 Igor Valentim Barreto1†Flávia Melo Cunha de Pinho Pessoa1†
Igor Valentim Barreto1†Flávia Melo Cunha de Pinho Pessoa1† Caio Bezerra Machado1Laudreísa da Costa Pantoja2,3Rodrigo Monteiro Ribeiro4Germison Silva Lopes5Maria Elisabete Amaral de Moraes1Manoel Odorico de Moraes Filho1
Caio Bezerra Machado1Laudreísa da Costa Pantoja2,3Rodrigo Monteiro Ribeiro4Germison Silva Lopes5Maria Elisabete Amaral de Moraes1Manoel Odorico de Moraes Filho1 Lucas Eduardo Botelho de Souza6
Lucas Eduardo Botelho de Souza6 Rommel Mário Rodriguez Burbano3André Salim Khayat3Caroline Aquino Moreira-Nunes1,3,7*
Rommel Mário Rodriguez Burbano3André Salim Khayat3Caroline Aquino Moreira-Nunes1,3,7*- 1Department of Medicine, Pharmacogenetics Laboratory, Drug Research and Development Center (NPDM), Federal University of Ceará, Fortaleza, Brazil
- 2Department of Pediatrics, Octávio Lobo Children’s Hospital, Belém, Brazil
- 3Department of Biological Sciences, Oncology Research Center, Federal University of Pará, Belém, Brazil
- 4Department of Hematology, Fortaleza General Hospital (HGF), Fortaleza, Brazil
- 5Department of Hematology, César Cals General Hospital, Fortaleza, Brazil
- 6Center for Cell-based Therapy, Regional Blood Center of Ribeirão Preto, University of São Paulo, São Paulo, Brazil
- 7Ceará State University, Northeast Biotechnology Network (RENORBIO), Fortaleza, Brazil
Hematopoietic stem cells (HSCs) are known for their ability to proliferate and self-renew, thus being responsible for sustaining the hematopoietic system and residing in the bone marrow (BM). Leukemic stem cells (LSCs) are recognized by their stemness features such as drug resistance, self-renewal, and undifferentiated state. LSCs are also present in BM, being found in only 0.1%, approximately. This makes their identification and even their differentiation difficult since, despite the mutations, they are cells that still have many similarities with HSCs. Although the common characteristics, LSCs are heterogeneous cells and have different phenotypic characteristics, genetic mutations, and metabolic alterations. This whole set of alterations enables the cell to initiate the process of carcinogenesis, in addition to conferring drug resistance and providing relapses. The study of LSCs has been evolving and its application can help patients, where through its count as a biomarker, it can indicate a prognostic factor and reveal treatment results. The selection of a target to LSC therapy is fundamental. Ideally, the target chosen should be highly expressed by LSCs, highly selective, absence of expression on other cells, in particular HSC, and preferentially expressed by high numbers of patients. In view of the large number of similarities between LSCs and HSCs, it is not surprising that current treatment approaches are limited. In this mini review we seek to describe the immunophenotypic characteristics and mechanisms of resistance presented by LSCs, also approaching possible alternatives for the treatment of patients.
Introduction
Hematopoietic stem cells (HSCs) are located in the bone marrow (BM) and are responsible for sustaining and regenerating the hematological system. It is estimated that in a human organism, 1x106 blood cells are produced every second. This feature comes from the ability of self-renewal together with a high proliferative rate and pluripotency of these cells. It is also worth mentioning the ability to resist apoptosis, necrosis and genotoxicity produced by reactive oxygen species (ROS) that HSCs have (1–9).
Most of the time, the HSCs are quiescent, at G0 phase of the cell cycle, depending on glucose to carry out their metabolic activities. However, these cells, when receiving the stimuli through severe situations, can quickly enter the cell cycle through activation of genetic factors. It begins with a positive control carried out in part by mTORC1 under the action of CDK6, which in the G0 phase is in low expression or accompanied by inhibitors such as p57 or p18 (8, 10–13).
HSCs enters the cell cycle, and therefore, their metabolic activities start to have mitochondrial oxidative phosphorylation as a source of energy due to the increase in energy demand. This metabolic alteration consequently triggers a series of proteins, such as histone and DNA modifying enzymes, which are fundamental for the epigenetic changes carried out by the modulation of key transcription factor activity. After their activation, HSCs generate multipotent progenitors that are then committed to a cell lineage and gradually differentiate until they become mature and specialized cells (8, 10–12, 14–16).
Due to aging, HSCs lose their regenerative ability and may undergo a process called age-related clonal hematopoiesis (ARCH). In this process, mutations acquired over time continue to be transmitted to their successors, giving rise to cells with mutations. Patients with ARCH are more likely to develop leukemias, however not all cells in this process will be related to the leukemic process. It is known that the presence of certain mutations is related to the severity factor of this cell, as mutations in TP53 and U2AF1 genes are associated with pre-leukemic stem cells, and mutations in DNMT3A and TET2 genes have a lower risk regarding transformation of malignancy (12, 17–19).
In this study, we investigated clinical trials in extended literature that focused their efforts on the identification of LSCs in different types of leukemia and we discussed their clinical outcome and the perspectives of new therapies.
Pre-Leukemic Stem Cell and Leukemic Stem Cell
The constant accumulation of mutations occurring in HSCs due to ARCH or other agents can stimulate the transformation of HSC into a pre-leukemic tumor cell (pre-LSCs). Although mutations are present in these cells, it is still possible for them to continue to give rise to healthy cells. However, pre-LSCs continue to accumulate mutations for years as well as significant clonal expansion until, after a long period, this cell acquires malignant characteristics, becoming a leukemic stem cell (LSC) (20–23). Despite the similarities between these two types of cells, it is still possible to make differentiations, mainly genetic differentiations, where it is observed that pre-LSCs do not have mutations associated with leukemia (12, 24, 25).
Mutations that occur in pre-LSCs are related to epigenetic genes that are responsible for histone modification, DNA methylation and chromatin looping. In pre-LSCs mutations can be found in AML1, ASXL1, CTCF, DNMT3A, E2H2, FOXO1, IDH1, IDH2, IKZF1, JAK2, NPM1, MED12, SMC1A, STAT5B, TET2 and WT1. These mutations alone are incapable of inducing leukemia and appear as precursor events to late mutations that transform pre-LSCs into LSCs, which is shown in Figure 1. Therefore, the genetic alterations present in LSCs are related to proliferation and active signaling (26–33).
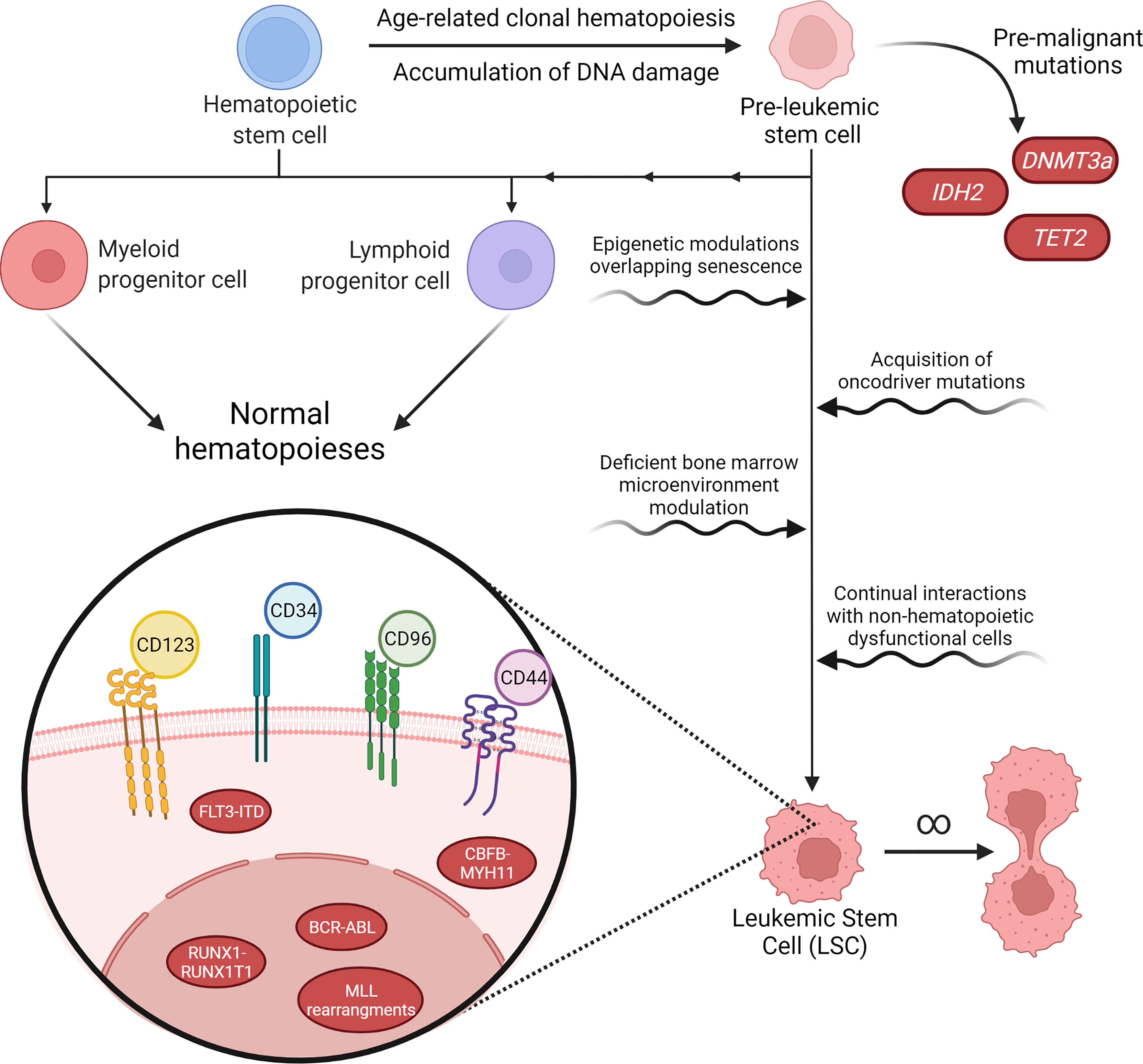
Figure 1 Pathways of malignancy in hematopoiesis and its characteristics. Aging and exposure to hazardous environmental agents lead to accumulation of DNA damage and mutations in hematological precursor cells, inducing a pre-leukemic stem cell (pre-LSC) phenotype. Pre-LSCs acquire proliferation advantages over normal hematopoietic stem cells (HSCs) due to mutations in genes such as DNA methyltransferase 3 alpha (DNMT3a), but still retain their capacity to promote normal hematopoiesis. However, further malignant characteristics acquired over the years may tip these cells into a proper leukemic stage. The transformation of pre-LSCs may happen through cell-specific processes, such as epigenetic modulation or new acquired mutations, or through interactions between these cells and their microenvironment, through changes in the normal growth and survival signaling pathways or due to interactions with dysfunctional stromal or mesenchymal cells that are also present in the bone marrow. After malignancy onset, leukemic stem cells (LSC) may present a variety of karyotype rearrangements, such as BCR-ABL or FLT3-ITD, that determine their malignant characteristics and tend to present immunophenotyping profiles that still resemble normal HSCs, such as CD34+38-, while also overexpressing a cohort of cell-surface antigens that are highly variable between patients and even among different cell populations in the same patient.
LSCs are recognized by their stemness features such as drug resistance, self-renewal, and undifferentiated state. They were initially pointed out by Lapidot and colleagues in 1994 (34). This rare population of resistant cells is believed to be at the origin of leukemia relapses. Their quiescent state and their self-renewal capacity makes it possible to leukemia repopulating cells, despite their low frequency (35–39).
LSCs are present at low levels in BM, being found in only 0.1%, approximately. This makes their identification and even their differentiation difficult since, despite the mutations, they are cells that still have many similarities with HSCs (20). Although the common characteristics, LSCs are heterogeneous cells and have different phenotypic characteristics, genetic mutations, and metabolic alterations. This whole set of alterations enables the cell to initiate the process of carcinogenesis, in addition to conferring drug resistance and inducing relapses (36, 37, 40).
During tumor progression, cancer cells continuously acquire genetic changes, and the fittest, most proliferative cells are selected for giving rise to distinct tumor subclones, which is known as clonal hematopoiesis (CH) (41–44). The clonal hematopoiesis of indeterminate potential (CHIP) refers to the presence of at least one driver mutation in hematopoietic cells of peripheral blood, without hematological malignancy. It is associated with increased risk of cancers, particularly in myeloid neoplasms, and chronic inflammatory diseases. The phenomenon of CHIP becomes very common in the population of people aged ≥80 years. That is explained by the accumulation of somatic mutations in HSCs, which occurs in an age-dependent manner (45, 46).
Clones evolve through the interaction of selectively advantageous ‘driver’ lesions, selectively neutral ‘passenger’ lesions and deleterious lesions. Driver lesions or mutations are the mutations that increase fitness and confer a clonal growth advantage. The neutral mutations, also known as passenger mutations, are accumulated in these cells but do not confer any fitness advantage. Neutral evolution of these passenger mutations can also shape clonal evolution, notably by a phenomenon called genetic drift, in which the allele frequencies of a mutation change over time. In addition, when both driver and passenger mutations occur in the same cell, the passenger mutations increase their allele frequency with the driver mutations, which is a phenomenon called hitchhiking that also participate in clonal evolution (47, 48).
The recognition of the important role of clonal hematopoiesis and clonal evolution in tumor initiation, disease progression and relapse have profound implications for the diagnosis and treatment of these malignancies. Additionally, the advent of new technologies may facilitate the definition of the molecular determinants and underlying mechanisms of clonal evolution in leukemia, which could provide targeted, individualized therapies for leukemia patients (49, 50).
Characterization of Leukemic Stem Cells
A major challenge in studying LSCs is identifying a possible unique cell surface antigen phenotype, from which it would be possible to develop a targeted and much more specific treatment than the treatments currently used. Furthermore, due to the heterogeneity of the different types of leukemias, there is a lot of variation in the antigens found on the surface of these cells, becoming even more difficult to identify a specific marker that is not expressed in normal cells, or has a different expression pattern, density, or distribution (51–54).
Some common stem cell’s markers are CD34, CD117 and HLA-DR, which expressions predict lower rate of complete remission (CR). In addition, the principal surface antigens of the myeloid lineage are CD13, CD33, CD14, CD15 and CD11b. Mostly, the expression of these markers have not yet showed any prognostic significance or are associated with a poorer outcome, such as reduction of CR, period of remission and survival. Although, cells that expresses CD15 usually presents a higher CR rate. Another marker found in myeloblasts is CD56, which expression is also reported as a poorer prognostic factor (55)
As an example, we can point out that normal HSC constitutively expresses CD34+ and CD38- antigens, in addition to others such as Thy-1+, c-kit+ and IL-3Rα. Much of the LSC population immunophenotypically resemble certain normal hematopoietic progenitor populations by also expressing CD34+ and CD38- besides others surface markers (56–58)
Despite these difficulties, a great number of cell surface markers have been identified that are upregulated on CD34+CD38- LSCs compared with normal CD34+CD38- HSPCs, for example, it has been revealed that CD90 and CD117 are deficient in acute myeloid leukemia (AML) LSCs, while CD123, TIM3, CD47, CD96, CLL-1, and IL-1 receptor accessory protein (IL1RAP), G protein-coupled receptor 56 (GPR56), CD93, CD44 and CD99 are highly expressed in AML LSCs. Targeting these surface markers might be a promising strategy for eradicating AML LSCs (35, 56, 59–62).
Some studies addressing chronic myeloid leukemia (CML) LSCs demonstrated that differentially expressed antigens include CD25, CD26, IL-1RAP, which is associated with the activation of NF-kβ and AKT signaling pathways, increasing proliferation of CML LSCs. In addition, the overexpression of the antigen CD25 is reported to reduce proliferation capacity of CML LSCs. Some data suggest CD25 and IL-1RAP expression are unique to LSCs of this type of leukemia (54, 63, 64).
The presence of LSCs is related to the rates of complete remission (CR) and general survival (OS) of patients, besides that, depending on the remaining amount, they may predispose to relapse of patients with leukemias (65). The identification of certain surface markers and molecular changes in these cells may influence the prognosis of patients, but the results of studies are still somewhat controversial. Bradstock et al. (66) pointed out that patients who expressed CD9, CD14 and CD2 in their CSLs had lower CR rates. The CD9, CD10 and CD11b markers were associated with lower OS rates, and CD11b was also related with a shorter duration of CR.
A study by Béné et al. (67) demonstrated that the expression of CD10, CD14 and CD15 was associated with lower survival rates. Nomdedeu et al. (68) found that patients who expressed CD34, CD45, CD117 and CD123 had worse prognoses with lower OS. Other studies, in turn, pointed out the markers CD2, CD7, CD11b, CD22, CD133, CD135, CD262 and CD120a as markers that confer worse prognosis to patients (69, 70).
The relationship between the molecular alterations observed in patients and their prognosis is also somewhat controversial among studies. Nomdedeu et al. (68) demonstrated that the FLT3-ITD mutation had a significant influence on OS, where patients affected by this alteration had lower rates compared to those who did not have the mutation (17.9 vs 41 months). On the other, Béné et al. (67) did not find a difference in survival between patients with and without molecular changes. However, other articles report the relationship of FLT3-ITD, MLL-PTD, RUNX1-RUNX1T1 and CBFB-MYH11 mutations with a poor outcome (71, 72).
In addition, studies demonstrated a correlation between white blood cells (WBC) count and poorer prognosis. Patients with a higher WBC count were less likely to achieve CR and presented a shorter survival rate (67, 73). Besides, a higher platelet count was associated with a longer survival time (67). These general findings corroborate with data found in other articles (74–77).
Presence of LSCS and Relapse
Even with the course of the disease, it is possible to observe that pre-LSCs and LSCs continue to evolve throughout the process, in addition to the fact that treatment often fails to reach these cells. LSCs play a key role from development to disease relapse. Thus, its analysis and quantification can be of great importance as a prognostic factor for patients. Such processes can also assist in choosing a more targeted treatment. The identification of these cells can be performed through immunophenotyping, where it is possible to differentiate HSCs from LSCs. From this differentiation, counting methods are performed (78–80).
Recent studies point to two classifications of relapse related LSCs: the first classification is known as LSCs of committed relapse origin, where these cells are most like the diagnostic cell type and were able to evolve similarly to the diagnostic dominant clone. The second classification is the LSCs of primitive relapse origin, which are rare cells at the diagnosis of the disease and do not usually form blasts. However, these cells may show greater resistance to treatment and later clonal evolutions, causing the patient to relapse, which usually continues with the increase in the amount of LSCs and with greater heterogeneity (81, 82).
Studies reveals that LSCs levels correspond to the clinical and laboratory characteristics of the patient. Due to their insufficient morphological and biochemical characterization, LSCs cannot be reliably measured in patient samples. However, as a consequence of the finite capacity of the joined stem cell niche, HSCs can act as a biomarker for LSCs numbers and help identify patients with an adverse clinical outcome (74, 83, 84).
This is noticeable through the LSCs and the HSCs count, considering that the BM may have a limited number of cells. Therefore, LSCs and HSCs compete for niches, their values being inversely proportional, that is, the more LSCs the less HSCs. Therefore, patients who had a low LSCs load also had lower blast, platelet, and leukocyte counts. In addition to clinical features, the cells count can represent how it might respond molecularly. This is important when evaluating the chosen treatment. Therefore, lower levels of LSCs are associated with a better molecular response to treatment (85, 86).
Still on the quantification of LSCs, its functionality also applies to the assessment of measurable residual disease (MRD) and is considered an effective biomarker for predicting relapses. So, in addition to being used in the diagnosis to choose the treatment, the immunophenotyping test can be used in the post-treatment phase to evaluate its effectiveness and predict the patient’s survival. High values of LSCs would be associated with worse survival and low efficacy. This analysis is then performed using molecular methodologies such as quantitative real-time polymerase chain reaction (RT-qPCR) or next-generation sequencing (NGS), and flow cytometry. It is important to remember that all these methodologies for prognostic analysis and MRD are still being carried out in studies and better clarification and standardization are needed for clinical application (83, 84, 87, 88).
Insights Into Clinical Investigations
Table 1 is comprised of clinical trials from the past 10 years that aimed to identify biomarkers specific to LSCs that could serve as targets for targeted therapies or could be used as prognostic factors (89–98). Most of the reported studies aim at AML treatment since LSC presence and complexity is an established risk factor for disease severity (51, 56).
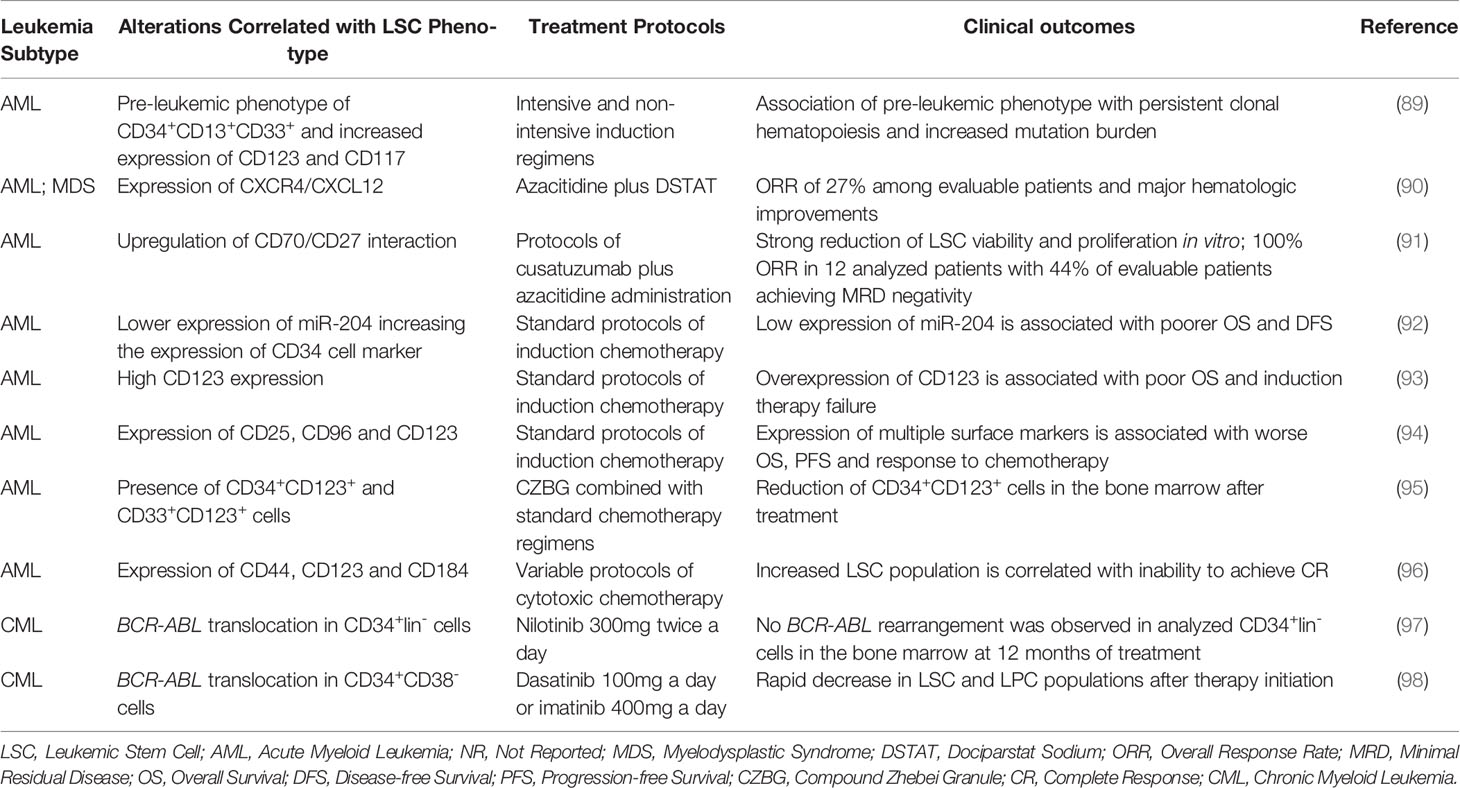
Table 1 Studies of the past 10 years indicating biomarkers for stem cells and “stemness” properties in leukemia and the respective prognostic relevance after treatment.
Studies utilizing standard chemotherapy protocols and induction therapies confirm that identified LSC markers, mainly transmembrane antigens such as CD34 and CD123, correlate with a worse patient prognosis. Increased mutation burden, lower response rates, inability to achieve CR, worst response to chemotherapy and higher incidence of relapse are some of the reported factors associated with increased presence of LSCs (89, 92–94, 96).
Although resistant to most treatment strategies, the use of novel agents targeting specifically LSCs molecular pathways combined with standard treatment protocols showed some promising results for AML patients (90, 91, 95). Huselton et al. (90) combined dociparstat sodium (DSTAT), a drug capable of inhibiting CXCR4/CXCL12 cell adhesion molecules, with hypomethylating agent (HMA) azacitidine in an attempt to disrupt bone marrow niches where LSCs remain in a quiescent state and was able to achieve CR in patients who were previously unresponsive to treatments with HMA alone.
Riether et al. (91) identified WNT pathway as being hyperactivated in AML LSCs due to increased expression and interaction of CD70/CD27 molecules. In vitro, the use of monoclonal antibodies targeting CD70 in combination with HMAs was demonstrated to have an additive effect in inhibiting LSC growth since the use of HMAs seem to increase LSCs dependency on CD70/CD27 pathway and concurrent CD70 inhibition was able to further reduce LSCs burden when compared to monotherapies of either agent. In previously untreated AML patients, protocols combining cusatuzumab plus azacitidine induced responses in all treated patients and transcriptome analysis after treatment revealed increased expression of genes involved in pathways of inflammation, differentiation and apoptosis (91).
Lastly, Wang et al. (95) utilized compound zhebei granule (CZBG), a herbal concoction with oncologic uses in traditional Chinese medicine, that acts through mechanism such as apoptosis induction and inhibition of resistance-related drug efflux proteins, combined with standard chemotherapy to treat AML patients and a significant decrease in CD34+CD123+ cells was observed in bone marrow niches. CZBG was also demonstrated to increase response rates in AML patients in combination with chemotherapy when compared to chemotherapy alone and to be able to reduce LSCs markers in tumor cell xenografts when combined with doxorubicin treatment (95, 99, 100).
In CML patients, in vitro studies indicate BCR-ABL tyrosine kinase inhibitors (TKIs) to have no efficacy over LSCs and leukemic progenitor cells (LPCs) and, while imatinib initial response rates are overwhelmingly positive, disease recurrence is usually the standard for patients after therapy discontinuation due to remaining Philadelphia-positive (Ph+) CD34+ cells (101, 102). The use of next-generation TKIs, however, seem to be more effective in reducing stem and progenitor cells in CML than imatinib and may point towards a choice for more intensive treatment options in accordance with increased LSCs and LPCs burden (103).
Pungolino et al. (97) utilized nilotinib, a second-generation BCR-ABL inhibitor, to treat newly diagnosed CML patients and observed a rapid decrease in CD34+lin-Ph+ cells in the bone marrow, with total clearance of the analyzed samples at 12 months of treatment. Mustjoki et al. (98) compared imatinib and dasatinib efficiency at decreasing stem and progenitor cell burden and, while both treatments had similar results at LSC inhibition, dasatinib showed increased activity over LPCs levels at 3 months analysis.
Treatment Perspectives
Most conventional treatments for leukemias seek to eradicate blasts, reaching cells that are in their active cycle. However, LSCs are usually in a quiescent state or protected through their molecular resistance mechanisms causing this cell to resist therapy, leading to patient relapse (104–111). One of the major known factors that confer drug resistance is the overexpression of drug efflux pumps, such as the ATP-binding cassette (ABC) transporter family proteins by CSCs. Moreover, drug efflux increase is often combined with the upregulation of enzymes involved in the metabolism of anticancer agents. Therefore, enzymes and efflux transporters expressed by LSCs appear to be crucial not only for their proliferation, but also for their resistance to clinical treatments (108, 112–115).
The selection of a target to LSCs therapy is fundamental. Ideally, the target chosen should be highly expressed by LSCs, highly selective, absence of expression on other cells, in particular HSCs, and preferentially expressed by a high number of patients. In view of the large number of similarities between LSCs and HSCs, it is not surprising that current treatment approaches are limited (20, 81, 116).
Currently, different treatment methodologies have been tested and addressed, such as the use of binding antibodies associated with different toxins to form a specific delivery vehicle for LSCs. Examples of such therapies are the use of Gemtuzumab Ozogamicin for the treatment of AML, a compound that uses an antigen against CD33, associated with a cytotoxic agent; and the inhibition of the SIRP1-α interaction with CD47 that activates innate immunity increasing the death of LSCs. In addition, CD244, CD123, LLC1 or TIM3 targets are also studied and demonstrate antileukemic efficacy in AML patients. However, one of the main difficulties in the treatment of LSCs is due to the low proliferation rate, which makes it difficult to identify the cell to start the therapy (62, 81, 117–119).
Several new strategies are under development to eliminating LSCs, which may result in a better patient response. Many of the studies are associated with the use of TKIs, which to improve their effectiveness can be combined with other agents. TNF-α inhibitors combined with TKIs, for example, have shown positive results in the elimination of LSCs. Blocking of IL-1 signaling may also be a combination with TKIs, as well as blocking of signal pathways such as Wnt/β-catenin, Hedgehog, MAPK/MNK1/2, mTOR, PTEN, PP2A, Alox5 and JAK/STAT. The action of HIF-1 inhibitors associated with TKIs has been shown to reduce the survival and growth of cells in CML in murine models. The HIF-1 deletion has also been tested in in vivo and in vitro models and has been shown to inhibit CML proliferation, both without serious effects on HSCs (120–122).
Activations and gene dissections can also be used, as in the case of p53 activation and EZH2 deletion. Both technologies demonstrate promising results that enhance the eradication of LSCs when combined with TKIs (123, 124). The combination of TKIs with cytarabine was also performed, demonstrating good results in AML patients. Despite the large number of tests involving TKIs, medications and methodologies have also been developing as the case of Bortezomibe. Its function is based on decreasing the expression of CDK6, an important agent in the proliferation of LSCs (120, 125, 126).
Undoubtedly, one of the greatest difficulties in eliminating LSCs is their resistance mechanisms. As a result, studies have specialized in finding drugs and technologies that help to overcome the resistance present in LSCs. In this scenario, research diverges in different areas such as transport proteins and signaling pathways, taking as an example Notch, Hedgehog, and Wnt/β-catenin that are describes also as responsible for drug resistance (104, 127–129). It is also worth mentioning studies focused on the epithelial-mesenchymal transition (EMT), histone acetylation, hypoxia and the BM niche (104, 130, 131).
The tumor microenvironment (TME) creates a niche for itself that influences not only the proliferation and differentiation of LSCs but also the response to drugs. A key factor that modulates the microenvironment and drug resistance is hypoxia, which signaling contributes to chemoresistance of CSCs by increasing the expression of ABC transporters and Aldehyde dehydrogenases, a family of intracellular enzymes, which can be used as molecular markers to identify normal stem cells (NSCs) and CSCs (104, 128, 132–134). An example was demonstrated by Giuntoli et al. (134), when CML cells were grown in low oxygen concentrations and became resistant to Imatinib.
The EMT process is already known in solid tumors and has recently been explored in hematological neoplasms as well as its treatment possibilities (104, 127, 134, 135). Thus, one of the ways is to look for drugs that can act on genes such as TRPS1, ETS2 and LSP, known to belong to this process in AML (136, 137). Competition for the BM niche between LSCs and HSCs has also become a therapeutic target, transforming the environment in a more favorable way for HSCs or increasing their hematopoietic reserve. E-selectin inhibition is an example, being able to promote the displacement of HSCs and LSCs. Regarding hypoxia, the use of hypoxia-activated prodrugs such as TH-302 has already been shown to reduce the population of LSCs in an AML model (138, 139).
In addition to those already mentioned, the combination of Venetoclax with Azacitidine demonstrates potential for the treatment of LSCs in AML, as it suppresses OXPHOS. Regarding gene deletion, it was observed that FOXO1 deletion through genetic or pharmacological pathways is able to inhibit the proliferation of malignant cells, which is present in LSCs and pre-LSCs, becoming a potential target. Studies aimed at the use of microRNAs mimics were also carried out and have potential, such as miR-15a/16-1 acting as a tumor suppressor acting negatively on the WT1 gene (28, 81, 140, 141).
Conclusion
Foremost is important to better define the molecular and cellular biologic features of normal HSCs and LSCs, for improve the identification of possible therapeutic targets to eradicate the LSCs, that are responsible for treatment resistance and clinical relapse for most patients. It is necessary to carry out studies that correlate the quantification and immunophenotypic characterization of LSCs with clinical data and prognosis presented by patients, regarding the significance of this information pointed in the studies here presented, and due to the lack of this type of study in the literature.
Author Contributions
IB, FP and CM wrote the manuscript. All authors contributed to the manuscript and were involved in revisions and proof-reading. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Brazilian funding agencies: Coordination for the Improvement of Higher Education Personnel (CAPES; to CM), National Council of Technological and Scientific Development (CNPq grant number 404213/2021-9 to CM-N; and Productivity in Research PQ Scholarships to MM-F, MM, RB and AK), Cearense Foundation of Scientific and Technological Support (FUNCAP grant number P20-0171-00078.01.00/20 to FP, MM and CM-N) and we also thank PROPESP/UFPA for publication payment.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wei Q, Frenette PS. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity (2018) 48(4):632–648. doi: 10.1016/j.immuni.2018.03.024
2. Yamamoto R, Wilkinson AC, Nakauchi H. Changing Concepts in Hematopoietic Stem Cells. Sci (1979) (2018) 362(6417):895–896. doi: 10.1126/science.aat7873
3. Bigas A, Espinosa L. Hematopoietic Stem Cells: To be or Notch to be. Blood (2012) 119(14):3226–35. doi: 10.1182/blood-2011-10-355826
4. Ema H, Morita Y, Suda T. Heterogeneity and Hierarchy of Hematopoietic Stem Cells. Exp Hematol (2014) 42(2):74–82.e2. doi: 10.1016/j.exphem.2013.11.004
5. Mosaad YM. Hematopoietic Stem Cells: An Overview. Transfusion Apheresis Sci (2014) 51(3):68–82. doi: 10.1016/j.transci.2014.10.016
6. Anthony BA, Link DC. Regulation of Hematopoietic Stem Cells by Bone Marrow Stromal Cells. Trends Immunol (2014) 35(1):32–7. doi: 10.1016/j.it.2013.10.002
7. Szade K, Gulati GS, Chan CKF, Kao KS, Miyanishi M, Marjon KD, et al. Where Hematopoietic Stem Cells Live: The Bone Marrow Niche. Antioxidants Redox Signaling (2018) 29(2):191–204. doi: 10.1089/ars.2017.7419
8. Yamashita M, Dellorusso P v., Olson OC, Passegué E. Dysregulated Haematopoietic Stem Cell Behaviour in Myeloid Leukaemogenesis. Nat Rev Cancer (2020) 20(7):365–382. doi: 10.1038/s41568-020-0260-3
9. Nagasawa T. Bone and Stem Cells. Bone Marrow Microenvironment Niches for Hematopoietic Stem and Progenitor Cells. Clin Calcium (2014) 24:517–526.
10. Boisset JC, Robin C. On the Origin of Hematopoietic Stem Cells: Progress and Controversy. Stem Cell Res (2012) 8(1):1–13. doi: 10.1016/j.scr.2011.07.002
11. Sawai CM, Babovic S, Upadhaya S, Knapp DJHF, Lavin Y, Lau CM, et al. Hematopoietic Stem Cells are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity (2016) 45(3):597–609. doi: 10.1016/j.immuni.2016.08.007
12. Abelson S, Collord G, Ng SWK, Weissbrod O, Cohen NM, Niemeyer E, et al. Prediction of Acute Myeloid Leukaemia Risk in Healthy Individuals. Nature (2018) 559(7714):400–404. doi:–10.1038/s41586-018-0317-6
13. Nakamura-Ishizu A, Takizawa H, Suda T. The Analysis, Roles and Regulation of Quiescence in Hematopoietic Stem Cells. Dev (Cambridge) (2014) 141(24):4656–66. doi: 10.1242/dev.106575
14. Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell (2016) 167(5):1296–1309.e10. doi: 10.1016/j.cell.2016.10.022
15. Slukvin II. Hematopoietic Specification From Human Pluripotent Stem Cells: Current Advances and Challenges Toward De Novo Generation of Hematopoietic Stem Cells. Blood (2013) 122(25):4035–46. doi: 10.1182/blood-2013-07-474825
16. Demirci S, Leonard A, Tisdale JF. Hematopoietic Stem Cells From Pluripotent Stem Cells: Clinical Potential, Challenges, and Future Perspectives. Stem Cells Trans Med (2020) 9(12):1549–1557. doi: 10.1002/sctm.20-0247
17. Bowman RL, Busque L, Levine RL. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell (2018) 22(2):157–170. doi: 10.1016/j.stem.2018.01.011
18. Verovskaya E v., Dellorusso P v., Passegué E. Losing Sense of Self and Surroundings: Hematopoietic Stem Cell Aging and Leukemic Transformation. Trends Mol Med (2019) 25(6):494–515. doi: 10.1016/j.molmed.2019.04.006
19. de Haan G, Lazare SS. Aging of Hematopoietic Stem Cells. Blood (2018) 131(5):479–487. doi: 10.1182/blood-2017-06-746412
20. Velten L, Story BA, Hernández-Malmierca P, Raffel S, Leonce DR, Milbank J, et al. Identification of Leukemic and Pre-Leukemic Stem Cells by Clonal Tracking From Single-Cell Transcriptomics. Nat Commun (2021) 12(1):1366. doi: 10.1038/s41467-021-21650-1
21. Corces MR, Chang HY, Majeti R. Preleukemic Hematopoietic Stem Cells in Human Acute Myeloid Leukemia. Front Oncol (2017) 7:263. doi: 10.3389/fonc.2017.00263
22. Pandolfi A, Barreyro L, Steidl U. Concise Review: Preleukemic Stem Cells: Molecular Biology and Clinical Implications of the Precursors to Leukemia Stem Cells. Stem Cells Trans Med (2013) 2(2):143–50. doi: 10.5966/sctm.2012-0109
23. Jordan CT, Guzman ML. Mechanisms Controlling Pathogenesis and Survival of Leukemic Stem Cells. Oncogene (2004) 23(43):7178–87. doi: 10.1038/sj.onc.1207935
24. Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal Evolution of Preleukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Sci Trans Med (2012) 4(149):149ra118. doi: 10.1126/scitranslmed.3004315
25. Corces-Zimmerman MR, Majeti R. Pre-Leukemic Evolution of Hematopoietic Stem Cells: The Importance of Early Mutations in Leukemogenesis. Leukemia (2014) 28(12):2276–82. doi: 10.1038/leu.2014.211
26. Saeed BR, Manta L, Raffel S, Pyl PT, Buss EC, Wang W, et al. Analysis of Nonleukemic Cellular Subcompartments Reconstructs Clonal Evolution of Acute Myeloid Leukemia and Identifies Therapy-Resistant Preleukemic Clones. Int J Cancer (2021) 148(11):2825–2838. doi: 10.1002/ijc.33461
27. Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic Mutations in Human Acute Myeloid Leukemia Affect Epigenetic Regulators and Persist in Remission. Proc Natl Acad Sci U.S.A. (2014) 111(7):2548–53. doi: 10.1073/pnas.1324297111
28. Lin S, Ptasinska A, Chen X, Shrestha M, Assi SA, Chin PS, et al. A FOXO1-Induced Oncogenic Network Defines the AML1-ETO Preleukemic Program. Blood (2017) 130(10):1213–1222. doi: 10.1182/blood-2016-11-750976
29. de Bie J, Demeyer S, Alberti-Servera L, Geerdens E, Segers H, Broux M, et al. Single-Cell Sequencing Reveals the Origin and the Order of Mutation Acquisition in T-Cell Acute Lymphoblastic Leukemia. Leukemia (2018) 32(6):1358–1369. doi: 10.1038/s41375-018-0127-8
30. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of Pre-Leukaemic Haematopoietic Stem Cells in Acute Leukaemia. Nature (2014) 506:328–33. doi: 10.1038/nature13038
31. Höllein A, Meggendorfer M, Dicker F, Jeromin S, Nadarajah N, Kern W, et al. NPM1 Mutated AML can Relapse With Wild-Type NPM1: Persistent Clonal Hematopoiesis can Drive Relapse. Blood Adv (2018) 2(22):3118–3125. doi: 10.1182/bloodadvances.2018023432
32. Potter N, Miraki-Moud F, Ermini L, Titley I, Vijayaraghavan G, Papaemmanuil E, et al. Single Cell Analysis of Clonal Architecture in Acute Myeloid Leukaemia. Leukemia (2019) 33:1113–1123. doi: 10.1038/s41375-018-0319-2
33. Wang X, Hoffman R. What are the Molecular Mechanisms Driving the Switch From Mpns to Leukemia? Best Pract Res: Clin Haematol (2021) 34(1):101254. doi: 10.1016/j.beha.2021.101254
34. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (1994) 367:645–648. doi: 10.1038/367645a0
35. Wang X, Huang S, Chen JL. Understanding of Leukemic Stem Cells and Their Clinical Implications. Mol Cancer (2017) 16(1):2. doi: 10.1186/s12943-016-0574-7
36. Touzet L, Dumezy F, Roumier C, Berthon C, Bories C, Quesnel B, et al. CD9 in Acute Myeloid Leukemia: Prognostic Role and Usefulness to Target Leukemic Stem Cells. Cancer Med (2019) 8(3):1279–1288. doi: 10.1002/cam4.2007
37. Park SM, Cho H, Thornton AM, Barlowe TS, Chou T, Chhangawala S, et al. IKZF2 Drives Leukemia Stem Cell Self-Renewal and Inhibits Myeloid Differentiation. Cell Stem Cell (2019) 24(1):153–165.e7. doi: 10.1016/j.stem.2018.10.016
38. Neering SJ, Bushnell T, Sozer S, Ashton J, Rossi RM, Wang PY, et al. Leukemia Stem Cells in a Genetically Defined Murine Model of Blast-Crisis CML. Blood (2007) 110(7):2578–85. doi: 10.1182/blood-2007-02-073031
39. Lane SW, Gilliland DG. Leukemia Stem Cells. Semin Cancer Biol (2010) 20(2):71–6. doi: 10.1016/j.semcancer.2009.12.001
40. Carroll D, St Clair DK. Hematopoietic Stem Cells: Normal Versus Malignant. Antioxidants Redox Signaling (2018) 29(16):1612–1632. doi: 10.1089/ars.2017.7326
41. Döhner K, Döhner H. Molecular Characterization of Acute Myeloid Leukemia. Haematologica (2008) 93(7):976–82. doi: 10.3324/haematol.13345
42. Desai RH, Zandvakili N, Bohlander SK. Dissecting the Genetic and non-Genetic Heterogeneity of Acute Myeloid Leukemia Using Next-Generation Sequencing and In Vivo Models. Cancers (Basel) (2022) 14:2182. doi: 10.3390/cancers14092182
43. Ye B, Sheng Y, Zhang M, Hu Y, Huang H. Early Detection and Intervention of Clonal Hematopoiesis for Preventing Hematological Malignancies. Cancer Lett (2022) 538:215691. doi: 10.1016/j.canlet.2022.215691
44. Alagpulinsa DA, Toribio MP, Alhallak I, Shmookler Reis RJ. Advances in Understanding the Molecular Basis of Clonal Hematopoiesis. Trends Mol Med (2022) 28(5):360–377. doi: 10.1016/j.molmed.2022.03.002
45. Rossi M, Meggendorfer M, Zampini M, Tettamanti M, Riva E, Travaglino E, et al. Clinical Relevance of Clonal Hematopoiesis in Persons Aged ≥80 Years. Blood (2021) 138(21):2093–2105. doi: 10.1182/blood.2021011320
46. Asada S, Kitamura T. Clonal Hematopoiesis and Associated Diseases: A Review of Recent Findings. Cancer Sci (2021) 112(10):3962–3971. doi: 10.1111/cas.15094
47. Lyne AM, Laplane L, Perié L. To Portray Clonal Evolution in Blood Cancer, Count Your Stem Cells. Blood (2021) 137(14):1862–1870. doi: 10.1182/blood.2020008407
48. Greaves M, Maley CC. Clonal Evolution in Cancer. Nature (2012) 481:306–13. doi: 10.1038/nature10762
49. Cooper JN, Young NS. Clonality in Context: Hematopoietic Clones in Their Marrow Environment. Blood (2017) 130(22):2363–2372. doi: 10.1182/blood-2017-07-794362
50. Ferrando AA, López-Otín C. Clonal Evolution in Leukemia. Nat Med (2017) 23(10):1135–1145. doi: 10.1038/nm.4410
51. Chopra M, Bohlander SK. The Cell of Origin and the Leukemia Stem Cell in Acute Myeloid Leukemia. Genes Chromosomes Cancer (2019) 58(12):850–858. doi: 10.1002/gcc.22805
52. Mastelaro de Rezende M, Ferreira AT, Paredes-Gamero EJ. Leukemia Stem Cell Immunophenotyping Tool for Diagnostic, Prognosis, and Therapeutics. J Cell Physiol (2020) 235(6):4989–4998. doi: 10.1002/jcp.29394
53. Lutz C, Hoang VT, Buss E, Ho AD. Identifying Leukemia Stem Cells - is it Feasible and Does it Matter? Cancer Lett (2013) 338(1):10–4. doi: 10.1016/j.canlet.2012.07.014
54. Houshmand M, Simonetti G, Circosta P, Gaidano V, Cignetti A, Martinelli G, et al. Chronic Myeloid Leukemia Stem Cells. Leukemia (2019) 33:1543–56. doi: 10.1038/s41375-019-0490-0
55. Mason KD, Juneja SK, Szer J. The Immunophenotype of Acute Myeloid Leukemia: Is There a Relationship With Prognosis? Blood Rev (2006) 20(2):71–82. doi: 10.1016/j.blre.2005.08.002
56. Thomas D, Majeti R. Biology and Relevance of Human Acute Myeloid Leukemia Stem Cells. Blood (2017) 129(12):1577–1585. doi: 10.1182/blood-2016-10-696054
57. Wang JCY, Dick JE. Cancer Stem Cells: Lessons From Leukemia. Trends Cell Biol (2005) 15:494–501. doi: 10.1016/j.tcb.2005.07.004
58. Roboz GJ, Guzman M. Acute Myeloid Leukemia Stem Cells: Seek and Destroy. Expert Rev Hematol (2009) 2(6):663–672. doi: 10.1586/ehm.09.53
59. Horton SJ, Huntly BJP. Recent Advances in Acute Myeloid Leukemia Stem Cell Biology. Haematologica (2012) 97(7):966–74. doi: 10.3324/haematol.2011.054734
60. Majeti R. Monoclonal Antibody Therapy Directed Against Human Acute Myeloid Leukemia Stem Cells. Oncogene (2011) 30(9):1009–19. doi: 10.1038/onc.2010.511
61. Guzman ML, Allan JN. Concise Review: Leukemia Stem Cells in Personalized Medicine. Stem Cells (2014) 32(4):829–843. doi: 10.1002/stem.1597
62. Haubner S, Perna F, Köhnke T, Schmidt C, Berman S, Augsberger C, et al. Coexpression Profile of Leukemic Stem Cell Markers for Combinatorial Targeted Therapy in AML. Leukemia (2019) 33:64–74. doi: 10.1038/s41375-018-0180-3
63. Sadovnik I, Hoelbl-Kovacic A, Herrmann H, Eisenwort G, Cerny-Reiterer S, Warsch W, et al. Identification of CD25 as STAT5-Dependent Growth Regulator of Leukemic Stem Cells in Ph+ CML. Clin Cancer Res (2016) 22(8):2051–61. doi: 10.1158/1078-0432.CCR-15-0767
64. Ågerstam H, Hansen N, von Palffy S, Sanden C, Reckzeh K, Karlsson C, et al. IL1RAP Antibodies Block IL-1-Induced Expansion of Candidate CML Stem Cells and Mediate Cell Killing in Xenograft Models. Blood (2016) 128(23):2683–2693. doi: 10.1182/blood-2015-11-679985
65. Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the Origins of Relapse in Acute Myeloid Leukaemia to Stem Cells. Nature (2017) 547:104–108. doi: 10.1038/nature22993
66. Bradstock K, Matthews J, Benson E, Page F, Bishop J. Prognostic Value of Immunophenotyping in Acute Myeloid Leukemia. Blood (1994) 25(4):367–74. doi: 10.1182/blood.v84.4.1220.1220
67. Béné MC, Bernier M, Casasnovas RO, Castoldi G, Doekharan D, van der Holt B, et al. Acute Myeloid Leukaemia M0: Haematological, Immunophenotypic and Cytogenetic Characteristics and Their Prognostic Significance: An Analysis in 241 Patients. Br J Haematol (2001) 113(3):737–45. doi: 10.1046/j.1365-2141.2001.02801.x
68. Nomdedeu J, Bussaglia E, Villamor N, Martinez C, Esteve J, Tormo M, et al. Immunophenotype of Acute Myeloid Leukemia With NPM Mutations: Prognostic Impact of the Leukemic Compartment Size. Leukemia Res (2011) 35(2):163–8. doi: 10.1016/j.leukres.2010.05.015
69. Costa AFO, Menezes DL, Pinheiro LHS, Sandes AF, Nunes MAP, Lyra Junior DP, et al. Role of New Immunophenotypic Markers on Prognostic and Overall Survival of Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. Sci Rep (2017) 7(1):4138. doi: 10.1038/s41598-017-00816-2
70. van Solinge TS, Zeijlemaker W, Ossenkoppele GJ, Cloos J, Schuurhuis GJ. The Interference of Genetic Associations in Establishing the Prognostic Value of the Immunophenotype in Acute Myeloid Leukemia. Cytomet Part B - Clin Cytomet (2018) 94(1):151–158. doi: 10.1002/cyto.b.21539
71. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic Relevance of Integrated Genetic Profiling in Acute Myeloid Leukemia. New Engl J Med (2012) 366:1079–89. doi: 10.1056/nejmoa1112304
72. Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, et al. Comprehensive Analysis of Genetic Alterations and Their Prognostic Impacts in Adult Acute Myeloid Leukemia Patients. Leukemia (2014) 28:1586–1595. doi: 10.1038/leu.2014.55
73. Geller RB, Zahurak M, Hurwitz CA, Burke PJ, Karp JE, Piantadosi S, et al. Prognostic Importance of Immunophenotyping in Adults With Acute Myelocytic Leukaemia: The Significance of the Stem-Cell Glycoprotein CD34 (My 10). Br J Haematol (1990) 76:340–347. doi: 10.1111/j.1365-2141.1990.tb06365.x
74. Pfirrmann M, Baccarani M, Saussele S, Guilhot J, Cervantes F, Ossenkoppele G, et al. Prognosis of Long-Term Survival Considering Disease-Specific Death in Patients With Chronic Myeloid Leukemia. Leukemia (2016) 30:48–56. doi: 10.1038/leu.2015.261
75. Wattad M, Weber D, Döhner K, Krauter J, Gaidzik VI, Paschka P, et al. Impact of Salvage Regimens on Response and Overall Survival in Acute Myeloid Leukemia With Induction Failure. Leukemia (2017) 31:1306–1313. doi: 10.1038/leu.2017.23
76. Padilha SL, Souza EJ dos S, Matos MCC, Domino NR. Acute Myeloid Leukemia: Survival Analysis of Patients at a University Hospital of Paraná. Rev Bras Hematologia e Hemoterapia (2013) 37(1):21–7. doi: 10.1016/j.bjhh.2014.11.008
77. Hoyos M, Nomdedeu JF, Esteve J, Duarte R, Ribera JM, Llorente A, et al. Core Binding Factor Acute Myeloid Leukemia: The Impact of Age, Leukocyte Count, Molecular Findings and Minimal Residual Disease. Eur J Haematol (2013) 91(3):209–18. doi: 10.1111/ejh.12130
78. Zeijlemaker W, Kelder A, Cloos J, Schuurhuis GJ. Immunophenotypic Detection of Measurable Residual (Stem Cell) Disease Using LAIP Approach in Acute Myeloid Leukemia. Curr Protoc Cytomet (2019) 91(1):e66. doi: 10.1002/cpcy.66
79. Heo SK, Noh EK, Ju LJ, Sung JY, Jeong YK, Cheon J, et al. CD45dimCD34+CD38-CD133+ Cells Have the Potential as Leukemic Stem Cells in Acute Myeloid Leukemia. BMC Cancer (2020) 20(1):285. doi: 10.1186/s12885-020-06760-1
80. Felipe Rico J, Hassane DC, Guzman ML. Acute Myelogenous Leukemia Stem Cells: From Bench to Bedside. Cancer Lett (2013) 338(1):4–9. doi: 10.1016/j.canlet.2012.05.034
81. Pollyea DA, Jordan CT. Therapeutic Targeting of Acute Myeloid Leukemia Stem Cells. Blood (2017) 129(12):1627–1635. doi: 10.1182/blood-2016-10-696039
82. Stauber J, Greally JM, Steidl U. Preleukemic and Leukemic Evolution at the Stem Cell Level. Blood (2021) 137(8):1013–1018. doi: 10.1182/blood.2019004397
83. Wouters R, Cucchi D, Kaspers GJL, Schuurhuis GJ, Cloos J. Relevance of Leukemic Stem Cells in Acute Myeloid Leukemia: Heterogeneity and Influence on Disease Monitoring, Prognosis and Treatment Design. Expert Rev Hematol (2014) 7(6):791–805. doi: 10.1586/17474086.2014.959921
84. Reinisch A, Chan SM, Thomas D, Majeti R. Biology and Clinical Relevance of Acute Myeloid Leukemia Stem Cells. Semin Hematol (2015) 52(3):150–64. doi: 10.1053/j.seminhematol.2015.03.008
85. Wang W, Stiehl T, Raffel S, Hoang VT, Hoffmann I, Poisa-Beiro L, et al. Reduced Hematopoietic Stem Cell Frequency Predicts Outcome in Acute Myeloid Leukemia. Haematologica (2017) 102(9):1567–1577. doi: 10.3324/haematol.2016.163584
86. Thielen N, Richter J, Baldauf M, Barbany G, Fioretos T, Giles F, et al. Leukemic Stem Cell Quantification in Newly Diagnosed Patients With Chronic Myeloid Leukemia Predicts Response to Nilotinib Therapy. Clin Cancer Res (2016) 22(16):4030–8. doi: 10.1158/1078-0432.CCR-15-2791
87. Bruserud Ø, Aasebø E, Hernandez-Valladares M, Tsykunova G, Reikvam H. Therapeutic Targeting of Leukemic Stem Cells in Acute Myeloid Leukemia–the Biological Background for Possible Strategies. Expert Opin Drug Discovery (2017) 12(10):1053–1065. doi: 10.1080/17460441.2017.1356818
88. Cloos J, Harris JR, Janssen JJWM, Kelder A, Huang F, Sijm G, et al. Comprehensive Protocol to Sample and Process Bone Marrow for Measuring Measurable Residual Disease and Leukemic Stem Cells in Acute Myeloid Leukemia. J Visualized Experiments (2018) 2018(133):56386. doi: 10.3791/56386
89. Loghavi S, DiNardo CD, Furudate K, Takahashi K, Tanaka T, Short NJ, et al. Flow Cytometric Immunophenotypic Alterations of Persistent Clonal Haematopoiesis in Remission Bone Marrows of Patients With NPM1-Mutated Acute Myeloid Leukaemia. Br J Haematol (2021) 192(6):1054–1063. doi: 10.1111/bjh.17347
90. Huselton E, Rettig MP, Campbell K, Cashen AF, DiPersio JF, Gao F, et al. Combination of Dociparstat Sodium (DSTAT), a CXCL12/CXCR4 Inhibitor, With Azacitidine for the Treatment of Hypomethylating Agent Refractory AML and MDS. Leukemia Res (2021) 110:106713. doi: 10.1016/j.leukres.2021.106713
91. Riether C, Pabst T, Höpner S, Bacher U, Hinterbrandner M, Banz Y, et al. Targeting CD70 With Cusatuzumab Eliminates Acute Myeloid Leukemia Stem Cells in Patients Treated With Hypomethylating Agents. Nat Med (2020) 26(9):1459–1467. doi: 10.1038/s41591-020-0910-8
92. Abdelhafiz AS, Elsayed GM, Saber MM, Gameel A, Hamdy N. Low Expression of Mir-204 is Associated With Expression of CD34 and Poor Performance Status in Denovo AML. Int J Lab Hematol (2020) 42(3):263–269. doi: 10.1111/ijlh.13161
93. Arai N, Homma M, Abe M, Baba Y, Murai S, Watanuki M, et al. Impact of CD123 Expression, Analyzed by Immunohistochemistry, on Clinical Outcomes in Patients With Acute Myeloid Leukemia. Int J Hematol (2019) 109(5):539–544. doi: 10.1007/s12185-019-02616-y
94. Yabushita T, Satake H, Maruoka H, Morita M, Katoh D, Shimomura Y, et al. Expression of Multiple Leukemic Stem Cell Markers is Associated With Poor Prognosis in De Novo Acute Myeloid Leukemia. Leukemia Lymphoma (2018) 59(9):2144–2151. doi: 10.1080/10428194.2017.1410888
95. Wang J, Lai Z l, Chen X y, Li D y, Zhang Y y, Ma W, et al. Effect of Compound Zhebei Granule (复方浙贝颗粒) Combined With Chemotherapy on Surface Markers of Leukemia Stem Cell in Patients With Acute Myeloid Leukemia. Chin J Integr Med (2016) 22(6):438–44. doi: 10.1007/s11655-015-2117-2
96. Hwang K, Park CJ, Jang S, Chi HS, Kim DY, Lee JH, et al. Flow Cytometric Quantification and Immunophenotyping of Leukemic Stem Cells in Acute Myeloid Leukemia. Ann Hematol (2012) 91(10):1541–6. doi: 10.1007/s00277-012-1501-7
97. Pungolino E, D’adda M, de Canal G, Trojani A, Perego A, Elena C, et al. Nilotinib-Induced Bone Marrow CD34+/Lin-Ph+ Cells Early Clearance in Newly Diagnosed CP-Chronic Myeloid Leukemia: Final Report of the Philosophi34 Study. Eur J Haematol (2021) 107(4):436–448. doi: 10.1111/ejh.13680
98. Mustjoki S, Richter J, Barbany G, Ehrencrona H, Fioretos T, Gedde-Dahl T, et al. Impact of Malignant Stem Cell Burden on Therapy Outcome in Newly Diagnosed Chronic Myeloid Leukemia Patients. Leukemia (2013) 27(7):1520–6. doi: 10.1038/leu.2013.19
99. Zhang Y, Hou L, Chen X y. Effects of Compound Zhebei Granule () Combined With Doxorubicin on Expression of Specific Surface Antigens in Mice With Transplanted KG-1a Cells. Chin J Integr Med (2018) 24(3):213–217. doi: 10.1007/s11655-017-2963-1
100. Hou L, Yang S, Yang W, Zhou Y, Liu F, Yang H, et al. Compound Zhebei Granules Combined With Chemotherapy for the Treatment of Refractory Acute Leukemia: A Randomized Clinical Trial. J Tradit Chin Med (2016) 36(5):606–12. doi: 10.1016/s0254-6272(16)30079-6
101. Vetrie D, Helgason GV, Copland M. The Leukaemia Stem Cell: Similarities, Differences and Clinical Prospects in CML and AML. Nat Rev Cancer (2020) 20(3):158–173. doi: 10.1038/s41568-019-0230-9
102. Bocchia M, Ippoliti M, Gozzetti A, Abruzzese E, Calabrese S, Amabile M, et al. CD34+/Ph+ Cells are Still Detectable in Chronic Myeloid Leukemia Patients With Sustained and Prolonged Complete Cytogenetic Remission During Treatment With Imatinib Mesylate [2]. Leukemia (2008) 22(2):426–8. doi: 10.1038/sj.leu.2404893
103. Tusa I, Cheloni G, Poteti M, Silvano A, Tubita A, Lombardi Z, et al. In Vitro Comparison of the Effects of Imatinib and Ponatinib on Chronic Myeloid Leukemia Progenitor/Stem Cell Features. Targeted Oncol (2020) 15(5):659–671. doi: 10.1007/s11523-020-00741-x
104. Cho Y, Kim YK. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front Oncol (2020) 10:764. doi: 10.3389/fonc.2020.00764
105. Moitra K. Overcoming Multidrug Resistance in Cancer Stem Cells. BioMed Res Int (2015) 2015:635745. doi: 10.1155/2015/635745
106. Zhang Q, Feng Y, Kennedy D. Multidrug-Resistant Cancer Cells and Cancer Stem Cells Hijack Cellular Systems to Circumvent Systemic Therapies, can Natural Products Reverse This? Cell Mol Life Sci (2017) 74(5):777–801. doi: 10.1007/s00018-016-2362-3
107. Zhang Z, Zhao Y, Jiang L, Miao X, Zhou H, Jia L. Glycomic Alterations are Associated With Multidrug Resistance in Human Leukemia. Int J Biochem Cell Biol (2012) 44(8):1244–53. doi: 10.1016/j.biocel.2012.04.026
108. Dalpiaz A, Paganetto G, Botti G, Pavan B. Cancer Stem Cells and Nanomedicine: New Opportunities to Combat Multidrug Resistance? Drug Discovery Today (2020) 25(9):1651–1667. doi: 10.1016/j.drudis.2020.07.023
109. Daflon-Yunes N, Pinto-Silva FE, Vidal RS, Novis BF, Berguetti T, Lopes RRS, et al. Characterization of a Multidrug-Resistant Chronic Myeloid Leukemia Cell Line Presenting Multiple Resistance Mechanisms. Mol Cell Biochem (2013) 383(1-2):123–35. doi: 10.1007/s11010-013-1761-0
110. de Moraes ACR, Maranho CK, Rauber GS, Santos-Silva MC. Importance of Detecting Multidrug Resistance Proteins in Acute Leukemia Prognosis and Therapy. J Clin Lab Anal (2013) 27(1):62–71. doi: 10.1002/jcla.21563
111. Patel C, Stenke L, Varma S, Lindberg ML, Björkholm M, Sjöberg J, et al. Multidrug Resistance in Relapsed Acute Myeloid Leukemia: Evidence of Biological Heterogeneity. Cancer (2013) 119(16):3076–83. doi: 10.1002/cncr.28098
112. Moitra K, Lou H, Dean M. Multidrug Efflux Pumps and Cancer Stem Cells: Insights Into Multidrug Resistance and Therapeutic Development. Clin Pharmacol Ther (2011) 89(4):491–502. doi: 10.1038/clpt.2011.14
113. Drewa T, Styczynski J, Szczepanek J. Is the Cancer Stem Cell Population “A Player” in Multi-Drug Resistance? Acta Poloniae Pharm - Drug Res (2008) 65.
114. Marques DS, Sandrini JZ, Boyle RT, Marins LF, Trindade GS. Relationships Between Multidrug Resistance (MDR) and Stem Cell Markers in Human Chronic Myeloid Leukemia Cell Lines. Leukemia Res (2010) 34(6):757–62. doi: 10.1016/j.leukres.2009.11.004
115. Moreira-Nunes CF, Azevedo TC, Beltrão AC, Francês LT, Sousa RG, Silva IT, et al. Differentially Expressed Genes Responsible for Insensitivity of CD34+ Cells to Kinase Inhibitors in Patients With Chronic Myeloid Leukemia. BMC Proc (2013) 7(2):01. doi: 10.1186/1753-6561-7-s2-o1
116. Hanekamp D, Cloos J, Schuurhuis GJ. Leukemic Stem Cells: Identification and Clinical Application. Int J Hematol (2017) 105(5):549–557. doi: 10.1007/s12185-017-2221-5
117. Theocharides APA, Jin L, Cheng PY, Prasolava TK, Malko A v, Ho JM, et al. Disruption of Sirpα Signaling in Macrophages Eliminates Human Acute Myeloid Leukemia Stem Cells in Xenografts. J Exp Med (2012) 209(10):1883–99. doi: 10.1084/jem.20120502
118. Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute Myeloid Leukemia Stem Cells and CD33-Targeted Immunotherapy. Blood (2012) 119(26):6198–208. doi: 10.1182/blood-2011-11-325050
119. Jiang YP, Liu BY, Zheng Q, Panuganti S, Chen R, Zhu J, et al. CLT030, a Leukemic Stem Cell-Targeting CLL1 Antibody-Drug Conjugate for Treatment of Acute Myeloid Leukemia. Blood Adv (2018) 2(14):1738–1749. doi: 10.1182/bloodadvances.2018020107
120. Chen Y, Zou J, Cheng F, Li W. Treatment-Free Remission in Chronic Myeloid Leukemia and New Approaches by Targeting Leukemia Stem Cells. Front Oncol (2021) 11:769730. doi: 10.3389/fonc.2021.769730
121. Cheloni G, Tanturli M, Tusa I, DeSouza NH, Shan Y, Gozzini A, et al. Targeting Chronic Myeloid Leukemia Stem Cells With the Hypoxia-Inducible Factor Inhibitor Acriflavine. Blood (2017) 130(5):655–665. doi: 10.1182/blood-2016-10-745588
122. Zhang H, Li H, Xi HS, Li S. Hif1α is Required for Survival Maintenance of Chronic Myeloid Leukemia Stem Cells. Blood (2012) 119(11):2595–607. doi: 10.1182/blood-2011-10-387381
123. Carter BZ, Mak PY, Mak DH, Ruvolo VR, Schober W, McQueen T, et al. Synergistic Effects of P53 Activation via MDM2 Inhibition in Combination With Inhibition of Bcl-2 or Bcr-Abl in CD34+ Proliferating and Quiescent Chronic Myeloid Leukemia Blast Crisis Cells. Oncotarget (2015) 6(31):30487–99. doi: 10.18632/oncotarget.5890
124. Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, et al. Chronic Myelogenous Leukemia-Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discovery (2016) 6(11):1237–1247. doi: 10.1158/2159-8290.CD-15-1439
125. Zhou B, Qin Y, Zhou J, Ruan J, Xiong F, Dong J, et al. Bortezomib Suppresses Self-Renewal and Leukemogenesis of Leukemia Stem Cell by NF-ĸb-Dependent Inhibition of CDK6 in MLL-Rearranged Myeloid Leukemia. J Cell Mol Med (2021) 25(6):3124–3135. doi: 10.1111/jcmm.16377
126. Polak A, Bialopiotrowicz E, Krzymieniewska B, Wozniak J, Stojak M, Cybulska M, et al. SYK Inhibition Targets Acute Myeloid Leukemia Stem Cells by Blocking Their Oxidative Metabolism. Cell Death Dis (2020) 11(11):956. doi: 10.1038/s41419-020-03156-8
127. Di C, Zhao Y. Multiple Drug Resistance Due to Resistance to Stem Cells and Stem Cell Treatment Progress in Cancer (Review). Exp Ther Med (2015) 9(2):289–293. doi: 10.3892/etm.2014.2141
128. Misaghian N, Ligresti G, Steelman LS, Bertrand FE, Bäsecke J, Libra M, et al. Targeting the Leukemic Stem Cell: The Holy Grail of Leukemia Therapy. Leukemia (2009) 23(1):25–42. doi: 10.1038/leu.2008.246
129. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt Pathways in Cancer Stem Cells: Clinical Update. Nat Rev Clin Oncol (2015) 12(8):445–64. doi: 10.1038/nrclinonc.2015.61
130. Liesveld J. Targeting Myelogenous Leukemia Stem Cells: Role of the Circulation. Front Oncol (2012) 2:86. doi: 10.3389/fonc.2012.00086. 2 AUG.
131. Marchand T, Pinho S. Leukemic Stem Cells: From Leukemic Niche Biology to Treatment Opportunities. Front Immunol (2021) 12:775128. doi: 10.3389/fimmu.2021.775128
132. Garcia-Mayea Y, Mir C, Masson F, Paciucci R, LLeonart ME. Insights Into New Mechanisms and Models of Cancer Stem Cell Multidrug Resistance. Semin Cancer Biol (2020) 60:166–180. doi: 10.1016/j.semcancer.2019.07.022
133. Bukowski K, Kciuk M, Kontek R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int J Mol Sci (2020) 21(9):3233. doi: 10.3390/ijms21093233
134. Giuntoli S, Rovida E, Barbetti V, Cipolleschi MG, Olivotto M, dello Sbarba P. Hypoxia Suppresses BCR/Abl and Selects Imatinib-Insensitive Progenitors Within Clonal CML Populations [2]. Leukemia (2006) 20(7):1291–3. doi: 10.1038/sj.leu.2404224
135. Chen SC, Liao TT, Yang MH. Emerging Roles of Epithelial-Mesenchymal Transition in Hematological Malignancies. J Biomed Sci (2018) 25(1):37. doi: 10.1186/s12929-018-0440-6
136. Kahlert UD, Joseph J v., Kruyt FAE. EMT- and MET-Related Processes in Nonepithelial Tumors: Importance for Disease Progression, Prognosis, and Therapeutic Opportunities. Mol Oncol (2017) 11(7):860–877. doi: 10.1002/1878-0261.12085
137. Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell (2016) 30(1):43–58. doi: 10.1016/j.ccell.2016.05.011
138. Benito J, Ramirez MS, Millward NZ, Velez J, Harutyunyan KG, Lu H, et al. Hypoxia-Activated Prodrug TH-302 Targets Hypoxic Bone Marrow Niches in Preclinical Leukemia Models. Clin Cancer Res (2016) 22(7):1687–98. doi: 10.1158/1078-0432.CCR-14-3378
139. Bernasconi P, Borsani O. Targeting Leukemia Stem Cell-Niche Dynamics: A New Challenge in AML Treatment. J Oncol (2019) 2019:8323592. doi: 10.1155/2019/8323592
140. Arrigoni E, del Re M, Galimberti S, Restante G, Rofi E, Crucitta S, et al. Concise Review: Chronic Myeloid Leukemia: Stem Cell Niche and Response to Pharmacologic Treatment. Stem Cells Trans Med (2018) 7(3):305–314. doi: 10.1002/sctm.17-0175
Keywords: hematopoietic stem cells, leukemia stem cell, molecular biomarkers, clinical relapse, drug resistance
Citation: Barreto IV, Pessoa FMCP, Machado CB, Pantoja LC, Ribeiro RM, Lopes GS, Amaral de Moraes ME, de Moraes Filho MO, de Souza LEB, Burbano RMR, Khayat AS and Moreira-Nunes CA (2022) Leukemic Stem Cell: A Mini-Review on Clinical Perspectives. Front. Oncol. 12:931050. doi: 10.3389/fonc.2022.931050
Received: 28 April 2022; Accepted: 25 May 2022;
Published: 24 June 2022.
Edited by:
Leonardo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilReviewed by:
Avik Choudhuri, Harvard University, United StatesCopyright © 2022 Barreto, Pessoa, Machado, Pantoja, Ribeiro, Lopes, Amaral de Moraes, de Moraes Filho, de Souza, Burbano, Khayat and Moreira-Nunes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Aquino Moreira-Nunes, Y2Fyb2xmYW1AZ21haWwuY29t
†These authors have contributed equally to this work and share first authorship