 Xiaozhuang Zhou
Xiaozhuang Zhou Shruthi Kandalai
Shruthi Kandalai Farzana Hossain
Farzana Hossain Qingfei Zheng
Qingfei Zheng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 25 July 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.933407
This article is part of the Research Topic Rising Stars in Cancer Metabolism 2022 View all 10 articles
Accumulating recent evidence indicates that the human microbiome plays essential roles in pathophysiological states, including cancer. The tumor microbiome, an emerging concept that has not yet been clearly defined, has been proven to influence both cancer development and therapy through complex mechanisms. Small molecule metabolites produced by the tumor microbiome through unique biosynthetic pathways can easily diffuse into tissues and penetrate cell membranes through transporters or free diffusion, thus remodeling the signaling pathways of cancer and immune cells by interacting with biomacromolecules. Targeting tumor microbiome metabolism could offer a novel perspective for not only understanding cancer progression but also developing new strategies for the treatment of multiple cancer types. Here, we summarize recent advances regarding the role the tumor microbiome plays as a game changer in cancer biology. Specifically, the metabolites produced by the tumor microbiome and their potential effects on the cancer development therapy are discussed to understand the importance of the microbial metabolism in the tumor microenvironment. Finally, new anticancer therapeutic strategies that target tumor microbiome metabolism are reviewed and proposed to provide new insights in clinical applications.
The human microbiota is a broad category consisting of diverse bacteria, fungi, protists, archaea, and viruses that occur in and on the human body (1). The total number of these microbes is believed to be more than 100 trillion, which amounts to 2 kg in mass (2). Due to its important pathophysiological role in human health and disease, the microbiome has also been referred to as “the last human organ under active research” (3) and “the second brain” (4). Moreover, the number of unique genes from the microbiome is estimated to be 100-fold higher than that from human cells, as noted by the NIH Human Microbiome Project (5, 6). The proteins encoded by these genes and the metabolites biosynthesized by these microbes are able to influence not only their own microbial communities, but also the biological functions of host cells (7, 8). Notably, small molecule metabolites secreted by the human microbiome affect local and systemic bodily functions, including energy generation, metabolism of dietary components, biosynthesis of vitamins, immune responses, behavior, and even mood (9–11).
While microbes were implicated in diseases long ago, the contributions of the tumor microbiome to carcinogenesis, cancer progression, metastasis, and treatment have been poorly understood until recently (12–14). Previous studies have shown that microbes belonging to the genera Salmonella and Helicobacter affect cellular dysplasia and carcinogenesis (15, 16). Microbiota homeostasis can also play a role in cancer development (17). For instance, dysbiosis is associated with the carcinogenesis of gastrointestinal (GI) and non-GI tumors while also acting as an oncogenic driver of colorectal cancer (CRC) (18). Current research indicates that human-associated microbes interact with host cells and affect disease states, especially cancer, via diverse mechanisms (19, 20). One key mechanism is microbial metabolites serving as small molecule messengers to mediate crosstalk between microbes and host cells (21). Specifically, microbial metabolites can alter the tumor microenvironment (TME) (22), which includes inflammatory mediators, recruited immune cells, fibroblasts, adipocytes, endothelial cells, and pericytes (22, 23), thereby directly influencing cancer progression (23, 24) and the efficacy of immunotherapy (1, 23). One well-studied example of this is the genotoxic metabolite colibactin, produced by pathogenic Escherichia coli, that can directly induce DNA double-strand breaks (DSBs) (25), thus motivating CRC development (26).
As the tumor microbiome metabolism exhibits direct and indirect impacts on cancer development, novel therapy strategies may be developed by targeting these unique metabolic pathways (27, 28). Chemical biology, synthetic biology, and biomedical engineering approaches facilitate the remodeling of the microbiome-containing TME and will provide new opportunities for the future development of bacterial, viral, chemical, and immunological therapies.
In this review, we intend to highlight the tumor microbiome and how it affects cancer development and therapy as a new game changer. Among the multiple crosstalk mechanisms between microbes and cancer cells, we specifically focus on the unique metabolites produced by the tumor microbiome. The chemical structures and biochemical mechanisms through which tumor microbiome metabolism affects cancer biology are addressed. Finally, yet importantly, the potential clinical applications of targeting tumor microbiome metabolism through multidisciplinary methods for future cancer therapy have been proposed and discussed.
The tumor microbiome is an emerging concept that has yet to be clearly defined. It broadly refers to all microorganisms located within the TME (Figure 1) and encompasses bacteria, fungi, archaea, viruses, and other microbes (29) that contribute to the reshaping of the microenvironment. These microbes are widespread in the TME and inhabit inside or outside the tumor cells and immune cells. It has long been in debate whether these microbes constitute a predetermined niche or rather represent a transient stochastic colonization (29).
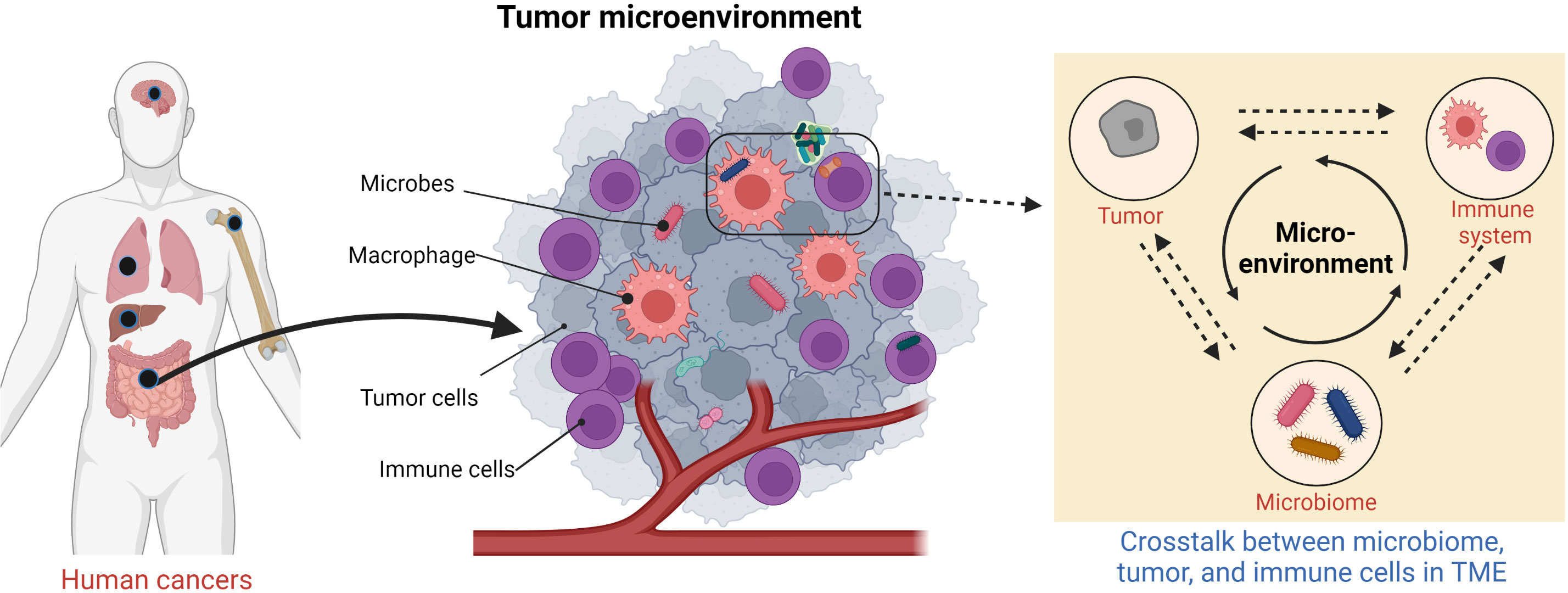
Figure 1 Schematic of human tumor microenvironment that contains tumor microbiome.
Within cancer biology, intratumoral bacteria and their effects are a newly raised concept (30). While bacteria were observed in tumor isolates previously, it was assumed that these were contaminants and were not associated with cancer cells (31). Recently, a large-scale analysis of over 1,500 clinical samples indicated that the majority of the tumor microbiome is intracellular bacteria that exhibit tumor-site-specific properties (32). Intratumoral bacteria and host cancer cells mutually influence each other through the transcriptome and metabolome (33). Since these intracellular bacteria inhabit cancer cells, direct crosstalk between host and microbes is easily mediated by biomacromolecules and small molecule metabolites. However, this still leads to a chicken-and-egg situation—is the accumulation of intratumoral bacteria a cause or effect of cancer? Further investigations are required to address this question. Intracellular microbes hiding inside other type of cells, such as macrophages and fibroblasts, have also been shown to remodel the TME (34, 35) and thus affect cancer development and treatment (36, 37).
On the other hand, viruses that directly cause cancer (also known as oncoviruses) have been thoroughly studied. These viruses currently include hepatitis B virus (HBV), hepatitis C virus (HCV), human papillomaviruses (HPVs), Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8), human T-lymphotropic virus (HTLV), Merkel cell polyomavirus (MCV), and Epstein–Barr virus (EBV) (38). They induce cancer through diverse mechanisms, such as the integration of viral DNA into the host genome (39) and the inactivation of tumor suppressor genes like p53 and Rb (40). Globally, these oncoviruses are associated with approximately 10%–16% of cancer cases (41, 42). It has also been suggested that other viruses, similar to the bacteria mentioned previously, may play a role in carcinogenesis, without directly causing cancer (37). Other microbes, such as fungi, have also been implicated in cancer (43, 44), although this is less studied.
Extracellular microorganisms in the TME, such as those in the gut microbiota, oral microbiota, vaginal flora, and skin flora, also play essential roles in cancer development (45–47) and have significant impacts on curative outcomes (48). For instance, it has long been known that the colonization by Helicobacter pylori in stomach can directly cause gastric cancer (49), as well as gastric mucosa–associated lymphoid tissue (MALT) lymphoma (50). As a result, H. pylori is associated with approximately 5% of cancers worldwide (42). Multiple studies have shown that the gut microbiota interacts with the host by producing of a diverse set of metabolites and toxins from exogenous dietary substrates and endogenous host cellular compounds (51). Host metabolic disorders are systematically associated with alterations in the composition and function of the gut microbiota (52). Specific classes of microbiota-derived metabolites, notably bile acids (BAs), short-chain fatty acids (SCFAs), branched-chain amino acids, trimethylamine N-oxide, and tryptophan and indole derivatives, have been implicated in the pathogenesis of host cell metabolic disorders, some of which directly relate to carcinogenesis (53). In addition, the gut microbiome is essential in shaping the development of innate and adaptive immunity (54) and plays an essential role in the clinical efficiency of cancer immunotherapy (55).
The crosstalk between the tumor microbiome and cancer cells is diverse and complex, involving cell–cell direct interactions and messenger molecule-mediated effects (Figure 2). With respect to host cell–microbe direct interactions, intracellular microbe–induced autophagy and extracellular microbe–caused inflammation are two well-studied examples. For instance, it has been shown that Fusobacterium nucleatum modulates the autophagy pathways of CRC cells by targeting TLR4 and MYD88 innate immune signaling and specific microRNAs, thereby promoting CRC chemoresistance and migration (56). Moreover, it has been accepted for decades that inflammation is a critical component of tumor progression (57). Inflammatory cells significantly influence the TME, thereby affecting neoplastic processes and fostering the proliferation, survival, and migration of cancer cells (58). Chronic, dysregulated, persistent, and unresolved inflammation is associated with an increased risk of malignancies, as well as the malignant progression of most types of cancer (58). As microorganisms are one of the major causes of inflammation, the tumor microbiome can manipulate cancer development by remodeling the TME through the recruitment of inflammatory cells. In fact, it has been pointed out that bacterial infections can trigger chronic inflammation that leads to host cell proliferation and tumor development (59).
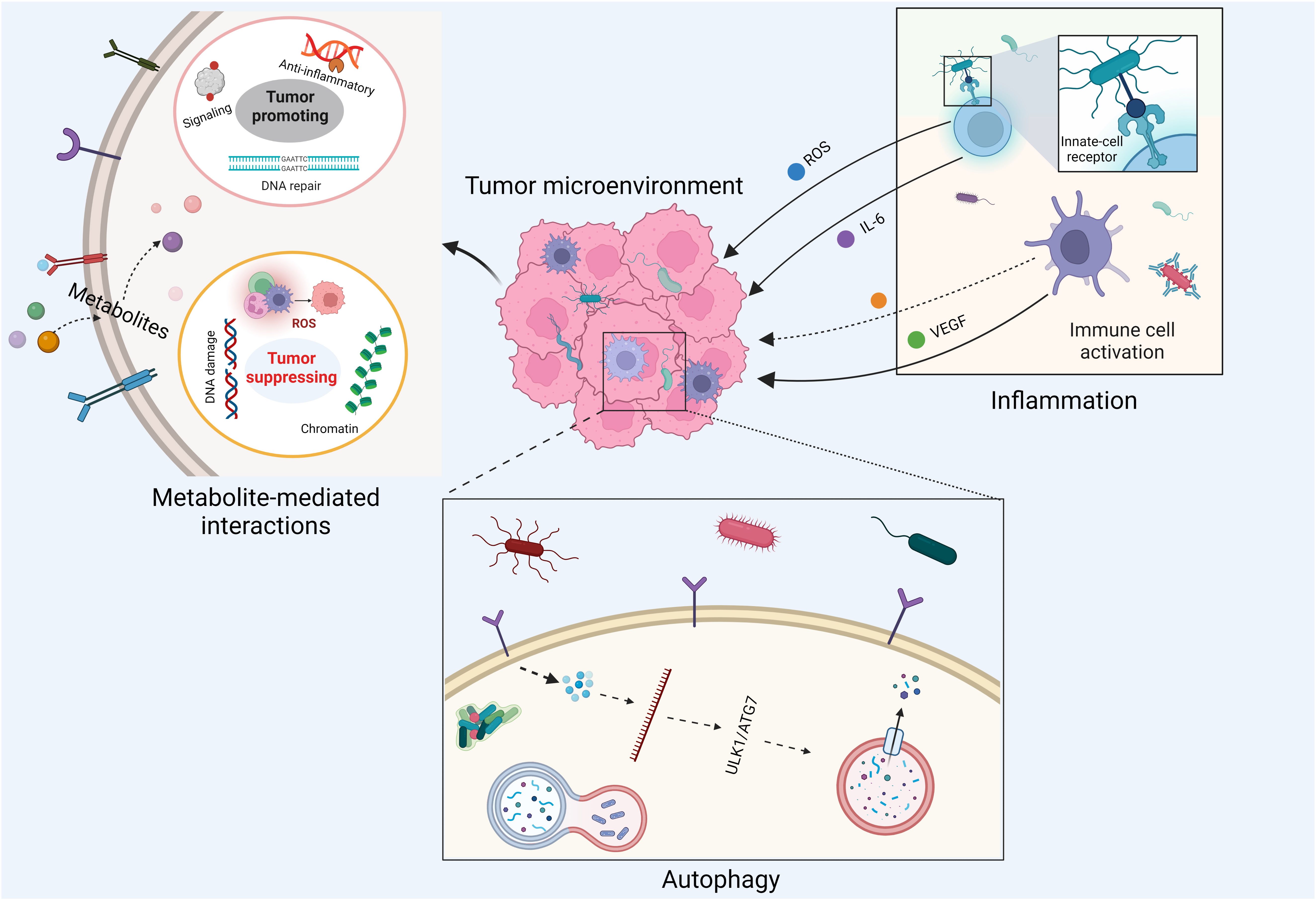
Figure 2 Impacts of tumor microbiome on cancer development.
Messenger molecule–mediated interactions between host cells and microbes are another key machinery linking the tumor microbiome to cancer progression. These messenger molecules involve secreted proteins, peptide toxins, and small-molecule metabolites. For example, the virulence factor cytolethal-distending toxin produced by Campylobacter jejuni is one of the major causes for infectious diarrhea worldwide and has been shown to induce carcinogenesis in vivo (60, 61). Moreover, tumor microbiome–derived small molecule metabolites can reach remote tumor entities through systemic circulation, free diffusion, and active transport (such as the transport of lactate and pyruvate by proton-coupled monocarboxylate transporters) (62). These metabolites are able to stimulate antitumoral or carcinogenic innate immune responses (22) via non-covalent interactions. For instance, evolutionary conserved pathogen-associated molecular patterns (PAMPs) from commensal microbes or pathogens can be systematically sensed by the innate immune system via pattern recognition receptors, such as Toll-like receptors and NOD-like receptors, leading to the host’s innate immune responses (63). There is evidence showing that bacterial PAMPs can boost antitumor immunity by augmenting Toll-like receptor signaling and serving as cancer vaccine adjuvants (64–66). Additionally, commensal gut bacteria can recruit natural killer T immune cells to control the growth of liver tumors via their unique microbial metabolism of BAs (67). Moreover, chemically reactive metabolites from the tumor microbiome can promote or inhibit tumor growth through the covalent modifications of DNA, RNA, histones, and other essential enzymes involved in host signaling transduction pathways (68). These modifications can be enzymatic or non-enzymatic and are capable of inducing cancer-causing and cancer-promoting epigenetic changes of host cells (69). As a result of this complex crosstalk between the host and tumor microbiome, both cancer and immune cells change their own metabolic status to adapt to the reshaped TME (70).
Furthermore, due to its novel metabolic and catabolic pathways, the gut microbiome is capable of converting human-ingested nutrients into functional microbial metabolites that closely link diet, cancer, and other metabolic diseases (19, 71, 72). These microbial metabolites produced by microbes from diet, such as BAs and SCFAs, have significant impacts on cancer and immune cells (73–78), thereby affecting cancer development and immunotherapies through complex mechanisms (79–81). Based on the important role of the microbiome in connecting diet and different types of cancer, recent research advances have suggested that gut microbiota modulation would become a novel strategy for prevention and treatment of CRC (82). As diet and microbial communities affect one another, dietary interventions have proven to be an efficient approach to modulate the intestinal microbiota, which is in line with the growing recognition of significant impacts of diet and lifestyle on human health through microbiome regulation (83).
The consequence of metabolism is the production of small molecule metabolites, which are typically classified into two categories: primary metabolites and secondary metabolites. Primary metabolites are compounds that are directly involved in an organism’s growth and development, while secondary metabolites are not directly involved in these processes and tend to vary more by species (84). There are a number of primary metabolites produced by microbes that contribute to cancer development or suppression, such as methylglyoxal (MGO), SCFAs, BAs, reactive oxygen species (ROS), amines, and methane (CH4) (85–87). These molecules are biosynthesized by diverse human-associated microorganisms, including archaea (88), bacteria (89, 90), fungi (90) protists (91) and parasites (91, 92).
There are several examples of secondary metabolites with well-established functions, such as colibactin, peptide aldehyde, and thiopeptide, that have been known to affect cancer development, and these metabolites have diverse chemical structures (Figure 3). As a well-studied secondary metabolite molecule, colibactin is a cytotoxin mainly produced by pathogenic Escherichia coli, as well as other members of the family Enterobacteriaceae. The production of colibactin was shown to have a direct and significant association with CRC via the induction of DNA DSBs (25, 26). Peptide aldehydes were discovered as metabolites from a variety of microbes (including E. coli, Bacillus subtilis, and Streptomyces species) and are known to inhibit protease functions (93, 94), which may increase carcinogenicity. Thiopeptides have complex structures and strong antibacterial activities (95, 96), which can affect the distribution of human flora (97). In addition to being isolated from multiple environmental microbes, thiopeptides have been discovered from many microbial species in various parts of the body, including Lactobacillus gasseri in the urogenital tract, Propionibacterium acnes on the skin, Streptococcus downei in the oral cavity, and Enterococcus faecalis in the gut (98). Moreover, emerging studies have suggested that thiopeptides may also serve as anticancer agents by targeting proteasomes and transcription factor FOXM1 (99).
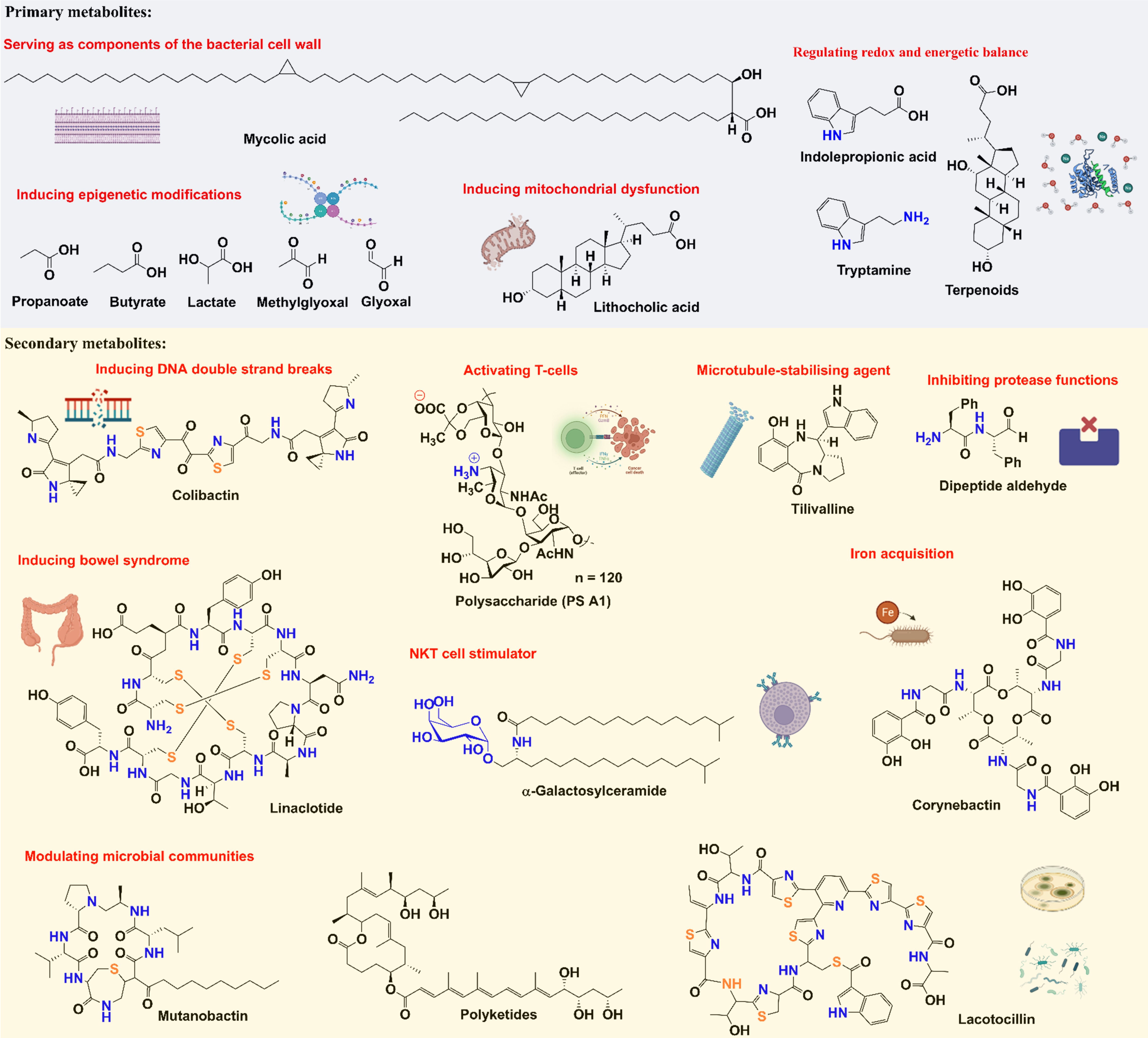
Figure 3 Chemical structures and functions of representative metabolites from tumor microbiome.
Since small molecule metabolites from tumor microbiome play essential roles in cancer development, we would like to summarize some examples in this section to emphasize the neglected but significant impacts of tumor microbiome metabolism on the TME (Figure 3). As stated above, colibactin’s ability to cause DNA DSBs allows it to promote tumorigenesis (100). Recently, it has been shown that colibactin also targets bacteria by triggering prophage induction (101), which may explain how this metabolite further affects the communities in the tumor microbiome.
SCFAs are mainly bacterial fermentation products from starch and other polysaccharides (102) and include a wide range of molecules including acetate, propionate, butyrate, and lactate (89). Among these, butyrate has been shown to potently inhibit the activity of histone deacetylases (103–105), whereas propionate does so moderately and acetate has no effect (106, 107). Lactate is known to play significant roles in the Warburg effect and reverse Warburg effect (108–110), as well as affect chromatin biology through histone modification (111, 112). It has also been shown that SCFAs can: 1) modulate macrophage functions by promoting the production of nitric oxide, IL-6, IL-12 (113), and IL-22 (114); 2) induce the differentiation of Treg cells (115–117); and 3) regulate the migration of neutrophils (118). There are many connections between SCFAs and cancer, where SCFAs function as a double-edged sword in tumorigenesis. SCFAs have been implicated to have cancer-promoting or cancer-suppressing effects that vary under different conditions and with different types of cancer. Previous research has shown that SCFAs are able to: inhibit human colon cancer invasion (119, 120), inhibit the migration and invasion of fibrosarcoma cells (121), increase IGF1 levels to promote the proliferation of prostate cancer cells (122), upregulate proapoptotic protein BAK (123), downregulate adhesion protein α2β1 integrin (124), induce cell stress responses and apoptosis in colorectal cells (125), inhibit proliferation and increase differentiation and apoptosis of adenocarcinoma cells (126), impair hypoxia-induced angiogenesis (127), and regulate p53 expression (128, 129).
BAs are steroid derivatives that play essential regulatory roles in the GI system and cancer development. While primary BAs are produced by the liver, secondary BAs, mainly deoxycholic acid and lithocholic acid, occur when primary BAs are further metabolized by gut bacteria. Secondary BAs have long been proposed to promote tumors (130). In addition, further derivatives of secondary bile salts can cause apoptosis, increase ROS production, and lessen pro-apoptotic effects (131). Deoxycholic acid is believed to be associated with oncogenic mutations of proto-oncogene KRAS (132) and can lead to DNA DSBs and apoptosis (133). Lithocholic acid has been shown to modulate Th17 and Treg cells (73), inhibit HLA class I genes (134), and induce endoplasmic reticulum stress and mitochondrial dysfunction in human prostate cancer cells (135). Moreover, CRC cells can obtain resistance to apoptosis after being exposed to specific bile salts (136, 137).
Polyamines are small molecule metabolites with two or more amino groups, which exhibit a variety of functions. The most common polyamines, putrescine, cadaverine, spermidine, and spermine, are metabolized from arginine (138) but can also be produced by gut bacteria (139, 140). Polyamines are known to protect cells from ROS (141) due to their reducing activities and have been significantly correlated with CRC (142, 143). Polyamines have been shown to be associated with inhibiting the growth of prostate cancer cells (144–146), downregulating estrogen receptor α in breast cancer cells (147), serving as a downstream effector from H. pylori, leading to DNA damage and immune cell apoptosis in stomach cancer (148–151), and increasing the risk for development of skin cancer in mouse models (152, 153). Moreover, microbial polyamines exhibit unique activities in the regulation of macrophage polarization and function, thereby affecting host immune responses (154).
MGO is a chemically reactive dicarbonyl metabolite of glucose metabolism (155, 156). In mammalian cells, MGO is mainly generated as a byproduct through a non-enzymatic dephosphorylation process during glycolysis, although it can also be produced by tumor microbes that contain microorganism-specific methylglyoxal synthases (88, 157). MGO can react with nucleophilic groups of biomacromolecules, such as lysine and arginine residues in proteins (158), as well as guanine residues in DNA and RNA (159). This MGO-induced non-enzymatic covalent modification (glycation) can result in the formation of advanced glycation end products (AGEs) (160–162) and changes in the three-dimensional chromatin architecture (163–165). It has been shown that elevated levels of MGO in the TME lead to the overexpression of an MGO detoxifier, glyoxalase I (Glo1), in cancer cells (166, 167). There is evidence showing that low concentrations of MGO are beneficial for cancer cell growth, while high levels of MGO contribute negatively to cell survival by disrupting multiple signaling pathways (168, 169). The biphasic model proposed recently is a convincing explanation for the function of MGO-induced glycation in manipulating chromatin damage and cancer cell survival (166). Moreover, the recently identified histone MGO-glycation eraser and rewriter enzymes, DJ-1 and PAD4, have been recognized to possess cancer-promoting effects as oncoproteins (163, 164). Thus, developing deglycase activity–oriented high-throughput screening assays for identifying DJ-1 and PAD4 inhibitors will provide new insights for the mechanistic studies of host deglycation pathways, as well as clinical applications (170).
As noted above, due to the inseparable connections between microbes, host immune cells, and cancer cells, targeting the tumor microbiome seems to be a practical tactic for cancer therapy (Figure 4). Specifically, strategies include the development of wild-type and/or engineered microbes for bacterial and viral therapies and the application of chemical biology, synthetic biology, and biomedical engineering to target the tumor microbiome metabolism for reshaping TME. Ideally, with a deeper understanding of the tumor microbiome’s function in the TME and cancer development, we could build up an artificial ecosystem of microorganisms in the TME to prevent cancer cells from spreading and enhance the efficiency of immunotherapy.
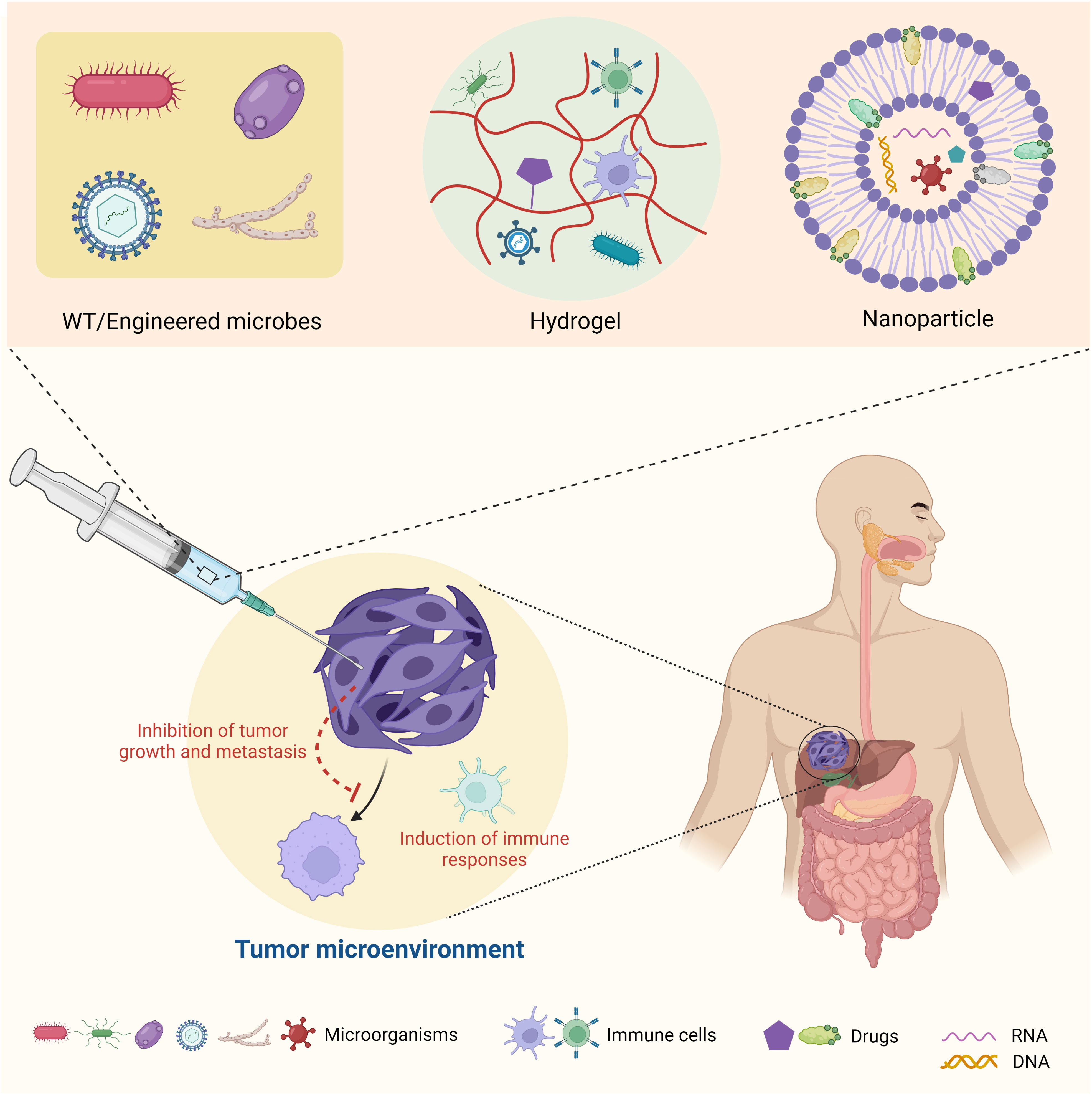
Figure 4 Summary of therapeutic strategies targeting tumor microbiome metabolism.
Based on their functions in suppressing or promoting cancer progression, microbes within the TME can be classified to “good bugs” or “bad bugs” for cancer therapies (171). A straightforward treatment strategy is to take advantage of “good bugs” and get rid of “bad bugs” in the TME. For example, Enterococcus species have been noted to promote responses to immune checkpoint immunotherapy (ICI) (172). Bifidobacterium pseudolongum and Akkermansia muciniphila were observed to produce the metabolite inosine, which enhances ICI through Th1 activation (173). Following biomaterial modulation, mice with increased levels of Peptostreptococcus anaerobius and reduced levels of other bacterial species responded better to oral squamous cell carcinoma ICI (174). Bacteria belonging to the Gammaproteobacteria family have been found to inactivate the chemotherapy drug gemcitabine, which is often used for the treatment of pancreatic ductal adenocarcinoma (175). Overall, modulating the microbial communities in the TME can provide new opportunities for cancer therapies (176). Accordingly, synthetic biology approaches have been applied to engineer specific tumor microbiome species to develop enhanced bacteria-based cancer therapies. For instance, as low concentrations of L-arginine can cause poor responses to PD-L1 ICI, probiotic strain E. coli Nissle 1917 was engineered to convert ammonia to L-arginine, thereby increasing T-cell infiltration and enhancing ICI (177). Additionally, Nissle 1917 and other E. coli strains were engineered to release nanobodies with diverse functions to motivate T-cell infiltration and tumor shrinkage (178, 179). There are also a number of clinical trials in various phases regarding the applications of engineered bacteria for cancer therapies, some of which have shown promising results (180) (Table 1).
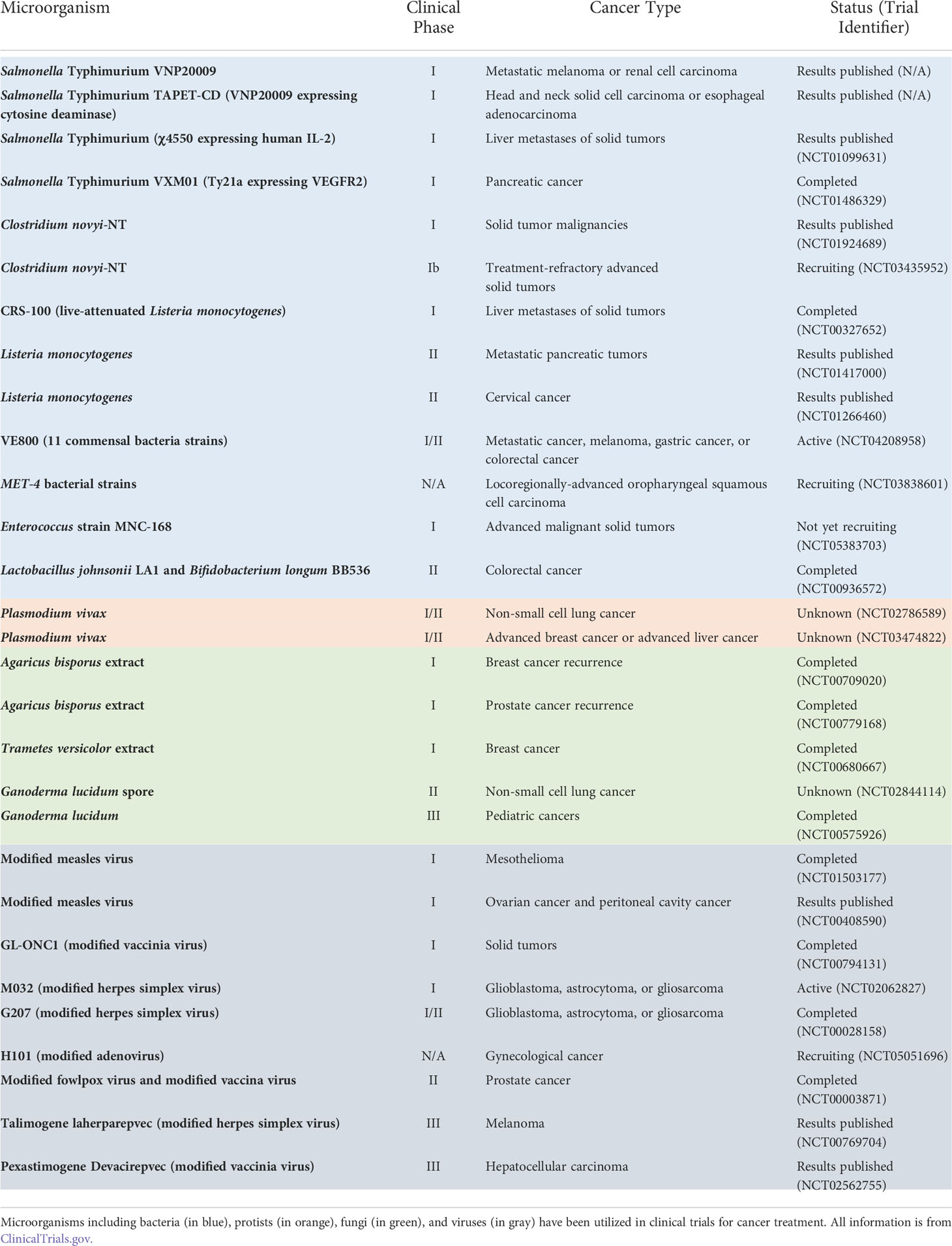
Table 1 Representative microorganisms applied for cancer therapy.
Similarly, oncolytic virotherapy has also been applied as an immunotherapy for cancer treatment (181–183). For example, alphavirus M1 was identified for such use, as it specifically targets cancer cells deficient in zinc-finger antiviral protein (184). Engineered oncolytic viruses expressing PD-L1 inhibitors have clinical potentials for curing cancers resistant to PD-1/PD-L1 ICI, as they are able to activate tumor neoantigen–specific T-cell responses (185). Notably, virotherapy has been approved in some countries for use against cancer. Imlygic, which is engineered from herpes simplex virus I (HSV1) and contains granulocyte-macrophage colony-stimulating factor, was approved in 2015 by the US Food and Drug Administration and European Medical Agency for the treatment of melanoma (186). G47Δ, which is engineered from HSV1, was approved in 2021 by Japan Ministry of Health, Labor and Welfare for the treatment of malignant glioma and other brain cancers (187). Oncorine, which is engineered from adenovirus, was approved in 2005 by the China Food and Drug Administration Department in combination with chemotherapy for the treatment of nasopharyngeal carcinoma (186). Moreover, there are other oncolytic virotherapies engineered from HSV1, adenovirus, and measles virus currently in various phases of clinical trials (186) (Table 1).
The toxins and chemicals extracted from microbes can also be used for cancer treatment. This strategy dates back to the late 19th century when Coley’s toxins (a mixture of toxins filtered from killed Streptococcus pyogenes and Serratia marcescens) were utilized to cure cancer (188). Although this was an unstable approach with poor repeatability, the application of Coley’s toxins led to milestone breakthroughs in immuno-oncology, such as the discovery of tumor necrosis factor α (TNF-α) (189). TNF-α has since been identified to suppress tumor growth and improve the efficacy of immunotherapy by activating cell death pathways (190, 191). Commensal bacteria have been found to play significant roles in CpG-oligodeoxynucleotide immunotherapy, which depend on the increased production of TNF-α (192). Microbial SCFAs have also been shown to improve CAR-T cell therapy by enhancing the levels of TNF-α in different cancer models (193).
Last but not least, recent advances in biomedical engineering have provided new opportunities for cancer treatment by targeting the tumor microbiome. For example, the utilization of biomaterials, such as nanoparticles (194, 195) and hydrogels (174), to modulate and deliver microbial communities to specific sites of the TME opens a new door for future cancer therapies (Figure 4). These novel materials can be designed to be stimuli responsive (196) and utilized for the controlled and targeted release of toxic chemotherapy drugs (197), therapeutic antibodies (198, 199), CAR-T cells (200, 201), or live microbes to reshape the TME (202–204). These applications of new biomaterials will offer a promising platform for basic and translational research and will accelerate clinical outcomes of drugs that may have poor solubility and high toxicity.
In this review, we have summarized the research process of the tumor microbiome, mainly focusing on the impacts of its unique microbial metabolism on cancer development and therapy. Over the past few decades, microorganisms have been regarded only as a cause of infectious disease. The pathophysiological functions of human-associated microbes have long been neglected until recently when the microbiome was identified to manipulate and affect diverse disease states, as well as therapeutic efficacy. The impacts of the human microbiome are so broad that research papers on the topic have exploded in the past few years. Accordingly, a number of new concepts have been raised to describe the omnipotent human microbiota, including the “brain-gut axis” and “second brain.” Despite these, the tumor microbiome still lacks a precise definition. Nevertheless, the tumor microbiome plays constructive roles in cancer biology, some of which are still elusive. Among these macro- and micropathophysiological effects induced by the tumor microbiome, small molecule metabolite–mediated crosstalk appears to be particularly important due to the free diffusion of metabolites that can easily impact local and distant tumor tissues via covalent modifications and/or non-covalent interactions. Here, we have provided representative examples to emphasize the role of tumor microbiome metabolism as a game changer in cancer biology and clinical treatment, as well as its broad biomedical effects that were once disregarded.
Targeting the pathways of microbial metabolism and crosstalk between host and microbes will provide future avenues for cancer diagnosis, treatment, and recovery. Accordingly, therapy strategies have been developed at distinct levels to target tumor microbiome metabolism: 1) directly applying wild-type or engineered live microbes in immuno-oncology; 2) utilizing the microbial-extracted fractions or synthetic chemicals that interfere with corresponding metabolic pathways for cancer treatment; and 3) utilizing rationally designed biomaterials to rebuild a benign TME by modulating the microbial ecosystem. All in all, after having a deeper understanding of the close correlation between the tumor microbiome and human cancer, we would change our perception of these microorganisms’ identities in tumor tissues from “short-term tenants” to “permanent residents.”
QZ proposed the conception, wrote, and edited the manuscript. XZ drafted the manuscript and figures. SK participated drafting and editing the manuscript as well as references. FH drafted and edited the chemical structures. All authors listed in the paper have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study is supported by OSUCCC startup funds for QZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Berg G, Rybakova D, Fischer D, Cernava T, Vergès MC, Charles T, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome (2020) 8(1):103. doi: 10.1186/s40168-020-00875-0
2. Flint HJ. The impact of nutrition on the human microbiome. Nutr Rev (2012) 70 Suppl 1:S10–3. doi: 10.1111/j.1753-4887.2012.00499.x
3. Baquero F, Nombela C. The microbiome as a human organ. Clin Microbiol Infect (2012) 18 Suppl 4:2–4. doi: 10.1111/j.1469-0691.2012.03916.x
4. Ochoa-Repáraz J, Kasper LH. The second brain: Is the gut microbiota a link between obesity and central nervous system disorders? Curr Obes Rep (2016) 5(1):51–64. doi: 10.1007/s13679-016-0191-1
5. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature (2010) 464(7285):59–65. doi: 10.1038/nature08821
6. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature (2007) 449(7164):804–10. doi: 10.1038/nature06244
7. Koren O. Moody microbes: Do microbes influence our behavior? Eur Neuropsychopharmacol (2017) 27:S478. doi: 10.1016/j.euroneuro.2016.09.561
8. Martin AM, Sun EW, Rogers GB, Keating DJ. The influence of the gut microbiome on host metabolism through the regulation of gut hormone release. Front Physiol (2019) 10:428. doi: 10.3389/fphys.2019.00428
9. Van Treuren W, Dodd D. Microbial contribution to the human metabolome: Implications for health and disease. Annu Rev Pathol (2020) 15:345–69. doi: 10.1146/annurev-pathol-020117-043559
10. Huang TT, Lai JB, Du YL, Xu Y, Ruan LM, Hu SH. Current understanding of gut microbiota in mood disorders: An update of human studies. Front Genet (2019) 10:98. doi: 10.3389/fgene.2019.00098
11. Merchak A, Gaultier A. Microbial metabolites and immune regulation: New targets for major depressive disorder. Brain Behav Immun Health (2020) 9:100169. doi: 10.1016/j.bbih.2020.100169
12. Bultman SJ. Emerging roles of the microbiome in cancer. Carcinogenesis (2014) 35(2):249–55. doi: 10.1093/carcin/bgt392
13. von Frieling J, Fink C, Hamm J, Klischies K, Forster M, Bosch TCG, et al. Grow with the challenge - microbial effects on epithelial proliferation, carcinogenesis, and cancer therapy. Front Microbiol (2018) 9:2020. doi: 10.3389/fmicb.2018.02020
14. Al-Hilu SA, Al-Shujairi WH. Dual role of bacteria in carcinoma: Stimulation and inhibition. Int J Microbiol (2020) 2020:4639761. doi: 10.1155/2020/4639761
15. Mager DL. Bacteria and cancer: cause, coincidence or cure? a review. J Transl Med (2006) 4:14. doi: 10.1186/1479-5876-4-14
16. Díaz P, Valenzuela Valderrama M, Bravo J, Quest AFG. And gastric cancer: Adaptive cellular mechanisms involved in disease progression. Front Microbiol (2018) 9:5. doi: 10.3389/fmicb.2018.00005
17. Li Y, Ye Z, Zhu J, Fang S, Meng L, Zhou C. Effects of gut microbiota on host adaptive immunity under immune homeostasis and tumor pathology state. Front Immunol (2022) 13:844335. doi: 10.3389/fimmu.2022.844335
18. Sheflin AM, Whitney AK, Weir TL. Cancer-promoting effects of microbial dysbiosis. Curr Oncol Rep (2014) 16(10):406. doi: 10.1007/s11912-014-0406-0
19. Thomas S, Izard J, Walsh E, Batich K, Chongsathidkiet P, Clarke G, et al. The host microbiome regulates and maintains human health: A primer and perspective for non-microbiologists. Cancer Res (2017) 77(8):1783–812. doi: 10.1158/0008-5472.CAN-16-2929
20. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med (2018) 24(4):392–400. doi: 10.1038/nm.4517
21. Levy M, Thaiss CA, Elinav E. Metabolites: messengers between the microbiota and the immune system. Genes Dev (2016) 30(14):1589–97. doi: 10.1101/gad.284091.116
22. Hanus M, Parada-Venegas D, Landskron G, Wielandt AM, Hurtado C, Alvarez K, et al. Immune system, microbiota, and microbial metabolites: The unresolved triad in colorectal cancer microenvironment. Front Immunol (2021) 12:612826. doi: 10.3389/fimmu.2021.612826
23. Rossi T, Vergara D, Fanini F, Maffia M, Bravaccini S, Pirini F. Microbiota-derived metabolites in tumor progression and metastasis. Int J Mol Sci (2020) 21(16):5786. doi: 10.3390/ijms21165786
24. Krautkramer KA, Fan J, Bäckhed F. Gut microbial metabolites as multi-kingdom intermediates. Nat Rev Microbiol (2021) 19(2):77–94. doi: 10.1038/s41579-020-0438-4
25. Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science (2006) 313(5788):848–51. doi: 10.1126/science.1127059
26. Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A (2010) 107(25):11537–42. doi: 10.1073/pnas.1001261107
27. Mikó E, Kovács T, Sebő É, Tóth J, Csonka T, Ujlaki G, et al. Microbiome-microbial metabolome-cancer cell interactions in breast cancer-familiar, but unexplored. Cells (2019) 8(4):293. doi: 10.3390/cells8040293
28. Ting NL, Lau HC, Yu J. Cancer pharmacomicrobiomics: targeting microbiota to optimise cancer therapy outcomes. Gut (2022) 71:1412–25. doi: 10.1136/gutjnl-2021-326264
29. Oliva M, Mulet-Margalef N, Ochoa-De-Olza M, Napoli S, Mas J, Laquente B, et al. Tumor-associated microbiome: Where do we stand? Int J Mol Sci (2021) 22(3):1446. doi: 10.3390/ijms22031446
30. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature (2020) 579(7800):567–74. doi: 10.1038/s41586-020-2095-1
31. Robinson KM, Crabtree J, Mattick JS, Anderson KE, Dunning Hotopp JC. Distinguishing potential bacteria-tumor associations from contamination in a secondary data analysis of public cancer genome sequence data. Microbiome (2017) 5(1):9. doi: 10.1186/s40168-016-0224-8
32. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science (2020) 368(6494):973–80. doi: 10.1126/science.aay9189
33. Qiu Q, Lin Y, Ma Y, Li X, Liang J, Chen Z, et al. Exploring the emerging role of the gut microbiota and tumor microenvironment in cancer immunotherapy. Front Immunol (2020) 11:612202. doi: 10.3389/fimmu.2020.612202
34. Ma J, Huang L, Hu D, Zeng S, Han Y, Shen H. The role of the tumor microbe microenvironment in the tumor immune microenvironment: bystander, activator, or inhibitor? J Exp Clin Cancer Res (2021) 40(1):327. doi: 10.1186/s13046-021-02128-w
35. Mola S, Pandolfo C, Sica A, Porta C. The macrophages-microbiota interplay in colorectal cancer (CRC)-related inflammation: Prognostic and therapeutic significance. Int J Mol Sci (2020) 21(18):6886. doi: 10.3390/ijms21186866
36. Livyatan I, Nejman D, Shental N, Straussman R. Characterization of the human tumor microbiome reveals tumor-type specific intra-cellular bacteria. Oncoimmunology (2020) 9(1):1800957. doi: 10.1080/2162402X.2020.1800957
37. Jain T, Sharma P, Are AC, Vickers SM, Dudeja V. New insights into the cancer-Microbiome-Immune axis: Decrypting a decade of discoveries. Front Immunol (2021) 12:622064. doi: 10.3389/fimmu.2021.622064
38. American Cancer Society. Infections that can lead to cancer n.d . Available at: https://www.cancer.org/cancer/cancer-causes/infectious-agents/infections-that-can-lead-to-cancer.html.
39. Fujii YR. Oncoviruses and pathogenic MicroRNAs in humans. Open Virol J (2009) 3:37–51. doi: 10.2174/1874357900903010037
40. Tornesello ML, Annunziata C, Tornesello AL, Buonaguro L, Buonaguro FM. Human oncoviruses and p53 tumor suppressor pathway deregulation at the origin of human cancers. Cancers (Basel) (2018) 10(7):213. doi: 10.3390/cancers10070213
41. Zapatka M, Borozan I, Brewer DS, Iskar M, Grundhoff A, Alawi M, et al. The landscape of viral associations in human cancers. Nat Genet (2020) 52(3):320–30. doi: 10.1038/s41588-019-0558-9
42. de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health (2020) 8(2):e180–e90. doi: 10.1016/S2214-109X(19)30488-7
43. Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature (2019) 574(7777):264–7. doi: 10.1038/s41586-019-1608-2
44. Vallianou N, Kounatidis D, Christodoulatos GS, Panagopoulos F, Karampela I, Dalamaga M. Mycobiome and cancer: What is the evidence? Cancers (Basel) (2021) 13(13):3149. doi: 10.3390/cancers13133149
45. Zitvogel L, Galluzzi L, Viaud S, Vétizou M, Daillère R, Merad M, et al. Cancer and the gut microbiota: an unexpected link. Sci Transl Med (2015) 7(271):271ps1. doi: 10.1126/scitranslmed.3010473
46. Gaonkar PP, Patankar SR, Tripathi N, Sridharan G. Oral bacterial flora and oral cancer: The possible link? J Oral Maxillofac Pathol (2018) 22(2):234–8. doi: 10.4103/jomfp.JOMFP_89_16
47. Kyrgiou M, Mitra A, Moscicki AB. Does the vaginal microbiota play a role in the development of cervical cancer? Transl Res (2017) 179:168–82. doi: 10.1016/j.trsl.2016.07.004
48. Cheng WY, Wu CY, Yu J. The role of gut microbiota in cancer treatment: friend or foe? Gut (2020) 69(10):1867–76. doi: 10.1136/gutjnl-2020-321153
49. McNamara D, O'Morain C. Helicobacter pylori and gastric cancer. Ital J Gastroenterol Hepatol (1998) 30 Suppl 3:S294–8.
50. Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary b-cell gastric lymphoma. Lancet (1991) 338(8776):1175–6. doi: 10.1016/0140-6736(91)92035-Z
51. Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol (2016) 16(6):341–52. doi: 10.1038/nri.2016.42
52. Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A. Gut microbiota and metabolic syndrome. World J Gastroenterol (2014) 20(43):16079–94. doi: 10.3748/wjg.v20.i43.16079
53. Fang Y, Yan C, Zhao Q, Xu J, Liu Z, Gao J, et al. The roles of microbial products in the development of colorectal cancer: a review. Bioengineered (2021) 12(1):720–35. doi: 10.1080/21655979.2021.1889109
54. Wu HJ, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes (2012) 3(1):4–14. doi: 10.4161/gmic.19320
55. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell (2018) 33(4):570–80. doi: 10.1016/j.ccell.2018.03.015
56. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell (2017) 170(3):548–63.e16. doi: 10.1016/j.cell.2017.07.008
57. Coussens LM, Werb Z. Inflammation and cancer. Nature (2002) 420(6917):860–7. doi: 10.1038/nature01322
58. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
59. Khatun S, Appidi T, Rengan AK. The role played by bacterial infections in the onset and metastasis of cancer. Curr Res Microb Sci (2021) 2:100078. doi: 10.1016/j.crmicr.2021.100078
60. Lai CK, Chen YA, Lin CJ, Lin HJ, Kao MC, Huang MZ, et al. Molecular mechanisms and potential clinical applications of campylobacter jejuni cytolethal distending toxin. Front Cell Infect Microbiol (2016) 6:9. doi: 10.3389/fcimb.2016.00009
61. Graillot V, Dormoy I, Dupuy J, Shay JW, Huc L, Mirey G, et al. Genotoxicity of cytolethal distending toxin (CDT) on isogenic human colorectal cell lines: Potential promoting effects for colorectal carcinogenesis. Front Cell Infect Microbiol (2016) 6:34. doi: 10.3389/fcimb.2016.00034
62. Sun X, Wang M, Yao L, Li X, Dong H, Li M, et al. Role of proton-coupled monocarboxylate transporters in cancer: From metabolic crosstalk to therapeutic potential. Front Cell Dev Biol (2020) 8:651. doi: 10.3389/fcell.2020.00651
63. Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol (2001) 2(8):675–80. doi: 10.1038/90609
64. Heeg K, Dalpke A, Peter M, Zimmermann S. Structural requirements for uptake and recognition of CpG oligonucleotides. Int J Med Microbiol (2008) 298(1-2):33–8. doi: 10.1016/j.ijmm.2007.07.007
65. Tosch C, Geist M, Ledoux C, Ziller-Remi C, Paul S, Erbs P, et al. Adenovirus-mediated gene transfer of pathogen-associated molecular patterns for cancer immunotherapy. Cancer Gene Ther (2009) 16(4):310–9. doi: 10.1038/cgt.2008.85
66. Wolska A, Lech-Marańda E, Robak T. Toll-like receptors and their role in carcinogenesis and anti-tumor treatment. Cell Mol Biol Lett (2009) 14(2):248–72. doi: 10.2478/s11658-008-0048-z
67. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science (2018) 360(6391):5931. doi: 10.1126/science.aan5931
68. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell (2022) 13(12):877–919. doi: 10.1007/s13238-021-00846-7
69. Park JW, Han JW. Targeting epigenetics for cancer therapy. Arch Pharm Res (2019) 42(2):159–70. doi: 10.1007/s12272-019-01126-z
70. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
71. Newman TM, Vitolins MZ, Cook KL. From the table to the tumor: The role of Mediterranean and Western dietary patterns in shifting microbial-mediated signaling to impact breast cancer risk. Nutrients (2019) 11(11):2565. doi: 10.3390/nu11112565
72. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer (2013) 13(11):800–12. doi: 10.1038/nrc3610
73. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control T. Nature (2019) 576(7785):143–8. doi: 10.1038/s41586-019-1785-z
74. Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, et al. Microbial bile acid metabolites modulate gut RORγ. Nature (2020) 577(7790):410–5. doi: 10.1038/s41586-019-1865-0
75. Nguyen TT, Ung TT, Kim NH, Jung YD. Role of bile acids in colon carcinogenesis. World J Clin Cases (2018) 6(13):577–88. doi: 10.12998/wjcc.v6.i13.577
76. Fu J, Yu M, Xu W, Yu S. Research progress of bile acids in cancer. Front Oncol (2021) 11:778258. doi: 10.3389/fonc.2021.778258
77. Corrêa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MA. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunol (2016) 5(4):e73. doi: 10.1038/cti.2016.17
78. Mirzaei R, Afaghi A, Babakhani S, Sohrabi MR, Hosseini-Fard SR, Babolhavaeji K, et al. Role of microbiota-derived short-chain fatty acids in cancer development and prevention. BioMed Pharmacother (2021) 139:111619. doi: 10.1016/j.biopha.2021.111619
79. Sipe LM, Chaib M, Pingili AK, Pierre JF, Makowski L. Microbiome, bile acids, and obesity: How microbially modified metabolites shape anti-tumor immunity. Immunol Rev (2020) 295(1):220–39. doi: 10.1111/imr.12856
80. Singh V, Yeoh BS, Vijay-Kumar M. Feed your gut with caution! Transl Cancer Res (2016) 5(Suppl 3):S507–S13. doi: 10.21037/tcr.2016.09.13
81. Golonka RM, Vijay-Kumar M. Atypical immunometabolism and metabolic reprogramming in liver cancer: Deciphering the role of gut microbiome. Adv Cancer Res (2021) 149:171–255. doi: 10.1016/bs.acr.2020.10.004
82. Fong W, Li Q, Yu J. Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer. Oncogene (2020) 39(26):4925–43. doi: 10.1038/s41388-020-1341-1
83. Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients (2014) 7(1):17–44. doi: 10.3390/nu7010017
84. Cavalier-Smith T. Origins of secondary metabolism. In: Chadwick DJ, Whelan J, editors. Secondary metabolites: Their function and evolution (Chichester, Wiley) (1992). p. 64–80.
85. Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK. Specialized metabolites from the microbiome in health and disease. Cell Metab (2014) 20(5):719–30. doi: 10.1016/j.cmet.2014.10.016
86. Postler TS, Ghosh S. Understanding the holobiont: How microbial metabolites affect human health and shape the immune system. Cell Metab (2017) 26(1):110–30. doi: 10.1016/j.cmet.2017.05.008
87. Ferguson GP, Tötemeyer S, MacLean MJ, Booth IR. Methylglyoxal production in bacteria: suicide or survival? Arch Microbiol (1998) 170(4):209–18. doi: 10.1007/s002030050635
88. Cai M, Kandalai S, Tang X, Zheng Q. Contributions of human-associated archaeal metabolites to tumor microenvironment and carcinogenesis. Microbiol Spectr (2022) 10(2):e0236721. doi: 10.1128/spectrum.02367-21
89. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol (2014) 12(10):661–72. doi: 10.1038/nrmicro3344
90. Ernst L, Steinfeld B, Barayeu U, Klintzsch T, Kurth M, Grimm D, et al. Methane formation driven by reactive oxygen species across all living organisms. Nature (2022) 603:482–7. doi: 10.1038/s41586-022-04511-9
91. Gao FF, Quan JH, Lee MA, Ye W, Yuk JM, Cha GH, et al. Trichomonas vaginalis induces apoptosis via ROS and ER stress response through ER-mitochondria crosstalk in SiHa cells. Parasit Vectors (2021) 14(1):603. doi: 10.1186/s13071-021-05098-2
92. Ohnishi S, Ma N, Thanan R, Pinlaor S, Hammam O, Murata M, et al. DNA Damage in inflammation-related carcinogenesis and cancer stem cells. Oxid Med Cell Longev (2013) 2013:387014. doi: 10.1155/2013/387014
93. Aoyagi T, Takeuchi T, Matsuzaki A, Kawamura K, Kondo S. Leupeptins, new protease inhibitors from actinomycetes. J Antibiot (Tokyo) (1969) 22(6):283–6. doi: 10.7164/antibiotics.22.283
94. Guo CJ, Chang FY, Wyche TP, Backus KM, Acker TM, Funabashi M, et al. Discovery of reactive microbiota-derived metabolites that inhibit host proteases. Cell (2017) 168(3):517–26.e18. doi: 10.1016/j.cell.2016.12.021
95. Zheng Q, Wang Q, Wang S, Wu J, Gao Q, Liu W. Thiopeptide antibiotics exhibit a dual mode of action against intracellular pathogens by affecting both host and microbe. Chem Biol (2015) 22(8):1002–7. doi: 10.1016/j.chembiol.2015.06.019
96. Wang S, Zheng Q, Wang J, Zhao Z, Li Q, Yu Y, et al. Target-oriented design and biosynthesis of thiostrepton-derived thiopeptide antibiotics with improved pharmaceutical properties. Org Chem Front (2015) 2:106–9. doi: 10.1039/C4QO00288A
97. Bagley MC, Dale JW, Merritt EA, Xiong X. Thiopeptide antibiotics. Chem Rev (2005) 105(2):685–714. doi: 10.1021/cr0300441
98. Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, et al. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell (2014) 158(6):1402–14. doi: 10.1016/j.cell.2014.08.032
99. Vinogradov AA, Suga H. Introduction to thiopeptides: Biological activity, biosynthesis, and strategies for functional reprogramming. Cell Chem Biol (2020) 27(8):1032–51. doi: 10.1016/j.chembiol.2020.07.003
100. Secher T, Samba-Louaka A, Oswald E, Nougayrède JP. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS One (2013) 8(10):e77157. doi: 10.1371/journal.pone.0077157
101. Silpe JE, Wong JWH, Owen SV, Baym M, Balskus EP. The bacterial toxin colibactin triggers prophage induction. Nature (2022) 603:315–20. doi: 10.1038/s41586-022-04444-3
102. Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev (2001) 81(3):1031–64. doi: 10.1152/physrev.2001.81.3.1031
103. Boffa LC, Vidali G, Mann RS, Allfrey VG. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J Biol Chem (1978) 253(10):3364–6. doi: 10.1016/S0021-9258(17)34804-4
104. Cousens LS, Gallwitz D, Alberts BM. Different accessibilities in chromatin to histone acetylase. J Biol Chem (1979) 254(5):1716–23. doi: 10.1016/S0021-9258(17)37831-6
105. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem (2008) 19(9):587–93. doi: 10.1016/j.jnutbio.2007.08.002
106. Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr (2002) 132(5):1012–7. doi: 10.1093/jn/132.5.1012
107. Kiefer J, Beyer-Sehlmeyer G, Pool-Zobel BL. Mixtures of SCFA, composed according to physiologically available concentrations in the gut lumen, modulate histone acetylation in human HT29 colon cancer cells. Br J Nutr (2006) 96(5):803–10. doi: 10.1017/BJN20061948
108. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
109. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell (2017) 168(4):657–69. doi: 10.1016/j.cell.2016.12.039
110. Goodwin ML, Gladden LB, Nijsten MW, Jones KB. Lactate and cancer: revisiting the warburg effect in an era of lactate shuttling. Front Nutr (2014) 1:27. doi: 10.3389/fnut.2014.00027
111. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
112. Chen AN, Luo Y, Yang YH, Fu JT, Geng XM, Shi JP, et al. Lactylation, a novel metabolic reprogramming code: Current status and prospects. Front Immunol (2021) 12:688910. doi: 10.3389/fimmu.2021.688910
113. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A (2014) 111(6):2247–52. doi: 10.1073/pnas.1322269111
114. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun (2020) 11(1):4457. doi: 10.1038/s41467-020-18262-6
115. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
116. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
117. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science (2013) 341(6145):569–73. doi: 10.1126/science.1241165
118. Vinolo MA, Hatanaka E, Lambertucci RH, Newsholme P, Curi R. Effects of short chain fatty acids on effector mechanisms of neutrophils. Cell Biochem Funct (2009) 27(1):48–55. doi: 10.1002/cbf.1533
119. Emenaker NJ, Basson MD. Short chain fatty acids inhibit human (SW1116) colon cancer cell invasion by reducing urokinase plasminogen activator activity and stimulating TIMP-1 and TIMP-2 activities, rather than via MMP modulation. J Surg Res (1998) 76(1):41–6. doi: 10.1006/jsre.1998.5279
120. Emenaker NJ, Calaf GM, Cox D, Basson MD, Qureshi N. Short-chain fatty acids inhibit invasive human colon cancer by modulating uPA, TIMP-1, TIMP-2, mutant p53, bcl-2, bax, p21 and PCNA protein expression in an in vitro cell culture model. J Nutr (2001) 131(11 Suppl):3041S–6S. doi: 10.1093/jn/131.11.3041S
121. Zeng H, Briske-Anderson M. Prolonged butyrate treatment inhibits the migration and invasion potential of HT1080 tumor cells. J Nutr (2005) 135(2):291–5. doi: 10.1093/jn/135.2.291
122. Matsushita M, Fujita K, Hayashi T, Kayama H, Motooka D, Hase H, et al. Gut microbiota-derived short-chain fatty acids promote prostate cancer growth via IGF1 signaling. Cancer Res (2021) 81(15):4014–26. doi: 10.1158/0008-5472.CAN-20-4090
123. Chirakkal H, Leech SH, Brookes KE, Prais AL, Waby JS, Corfe BM. Upregulation of BAK by butyrate in the colon is associated with increased Sp3 binding. Oncogene (2006) 25(54):7192–200. doi: 10.1038/sj.onc.1209702
124. Buda A, Qualtrough D, Jepson MA, Martines D, Paraskeva C, Pignatelli M. Butyrate downregulates alpha2beta1 integrin: a possible role in the induction of apoptosis in colorectal cancer cell lines. Gut (2003) 52(5):729–34. doi: 10.1136/gut.52.5.729
125. Fung KY, Brierley GV, Henderson S, Hoffmann P, McColl SR, Lockett T, et al. Butyrate-induced apoptosis in HCT116 colorectal cancer cells includes induction of a cell stress response. J Proteome Res (2011) 10(4):1860–9. doi: 10.1021/pr1011125
126. Comalada M, Bailón E, de Haro O, Lara-Villoslada F, Xaus J, Zarzuelo A, et al. The effects of short-chain fatty acids on colon epithelial proliferation and survival depend on the cellular phenotype. J Cancer Res Clin Oncol (2006) 132(8):487–97. doi: 10.1007/s00432-006-0092-x
127. Zgouras D, Wächtershäuser A, Frings D, Stein J. Butyrate impairs intestinal tumor cell-induced angiogenesis by inhibiting HIF-1alpha nuclear translocation. Biochem Biophys Res Commun (2003) 300(4):832–8. doi: 10.1016/S0006-291X(02)02916-9
128. Terui T, Murakami K, Takimoto R, Takahashi M, Takada K, Murakami T, et al. Induction of PIG3 and NOXA through acetylation of p53 at 320 and 373 lysine residues as a mechanism for apoptotic cell death by histone deacetylase inhibitors. Cancer Res (2003) 63(24):8948–54.
129. Gope R, Gope ML. Effect of sodium butyrate on the expression of retinoblastoma (RB1) and P53 gene and phosphorylation of retinoblastoma protein in human colon tumor cell line HT29. Cell Mol Biol (Noisy-le-grand) (1993) 39(6):589–97.
130. Cook JW, Kennaway EL, Kennaway NM. Production of tumours in mice by deoxycholic acid. Nature (1940) 145(3677):627. doi: 10.1038/145627a0
131. Májer F, Sharma R, Mullins C, Keogh L, Phipps S, Duggan S, et al. New highly toxic bile acids derived from deoxycholic acid, chenodeoxycholic acid and lithocholic acid. Bioorg Med Chem (2014) 22(1):256–68. doi: 10.1016/j.bmc.2013.11.029
132. Narahara H, Tatsuta M, Iishi H, Baba M, Uedo N, Sakai N, et al. K-Ras point mutation is associated with enhancement by deoxycholic acid of colon carcinogenesis induced by azoxymethane, but not with its attenuation by all-trans-retinoic acid. Int J Cancer (2000) 88(2):157–61. doi: 10.1002/1097-0215(20001015)88:2<157::AID-IJC2>3.0.CO;2-B
133. Powolny A, Xu J, Loo G. Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53. Int J Biochem Cell Biol (2001) 33(2):193–203. doi: 10.1016/S1357-2725(00)00080-7
134. Arvind P, Papavassiliou ED, Tsioulias GJ, Duceman BW, Lovelace CI, Geng W, et al. Lithocholic acid inhibits the expression of HLA class I genes in colon adenocarcinoma cells. differential effect on HLA-a, -b and -c loci. Mol Immunol (1994) 31(8):607–14. doi: 10.1016/0161-5890(94)90168-6
135. Gafar AA, Draz HM, Goldberg AA, Bashandy MA, Bakry S, Khalifa MA, et al. Lithocholic acid induces endoplasmic reticulum stress, autophagy and mitochondrial dysfunction in human prostate cancer cells. PeerJ (2016) 4:e2445. doi: 10.7717/peerj.2445
136. Martinez JD, Stratagoules ED, LaRue JM, Powell AA, Gause PR, Craven MT, et al. Different bile acids exhibit distinct biological effects: the tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferation. Nutr Cancer (1998) 31(2):111–8. doi: 10.1080/01635589809514689
137. Crowley-Weber CL, Payne CM, Gleason-Guzman M, Watts GS, Futscher B, Waltmire CN, et al. Development and molecular characterization of HCT-116 cell lines resistant to the tumor promoter and multiple stress-inducer, deoxycholate. Carcinogenesis (2002) 23(12):2063–80. doi: 10.1093/carcin/23.12.2063
138. Wu G, Morris SM. Arginine metabolism: nitric oxide and beyond. Biochem J (1998) 336(Pt 1):1–17. doi: 10.1042/bj3360001
139. Tabor CW, Tabor H. Polyamines in microorganisms. Microbiol Rev (1985) 49(1):81–99. doi: 10.1128/mr.49.1.81-99.1985
140. Di Martino ML, Campilongo R, Casalino M, Micheli G, Colonna B, Prosseda G. Polyamines: emerging players in bacteria-host interactions. Int J Med Microbiol (2013) 303(8):484–91. doi: 10.1016/j.ijmm.2013.06.008
141. Khan AU, Mei YH, Wilson T. A proposed function for spermine and spermidine: protection of replicating DNA against damage by singlet oxygen. Proc Natl Acad Sci U S A (1992) 89(23):11426–7. doi: 10.1073/pnas.89.23.11426
142. Linsalata M, Caruso MG, Leo S, Guerra V, D'Attoma B, Di Leo A. Prognostic value of tissue polyamine levels in human colorectal carcinoma. Anticancer Res (2002) 22(4):2465–9.
143. Linsalata M, Giannini R, Notarnicola M, Cavallini A. Peroxisome proliferator-activated receptor gamma and spermidine/spermine N1-acetyltransferase gene expressions are significantly correlated in human colorectal cancer. BMC Cancer (2006) 6:191. doi: 10.1186/1471-2407-6-191
144. Smith RC, Litwin MS, Lu Y, Zetter BR. Identification of an endogenous inhibitor of prostatic carcinoma cell growth. Nat Med (1995) 1(10):1040–5. doi: 10.1038/nm1095-1040
145. Kee K, Foster BA, Merali S, Kramer DL, Hensen ML, Diegelman P, et al. Activated polyamine catabolism depletes acetyl-CoA pools and suppresses prostate tumor growth in TRAMP mice. J Biol Chem (2004) 279(38):40076–83. doi: 10.1074/jbc.M406002200
146. Kee K, Vujcic S, Merali S, Diegelman P, Kisiel N, Powell CT, et al. Metabolic and antiproliferative consequences of activated polyamine catabolism in LNCaP prostate carcinoma cells. J Biol Chem (2004) 279(26):27050–8. doi: 10.1074/jbc.M403323200
147. Huang Y, Keen JC, Pledgie A, Marton LJ, Zhu T, Sukumar S, et al. Polyamine analogues down-regulate estrogen receptor alpha expression in human breast cancer cells. J Biol Chem (2006) 281(28):19055–63. doi: 10.1074/jbc.M600910200
148. Xu H, Chaturvedi R, Cheng Y, Bussiere FI, Asim M, Yao MD, et al. Spermine oxidation induced by helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Res (2004) 64(23):8521–5. doi: 10.1158/0008-5472.CAN-04-3511
149. Chaturvedi R, Cheng Y, Asim M, Bussière FI, Xu H, Gobert AP, et al. Induction of polyamine oxidase 1 by helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J Biol Chem (2004) 279(38):40161–73. doi: 10.1074/jbc.M401370200
150. Bussière FI, Chaturvedi R, Cheng Y, Gobert AP, Asim M, Blumberg DR, et al. Spermine causes loss of innate immune response to helicobacter pylori by inhibition of inducible nitric-oxide synthase translation. J Biol Chem (2005) 280(4):2409–12. doi: 10.1074/jbc.C400498200
151. Gobert AP, Chaturvedi R, Wilson KT. Methods to evaluate alterations in polyamine metabolism caused by helicobacter pylori infection. Methods Mol Biol (2011) 720:409–25. doi: 10.1007/978-1-61779-034-8_26
152. Wang X, Feith DJ, Welsh P, Coleman CS, Lopez C, Woster PM, et al. Studies of the mechanism by which increased spermidine/spermine N1-acetyltransferase activity increases susceptibility to skin carcinogenesis. Carcinogenesis (2007) 28(11):2404–11. doi: 10.1093/carcin/bgm162
153. Coleman CS, Pegg AE, Megosh LC, Guo Y, Sawicki JA, O'Brien TG. Targeted expression of spermidine/spermine N1-acetyltransferase increases susceptibility to chemically induced skin carcinogenesis. Carcinogenesis (2002) 23(2):359–64. doi: 10.1093/carcin/23.2.359
154. Latour YL, Gobert AP, Wilson KT. The role of polyamines in the regulation of macrophage polarization and function. Amino Acids (2020) 52(2):151–60. doi: 10.1007/s00726-019-02719-0
155. Vander Jagt DL, Robinson B, Taylor KK, Hunsaker LA. Reduction of trioses by NADPH-dependent aldo-keto reductases. aldose reductase, methylglyoxal, and diabetic complications. J Biol Chem (1992) 267(7):4364–9. doi: 10.1016/S0021-9258(18)42844-X
156. Speer O, Morkunaite-Haimi S, Liobikas J, Franck M, Hensbo L, Linder MD, et al. Rapid suppression of mitochondrial permeability transition by methylglyoxal. role of reversible arginine modification. J Biol Chem (2003) 278(37):34757–63. doi: 10.1074/jbc.M301990200
157. Baskaran S, Rajan DP, Balasubramanian KA. Formation of methylglyoxal by bacteria isolated from human faeces. J Med Microbiol (1989) 28(3):211–5. doi: 10.1099/00222615-28-3-211
158. Richarme G, Mihoub M, Dairou J, Bui LC, Leger T, Lamouri A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J Biol Chem (2015) 290(3):1885–97. doi: 10.1074/jbc.M114.597815
159. Richarme G, Liu C, Mihoub M, Abdallah J, Leger T, Joly N, et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science (2017) 357(6347):208–11. doi: 10.1126/science.aag1095
160. Zheng Q, Maksimovic I, Upad A, David Y. Non-enzymatic covalent modifications: a new link between metabolism and epigenetics. Protein Cell (2020) 11(6):401–16. doi: 10.1007/s13238-020-00722-w
161. Zheng Q, Prescott NA, Maksimovic I, David Y. (De)Toxifying the epigenetic code. Chem Res Toxicol (2019) 32(5):796–807. doi: 10.1021/acs.chemrestox.9b00013
162. Zheng Q, Maksimovic I, Upad A, Guber D, David Y. Synthesis of an alkynyl methylglyoxal probe to investigate nonenzymatic histone glycation. J Org Chem (2020) 85(3):1691–7. doi: 10.1021/acs.joc.9b02504
163. Zheng Q, Omans ND, Leicher R, Osunsade A, Agustinus AS, Finkin-Groner E, et al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat Commun (2019) 10(1):1289. doi: 10.1038/s41467-019-09192-z
164. Zheng Q, Osunsade A, David Y. Protein arginine deiminase 4 antagonizes methylglyoxal-induced histone glycation. Nat Commun (2020) 11(1):3241. doi: 10.1038/s41467-020-17066-y
165. Ray DM, Jennings EQ, Maksimovic I, Chai X, Galligan JJ, David Y, et al. Chemical labeling and enrichment of histone glyoxal adducts. ACS Chem Biol (2022) 17(4):756–61. doi: 10.1021/acschembio.1c00864
166. Schalkwijk CG, Stehouwer CDA. Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol Rev (2020) 100(1):407–61. doi: 10.1152/physrev.00001.2019
167. Bellahcène A, Nokin MJ, Castronovo V, Schalkwijk C. Methylglyoxal-derived stress: An emerging biological factor involved in the onset and progression of cancer. Semin Cancer Biol (2018) 49:64–74. doi: 10.1016/j.semcancer.2017.05.010
168. Nokin MJ, Durieux F, Peixoto P, Chiavarina B, Peulen O, Blomme A, et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. Elife (2016) 5:e19375. doi: 10.7554/eLife.19375
169. Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature (2018) 562(7728):600–4. doi: 10.1038/s41586-018-0622-0
170. Maksimovic I, Finkin-Groner E, Fukase Y, Zheng Q, Sun S, Michino M, et al. Deglycase-activity oriented screening to identify DJ-1 inhibitors. RSC Med Chem (2021) 12(7):1232–8. doi: 10.1039/D1MD00062D
171. Garrett WS. Cancer and the microbiota. Science (2015) 348(6230):80–6. doi: 10.1126/science.aaa4972
172. Griffin ME, Espinosa J, Becker JL, Luo JD, Carroll TS, Jha JK, et al. Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science (2021) 373(6558):1040–6. doi: 10.1126/science.abc9113
173. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science (2020) 369(6510):1481–9. doi: 10.1126/science.abc3421
174. Zheng DW, Deng WW, Song WF, Wu CC, Liu J, Hong S, et al. Biomaterial-mediated modulation of oral microbiota synergizes with PD-1 blockade in mice with oral squamous cell carcinoma. Nat BioMed Eng (2022) 6(1):32–43. doi: 10.1038/s41551-021-00807-9
175. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (2017) 357(6356):1156–60. doi: 10.1126/science.aah5043
176. Kramer MG, Masner M, Ferreira FA, Hoffman RM. Bacterial therapy of cancer: Promises, limitations, and insights for future directions. Front Microbiol (2018) 9:16. doi: 10.3389/fmicb.2018.00016
177. Canale FP, Basso C, Antonini G, Perotti M, Li N, Sokolovska A, et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature (2021) 598(7882):662–6. doi: 10.1038/s41586-021-04003-2
178. Chowdhury S, Castro S, Coker C, Hinchliffe TE, Arpaia N, Danino T. Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat Med (2019) 25(7):1057–63. doi: 10.1038/s41591-019-0498-z
179. Gurbatri CR, Lia I, Vincent R, Coker C, Castro S, Treuting PM, et al. Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci Transl Med (2020) 12(530):eaax0876. doi: 10.1126/scitranslmed.aax0876
180. Zhou S, Gravekamp C, Bermudes D, Liu K. Tumour-targeting bacteria engineered to fight cancer. Nat Rev Cancer (2018) 18(12):727–43. doi: 10.1038/s41568-018-0070-z
181. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol (2012) 30(7):658–70. doi: 10.1038/nbt.2287
182. Sze DY, Reid TR, Rose SC. Oncolytic virotherapy. J Vasc Interv Radiol (2013) 24(8):1115–22. doi: 10.1016/j.jvir.2013.05.040
183. Li L, Liu S, Han D, Tang B, Ma J. Delivery and biosafety of oncolytic virotherapy. Front Oncol (2020) 10:475. doi: 10.3389/fonc.2020.00475
184. Lin Y, Zhang H, Liang J, Li K, Zhu W, Fu L, et al. Identification and characterization of alphavirus M1 as a selective oncolytic virus targeting ZAP-defective human cancers. Proc Natl Acad Sci U S A (2014) 111(42):E4504–12. doi: 10.1073/pnas.1408759111
185. Wang G, Kang X, Chen KS, Jehng T, Jones L, Chen J, et al. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat Commun (2020) 11(1):1395. doi: 10.1038/s41467-020-15229-5
186. Mondal M, Guo J, He P, Zhou D. Recent advances of oncolytic virus in cancer therapy. Hum Vaccin Immunother (2020) 16(10):2389–402. doi: 10.1080/21645515.2020.1723363
187. Fukuhara H, Takeshima Y, Todo T. Triple-mutated oncolytic herpes virus for treating both fast- and slow-growing tumors. Cancer Sci (2021) 112(8):3293–301. doi: 10.1111/cas.14981
189. Wiemann B, Starnes CO. Coley's toxins, tumor necrosis factor and cancer research: a historical perspective. Pharmacol Ther (1994) 64(3):529–64. doi: 10.1016/0163-7258(94)90023-X
190. Salomon BL, Leclerc M, Tosello J, Ronin E, Piaggio E, Cohen JL. Tumor necrosis factor α and regulatory T cells in oncoimmunology. Front Immunol (2018) 9:444. doi: 10.3389/fimmu.2018.00444
191. Chen AY, Wolchok JD, Bass AR. TNF in the era of immune checkpoint inhibitors: friend or foe? Nat Rev Rheumatol (2021) 17(4):213–23. doi: 10.1038/s41584-021-00584-4
192. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (2013) 342(6161):967–70. doi: 10.1126/science.1240527
193. Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A, et al. Microbial short-chain fatty acids modulate CD8. Nat Commun (2021) 12(1):4077. doi: 10.1038/s41467-021-24331-1
194. Zheng DW, Dong X, Pan P, Chen KW, Fan JX, Cheng SX, et al. Phage-guided modulation of the gut microbiota of mouse models of colorectal cancer augments their responses to chemotherapy. Nat BioMed Eng (2019) 3(9):717–28. doi: 10.1038/s41551-019-0423-2
195. Song W, Tiruthani K, Wang Y, Shen L, Hu M, Dorosheva O, et al. Trapping of lipopolysaccharide to promote immunotherapy against colorectal cancer and attenuate liver metastasis. Adv Mater (2018) 30(52):e1805007. doi: 10.1002/adma.201805007
196. Li L, Yang Z, Chen X. Recent advances in stimuli-responsive platforms for cancer immunotherapy. Acc Chem Res (2020) 53(10):2044–54. doi: 10.1021/acs.accounts.0c00334
197. Seitz I, Shaukat A, Nurmi K, Ijäs H, Hirvonen J, Santos HA, et al. Prospective cancer therapies using stimuli-responsive DNA nanostructures. Macromol Biosci (2021) 21(12):e2100272. doi: 10.1002/mabi.202100272
198. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discovery (2008) 7(9):771–82. doi: 10.1038/nrd2614
199. Ye QN, Wang Y, Shen S, Xu CF, Wang J. Biomaterials-based delivery of therapeutic antibodies for cancer therapy. Adv Healthc Mater (2021) 10(11):e2002139. doi: 10.1002/adhm.202002139
200. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol (2017) 12(8):813–20. doi: 10.1038/nnano.2017.57
201. Grosskopf AK, Labanieh L, Klysz DD, Roth GA, Xu P, Adebowale O, et al. Delivery of CAR-T cells in a transient injectable stimulatory hydrogel niche improves treatment of solid tumors. Sci Adv (2022) 8(14):eabn8264. doi: 10.1126/sciadv.abn8264
202. Pan H, Zheng M, Ma A, Liu L, Cai L. Cell/Bacteria-based bioactive materials for cancer immune modulation and precision therapy. Adv Mater (2021) 33(50):e2100241. doi: 10.1002/adma.202100241
203. Liu Y, Li Z, Wu Y, Jing X, Li L, Fang X. Intestinal bacteria encapsulated by biomaterials enhance immunotherapy. Front Immunol (2020) 11:620170. doi: 10.3389/fimmu.2020.620170
Keywords: tumor microbiome, metabolism, cancer therapy, cancer development, immune response
Citation: Zhou X, Kandalai S, Hossain F and Zheng Q (2022) Tumor microbiome metabolism: A game changer in cancer development and therapy. Front. Oncol. 12:933407. doi: 10.3389/fonc.2022.933407
Received: 30 April 2022; Accepted: 28 June 2022;
Published: 25 July 2022.
Edited by:
Domenica Scumaci, Magna Græcia University of Catanzaro, ItalyReviewed by:
Liza Makowski, University of Tennessee Health Science Center (UTHSC), United StatesCopyright © 2022 Zhou, Kandalai, Hossain and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingfei Zheng, UWluZ2ZlaS5aaGVuZ0Bvc3VtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.