 Hayle Scanlan
Hayle Scanlan Zachary Coffman2
Zachary Coffman2 Timothy Shipley
Timothy Shipley Debra E. Bramblett
Debra E. Bramblett
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 July 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.940019
This article is part of the Research TopicWomen in Cancer Molecular Targets and Therapeutics, Volume II: 2022View all 23 articles
The need for efficacious and non-toxic cancer therapies is paramount. Oncolytic viruses (OVs) are showing great promise and are introducing new possibilities in cancer treatment with their ability to selectively infect tumor cells and trigger antitumor immune responses. Herpes Simplex Virus 1 (HSV-1) is a commonly selected OV candidate due to its large genome, relative safety profile, and ability to infect a variety of cell types. Talimogene laherparevec (T-VEC) is an HSV-1-derived OV variant and the first and only OV therapy currently approved for clinical use by the United States Food and Drug Administration (FDA). This review provides a concise description of HSV-1 as an OV candidate and the genomic organization of T-VEC. Furthermore, this review focuses on the advantages and limitations in the use of T-VEC compared to other HSV-1 OV variants currently in clinical trials. In addition, approaches for future directions of HSV-1 OVs as cancer therapy is discussed.
Nearly 40% of people in the United States will be diagnosed with cancer during their lifetime (1). In 2018, the CDC attributed 21.1% of total deaths to malignant neoplasms which can become unresponsive to treatment (refractory) (2). Current anticancer drugs are toxic and often not entirely effective. Therefore, there is an urgent need for novel therapies that are efficacious and non-toxic. New treatments involving the use of oncolytic virus (OV) therapies, many of which are currently undergoing clinical trials, are showing great promise.
Herpesviridae is a large family of enveloped, double-stranded DNA viruses that can undergo both lytic and latent lifecycles, depending on cell type infected by the virus. Cell-type specificity of the Herpesviridae is defined by surface glycoproteins on individual virions that interact with cell-surface receptors. Upon binding, fusion of the viral envelope with the cell membrane leads to the release of the viral capsid into the cytoplasm. The viral DNA is then transported to the cell nucleus within the now-naked nucleocapsid. The nucleocapsid then attaches to the host cell’s nuclear membrane enabling insertion of the viral genome into the nucleus through a nuclear pore. After circularization, the virus genome can be transcribed, leading to productive virus replication in permissive cells or latency in non-permissive cell types. The virus’ life cycle is complete upon budding of new virions which go on to infect neighboring cells (Figure 1).
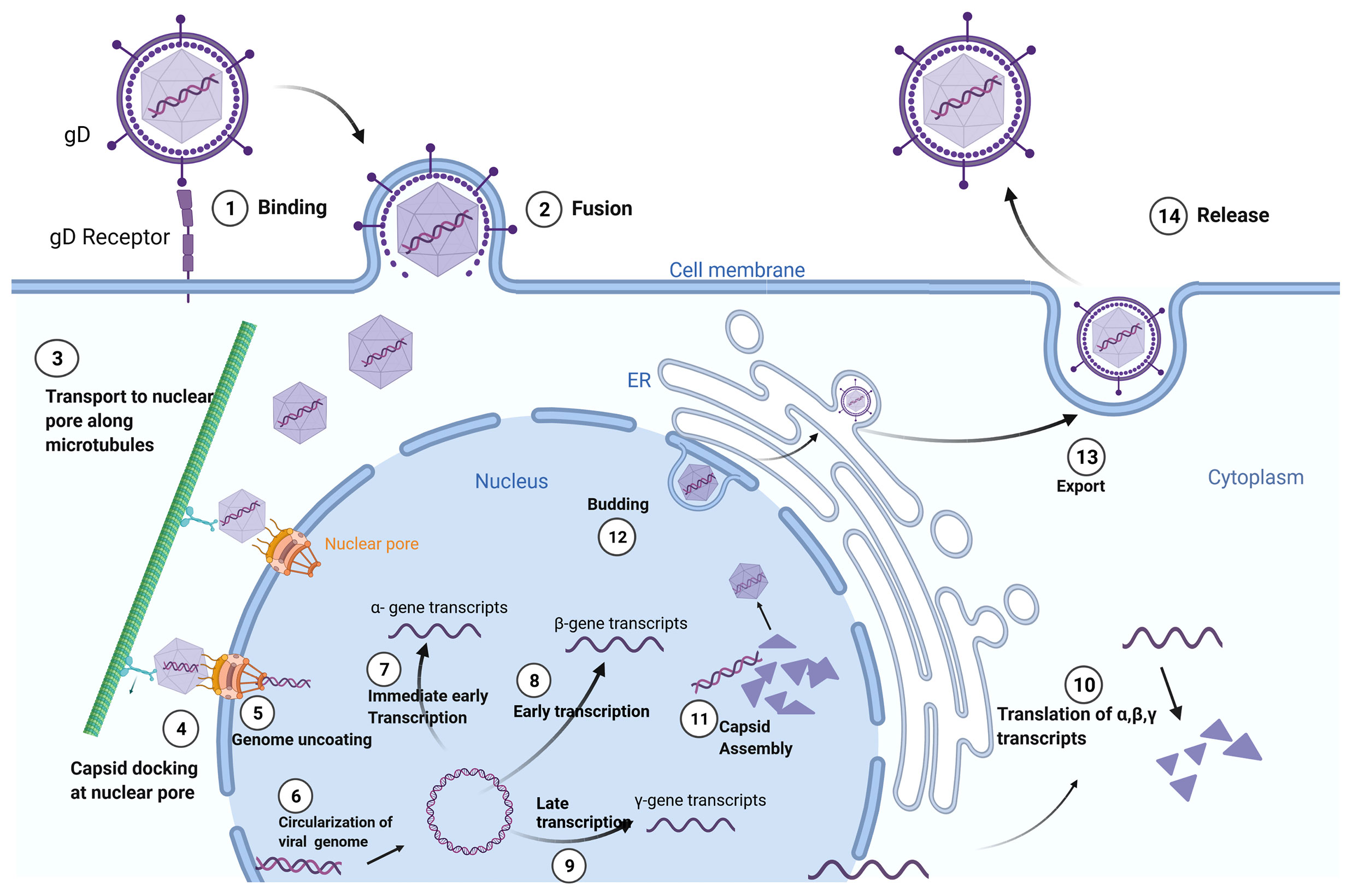
Figure 1 Herpes Simplex Virus-1 (HSV-1) Life Cycle: Adsorption and fusion of HSV-1 to its target cell is initiated by viral glycoprotein D (gD) to cell specific gD receptors (1). Fusion of the viral envelope with cell membrane allows for capsid entry into the cytoplasm and release of tegument proteins (2). The naked viral capsid is transported (3) to nuclear pore complexes in the nuclear envelope (4), through which the viral genome is extruded into the nucleus (5). The linear viral genome is circularized (6). Herpes viruses have three rounds of transcription: immediate early (α-genes) (7), early (β-genes) (8), and late (γ-genes) (9). Translation of the structural proteins from γ- transcripts occurs only after the initiation of viral genome replication, which is dependent on β-proteins. Viral transcripts leave the nucleus to be translated (10) in either the cytoplasm or in the context of the endoplasmic reticulum (ER). Capsids are assembled in the nucleus, encasing the viral genome in an icosahedral protein coat (11). The newly generated viral capsid acquires an envelope by budding into the inner nuclear membrane (12). The completed virus translocates through the ER and matures in the Golgi apparatus prior to exiting the cell by exocytosis (13). Created with BioRender.com.
Production of herpesvirus messenger RNA by an infected cell leads to expression of a variety of proteins that perform a wide array of functions, ranging from virion assembly to suppression of host cell antiviral responses. Gene expression occurs in three phases of transcription, and these genes have been designated by the kinetic classes, α, β, and γ, which correspond to immediate early, early, and late phases of gene expression, respectively. Proteins expressed during the α phase impact cellular and viral gene expression, including β gene expression. During the β phase of transcription, proteins involved in DNA replication are expressed, including viral DNA polymerase. Most of the γ genes encode structural proteins. Of significance to this review, gene names correspond to the region of the genome in which it resides, with the HSV-1 genome being organized into a unique long region (UL) and a unique short region (US) (Table 1 and Figure 2). The designation “infected cell protein” (ICP) is given for viral proteins that are not structural.

Table 1 HSV-1 genes modified in OV candidates.
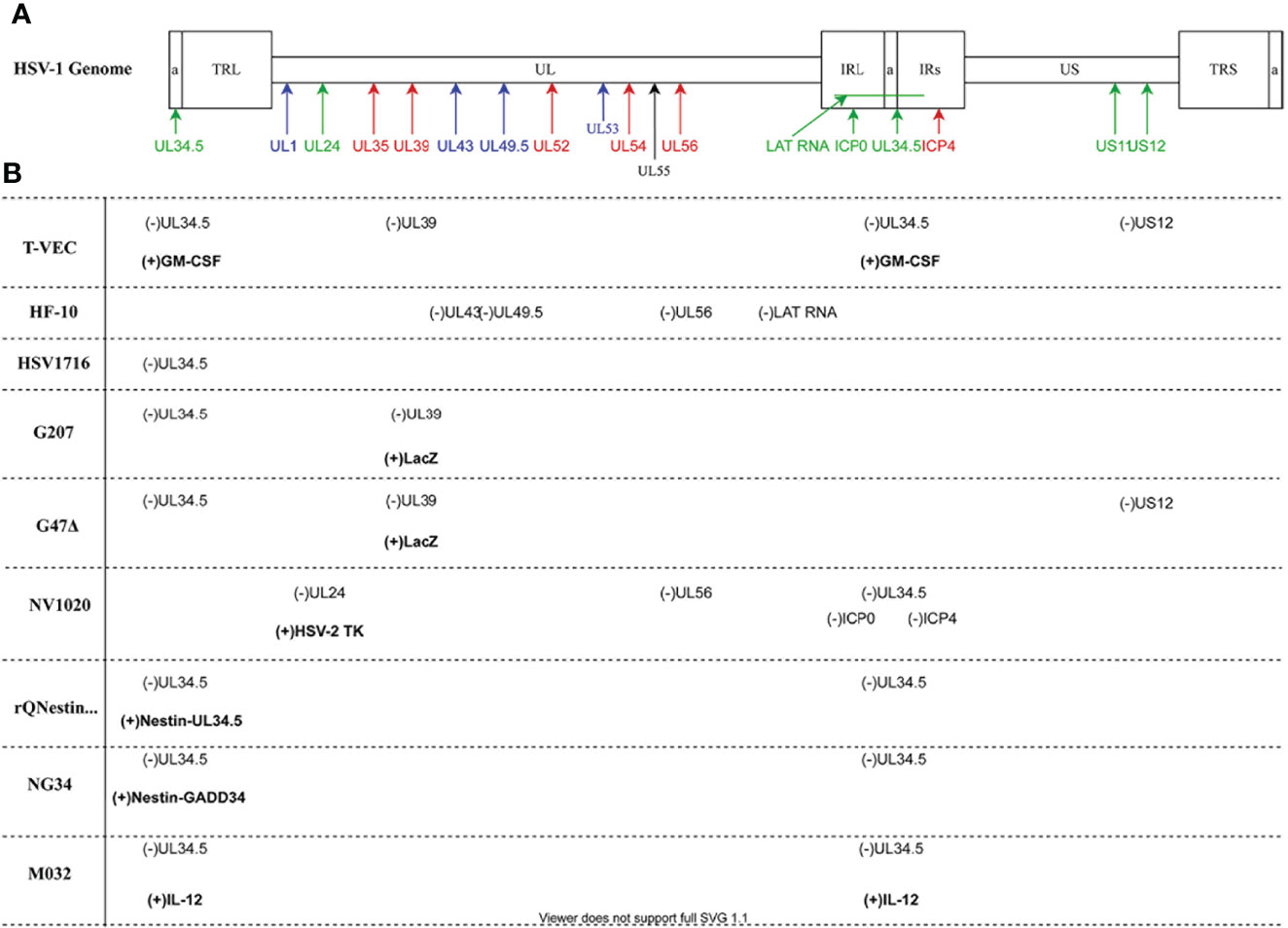
Figure 2 HSV-1 Genomic map and oncolytic virus modifications (A) The HSV-1 genome has two covalently joined segments long (L) and short (S) each of which has a unique sequence (UL and US) flanked by a pair of repeat sequences, the terminal and internal long repeats (TLL, IRL, TRS and IRS). There is also a 400 base pair terminal repeat at each end of the genome and internally at the joint between the L and S segments which is called (a) Genes are coded according to functional group: blue- structural; red- replication; green- virulence. (B) Oncolytic viruses mentioned in the paper. Deleted viral genes are indicated as (-) while transgenes or viral genes that have been inserted are in bold and indicated by (+). Diagram prepared with DRAWIO.
Researchers have taken advantage of the cell-killing attributes of viruses to target tumor cells specifically. One of the main advantages of OV therapy over conventional chemotherapy is that viruses are capable of self-propagation allowing them to replicate and infect more tumor cells, ideally until all cells of a malignant mass have been killed. Many virus types such as Adenovirus, Poxvirus, Coxsackievirus and Herpesvirus are being used in OV therapy development (3–12). Tumor destruction by OV therapy is accomplished by direct viral lysis of malignant cells or by indirect mechanisms involving elicitation of enhanced anti-tumor responses as the host immune system attacks the virus-infected cells, or by a combination of these two mechanisms. Advances in recombinant DNA technology and better understanding of viral pathogenesis have allowed manipulation of viral genomes, leading to improved OV specificity for tumor cell targets and greater ability to direct host responses.
The potential impact of OV therapy on patients stands in stark contrast to conventional tumor treatments. Treatment by radiation and chemotherapy typically exhibit minimal cell specificity, resulting in destruction of normal tissues in addition to the tumor targets, thus causing multiple adverse effects in patients. OV therapies offer significant advantages due to decreased toxicity stemming from their ability to specifically target malignant cells. Furthermore, OV treatments are able to circumvent resistance to conventional therapies frequently acquired by tumor cells (13).
Currently, in all stages of testing, from pre-clinical testing to phase III clinical trials, there are several Herpes Simplex Virus (HSV) variants that are being investigated for use as OV therapies (14). HSV is the most commonly investigated virus for OV development because 1) its genome is relatively easy to manipulate, 2) its surface glycoproteins can be altered to target specific cellular receptors, and 3) its replication can be controlled with herpesvirus-specific drugs such as acyclovir. One oncolytic herpesvirus therapy, Talimogene laherparevec (T-VEC; also known as OncoVEX^GM-CSF and IMLYGIC™), has received approval from the United States Food and Drug Administration (FDA) for treatment of inoperable melanoma, and investigations for its use in a wide variety of malignant conditions are underway (15).
Development of HSV-1 OVs has led to progressively more sophisticated constructs that are more refined in their oncolytic abilities. The primary focus in early studies with OVs was safety. As one of the earliest OVs brought to the clinic, G207 had few modifications to wild-type HSV-1. Importantly, its safety has been demonstrated in multiple clinical trials for treatment of many different cancer types. Later, the OV therapeutic agent, NV1020, which was first studied as a potential vaccine against HSV-2, was repurposed as an OV that could more efficiently replicate in tumor cells due to further modifications to the HSV-1 genome while maintaining minimal toxicity. In subsequent years, HSV1716, perhaps the least sophisticated of the HSV-1 OVs with respect to genomic alterations, has been used in several clinical trials with the primary outcome being demonstration of a high safety profile that ensuing OVs have striven to duplicate. With insertion of a transgene in addition to genomic modifications already in use by earlier OVs, T-VEC represents the next level of sophistication for OV-based cancer treatments. Both its efficacy and safety were such that T-VEC became the first OV treatment approved for clinical use against advanced melanoma. Currently, a plethora of trials examining the use of T-VEC in combination with other drugs and against other cancers are underway. HF10, with the greatest number of genomic alterations of the HSV-1 OVs discussed herein, was developed simultaneously with HSV1716 and T-VEC. Although genomic alterations in HF10 occurred naturally, they serendipitously imbued the OV with considerable safety and efficacy against many cancer types. The HSV-1 OV, M032, is a more recent OV variant that mirrors the development of T-VEC with the incorporation of a transgene to boost efficacy. It also shares other genomic alterations with T-VEC to minimize toxicity. Its first clinical trials are just underway. Most recently, the HSV-OV G47Δ, a derivative of G207, has been determined to be safe and effective in clinical trials in Japan. Finally, a new pinnacle of genomic engineering has been demonstrated with the OVs, rQNestin and NG34, possibly representing the future evolution of OV therapies that are specifically targeted to tumor cell type. All of these OVs will be discussed in detail in the following sections.
The first step of cell infection by HSV-1 is adsorption and fusion between the virion and the plasma membrane of target cells, which are mucoepithelial cells and sensory neurons. Viral fusion with these cells is specifically mediated by viral glycoprotein D (gD), initially by low-affinity adherence to cell specific heparin sulfate proteoglycans (HSPGs). This is followed by high affinity binding to either nectin-1 or herpesvirus entry mediator (HVEM), the former of which is found on both epithelial and neural cells, and the latter of which is found only on epithelial cells (16).
During productive infections with HSV-1, copies of the viral genome are encased by protein nucleocapsids that leave the cell by budding. In lytic infections, cytolysis is caused by the combination of inhibition of host cell macromolecules, disruption of the host cell cytoskeleton, and induction of nuclear DNA fragmentation. The virus also induces increased membrane permeability, ultimately leading to cell death.
During acute, active infections, intracellular virus replication is followed by budding from the host cell surface, allowing the virus to infect neighboring cells. Infection of cells in oral or genital mucosae by HSV-1 often results in characteristic painful vesicular lesions, a relatively benign condition. In addition, acute infection with HSV-1 typically leads to seeding of ganglia innervating the area of the primary infection and, eventually, latency within the regional ganglia. During latency, the genome remains quiescent in the host cell, so production of new virions does not take place. Latency is established and maintained by the latency-associated transcript (LAT) (17). MicroRNAs, expressed from LAT, also block the expression of host cell genes whose expression would otherwise induce antiviral responses (18, 19). Reactivation from latency may be induced in response to a variety of triggering events, such as hormonal fluctuation, trauma, UV light, and immunosuppression, thus leading to damage of healthy tissues upon reactivation. Although mucoepithelial infections are normally benign, HSV-1 can cause herpes simplex encephalitis, an acute illness characterized by general and focal signs of cerebral dysfunction. This can lead to permanent brain damage and even death, thus presenting a significant concern for use of HSV-1 as a therapeutic agent.
In addition to the risk of neuroinvasion, there are other shortcomings associated with use of HSV-1 for OV therapy. Although intravenous delivery of cancer therapeutics is the desired delivery mode so that all metastases can be reached, treatment with HSV-1 OV is currently restricted to application by intratumoral injection. Intravenous delivery of an HSV-1 OV, however, may result in reduced amounts of virus reaching tumor sites due to virus sequestration by the large numbers of nontumor epithelial cells and/or sensory neurons displaying the herpes virus receptors, nectin-1 and herpesvirus entry mediator (HVEM). Hence, intravenous delivery of an HSV-1 OV would lead to too much of the virus non-specifically targeting healthy tissues instead of tumor tissues. Moreover, the World Health Organization (WHO) estimates that two-thirds of the world’s population under the age of 50 have been infected with HSV-1 (20). Therefore, systemic delivery of HSV-1 might also be limited by the prevalence of HSV-1-specific antibodies in the general population, which would rapidly neutralize HSV-1 OV administered intravenously.
Despite its pathogenicity and other limitations, HSV-1 has many properties that make it an attractive candidate for use in OV therapy. First, it is a DNA virus containing a large genome that does not integrate into the host genome. The virus’ genome contains several redundant elements and genes not required for infection, which along with its size, conveys the ability to engineer the viral genome for incorporation of large or multiple therapeutic gene cassettes using standard molecular cloning techniques to increase tumor specificity and improve safety. Another aspect contributing to its viability as an OV therapy is that the pathology of wild-type HSV-1 is typically mild compared to other viruses under development. Moreover, HSV-1 replication can be inhibited by members of the antiviral acyclovir drug class based on the virus’ dependence on thymidine kinase (TK), thus providing an extra layer of protection.
Other advantages of HSV-1 include its use of an envelope, which facilitates retargeting of the virus via genetic engineering. This is in contrast to non-enveloped icosahedral viruses such as the Adenoviridae, which have stringent structural constraints with respect to particle assembly. Furthermore, early and late gene expression by HSV-1 allows for sequential gene expression, providing the possibility to enhance OV efficacy. Additionally, HSV-1 utilizes multiple genes to manipulate signaling pathways to circumvent common host defense mechanisms. For example, HSV-1 inhibits innate anti-viral responses such as the interferon (IFN) response, which would otherwise result in a dramatic reduction of progeny virions produced during infection, thus limiting the effectiveness of using it as an OV. The IFN response, mediated by type I IFN and produced in response to virus infection of human cells, induces a network of host cell IFN-stimulated genes (ISGs), such as RNA-dependent protein kinase R (PKR), 2’-5’ oligoadenylate synthetase (OAS) and others that mediate cessation of replication of the virus in neighboring cells (21). Alternatively, if mounting a robust immune response against infected cells is desired in lieu of allowing HSV-1’s lytic lifecycle to run its course, manipulation of genes controlling these pathways can be readily performed in order to develop OV agents less able to evade host defenses. A summary of genes related to immune evasion abilities exhibited by wild-type HSV-1 and how they have been modulated in various strains of HSV-1 OVs is found in Table 2. Additionally, transgenes inserted to enhance immune responses against HSV-1 OV-infected cells are included.
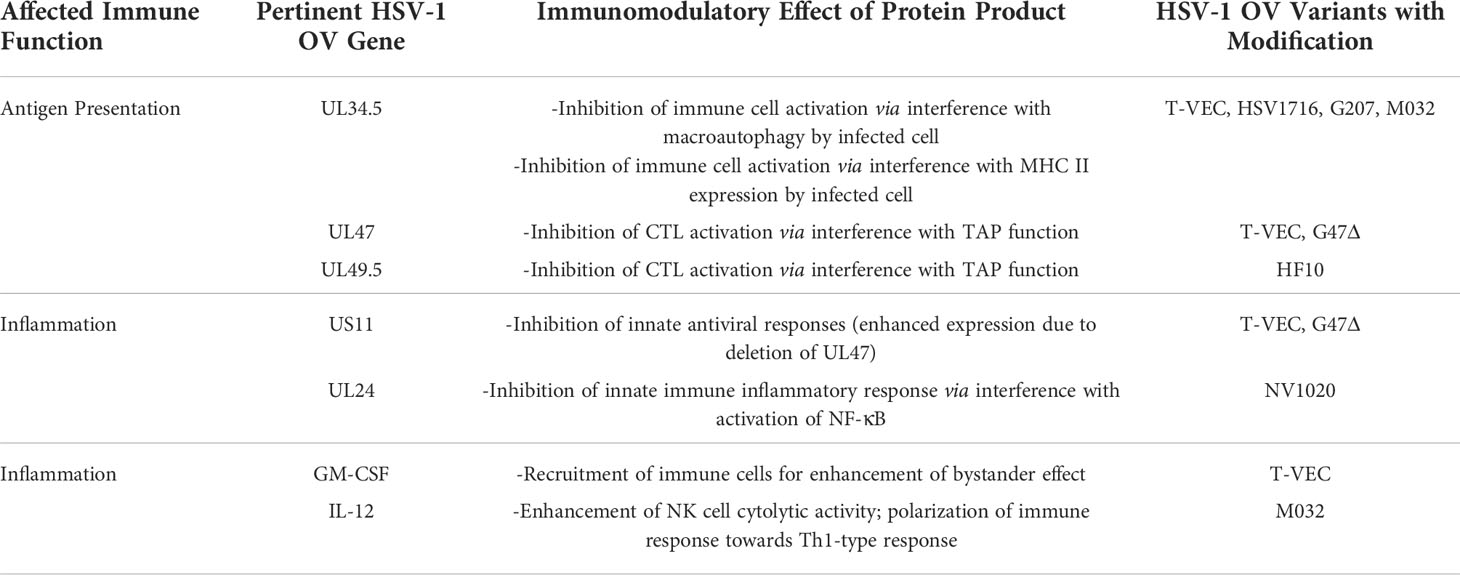
Table 2 Genes responsible for immunomodulatory functions found in OV variants.
In the following sections, we review the clinically available T-VEC and compare and contrast it to other HSV OV therapies currently in clinical testing.
In 2015, the FDA approved T-VEC for treatment of patients with unresectable metastatic melanoma via intratumoral injection (12). T-VEC is a JS1 strain of HSV-1, which was selected for its ability to specifically replicate in and kill human tumor cells (22, 23). In addition to functional deletion of the wild-type HSV-1 genes, UL34.5 and US12 (Figure 2), T-VEC contains a human transgene coding for the cytokine, granulocyte macrophage-colony stimulating factor (GM-CSF) (12, 22). Preclinical studies conducted with T-VEC have demonstrated improved tumor shrinkage and clearance, and clinical studies have shown an enhanced durable response rate for advanced melanoma treatment when compared to use of GM-CSF alone (12, 22).
HSV-1 ICP34.5 is a multifactorial virulence factor encoded by UL34.5, which enhances virulence of the wild-type virus. In an infection by wild-type HSV-1, ICP34.5 is able to complex with protein phosphatase 1 alpha (PP1α) to dephosphorylate the alpha subunit of eukaryotic initiation factor 2 (eIF2), thus blocking the action of double-stranded RNA-activated protein kinase R (PKR) (24), which inhibits protein production. Deletion of UL34.5, therefore, suppresses T-VEC replication in non-tumor cells while simultaneously maintaining replication capacity in tumor cells due to the absence of PKR activity in most tumor cells (25). Thus, the deletion of UL34.5 conveys tumor-specificity. In addition, ICP34.5 is required for the neurovirulence associated with HSV-1 (26, 27). Neurons and other cells deploy autophagy in defense against invading microorganisms (28). In this process, the host cell protein, beclin 1, stimulates autophagy in response to the PKR signaling pathway triggered by HSV-1 infection (29, 30). However, ICP34.5 binds to beclin 1, inhibiting this protective autophagy response in neurons (31). Therefore, reduced neurovirulence of T-VEC is brought about by the loss of ICP34.5 (31). ICP34.5 has also been reported to interfere with activation of adaptive immune responses in numerous ways, including inhibition of dendritic cell (DC) maturation and antigen presentation (32, 33). Specifically, ICP34.5 has been observed to block cell surface expression of MHC II antigen presentation proteins (34). In sum, ICP34.5 contributes to both neurovirulence and inhibition of immune-mediated killing of infected cells.
The efficacy of OVs can be enhanced by arming the virus so that neighboring uninfected tumor cells are killed (bystander effect) (35). This has been successfully accomplished in several virotherapies, including T-VEC, by expression of an immunostimulatory transgene. As with all OVs, productive T-VEC replication in tumor cells results in lysis of the infected cells, leading to necrotic cell death. Subsequent engulfment of the tumor antigen-containing cellular debris by host antigen presenting cells (APC), such as DCs and macrophages, leads to enhanced anti-tumor responses because the APCs can subsequently induce an adaptive immune response. This is further amplified by T-VEC because it carries the transgene, GM-CSF, which, when expressed, recruits an array of immune cells to the infection site and promotes maturation of DCs, macrophages, and granulocytes.
Another mechanism used by HSV-1 to block responses to viral infection involves ICP47, which inhibits adaptive immune responses by interfering with viral antigen processing by the host cell. Specifically, ICP47 is a high affinity competitor for the peptide-binding site on a host cell’s transporter-associated protein (TAP) (36, 37). TAP normally directs pathogen peptides into the host cell endoplasmic reticulum for loading onto MHC I molecules that will be transported to the cell surface for presentation to and activation of cytolytic T lymphocytes (CTL). Fully functional ICP47, therefore, restricts the immune response against the virus because viral antigen presentation by infected cells is reduced (38). Hence, deletion of the ICP47 gene (US12) in T-VEC enables robust CTL responses to be mounted against virus-infected cells.
A secondary consequence of deleting US12 is the shifting of the US11 coding region closer to the promoter that normally regulates the expression of US12. This shift alters its regulation so that the US11 protein product is expressed as an early rather than a late gene and results in increased transcription/expression of US11 (22, 23, 39). The product of US11 is a component of the viral tegument and has multiple functions, all of which promote HSV-1 infection. US11 expression leads to the inhibition of antiviral response mediators that include retinoic acid-inducible gene 1 (RIG-1), heat shock protein 90, nucleophosmin, OAS, and PKR, thus enhancing viral expansion (40–45). In summary, the enhanced expression of US11, together with the deletion of UL34.5 and US12, allows T-VEC to replicate successfully in tumor cells by suppressing cellular antiviral response, while simultaneously promoting a robust immune response to infection, resulting in tumor-specific killing properties of this OV.
Although T-VEC is the first and only viral therapy currently approved by the FDA for treatment of unresectable melanoma lesions recurrent after initial surgery, there are limitations to its use and efficacy as a cancer treatment (46). These limitations derive from how it is administered and dosed, median time to response, effect on distant and visceral metastases, and the current requirement that it only be used as an adjuvant treatment.
When used as intended by the manufacturer, T-VEC must be injected directly into cutaneous, subcutaneous, and lymph node lesions multiple times for optimal activity (46). Moreover, administration of T-VEC is intended to be used on the largest lesions first, followed by the smaller lesions, until all lesions have been injected or the maximum injection volume of four milliliters is reached. As a result, patients may see evidence of regression of injected lesions, but disease progression may take place in untreated lesions (23). Additionally, realization of treatment benefits seems to be delayed with therapeutic responses occurring months after the prescribed dosing regimen is used. Because of this extended time-to-response period, many patients may see disease progression rather than regression (12). Thus, the combination of a maximum dosing volume and prolonged median time-to-response represent shortcomings of T-VEC therapy for melanoma.
In this light, the application of T-VEC for more generalized cancer therapy is under investigation by several groups (Table 3). As reviewed by Grigg et al, profound systemic regressions did not occur in T-VEC clinical trials involving visceral metastases, therefore, subjects with more advanced disease derived considerably less benefit from T-VEC therapy (23). Even so, 15% of measurable visceral metastases reduced in size by at least 50% in T-VEC-treated patients in a separate phase III clinical trial (12, 47).
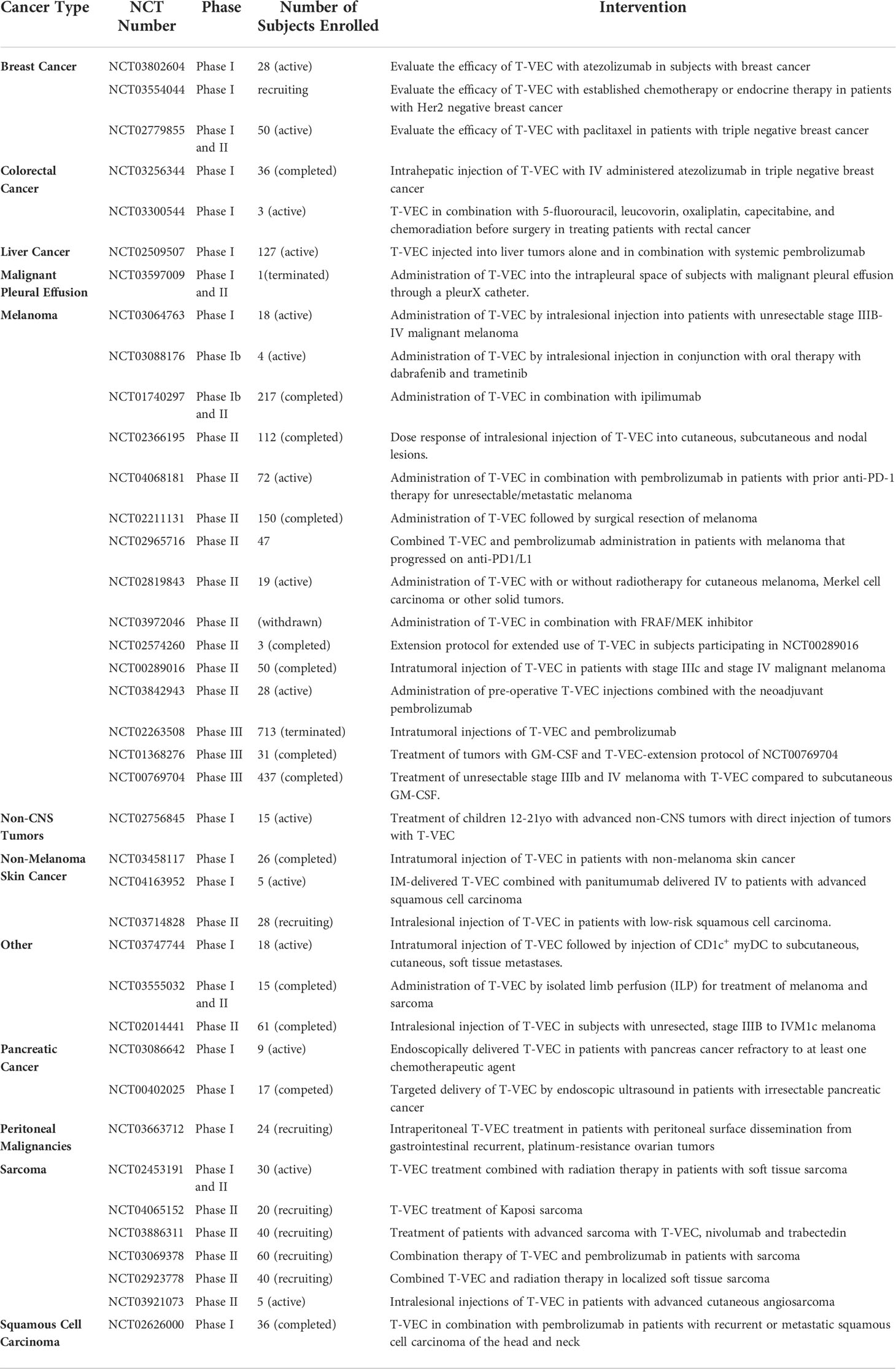
Table 3 Current T-VEC Clinical Trials.
Lastly, findings by Kaufman et al. demonstrated that T-VEC therapy yielded significantly higher durable response rates across all disease stages compared to patients in the control group (47). This was particularly true for treatment-naïve patients who had not received any sort of prior treatment (12). This is significant because given that T-VEC is currently considered a novel immunotherapy, it is unlikely that patients will receive it as a first-line treatment. Therefore, in many patients under current treatment guidelines, diminished therapeutic outcomes using T-VEC are likely.
While there is evidence that T-VEC effectively promotes regression and necrosis of superficial melanoma lesions, it does not represent a complete therapy and has limited efficacy in patients who have visceral metastases. Hence, there is a need for continued research and development of other virus-based immunotherapies that are more broadly applicable. Many HSV-1 OV therapies are currently in clinical trials and may provide solutions to the limitations associated with T-VEC.
HF10 is a naturally occurring mutant of HSV-1 undergoing clinical trials for use as an OV cancer treatment (48). Despite HF10’s ability to replicate efficiently in cells, its pathogenicity is highly attenuated in humans (49, 50). The genome of HF10 has insertions, deletions, and frameshift mutations affecting several genes. HF10 lacks functional expression of UL43, UL49.5 and has only a single copy of both UL56, and LAT for which there are two in the wild-type virus. UL52, UL53, UL54, UL55, and UL56, are removed from their original positions, inverted, and reinserted later in the genome (Figure 2) (48). Because these mutations were not deliberate and methodical, it is difficult to ascertain their cumulative effect.
Individual consideration of each of the four genes that are functionally deleted provides clarification as to why HF10 may be superior to T-VEC for use as an OV therapeutic agent. For example, studies have shown that the bovine herpesvirus-1 (BHV-1) version of UL49.5 encodes a protein that normally binds and degrades TAP in virus-infected cells leading to downregulation of MHC class I antigen presentation (51). Therefore, the loss of functional UL49.5 in HF10 may similarly allow for maintenance of MHC I antigen presentation in infected human cells, resulting in a more robust adaptive immune response, similar to the effects derived from deletion of US12 in T-VEC.
LAT, which encodes a long, noncoding RNA transcript, is functionally absent from the HF10 genome. Although LAT limits both establishment and reactivation from latency specifically in neurons, it appears to be less important for peripheral infections (17, 52). Mutational analysis of HSV-1 shows that viruses lacking LAT expression either fail to efficiently establish latency, or they cannot readily reactivate from latency, further increasing the safety of HF10 (53–55).
The absence of UL56 results in reduction of the neuroinvasiveness and pathogenicity of HF10 compared to wild-type HSV-1 (56, 57). While UL56 is not necessary for viral replication, viral strains lacking UL56 are substantially less neuroinvasive and are unable to penetrate the central nervous system (CNS) (57, 58). UL56 functionally increases the pathogenicity of HSV-1 by promoting the axonal transport of vesicles containing viral envelope glycoproteins through its interaction with KIF1A, a neuron specific kinesin. KIF1A plays an important role in the transport of synaptic vesicle precursors and in the axonal transport of pre-synaptic vesicles (59). The binding of UL56 to KIF1A leads to neuronal cell dysfunction and, therefore, is partially responsible for the neuropathology of HSV-1 infection (56). Together, mutations in LAT and UL56 in HF10 work to reduce viral neurovirulence and the likelihood of reactivation from latency, further enhancing the long-term safety of HF10 (60).
Unlike all other HSV-1 OVs that have entered clinical trials, UL34.5 remains functionally intact in the HF10 genome (48). A potential drawback of the IL34.5 loss-of-function mutants is that they replicate less efficiently, giving rise to lower viral yields as compared to wild-type virus, which could account for the limited efficacy demonstrated by T-VEC in clinical trials (12, 61, 62). It seems likely that functional ICP34.5 leads to high viral replication and the stronger antitumor effects of HF10 seen in clinical trials (63). Additionally, the duplications of UL53, UL54, and UL55, all of which are essential virus life cycle genes, could contribute to the high replication rate of HF10 (Table 1). Therefore, HF10 maintains tumor specificity and high-level replication.
High mitotic rates and the weakened interferon responses of tumor cells potentiate HF10’s candidacy as a cancer therapeutic. This is due to HF10’s superior ability to replicate and spread, as compared to wild-type HSV-1 strains (64, 65). Indeed, it has been shown to provoke a complete cytopathic effect and elicit a potent antitumor effect against a broad range of malignancies (66–68). Specifically, HF10 is able to produce a more vigorous bystander effect as compared to other HSV-1 variants (69). This is in alignment with studies that have shown the importance of intercellular trafficking and gap junctions for the production of this effect (69). Cancer cells typically have a dramatically reduced number of functional gap junctions allowing for more efficient tumor progression but increasing the difficulty for many OVs to produce the bystander phenomenon (70). Cancer cells decrease their gap junctions by altering their connexin expression, including Connexin 43 (Cx43). Cx43 is a protein that links adjacent cells’ cytoplasm. If the expression of Cx43 is suppressed, then gap junction activity is reduced or abrogated (71). However, HF10 appears to upregulate expression of Cx43 (72). This may allow HF10 to produce a more potent bystander effect through the formation of gap junctions.
HF10 has been shown to be safe in many dose-escalation phase I trials involving a variety of cancer types including recurrent and metastatic breast cancer, recurrent head and neck squamous cell carcinoma, unresectable pancreatic cancer, and melanoma (49, 50, 65, 72–74). A phase II trial has also been conducted to assess efficacy and safety of intratumoral injection of HF10 in combination with intravenous infusions of ipilimumab, a drug designed to boost T-cell responses. This trial revealed that HF10 in combination with ipilimumab is safe and well tolerated, with high antitumor efficacy (73). Additional phase II trials have been completed (NCT03153085, NCT02428036, NCT01017185, NCT02272855) or are currently underway (NCT03259425) for use of HF10 against melanoma. In total, the combined data from these clinical trials and preclinical studies illustrate multiple characteristics that make HF10 a superior OV therapy candidate when compared to other OVs that can be summarized in five key points: 1) high tumor selectivity, 2) high viral replication, 3) strong cytopathic effect, 4) potent bystander effect, and 5) vigorous antitumor effect against a variety of malignancies (66–69, 75–77). Therefore, HF10 shows great promise as a virus-based therapy and is likely to be broadly applicable, providing solutions to many limitations associated with T-VEC.
Having a relatively simple platform, HSV1716 was derived from a naturally occurring strain of HSV-1 containing only a spontaneous mutation resulting in the loss of functional neurovirulence-related UL34.5, making it similar to T-VEC (27, 78). In like fashion, the mutation renders HSV1716 incapable of replicating in the CNS and yet capable of replicating in and lysing dividing tumor cells (78).
The safety of this OV strain has been assessed in phase I and IIa trials for high-grade glioma (HGG), stage IV melanoma and mesothelioma (26, 79–81). HGG patients who experience recurrence of disease typically exhibit new lesions within approximately two centimeters of the original site of cancer growth. Harrow et al. injected HSV1716 into sites of healthy brain tissue directly adjacent to the region where HGG had been resected (26). In this way, lytic replication of HSV1716 could contain tumor spread. Although disease progression varied, no toxicity from the HSV1716 was observed in any of the study’s 12 subjects. Importantly, there was a significant increase in long-term survival post-resection in the OV treatment group. The potential of HSV1716 for increasing HGG survival is encouraging, but more trials need to be completed.
In a different application of HSV1716, Mackie et al. injected melanoma nodules with the OV, and performed immunohistochemical staining demonstrating that viral replication was restrained to tumor cells (80). Furthermore, in the three patients that received two or more doses of HSV1716, microscopic tumor necrosis was detectable. None of the patients exhibited any toxic effects due to the treatment, demonstrating an acceptable safety profile for HSV1716 and making it a potentially viable treatment for advanced melanoma.
In yet another phase I trial of HSV1716, Streby et al. investigated the safety of intratumoral injection of the OV in pediatric patients with non-CNS solid tumors (20). It was determined that single-dose intratumoral administration of HSV1716 is safe and well-tolerated in pediatric subjects with refractory non-CNS solid tumors. However, none of the subjects had a clinically measurable outcome. Therefore, the group suggested that this OV treatment should be used in a combination therapy or administered earlier in disease progression to allow it to develop an antitumor immune response (20). In fact, many pre-clinical trials have used HSV1716 in combination therapy showing synergistic effect (82–86). Uniquely, most of the subjects in this trial were HSV-1 seronegative, possibly because all of the subjects were children and had not been previously exposed to the virus. Since adults tend to be HSV-1 seropositive at a greater rate than this trial’s study population, the data could not be extrapolated to the general population.
A more recent study by Streby et al. explored the safety of HSV1716 applied intravenously. In this study they were unable to detect the OV in tumor biopsies, likely because the doses used were too low. Nevertheless, because it has the potential to reach all metastatic sites, the investigators remain optimistic about intravenous HSV1716 application at higher doses because no dose-limiting toxicities were observed (87). The absence of any observable anti-tumoral effect from the intravenous OV inoculation prompted the investigators to speculate that patients might receive greater benefit from combined intravenous and intratumoral administration, as such a dosing regimen could boost local immune responses within tumors.
In addition to these phase I trials, HSV1716 has also been used in a phase I/IIa trial investigating use of the OV for treatment of mesothelioma (MPM) patients (81). In this study, HSV1716 was well tolerated, with 50% of participants exhibiting disease stabilization and four out of twelve patients developing anti-tumor IgG. Intrapleural HSV1716 was well-tolerated and demonstrated an anti-tumor immune response in MPM patients. These results provide a rationale for further studies with this agent in MPM and in combination with other therapies
As one of the first OVs to be developed, HSV1716 has been used in multiple clinical trials, animal studies and in vitro studies that continue to generate invaluable data, which influence the design of other more sophisticated OVs such as T-VEC. Indeed, the safety profile of HSV1716 is excellent even if robust efficacy has not yet been demonstrated, thus providing a benchmark for future OV development either as standalone or part of combination cancer therapy. The repeated demonstration of HSV1716’s safety paves the way for further studies utilizing this OV to be performed, likely enabling the development of next-generation OVs.
G207 is the first HSV-1 strain genetically engineered for treatment of intracerebral cancer to be used in clinical trials in North America (88). In contrast to T-VEC which was derived from wild-type HSV-1 strain JS1, this OV was derived from wild-type HSV-1 strain F (89). While G207 is modified differently than T-VEC, it does share the similarity that both copies of UL34.5 are deleted, resulting in reduced neurotoxicity (89). Deletion of UL34.5 in this HSV-1 variant also results in the alteration of LAT expression, leading to in the inability of the virus to establish latency (90).
G207 also has a lacZ insertion in UL39, leading to the inactivation of ICP6, a large subunit of the viral ribonucleotide reductase (Figure 2). ICP6 catalyzes conversion of ribonucleotides to deoxyribonucleotides, which is important for viral DNA replication. Therefore, it is required for efficient growth of the virus in nondividing cells, which do not produce a functionally equivalent enzyme like that produced in proliferating host cells (91–93). Consequently, functional deletion of ICP6 likely curtails G207 replication in quiescent cells, thereby preventing destruction of tissues adjacent to the tumor (91, 94). This makes the utility of this OV in cancer therapy clear (90). Other functions attributed to ICP6 include inhibition of apoptosis, establishment of latent infections, and reactivation from latency. Thus, loss of ICP6 as in this OV variant, contributes to both its safety and its efficacy in multiple ways (95, 96).
Markert et al. conducted multiple phase I trials to test the safety of G207 in the context of progressive, recurrent malignant glioma. Their original study was a dose escalation study in which the maximal tolerated dose was not achieved (88). In a follow-up study, they determined that intratumoral delivery of the OV before tumor resection followed by delivery into brain tissue surrounding the resection cavity, within one week of the first dose, was also safe (97). In a third trial, G207 OV therapy showed an excellent safety profile with none of the patients developing HSV encephalitis following intratumoral injections combined with radiation treatment (98). Additionally, these studies exhibited potential for clinical response in patients with progressive, recurrent, malignant glioma. Other phase I trials investigating the use of G207 in children are ongoing (NCT03911388, NCT02457845, NCT04482933). All three studies will investigate the use of G207 in children with recurrent or progressive brain tumors with or without radiation to enhance viral replication and an associated anti-tumor immune response.
In short, there is potential for G207 to play a vital role in combination therapy by intratumoral injection in glioblastoma, having demonstrated an acceptable safety profile in multiple trials. Compared to T-VEC and other OV therapies, its genetic composition differs due to the insertion of lacZ within UL39. The advantage of lacZ expression is that it potentially allows detection of viral replication in treated tumor tissues and any spread of the virus.
A third generation OV, G47Δ, was derived from G207 by deleting the gene encoding ICP47 (Figure 2), which prevents downregulation of MHC I, thus enhancing antitumor immune responses similar to T-VEC (Table 2) (99). This deletion also places US11 under the control of the US12 promoter, which may allow for higher replication capacity than OVs lacking UL34.5. Many studies utilizing animal or in vitro models have suggested great potential for G47Δ efficacy in killing tumor cells (100–106). To date, all clinical trials involving G47Δ have been conducted in Japan. In addition to demonstration of safety, this OV exhibited strong antitumor efficacy in patients with glioblastoma when used in a phase I-IIa trial (UMIN000002661) and a phase II trial (UMIN000015995) (107). Furthermore, clinical trials to investigate the safety of G47Δ in patients with recurrent or progressive olfactory neuroblastoma (UMIN000011636) and progressive malignant pleural mesothelioma (UMIN000034063) are underway. Significantly, a clinical trial using G47Δ to treat prostate cancer has also been completed (108).
The modifications to the G47Δ genome have given it a higher replication capacity than T-VEC and higher antitumor activity than its G207 parent virus. With improved therapeutic efficacy arising from increased replication and spread, this OV may represent the next stage in cancer therapy. Indeed, the architects of G47Δ, now called Teserpaturev, are seeking approval for Malignant Glioma therapy from Japan’s Ministry of Health (109).
NV1020, previously R7020, is a first-generation HSV-1 OV variant that is highly attenuated, having originally been developed as a herpes vaccine, albeit unsuccessfully (110). The construction of NV1020, described by Meignier, has a 15-kb deletion at the junction of the UL and US regions of the HSV-1 genome (Figure 2) where one of two copies of each of the genes encoding ICP0, ICP4, and ICP34.5 are located (111). In addition, this deletion removed one copy of UL56. ICP0 is a dispensable gene product, but at least one copy of the gene encoding ICP4, which blocks apoptosis and positively regulates several other HSV-1 genes, must be retained for viral replication. NV1020 also contains a 700-bp deletion that encompasses TK and the UL24 promoter. HSV-1 UL24 has been shown to inhibit the activation of NF-ĸB, which together with interferon-response factor 3 (IRF3), triggers the host antivirus response via the cyclic GMP-AMP synthase viral DNA recognition pathway. Therefore, deletion of UL24 likely results in a greater innate immune response to this OV (Table 2) (112, 113). In place of the deletion at the junction of the UL and US regions, a 5.2-kb fragment of HSV-2 DNA and an exogenous TK have been inserted. Addition of the TK sequence guarantees that any potential NV1020 infection can be contained by TK-converted prodrugs such as acyclovir, adding a level of safety to this OV treatment.
In contrast to G207, NV1020 retains ICP6 and one copy of UL34.5, and for this reason was predicted to replicate more efficiently in tumor cells that have variable capacity to compensate for the loss of the ribonucleotide reductase (114). Further, investigators predicted that the maintenance of one copy of UL34.5 in NV1020 likely allows for greater viral replication than G207. Indeed, it was determined in cell lines and in vivo animal experiments that NV1020, with improved therapeutic efficacy due to increased replication and spread, exerts greater cytotoxicity at lower multiplicities of infection (MOIs). The paradox of NV1020’s increased neurotoxicity potential set against an increased survival advantage at low MOIs of this OV led investigators to project that NV1020 might be advantageous for patients with more advanced cancer and larger tumors (114).
To date, NV1020 has been tested in at least two clinical trials, a phase I trial in colorectal cancer (NCT00012155) and a combined phase I/II trial (NCT00149396), both of which utilized hepatic artery infusion for liver-metastasized colorectal cancer patients who had failed their first line of chemotherapy (115, 116). Although adverse events were observed after administration in the phase I trial, most were mild to moderate and self-limiting, leading the investigators to conclude that NV1020 can be safely administered into the hepatic artery without significant effects on normal liver function (115). The second trial also integrated a dose escalation study, but it was followed by two cycles of chemotherapy for the cohort of subjects determined to have received the optimal dose. Once again, it was demonstrated that the treatment was minimally toxic, and a significant number of the study participants showed at least a partial response and/or stable disease. It was, therefore, concluded that the treatment may sensitize metastases to salvage chemotherapy, thus justifying performance of a phase II/III trial (116).
NV1020 retains one copy of UL34.5, suggesting a greater replication capacity than T-VEC. The fact that NV1020 was found to have greater replication and killing capacity than G207 in in vitro and animal studies justified pursuit of phase I and II trials (114). Although retention of one copy of UL34.5 likely allows for increased cytolytic activity over T-VEC, given the role of ICP34.5 in neurovirulence, the retention of this gene also suggests a potential risk of neurotoxicity when administered systemically or intracerebrally. However, the lack of NV1020-related adverse events in phase I and II clinical trials, including those measured by neurological examination suggests low risk of neurotoxicity and supports phase II/III trials (116).
One of the downsides of the OV therapies utilizing HSV-1 variants that has been pointed out several times in this review is that they often have severely attenuated replication due to the loss of ICP34.5 expression through deletion of both gene copies. Recognizing this issue, Kambara et al. engineered an OV that safely retains UL34.5. In rQNestin34.5, UL34.5 has been reintroduced into the HSV-1 genome under the control of the glioblastoma multiforme (GBM) specific promoter, nestin (Figure 2) (117). Nestin, normally active during embryogenesis, is shut down in the adult brain but is active in glioblastoma cells. Consequently, robust OV expression should occur only in GBM cells.
Researchers are currently recruiting for a phase I clinical trial using rQNestin34.5 in GBM patients (NCT03152318). Despite carrying a single copy of UL34.5, researchers hope that its re-engineered genome will confine expression of the gene to cancer cells and that neurotoxicity will be highly attenuated, thus mirroring the safety profile of UL34.5-lacking T-VEC.
In a resourceful workaround that could be applied more generally for the ICP34.5 neurotoxicity issue, the nestin promoter-controlled UL34.5 was switched out with the human ortholog, GADD34. The new protein product, GADD34, expressed by this OV variant, known as NG34, mimics ICP34.5’s ability to dephosphorylate eIF2α by association with PP1, but it lacks a beclin 1 binding domain, effectively eliminating beclin 1-mediated neurotoxicity. To date, only animal studies have been performed with NG34, but greater tolerability compared to rQNestin34.5 has been demonstrated and could represent a viable evolutionary path of the parental OV if it performs well in clinical trials (118).
M032 is a second-generation oncolytic HSV variant that has been modified not only to take advantage of direct oncolytic activity but, also, to recruit inflammatory cells. This latter ability was imbued by incorporating interleukin-12 (IL-12) into both sites where the gene encoding ICP34.5 was deleted to induce significant expression of this cytokine (Figure 2 and Table 2). Transgene expression of IL-12 by an OV complements work performed with other vectors that yielded promising results in numerous other cancer therapy preclinical and clinical trials (119–124). Preclinical studies using an identical virus, except that it expresses murine IL-12, have shown this OV to be both safe and superior in efficacy to non-cytokine-expressing parental strains in various brain cancer models (125). This sets the stage for a phase I clinical trial (NCT02062827) that is currently recruiting for the treatment of recurrent/progressive glioblastoma.
With the insertion of a cytokine gene to enhance immune responses against OV-infected cells, M032’s design parallels that of T-VEC. The design similarities extend to deletion of the gene encoding ICP34.5 in both OVs, but the similarities end with the deletion of the gene encoding ICP47 in T-VEC. Therefore, it is difficult to project whether M032 will exhibit superior performance to T-VEC with respect to the treatment of malignancies. Nevertheless, expression of IL-12 could make this a more effective OV-based cancer treatment because of its ability to create an antitumor environment through enhancement of natural killer cell cytolytic activity. Additionally, IL-12 mediates production of IFN-γ, polarizing T helper cells towards a cell-mediated response. Moreover, it has been demonstrated that IL-12 is antiangiogenic, thereby inhibiting tumor growth (119, 122, 126).
There is an ongoing need for development of efficacious cancer therapies that will increase survival of patients with resistant malignancies, and OV treatment shows considerable promise. As a component of combination therapy regimens utilizing standard-of-care (SOC) treatments, there is mounting preclinical and clinical evidence that OV treatments can boost overall therapeutic efficacy against a variety of malignancies (73, 82–84, 127, 128). Incorporation of OVs into treatment regimens introduces the potential for significant dose reductions without compromising tumor cell-killing capacity. In turn, toxicity associated with chemo- and radiotherapy can be minimized. In this review of the many OV therapies currently in development, only HSV-1 variants in clinical trials or approved for clinical use were discussed.
OV treatments display potential for extending life expectancy in patients with various refractory cancers, however, completion of subsequent phases of these trials must be performed in order to determine efficacy and establish patient safety. Furthermore, many aspects of OV therapy require further research to optimize mode of adminstration to establish efficacy against different types of malignancies and to hone the specificity and safety properties of each engineered OV variant.
Use of OVs for cancer treatment ideally results in two separate but related therapeutic effects. The first is that infection of tumor cells by an OV leads to direct death of the cancer cells as a part of their lytic lifecycle. The viral progeny released upon lytic destruction can subsequently infect other cancer cells to perpetuate killing of more cells. However, solid tumors are known to extensively manufacture extracellular matrix (ECM) (129). Therefore, ECM-mediated inhibition of virus spread throughout tumors is an area of active exploration within the OV therapy field (130, 131). Several groups have incorporated genes for ECM-remodeling proteins into various OVs that have shown promise in preclinical models (132–136). Improved antitumor efficacy has been observed when OVs expressing chondroitinase, hyaluronidase, relaxin, and decorin were used (132–137). Since studies using OVs containing a single transgene that regulates connective tissue remodeling have shown some success, it is possible that delivery of a combination of these genes by OVs will be required for maximum tissue penetration and tumor regression.
Release of cancer antigens from lysed cells leads to the second therapeutic effect- the induction of an adaptive immune response that kills residual local and distant tumor cells. To this end, establishment of highly robust immune responses against cancer cells has been the focus of many OV engineering efforts. Earliest efforts using HSV-1 as an OV were directed at modifying the virus so that its capacity to evade host immune responses would be attenuated. The deletion of UL34.5 from many HSV-1 OV strains is an example. This deletion reduces neurovirulence and allows enhanced immune responses due to the fact that wild-type virus expression of ICP34.5 inhibits DC maturation and antigen presentation (26, 27, 32–34). Other gene deletions in HSV-1 OVs that result in more complete immune responses against virus-infected cells include removal of US12 and UL49.5, both of which negatively affect the function of TAP (51).
Excellent safety profiles of early HSV-1 OVs have been clinically established, so recent efforts are aimed at increasing efficacy by addition of transgenes that enhance physiological responses directed towards OV-infected cells. For example, HSV-1 has been engineered to include transgenes that allow the virus to synergize with SOC treatments such as chemotherapy and radiation. To address inadequate chemotherapeutic responses Braidwood et al, developed an HSV-1 OV that contains the enzyme, nitroreductase, which converts the prodrug, CB1954, into an active alkylating chemotherapeutic agent. Use of this OV together with CB1954 enhanced tumor cell killing in vitro and improved survival in preclinical cancer models (138).
Another SOC treatment commonly used for treating cancer is radiation therapy. Observations have been made wherein radiation positively affects viral replication and, therefore, has the potential to contribute to OV therapeutics. Conversely, radiation therapy can be enhanced with OV treatment. This approach was taken by Quigg, et. Al. wherein they inserted a transgene coding for the noradrenaline transporter into an HSV-1 OV (139). This modification enabled targeted radiotherapy of HSV-infected cells expressing the membrane symporter because they accumulated the iodine-131-labeled noradrenaline analog, metaiodobenzylguanidine (MIBG). The combination of the OV and radiolabeled MIBG led to decreased tumor growth and increased survival in an animal model relative to either agent given alone (140).
Both approaches result in concentration of conventional therapeutic agents in and around infected cells with the goal of killing tumor cells with high precision while minimizing toxicity to healthy cells. However, replacement of toxic therapeutic agents by Ovs that have been engineered to promote physiological responses robust enough to destroy the tumor is the ultimate goal. One way this might be accomplished is to boost immune responses against tumor cells by modifying Ovs to deliver therapeutic payloads. Currently, insertion of cytokine genes that promote inflammation and subsequent bystander killing of tumor cells into Ovs is a common approach. The incorporation of GM-CSF into T-VEC, and IL-12 into M032 are notable examples. Numerous other cytokine transgenes have been incorporated into Ovs as well as inhibitory receptors and bispecific T cell engagers with varying degrees of success (141–145). Given the large HSV-1 genome containing multiple accessory genes and the seemingly endless array of possible transgenes that could be incorporated, this approach represents a significant source of as yet untapped potential.
T-VEC’s reduced replication and restricted mode of delivery by intra-tumoral application has led investigators to take a step back and consider the possible use of unattenuated viruses. Replication-competent viruses might be safely used if they can be engineered to infect only specific cancer cells, sparing non-cancerous surrounding cells. Therefore, reliance on specificity rather than attenuation for OV therapy may be a superior strategy.
By and large, retargeting is accomplished by modification of the viral receptor, gD. Cancer cells employ a variety of strategies to enhance their own survival. For example, many cancer cells express the IL-13 receptor to promote an anti-inflammatory environment. Zhou et al. created an HSV-1 OV that expresses a chimeric gD receptor that contains IL-13 sequences to target cells expressing the IL-13 receptor (146). In an alternative approach, multiple groups have replaced portions of the gD gene sequence with that of single-chain immunoglobulin receptors. These were directed against tumor associated antigens such as human epidermal growth factor receptor (EGFR), epithelial cell adhesion molecule (EpCAM) and human epithelial growth factor receptor 2 (HER2), thereby retargeting Ovs to recognize a variety of tumor cell types (147–150). In each of these retargeted HSV-1 Ovs, gD was modified but UL34.5 was retained, preserving full replication capacity while providing greater specificity. This strategy parallels the previously mentioned rQNestin34.5, which has UL34.5 under a nestin promoter to specifically kill glioblastoma cells.
Avoiding immune responses is another area of OV development being actively researched. Although OVs can be modified to specifically target tissues, administration of unaccompanied virus has one significant downside in which neutralizing antibodies that either already exist in circulation or arise as a result of treatment can drastically reduce viral titers. Viral clearance by neutralizing antibodies has hampered the use of T-VEC and other OVs in clinical trials for treatment of visceral metastases by systemic or intravenous application (12, 23, 73). Consequently, a number of ‘ghosting’ techniques have been developed that allow delivery of the virus in a manner that prevents it from being exposed to the body’s immune defenses.
Ghosting OVs can involve ‘Trojan horse’ or ‘hitchhiking’ cell-based delivery methods. Systemic administration of OVs by Trojan horse is one of the most commonly explored techniques, likely because it mimics naturally occurring microbial strategies to avoid the immune system, such as that used by Flaviviruses. It is well established that tumor cells tend to traffic in vivo to sites of origin or tumor cell distribution, making them attractive potential delivery vehicles (151). To avoid seeding de novo tumor growth by these carriers, strategies have employed: lethal irradiation of tumor cells prior to delivery, use of allogeneic or xenogeneic cells, and/or use of inducible suicide programs (152, 153). Similarly, mesenchymal stem cells (MSCs) can serve as cell-based OV delivery vehicles since they readily migrate to sites of inflammation, injury, ischemia, and home to tumor microenvironments (154–158). Additional advantages gained from use of MSCs include ease of isolation from patients and growth in culture, reduced immunogenicity, and high metabolic activity that allows for increased viral replication and subsequent viral yields at tumor sites (155, 158). These cell-based delivery mechanisms avoid immune depletion of virus as demonstrated by higher viral titers and improved tumor killing in animal models.
Hitchhiking commonly involves the use of immune cells, which naturally home to tumors as a part of host defense. HSV-1 OVs adsorbed onto the surface of tumor-specific lymphocytes followed by systemic injection resulted in greater cytotoxicity of tumor cells and increased survival in a disseminated tumor mouse model, supporting the use of hitchhiking as a second viable mechanism for OV delivery (159).
As an alternative to cell-based carriers, the viral envelope can be modified to mask it from host immune defenses. This has been done in multiple ways, including addition of inhibitory regulators of phagocytosis or biodegradable polymers to OV envelopes (160, 161). Both of these techniques enable OVs to evade host defenses, thus allowing the virus to persist longer so as to reach more tumor cells.
Since only a small portion of OV carriers typically reach their intended destination, researchers have gone a step further by combining directed targeting with Trojan horse strategies. Using a mouse model, Muthana et al. employed magnetic resonance imaging (MRI) to steer systemically injected macrophages, loaded with magnetic beads and a payload of OV (HSV1716), from the blood to specific tumor sites. This combination strategy resulted in increased tumor macrophage infiltration and a reduction in tumor burden and metastases (162). This work supports the possibility for real-time image-guided trafficking of carrier cells containing targeted OVs to ensure viral delivery at tumor sites and improve clinical outcomes.
OV therapies show great promise towards the goal of defeating cancer with minimal collateral pathology. In particular, patients who are refractory to current SOC treatments may derive enormous benefit from OV therapy. Of the many viruses being studied for OV development, HSV-1 is particularly suitable for the task. The HSV-1 OV, T-VEC, is the only FDA-approved OV therapy in the United States and has already been shown to be safe and effective in the treatment of melanoma. However, there is room for improvement of this or any of the other HSV-1 OV therapies currently being developed. New research has made significant progress in creating OV technologies that can be applied to the HSV-1 platform to provide greater cancer specificity and augment tumor killing with minimal toxicity.
HS: Originated the idea for the review, wrote the paper outline, the Introduction and other several sections, prepared the final format for Figure 1; ZC: Wrote several sections, prepared Table 3; B: Wrote several sections, and performed reference research well as reference list assembly; TS: Wrote several sections, created Table 2 and oversaw editing; DB: Wrote several sections, created Table 1 and oversaw editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Howlader N NA, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, et al. SEER cancer statistics review, 1975-2016, national cancer institute. Bethesda, MD (2019).
2. Murphy SL, Xu J, Kochanek KD, Arias E, Tejada-Vera B. Deaths: Final data for 2018. Natl Vital Stat Rep (2021) 69:1–83.
3. Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst (2006) 98:298–300. doi: 10.1093/jnci/djj111
4. DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, Goemann M, et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res (2001) 61:7464–72.
5. Chang J, Zhao X, Wu X, Guo Y, Guo H, Cao J, et al. A Phase I study of KH901, a conditionally replicating granulocyte-macrophage colony-stimulating factor: armed oncolytic adenovirus for the treatment of head and neck cancers. Cancer Biol Ther (2009) 8:676–82. doi: 10.4161/cbt.8.8.7913
6. Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther (2012) 20:749–58. doi: 10.1038/mt.2011.276
7. Gulley JL, Arlen PM, Tsang KY, Yokokawa J, Palena C, Poole DJ, et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res (2008) 14:3060–9. doi: 10.1158/1078-0432.CCR-08-0126
8. Scholl SM, Balloul JM, Le Goc G, Bizouarne N, Schatz C, Kieny MP, et al. Recombinant vaccinia virus encoding human MUC1 and IL2 as immunotherapy in patients with breast cancer. J Immunother (2000) 23:570–80. doi: 10.1097/00002371-200009000-00007
9. Shafren DR, Sylvester D, Johansson ES, Campbell IG, Barry RD. Oncolysis of human ovarian cancers by echovirus type 1. Int J Cancer (2005) 115:320–8. doi: 10.1002/ijc.20866
10. Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res (2012) 72:2609–21. doi: 10.1158/0008-5472.CAN-11-3185
11. Andtbacka RHI, Curti BD, Kaufman H, Daniels GA, Nemunaitis JJ, Spitler LE, et al. Final data from CALM: A phase II study of Coxsackievirus A21 (CVA21) oncolytic virus immunotherapy in patients with advanced melanoma. J Clin Oncol (2015) 33:9030–0. doi: 10.1200/jco.2015.33.15_suppl.9030
12. Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol (2015) 33:2780–8. doi: 10.1200/JCO.2014.58.3377
13. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer (2013) 13:714–26. doi: 10.1038/nrc3599
14. Pol JG, Levesque S, Workenhe ST, Gujar S, Le Boeuf F, Clements DR, et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology (2018) 7:e1503032. doi: 10.1080/2162402X.2018.1503032
15. (2015) 20993-0002. U.S.F.a.D. Administration, BLA Approval for Talimogene laherparepvec in: C.f.B.E.a. Research, (Ed.), Silver Spring, MD.
16. Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell (1996) 87:427–36. doi: 10.1016/S0092-8674(00)81363-X
17. Watson ZL, Washington SD, Phelan DM, Lewin AS, Tuli SS, Schultz GS, et al. In Vivo Knockdown of the Herpes Simplex Virus 1 Latency-Associated Transcript Reduces Reactivation from Latency. J Virol (2018) 92. doi: 10.1128/JVI.00812-18
18. Du T, Han Z, Zhou G, Roizman B. Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc Natl Acad Sci USA (2015) 112:E49–55. doi: 10.1073/pnas.1422657112
19. Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature (2008) 454:780–3. doi: 10.1038/nature07103
20. Streby KA, Geller JI, Currier MA, Warren PS, Racadio JM, Towbin AJ, et al. Intratumoral injection of hsv1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin Cancer Res (2017) 23:3566–74. doi: 10.1158/1078-0432.CCR-16-2900
21. Su C, Zhan G, Zheng C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol J (2016) 13:38. doi: 10.1186/s12985-016-0495-5
22. Liu J, Lewin AS, Tuli SS, Ghivizzani SC, Schultz GS, Bloom DC. Reduction in severity of a herpes simplex virus type 1 murine infection by treatment with a ribozyme targeting the UL20 gene RNA. J Virol (2008) 82:7467–74. doi: 10.1128/JVI.02720-07
23. Grigg C, Blake Z, Gartrell R, Sacher A, Taback B, Saenger Y. Talimogene laherparepvec (T-Vec) for the treatment of melanoma and other cancers. Semin Oncol (2016) 43:638–46. doi: 10.1053/j.seminoncol.2016.10.005
24. He B, Chou J, Brandimarti R, Mohr I, Gluzman Y, Roizman B. Suppression of the phenotype of gamma(1)34.5- herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J Virol (1997) 71:6049–54. doi: 10.1128/jvi.71.8.6049-6054.1997
25. Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther (2003) 10:292–303. doi: 10.1038/sj.gt.3301885
26. Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther (2004) 11:1648–58. doi: 10.1038/sj.gt.3302289
27. Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science (1990) 250:1262–6. doi: 10.1126/science.2173860
28. Levine. Eating oneself B. and uninvited guests: autophagy-related pathways in cellular defense. Cell (2005) 120:159–62. doi: 10.1016/j.cell.2005.01.005
29. Talloczy Z, Jiang W, H.W.t. Virgin DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA (2002) 99:190–5. doi: 10.1073/pnas.012485299
30. Talloczy Z, Virgin H. and B. Levine. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy (2006) 2:24–9. doi: 10.4161/auto.2176
31. Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe (2007) 1:23–35. doi: 10.1016/j.chom.2006.12.001
32. Ma Y, He B. Recognition of herpes simplex viruses: toll-like receptors and beyond. J Mol Biol (2014) 426:1133–47. doi: 10.1016/j.jmb.2013.11.012
33. Gobeil PA, Leib DA. Herpes simplex virus gamma34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. MBio (2012) 3:e00267–12. doi: 10.1128/mBio.00267-12
34. Trgovcich J, Johnson D, Roizman B. Cell surface major histocompatibility complex class II proteins are regulated by the products of the gamma(1)34.5 and U(L)41 genes of herpes simplex virus 1. J Virol (2002) 76:6974–86. doi: 10.1128/jvi.76.14.6974-6986.2002
35. Neschadim A, Wang JCM, Lavie A, Medin JA. Bystander killing of malignant cells via the delivery of engineered thymidine-active deoxycytidine kinase for suicide gene therapy of cancer. Cancer Gene Ther (2012) 19:320–7. doi: 10.1038/cgt.2012.4
36. Tomazin R, Hill AB, Jugovic P, York I, van Endert P, Ploegh HL, et al. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J (1996) 15:3256–66. doi: 10.1002/j.1460-2075.1996.tb00690.x
37. Ahn K, Meyer TH, Uebel S, Sempe P, Djaballah H, Yang Y, et al. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J (1996) 15:3247–55. doi: 10.1002/j.1460-2075.1996.tb00689.x
38. York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell (1994) 77:525–35. doi: 10.1016/0092-8674(94)90215-1
39. Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med (1998) 187:341–8. doi: 10.1084/jem.187.3.341
40. Sanchez R, Mohr I. Inhibition of cellular 2’-5’ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J Virol (2007) 81:3455–64. doi: 10.1128/JVI.02520-06
41. Cassady KA, Gross M, Roizman B. The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J Virol (1998) 72:8620–6. doi: 10.1128/JVI.72.11.8620-8626.1998
42. Ishioka K, Ikuta K, Sato Y, Kaneko H, Sorimachi K, Fukushima E, et al. Herpes simplex virus type 1 virion-derived US11 inhibits type 1 interferon-induced protein kinase R phosphorylation. Microbiol Immunol (2013) 57:426–36. doi: 10.1111/1348-0421.12048
43. Nouri K, Moll JM, Milroy LG, Hain A, Dvorsky R, Amin E, et al. Biophysical characterization of nucleophosmin interactions with human immunodeficiency virus rev and herpes simplex virus us11. PloS One (2015) 10:e0143634. doi: 10.1371/journal.pone.0143634
44. Liu X, Main D, Ma Y, He B. Herpes Simplex Virus 1 Inhibits TANK-Binding Kinase 1 through Formation of the Us11-Hsp90 Complex. doi: 10.1128/JVI.00402-18
45. Nikonov A, Molder T, Sikut R, Kiiver K, Mannik A, Toots U, et al. RIG-I and MDA-5 detection of viral RNA-dependent RNA polymerase activity restricts positive-strand RNA virus replication. PloS Pathog (2013) 9:e1003610. doi: 10.1371/journal.ppat.1003610
46. I. BioVex, a subsidiary of Amgen, Inc., IMLYGIC (talimogene laherparepvec) Prescribing Information, Author, Thousand Oaks, California, 10/01/2019.
47. Kaufman HL, Ruby CE, Hughes T, Slingluff CL Jr.. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J Immunother Cancer (2014) 2:11. doi: 10.1186/2051-1426-2-11
48. Eissa IR, Naoe Y, Bustos-Villalobos I, Ichinose T, Tanaka M, Zhiwen W, et al. Genomic signature of the natural oncolytic herpes simplex virus hf10 and its therapeutic role in preclinical and clinical trials. Front Oncol (2017) 7:149. doi: 10.3389/fonc.2017.00149
49. Kimata H, Imai T, Kikumori T, Teshigahara O, Nagasaka T, Goshima F, et al. Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann Surg Oncol (2006) 13:1078–84. doi: 10.1245/ASO.2006.08.035
50. Nakao A, Kasuya H, Sahin TT, Nomura N, Kanzaki A, Misawa M, et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther (2011) 18:167–75. doi: 10.1038/cgt.2010.65
51. Pannhorst K, Wei H, Yezid H, He J, Chowdhury SI. Bovine herpesvirus 1 ul49.5 interacts with gm and vp22 to ensure virus cell-to-cell spread and virion incorporation: novel role for vp22 in gm-independent ul49.5 virion incorporation. J Virol (2018) 92. doi: 10.1128/JVI.00240-18
52. Jin L, Perng GC, Mott KR, Osorio N, Naito J, Brick DJ, et al. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol (2005) 79:12286–95. doi: 10.1128/JVI.79.19.12286-12295.2005
53. Perng GC, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, et al. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol (2002) 76:1224–35. doi: 10.1128/jvi.76.3.1224-1235.2002
54. Henderson G, Peng W, Jin L, Perng GC, Nesburn AB, Wechsler SL, et al. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J Neurovirol (2002) 8 Suppl 2:103–11. doi: 10.1080/13550280290101085
55. Branco FJ, Fraser NW. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol (2005) 79:9019–25. doi: 10.1128/JVI.79.14.9019-9025.2005
56. Koshizuka T, Kawaguchi Y, Nishiyama Y. Herpes simplex virus type 2 membrane protein UL56 associates with the kinesin motor protein KIF1A. J Gen Virol (2005) 86:527–33. doi: 10.1099/vir.0.80633-0
57. Berkowitz C, Moyal M, Rosen-Wolff A, Darai G, Becker Y. Herpes simplex virus type 1 (HSV-1) UL56 gene is involved in viral intraperitoneal pathogenicity to immunocompetent mice. Arch Virol (1994) 134:73–83. doi: 10.1007/BF01379108
58. Peles E, Rosen H, Darai G, Rosen-Wolff A, Becker Y. Importance of the HpaI-P sequence for herpes simplex virus-1 replication in the adrenal glands. Arch Virol (1990) 113:151–63. doi: 10.1007/bf01316669
59. Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell (1995) 81:769–80. doi: 10.1016/0092-8674(95)90538-3
60. Jones C, Inman M, Peng W, Henderson G, Doster A, Perng GC, et al. The herpes simplex virus type 1 locus that encodes the latency-associated transcript enhances the frequency of encephalitis in male BALB/c mice. J Virol (2005) 79:14465–9. doi: 10.1128/JVI.79.22.14465-14469.2005
61. Adusumilli PS, Eisenberg DP, Stiles BM, Hendershott KJ, Stanziale SF, Chan MK, et al. Virally-directed fluorescent imaging (VFI) can facilitate endoscopic staging. Surg Endosc (2006) 20:628–35. doi: 10.1007/s00464-005-0259-6
62. McKie EA, MacLean AR, Lewis AD, Cruickshank G, Rampling R, Barnett SC, et al. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours–evaluation of a potentially effective clinical therapy. Br J Cancer (1996) 74:745–52. doi: 10.1038/bjc.1996.431
63. Hirooka Y, Kasuya H, Ishikawa T, Kawashima H, Ohno E, Villalobos IB, et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer (2018) 18:596. doi: 10.1186/s12885-018-4453-z
64. Matveeva OV, Chumakov PM. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev Med Virol (2018) 28:e2008. doi: 10.1002/rmv.2008
65. Kasuya H, Kodera Y, Nakao A, Yamamura K, Gewen T, Zhiwen W, et al. phase i dose-escalation clinical trial of hf10 oncolytic herpes virus in 17 japanese patients with advanced cancer. Hepatogastroenterology (2014) 61:599–605.
66. Simpson GR, Han Z, Liu B, Wang Y, Campbell G, Coffin RS. Combination of a fusogenic glycoprotein, prodrug activation, and oncolytic herpes simplex virus for enhanced local tumor control. Cancer Res (2006) 66:4835–42. doi: 10.1158/0008-5472.CAN-05-4352
67. Watanabe D, Goshima F, Mori I, Tamada Y, Matsumoto Y, Nishiyama Y. Oncolytic virotherapy for malignant melanoma with herpes simplex virus type 1 mutant HF10. J Dermatol Sci (2008) 50:185–96. doi: 10.1016/j.jdermsci.2007.12.001
68. Takakuwa H, Goshima F, Nozawa N, Yoshikawa T, Kimata H, Nakao A, et al. Oncolytic viral therapy using a spontaneously generated herpes simplex virus type 1 variant for disseminated peritoneal tumor in immunocompetent mice. Arch Virol (2003) 148:813–25. doi: 10.1007/s00705-002-0944-x
69. Luo C, Mori I, Goshima F, Ushijima Y, Nawa A, Kimura H, et al. Replication-competent, oncolytic herpes simplex virus type 1 mutants induce a bystander effect following ganciclovir treatment. J Gene Med (2007) 9:875–83. doi: 10.1002/jgm.1085
70. Yamasaki H, Mesnil M, Omori Y, Mironov N, Krutovskikh V. Intercellular communication and carcinogenesis. Mutat Research/Fundamental Mol Mech Mutagenesis (1995) 333:181–8. doi: 10.1016/0027-5107(95)00144-1
71. Haass NK, Smalley KSM, Herlyn M. The role of altered cell–cell communication in melanoma progression. J Mol Histol (2004) 35:309–18. doi: 10.1023/B:HIJO.0000032362.35354.bb
72. Nakao A, Kimata H, Imai T, Kikumori T, Teshigahara O, Nagasaka T, et al. Intratumoral injection of herpes simplex virus HF10 in recurrent breast cancer. Ann Oncol (2004) 15:988–9. doi: 10.1093/annonc/mdh225
73. Andtbacka RH, Agarwala SS, Ollila DW, Hallmeyer S, Milhem M, Amatruda T, et al. Cutaneous head and neck melanoma in OPTiM, a randomized phase 3 trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor for the treatment of unresected stage IIIB/IIIC/IV melanoma. Head Neck (2016) 38:1752–8. doi: 10.1002/hed.24522
74. Fujimoto Y, Mizuno T, Sugiura S, Goshima F, Kohno S, Nakashima T, et al. Intratumoral injection of herpes simplex virus HF10 in recurrent head and neck squamous cell carcinoma. Acta Otolaryngol (2006) 126:1115–7. doi: 10.1080/00016480600702100
75. Tan G, Kasuya H, Sahin TT, Yamamura K, Wu Z, Koide Y, et al. Combination therapy of oncolytic herpes simplex virus HF10 and bevacizumab against experimental model of human breast carcinoma xenograft. Int J Cancer (2015) 136:1718–30. doi: 10.1002/ijc.29163
76. Shimoyama S, Goshima F, Teshigahara O, Kasuya H, Kodera Y, Nakao A, et al. Enhanced efficacy of herpes simplex virus mutant HF10 combined with paclitaxel in peritoneal cancer dissemination models. Hepatogastroenterology (2007) 54:1038–42.
77. Sugiura S, Goshima F, Takakuwa H, Sata T, Nakashima T, Nishiyama Y. Treatment of solid sarcomas in immunocompetent mice with novel, oncolytic herpes simplex viruses. Otolaryngol Head Neck Surg (2004) 130:470–8. doi: 10.1016/j.otohns.2004.01.001
78. MacLean AR, M. ul-Fareed L, Harland J, Brown SM. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the A’’ sequence. J Gen Virol (1991) 72(Pt 3):631–9. doi: 10.1099/0022-1317-72-3-631
79. Papanastassiou V, Rampling R, Fraser M, Petty R, Hadley D, Nicoll J, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther (2002) 9:398–406. doi: 10.1038/sj.gt.3301664
80. MacKie RM, Stewart B, Brown SM. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet (2001) 357:525–6. doi: 10.1016/S0140-6736(00)04048-4
81. Danson SJ, Conner J, Edwards JG, Blyth KG, Fisher PM, Muthana M, et al. Oncolytic herpesvirus therapy for mesothelioma - A phase I/IIa trial of intrapleural administration of HSV1716. Lung Cancer (2020) 150:145–51. doi: 10.1016/j.lungcan.2020.10.007
82. Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC, McCart JA. Intelligent design: combination therapy with oncolytic viruses. Mol Ther (2010) 18:251–63. doi: 10.1038/mt.2009.283
83. Kumar S, Gao L, Yeagy B, Reid T. Virus combinations and chemotherapy for the treatment of human cancers. Curr Opin Mol Ther (2008) 10:371–9.
84. Kanai R, Wakimoto H, Cheema T, Rabkin SD. Oncolytic herpes simplex virus vectors and chemotherapy: are combinatorial strategies more effective for cancer? Future Oncol (2010) 6:619–34. doi: 10.2217/fon.10.18
85. Mace AT, Harrow SJ, Ganly I, Brown SM. Cytotoxic effects of the oncolytic herpes simplex virus HSV1716 alone and in combination with cisplatin in head and neck squamous cell carcinoma. Acta Otolaryngol (2007) 127:880–7. doi: 10.1080/00016480601075381
86. Currier MA, Sprague L, Rizvi TA, Nartker B, Chen CY, Wang PY, et al. Aurora A kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation. Oncotarget (2017) 8:17412–27. doi: 10.18632/oncotarget.14885
87. Streby KA, Currier MA, Triplet M, Ott K, Dishman DJ, Vaughan MR, et al. First-in-human intravenous seprehvir in young cancer patients: A phase 1 clinical trial. Mol Ther (2019) 27:1930–8. doi: 10.1016/j.ymthe.2019.08.020
88. Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther (2000) 7:867–74. doi: 10.1038/sj.gt.3301205
89. Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med (1995) 1:938–43. doi: 10.1038/nm0995-938
90. Waters AM, Johnston JM, Reddy AT, Fiveash J, Madan-Swain A, Kachurak K, et al. Rationale and design of a phase 1 clinical trial to evaluate hsv g207 alone or with a single radiation dose in children with progressive or recurrent malignant supratentorial brain tumors. Hum Gene Ther Clin Dev (2017) 28:7–16. doi: 10.1089/humc.2017.002
91. Goldstein DJ, Weller SK. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology (1988) 166:41–51. doi: 10.1016/0042-6822(88)90144-4
92. Jacobson JG, Leib DA, Goldstein DJ, Bogard CL, Schaffer PA, Weller SK, et al. A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virology (1989) 173:276–83. doi: 10.1016/0042-6822(89)90244-4
93. Preston VG, Palfreyman JW, Dutia BM. Identification of a herpes simplex virus type 1 polypeptide which is a component of the virus-induced ribonucleotide reductase. J Gen Virol 65 (Pt (1984) 9):1457–66. doi: 10.1099/0022-1317-65-9-1457
94. Mineta T, Rabkin SD, Martuza RL. Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res (1994) 54:3963–6.
95. Mostafa HH, Thompson TW, Konen AJ, Haenchen SD, Hilliard JG, Macdonald SJ, et al. Herpes Simplex Virus 1 Mutant with Point Mutations in UL39 Is Impaired for Acute Viral Replication in Mice, Establishment of Latency, and Explant-Induced Reactivation. J Virol (2018) 92. doi: 10.1128/JVI.01654-17
96. Dufour F, Bertrand L, Pearson A, Grandvaux N, Langelier Y. The ribonucleotide reductase R1 subunits of herpes simplex virus 1 and 2 protect cells against poly(I. C)-induced apoptosis. J Virol (2011) 85:8689–701. doi: 10.1128/JVI.00362-11
97. Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther (2009) 17:199–207. doi: 10.1038/mt.2008.228
98. Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther (2014) 22:1048–55. doi: 10.1038/mt.2014.22
99. Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA (2001) 98:6396–401. doi: 10.1073/pnas.101136398
100. Wang JN, Hu P, Zeng MS, Liu RB. Anti-tumor effect of oncolytic herpes simplex virus G47delta on human nasopharyngeal carcinoma. Chin J Cancer (2011) 30:831–41. doi: 10.5732/cjc.011.10301
101. Messerli SM, Prabhakar S, Tang Y, Mahmood U, Giovannini M, Weissleder R, et al. Treatment of schwannomas with an oncolytic recombinant herpes simplex virus in murine models of neurofibromatosis type 2. Hum Gene Ther (2006) 17:20–30. doi: 10.1089/hum.2006.17.20
102. Liu R, Martuza RL, Rabkin SD. Intracarotid delivery of oncolytic HSV vector G47Δ to metastatic breast cancer in the brain. Gene Ther (2005) 12:647–54. doi: 10.1038/sj.gt.3302445
103. Wang J, Hu P, Zeng M-S, Rabkin S, Liu R. Oncolytic herpes simplex virus treatment of metastatic breast cancer. Int J Oncol (2011) 40:757–63. doi: 10.3892/ijo.2011.1266
104. Wang J, Xu L, Zeng W, Hu P, Zeng M, Rabkin SD, et al. Treatment of human hepatocellular carcinoma by the oncolytic herpes simplex virus G47delta. Cancer Cell Int (2014) 14:83. doi: 10.1186/s12935-014-0083-y
105. Wang JN, Xu LH, Zeng WG, Hu P, Rabkin SD, Liu RR. Treatment of human thyroid carcinoma cells with the g47delta oncolytic herpes simplex virus. Asian Pac J Cancer Prev (2015) 16:1241–5. doi: 10.7314/apjcp.2015.16.3.1241
106. Sugawara K, Iwai M, Yajima S, Tanaka M, Yanagihara K, Seto Y, et al. Efficacy of a third-generation oncolytic herpes virus g47δ in advanced stage models of human gastric cancer. Mol Ther - Oncolytics (2020) 17:205–15. doi: 10.1016/j.omto.2020.03.022
107. Taguchi S, Fukuhara H, Homma Y, Todo T. Current status of clinical trials assessing oncolytic virus therapy for urological cancers. Int J Urol (2017) 24:342–51. doi: 10.1111/iju.13325
108. Fukuhara H, Martuza RL, Rabkin SD, Ito Y, Todo T. Oncolytic herpes simplex virus vector g47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin Cancer Res (2005) 11:7886–90. doi: 10.1158/1078-0432.CCR-05-1090
109. Daiichi-Sankyo. Daiichi sankyo submits application for oncolytic virus teserpaturev (g47δ) for treatment of patients with malignant glioma in japan. (2021).
110. C.M.M.M.S.J.M.M.M.R.B.C.M. P. Phase 1 trial of R7020: A live attenuated recombinant herpes simplex (hsv) candidate vaccine, 32nd interscience conference on antimicrobial agents and chemotherapy. Anaheim, CA (1992).
111. Meignier B, Longnecker R, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: Construction and evaluation in rodents. J Infect Dis (1988) 158:602–14. doi: 10.1093/infdis/158.3.602
112. Roizman B. The function of herpes simplex virus genes: a primer for genetic engineering of novel vectors. Proc Natl Acad Sci USA (1996) 93:11307–12. doi: 10.1073/pnas.93.21.11307
113. Xu H, Su C, Pearson A, Mody CH, Zheng C. Herpes simplex virus 1 ul24 abrogates the dna sensing signal pathway by inhibiting nf-κb activation. J Virol (2017) 91. doi: 10.1128/jvi.00025-17
114. Bennett JJ, Delman KA, Burt BM, Mariotti A, Malhotra S, Zager J, et al. Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene Ther (2002) 9:935–45. doi: 10.1038/sj.cgt.7700510
115. Kemeny N, Brown K, Covey A, Kim T, Bhargava A, Brody L, et al. open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther (2006) 17:1214–24. doi: 10.1089/hum.2006.17.1214
116. Geevarghese SK, Geller DA, de Haan HA, Horer M, Knoll AE, Mescheder A, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther (2010) 21:1119–28. doi: 10.1089/hum.2010.020
117. Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res (2005) 65:2832–9. doi: 10.1158/0008-5472.CAN-04-3227
118. Nakashima H, Nguyen T, Kasai K, Passaro C, Ito H, Goins WF, et al. Toxicity and efficacy of a novel gadd34-expressing oncolytic hsv-1 for the treatment of experimental glioblastoma. Clin Cancer Res (2018) 24:2574–84. doi: 10.1158/1078-0432.CCR-17-2954
119. Cicchelero L, Denies S, Haers H, Vanderperren K, Stock E, Van Brantegem L, et al. Intratumoural interleukin 12 gene therapy stimulates the immune system and decreases angiogenesis in dogs with spontaneous cancer. Vet Comp Oncol (2017) 15:1187–205. doi: 10.1111/vco.12255
120. Chuang TF, Lee SC, Liao KW, Hsiao YW, Lo CH, Chiang BL, et al. Electroporation-mediated IL-12 gene therapy in a transplantable canine cancer model. Int J Cancer (2009) 125:698–707. doi: 10.1002/ijc.24418
121. Caruso M, Pham-Nguyen K, Kwong YL, Xu B, Kosai KI, Finegold M, et al. Adenovirus-mediated interleukin-12 gene therapy for metastatic colon carcinoma. Proc Natl Acad Sci USA (1996) 93:11302–6. doi: 10.1073/pnas.93.21.11302
122. Heinzerling L, Burg G, Dummer R, Maier T, Oberholzer PA, Schultz J, et al. Intratumoral injection of DNA encoding human interleukin 12 into patients with metastatic melanoma: clinical efficacy. Hum Gene Ther (2005) 16:35–48. doi: 10.1089/hum.2005.16.35
123. Cutrera J, King G, Jones P, Kicenuik K, Gumpel E, Xia X, et al. Safety and efficacy of tumor-targeted interleukin 12 gene therapy in treated and non-treated, metastatic lesions. Curr Gene Ther (2015) 15:44–54. doi: 10.2174/1566523214666141127093654
124. Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK, et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol (2008) 26:5896–903. doi: 10.1200/JCO.2007.15.6794
125. Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. design of a phase i clinical trial to evaluate m032, a genetically engineered hsv-1 expressing il-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum Gene Ther Clin Dev (2016) 27:69–78. doi: 10.1089/humc.2016.031
126. Voest EE, Kenyon BM, O’Reilly MS, Truitt G, D’Amato RJ, Folkman J. Inhibition of angiogenesis in vivo by interleukin 12. J Natl Cancer Inst (1995) 87:581–6. doi: 10.1093/jnci/87.8.581
127. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage iiib-iv melanoma. J Clin Oncol (2016) 34:2619–26. doi: 10.1200/JCO.2016.67.1529
128. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-pd-1 immunotherapy. Cell (2017) 170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027
129. McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res (2006) 66:2509–13. doi: 10.1158/0008-5472.CAN-05-2242
130. Kuriyama N, Kuriyama H, Julin CM, Lamborn K, Israel MA. Pretreatment with protease is a useful experimental strategy for enhancing adenovirus-mediated cancer gene therapy. Hum Gene Ther (2000) 11:2219–30. doi: 10.1089/104303400750035744
131. Haseley A, Boone S, Wojton J, Yu L, Yoo JY, Yu J, et al. Extracellular matrix protein CCN1 limits oncolytic efficacy in glioma. Cancer Res (2012) 72:1353–62. doi: 10.1158/0008-5472.CAN-11-2526
132. Dmitrieva N, Yu L, Viapiano M, Cripe TP, Chiocca EA, Glorioso JC, et al. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res (2011) 17:1362–72. doi: 10.1158/1078-0432.CCR-10-2213
133. Martinez-Quintanilla J, He D, Wakimoto H, Alemany R, Shah K. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol Ther (2015) 23:108–18. doi: 10.1038/mt.2014.204
134. Jung KH, Choi IK, Lee HS, Yan HH, Son MK, Ahn HM, et al. Corrigendum to “Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett (2017) 396:155–66. doi: 10.1016/j.canlet.2017.06.019
135. Li Y, Hong J, Oh JE, Yoon AR, Yun CO. Potent antitumor effect of tumor microenvironment-targeted oncolytic adenovirus against desmoplastic pancreatic cancer. Int J Cancer (2018) 142:392–413. doi: 10.1002/ijc.31060
136. Choi IK, Lee YS, Yoo JY, Yoon AR, Kim H, Kim DS, et al. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther (2010) 17:190–201. doi: 10.1038/gt.2009.142
137. Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther (2010) 18:1275–83. doi: 10.1038/mt.2010.79
138. Braidwood L, Dunn PD, Hardy S, Evans TR, Brown SM. Antitumor activity of a selectively replication competent herpes simplex virus (HSV) with enzyme prodrug therapy. Anticancer Res (2009) 29:2159–66.
139. Quigg M, Mairs RJ, Brown SM, Harland J, Dunn P, Rampling R, et al. Assessment in vitro of a novel therapeutic strategy for glioma, combining herpes simplex virus HSV1716-mediated oncolysis with gene transfer and targeted radiotherapy. Med Chem (2005) 1:423–9. doi: 10.2174/1573406054864124
140. Sorensen A, Mairs RJ, Braidwood L, Joyce C, Conner J, Pimlott S, et al. In vivo evaluation of a cancer therapy strategy combining hsv1716-mediated oncolysis with gene transfer and targeted radiotherapy. J Nucl Med (2012) 53:647–54. doi: 10.2967/jnumed.111.090886
141. de Graaf JF, de Vor L, Fouchier RAM, van den Hoogen BG. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth factor Rev (2018) 41:28–39. doi: 10.1016/j.cytogfr.2018.03.006
142. Pearl TM, Markert JM, Cassady KA, Ghonime MG. oncolytic virus-based cytokine expression to improve immune activity in brain and solid tumors. Mol Ther oncolytics (2019) 13:14–21. doi: 10.1016/j.omto.2019.03.001
143. Torres-Domínguez LE, McFadden G. Poxvirus oncolytic virotherapy. Expert Opin Biol Ther (2019) 19:561–73. doi: 10.1080/14712598.2019.1600669
144. Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic viruses: Priming time for cancer immunotherapy. BioDrugs: Clin immunotherapeutics biopharmaceuticals Gene Ther (2019) 33:485–501. doi: 10.1007/s40259-019-00367-0
145. Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer (2018) 18:419–32. doi: 10.1038/s41568-018-0009-4
146. Zhou G, Roizman B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc Natl Acad Sci USA (2006) 103:5508–13. doi: 10.1073/pnas.0601258103
147. Uchida H, Marzulli M, Nakano K, Goins WF, Chan J, Hong CS, et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther (2013) 21:561–9. doi: 10.1038/mt.2012.211
148. Shibata T, Uchida H, Shiroyama T, Okubo Y, Suzuki T, Ikeda H, et al. Development of an oncolytic HSV vector fully retargeted specifically to cellular EpCAM for virus entry and cell-to-cell spread. Gene Ther (2016) 23:479–88. doi: 10.1038/gt.2016.17
149. Nanni P, Gatta V, Menotti L, De Giovanni C, Ianzano M, Palladini A, et al. Preclinical therapy of disseminated HER-2(+) ovarian and breast carcinomas with a HER-2-retargeted oncolytic herpesvirus. PloS Pathog (2013) 9:e1003155. doi: 10.1371/journal.ppat.1003155
150. Menotti L, Cerretani A, Hengel H, Campadelli-Fiume G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J Virol (2008) 82:10153–61. doi: 10.1128/JVI.01133-08
151. Willmon C, Harrington K, Kottke T, Prestwich R, Melcher A, Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol therapy: J Am Soc Gene Ther (2009) 17:1667–76. doi: 10.1038/mt.2009.194
152. Coukos G, Makrigiannakis A, Kang EH, Caparelli D, Benjamin I, Kaiser LR, et al. Use of carrier cells to deliver a replication-selective herpes simplex virus-1 mutant for the intraperitoneal therapy of epithelial ovarian cancer. Clin Cancer Res (1999) 5:1523–37.
153. Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther (2007) 15:123–30. doi: 10.1038/sj.mt.6300039
154. Shah K. Mesenchymal stem cells engineered for cancer therapy. Advanced Drug Deliv Rev (2012) 64:739–48. doi: 10.1016/j.addr.2011.06.010
155. Du W, Seah I, Bougazzoul O, Choi G, Meeth K, Bosenberg MW, et al. Stem cell-released oncolytic herpes simplex virus has therapeutic efficacy in brain metastatic melanomas. Proc Natl Acad Sci (2017) 114:E6157–65. doi: 10.1073/pnas.1700363114
156. Duebgen M, Martinez-Quintanilla J, Tamura K, Hingtgen S, Redjal N, Wakimoto H, et al. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J Natl Cancer Inst (2014) 106:dju090. doi: 10.1093/jnci/dju090
157. Leoni V, Gatta V, Palladini A, Nicoletti G, Ranieri D, Dall’Ora M, et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget (2015) 6:34774–87. doi: 10.18632/oncotarget.5793
158. Mahasa KJ, de Pillis L, Ouifki R, Eladdadi A, Maini P, Yoon AR, et al. Mesenchymal stem cells used as carrier cells of oncolytic adenovirus results in enhanced oncolytic virotherapy. Sci Rep (2020) 10:425. doi: 10.1038/s41598-019-57240-x
159. Kanzaki A, Kasuya H, Yamamura K, Sahin TT, Nomura N, Shikano T, et al. Antitumor efficacy of oncolytic herpes simplex virus adsorbed onto antigen-specific lymphocytes. Cancer Gene Ther (2012) 19:292–8. doi: 10.1038/cgt.2011.91
160. Fu X, Tao L, Zhang X. Genetically coating oncolytic herpes simplex virus with CD47 allows efficient systemic delivery and prolongs virus persistence at tumor site. Oncotarget (2018) 9:34543–53. doi: 10.18632/oncotarget.26167
161. Khalil IR, Khechara MP, Kurusamy S, Armesilla AL, Gupta A, Mendrek B, et al. Gamma-Glutamic Acid (γ-PGA)-Based Encapsulation of Adenovirus to Evade Neutralizing Antibodies. Molecules (Basel Switzerland) (2018) 23. doi: 10.3390/molecules23102565
Keywords: oncolytic virus, Herpes simplex virus 1, cancer, T-VEC, virotherapy
Citation: Scanlan H, Coffman Z, Bettencourt J, Shipley T and Bramblett DE (2022) Herpes simplex virus 1 as an oncolytic viral therapy for refractory cancers. Front. Oncol. 12:940019. doi: 10.3389/fonc.2022.940019
Received: 09 May 2022; Accepted: 27 June 2022;
Published: 27 July 2022.
Edited by:
Ioana Berindan Neagoe, Iuliu Haţieganu University of Medicine and Pharmacy, RomaniaReviewed by:
Dat Ha, University of Southern California, United StatesCopyright © 2022 Scanlan, Coffman, Bettencourt, Shipley and Bramblett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Debra E. Bramblett, ZGJyYW1ibGV0dEBidXJyZWxsLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.