 Ling Zhang1*
Ling Zhang1* Bin Zhang
Bin Zhang- 1School of Nursing, Jining Medical University, Jining, China
- 2School of Public Health, North China University of Science and Technology, Tangshan, China
- 3Department of Laboratory Medicine, Affiliated Hospital of Jining Medical University, Jining Medical University, Jining, China
- 4Institute of Forensic Medicine and Laboratory Medicine, Jining Medical University, Jining, China
Cyclophilin D (CypD) is a peptide-proline cis-trans isomerase (PPIase) distributed in the mitochondrial matrix. CypD regulates the opening of the mitochondrial permeability conversion pore (mPTP) and mitochondrial bioenergetics through PPIase activity or interaction with multiple binding partners in mitochondria. CypD initially attracted attention due to its regulation of mPTP overopening-mediated cell death. However, recent studies on the effects of CypD on tumors have shown conflicting results. Although CypD has been proven to promote the aerobic glycolysis in tumor cells, its regulation of malignant characteristics such as the survival, invasion and drug resistance of tumor cells remains controversial. Here, we elaborate the main biological functions of CypD and its relationships with tumor progression identified in recent years, focusing on the dual role of CypD in tumors.
Introduction
Cyclophilin D (CypD) is a cyclophilin distributed in the mitochondrial matrix that acts as the gatekeeper of mitochondria (1, 2). Most previous studies considered that CypD plays a vital role in regulating cell apoptosis or necrosis by regulating mitochondrial permeability. Early studies on CypD were mostly limited to ischaemia-reperfusion injury, neurodegeneration, ageing, diabetes, etc (3–6). With the in-depth research on CypD function, the relationship between CypD and tumors has gradually become a research hotspot in recent years. It remains controversial whether CypD promotes or inhibits tumor progression. Many studies on the regulation by CypD of tumor cell survival, invasion and drug resistance have reported inconsistent conclusions. It has been reported that CypD can affect the malignant characteristics of tumor progression by regulating the bioenergetics and mitochondrial permeability of tumor cells. Here, we review the biological functions of CypD and its role in tumor progression. In particular, the question of whether CypD is the guardian of tumor progression or the executioner of tumor treatment is fully expounded upon this review.
Characteristics of CypD
CypD is a peptide-proline cis-trans isomerase (PPIase) widely expressed in all mammalian tissues. CypD is encoded by the nuclear gene Ppif (located at 10q22-10q23), and its mRNA is 2,213 bases in length and encodes 207 amino acids (aa). After shearing, it is translocated to the mitochondrial matrix and transformed into a 178-aa mature peptide. CypD was originally named mitochondrial cyclophilin (CYP-M) and is now sometimes referred to as cyclophilin F or cyclophilin 3 (7). It should be noted, however, that CypD has been used in the past to refer to a 370-aa cytoplasmic cyclophilin protein encoded by the Ppid gene. Under electron microscopy, CypD shows a spindle or rod structure with a diameter of 0.5-1.0 μm. CypD isomers are rarely reported.
The maintenance of normal mitochondrial permeability is critical to mitochondrial function and is mainly controlled by the status of the mitochondrial permeability transition pore (mPTP) (8, 9). mPTP, also known as the mitochondrial giant channel, is a calcium-dependent nonselective highly conductive channel. As early as the 1990s, CypD attracted attention due to its regulatory role in mPTP. The binding partners of CypD are mostly located in the mitochondrial inner membrane (MIM), including adenine nucleotide transporter (ANT), phosphate carrier (PiC), and oligomycin-sensitive binding protein (OSCP, a subunit of F1FO ATP synthase) (9). CypD binding with its binding partners can induce a transient low level of mPTP opening, promoting ROS excretion out of mitochondria and maintaining mitochondrial calcium homeostasis. Upregulated expression or activity of CypD can provoke mPTP overopening and lead to the influx of a large number of substances with molecular weights less than 1.5 kDa into the mitochondrial matrix, resulting in mitochondrial membrane depolarization, oxidative phosphorylation (OXPHOS) uncoupling, ATP depletion, and the release of proapoptotic factors, subsequently inducing apoptosis or necrosis (10). Thus, abnormal activation of the CypD-mPTP axis is considered to be the executioner in various diseases, such as ischaemia/reperfusion injury, ageing and neurodegeneration. However, it has been reported that CypD overexpression inhibits ANT-mediated apoptosis, enhances cell resistance to harmful stimuli, and promotes cell survival (11). Therefore, for mitochondria and cells, the identity of CypD remains a mystery.
The Major Biological Functions of CypD
Regulation of Mitochondrial Permeability
mPTP is regarded as a key effector of cell death, where many signals that regulate cell death converge. The physiological and pathological roles of mPTP have been well studied, but its molecular identity and necessary regulatory factors remain controversial. mPTP was initially thought to be mainly composed of voltage-dependent anion channels [VDAC, located in the mitochondrial outer membrane (MOM)], ANT and CypD and is regulated by hexokinase II (HK II), PiC, mitochondrial creatine kinase (mtCK) and other proteins (9, 12) (Figure 1). However, with further research, the credibility of the classical mPTP model has been questioned in a series of genetic knockout studies (13, 14). In the absence of ANT or VDAC, increased Ca2+ still activates mPTP, and does not prevent cell death caused by mPTP overopening. These results suggest that CypD may also associate with other binding partners to promote mPTP opening and influence mitochondrial permeability. It is worth to mention that CypD is the only genetically proven indispensable mPTP component (15).
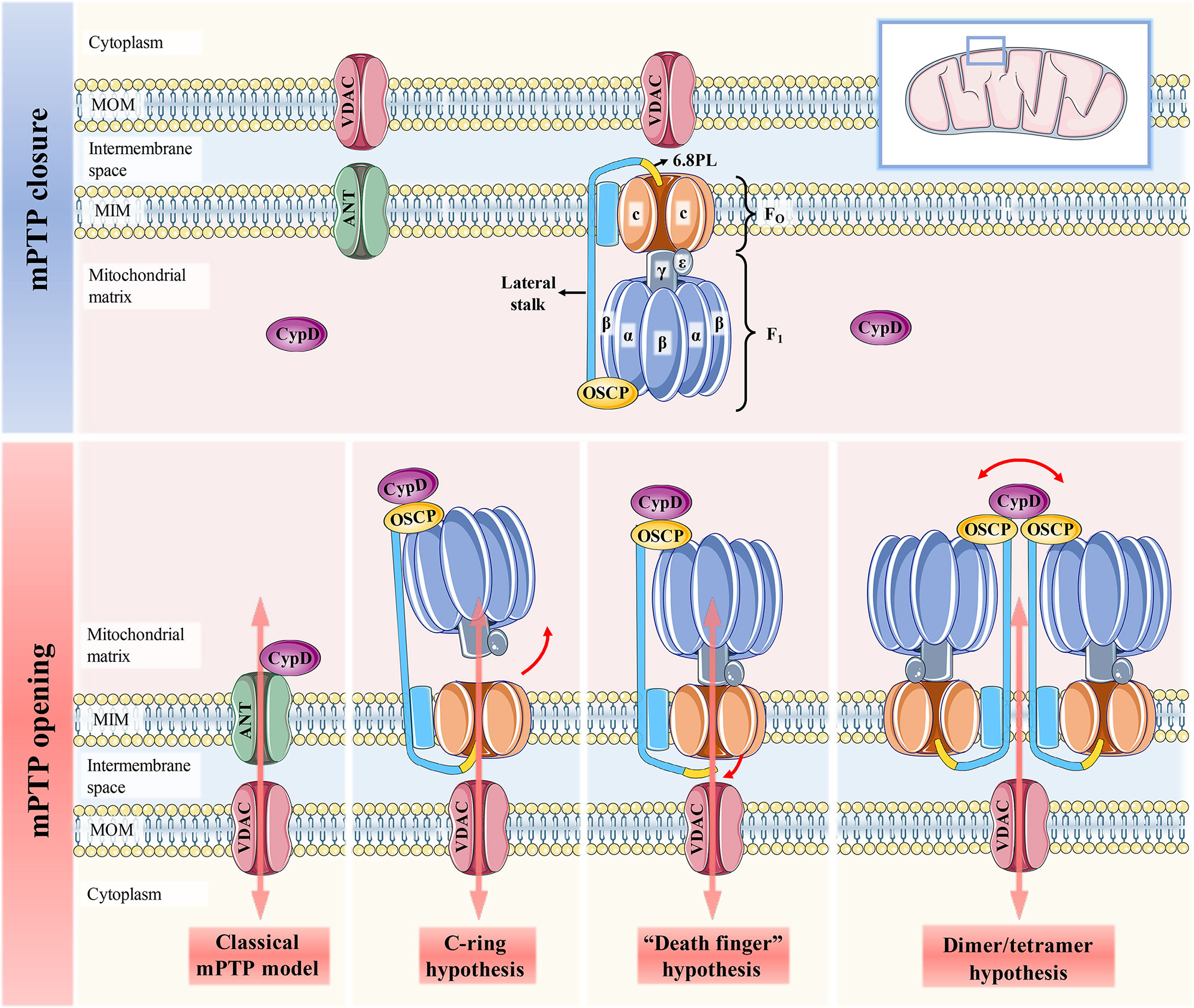
Figure 1 Schematic representation of the four mPTP models. When CypD is free in the mitochondrial matrix, mPTP is not activated and remains completely closed. The classical mPTP model holds that when CypD translocates from the matrix to the MIM and interacts with ANT, which can further induce the binding of ANT and VDAC to open the mPTP. The c-ring hypothesis suggests that conformational changes of F1 caused by CypD-OSCP association abolish the sealing effect of F1 on the c-ring, thereby promoting the central hole of the c-ring to form mPTP. Both the “dead finger” hypothesis and the dimer/tetramer hypothesis are caused by the conformation of the lateral stalk induced by the CypD-OSCP association, but the mPTP formation sites of the two are different. The former considers disassociating 6.8PL from c-ring to form mPTP. mPTP is formed by the lateral stalks of all F1FO ATP synthases that make up the dimer/tetramers. The former suggests that mPTP is activated by the removal of 6.8PL from the c-ring, while the latter suggests that mPTP is formed by the surrounding lateral stalks of all F1FO ATP synthases in the dimers/tetramers.
It has been reported that mPTP is more likely to be located in the F1FO ATP synthase (complex V of OXPHOS). At present, there are three hypotheses about the formation of mPTP by F1FO ATP synthase, namely, the c-ring hypothesis, “death finger” hypothesis and dimer/tetramer hypothesis (16) (Figure 1). The initiation signal of these three hypotheses is the mechanical combination of CypD and OSCP located in the crown of F1, but its downstream effect events are quite different (1): The c-ring hypothesis suggests that CypD-OSCP association induces the conformational changes in the F1 β subunit, decouples F1 and FO, and exposes the central hole of the c-ring to form mPTP (17) (2). The “death finger” hypothesis suggests that the mechanical force generated by CypD-OSCP association can be transmitted along the lateral stalk to the base of FO and remove the sealing effect of the 6.8PL subunit on the c-ring (18, 19) (3). The dimer/tetramer hypothesis also derives from CypD-mediated lateral stalk conformational changes, which allow adjacent e and g subunits on the dimer/tetramer to form mPTP together (20). It has been reported that the inhibition of the β subunit on the c-ring can be relieved even without Ca2+ overload. Therefore, the pore formed in the c-ring hypothesis is not true mPTP, given that the formation of mPTP strictly requires Ca2+ for activation (21). In contrast, the status of mPTP in the other two hypotheses can be affected by Ca2+ concentration (22). The CypD-OSCP association not only induces conformational changes in F1FO ATP synthase to form mPTP, but also enhances the affinity of Ca2+ to the binding site on the β subunit, and significantly downregulates the Ca2+ threshold that makes mPTP open (23). If either hypothesis is true, it means that mPTP is located only in the MIM. This is also recognized by most researchers, and they believe that mPTP is actually a large nonselective channel formed in MIM, which is apparently different from the traditional definition of mPTP (8, 10). However, the results of Carroll et al. cast doubt on the credibility of these three hypotheses (24). This is because the pore survives and can be induced to open by Ca2+ overload in mitochondria even after the removal of different subunits of the c-ring or lateral stalk. Thus, they concluded that any form or component of F1FO ATP synthase was unlikely to form mPTP. The reason for this discrepancy may be that the purity, stability and functionality of F1FO ATP synthase may differ to varying degrees from those in vivo when isolated from mitochondria and reconstructed into extracorporeal membrane systems for independent study (19). Regardless of the specific location of mPTP, the excessive activation of the CypD-mPTP axis always triggers the permeability of MIM and MOM sequentially and ultimately initiates the cascade of cell death signals.
Additionally, the cell fate regulation by CypD is controversial. It has been suggested that CypD is a signalling molecule that promotes cell survival. This is because the overexpression of CypD delays the occurrence of mitochondrial membrane potential (MMP) collapse in HEK293 and B50 cells induced by oxidative stress and astrosporin, preserves the integrity of the mitochondrial membrane and promotes cell survival. This difference may be caused by the fact that PPIase activity is required for the protective effect of CypD, while the CypD-mediated mPTP opening is independent of PPIase activity. Thus, CypD may maintain physiological mitochondrial permeability and reverse the fate of apoptosis by binding to its binding partners other than ANT or F1FO ATP synthase. The specific identity and regulatory mechanisms of the binding partners involved in CypD-mediated protection need to be further explored.
Regulation of Mitochondrial Energy Metabolism
It should be noted that F1FO ATP synthase is also a key site for catalysing ATP production from ADP and inorganic phosphate (Pi). The conformation of the catalytic site on the β subunit can be altered when the protons generated by electron transport flows through the c-ring to drive ATP synthesis. The CypD-OSCP association not only induces the formation of mPTP, but also blocks the driving effect of the c-ring on F1 and interferes with ATP metabolism (8, 25, 26). To improve energy metabolism efficiency, F1FO ATP synthase also combines with ANT and PiC to assemble a bioactive unit with a molecular weight of at least 700,000, called the ATP synthasome (27, 28). Compared with wild-type mice, the formation of the ATP synthasome was enhanced in the heart mitochondria of CypD-/- mice, as well as in the brain and liver. In tissues with higher energy requirements (such as the heart and brain), F1FO ATP synthase is more oligomerized, and CypD expression is lower (8). This is because CypD promotes the disassembly of the ATP synthasome and reduction to monomer or dimer forms, which is beneficial for mPTP opening. Therefore, no matter what the specific molecular nature of mPTP is, the regulatory role of CypD on mPTP opening cannot be overcome. The influences of the CypD-mPTP axis on mitochondrial permeability and energy metabolism modulate and promote each other, jointly determining cells fate.
CypD activity is regulated by posttranslational modifications (PTMs) on multiple specific residues (29–36). The same PTM at different residues or different PTMs at the same residue may have different effects on CypD activity, translocation and its ability to regulate mPTP opening (Figure 2). Additionally, CypD can bind to multiple direct or indirect binding partners in the mitochondrial matrix to activate or inhibit the CypD-mPTP axis, as described in the review by Porter et al. (7) (Figure 3).
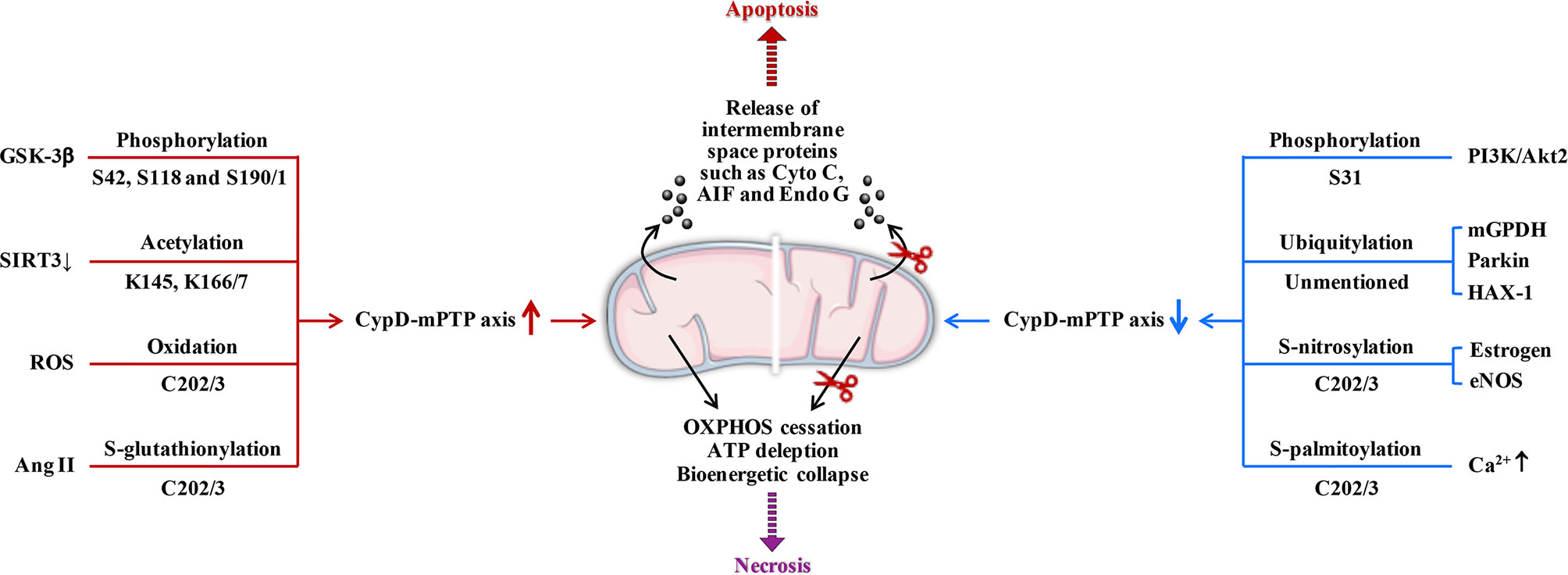
Figure 2 Posttranslational modifications of CypD. Overall, phosphorylation (S42 and S191), S-acetylation, oxidation, and S-glutathionylation (several sites) can upregulate CypD activity and sensitize mPTP to induce cell death. In contrast, phosphorylation (S31), ubiquitination, S-nitrosylation, S-palmitoylation, and S-glutathionylation (other sites) may downregulate CypD activity and provide cellular protection by desensitizing mPTP. S190/1, K166/7, and C202/3 represent homologous serine, lysine, and cysteine residues in the mouse/human form of CypD, respectively. GSK-3β, glycogen synthase kinase-3β; SIRT3, sirtuin3; ROS, reactive oxygen species; Ang II, angiotensin II; PI3K, phosphatidylinositol 3-hydroxy kinase; Akt2, Ser/Thr kinase; mGPDH, mitochondrial glycerol 3-phosphate dehydrogenase; HAX-1, haematopoietic-substrate-1 associated protein X-1; eNOS, endothelial nitric oxide synthase; AIF, apoptosis inducing factor; Endo G, endonuclease G; ↑, upregulation; ↓, downregulation.
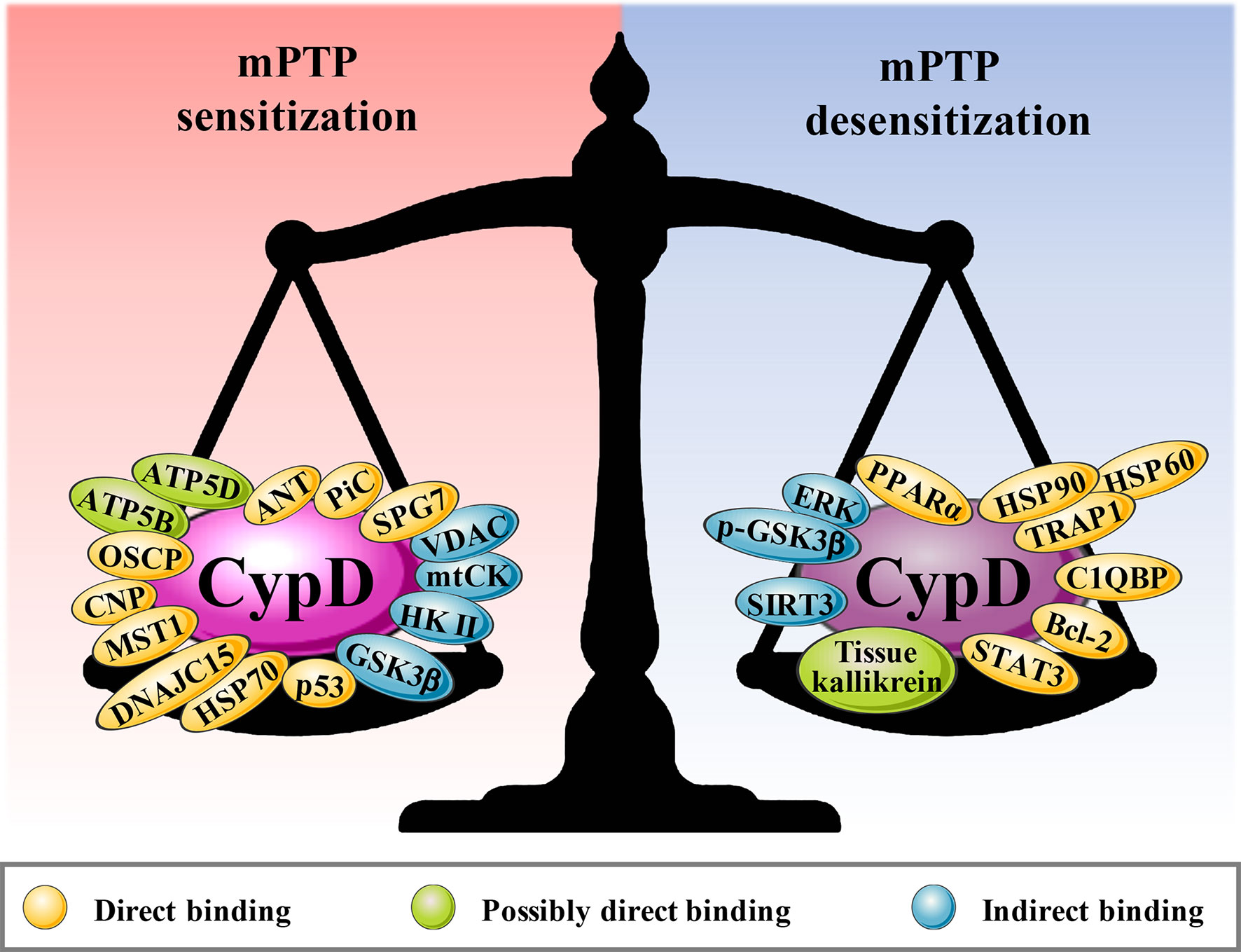
Figure 3 The binding partners of CypD. Red areas are the binding partners that sensitize mPTP, and blue areas are the binding partners that desensitize mPTP.
Positive effect of CypD on Tumor Progression
Promoting Tumorigenesis
The oncogene Ras was reported to enhance CypD expression through the Raf-1/MEK/ERK pathway (37). Upregulated expression of CypD could antagonize the inhibitory effect of the p53-p21 pathway on tumor cell growth and induce Ras-mediated tumorigenesis. The study also found that the oncogenic activity of CypD is p53 dependent. Inhibition or knockout of CypD can effectively prevent Ras-dependent lung cancer and Erbb2-mediated breast cancer formation.
Maintaining Aerobic Glycolysis
Different from normal cells, tumor cells still have strong glycolysis activity and high acid metabolites even under the condition of adequate oxygen, which is known as the Warburg effect (38). The metabolic transition from OXPHOS to aerobic glycolysis reduces the dependence of tumor cells on oxygen availability and is beneficial to the survival and invasion of tumor cells in the hypoxic microenvironment. The enhancement of glycolytic activity in tumor cells is closely related to irreversible damage to OXPHOS. The interaction between CypD and different components of the ATP synthasome can interfere with the progression of OXPHOS in tumor cells to a certain extent but does not cause ATP depletion. The initiation of aerobic glycolysis in tumor cells requires ATP produced by OXPHOS as the substrate, which is different from traditional anaerobic glycolysis (39). Hexokinase (HK), as the first rate-limiting enzyme of glycolysis, consists of four subtypes. It is worth noting that HK II is widely expressed in embryonic tissues and invasive tumors but is rarely expressed in normal tissues and only sparingly expressed in insulin-sensitive tissues (fat, muscle, heart) (40, 41). HK II can translocate from the cytoplasm to the MOM and bind with VDAC, which is called mitochondrial HK II (mtHK II) (42). mtHK II limits the movement of the N-terminal spiral of VDAC and keeps VDAC open. This ensures that ATP produced within the mitochondrial matrix is preferentially and continuously transported to mtHK II for aerobic glycolysis (43).
It has been reported that CypD overexpression can promote the formation of mtHK II, while inhibition or knockdown of CypD inhibits the level of HK II binding to the MOM (42, 44). Upregulated sirtuin 3 (SIRT3) was confirmed to inhibit the acetylation degree and PPIase activity of CypD and lead to the separation of CypD from ANT in breast cancer cells treated with oroxylin A (45). Simultaneously, the deacetylation of CypD triggered the dissociation of mtHK II from the MOM and inhibited the aerobic glycolysis activity of breast cancer cells. Similarly, ganoderic acid D (GAD) inhibited mtHK II formation and energy reprogramming in colon cancer cells by inducing SIRT3-mediated CypD deacetylation (46). Inhibition of SIRT3 activity effectively reversed mitochondrial cytotoxicity and reduced aerobic glycolysis induced by the abovementioned antitumor drugs. Intriguingly, overexpression of mutant CypD did not significantly change the level of mtHK II impaired by oroxylin A. Moreover, glioma cells expressing PPIase-deficient CypD were not effective against Bax-induced apoptosis compared to cells overexpressing wild-type CypD (47). Therefore, it is reasonable to assume that CypD relies on its PPIase activity to stabilize the association of ANT-VDAC-mtHK II.
An important question is why CypD-ANT binding is inconsistent in its dependence on PPIase activity in tumor and nontumor cells, which has been attributed to different ANT subtypes. There are four different subtypes of human ANT, including ANT1, ANT2, ANT3, and ANT4. Compared with the other three, ANT2 is widely expressed in rapidly growing cells, and only ANT2 can be highly induced in tumor cells (48, 49). The ANT2-VDAC-mtHK II association in tumor cells has been reported to play an important role in the maintenance of aerobic glycolysis and other carcinogenic effects (50, 51). Since ATP is continuously exported to the cytoplasm, it is not conducive to the maintenance of the essential intramitochondrial enzymatic pathways in tumor cells. Thus, mtHK II can reversely catalyse ATP production from G6P when the cytoplasmic ATP concentration reaches a certain threshold. Only ANT2 precisely allows ATP to be transported inwards into the mitochondrial matrix to maintain tumor cell survival and apoptotic resistance (52) (Figure 4). However, other ANTs, mainly located in nontumor cells, can continuously export mitochondrial ATP to the cytoplasm, which is detrimental to tumor cells with mitochondrial dysfunction. Given that the major ANT subtypes are different in tumor cells and nontumor cells, there may be discrepancies in the mechanisms that mediate CypD-ANT binding and the subsequent downstream effects.
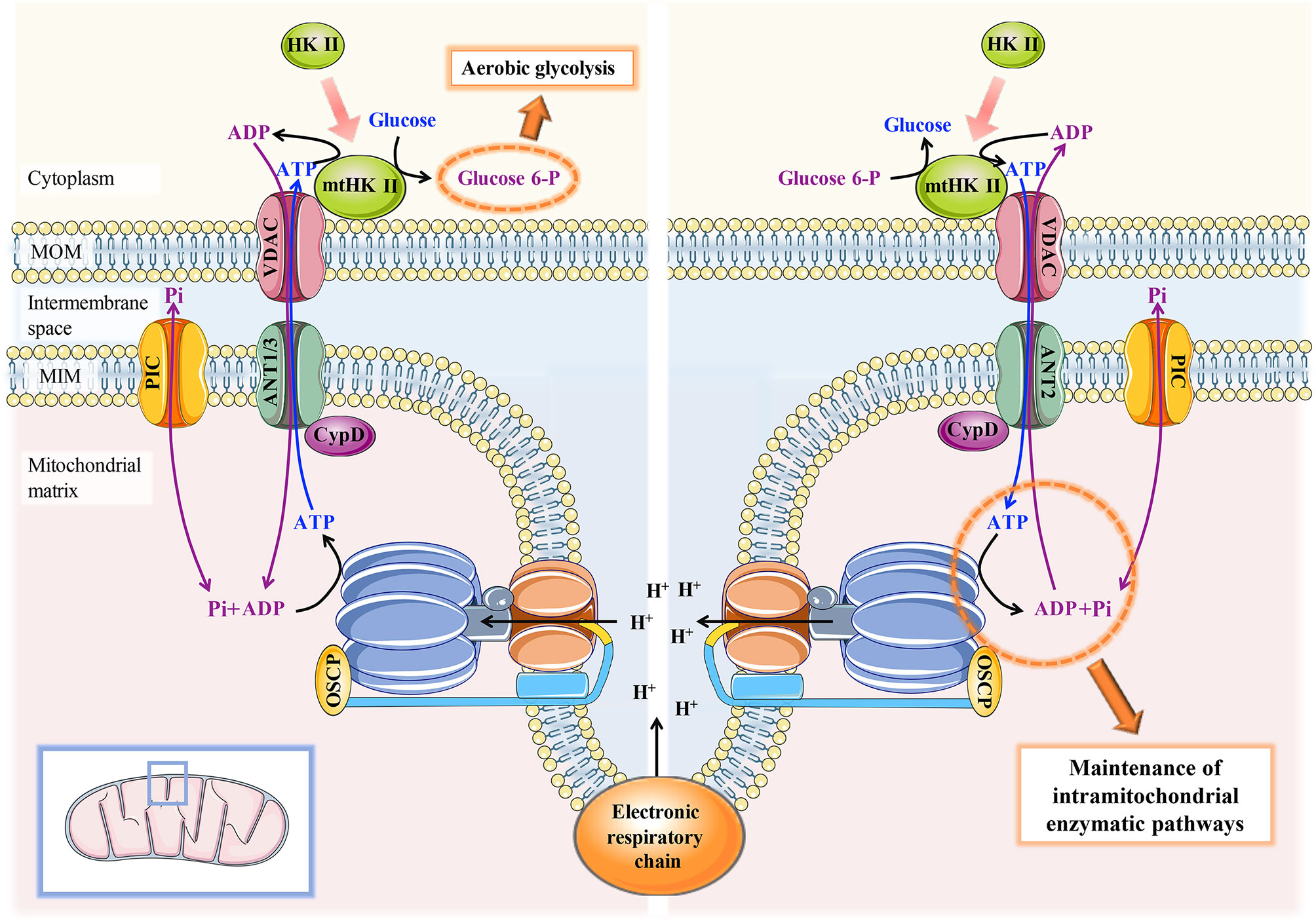
Figure 4 Schematic diagram of the mechanism of the CypD-ANT-VDAC-mtHK II association in the maintenance of aerobic glycolysis and mitochondrial function in tumor cells. Left: CypD-ANT1/3 association helps stabilize the binding of mtHKII to VDAC and thereby moderately activates mPTP, which ensures that ATP generated by OXPHOS can be preferentially and continuously transported to mtHK II for aerobic glycolysis. Right: conversely, mtHK II can reverse catalyse ATP production from G6P to maintain the essential intramitochondrial enzymatic pathways in tumor cells when the ATP concentration in the cytoplasm drops to a certain threshold. The maintenance of the latter process requires the CypD-ANT2-VDAC-mtHKII association.
Inhibiting Tumor Cell Apoptosis
Inhibition of tumor cell apoptosis is an important prerequisite and marker of tumorigenesis. CypD overexpression has been reported to attenuate apoptosis. As early as 2004, Schubert A et al. found that CypD was significantly upregulated in various tumors of reproductive tissues (breast, uterus and ovary) and inhibited apoptosis of tumor cells (11). This study found that CypD may attenuate the activation of mPTP triggered by other proapoptotic stimuli and the release of pro-apoptotic factors in a manner independent of PPIase activity, thus promoting tumor cell survival. Unfortunately, the protective mechanism of CypD on tumor cells was not specifically elucidated in this study. The formation of mtHK II is known to activate metabolic pathways such as aerobic glycolysis and pentose phosphate pathways to produce sufficient ATP and metabolic intermediates (such as NADPH) to rapidly provide energy and biosynthetic substrates for tumor cell growth and invasion. Furthermore, mtHK II can competitively bind VDAC with the proapoptotic factor Bax to inhibit the translocation of Bax to mitochondria, thus antagonizing Bax-mediated apoptosis (53, 54). Intriguingly, Machida et al. found that mtHK II formation is necessary for CypD to inhibit apoptosis of HeLa cells (human cervical cancer cell line) and C6 cells (rat glioma cell line), while PPIase activity is essential for CypD to stabilize mtHK II (47). This is inconsistent with the results of Schubert et al., which may be attributed to different tissue sources of tumor cells or diverse upstream signalling pathways that regulate CypD. In addition, the binding of CypD and Bcl-2 enhances the limiting effect of Bcl-2 on cytochrome C (Cyto C) release and improves the antiapoptotic effect of various tumor cell lines (human osteosarcoma cell line Saos2 and human acute leukemia cell line HL60) (55). Overexpression or knockdown of CypD can upregulate or downregulate the resistance of tumor cells to apoptotic stimulation, respectively (56). Coincidentally, downregulated miRNA-27b-3p in oral mucosal basal cells of patients with oral lichen planus (a typical precancerous lesion) also enhances the interaction of CypD-Bcl2, inhibits the release of proapoptotic factors and accelerates deterioration (57).
As previously mentioned, the association of CypD-ANT2-VDAC1-mtHK II contributes to mPTP opening to ensure either continuous ATP output for mtHK II utilization or ATP input for the maintenance of intramitochondrial enzymatic pathways. How do tumor cells avoid mPTP-mediated cell apoptosis or necrosis while maintaining the essential aerobic glycolysis through the CypD-mPTP axis? We hypothesize that the degree of mPTP opening for maintaining ATP flow is confined, and that cytochrome C and apoptosis-inducing factor accompanying ATP output are not sufficient to initiate the caspase cascade-mediated mitochondrial apoptosis pathway. Indeed, although some degree of mitochondrial swelling and decreased MMP has been reported in LM7 and 143B osteosarcoma cells, cytochrome C levels in the cytoplasm do not exceed those in noncancerous hFOB cells (44). In addition, although mPTP opening can interfere with OXPHOS, tumor cells mainly rely on aerobic glycolysis to produce ATP. Aerobic glycolysis can fully improve the utilization rate of glucose, rapidly supply energy to tumor cells, and improve mitochondrial dysfunction by reversing the delivery of ATP to mitochondria. Thus, tumor cells maintain the essential MMP and ATP levels in a virtuous cycle to avoid cell necrosis induced by ATP depletion (Figure 4).
Inducing Tumor Metastasis and Invasion
p53 is a typical tumor suppressor gene. Different forms of p53 mutations can affect the tumor suppressor or transcriptional activity of p53 and may even promote tumor progression. TP53 truncating mutations are common in human tumors, especially TP53 exon-6 truncating mutations. p53 exon-6 truncating mutants are similar to the naturally occurring selective p53 splice variant (p53-psi), which lacks transcriptional activity and responds to DNA damage. However, they can translocate into mitochondria, bind to CypD and activate mPTP opening (58). Traditionally, mPTP opening suppresses mitochondrial function and triggers apoptosis. Interestingly, however, the CypD-mPTP axis activated by p53 exon-6 truncating mutants or p53-psi could promote tumorigenesis and present malignant features such as downregulation of E-cadherin expression. Likewise, p53ψ derived from the use of alternative 3’ splice site in p53 intron 6 could also enhance the motility and invasive capacity of multiple lung cancer and breast cancer cell lines by activating the CypD-mPTP axis (59). Elevated ROS levels provoked by mPTP overopening played a pivotal role in p53ψ-induced epithelial-mesenchymal transformation (EMT).
Considering that the vast majority of p53 mutations that occur in human cancers are missense, the role of p53 missense mutants in regulating CypD is of interest to us. So far, only one article has mentioned both p53 missense mutants and CypD (60). It was found that mitochondrial wild-type p53 protein severely damaged the integrity of MOM and MIM by inducing the oligomerization of Bax, Bak and VDAC and the endogenous complex formation with CypD, respectively, thereby promoting the release of apoptotic factors. However, tumor-derived p53 missense mutants lost the ability to activate the Bax/Bak lipid pore. Unfortunately, the association between p53 missense mutants and CypD has not been further studied. Therefore, do missense mutations of p53 affect the association with CypD? If so, what are the downstream effects of this association? Are all or only some forms of p53 missense mutations associated with CypD? These seem to be very promising questions.
Facilitating Tumor Resistance
The phosphatidylinositol-3 kinase (PI3K) pathway is a convergence point that regulates cell proliferation, survival and bioenergetics and is often used in tumor therapy. It has been reported that Akt2 abnormally activated by PI3K small molecule inhibitors (PI3Ki) in glioblastoma cells can translocate to mitochondria and subsequently phosphorylate CypD at S31 (61). Phosphorylated CypD supported mitochondrial bioenergetics, inhibited tumor cell apoptosis, and thereby mediated resistance to PI3K therapy. When combined with gamitrinib (a mitochondrial homeostasis inhibitor), PI3Ki effectively eliminated PI3K/Akt2/CypD pathway-mediated tumor resistance and significantly induced glioblastoma cell apoptosis.
Negative Effect of CypD on Tumor Progression
As shown in Figure 5, the role of CypD in tumor development is dual. Even for the same dimension of tumor development, CypD provoked by disparate upstream signals will exhibit bidirectional influence on tumors.
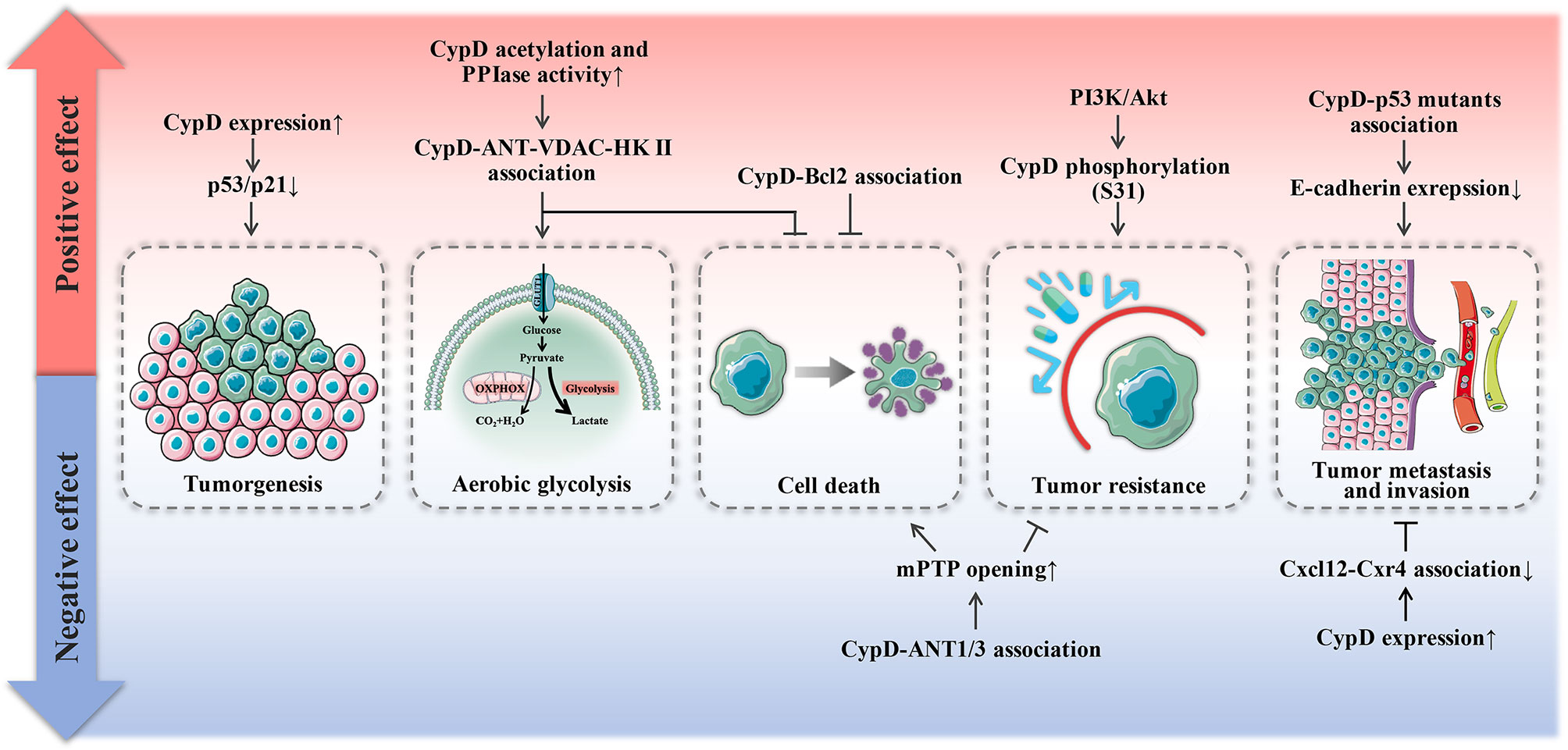
Figure 5 Dual effect of CypD on tumor progression. Red areas represent promotional mechanisms, and blue areas represent inhibitory mechanisms.
Promotion of Tumor Cell Death
As shown in Figure 6, most antitumor drugs can induce tumor cell necrosis by activating the CypD-mPTP axis (62–70). For example, icaritin could induce necrosis of colon tumor cells, but not apoptosis, which is dependent on activation of the JNK pathway (71). It was found that CypD-ANT1 association enhanced mPTP opening, resulting in increased mitochondrial depolarization and the release of lactate dehydrogenase into the cytoplasm. Indeed, CypD-ANT1 association was also involved in the therapeutic mechanisms of several other antitumor drugs (72–74). Furthermore, both bishonokiol A and 1, 2-Diarachidonoyl-Sn-glycero-3-phosphoethanolamine (DAPE) upregulated CypD expression by activating the RIP1/RIP3/MLKL necrosis cascade, thus promoting mPTP-mediated necrosis of breast cancer and malignant pleural mesothelioma cells (75, 76). Similarly, bromocriptine could also promote the phosphorylation of CypD through the RIP3/MLKL pathway, thus inducing necrosis of prolactinoma cells (77). Moreover, CypD-p53 association has also been reported to be involved in the process of tumor cell necrosis induced by various antitumor drugs (78–83).
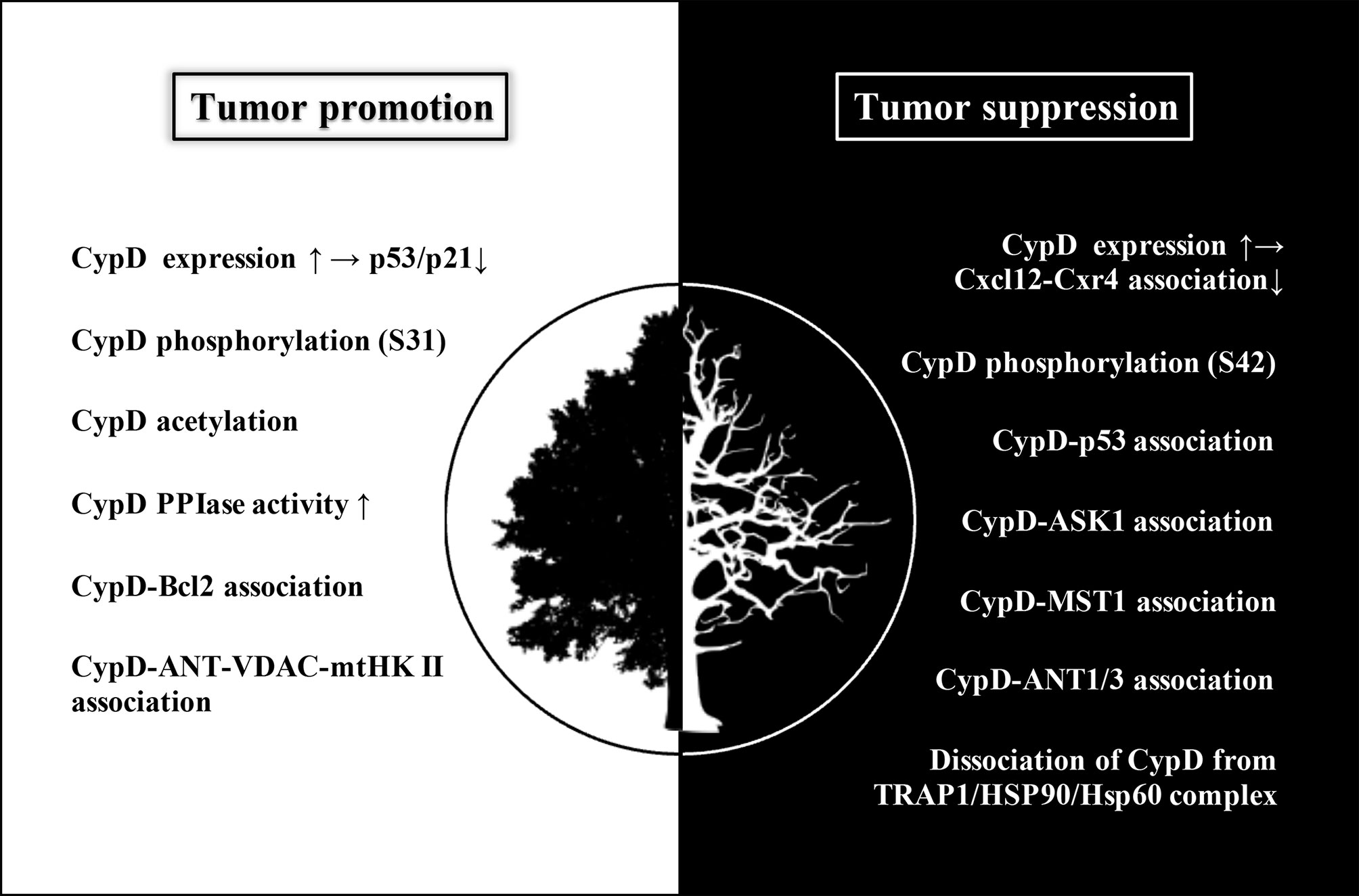
Figure 6 The white area (left) shows the modulations of CypD that promote tumor progression, mainly including inhibition of p53/p21 pathway mediated by upregulated CypD expression, CypD phosphorylation (S31), CypD acetylation, increased CypD PPIase activity, CypD-Bcl2 association and CypD-Ant-VDAC-mtHK II association. The black area (right) shows the modulations of CypD that suppress tumor progression, mainly including inhibition of Cxcl12-Cxr4 association mediated by upregulation of CypD expression, CypD phosphorylation (S42), association of CypD with several binding partners (p53, ASK1, MST1, ANT1/3), and dissociation of CypD from TRAP1/HSP90/HSP60 complex. Although increased CypD expression may occur in both tumor promotion and suppression, its downstream pathways are different, subsequently leading to two distinct cellular events. Elevated CypD expression actuates tumorigenesis by inhibiting p53/p21 pathway on the one hand, and hampers tumor metastasis and invasion by interfering Cxcl12-Cxr4 association on the other hand.
Moreover, CypD can also mediate the apoptosis process of tumor cells induced by antitumor drugs (84–93). For example, phosphorylated GSK3β is known to competitively bind ANT1 with CypD, thereby inhibiting mPTP opening. However, hirsutine dephosphorylated GSK3β in lung cancer cells through the ROCK1/PTEN/PISK/AKT pathway and activated the CypD-mPTP axis, thereby resulting in MMP decline, ATP dissipation, and caspase cascade-triggered apoptosis (94). Consistently, esculetin could activate the CypD-mPTP axis and induce apoptosis of gastric cancer cells by upregulating intracellular oxidative stress levels (69). In addition, tumor necrosis factor receptor-associated protein (TRAP1), a major member of the mitochondrial heat shock protein 90 (HSP90) family, can trap CypD in the TRAP1/HSP9/HSP60 multichaperone complex, thus limiting the translocation of CypD to the MIM. Treatment with HSP60 siRNA could promote the escape of CypD from the above complex, restore its activity and activate the excessive opening of mPTP, resulting in multiple tumor cell apoptosis (95). It should be noted that antitumor drugs do not induce just one type of cell death when treating a given tumor. For example, the enhanced association of p53-CypD-ANT1 led to both apoptosis and necrosis in non-small-cell lung cancer cells after treatment with ASP4132 (96). Inhibition or knockdown of CypD reversed cell necrosis induced by ASP4132 without affecting cell apoptosis. As mentioned above, the endogenous complex formed by wild-type p53 and CypD can participate in the destruction of MIM’s integrity. P53 is not only a typical tumor suppressor gene, but also regarded as a stress sensor that can sense multiple insults. For example, in response to oxidative stress during ischemia-reperfusion injury, p53 translocated into the mitochondrial matrix and triggered brain tissue necrosis by forming a robust complex with CypD (97).
In addition to the above two common forms of tumor cell death, CypD-induced autophagic cell death has been observed in liver cancer cells treated with andrographolide (98). This is because the activation of the CypD-mPTP axis by andrographolide leads to an increase in LC3 II and autophagosome in hepatoma carcinoma cells. Activation of CypD-mPTP axis is a common mechanism of tumor cell death induced by most antitumor drugs which is primarily achieved by upregulating the CypD-ANT1/3 association. As previously described, ANT1/3 can continuously export ATP into the cytoplasm, causing bioenergetic collapse of dysfunctional mitochondria in tumor cells and subsequently triggering cell death (99).
Attenuating Tumor Metastasis and Invasion
In contrast, Tavecchio et al. found that CypD expression was reduced or even lost in a variety of tumor cell lines (human glioma cell line LN229, human breast cancer cell line MCF-7 and human pancreatic cancer cell line MiaPACA), which could obviously activate interorganelle signalling and the pleiotropic inflammatory mediator STAT3 (100). On the one hand, activated STAT3 enhanced tumor cell proliferation by accelerating entry into S-phase. On the other hand, the Cxcl12-Cxcr4 association mediated chemokine-dependent autocrine/paracrine cell motility after CypD ablation, which was manifested as enhanced tumor cell migration and invasion. This finding indicates that CypD can inhibit tumor metastasis to some extent.
Suppressing Tumor Resistance
Wu et al. found that mortalin reduced mitochondrial permeability by blocking CypD-ANT3 binding and promoted the proliferation of human B-RafV600E melanoma cells and their resistance to vemurafenib (101, 102). Moreover, CypD expression and its association with mammalian sterile 20-like kinase 1 (MST1) were significantly downregulated, mediating gemcitabine resistance in pancreatic tumor cell lines (103). These data suggest that the association of CypD with its binding partners may be involved in the chemoresistance of some tumors.
Conclusions and Perspectives
CypD is recognized as a gate that regulates cell fate and energy metabolism by many mechanisms. It has been demonstrated that CypD can affect tumor progression in multiple ways and seems to play a dual role in tumor cell fate (Table 1 and 2). CypD-mediated transient opening of mPTP helps regulate calcium homeostasis and attenuate ROS accumulation in mitochondria to maintain mitochondrial activity. Moderate levels of mPTP opening continuously provide substrates for aerobic glycolysis and essential intramitochondrial enzymatic pathways in tumor cells. The modestly activated CypD-mPTP axis can also promote the survival, metastasis and invasion of tumor cells. However, excessive mPTP opening inhibits the bioenergetics and malignant characteristics of tumor cells and triggers various forms of tumor cell death. This regulatory feature of the CypD-mPTP axis has been used to develop a variety of antitumor drugs. Therefore, the complex regulatory mechanisms of CypD make it a promoter of tumor progression, but also a weapon by which antitumour drugs may kill tumor cells (Figure 6). Rational utilization of the biological functions of CypD may make CypD a hot target for the treatment of various diseases, including tumors, in the future.
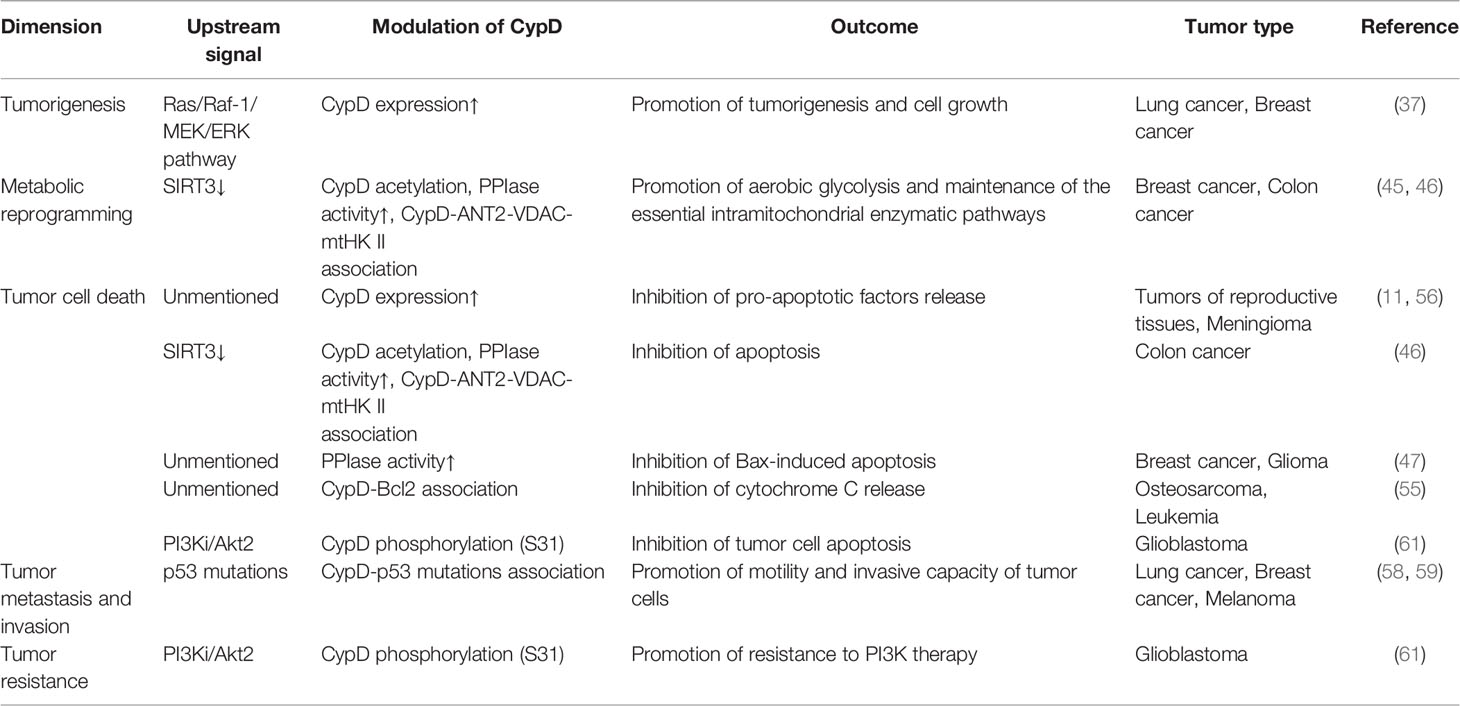
Table 1 Positive effect of CypD on tumor progression.
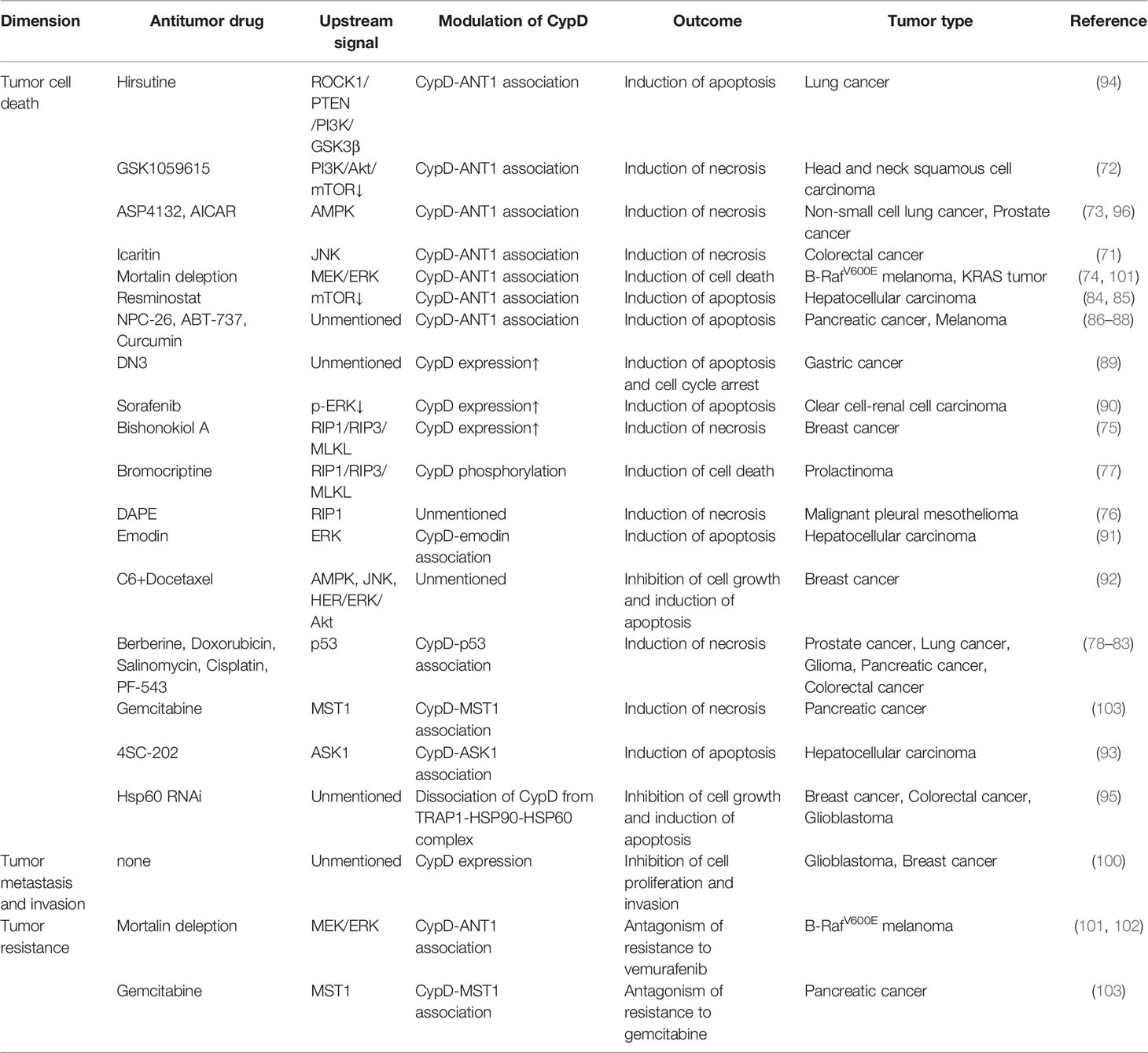
Table 2 Negative effect of CypD on tumor progression.
Author Contributions
LZ is responsible for drafting of the paper and drawing the figures. BZ is responsible for the conception and review of the paper. YL, RZ, BH and WW are responsible for the writing, review and revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 82173371), Tai Shan Young Scholar Foundation of Shandong Province (No.tsqn201909192) and Natural Science Foundation of Shandong Province (No. ZR2021QH294).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The work was supported by the members of Zhang Lab, and I would like to show great gratitude to them all.
References
1. Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of Cyclophilin With the Mitochondrial Inner Membrane and Regulation of the Permeability Transition Pore, and Cyclosporin a-Sensitive Channel. J Biol Chem (1996) 271(4):2185–92. doi: 10.1074/jbc.271.4.2185
2. Scorrano L, Nicolli A, Basso E, Petronilli V, Bernardi P. Two Modes of Activation of the Permeability Transition Pore: The Role of Mitochondrial Cyclophilin. Mol Cell Biochem (1997) 174(1-2):181–4. doi: 10.1023/A:1006887921810
3. Teodoro JS, Varela AT, Duarte FV, Gomes AP, Palmeira CM, Rolo AP. Indirubin and Nad(+) Prevent Mitochondrial Ischaemia/Reperfusion Damage in Fatty Livers. Eur J Clin Invest (2018) 48(6):e12932. doi: 10.1111/eci.12932
4. Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, et al. Regulation of the Mptp by Sirt3-Mediated Deacetylation of Cypd at Lysine 166 Suppresses Age-Related Cardiac Hypertrophy. Aging (Albany NY) (2010) 2(12):914–23. doi: 10.18632/aging.100252
5. Sun F, Si Y, Bao H, Xu Y, Pan X, Zeng L, et al. Regulation of Sirtuin 3-Mediated Deacetylation of Cyclophilin D Attenuated Cognitive Dysfunction Induced by Sepsis-Associated Encephalopathy in Mice. Cell Mol Neurobiol (2017) 37(8):1457–64. doi: 10.1007/s10571-017-0476-2
6. Naguib M, Abou Elfotouh M, Wifi MN. Elevated Serum Cyclophilin D Level Is Associated With Nonalcoholic Fatty Liver Disease and Higher Fibrosis Scores in Patients With Diabetes Mellitus. Int J Gen Med (2021) 14:4665–75. doi: 10.2147/ijgm.S322986
7. Porter GA Jr., Beutner G, Cyclophilin D. Somehow a Master Regulator of Mitochondrial Function. Biomolecules (2018) 8(4):176. doi: 10.3390/biom8040176
8. Beutner G, Alanzalon RE, Porter GA Jr. Cyclophilin D Regulates the Dynamic Assembly of Mitochondrial Atp Synthase Into Synthasomes. Sci Rep (2017) 7(1):14488. doi: 10.1038/s41598-017-14795-x
9. Cui Y, Pan M, Ma J, Song X, Cao W, Zhang P. Recent Progress in the Use of Mitochondrial Membrane Permeability Transition Pore in Mitochondrial Dysfunction-Related Disease Therapies. Mol Cell Biochem (2021) 476(1):493–506. doi: 10.1007/s11010-020-03926-0
10. Bonora M, Patergnani S, Ramaccini D, Morciano G, Pedriali G, Kahsay AE, et al. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules (2020) 10(7):998. doi: 10.3390/biom10070998
11. Schubert A, Grimm S, Cyclophilin D. a Component of the Permeability Transition-Pore, Is an Apoptosis Repressor. Cancer Res (2004) 64(1):85–93. doi: 10.1158/0008-5472.can-03-0476
12. Jia K, Du H. Mitochondrial Permeability Transition: A Pore Intertwines Brain Aging and Alzheimer's Disease. Cells (2021) 10(3):649. doi: 10.3390/cells10030649
13. Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, et al. The Adp/Atp Translocator Is Not Essential for the Mitochondrial Permeability Transition Pore. Nature (2004) 427(6973):461–5. doi: 10.1038/nature02229
14. Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential Role of Voltage-Dependent Anion Channel in Various Forms of Apoptosis in Mammalian Cells. J Cell Biol (2001) 152(2):237–50. doi: 10.1083/jcb.152.2.237
15. Galluzzi L, Kroemer G. Mitochondrial Apoptosis Without Vdac. Nat Cell Biol (2007) 9(5):487–9. doi: 10.1038/ncb0507-487
16. Carraro M, Carrer A, Urbani A, Bernardi P. Molecular Nature and Regulation of the Mitochondrial Permeability Transition Pore(S), Drug Target(S) in Cardioprotection. J Mol Cell Cardiol (2020) 144:76–86. doi: 10.1016/j.yjmcc.2020.05.014
17. Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An Uncoupling Channel Within the C-Subunit Ring of the F1fo Atp Synthase Is the Mitochondrial Permeability Transition Pore. Proc Natl Acad Sci USA (2014) 111(29):10580–5. doi: 10.1073/pnas.1401591111
18. Mnatsakanyan N, Llaguno MC, Yang Y, Yan Y, Weber J, Sigworth FJ, et al. A Mitochondrial Megachannel Resides in Monomeric F(1)F(O) Atp Synthase. Nat Commun (2019) 10(1):5823. doi: 10.1038/s41467-019-13766-2
19. Gerle C. Mitochondrial F-Atp Synthase as the Permeability Transition Pore. Pharmacol Res (2020) 160:105081. doi: 10.1016/j.phrs.2020.105081
20. Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of Mitochondrial Atp Synthase Form the Permeability Transition Pore. Proc Natl Acad Sci USA (2013) 110(15):5887–92. doi: 10.1073/pnas.1217823110
21. Giorgio V, Guo L, Bassot C, Petronilli V, Bernardi P. Calcium and Regulation of the Mitochondrial Permeability Transition. Cell calcium (2018) 70:56–63. doi: 10.1016/j.ceca.2017.05.004
22. He J, Carroll J, Ding S, Fearnley IM, Walker JE. Permeability Transition in Human Mitochondria Persists in the Absence of Peripheral Stalk Subunits of Atp Synthase. Proc Natl Acad Sci USA (2017) 114(34):9086–91. doi: 10.1073/pnas.1711201114
23. Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, et al. Cyclophilin D Is a Component of Mitochondrial Permeability Transition and Mediates Neuronal Cell Death After Focal Cerebral Ischemia. Proc Natl Acad Sci U States America (2005) 102(34):12005–10. doi: 10.1073/pnas.0505294102
24. Carroll J, He J, Ding S, Fearnley IM, Walker JE. Persistence of the Permeability Transition Pore in Human Mitochondria Devoid of an Assembled Atp Synthase. Proc Natl Acad Sci USA (2019) 116(26):12816–21. doi: 10.1073/pnas.1904005116
25. Gauba E, Chen H, Guo L, Du H. Cyclophilin D Deficiency Attenuates Mitochondrial F1fo Atp Synthase Dysfunction Via Oscp in Alzheimer's Disease. Neurobiol Dis (2019) 121:138–47. doi: 10.1016/j.nbd.2018.09.020
26. Gauba E, Guo L, Du H. Cyclophilin D Promotes Brain Mitochondrial F1fo Atp Synthase Dysfunction in Aging Mice. J Alzheimer's Dis (2017) 55(4):1351–62. doi: 10.3233/jad-160822
27. Ko YH, Delannoy M, Hullihen J, Chiu W, Pedersen PL. Mitochondrial Atp Synthasome. Cristae-Enriched Membranes and a Multiwell Detergent Screening Assay Yield Dispersed Single Complexes Containing the Atp Synthase and Carriers for Pi and Adp/Atp. J Biol Chem (2003) 278(14):12305–9. doi: 10.1074/jbc.C200703200
28. Chen C, Ko Y, Delannoy M, Ludtke SJ, Chiu W, Pedersen PL. Mitochondrial Atp Synthasome: Three-Dimensional Structure by Electron Microscopy of the Atp Synthase in Complex Formation With Carriers for Pi and Adp/Atp. J Biol Chem (2004) 279(30):31761–8. doi: 10.1074/jbc.M401353200
29. Li A, Li X, Yi J, Ma J, Zhou J. Butyrate Feeding Reverses Cypd-Related Mitoflash Phenotypes in Mouse Myofibers. Int J Mol Sci (2021) 22(14):7412. doi: 10.3390/ijms22147412
30. Hurst S, Gonnot F, Dia M, Crola Da Silva C, Gomez L, Sheu SS. Phosphorylation of Cyclophilin D at Serine 191 Regulates Mitochondrial Permeability Transition Pore Opening and Cell Death After Ischemia-Reperfusion. Cell Death Dis (2020) 11(8):661. doi: 10.1038/s41419-020-02864-5
31. Parks RJ, Menazza S, Holmström KM, Amanakis G, Fergusson M, Ma H, et al. Cyclophilin D-Mediated Regulation of the Permeability Transition Pore Is Altered in Mice Lacking the Mitochondrial Calcium Uniporter. Cardiovasc Res (2019) 115(2):385–94. doi: 10.1093/cvr/cvy218
32. Wang NN, Xu HH, Zhou W, Yang HX, Wang J, Ma ZC, et al. Aconitine Attenuates Mitochondrial Dysfunction of Cardiomyocytes Via Promoting Deacetylation of Cyclophilin-D Mediated by Sirtuin-3. J Ethnopharmacol (2021) 270:113765. doi: 10.1016/j.jep.2020.113765
33. Wang X, Mao J, Zhou X, Li Q, Gao L, Zhao J. Thyroid Stimulating Hormone Triggers Hepatic Mitochondrial Stress Through Cyclophilin D Acetylation. Oxid Med Cell Longevity (2020) 2020:1249630. doi: 10.1155/2020/1249630
34. Sun T, Ding W, Xu T, Ao X, Yu T, Li M, et al. Parkin Regulates Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury by Targeting Cyclophilin-D. Antioxidants Redox Signaling (2019) 31(16):1177–93. doi: 10.1089/ars.2019.7734
35. Bibli SI, Papapetropoulos A, Iliodromitis EK, Daiber A, Randriamboavonjy V, Steven S, et al. Nitroglycerine Limits Infarct Size Through S-Nitrosation of Cyclophilin D: A Novel Mechanism for an Old Drug. Cardiovasc Res (2019) 115(3):625–36. doi: 10.1093/cvr/cvy222
36. Amanakis G, Sun J, Fergusson MM, McGinty S, Liu C, Molkentin JD, et al. Cysteine 202 of Cyclophilin D Is a Site of Multiple Post-Translational Modifications and Plays a Role in Cardioprotection. Cardiovasc Res (2021) 117(1):212–23. doi: 10.1093/cvr/cvaa053
37. Bigi A, Beltrami E, Trinei M, Stendardo M, Pelicci PG, Giorgio M. Cyclophilin D Counteracts P53-Mediated Growth Arrest and Promotes Ras Tumorigenesis. Oncogene (2016) 35(39):5132–43. doi: 10.1038/onc.2016.42
38. Vaupel P, Schmidberger H, Mayer A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. Int J Radiat Biol (2019) 95(7):912–9. doi: 10.1080/09553002.2019.1589653
39. Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, et al. Increased Demand for Nad(+) Relative to Atp Drives Aerobic Glycolysis. Mol Cell (2021) 81(4):691–707.e6. doi: 10.1016/j.molcel.2020.12.012
40. Wilson JE. Isozymes of Mammalian Hexokinase: Structure, Subcellular Localization and Metabolic Function. J Exp Biol (2003) 206(Pt 12):2049–57. doi: 10.1242/jeb.00241
41. Roberts DJ, Miyamoto S. Hexokinase Ii Integrates Energy Metabolism and Cellular Protection: Akting on Mitochondria and Torcing to Autophagy. Cell Death Differ (2015) 22(2):364. doi: 10.1038/cdd.2014.208
42. Nederlof R, van den Elshout MAM, Koeman A, Uthman L, Koning I, Eerbeek O, et al. Cyclophilin D Ablation Is Associated With Increased End-Ischemic Mitochondrial Hexokinase Activity. Sci Rep (2017) 7(1):12749. doi: 10.1038/s41598-017-13096-7
43. Shangguan X, He J, Ma Z, Zhang W, Ji Y, Shen K, et al. Sumoylation Controls the Binding of Hexokinase 2 to Mitochondria and Protects Against Prostate Cancer Tumorigenesis. Nat Commun (2021) 12(1):1812. doi: 10.1038/s41467-021-22163-7
44. Giang AH, Raymond T, Brookes P, de Mesy Bentley K, Schwarz E, O'Keefe R, et al. Mitochondrial Dysfunction and Permeability Transition in Osteosarcoma Cells Showing the Warburg Effect. J Biol Chem (2013) 288(46):33303–11. doi: 10.1074/jbc.M113.507129
45. Wei L, Zhou Y, Dai Q, Qiao C, Zhao L, Hui H, et al. Oroxylin a Induces Dissociation of Hexokinase Ii From the Mitochondria and Inhibits Glycolysis by Sirt3-Mediated Deacetylation of Cyclophilin D in Breast Carcinoma. Cell Death Dis (2013) 4(4):e601. doi: 10.1038/cddis.2013.131
46. Liu Z, Li L, Xue B. Effect of Ganoderic Acid D on Colon Cancer Warburg Effect: Role of Sirt3/Cyclophilin D. Eur J Pharmacol (2018) 824:72–7. doi: 10.1016/j.ejphar.2018.01.026
47. Machida K, Ohta Y, Osada H. Suppression of Apoptosis by Cyclophilin D Via Stabilization of Hexokinase Ii Mitochondrial Binding in Cancer Cells. J Biol Chem (2006) 281(20):14314–20. doi: 10.1074/jbc.M513297200
48. Hubackova S, Davidova E, Rohlenova K, Stursa J, Werner L, Andera L, et al. Selective Elimination of Senescent Cells by Mitochondrial Targeting Is Regulated by Ant2. Cell Death Differ (2019) 26(2):276–90. doi: 10.1038/s41418-018-0118-3
49. Li T, Li Y, Liu T, Hu B, Li J, Liu C, et al. Mitochondrial Pak6 Inhibits Prostate Cancer Cell Apoptosis Via the Pak6-Sirt4-Ant2 Complex. Theranostics (2020) 10(6):2571–86. doi: 10.7150/thno.42874
50. Chevrollier A, Loiseau D, Chabi B, Renier G, Douay O, Malthièry Y, et al. Ant2 Isoform Required for Cancer Cell Glycolysis. J bioenergetics biomembranes (2005) 37(5):307–16. doi: 10.1007/s10863-005-8642-5
51. Shoshan-Barmatz V, Krelin Y, Shteinfer-Kuzmine A. Vdac1 Functions in Ca(2+) Homeostasis and Cell Life and Death in Health and Disease. Cell calcium (2018) 69:81–100. doi: 10.1016/j.ceca.2017.06.007
52. Zhao L, Tang M, Bode AM, Liao W, Cao Y. Ants and Cancer: Emerging Pathogenesis, Mechanisms, and Perspectives. Biochim Biophys Acta Rev Cancer (2021) 1875(1):188485. doi: 10.1016/j.bbcan.2020.188485
53. Pastorino JG, Shulga N, Hoek JB. Mitochondrial Binding of Hexokinase Ii Inhibits Bax-Induced Cytochrome C Release and Apoptosis. J Biol Chem (2002) 277(9):7610–8. doi: 10.1074/jbc.M109950200
54. Cai J, Yi M, Tan Y, Li X, Li G, Zeng Z, et al. Natural Product Triptolide Induces Gsdme-Mediated Pyroptosis in Head and Neck Cancer Through Suppressing Mitochondrial Hexokinase-Iι. J Exp Clin Cancer Res (2021) 40(1):190. doi: 10.1186/s13046-021-01995-7
55. Eliseev RA, Malecki J, Lester T, Zhang Y, Humphrey J, Gunter TE. Cyclophilin D Interacts With Bcl2 and Exerts an Anti-Apoptotic Effect. J Biol Chem (2009) 284(15):9692–9. doi: 10.1074/jbc.M808750200
56. Talari NK, Panigrahi MK, Madigubba S, Phanithi PB. Overexpression of Aryl Hydrocarbon Receptor (Ahr) Signalling Pathway in Human Meningioma. J Neurooncol (2018) 137(2):241–8. doi: 10.1007/s11060-017-2730-3
57. Chen J, Wang Y, Du G, Zhang W, Cao T, Shi L, et al. Down-Regulation of Mirna-27b-3p Suppresses Keratinocytes Apoptosis in Oral Lichen Planus. J Cell Mol Med (2019) 23(6):4326–37. doi: 10.1111/jcmm.14324
58. Shirole NH, Pal D, Kastenhuber ER, Senturk S, Boroda J, Pisterzi P, et al. Tp53 Exon-6 Truncating Mutations Produce Separation of Function Isoforms With Pro-Tumorigenic Functions. Elife (2016) 5:e17929. doi: 10.7554/eLife.17929
59. Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, Walsh AM, et al. P53ψ Is a Transcriptionally Inactive P53 Isoform Able to Reprogram Cells Toward a Metastatic-Like State. Proc Natl Acad Sci USA (2014) 111(32):E3287–96. doi: 10.1073/pnas.1321640111
60. Wolff S, Erster S, Palacios G, Moll UM. P53's Mitochondrial Translocation and Momp Action Is Independent of Puma and Bax and Severely Disrupts Mitochondrial Membrane Integrity. Cell Res (2008) 18(7):733–44. doi: 10.1038/cr.2008.62
61. Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, et al. Adaptive Mitochondrial Reprogramming and Resistance to Pi3k Therapy. J Natl Cancer Inst (2015) 107(3):dju502. doi: 10.1093/jnci/dju502
62. Lu CC, Huang BR, Liao PJ, Yen GC. Ursolic Acid Triggers Nonprogrammed Death (Necrosis) in Human Glioblastoma Multiforme Dbtrg-05mg Cells Through Mpt Pore Opening and Atp Decline. Mol Nutr Food Res (2014) 58(11):2146–56. doi: 10.1002/mnfr.201400051
63. Su J, Cheng H, Zhang D, Wang M, Xie C, Hu Y, et al. Synergistic Effects of 5-Fluorouracil and Gambogenic Acid on A549 Cells: Activation of Cell Death Caused by Apoptotic and Necroptotic Mechanisms Via the Ros-Mitochondria Pathway. Biol Pharm Bull (2014) 37(8):1259–68. doi: 10.1248/bpb.b13-00972
64. Li L, Han W, Gu Y, Qiu S, Lu Q, Jin J, et al. Honokiol Induces a Necrotic Cell Death Through the Mitochondrial Permeability Transition Pore. Cancer Res (2007) 67(10):4894–903. doi: 10.1158/0008-5472.Can-06-3818
65. Olivas-Aguirre M, Torres-López L, Gómez-Sandoval Z, Villatoro-Gómez K, Pottosin I, Dobrovinskaya O. Tamoxifen Sensitizes Acute Lymphoblastic Leukemia Cells to Cannabidiol by Targeting Cyclophilin-D and Altering Mitochondrial Ca(2+) Homeostasis. Int J Mol Sci (2021) 22(16):8688. doi: 10.3390/ijms22168688
66. Ying L, Chunxia Y, Wei L. Inhibition of Ovarian Cancer Cell Growth by a Novel Tak1 Inhibitor Lytak1. Cancer Chemother Pharmacol (2015) 76(3):641–50. doi: 10.1007/s00280-015-2822-8
67. Minjie S, Defei H, Zhimin H, Weiding W, Yuhua Z. Targeting Pancreatic Cancer Cells by a Novel Hydroxamate-Based Histone Deacetylase (Hdac) Inhibitor St-3595. Tumour Biol (2015) 36(11):9015–22. doi: 10.1007/s13277-015-3537-5
68. Huo H, Zhou Z, Qin J, Liu W, Wang B, Gu Y. Erastin Disrupts Mitochondrial Permeability Transition Pore (Mptp) and Induces Apoptotic Death of Colorectal Cancer Cells. PloS One (2016) 11(5):e0154605. doi: 10.1371/journal.pone.0154605
69. Pan H, Wang BH, Lv W, Jiang Y, He L. Esculetin Induces Apoptosis in Human Gastric Cancer Cells Through a Cyclophilin D-Mediated Mitochondrial Permeability Transition Pore Associated With Ros. Chem Biol Interact (2015) 242:51–60. doi: 10.1016/j.cbi.2015.09.015
70. Kai S, Lu JH, Hui PP, Zhao H. Pre-Clinical Evaluation of Cinobufotalin as a Potential Anti-Lung Cancer Agent. Biochem Biophys Res Commun (2014) 452(3):768–74. doi: 10.1016/j.bbrc.2014.08.147
71. Zhou C, Chen Z, Lu X, Wu H, Yang Q, Xu D. Icaritin Activates Jnk-Dependent Mptp Necrosis Pathway in Colorectal Cancer Cells. Tumour Biol (2016) 37(3):3135–44. doi: 10.1007/s13277-015-4134-3
72. Xie J, Li Q, Ding X, Gao Y. Gsk1059615 Kills Head and Neck Squamous Cell Carcinoma Cells Possibly Via Activating Mitochondrial Programmed Necrosis Pathway. Oncotarget (2017) 8(31):50814–23. doi: 10.18632/oncotarget.15135
73. Guo F, Liu SQ, Gao XH, Zhang LY. Aicar Induces Ampk-Independent Programmed Necrosis in Prostate Cancer Cells. Biochem Biophys Res Commun (2016) 474(2):277–83. doi: 10.1016/j.bbrc.2016.04.077
74. Wu PK, Hong SK, Starenki D, Oshima K, Shao H, Gestwicki JE, et al. Mortalin/Hspa9 Targeting Selectively Induces Kras Tumor Cell Death by Perturbing Mitochondrial Membrane Permeability. Oncogene (2020) 39(21):4257–70. doi: 10.1038/s41388-020-1285-5
75. Li HM, Li B, Ma H, Sun X, Zhu M, Dai Y, et al. Bishonokiol a Induces Multiple Cell Death in Human Breast Cancer Mcf-7 Cells. Asian Pac J Cancer Prev (2020) 21(4):1073–80. doi: 10.31557/apjcp.2020.21.4.1073
76. Kaku Y, Tsuchiya A, Kanno T, Nakano T, Nishizaki T. Diarachidonoylphosphoethanolamine Induces Necrosis/Necroptosis of Malignant Pleural Mesothelioma Cells. Cell signalling (2015) 27(9):1713–9. doi: 10.1016/j.cellsig.2015.05.007
77. Zhang SL, Tang HB, Hu JT, Zang ZL, Ding X, Li S, et al. Pgam5-Cypd Pathway Is Involved in Bromocriptine-Induced Rip3/Mlkl-Dependent Necroptosis of Prolactinoma Cells. Biomed pharmacother (2019) 111:638–48. doi: 10.1016/j.biopha.2018.12.128
78. Zhang LY, Wu YL, Gao XH, Guo F. Mitochondrial Protein Cyclophilin-D-Mediated Programmed Necrosis Attributes to Berberine-Induced Cytotoxicity in Cultured Prostate Cancer Cells. Biochem Biophys Res Commun (2014) 450(1):697–703. doi: 10.1016/j.bbrc.2014.06.039
79. Lu JH, Shi ZF, Xu H. The Mitochondrial Cyclophilin D/P53 Complexation Mediates Doxorubicin-Induced Non-Apoptotic Death of A549 Lung Cancer Cells. Mol Cell Biochem (2014) 389(1-2):17–24. doi: 10.1007/s11010-013-1922-1
80. Qin LS, Jia PF, Zhang ZQ, Zhang SM. Ros-P53-Cyclophilin-D Signaling Mediates Salinomycin-Induced Glioma Cell Necrosis. J Exp Clin Cancer Res (2015) 34(1):57. doi: 10.1186/s13046-015-0174-1
81. Chen B, Xu M, Zhang H, Wang JX, Zheng P, Gong L, et al. Cisplatin-Induced Non-Apoptotic Death of Pancreatic Cancer Cells Requires Mitochondrial Cyclophilin-D-P53 Signaling. Biochem Biophys Res Commun (2013) 437(4):526–31. doi: 10.1016/j.bbrc.2013.06.103
82. Ju T, Gao D, Fang ZY. Targeting Colorectal Cancer Cells by a Novel Sphingosine Kinase 1 Inhibitor Pf-543. Biochem Biophys Res Commun (2016) 470(3):728–34. doi: 10.1016/j.bbrc.2016.01.053
83. Wu L, Cao K, Ni Z, Wang S, Li W, Liu X, et al. Rhein Reverses Doxorubicin Resistance in Smmc-7721 Liver Cancer Cells by Inhibiting Energy Metabolism and Inducing Mitochondrial Permeability Transition Pore Opening. Biofactors (2019) 45(1):85–96. doi: 10.1002/biof.1462
84. Peng X, Zhang D, Li Z, Fu M, Liu H. Mtor Inhibition Sensitizes Human Hepatocellular Carcinoma Cells to Resminostat. Biochem Biophys Res Commun (2016) 477(4):556–62. doi: 10.1016/j.bbrc.2016.06.060
85. Fu M, Shi W, Li Z, Liu H. Activation of Mptp-Dependent Mitochondrial Apoptosis Pathway by a Novel Pan Hdac Inhibitor Resminostat in Hepatocellular Carcinoma Cells. Biochem Biophys Res Commun (2016) 477(4):527–33. doi: 10.1016/j.bbrc.2016.04.147
86. Dong YY, Zhuang YH, Cai WJ, Liu Y, Zou WB. The Mitochondrion Interfering Compound Npc-26 Exerts Potent Anti-Pancreatic Cancer Cell Activity in Vitro and in Vivo. Tumour Biol (2016) 37(11):15053–63. doi: 10.1007/s13277-016-5403-5
87. Yu T, Chen C, Sun Y, Sun H, Li TH, Meng J, et al. Abt-737 Sensitizes Curcumin-Induced Anti-Melanoma Cell Activity Through Facilitating Mptp Death Pathway. Biochem Biophys Res Commun (2015) 464(1):286–91. doi: 10.1016/j.bbrc.2015.06.144
88. Qiu Y, Yu T, Wang W, Pan K, Shi D, Sun H. Curcumin-Induced Melanoma Cell Death Is Associated With Mitochondrial Permeability Transition Pore (Mptp) Opening. Biochem Biophys Res Commun (2014) 448(1):15–21. doi: 10.1016/j.bbrc.2014.04.024
89. Ma YC, Zhu YL, Su N, Ke Y, Fan XX, Shi XJ, et al. A Novel Ent-Kaurane Diterpenoid Analog, Dn3, Selectively Kills Human Gastric Cancer Cells Via Acting Directly on Mitochondria. Eur J Pharmacol (2019) 848:11–22. doi: 10.1016/j.ejphar.2019.01.013
90. Hu W, Yuan Q, Liu XH, Zhu HC, Lv SQ, Wang XH. Cyclophilin D-Mediated Apoptosis Attributes to Sorafenib-Induced Cytotoxicity in Clear Cell-Renal Cell Carcinoma. Eur J Pharmacol (2015) 749:142–50. doi: 10.1016/j.ejphar.2014.12.025
91. Zhang L, He D, Li K, Liu H, Wang B, Zheng L, et al. Emodin Targets Mitochondrial Cyclophilin D to Induce Apoptosis in Hepg2 Cells. Biomed pharmacother (2017) 90:222–8. doi: 10.1016/j.biopha.2017.03.046
92. Yang L, Zheng LY, Tian Y, Zhang ZQ, Dong WL, Wang XF, et al. C6 Ceramide Dramatically Enhances Docetaxel-Induced Growth Inhibition and Apoptosis in Cultured Breast Cancer Cells: A Mechanism Study. Exp Cell Res (2015) 332(1):47–59. doi: 10.1016/j.yexcr.2014.12.017
93. Fu M, Wan F, Li Z, Zhang F. 4sc-202 Activates Ask1-Dependent Mitochondrial Apoptosis Pathway to Inhibit Hepatocellular Carcinoma Cells. Biochem Biophys Res Commun (2016) 471(2):267–73. doi: 10.1016/j.bbrc.2016.01.030
94. Zhang R, Li G, Zhang Q, Tang Q, Huang J, Hu C, et al. Hirsutine Induces Mptp-Dependent Apoptosis Through Rock1/Pten/Pi3k/Gsk3β Pathway in Human Lung Cancer Cells. Cell Death Dis (2018) 9(6):598. doi: 10.1038/s41419-018-0641-7
95. Ghosh JC, Siegelin MD, Dohi T, Altieri DC. Heat Shock Protein 60 Regulation of the Mitochondrial Permeability Transition Pore in Tumor Cells. Cancer Res (2010) 70(22):8988–93. doi: 10.1158/0008-5472.Can-10-2225
96. Xia YC, Zha JH, Sang YH, Yin H, Xu GQ, Zhen J, et al. Ampk Activation by Asp4132 Inhibits Non-Small Cell Lung Cancer Cell Growth. Cell Death Dis (2021) 12(4):365. doi: 10.1038/s41419-021-03655-2
97. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. P53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell (2012) 149(7):1536–48. doi: 10.1016/j.cell.2012.05.014
98. Chen W, Feng L, Nie H, Zheng X. Andrographolide Induces Autophagic Cell Death in Human Liver Cancer Cells Through Cyclophilin D-Mediated Mitochondrial Permeability Transition Pore. Carcinogenesis (2012) 33(11):2190–8. doi: 10.1093/carcin/bgs264
99. Brenner C, Subramaniam K, Pertuiset C, Pervaiz S. Adenine Nucleotide Translocase Family: Four Isoforms for Apoptosis Modulation in Cancer. Oncogene (2011) 30(8):883–95. doi: 10.1038/onc.2010.501
100. Tavecchio M, Lisanti S, Lam A, Ghosh JC, Martin NM, O'Connell M, et al. Cyclophilin D Extramitochondrial Signaling Controls Cell Cycle Progression and Chemokine-Directed Cell Motility. J Biol Chem (2013) 288(8):5553–61. doi: 10.1074/jbc.M112.433045
101. Wu PK, Hong SK, Park JI. Mortalin Depletion Induces Mek/Erk-Dependent and Ant/Cypd-Mediated Death in Vemurafenib-Resistant B-Raf(V600e) Melanoma Cells. Cancer Lett (2021) 502:25–33. doi: 10.1016/j.canlet.2020.12.044
102. Wu PK, Hong SK, Chen W, Becker AE, Gundry RL, Lin CW, et al. Mortalin (Hspa9) Facilitates Braf-Mutant Tumor Cell Survival by Suppressing Ant3-Mediated Mitochondrial Membrane Permeability. Sci Signal (2020) 13(622):eaay1478. doi: 10.1126/scisignal.aay1478
Keywords: cyclophilin D, mitochondrial permeability transition pore, tumor energy metabolism, tumor cell death, tumor metastasis and invasion, tumor resistance
Citation: Zhang L, Liu Y, Zhou R, He B, Wang W and Zhang B (2022) Cyclophilin D: Guardian or Executioner for Tumor Cells? Front. Oncol. 12:939588. doi: 10.3389/fonc.2022.939588
Received: 09 May 2022; Accepted: 09 June 2022;
Published: 04 July 2022.
Edited by:
Lucília Ataíde Saraiva, University of Porto, PortugalCopyright © 2022 Zhang, Liu, Zhou, He, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Zhang, emhiODYxMTA5QDE2My5jb20=; Ling Zhang, emx5ejg5MTAyM0AxNjMuY29t