
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 03 August 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.931209
This article is part of the Research TopicIdentification of Immune-Related Biomarkers for Cancer Diagnosis Based on Multi-Omics DataView all 31 articles
 Yansong Huo1†
Yansong Huo1† Leina Sun2†Jie Yuan3†
Leina Sun2†Jie Yuan3† Hua Zhang1†
Hua Zhang1† Zhenfa Zhang1
Zhenfa Zhang1 Lianmin Zhang1
Lianmin Zhang1 Wuhao Huang1Xiaoyan Sun1Zhe Tang1Yingnan Feng1Huilan Mo1Zuoquan Yang3
Wuhao Huang1Xiaoyan Sun1Zhe Tang1Yingnan Feng1Huilan Mo1Zuoquan Yang3 Chao Zhang3Zicheng Yu3
Chao Zhang3Zicheng Yu3 Dongsheng Yue1*
Dongsheng Yue1* Bin Zhang1*
Bin Zhang1* Changli Wang1*
Changli Wang1*Lung adenocarcinoma (LUAD) usually contains heterogeneous histological subtypes, among which the micropapillary (MIP) subtype was associated with poor prognosis while the lepidic (LEP) subtype possessed the most favorable outcome. However, the genomic features of the MIP subtype responsible for its malignant behaviors are substantially unknown. In this study, eight FFPE samples from LUAD patients were micro-dissected to isolate MIP and LEP components, then sequenced by whole-exome sequencing. More comprehensive analyses involving our samples and public validation cohorts on the two subtypes were performed to better decipher the key biological and evolutionary mechanisms. As expected, the LEP and MIP subtypes exhibited the largest disease-free survival (DFS) differences in our patients. EGFR was found with the highest mutation frequency. Additionally, shared mutations were observed between paired LEP and MIP components from single patients, and recurrent mutations were verified in the Lung-Broad, Lung-OncoSG, and TCGA-LUAD cohorts. Distinct biological processes or pathways were involved in the evolution of the two components. Besides, analyses of copy number variation (CNV) and intratumor heterogeneity (ITH) further discovered the possible immunosurveillance escape, the discrepancy between mutation and CNV level, ITH, and the pervasive DNA damage response and WNT pathway gene alternations in the MIP component. Phylogenetic analysis of five pairs of LEP and MIP components further confirmed the presence of ancestral EGFR mutations. Through comprehensive analyses combining our samples and public cohorts, PTP4A3, NAPRT, and RECQL4 were identified to be co-amplified. Multi-omics data also demonstrated the immunosuppression prevalence in the MIP component. Our results uncovered the evolutionary pattern of the concomitant LEP and MIP components from the same patient that they were derived from the same initiation cells and the pathway-specific mutations acquired after EGFR clonal mutation could shape the subtype-specificity. We also confirmed the immunosuppression prevalence in the MIP subtype by multi-omics data analyses, which may have resulted in its unfavorable prognosis.
Lung adenocarcinoma (LUAD) is the most common histological type of non-small-cell lung cancer (NSCLC) (1). Most cases of adenocarcinoma are composed of heterogeneous histological subtypes rather than a single one. In the year of 2015, the World Health Organization (WHO) proposed a novel definition of five LUAD subtypes to address the histologic heterogeneity, including the lepidic (LEP), acinar (ACI), papillary (PAP), micropapillary (MIP), and solid (SOL) pattern types. Patients presenting with MIP are prone to lymphovascular invasion and pleural invasion, as well as lymph node or intrapulmonary metastasis after surgical resection (2). Meanwhile, previous studies indicated that patients with a LEP growth pattern exhibited less aggressive behavior and had the most favorable outcomes among the predefined subtypes (3).
Aiming for the elucidation of the mechanisms beyond tumorigenesis and malignance discrepancy, several studies were conducted to evaluate the molecular and genetic features of LUAD subtypes, especially on MIP and LEP. As for the MIP subtype, a recent study observed the disruption of the catenin–cadherin complex (4), which possibly contributed to its poor intercellular adherence. At the genetic level, MIP/SOL tumors have a significantly higher tumor mutation burden (TMB) and fraction of the genome altered than other LUAD subtypes. Key oncogenes BRAF and EGFR were found with higher mutation frequency in LUAD with MIP in multiregional and multiracial cohorts (5). The gene and protein levels of c-MET were also found to be elevated in MIP and patients with a poor prognosis (5, 6). Although dysregulated oncogenes associated with poorer prognoses of MIP-predominant LUAD were identified, there remain key mechanisms that are uncharacterized. For example, the genetic association between subtypes and the evolutional trajectory of the relatively malignant MIP subtype was scarcely discussed.
Noticing the recent emergence of lung cancer immunotherapy, studies assessing the efficacy of immune-related therapies on MIP-predominant LUAD have emerged. Considering the abundance of programmed death-ligand 1 (PD-L1) and programmed cell death protein 1 (PD-1) as well as the tumor immunological microenvironment crucially influence the immunotherapy effectiveness, Francois et al. found the significant differences in PD-L1 expression levels between LUAD histological patterns (7), while Zhang et al. detected higher CD4+ and CD8+ T-cell infiltration as well as increased PD-L1 abundance in samples with a higher percentage of MIP components through the immunohistochemistry staining (8). Regarding the fact that both the studies focused on restricted components of the tumor microenvironment (TME), a more comprehensive analysis of the variation of TME in specific LUAD subtypes could fill the gap in optimal treatment determination, especially for MIP patients.
To address the abovementioned limitations, we retrospectively reviewed 286 patients with different histological subtype-predominance and compared their survival differences. Patients simultaneously possessing MIP and LEP components were further selected for whole-exome sequencing (WES) on both LEP and MIP components, and the genetic differences responsible for variated prognosis and the subtype-level genetic association was investigated. Multi-cohort analyses further discovered the genes specifically altered in MIP or LEP as well as the extent of immune infiltration. Our results expanded the evolution of cognition between the LUAD subtypes and offered therapeutic suggestions for MIP patients.
We retrospectively reviewed patients diagnosed with LUAD at the Tianjin Cancer Hospital from 2011 to 2014. Among patients who underwent tumor resection, those with an MIP component exceeding 5% of the area size were primarily selected. Patients receiving pre-surgery anticancer treatment, with stage IV disease or other malignancies were excluded. A total of 286 patients passed the selection criteria, and the resected tumors were restaged according to the eighth edition of the American Joint Committee on Cancer TNM staging system for lung cancer. For the LUAD histological subtyping, the formalin-fixed paraffin-embedded (FFPE) samples were first stained with hematoxylin and eosin (H&E) and reviewed by two pathologists. The percentage of each histological component was further calculated in 5% increments and the most dominant pattern was recorded. This study was approved by our institutional review board. Written informed consent was obtained from all patients.
Eight FFPE samples from LUAD patients who underwent surgery at the Tianjin Cancer Hospital between 2018 and 2020 were micro-dissected to isolate MIP and LEP components using a NIKON ECLIPSE TI2, Japan microscope. More specifically, 20 FFPE slides were cut into 10 um thick sections, baked for 1 h at 60 °C, stained with H&E, and immersed in xylene. The MIP and LEP areas were circumscribed electronically under the microscope and collected in a centrifugation tube with an adhesive-cap after ablating with a cold laser. Later, genomic DNA of the five pairs of MIP and LEP components plus one MIP component passing initial quality control was extracted by a Maxwell 16 FFPE Plus LEV DNA purification kit and fragmented by an ultra-sonicator UCD-200 (Diagenode, Seraing, Belgium) with length-based selection through Hieff NGS DNA selection beads. DNA quantity was assessed by a Qubit 2.0 Fluorometer with a Quanti-IT dsDNA HS Assay Kit (Thermo Fisher Scientific, MA, USA). The sequencing libraries were further constructed by a custom 53 M whole-exon capturing probe (IDT, IA, USA). The Geneplus-2000 sequencing platform (Geneplus, Beijing, China) further sequenced the libraries in a 100 bp paired-end manner.
Raw sequencing data were primarily filtered on the total read volume, GC content, Q30 percentage, and duplication rate. Later read alignment to the human genome (hg19) was performed by BWA (9) (version 0.7.10). After sample coverage filtration, MuTect (10) (version 1.1.4) from the GATK (version 4.0) pipeline identified single nucleotide variants (SNVs), small insertions and deletions (InDels), while segment-level somatic copy number alternations (SCNAs) were detected by GATK. Several rounds of filtration on SNVs were conducted, including (1) retaining variants with low frequency (≤0.01) in a population from the 1,000 Genomes Project (https://www.internationalgenome.org/), the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org), and the Exome Aggregation Consortium (ExAC); (2) keeping mutations with a number of supporting reads greater than 3; (3) keeping mutations with non-zero variant allele frequency (VAF) (≥0.01); and (4) only functional alternations were preserved. Cancer-associated genes and cancer driver genes were collected from the Cancer Gene Census in the COSMIC database (https://cancer.sanger.ac.uk/census) and two pan-cancer publications (11, 12) for further comparisons. As for SCNAs, an in-house script (13) employed statistical significance between tumor and normal tissues for focal level SCNA inference. Additionally, the mutational spectrum and absolute contribution of COSMIC v3 SBS (single base substitution) mutational signatures were derived by MutationalPatterns (14) on unfiltered somatic mutations, while Sigminer (15) quantified the absolute exposures of COSMIC v3 DBS (double base substitution) and ID (InDel) signatures.
We measured the intratumor heterogeneity (ITH) of samples on both SNV and SCNA. For filtered SNVs, the mutant-allele tumor heterogeneity (MATH) score (16) was calculated using VAF values. The ABSOLUTE (17) tool further estimated the cancer ploidy, tumor purity, rescaled copy ratio, and cancer cell fraction (CCF) by combining SNV and SCNA data. The clonal architectures of SNVs were derived from the higher clonal mutation probability and the CCF upper 95% confidence interval greater than 1. For SCNAs, copy-neutral LOH (CNLOH) segments were initially discarded, and clonal architectures were further annotated using allelic subclonal information from ABSOLUTE outputs. Additionally, by using an in-house script, we constructed the phylogenetic trees by identifying shared and private mutations or focal SCNAs in concomitant MIP and LEP components from one patient. Clonal mutations existing in both components constituted the trunk of the phylogenetic tree, while private mutations constituted the tree branches. Focal SCNA information was also considered in the tree construction procedure, i.e., marking the shared focal SCNA between MIP and LEP.
For the integrative analysis of SNVs and SCNAs, DNA damage repair (DDR) related genes were collected from a previous publication (18). The populational structures of mutations were identified on filtered SNVs and annotated SCNAs by PyClone-VI (19). These clone clusters were visualized by ClonEvol (20). The Enrichr (21) tool was used for pathway enrichment and Gene Oncology (GO) analysis. Enriched GO Biological Processes and Reactome (22) pathway entries were reported with P-values.
For multi-omics data comparison, SNV, SCNA, transcriptomic, and proteomic data were retrieved from multiple LUAD datasets. More specifically, SNV and SCNA data from four datasets (Lung-Broad (23), Lung-MSKCC, Lung-OncoSG (24), and TCGA-LUAD) were downloaded from the cBioPortal for Cancer Genomics database (25) or the UCSC Xena database (26) and only non-synonymous mutations were retained. Survival information is also downloaded if available. Additionally, transcriptomic data from the Lung-OncoSG, TCGA-LUAD, and one GEO dataset GSE148801 (27) containing good-prognosis (e.g., LEP, ACI, and PAP histological subtypes) and poor-prognosis (e.g., MIP and SOL) samples were collected while proteomics data from the TCGA-LUAD were similarly curated. Only data from LEP and MIP subtypes were used for further comparisons.
Immune infiltration analysis was conducted on curated transcriptomic samples for comparison between LEP and MIP components. Regarding the recognition, response, and killing of cancer cells by the immune system in a step-wise manner, i.e., the cancer immunity cycle (28), we applied the single sample Gene Set Enrichment Analysis (ssGSEA) method from the GSVA R package (http://bioconductor.org/packages/release/bioc/html/GSVA.html) to assess the activities of these steps in the cycle. More specifically, gene signature sets for steps of the cancer immunity cycle were first downloaded from the TIP database (http://biocc.hrbmu.edu.cn/TIP/). Later, the gsva function in the GSVA R package was applied in the step activity quantification procedure.
A two-sided Mann–Whitney test was used for evaluating group-level differences between LEP and MIP components. As for multiple comparisons, P-values were adjusted by the Benjamini–Hochberg method. When the comparisons were conducted on categorical data, Fisher’s exact test was used. As for the protein expression data, a one-sided Student’s t-test was used for comparison. For all tests, a P-value (adjusted P-value) <0.05 was considered statistically significant. The Kaplan–Meier (K-M) survival curves were generated by the survminer package (https://rpkgs.datanovia.com/survminer/) and the P-values were calculated using the log-rank test.
Among the 286 patients, 51 (17.8%) were LEP-predominant, 178 (62.2%) were ACI-predominant, 16 (5.6%) were PAP-predominant, 29 (10.1%) were MIP-predominant, and 12 (4.2%) were SOL-predominant adenocarcinoma. The clinicopathologic characteristics of all patients are summarized in Supplementary Table 1. As shown in Supplementary Table 2 and Supplementary Figure 1, patients with the MIP-predominant subtype exhibited a significantly worse DFS than those with other adenocarcinoma subtypes (P <0.05, Supplementary Figure 1A), and LEP and MIP subtypes exhibited the largest DFS difference, while SOL-predominant patients showed significantly worse OS (P <0.05, Supplementary Figure 1B).
Among eight micro-dissected samples, six cases passed quality control, but the quantity of LEP component in one case was inadequate. Six MIP and five LEP components were finally sequenced (Supplementary Table 3 and Figure 1A). A total of 2,035 and 2,757 SNVs and InDels were identified, while 684/791 and 257/284 mutations were retained after quality and cancer-related gene filtration (Supplementary Figure 2Aand Supplementary Table 4). Genes with the highest mutation frequency after quality filtering are shown in Figure 1B. EGFR was identified to be the most frequently mutated drive gene, in line with the finding that LEP and MIP components possess significantly higher EGFR mutation frequencies (29). Besides, several cancer-associated genes including TP53, TRIO, CEBPA, PCLO, and PDE4DIP were concomitantly mutated (Figure 1B), denoting p53, WNT-beta-catenin signaling, PI3K/AKT/mTOR signaling, and DNA repair pathways were affected. Interestingly, shared mutations were observed between paired LEP and MIP components from single patients (Figure 1B), raising the presumption that the paired LEP and MIP components could be homogeneous. We also compared the mutation frequency of these genes with public cohorts, including Lung-Broad, Lung-OncoSG, and TCGA-LUAD. Several cancer-associated genes, including EGFR, TP53, TRIO, PCLO, and PDE4DIP, were found recurrently mutated (Supplementary Figure 2B).
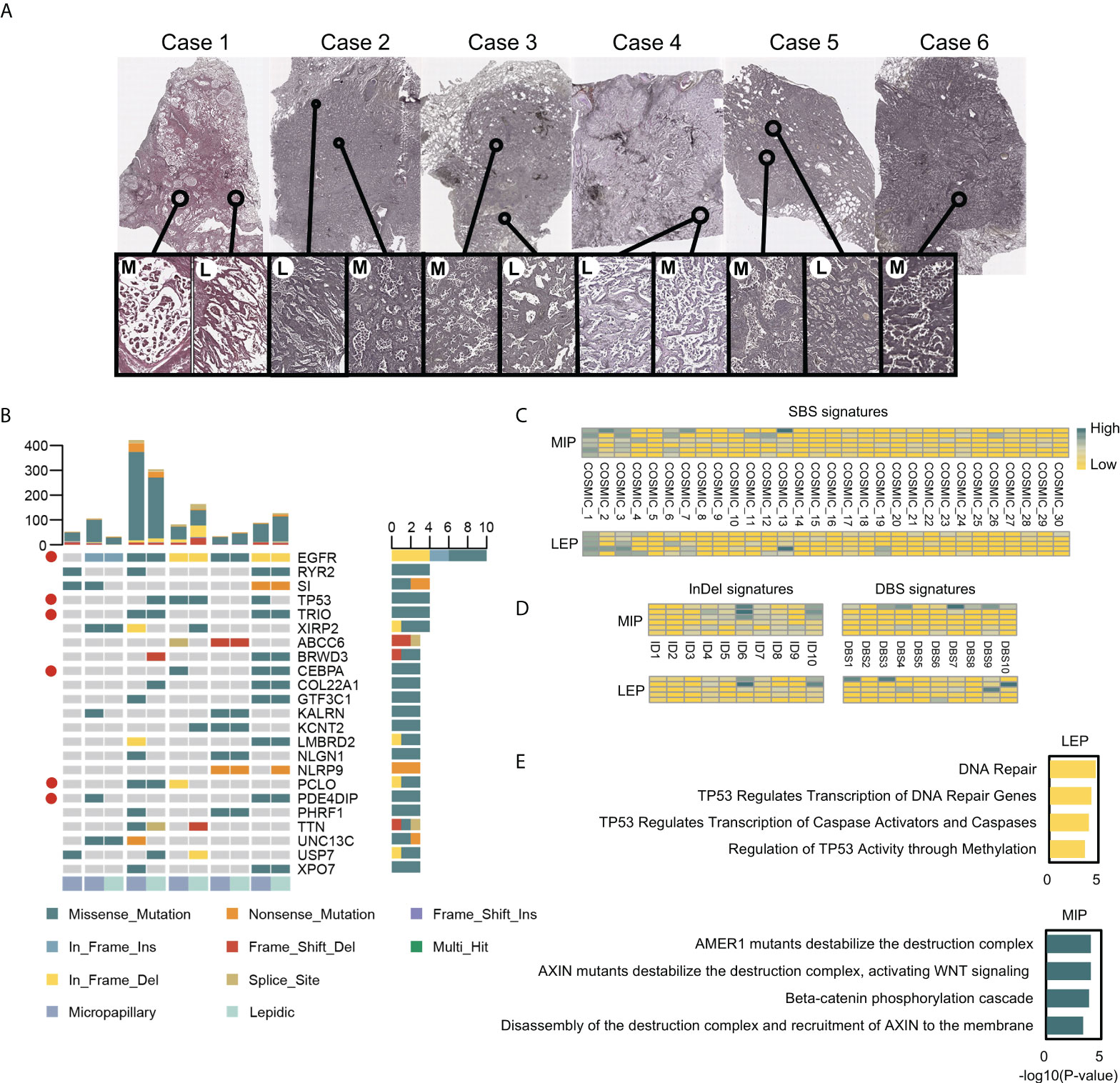
Figure 1 The H&E stained slides and the mutational landscape of MIP and LEP subtypes. (A) Scanner view (magnification ×20) of the H&E-stained MIP (M) and LEP (L) subtypes from six patients. (B) Genes with top mutation frequency in samples. Cancer-associated genes were marked with red circles. (C) Quantifications of the SBS signatures in two histological subtypes. (D) Quantifications on the InDel and DBS signatures in subtypes. (E) Pathways enriched in mutated tumor suppressor genes (TSGs) for LEP and MIP subtypes.
Mutation signature analysis was conducted on unfiltered mutations separately for LEP and MIP components. The point substitution spectrum plot displayed an insignificant difference between the two histological subtypes (Supplementary Figure 2C). Similarly, the SBS, InDel, and DBS signatures mapped to the COSMIC database (accessed in March 2021) were similar between the two subtypes (Figures 1C, D), indicating the histological differences between LEP and MIP components could be caused by alternations in specific key genes.
Of particular interest, mutated tumor suppressor genes (TSGs) were enriched in distinct pathways in the LEP and MIP subtypes (Figure 1E). TSGs from LEP components were enriched in DNA repair and TP53-related pathways, while mutated TSGs in MIP components were found to be enriched in pathways associated with beta-catenin destruction complex, AXIN mutation and WNT signaling, which followed report (4). When concerning the enriched pathways for mutated oncogenes, seven of the 10 top-enriched Reactome pathways from the two groups were identical, which were mainly associated with EGFR and PI3K signaling (Supplementary Figure 2D). By inspecting the mutated TSG pathway enrichment pattern in Lung-Broad (Supplementary Figures 3A, B), Lung-OncoSG (Supplementary Figures 3C, D), and TCGA-LUAD (Supplementary Figures 3E, F) cohorts, similar entries were identified in both LEP and MIP samples, while NOTCH1-related pathways were additionally found in TCGA-LUAD MIP samples, which was unsurprising since the cross-talk between NOTCH and WNT pathways was previously unveiled (30). As for the oncogenes mutated in three public cohorts, shared terms were found between LEP and MIP components in Lung-Broad (Supplementary Figures 4A, B), Lung-OncoSG (Supplementary Figures 4C, D), and TCGA-LUAD (Supplementary Figures 4E, F) cohorts but with lower overlapping proportion, which endorsed the possible existence of a common mutational ancestor in the paired components.
Through the segment-level copy number alternation identification procedures, multiple amplified and deleted segments were detected (Supplementary Figure 5A). Chromosomes including 3,4,5,10,15,17, and 18 exhibited different copy number alternation patterns between the two subtypes, and the MIP subtype showed both higher chromosome level (Supplementary Figure 5B) and arm-level copy number variation (CNV) burden (Supplementary Figure 5C), which followed the report (31). To further pinpoint the recurrent SCNAs at the focal level, we identified 1,116 genes with somatic copy number alternations through statistical testing on read coverages from all samples (details in Materials and methods), among which 159/80 genes were uniquely amplified/deleted in the LEP component, while 34/11 genes were uniquely amplified/deleted in the MIP component. By annotating the enriched pathways on these genes, 27 pathways were found to overlap between the enrichment results of uniquely amplified genes in LEP and deleted genes in MIP, which could be categorized into the immune system, innate immune system, interleukin signaling, SHC1 events, ERK activation, and FRS-mediated signaling pathways (Figure 2A). When inspecting the number of genes, pathways related to the immune system and innate immune system got the highest gene number variated (37 genes amplified in LEP and four genes deleted in MIP subtype), indicating that MIP LUADs tend to have induced immunosurveillance escape. Additionally, two pathways were identical between the enrichment results of uniquely deleted genes in LEP and amplified genes in MIP (Figure 2B), which were associated with homology directed repair (HDR) and mRNA fate regulation, but the variated gene number was limited (six genes for LEP and four genes for the MIP group).
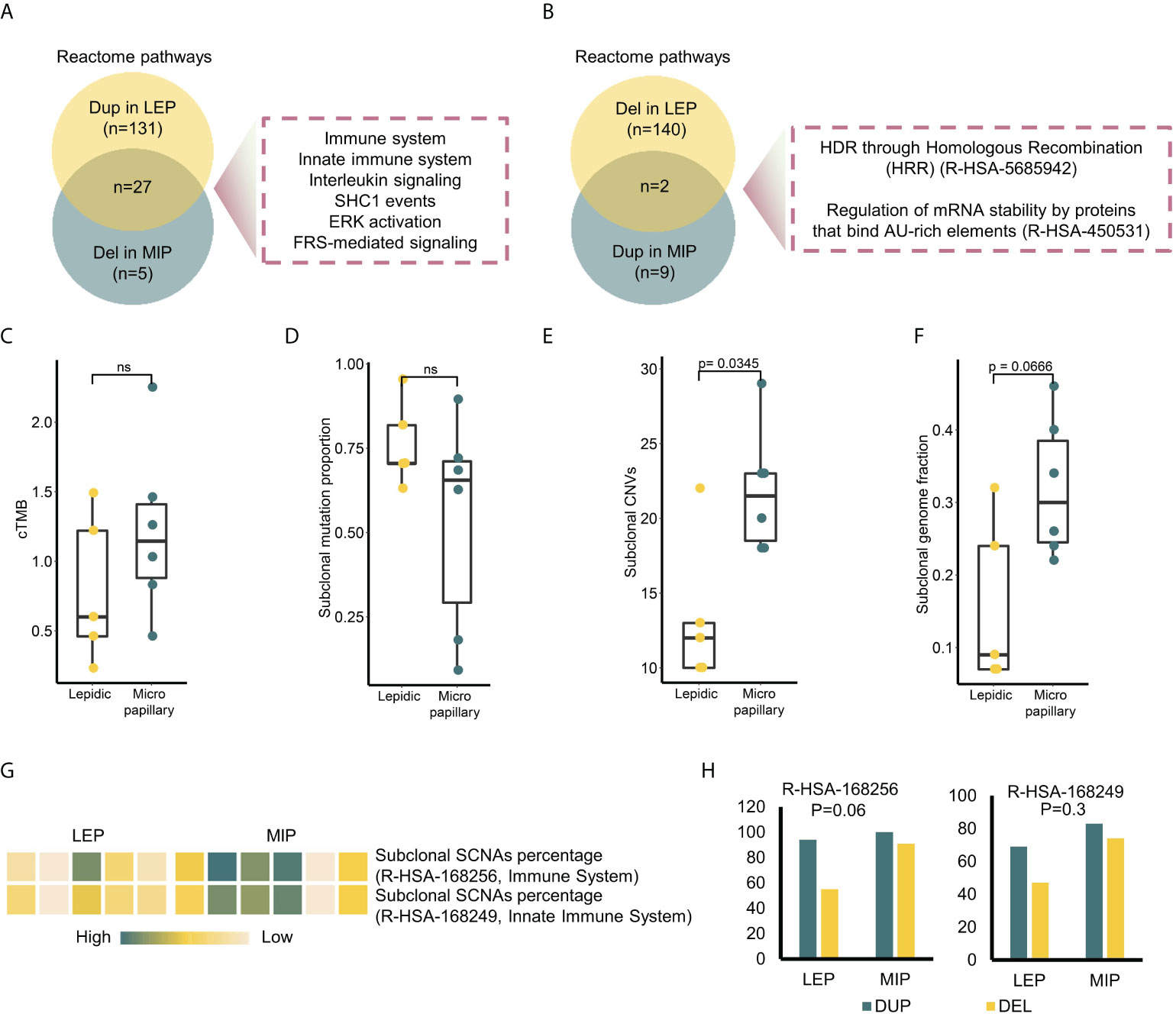
Figure 2 Multi-perspective investigation on intratumor heterogeneity (ITH) difference between two subtypes. (A) Intersection of the enriched pathways in genes uniquely amplified in LEP and deleted in MIP. (B) Intersection of the enriched pathways in genes uniquely deleted in LEP and amplified in MIP. (C–F) Clonal tumor mutation burden (cTMB), subclonal mutation proportion, subclonal CNV and subclonal genome fraction distribution in two subtypes. (G) Subclonal SCNA percentage of two Reactome immune pathways in sequenced samples. (H) Focal alternation number on the genes in two immune pathways. P-values on the alternation discrepancy between subtypes were calculated by Fisher’s exact test.
Intratumor heterogeneity (ITH) can depict the genetic and epigenetic tumor inner diversity and is proven to be closely related to cancer progression, therapeutic resistance, and recurrences. To compare the ITH of the two histological subtypes at both the mutational and copy number level, we annotated the mutations/SCNAs with clonality. As shown in Figure 2C, there was no significant difference in the clonal tumor mutation burden (cTMB) and subclonal mutation proportion between the MIP and LEP groups (Figure 2D). The MATH score, which is widely used to measure the mutational ITH, exhibited a similar trend (Supplementary Figure 5D). As for the copy number variations, the MIP group possessed a significantly higher proportion of subclonal SCNAs (Figure 2E) as well as a trend of higher subclonal genome fraction (Figure 2F). Interestingly, the frequency of clonal mutations in DNA Damage Response (DDR) and WNT pathway genes was higher in the MIP subtype (Supplementary Figure 5E), which may possibly partially increase the subclonal genome alternations in immune-related genes since the association between canonical WNT-beta-catenin signaling and carcinogenesis as well as immune suppression was clear (32). Indeed, six MIP components showed a trend of a higher percentage of subclonal SCNA (Figure 2G) as well as a higher number of focal deletions (Figure 2H) on the genes related to the two immune pathways. We confirmed the results that genes significantly deleted in the MIP subgroup exhibited association with immune-related terms in the Lung-Broad and Lung-OncoSG cohorts (Supplementary Figure 6).
To elaborate on the possible evolutionary process between LEP and MIP subtypes, we delineated the phylogenetic trees for each patient based on mutations as well as focal level SCNAs. As shown in Figure 3, all five patients possessed truncal mutations between paired LEP and MIP components, while no obvious bias on private mutation burden after truncal divergence was observed. Clonal mutations on cancer drivers including EGFR, TP53, and CEBPA were identified, and EGFR was the only gene coincident in five pairs, which confirmed the presence of ancestral mutations. The driver mutations private to LEP were enriched in chromatin organization, TP53-related and DNA double strand repair pathways (Supplementary Figure 7A), while mutations private to MIP were enriched in cellular signaling and beta-catenin-related pathways (Supplementary Figure 7B). We further annotated the shared mutations in Figure 3 with clonality to explore the clonal-subclonal transitions between the LEP and MIP subtypes. For the genes possessing mutations with increased clonality in MIP, GO terms related to neurogenesis were found enriched (Supplementary Figure 7C), denoting that tumor-induced neurogenesis and nerve-cancer crosstalk may account for the aggressiveness of the MIP subtype. Oppositely, genes with mutations switched from subclonal to clonal in LEP were associated with cell-cycle related GO biological processes (Supplementary Figure 7D). As for the truncal focal SCNAs, several driver genes including CSMD3, SPTAN1, BCORL1, CAMTA1, GRIN2A, MED12, and TRAF7 were concurrently amplified in the two subtypes (Figure 3), which were associated with developmental biology (R-HSA-1266738) and EGFR-related Reactome pathways (R-HSA-179812 and R-HSA-180336). Moreover, the deletion of TP53, MUC4, ARID5B, ANK1, PTEN, SFPQ, FANCA, MAF, and ZFHX3 were observed in the two subtypes. Interestingly, no driver gene showed concordant copy number variation in five pairs of samples, possibly due to the elevated SCNA-level ITH in the MIP group. We also scrutinized the genes with shared copy number variation in the paired samples. As shown in Supplementary Figure 8A, most shared deletions were on immune-related genes, while signal transduction and PI3K/AKT pathways, which abnormality is highly associated with tumor progression and therapeutic resistance, were found uniformly amplified (Supplementary Figure 8B). To further derive the mutational transitions and evolutionary trajectory, we used PyClone-VI to infer the mutational populations and their evolution among paired components. As shown in Supplementary Figure 9A, numerous clone clusters were identified in 5 patients, which exhibited dynamic variant allele frequency (VAF) alternation. Clusters with drastically increased VAF in the LEP subtype were mainly enriched in mRNA splicing pathways (Supplementary Figure 9B), while clusters with increased VAF in the MIP subtype were associated with ERBB2 functions (Supplementary Figure 9C). These data imply that LEP and MIP components from one patient were derived from the same initiation cells and the pathway-specific mutations acquired after the EGFR clonal mutation eventually shaped the subtype-specificity.
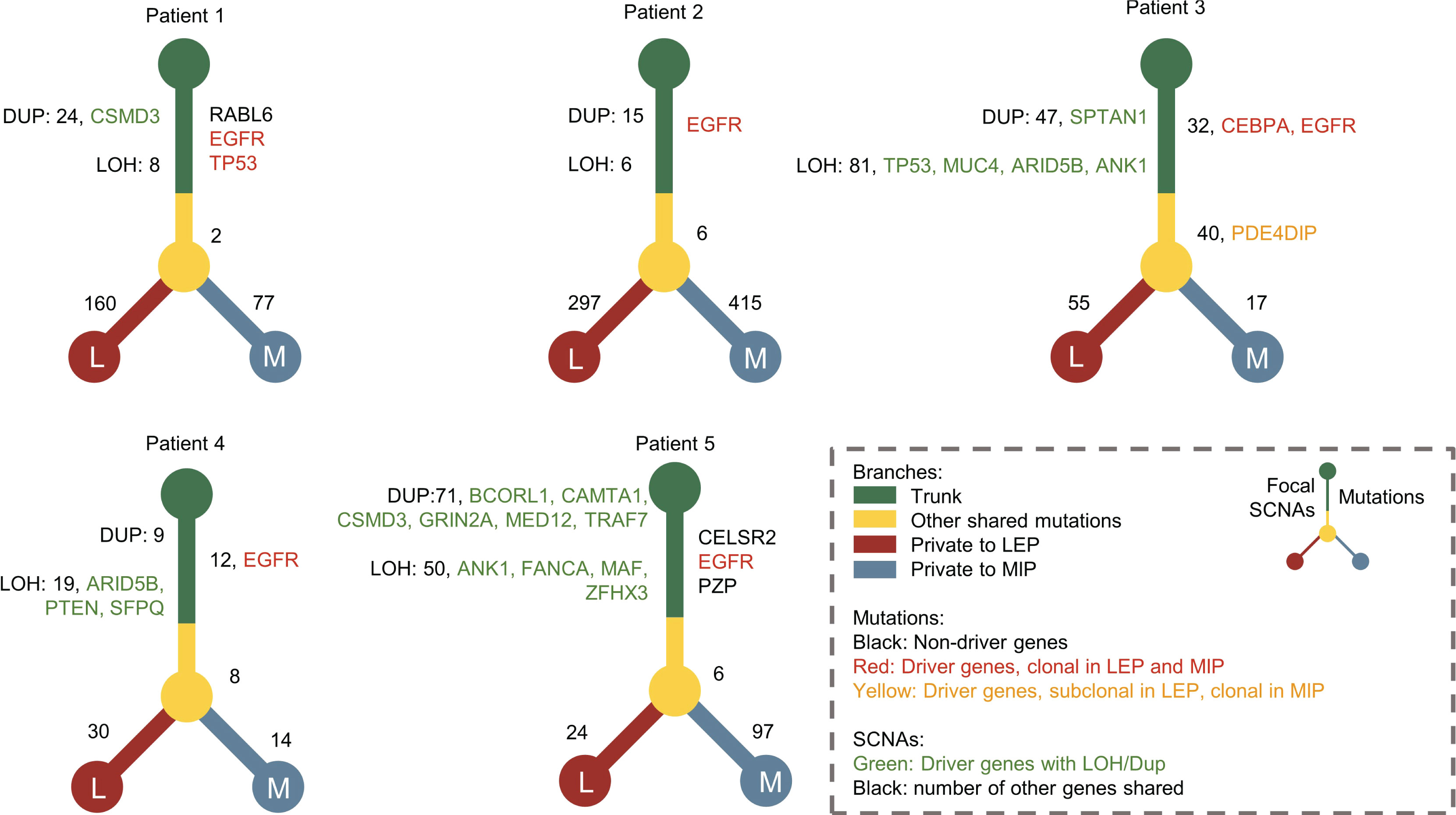
Figure 3 The phylogenetic trees constructed for patients with concomitant MIP and LEP components. Driver genes with mutations and focal CNV were marked with different colors. The numbers of shared mutations and focal CNVs were labeled beside the tree trunk, while mutation numbers private to MIP and LEP components were labeled beside the tree branches.
We next gathered SNV and SCNA data and identified the genes with an alternation frequency difference between the LEP and MIP groups. As shown in Table 1, mutation frequency difference was observed on nine genes, with three genes specifically mutated in LEP group. Additionally, five genes were found with a distinct copy number alternation pattern, with one gene specifically amplified in the LEP group (Table 1). Similarly, mutation frequencies on the above nine genes and the key gene identified in phylogenetic analyses (EGFR) were inspected in four public cohorts (Table 2), and EGFR was the only gene with a significant mutational difference in non-east Asian public cohorts. Moreover, the five SCNA genes showed an alternation frequency difference in three non-east Asian cohorts (Table 2). Interestingly, the altered sample proportion or the alternation frequency for PTP4A3, NAPRT, and RECQL4 was highly similar between our MIP group (Table 1) and public cohorts (Lung-Broad, Lung-OncoSG, and TCGA-LUAD, Table 2), implying the feasibility of their cooperative function through duplication in MIP components. Spearman’s correlation coefficient (SCC) on the SCNA pattern of our 11 samples confirmed the association of the co-amplified genes (Supplementary Figure10 A). Such strong association was also observed on SCNA data for LEP and MIP adenocarcinoma from Lung-Broad (Supplementary Figure 10B), Lung-OncoSG (Supplementary Figure 10C, left), and TCGA-LUAD (Supplementary Figure 10D, left) cohorts. Concerning the fact that SCNA is highly related to the consequent gene expression alternation, we calculated the expressional SCC of the five genes in cohorts with available transcriptomic data. When compared to all samples (Supplementary Figures 10C, D, middle), the expressional associations between PTP4A3, NAPRT, and RECQL4 transformed to a higher synergetic state for LEP and MIP samples in both the Lung-OncoSG (Supplementary Figure 10C, right) and TCGA-LUAD (Supplementary Figure 10D, right) cohorts. More explicitly, the correlation between SCNA and RNA expression was higher for the three genes in two public cohorts (Supplementary Figure 10E), and NAPRT as well as PTP4A3 exhibited significantly higher LEP/MIP group-specificity. As an exemplification, the correlation between the SCNA and RNA expression of the PTP4A3 gene increased from 0.372 to 0.693 when narrowed to only LEP/MIP samples in the TCGA-LUAD cohort (Supplementary Figures 10F, G). All three co-amplified genes were significantly overexpressed in tumor samples (Supplementary Figures 10H–J, left) and RECQL4 possessed significantly higher expression levels in the MIP subtype (Supplementary Figure 10J, right) in the TCGA-LUAD cohort. To conclude, our comprehensive analyses identified PTP4A3, NAPRT, and RECQL4 were co-amplified and co-expressed specifically in LEP/MIP adenocarcinoma, and RECQL4 was upregulated in the MIP group.
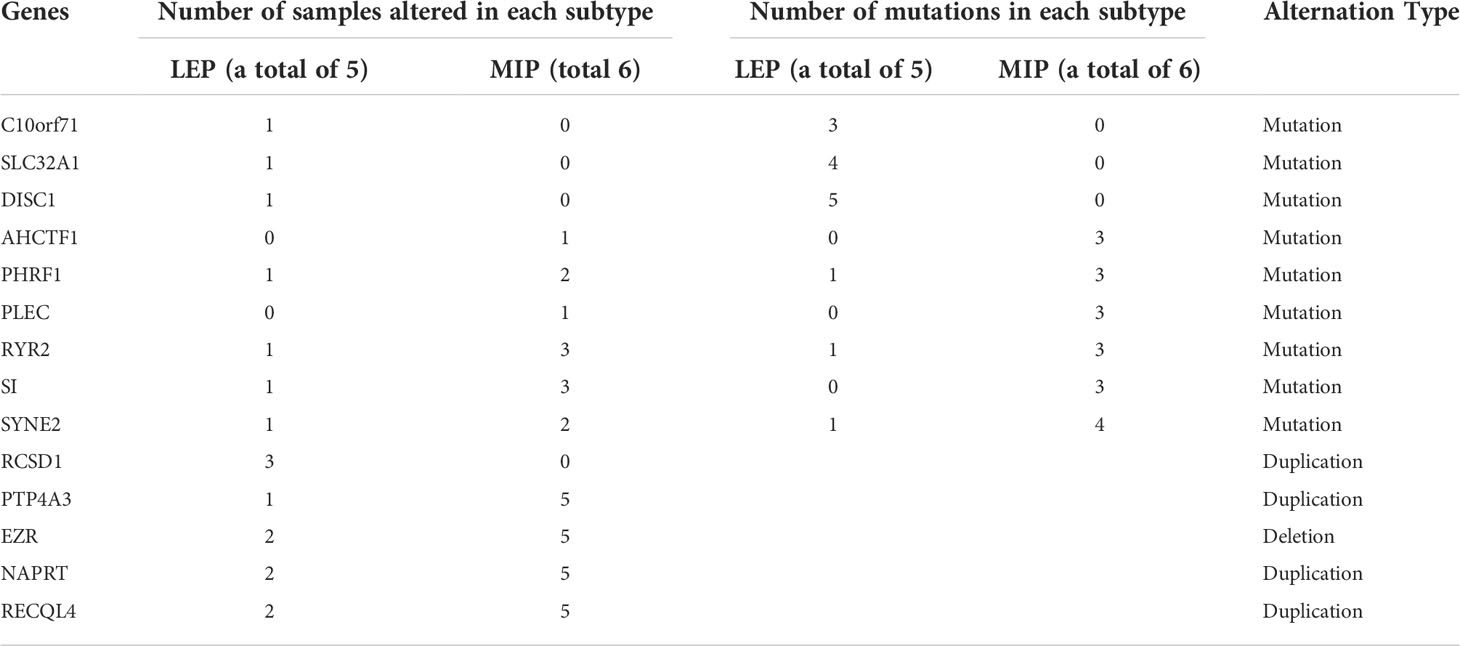
Table 1 Genes exhibited subtype-specific alternation frequency among the sequenced samples.
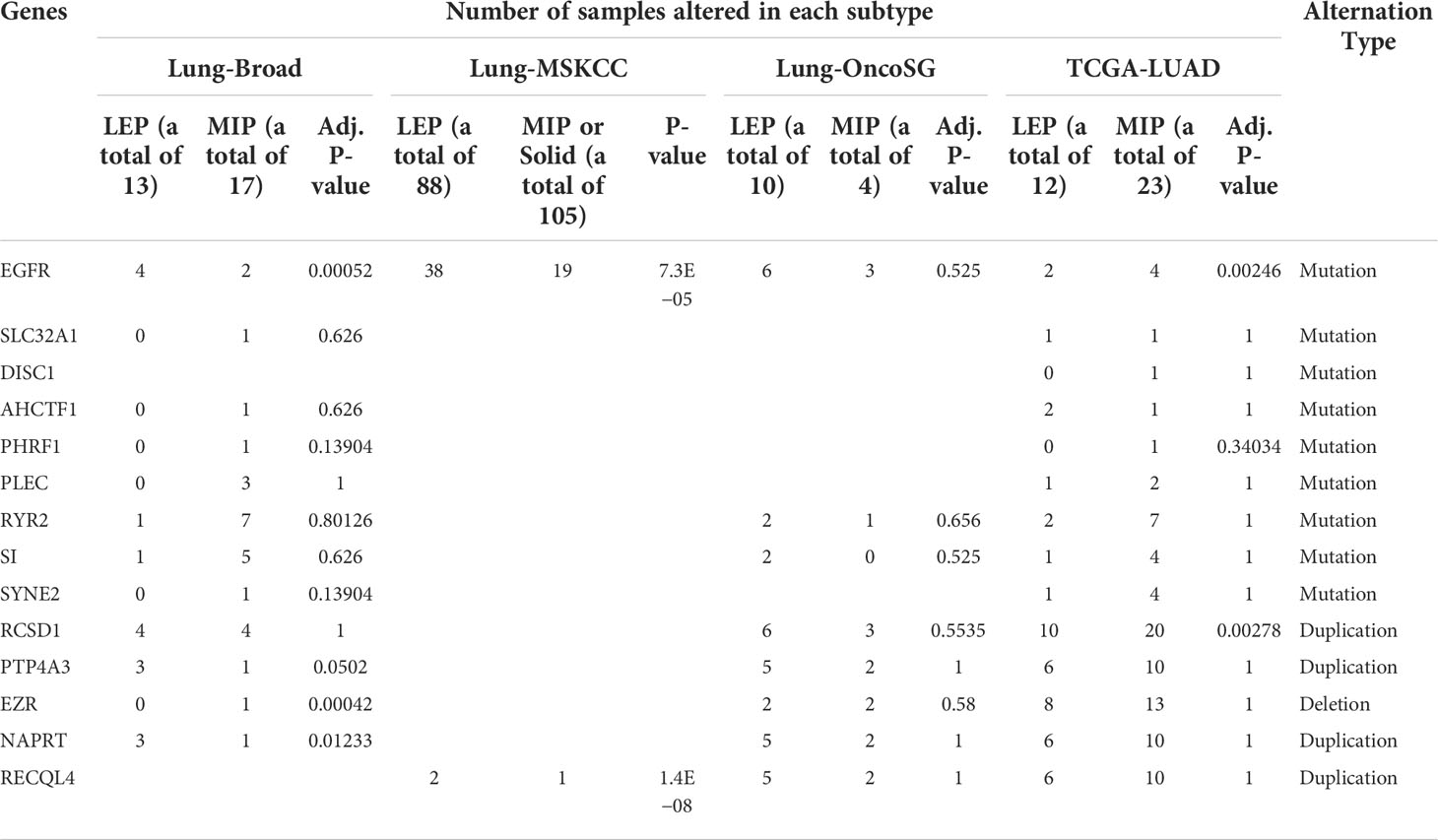
Table 2 Inspection on the alternation frequency of genes exhibited subtype-specific alternations using four public cohorts.
The disparity of cancer immunity cycle activity was examined in the Lung-OncoSG, GEO, and TCGA-LUAD cohorts as described in Materials and methods. Activities of recurrent cancer immunity steps including the release of cancer cell antigen, CD8+ T-cell recruiting, dendritic cell recruiting, macrophage recruiting, T-helper 17 (Th17) cell recruiting, T-cell infiltration into tumors and killing of cancer cells were significantly higher in MIP subtype (Supplementary Figures 11A–C). By further examining the differentially expressed proteins between LEP and MIP subtypes from the TCGA-LUAD dataset, the identified proteins with MIP-specific elevation (Supplementary Figure 12A) were significantly enriched in PD-L1 and PD-1 checkpoint pathways in cancer (Supplementary Figure 12B). These data suggest that the MIP subtype could exist in an immune-suppressive microenvironment.
Consistent with other studies, we confirmed the survival disparity between LUAD subtypes. By performing WES on micro-dissected LUAD tissue samples of MIP and LEP components, we explored the genetic features related to the LEP/MIP growth pattern and the evolutional connection between LUAD subtypes. Our results revealed that LEP and MIP subtypes could be derived from the same initiation cells with EGFR mutation and the ultimate histological dissimilitude was shaped by the pathway-specific mutations acquired along evolution. Our results showed that the EGFR trunk mutation arose between pre-invasive and invasive LUAD and LEP/MIP components were evolved by a branched evolution model.
Through comprehensive comparisons of genetic alternations, the biological characteristics of the two LUAD subtypes were elucidated. As for mutational comparisons, TSG mutations in LEP were associated with DNA repair and TP53 regulation, while genes related to WNT signaling and beta-catenin destruction complexes got both higher mutational frequency and clonality. Driver mutations private to MIP were also enriched in cellular signaling and beta-catenin-related pathways, while genes that possessed lower mutational heterogeneity in MIP were associated with neurogenesis and ERBB2 functions. Aberrant WNT signaling pathway activation caused by gene mutations of intracellular components is associated with a higher rate of recurrence in early-stage NSCLC. On the other hand, being the critical downstream effector in the canonical WNT pathway, excessive intracellular beta-catenin promotes lung cancer aggression. Liang et al. further confirmed that the intracellular beta-catenin expression in MIP-predominant LUAD was higher than LEP-predominant LUAD (4). Besides, neurogenesis induced by tumors shapes an immunosuppressive microenvironment (33). Although cancer-related neurogenesis is considered to be associated with solid tumor metastasis, its role in LUAD remains poorly understood. Our results suggest an inner association between the MIP aggressive phenotype and neurogenesis. The activation of well-known proto-oncogene ERBB2 signaling was associated with poor outcomes in NSCLC (34), coinciding with MIP characteristics. The copy number of genes related to the immune system, innate immune system, interleukin signaling, SHC1 events, ERK activation, and FRS-mediated signaling were also found to decrease in MIP. With the highest proportion of immune genes affected, the immunosuppression status in the MIP subtype was confirmed. Apart from specified genomic alternations, ITH provides crucial information for drug responsiveness and clinical prognosis. Discordance between SNV and SCNA ITH was particularly observed in the MIP subtype. Subclonal genetic instability possibly facilitated MIP neoplastic cell proliferation (35) and the clonal mutations on key MIP-specific pathways contributed to its aggressive behavior.
We discovered three genes with co-amplification tendency, both in our discovery cohort and in three public validation cohorts. Previous studies proved that the knockdown of PTP4A3 inhibited cell migration and invasion of lung cancer cell lines (36). It also induced microvascular and lymphatic vessel formation by increasing VEGF and VEGF-C expression in lung cancer tissues, which was in accordance with the clinical observations that the MIP component in LUAD increases the risk of distant and lymph node metastasis. A previous study also found that the loss of NAPRT promoted the epithelial–mesenchymal transition (EMT) by stabilizing beta-catenin (37). The elevated expression of NAPRT was conceivably associated with the disruption of the catenin–cadherin complex in MIP. Moreover, RECQL4 could coordinate and regulate cell proliferation and cell cycle progression by protecting chromosome stability (38), and its protein expression was remarkably higher in LUAD (39). The biological mechanisms of these three genes further verified our discoveries.
Interestingly, we observed numerous genetic alternations associated with the immune status in the LEP and MIP subtypes. For example, the enrichment of immune-related pathways, including immune system and innate immune system, was observed on genes uniquely amplified in LEP and deleted in the MIP category. Along with the observation of a higher percentage of focal deletions on the immune pathway genes in our cohort and the deficiency in immune-related pathways for MIP group-specific deleted genes in Lung-Broad and Lung-OncoSG cohorts, we suspected that the MIP subtype could possess an immuno-suppression microenvironment, in other words, an induced immuno-surveillance escape. Regarding the increasing enthusiasm for lung cancer immunotherapy and our hypothesis, we next assessed and compared the TME landscape between the MIP and LEP components using the stepwise activities of the cancer immunity cycle. Steps including CD8+ T-cell recruiting, T-helper 17 (Th17) cell recruiting, T-cell infiltration into tumors and killing of cancer cells were significantly higher in MIP samples. However, elevated T-cell infiltration does not always indicate better clinical outcomes for patients. For instance, elevated expression of PD-L1 and PD-1 could inhibit the activation of T cells, conferring an immuno-evasion and immuno-suppression tumor status. Unsurprisingly, proteomic analysis further confirmed the activation of the PD1/PD-L1 pathway in the MIP subtype, partially elucidating the deteriorative survival of MIP patients. Generally, our work revealed the comprehensive TME situation of LEP/MIP components and immuno-suppression features in MIP-predominant LUAD.
Our study has several limitations. Firstly, the cohort only included five pairs of LEP/MIP components detached from five LUAD patients and 11 samples. Further studies with a larger amount of patient involvement can better decipher the evolutionary trajectory between LUAD histological subtypes and identify subtype-specific genetic changes. Moreover, the three genes with co-amplification or co-expression tendency should be further experimentally validated, particularly on their protein expression status. Additionally, we portrayed the TME heterogeneity using bulk RNA-seq data. With the recent maturation of multiple advanced techniques, using methods including single-cell RNA-seq, spatial transcriptomics, and multiplexed immunohistochemistry could better dissect the TME in LUAD. Lastly, our analyses only focused on MIP and LEP subtypes. A more integrated study incorporating other LUAD histologic subtypes could better decode the disease.
To conclude, we identified subtype-specific genetic differences responsible for variated prognosis and the evolution trajectory of the MIP subtype. The subtype-specificity was possibly shaped by pathway-specific mutations acquired after the EGFR clonal mutation. The tumor microenvironment revealed the immunosuppression prevalence in MIP, which could contribute to its unfavorable prognosis. Immune checkpoint inhibitor treatments like anti-PD-1/anti-PD-L1 could maximize the therapeutic benefit for MIP-predominant LUAD patients.
The raw sequencing datasets presented in this study can be found in the China National GeneBank DataBase (https://db.cngb.org/) with project number CNP0003191.
The studies involving human participants were reviewed and approved by the Tianjin Medical University Cancer Institute and Hospital review board. The patients/participants provided their written informed consent to participate in this study.
CW, BZ, and DY contributed to the study design. LS, ZZ, LZ, YH, WH, XS, ZT, YF, and HM contributed to data collection. JY, HZ, ZuY, CZ, and ZiY performed statistical analysis and data interpretation. YH and JY drafted the manuscript. CW and BZ revised the manuscript. CW and DY provided financial support and study supervision. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was financially supported by the National Natural Science Foundation of China (grant number 81772484 to CW, grant number 82173038 to DY).
Authors JY, ZuY, CZ, and ZiY were employed by GenePlus-Shenzen.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.931209/full#supplementary-material
1. Bray F, Ferlay J, Laversanne M, Brewster DH, Gombe Mbalawa C, Kohler B, et al. Cancer incidence in five continents: Inclusion criteria, highlights from volume X and the global status of cancer registration. Int J Cancer (2015) 137(9):2060–71. doi: 10.1002/ijc.29670
2. Hung JJ, Yeh YC, Jeng WJ, Wu YC, Chou TY, Hsu WH. Factors predicting occult lymph node metastasis in completely resected lung adenocarcinoma of 3 Cm or smaller. Eur J Cardiothorac Surg (2016) 50(2):329–36. doi: 10.1093/ejcts/ezv485
3. Makinen JM, Laitakari K, Johnson S, Makitaro R, Bloigu R, Lappi-Blanco E, et al. Nonpredominant lepidic pattern correlates with better outcome in invasive lung adenocarcinoma. Lung Cancer (2015) 90(3):568–74. doi: 10.1016/j.lungcan.2015.10.014
4. Zhu L, Yang S, Zheng L, Zhang G, Cheng G. Wnt/Beta-catenin pathway activation Via Wnt1 overexpression and Axin1 downregulation correlates with cadherin-catenin complex disruption and increased lymph node involvement in micropapillary-predominant lung adenocarcinoma. J Thorac Dis (2020) 12(10):5906–15. doi: 10.21037/jtd-20-1495
5. Warth A, Penzel R, Lindenmaier H, Brandt R, Stenzinger A, Herpel E, et al. Egfr, kras, braf and alk gene alterations in lung adenocarcinomas: Patient outcome, interplay with morphology and immunophenotype. Eur Respir J (2014) 43(3):872–83. doi: 10.1183/09031936.00018013
6. Tsao MS, Marguet S, Le Teuff G, Lantuejoul S, Shepherd FA, Seymour L, et al. Subtype classification of lung adenocarcinoma predicts benefit from adjuvant chemotherapy in patients undergoing complete resection. J Clin Oncol (2015) 33(30):3439–46. doi: 10.1200/JCO.2014.58.8335
7. Ng Kee Kwong F, Laggner U, McKinney O, Croud J, Rice A, Nicholson AG. Expression of pd-L1 correlates with pleomorphic morphology and histological patterns of non-Small-Cell lung carcinomas. Histopathology (2018) 72(6):1024–32. doi: 10.1111/his.13466
8. Zhang S, Xu Y, Zhao P, Bao H, Wang X, Liu R, et al. Integrated analysis of genomic and immunological features in lung adenocarcinoma with micropapillary component. Front Oncol (2021) 11:652193. doi: 10.3389/fonc.2021.652193
9. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv (2013) 1303.3997. doi: 10.48550/arXiv.1303.3997
10. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnology (2013) 31(3):213–9. doi: 10.1038/nbt.2514
11. Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell (2018) 173(2):371–85. e18. doi: 10.1016/j.cell.2018.02.060
12. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science (2013) 339(6127):1546–58. doi: 10.1126/science.1235122
13. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific hla loss and immune escape in lung cancer evolution. Cell (2017) 171(6):1259–71.e11. doi: 10.1016/j.cell.2017.10.001
14. Blokzijl F, Janssen R, van Boxtel R, Cuppen E. Mutationalpatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med (2018) 10(1):33. doi: 10.1186/s13073-018-0539-0
15. Wang S, Tao Z, Wu T, Liu XS. Sigflow: An automated and comprehensive pipeline for cancer genome mutational signature analysis. Bioinformatics (2021) 37(11):1590–2. doi: 10.1093/bioinformatics/btaa895
16. Mroz EA, Rocco JW. Math, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral Oncol (2013) 49(3):211–5. doi: 10.1016/j.oraloncology.2012.09.007
17. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol (2012) 30(5):413–21. doi: 10.1038/nbt.2203
18. Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep (2018) 23(1):239–54 e6. doi: 10.1016/j.celrep.2018.03.076
19. Gillis S, Roth A. Pyclone-vi: Scalable inference of clonal population structures using whole genome data. BMC Bioinf (2020) 21(1):571. doi: 10.1186/s12859-020-03919-2
20. Dang HX, White BS, Foltz SM, Miller CA, Luo J, Fields RC, et al. Clonevol: Clonal ordering and visualization in cancer sequencing. Ann Oncol (2017) 28(12):3076–82. doi: 10.1093/annonc/mdx517
21. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res (2016) 44(W1):W90–7. doi: 10.1093/nar/gkw377
22. Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, et al. The reactome pathway knowledgebase. Nucleic Acids Res (2018) 46(D1):D649–55. doi: 10.1093/nar/gkx1132
23. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell (2012) 150(6):1107–20. doi: 10.1016/j.cell.2012.08.029
24. Chen J, Yang H, Teo ASM, Amer LB, Sherbaf FG, Tan CQ, et al. Genomic landscape of lung adenocarcinoma in East asians. Nat Genet (2020) 52(2):177–86. doi: 10.1038/s41588-019-0569-6
25. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci Signaling (2013) 6(269):pl1–pl. doi: 10.1126/scisignal.2004088
26. Goldman M, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nature Biotechnol (2020) 38(6):675–8.
27. Tone M, Tahara S, Nojima S, Motooka D, Okuzaki D, Morii E. Htr3a is correlated with unfavorable histology and promotes proliferation through erk phosphorylation in lung adenocarcinoma. Cancer Sci (2020) 111(10):3953–61. doi: 10.1111/cas.14592
28. Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
29. Li H, Pan Y, Li Y, Li C, Wang R, Hu H, et al. Frequency of well-identified oncogenic driver mutations in lung adenocarcinoma of smokers varies with histological subtypes and graduated smoking dose. Lung Cancer (2013) 79(1):8–13. doi: 10.1016/j.lungcan.2012.09.018
30. Krishnamurthy N, Kurzrock R. Targeting the Wnt/Beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev (2018) 62:50–60. doi: 10.1016/j.ctrv.2017.11.002
31. Caso R, Sanchez-Vega F, Tan KS, Mastrogiacomo B, Zhou J, Jones GD, et al. The underlying tumor genomics of predominant histologic subtypes in lung adenocarcinoma. J Thorac Oncol (2020) 15(12):1844–56. doi: 10.1016/j.jtho.2020.08.005
32. Chehrazi-Raffle A, Dorff T, Pal S, Lyou Y. Wnt/B-catenin signaling and immunotherapy resistance: Lessons for the treatment of urothelial carcinoma. Cancers (2021) 13:889. doi: 10.1038/s41392-020-0205-z
33. Cervantes-Villagrana RD, Albores-Garcia D, Cervantes-Villagrana AR, Garcia-Acevez SJ. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct Target Ther (2020) 5(1):99. doi: 10.1038/s41392-020-0205-z
34. Riudavets M, Sullivan I, Abdayem P, Planchard D. Targeting Her2 in non-Small-Cell lung cancer (Nsclc): A glimpse of hope? an updated review on therapeutic strategies in nsclc harbouring Her2 alterations. ESMO Open (2021) 6(5):100260. doi: 10.1016/j.esmoop.2021.100260
35. Dentro SC, Leshchiner I, Haase K, Tarabichi M, Wintersinger J, Deshwar AG, et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell (2021) 184(8):2239–54.e39. doi: 10.1016/j.cell.2021.03.009
36. Jian M, Nan L, Guocheng J, Qingfu Z, Xueshan Q, Enhua W. Downregulating prl-3 inhibit migration and invasion of lung cancer cell Via rhoa and Mdia1. Tumori (2012) 98(3):370–6. doi: 10.1700/1125.12407
37. Lee J, Kim H, Lee JE, Shin SJ, Oh S, Kwon G, et al. Selective cytotoxicity of the nampt inhibitor Fk866 toward gastric cancer cells with markers of the epithelial-mesenchymal transition, due to loss of naprt. Gastroenterology (2018) 155(3):799–814 e13. doi: 10.1053/j.gastro.2018.05.024
38. Fang H, Niu K, Mo D, Zhu Y, Tan Q, Wei D, et al. Recql4-aurora b kinase axis is essential for cellular proliferation, cell cycle progression, and mitotic integrity. Oncogenesis (2018) 7(9):68. doi: 10.1038/s41389-018-0080-4
Keywords: lung adenocarcinoma, histological subtypes, whole-exome sequencing, copy number alternation, intratumor heterogeneity
Citation: Huo Y, Sun L, Yuan J, Zhang H, Zhang Z, Zhang L, Huang W, Sun X, Tang Z, Feng Y, Mo H, Yang Z, Zhang C, Yu Z, Yue D, Zhang B and Wang C (2022) Comprehensive analyses unveil novel genomic and immunological characteristics of micropapillary pattern in lung adenocarcinoma. Front. Oncol. 12:931209. doi: 10.3389/fonc.2022.931209
Received: 28 April 2022; Accepted: 11 July 2022;
Published: 03 August 2022.
Edited by:
Liang Cheng, Harbin Medical University, ChinaReviewed by:
Shuyuan Wang, Harbin Medical University, ChinaCopyright © 2022 Huo, Sun, Yuan, Zhang, Zhang, Zhang, Huang, Sun, Tang, Feng, Mo, Yang, Zhang, Yu, Yue, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changli Wang, d2FuZ2NoYW5nbGlAdGptdWNoLmNvbQ==; Bin Zhang, emhhbmdiaW5fMDlAdG11LmVkdS5jbg==; Dongsheng Yue, eXVlZG9uZ3NoZW5nQHRtdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.