-5
5. Landgren O, Rajkumar SV. New Developments in Diagnosis, Prognosis, and Assessment of Response in Multiple Myeloma. Clin Cancer Res: Off J Am Assoc Cancer Res (2016) 22:5428–33. doi: 10.1158/1078-0432.CCR-16-0866
6. Rajkumar SV, Kumar S. Multiple Myeloma Current Treatment Algorithms. Blood Cancer J (2020) 10(9):94. doi: 10.1038/s41408-020-00359-2
7. Kumar SK, Therneau TM, Gertz MA, Lacy MQ, Dispenzieri A, Rajkumar SV, et al. Clinical Course of Patients With Relapsed Multiple Myeloma. Mayo Clin Proc (2004) 79(7):867–74. doi: 10.4065/79.7.867
8. Bladé J, Fernández de Larrea C, Rosiñol L, Cibeira MT, Jiménez R, Powles R. Soft-Tissue Plasmacytomas in Multiple Myeloma: Incidence, Mechanisms of Extramedullary Spread, and Treatment Approach. J Clin Oncol Off J Am Soc Clin Oncol (2011) 29(28):3805–12. doi: 10.1200/JCO.2011.34.9290
9. Witzig TE, Kyle RA, Greipp PR. Circulating Peripheral Blood Plasma Cells in Multiple Myeloma. Curr Top Microbiol Immunol (1992) 182:195–9. doi: 10.1007/978-3-642-77633-5_23
10. Nowakowski GS, Witzig TE, Dingli D, Tracz MJ, Gertz MA, Lacy MQ, et al. Circulating Plasma Cells Detected by Flow Cytometry as a Predictor of Survival in 302 Patients With Newly Diagnosed Multiple Myeloma. Blood (2005) 106(7):2276–9. doi: 10.1182/blood-2005-05-1858
11. Minnema MC, van de Donk NWCJ, Zweegman S, Hegenbart U, Schonland S, Raymakers R, et al. Extramedullary Relapses After Allogeneic non-Myeloablative Stem Cell Transplantation in Multiple Myeloma Patients do Not Negatively Affect Treatment Outcome. Bone Marrow Transplant (2008) 41(9):779–84. doi: 10.1038/sj.bmt.1705982
12. Mitsiades CS, McMillin DW, Klippel S, Hideshima T, Chauhan D, Richardson PG, et al. The Role of the Bone Marrow Microenvironment in the Pathophysiology of Myeloma and its Significance in the Development of More Effective Therapies. Hematol Oncol Clin North Am (2007) 21(6):1007–34, vii–viii. doi: 10.1016/j.hoc.2007.08.007
13. Fassas ABT, Ward S, Muwalla F, Van Hemert R, Schluterman K, Harik S, et al. Myeloma of the Central Nervous System: Strong Association With Unfavorable Chromosomal Abnormalities and Other High-Risk Disease Features. Leuk Lymphoma (2004) 45(2):291–300. doi: 10.1080/10428190310001597964
14. Wu P, Davies FE, Boyd K, Thomas K, Dines S, Saso RM, et al. The Impact of Extramedullary Disease at Presentation on the Outcome of Myeloma. Leuk Lymphoma (2009) 50(2):230–5. doi: 10.1080/10428190802657751
15. Varettoni M, Corso A, Pica G, Mangiacavalli S, Pascutto C, Lazzarino M. Incidence, Presenting Features and Outcome of Extramedullary Disease in Multiple Myeloma: A Longitudinal Study on 1003 Consecutive Patients. Ann Oncol Off J Eur Soc Med Oncol (2010) 21(2):325–30. doi: 10.1093/annonc/mdp329
16. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
17. García-Ortiz A, Rodríguez-García Y, Encinas J, Maroto-Martín E, Castellano E, Teixidó J, et al. The Role of Tumor Microenvironment in Multiple Myeloma Development and Progression. Cancers (Basel) (2021) 13(2):217. doi: 10.3390/cancers13020217
18. Forster S, Radpour R. Molecular Immunotherapy: Promising Approach to Treat Metastatic Colorectal Cancer by Targeting Resistant Cancer Cells or Cancer Stem Cells. Front Oncol (2020) 10:569017. doi: 10.3389/fonc.2020.569017
19. Radpour R. Tracing and Targeting Cancer Stem Cells: New Venture for Personalized Molecular Cancer Therapy. World J Stem Cells (2017) 9(10):169–78. doi: 10.4252/wjsc.v9.i10.169
20. Radpour R, Forouharkhou F. Single-Cell Analysis of Tumors: Creating New Value for Molecular Biomarker Discovery of Cancer Stem Cells and Tumor-Infiltrating Immune Cells. World J Stem Cells (2018) 10(11):160–71. doi: 10.4252/wjsc.v10.i11.160
21. Kim CH, Broxmeyer HE. In Vitro Behavior of Hematopoietic Progenitor Cells Under the Influence of Chemoattractants: Stromal Cell-Derived Factor-1, Steel Factor, and the Bone Marrow Environment. Blood (1998) 91(1):100–10. doi: 10.1182/blood.V91.1.100
22. Ito S, Sato T, Maeta T. Role and Therapeutic Targeting of SDF-1α/CXCR4 Axis in Multiple Myeloma. Cancers (Basel) (2021) 13(8):1793. doi: 10.3390/cancers13081793
23. Xu S, Menu E, De Becker A, Van Camp B, Vanderkerken K, Van Riet I. Bone Marrow-Derived Mesenchymal Stromal Cells are Attracted by Multiple Myeloma Cell-Produced Chemokine CCL25 and Favor Myeloma Cell Growth In Vitro and In Vivo. Stem Cells (2012) 30(2):266–79. doi: 10.1002/stem.787
24. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis Through Elevated SDF-1/CXCL12 Secretion. Cell (2005) 121(3):335–48. doi: 10.1016/j.cell.2005.02.034
25. Robak P, Jarych D, Mikulski D, Dróżdż I, Węgłowska E, Kotkowska A, et al. The Prognostic Value of Whole-Blood PSMB5, CXCR4, POMP, and RPL5 mRNA Expression in Patients With Multiple Myeloma Treated With Bortezomib. Cancers (Basel) (2021) 13(5):951. doi: 10.3390/cancers13050951
26. Roccaro AM, Mishima Y, Sacco A, Moschetta M, Tai Y-T, Shi J, et al. CXCR4 Regulates Extra-Medullary Myeloma Through Epithelial-Mesenchymal-Transition-Like Transcriptional Activation. Cell Rep (2015) 12(4):622–35. doi: 10.1016/j.celrep.2015.06.059
27. Stessman HAF, Mansoor A, Zhan F, Janz S, Linden MA, Baughn LB, et al. Reduced CXCR4 Expression is Associated With Extramedullary Disease in a Mouse Model Of Myeloma and Predicts Poor Survival in Multiple Myeloma Patients Treated With Bortezomib. Leukemia (2013) 27:2075–7. doi: 10.1038/leu.2013.148
28. Balleari E, Bruzzone A. Unusual Myeloma Relapse After Thalidomide Therapy: The Dark Side of the Moon? Leukemia Res (2009) 33:1164–5. doi: 10.1016/j.leukres.2009.04.020
29. Katodritou E, Gastari V, Verrou E, Hadjiaggelidou C, Varthaliti M, Georgiadou S, et al. Extramedullary (EMP) Relapse in Unusual Locations in Multiple Myeloma: Is There An Association With Precedent Thalidomide Administration and a Correlation of Special Biological Features With Treatment and Outcome? Leuk Res 2009 33(8):1137–40. doi: 10.1016/j.leukres.2009.01.036
30. Sanz-Rodríguez F, Hidalgo A, Teixidó J. Chemokine Stromal Cell-Derived Factor-1alpha Modulates VLA-4 Integrin-Mediated Multiple Myeloma Cell Adhesion to CS-1/Fibronectin and VCAM-1. Blood (2001) 97(2):346–51. doi: 10.1182/blood.V97.2.346
31. Martínez-Moreno M, Leiva M, Aguilera-Montilla N, Sevilla-Movilla S, Isern de Val S, Arellano-Sánchez N, et al. In Vivo Adhesion of Malignant B Cells to Bone Marrow Microvasculature is Regulated by α4β1 Cytoplasmic-Binding Proteins. Leukemia (2016) 30(4):861–72. doi: 10.1038/leu.2015.332
32. Hathi D, Chanswangphuwana C, Cho N, Fontana F, Maji D, Ritchey J, et al. Ablation of VLA4 in Multiple Myeloma Cells Redirects Tumor Spread and Prolongs Survival. Sci Rep (2022) 12(1):30. doi: 10.1038/s41598-021-03748-0
33. Jung O, Trapp-Stamborski V, Purushothaman A, Jin H, Wang H, Sanderson RD, et al. Heparanase-Induced Shedding of Syndecan-1/CD138 in Myeloma and Endothelial Cells Activates VEGFR2 and an Invasive Phenotype: Prevention by Novel Synstatins. Oncogenesis (2016) 5(2):e202. doi: 10.1038/oncsis.2016.5
34. Vande Broek I, Vanderkerken K, Van Camp B, Van Riet I. Extravasation and Homing Mechanisms in Multiple Myeloma. Clin Exp Metastasis (2008) 25(4):325–34. doi: 10.1007/s10585-007-9108-4
35. Klimienė I, Radzevičius M, Matuzevičienė R, Sinkevič-Belliot K, Kučinskienė ZA, Pečeliūnas V. Adhesion Molecule Immunophenotype of Bone Marrow Multiple Myeloma Plasma Cells Impacts the Presence of Malignant Circulating Plasma Cells in Peripheral Blood. Int J Lab Hematol (2021) 43(3):403–8. doi: 10.1111/ijlh.13387
36. Koumpis E, Tassi I, Malea T, Papathanasiou K, Papakonstantinou I, Serpanou A, et al. CD56 Expression in Multiple Myeloma: Correlation With Poor Prognostic Markers But No Effect on Outcome. Pathol Res Pract (2021) 225:153567. doi: 10.1016/j.prp.2021.153567
37. Sahara N, Takeshita A, Shigeno K, Fujisawa S, Takeshita K, Naito K, et al. Clinicopathological and Prognostic Characteristics of CD56-Negative Multiple Myeloma. Br J Haematol (2002) 117(4):882–5. doi: 10.1046/j.1365-2141.2002.03513.x
38. Van Driel M, Günthert U, van Kessel AC, Joling P, Stauder R, Lokhorst HM, et al. CD44 Variant Isoforms are Involved in Plasma Cell Adhesion to Bone Marrow Stromal Cells. Leukemia (2002) 16(1):135–43. doi: 10.1038/sj.leu.2402336
39. Fujita N, Takagi S. The Impact of Aggrus/podoplanin on Platelet Aggregation and Tumour Metastasis. J Biochem (2012) 152(5):407–13. doi: 10.1093/jb/mvs108
40. Labelle M, Begum S, Hynes RO. Platelets Guide the Formation of Early Metastatic Niches. Proc Natl Acad Sci U S A (2014) 111(30):E3053–61. doi: 10.1073/pnas.1411082111
41. Labelle M, Begum S, Hynes RO. Direct Signaling Between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell (2011) 20(5):576–90. doi: 10.1016/j.ccr.2011.09.009
42. Takagi S, Tsukamoto S, Park J, Johnson KE, Kawano Y, Moschetta M, et al. Platelets Enhance Multiple Myeloma Progression via IL-1β Upregulation. Clin Cancer Res (2018) 24(10):2430–9. doi: 10.1158/1078-0432.CCR-17-2003
43. Bianchi G, Czarnecki PG, Ho M, Roccaro AM, Sacco A, Kawano Y, et al. ROBO1 Promotes Homing, Dissemination, and Survival of Multiple Myeloma Within the Bone Marrow Microenvironment. Blood Cancer Discovery (2021) 2(4):338–53. doi: 10.1158/2643-3230.BCD-20-0164
44. Hideshima T, Anderson KC. Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers (Basel) (2021) 13(2):216. doi: 10.3390/cancers13020216
45. Yasui H, Hideshima T, Richardson PG, Anderson KC. Recent Advances in the Treatment of Multiple Myeloma. Curr Pharm Biotechnol (2006) 7(5):381–93. doi: 10.2174/138920106778521569
46. Golstein P, Griffiths GM. An Early History of T Cell-Mediated Cytotoxicity. Nat Rev Immunol (2018) 18(8):527–35. doi: 10.1038/s41577-018-0009-3
47. Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 Pathway From Discovery to Clinical Implementation. Front Immunol (2016) 7:550. doi: 10.3389/fimmu.2016.00550
48. Lee B-H, Park Y, Kim JH, Kang K-W, Lee SJ, Kim SJ, et al. PD-L1 Expression in Bone Marrow Plasma Cells as a Biomarker to Predict Multiple Myeloma Prognosis: Developing a Nomogram-Based Prognostic Model. Sci Rep (2020) 10(1):12641. doi: 10.1038/s41598-020-69616-5
49. Radpour R, Stucki M, Riether C, Ochsenbein AF. Epigenetic Silencing of Immune-Checkpoint Receptors in Bone Marrow- Infiltrating T Cells in Acute Myeloid Leukemia. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.663406
50. Tan J, Chen S, Huang J, Chen Y, Yang L, Wang C, et al. Increased Exhausted CD8(+) T Cells With Programmed Death-1, T-Cell Immunoglobulin and Mucin-Domain-Containing-3 Phenotype in Patients With Multiple Myeloma. Asia Pac J Clin Oncol (2018) 14(5):e266–74. doi: 10.1111/ajco.13033
51. Ribrag V, Avigan DE, Green DJ, Wise-Draper T, Posada JG, Vij R, et al. Phase 1b Trial of Pembrolizumab Monotherapy for Relapsed/Refractory Multiple Myeloma: KEYNOTE-013. Br J Haematol (2019) 186(3):e41–4. doi: 10.1111/bjh.15888
52. Mateos M-V, Blacklock H, Schjesvold F, Oriol A, Simpson D, George A, et al. Pembrolizumab Plus Pomalidomide and Dexamethasone for Patients With Relapsed or Refractory Multiple Myeloma (KEYNOTE-183): A Randomised, Open-Label, Phase 3 Trial. Lancet Haematol (2019) 6(9):e459–69. doi: 10.1016/S2352-3026(19)30110-3
53. Leone P, Berardi S, Frassanito MA, Ria R, De Re V, Cicco S, et al. Dendritic Cells Accumulate in the Bone Marrow of Myeloma Patients Where They Protect Tumor Plasma Cells From CD8+ T-Cell Killing. Blood (2015) 126(12):1443–51. doi: 10.1182/blood-2015-01-623975
54. Verkleij CPM, Jhatakia A, Broekmans MEC, Frerichs KA, Zweegman S, Mutis T, et al. Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination With a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. Cancers (Basel) (2020) 12(12):3713. doi: 10.3390/cancers12123713
55. He C, Sheng L, Pan D, Jiang S, Ding L, Ma X, et al. Single-Cell Transcriptomic Analysis Revealed a Critical Role of SPP1/CD44-Mediated Crosstalk Between Macrophages and Cancer Cells in Glioma. Front Cell Dev Biol (2021) 9:779319. doi: 10.3389/fcell.2021.779319
56. Valencia JC, Erwin-Cohen RA, Clavijo PE, Allen C, Sanford ME, Day C-P, et al. Myeloid-Derived Suppressive Cell Expansion Promotes Melanoma Growth and Autoimmunity by Inhibiting CD40/IL27 Regulation in Macrophages. Cancer Res (2021) 81(23):5977–90. doi: 10.1158/0008-5472.CAN-21-1148
57. Panneerselvam J, Madka V, Rai R, Morris KT, Houchen CW, Chandrakesan P, et al. Inflammatory Mediators and Gut Microbial Toxins Drive Colon Tumorigenesis by IL-23 Dependent Mechanism. Cancers (Basel) (2021) 13(20):5159. doi: 10.3390/cancers13205159
58. Seong JB, Kim B, Kim S, Kim MH, Park Y-H, Lee Y, et al. Macrophage Peroxiredoxin 5 Deficiency Promotes Lung Cancer Progression via ROS-Dependent M2-Like Polarization. Free Radic Biol Med (2021) 176:322–34. doi: 10.1016/j.freeradbiomed.2021.10.010
59. Song M, Yeku OO, Rafiq S, Purdon T, Dong X, Zhu L, et al. Tumor Derived UBR5 Promotes Ovarian Cancer Growth and Metastasis Through Inducing Immunosuppressive Macrophages. Nat Commun (2020) 11(1):6298. doi: 10.1038/s41467-020-20140-0
60. Akhmetzyanova I, Aaron T, Galbo P, Tikhonova A, Dolgalev I, Tanaka M, et al. Tissue-Resident Macrophages Promote Early Dissemination of Multiple Myeloma via IL-6 and Tnfα. Blood Adv (2021) 5(18):3592–608. doi: 10.1182/bloodadvances.2021005327
61. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MPJ, Donners MMPC. Anti-Inflammatory M2, But Not Pro-Inflammatory M1 Macrophages Promote Angiogenesis In Vivo. Angiogenesis (2014) 1):109–18. doi: 10.1007/s10456-013-9381-6
62. Sun M, Xiao Q, Wang X, Yang C, Chen C, Tian X, et al. Tumor-Associated Macrophages Modulate Angiogenesis and Tumor Growth in a Xenograft Mouse Model of Multiple Myeloma. Leuk Res (2021) 110:106709. doi: 10.1016/j.leukres.2021.106709
63. Tirier SM, Mallm J-P, Steiger S, Poos AM, Awwad MHS, Giesen N, et al. Subclone-Specific Microenvironmental Impact and Drug Response in Refractory Multiple Myeloma Revealed by Single-Cell Transcriptomics. Nat Commun (2021) 12(1):6960. doi: 10.1038/s41467-021-26951-z
64. Zhang D, Huang J, Wang F, Ding H, Cui Y, Yang Y, et al. BMI1 Regulates Multiple Myeloma-Associated Macrophage’s Pro-Myeloma Functions. Cell Death Dis (2021) 12(5):495. doi: 10.1038/s41420-021-00432-0
65. De Beule N, De Veirman K, Maes K, De Bruyne E, Menu E, Breckpot K, et al. Tumour-Associated Macrophage-Mediated Survival of Myeloma Cells Through STAT3 Activation. J Pathol (2017) 241(4):534–46. doi: 10.1002/path.4860
66. Wang H, Hu W-M, Xia Z-J, Liang Y, Lu Y, Lin S-X, et al. High Numbers of CD163+ Tumor-Associated Macrophages Correlate With Poor Prognosis in Multiple Myeloma Patients Receiving Bortezomib-Based Regimens. J Cancer (2019) 10(14):3239–45. doi: 10.7150/jca.30102
67. Ullah TR. The Role of CXCR4 in Multiple Myeloma: Cells’ Journey From Bone Marrow to Beyond. J Bone Oncol (2019) 17:100253. doi: 10.1016/j.jbo.2019.100253
68. Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 Antibodies Promote Phagocytosis and Inhibit the Growth of Human Myeloma Cells. Leukemia (2012) 26(12):2538–45. doi: 10.1038/leu.2012.141
69. Giuliani N, Colla S, Morandi F, Lazzaretti M, Sala R, Bonomini S, et al. Myeloma Cells Block RUNX2/CBFA1 Activity in Human Bone Marrow Osteoblast Progenitors and Inhibit Osteoblast Formation and Differentiation. Blood (2005) 106(7):2472–83. doi: 10.1182/blood-2004-12-4986
70. Roodman GD. Osteoblast Function in Myeloma. Bone (2011) 48(1):135–40. doi: 10.1016/j.bone.2010.06.016
71. Mundy GR, Raisz LG, Cooper RA, Schechter GP, Salmon SE. Evidence for the Secretion of an Osteoclast Stimulating Factor in Myeloma. N Engl J Med (1974) 291(20):1041–6. doi: 10.1056/NEJM197411142912001
72. Andersen TL, Søe K, Sondergaard TE, Plesner T, Delaisse J-M. Myeloma Cell-Induced Disruption of Bone Remodelling Compartments Leads to Osteolytic Lesions and Generation of Osteoclast-Myeloma Hybrid Cells. Br J Haematol (2010) 148(4):551–61. doi: 10.1111/j.1365-2141.2009.07980.x
73. Chen Z, Orlowski RZ, Wang M, Kwak L, McCarty N. Osteoblastic Niche Supports the Growth of Quiescent Multiple Myeloma Cells. Blood (2014) 123(14):2204–8. doi: 10.1182/blood-2013-07-517136
74. Heider U, Hofbauer LC, Zavrski I, Kaiser M, Jakob C, Sezer O. Novel Aspects of Osteoclast Activation and Osteoblast Inhibition in Myeloma Bone Disease. Biochem Biophys Res Commun (2005) 338(2):687–93. doi: 10.1016/j.bbrc.2005.09.146
75. Sezer O, Heider U, Jakob C, Zavrski I, Eucker J, Possinger K, et al. Immunocytochemistry Reveals RANKL Expression of Myeloma Cells. Blood (2002) 99:4646–7. doi: 10.1182/blood-2002-01-0148
76. McDonald MM, Fairfield H, Falank C, Reagan MR. Adipose, Bone, and Myeloma: Contributions From the Microenvironment. Calcif Tissue Int (2017) 100(5):433–48. doi: 10.1007/s00223-016-0162-2
77. Oyajobi BO, Franchin G, Williams PJ, Pulkrabek D, Gupta A, Munoz S, et al. Dual Effects of Macrophage Inflammatory Protein-1alpha on Osteolysis and Tumor Burden in the Murine 5TGM1 Model of Myeloma Bone Disease. Blood (2003) 102(1):311–9. doi: 10.1182/blood-2002-12-3905
78. Croucher PI, McDonald MM, Martin TJ. Bone Metastasis: The Importance of the Neighbourhood. Nat Rev Cancer (2016) 16(6):373–86. doi: 10.1038/nrc.2016.44
79. Abe M, Kido S, Hiasa M, Nakano A, Oda A, Amou H, et al. BAFF and APRIL as Osteoclast-Derived Survival Factors for Myeloma Cells: A Rationale for TACI-Fc Treatment in Patients With Multiple Myeloma. Leukemia (2006) 20:1313–5. doi: 10.1038/sj.leu.2404228
80. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts Control Reactivation of Dormant Myeloma Cells by Remodelling the Endosteal Niche. Nat Commun (2015) 6:8983. doi: 10.1038/ncomms9983
81. Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res (2016) 76(5):1089–100. doi: 10.1158/0008-5472.CAN-15-1703
82. Habibi H, Abroun S, Hajifathali A, Soleimani M, Kaviani S, Kalantari N, et al. Osteogenic Inhibition in Multiple Myeloma. Cell J (2013) 15(3):266–71.
83. Iyer SP, Beck JT, Stewart AK, Shah J, Kelly KR, Isaacs R, et al. A Phase IB Multicentre Dose-Determination Study of BHQ880 in Combination With Anti-Myeloma Therapy and Zoledronic Acid in Patients With Relapsed or Refractory Multiple Myeloma and Prior Skeletal-Related Events. Br J Haematol (2014) 167(3):366–75. doi: 10.1111/bjh.13056
84. Xu X, Zhang C, Trotter TN, Gowda PS, Lu Y, Ponnazhagan S, et al. Runx2 Deficiency in Osteoblasts Promotes Myeloma Progression by Altering the Bone Microenvironment at New Bone Sites. Cancer Res (2020) 80(5):1036–48. doi: 10.1158/0008-5472.CAN-19-0284
85. Akhmetzyanova I, McCarron MJ, Parekh S, Chesi M, Bergsagel PL, Fooksman DR. Dynamic CD138 Surface Expression Regulates Switch Between Myeloma Growth and Dissemination. Leukemia (2020) 34(1):245–56. doi: 10.1038/s41375-019-0519-4
86. Kawano Y, Kikukawa Y, Fujiwara S, Wada N, Okuno Y, Mitsuya H, et al. Hypoxia Reduces CD138 Expression and Induces an Immature and Stem Cell-Like Transcriptional Program in Myeloma Cells. Int J Oncol (2013) 43(6):1809–16. doi: 10.3892/ijo.2013.2134
87. Vandyke K, Zeissig MN, Hewett DR, Martin SK, Mrozik KM, Cheong CM, et al. HIF-2α Promotes Dissemination of Plasma Cells in Multiple Myeloma by Regulating CXCL12/CXCR4 and CCR1. Cancer Res (2017) 77(20):5452–63. doi: 10.1158/0008-5472.CAN-17-0115
88. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai Y-T, Reagan M, et al. BM Mesenchymal Stromal Cell-Derived Exosomes Facilitate Multiple Myeloma Progression. J Clin Invest (2013) 123(4):1542–55. doi: 10.1172/JCI66517
89. Colombo M, Giannandrea D, Lesma E, Basile A, Chiaramonte R. Extracellular Vesicles Enhance Multiple Myeloma Metastatic Dissemination. Int J Mol Sci (2019) 20(13):3236. doi: 10.3390/ijms20133236
90. Taraboletti G, D’Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the Matrix Metalloproteinases MMP-2, MMP-9, and MT1-MMP as Membrane Vesicle-Associated Components by Endothelial Cells. Am J Pathol (2002) 160(2):673–80. doi: 10.1016/S0002-9440(10)64887-0
91. Schillaci O, Fontana S, Monteleone F, Taverna S, Di Bella MA, Di Vizio D, et al. Exosomes From Metastatic Cancer Cells Transfer Amoeboid Phenotype to non-Metastatic Cells and Increase Endothelial Permeability: Their Emerging Role in Tumor Heterogeneity. Sci Rep (2017) 7(1):4711. doi: 10.1038/s41598-017-05002-y
92. Remiker AS, Palumbo JS. Mechanisms Coupling Thrombin to Metastasis and Tumorigenesis. Thromb Res (2018) 164 Suppl:S29–33. doi: 10.1016/j.thromres.2017.12.020
93. Bandari SK, Purushothaman A, Ramani VC, Brinkley GJ, Chandrashekar DS, Varambally S, et al. Chemotherapy Induces Secretion of Exosomes Loaded With Heparanase That Degrades Extracellular Matrix and Impacts Tumor and Host Cell Behavior. Matrix Biol (2018) 65:104–18. doi: 10.1016/j.matbio.2017.09.001
Keywords: multiple myeloma, tumor microenvironment, TME, targeted therapy, malignant plasma cells
Citation: Forster S and Radpour R (2022) Molecular Impact of the Tumor Microenvironment on Multiple Myeloma Dissemination and Extramedullary Disease. Front. Oncol. 12:941437. doi: 10.3389/fonc.2022.941437
Received: 11 May 2022; Accepted: 06 June 2022;
Published: 01 July 2022.
Edited by:
Mohamed A. Yassin, Hamad Medical Corporation, QatarReviewed by:
Hadi Goubran Messiha, University of Saskatchewan, CanadaIoannis Ntanasis-Stathopoulos, National and Kapodistrian University of Athens Medical School, Greece
Copyright © 2022 Forster and Radpour. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramin Radpour, cmFtaW4ucmFkcG91ckBkYm1yLnVuaWJlLmNo