 Meghri Katerji1
Meghri Katerji1 Antonella Bertucci2Valery Filippov1
Antonella Bertucci2Valery Filippov1 Marcelo Vazquez1,2Xin Chen1,3
Marcelo Vazquez1,2Xin Chen1,3 Penelope J. Duerksen-Hughes1*
Penelope J. Duerksen-Hughes1*- 1Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA, United States
- 2Department of Radiation Medicine, Loma Linda University Medical Center, Loma Linda, CA, United States
- 3Center for Genomics, Loma Linda University School of Medicine, Loma Linda, CA, United States
High-risk human papillomaviruses (HPVs) cause virtually all cervical cancer cases and are also associated with other types of anogenital and oropharyngeal cancers. Normally, HPV exists as a circular episomal DNA in the infected cell. However, in some instances, it integrates into the human genome in such a way as to enable increased expression of viral oncogenes, thereby leading to carcinogenesis. Since viral integration requires breaks in both viral and human genomes, DNA damage likely plays a key role in this critical process. One potentially significant source of DNA damage is exposure to elevated doses of ionizing radiation. Natural background radiation is ubiquitous; however, some populations, including radiological workers, radiotherapy patients, and astronauts, are exposed to significantly higher radiation doses, as well as to different types of radiation such as particle radiation. We hypothesize that ionizing radiation-induced DNA damage facilitates the integration of HPV into the human genome, increasing the risk of developing HPV-related cancers in the exposed population. To test this, we first determined the kinetics of DNA damage in keratinocytes exposed to ionizing radiation (protons) by assessing γ-H2AX foci formation using immunofluorescence (direct damage), and also measured ROS and 8-oxoG levels via DCFDA and Avidin-FITC (indirect damage).As anticipated, direct DNA damage was observed promptly, within 30 min, whereas indirect DNA damage was delayed due to the time required for ROS to accumulate and cause oxidative damage. Although radiation was lethal at high doses, we were able to establish an experimental system where radiation exposure (protons and X-rays) induced DNA damage dose-dependently without causing major cytotoxic effects as assessed by several cytotoxicity assays. Most importantly, we explored the impact of radiation exposure on integration frequency using a clonogenic assay and demonstrated that as predicted, proton-induced DNA damage promotes the integration of HPV-like foreign DNA in oral keratinocytes. Overall, the insights gained from this work enable us to better understand the contribution of radiation exposure and DNA damage to HPV-mediated carcinogenesis and direct us toward strategies aimed at preventing malignancies in HPV-infected individuals.
Introduction
Human papillomavirus (HPV) is the most common sexually transmitted infection. According to the Centers for Disease Control and Prevention, 79 million Americans are currently infected with HPV, with another 20 million new infections occurring each year (1). In about 90% of these cases, the infection is cleared by the immune system within two years. However, a relatively small subset of infections persists, and of these, some progress to malignancy (2–4). High-risk HPVs are the causative agents of virtually all cases of cervical cancer as well as a significant percentage of other anogenital and oropharyngeal cancers. In fact, current estimates indicate that HPV infection may be associated with as many as 93% of anal cancers, 63% of oropharyngeal cancers, 40% of penile cancers, 64% of vaginal cancers, and 51% of vulvar cancers (5). It is projected that 5.2% of all cancers worldwide can be attributed to HPV (6, 7).
The oncogenic properties of high-risk HPVs are encoded in two viral oncoproteins, E6 and E7. The E6 oncoprotein forms a complex with p53 and targets it to proteasomal degradation, resulting in evasion of apoptosis and perturbation of cell cycle control. On the other hand, the E7 oncoprotein binds to Rb and causes its degradation, leading to the inappropriate release of E2F transcription factor which stimulates unrestrained replication and cell division. Therefore, when these viral oncoproteins are over-expressed, the HPV-infected cells undergo uncontrolled cell proliferation and survival, and consequently develop HPV-induced malignancies (7–9).
Normally, the HPV genome is present inside the host cells in the circular episomal form. Under these circumstances, the viral E2 gene regulates viral transcription and genome replication, and thereby maintains the expression of the viral E6 and E7 oncogenes at low levels, insufficient to cause malignancy (10, 11). In the course of carcinogenesis, however, the extrachromosomal viral genome often becomes integrated into the host genome. This integration event functions as a critical biological driver of cellular transformation, since it typically results in the disruption and loss of the negative regulator E2, allowing persistent over-expression of the E6 and E7 oncogenes (12–15). Less commonly, elevated expression of the viral oncogenes can also be achieved by genetic or epigenetic modifications of the HPV genome or the presence of an increased number of episomal HPV copies (16–19). While a high episomal viral load, combined with an absence of HPV integration, is frequently detected in precancerous lesions, a high proportion of invasive HPV-associated cancers contain the viral DNA integrated into the host genome (18, 20). Analysis of samples from The Cancer Genome Atlas study indicates that HPV integration occurs in >80% of HPV-positive cervical cancers. Of these, 76% of HPV-16 positive samples contained integrated HPV, whereas integration was detected in all HPV18-positive samples (21, 22). The rate of HPV integration in other anogenital cancers is not as well documented, with one study reporting that almost 80% of anal cancers contain integrated HPV (23). In the case of HPV-positive oropharyngeal squamous cell carcinomas, the incidence of viral integration is relatively lower, with many tumors having either extrachromosomal or mixed extrachromosomal and integrated viral DNA (24–26).
It has been suggested that DNA damage plays an essential role in HPV integration, since the process of integration requires linearization of the viral genome, breakage of the host genome, and insertion of viral genome followed by re-ligation of the ends together (27–30). Determining which agents and events are likely to cause breaks in the viral and host DNA is therefore a reasonable approach to identify the factors that impact HPV integration frequency. One of the most common agents known to cause DNA damage are reactive oxygen species (ROS), generated in excessive amounts under conditions of oxidative stress (31–33). Our lab has previously demonstrated a high variability in the background levels of oxidative stress markers between non-cancerous cervical cells and tissues of different women, which provides a possible explanation for why some, but not most, HPV-infected individuals seem predisposed to develop cervical cancer (34). Importantly, we have also shown that chronic oxidative stress, caused by environmental or viral factors such as E6*, can induce DNA damage and increase the frequency of integration of HPV16 into the genome of cervical keratinocytes, and in that way contribute to HPV-mediated carcinogenesis (35). Supporting our data, environmental conditions known to cause oxidative stress, such as smoking and co-infection with STD-associated pathogens Chlamydia trachomatis and Neisseria gonorrhoeae, have been associated with increased incidence of HPV-mediated malignancies (36–38), and several publications have reported increased oxidative stress in patients with HPV-related cancers (39–42).
Another potentially significant source of DNA damage is exposure to elevated doses and different qualities of ionizing radiation. Natural background radiation is ubiquitous; hence, all are exposed. However, some populations, including radiological workers, military personnel, radiation therapy patients, commercial pilots, and astronauts, are exposed to significantly higher acute or chronic doses of ionizing radiation (43). One type of ionizing radiation is proton radiation which is characterized by its physical properties as a subatomic particle with a mass and a positive charge that can be accelerated naturally in space (by the sun) or artificially (by particle accelerators).
In space, proton radiation is part of the radiation environmental spectra and can pose a health risk for humans during long- and short-term spaceflights (44, 45). Solar particle events (SPEs) which accelerate protons and release unpredictable doses of radiation is one of the space radiation environment components. SPE radiation exposure poses a threat to astronauts in a spacecraft where shielding is available, and especially during an extravehicular activity (EVA), in which shielding is limited. SPE radiation consists predominantly of energetic proton particles with energies greater than 10 MeV. It has been estimated that the largest dose of SPE radiation recorded from a historically large SPE (in August 1972) was capable of delivering a 32.4 Gy dose to the skin, 1.38 Gy blood forming organs (BFO) dose and 0.42 Gy to the stomach (during an EVA), and a 2.7 Gy to the skin, 0.46 Gy BFO dose and 0.17 to the stomach (inside spacecraft) to the astronauts (46).
On the other hand, on Earth, we artificially exploit proton’s characteristics to treat cancer. As a charged particle, protons can penetrate a certain depth in tissues depending on the energy of the proton (>150 MeV). Proton has physical advantages over photon radiation (X-rays, gamma rays) by depositing most of its energy at the Bragg Peak, beyond which there is no energy/dose delivered (47). Therefore, normal tissues distal to the Bragg peak can be protected by averting radiation doses. However, considerable dose or radiation (>1 Gy) are still delivered in the plateau region of the Bragg curve in front of the tumor volume. Overall, comparing to the most advanced photon techniques such as intensity-modulated radiotherapy (IMRT), proton therapy can deliver similar or higher radiation doses to tumor volumes with a reduction in integral radiation dose (50%–60% reduction) (48, 49). With the development of pencil beam scanning technique, intensity-modulated proton therapy (IMPT) can also be performed which yields highly conformal dose distribution around the target volumes (50). Because of these advantages, proton therapy has become the preferred radiotherapy modality for pediatric cancer patients and head and neck cancers as well as other tumor types in adults.
Ionizing radiation can induce DNA damage by its direct interaction with the target macromolecule, resulting in the disruption of the molecular structure. Additionally, it can act indirectly through radiolysis of water molecules, generating ROS particles that cause oxidative damage to the DNA (51, 52). The ability of radiation-induced DNA damage to cause genomic instability and cancer is well established (53). In addition, the use of conventional radiation to induce DNA damage and enhance DNA integration has been explored previously (54–57). However, little is known about the effect of particle radiation-induced DNA damage on HPV integration enhancement, an event that is critical for HPV carcinogenesis. We thereby postulate that the impact of proton-induced DNA damage is amplified in the case of oncogenic DNA viruses such as HPV, because such damage has the additional effect of increasing HPV integration frequency. In particular, we predict that radiation-induced damage to the host DNA puts the exposed population at higher risk of developing HPV-related cancers by increasing the likelihood of HPV integration. To assess this possibility, we exposed oral keratinocytes to protons, as a source of ionizing radiation, and demonstrated that proton-induced DNA damage promotes the integration of foreign DNA into the host genome. Establishing that protons exposure increases the process of HPV integration enables the development of strategies designed specifically to prevent such integration in radiation-exposed populations, with an ultimate goal of reducing or eliminating these malignancies.
Materials and methods
Cell culture
Normal oral keratinocytes (NOK), non-transformed cells immortalized by Human Telomerase
Reverse Transcriptase (hTERT) that were kindly provided by Dr. Karl Münger (58), were grown in keratinocyte serum-free medium (Invitrogen, Carlsbad, CA), supplemented with penicillin (100 U/ml) and streptomycin (100 μg/ml) (Sigma-Aldrich, St. Louis, MO).
Exposure to protons
Cell irradiation with protons was completed at the James M. Slater Proton Treatment and Research Center, Loma Linda California. Exposures were done at room temperature. Two hundred and fifty MeV protons were modulated to generate a 5.0 cm wide spread-out Bragg peak (SOBP). The cells were located at a water equivalent depth of 29.6 cm, specified using CIRS plastic water blocks, which placed the cells in the uniform dose SOBP region of the proton dose profile. Irradiations were conducted with the beam incident on the underside of the flask to ensure accurate placement of the cell layer with respect to the proton depth dose profile. The proton field size employed for the irradiation of the NOK cells was circular with an 18 cm diameter. Protons were delivered from our synchrotron accelerator in a pulsed fashion, with a pulse duration of 0.125 seconds and a duty cycle of 2.2 seconds. This pulsed modality of beam delivery gave a dose rate of approximately 0.8 Gy/min. The integrated number of protons per spill or per exposure is measured by the transmission ionization chamber (TIC) and a secondary electron emission monitor. The beam position and beam profile are monitored using multiwire ion chambers (MWICs). The wire chamber resolution is 2 mm. A 25 × 25 cm2 ion chamber segmented into 400 square pads are placed after the range modulator to monitor the dose delivered to the target volume. The detector consists of a 20 × 20 array of pads (each 1.25 × 1.25 cm2) from a thin sheet of gold-plated Kapton. Any of the pads in the central region of the pad plane can be used to monitor and control the dose delivered to the target and the remaining pads are also used to study the transverse dose distribution (59). Depending on the experimental setting, cells were exposed to single doses of 0, 0.5, 1, 2, 4, 8 Gy.
γ-H2AX foci formation using immunofluorescence
Double-strand DNA breaks and the subsequent repair of DNA lesions were monitored by examining the formation of γ-H2AX foci using antibodies specific for the phosphorylation of Ser-139 at the C-terminal region. Thirty thousand cells were seeded into each well of 4-well chamber slides. After treatment, cells were fixed in methanol for 15 min, washed with PBS, and permeabilized in 0.5% Triton X-100 for 5 min. After blocking with blocking serum (5% goat serum-0.3% Triton X-100) for 30 min, cells were stained with anti-γH2AX mouse monoclonal antibody (1:200 dilution; Abcam, Cambridge, MA) for 1 h, and AlexaFluor-488 goat anti-mouse secondary antibody (1:600 dilution; Invitrogen, Carlsbad, CA) for another hour. Coverslips were mounted with Vectashield Mounting Medium containing DAPI (Vector Laboratories, Burlingame, CA), to counterstain cellular nuclei. The slides were observed with an Olympus BX51 fluorescence microscope and images were taken using Olympus CellSens Standard software.
Measurement of reactive oxygen species using dichlorodihydrofluorescin DiOxyQ
Two and a half million cells were centrifuged at 2000 rpm (450 xg) for 5 minutes and washed with 1 mL PBS. The pellet was resuspended in 500 µl PBS and homogenized for 2 minutes. The sample was then centrifuged at 10,000 rpm at 4°C for 5 minutes, and the supernatant was aliquoted in an eppendorf tube and stored at -80°C until use. The levels of reactive oxygen and nitrogen species (i.e. hydrogen peroxide, peroxyl radical, nitric oxide and peroxynitrite anion), generally referred to as “ROS”, were then measured in these cell homogenates using the OxiSelect In Vitro ROS/RNS assay kit (Cell BioLabs, San Diego, CA). The fluorescence intensity was detected at 480 nm excitation/530 nm emission using a SpectraMax i3X fluorometric plate reader (Molecular Devices). Protein concentrations were also measured with the Coomassie Plus Assay Reagent (Thermo Fisher Scientific, Rockford, IL) and were used for normalization. Therefore, ROS levels are expressed as Relative Fluorescence Unit (RFU) per μg of protein.
Assessment of 8-oxoG DNA damage using avidin-FITC
Oxidative DNA damage was determined via the direct binding of fluorescein isothiocyanate (FITC)-labeled avidin to 8-hydroxy-2’-deoxyguanosine (8-oxoG) residues in the genomic DNA. Briefly, 5 x 106 cells were collected, washed twice with PBS, and fixed with 4% paraformaldehyde for 15 minutes. Cells were then washed three times with PBS, permeabilized with 75% ethanol and stored at -20°C until ready for experimental use. All samples were washed, blocked, and incubated with 2 µg/ml Avidin-FITC (Thermo Fisher Scientific, Rockford, IL) for 1 h in the dark. After two washes, they were resuspended in PBS and analyzed by flow cytometry for fluorescence (excitation 495 nm, emission 515 nm) on a MACSQuant Analyzer 10 (Miltenyi Biotec Inc). A total of 10,000 events were measured per sample and data were analyzed using FlowJo software.
Trypan blue exclusion test
The short-term cytotoxicity of proton exposure was examined using the trypan blue exclusion test. While viable cells remain unstained, those with damaged membrane stain blue. Radiation-exposed cells, collected 48-72 hours after radiation exposure by trypsinization, were re-suspended in 1 ml of media, mixed with 0.4% trypan blue solution at a ratio of 1:1, and counted using a Biorad TC20 automated cell counter. The ratio of unstained cells to the total number of cells (stained and unstained) was used to determine the percentage of cell viability, while the total cell density relative to the control group was used to estimate the rate of cell proliferation.
Colony formation assay
The long-term cytotoxicity of proton exposure was determined using a clonogenic assay. Cells were trypsinized and seeded in 6-well plates at specific numbers (100 cells for 0 Gy; 200 cells for 1 Gy; 300 cells for 2 Gy; 400 cells for 4 Gy; and 800 cells for 8 Gy). After an overnight incubation, the cells were subjected to different doses of radiation. The cells were incubated at 37 °C for an additional 8 to 10 days, fixed with methanol:acetic acid (3:1 ratio) for 5 min, and stained with 1% crystal violet for 1 h. Plates were then rinsed with water and left to dry overnight at room temperature. Colonies with >50 cells were counted using ImageJ. Plating efficiency and surviving fraction for given treatments were calculated based on the survival of non-treated cells (60, 61).
Cell cycle analysis
The effect of radiation exposure on cell cycle progression was examined using propidium iodide (PI) DNA staining. The fluorescence intensity of PI-stained DNA reflects the DNA content of a cell, which determines the proportion of cells in the different cell cycle phases. Forty-eight to seventy-two hours after radiation exposure, 3 x 106 cells were washed with PBS, and fixed in ice cold 70% ethanol for at least 1h at -20°C. They were then allowed to warm at room temperature, washed with PBS, and resuspended in 0.5 mL of FxCycle PI/RNase A staining solution (Thermo Fisher Scientific, Rockford, IL) for 30 min, following which PI-elicited fluorescence was measured using the MACSQuant Analyzer 10 (Miltenyi Biotec Inc). Each sample was collected as 10,000 events, and the corresponding cell cycle distribution was then determined according to the DNA content on FlowJo software.
Apoptosis detection using annexin V/7AAD staining
Apoptosis was assessed using the Annexin V Apoptosis Detection Kit with 7AAD (BioLegend, San Diego, CA). Forty-eight to seventy-two hours after radiation exposure, 2 x 105 cells were added in triplicate into a 96 well plate, rinsed with PBS, re-suspended in 10 µl of 1X annexin V binding buffer, and labeled with 1 µl of Annexin V for 15 min. 1 µl of 7AAD was then added for 5 min for dead cell discrimination. Finally, 180 µl was added to Annexin V binding buffer, following which samples were analyzed using the MACSQuant Analyzer 10 (Miltenyi Biotec Inc). Annexin V-/7AAD− cell population was considered healthy, Annexin V-/7AAD+ was indicative of necrotic cells, whereas the Annexin V+/7AAD− and Annexin V+/7AAD+ were representative of early and late apoptotic cells, respectively.
Puromycin-resistance clonogenic assay in NOK cells
Five hundred thousand cells were seeded in 6-well plates, and 24 hr later exposed to radiation, following which they were transfected with pMetluc puro plasmid encoding the puromycin resistance gene. 48 hours later, the media was collected and the expression of secreted Metridia luciferase was measured using the ready-to-glow secreted luciferase reported assay (Takara Bio USA Inc) for normalization of the transfection efficiency. The integration frequency of this foreign circular DNA was then assessed using a clonogenic assay as previously described (35). Briefly, selection of cells resistant to puromycin at a concentration of 5 μg/ml was performed for 2-3 weeks. After antibiotic selection, the resulting colonies were fixed using 10% formaldehyde for 30-45 min. After rinsing with water and drying overnight at room temperature, cells were stained with 1% crystal violet for 1 hour. Plates were then rinsed with water and left to dry overnight at room temperature. Colonies were counted the following day and normalized for transfection efficiency by MetLuc activity. Integration frequency was compared between untreated cells and cells treated with the indicated doses of radiation.
Statistical analysis
Statistical significances were determined using the SPSS software. Student’s t test was applied and a p-value < 0.05 was regarded as significant. For each parameter tested, a set of three different experiments were performed. Data are represented as the mean ± standard error of the mean.
Results
Proton radiation induces dsDNA breaks promptly and dose-dependently in NOK cells
Radiation-induced DNA damage can be caused by its direct interaction with the DNA, thereby disrupting the molecular structure. To examine the kinetics of direct DNA damage following proton exposure, NOK cells were irradiated with a high dose of protons (4 Gy), fixed at different time-points (0, 0.5, 3, 6, 12 hr), and stained with γ-H2AX antibodies (Figure 1A). Our results show that foci formation, indicative of double-stranded DNA breaks, is at its highest intensity at 0.5 hr, and continuously decreases with time to be almost undetected at 12 hr.
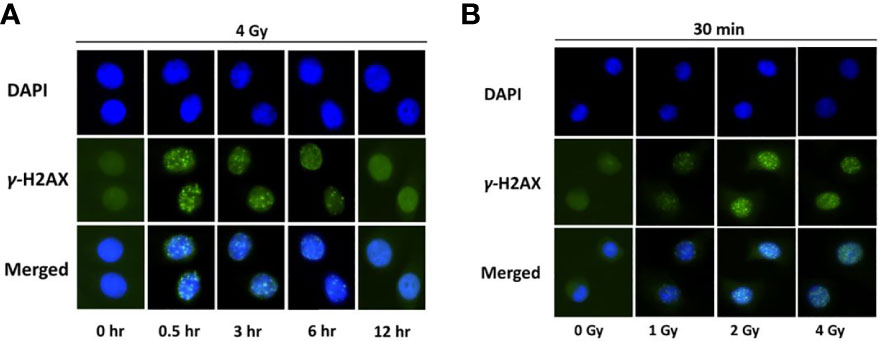
Figure 1 H2AX is phosphorylated in normal oral keratinocytes exposed to proton radiation. (A) Immunofluorescence of γ-H2AX at different time-points (0, 0.5, 3, 6, 12 hr) in normal oral keratinocytes irradiated with 4 Gy of protons. Nuclear DNA was counterstained with DAPI. (B) Immunofluorescence of γ-H2AX in NOK cells, 30 minutes after exposure to different doses of proton radiation (0, 1, 2, 4 Gy). Nuclear DNA was counterstained with DAPI. Images are representative of three independent experiments.
After detecting the time-point with the highest signal of γ-H2AX (0.5 hr), we examined the dose-dependent effect of proton exposure on direct DNA damage. NOK cells were treated with different doses of proton (0, 1, 2, and 4 Gy), and 30 min later were fixed and stained with γ-H2AX (Figure 1B). As expected, foci formation increased dose-dependently following exposure with different doses of ionizing radiation.
ROS-induced DNA damage is delayed but occurs dose-dependently in proton-exposed NOK cells
Radiation exposure can also act indirectly through radiolysis of water molecules, generating ROS particles that cause oxidative damage to the DNA. The kinetics of indirect DNA damage following proton exposure was assessed by measuring the levels of ROS and the resulting oxidative DNA damage 8-oxoG at different time-points (0, 1, 3, 6, 8, 11, 21 hr) in NOK cells irradiated with 4 Gy of protons (Figure 2A). As expected, indirect DNA damage was delayed due to the time needed for ROS particles to accumulate and cause oxidative DNA damage. An increase in ROS levels was detected as early as 1 hr and increased with time up until 11 hours, whereas oxidative DNA damage was observed at only 3, 6, 8 hrs.
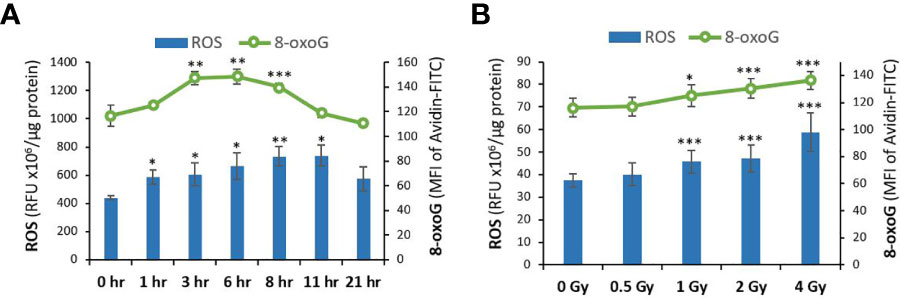
Figure 2 ROS and oxidative DNA damage levels are increased in normal oral keratinocytes exposed to proton radiation. (A) The levels of ROS and the resulting oxidative DNA damage biomarker 8-oxoG were analyzed using DCF and Avidin-FITC staining in NOK cells irradiated with 4 Gy of protons at different time-points (0, 1, 3, 6, 8, 11, 21 hr). (B) ROS and 8-oxoG were measured in NOK cells irradiated with different doses of proton radiation (0, 1, 2, 4 Gy) 6-8 hours after exposure. Asterisks on bars represent significance relative to the control group. (*), (**), and (***) correspond to p<0.05, 0.01, and 0.001, respectively.
Based on these results, 6-8 hr post-exposure was selected as the time-point to examine the dose-dependent effect of protons on ROS-induced DNA damage. Figure 2B represents the measured levels of ROS and 8-oxoG in NOK cells irradiated with different doses of proton (0, 0.5, 1, 2, 4 Gy). Our results show that indirect DNA damage, similar to direct DNA damage, also increases dose-dependently following exposure to 1-4 Gy of proton radiation.
Proton radiation is not cytotoxic at low doses, but is lethal at 8 Gy
Radiation exposure can be detrimental at high doses. Therefore, we examined the potential short- and long-term cytotoxicity of the selected doses of protons on normal oral keratinocytes. We assessed the acute cytotoxic effect of proton exposure on NOK cells at 48-72 hr using the trypan blue exclusion test (Figure 3A). Although the percentage of cell death in NOK cells exposed to 1-8 Gy of protons did not significantly increase in the treated groups (in gray), the cell density was significantly decreased at 4 Gy (81%) and 8 Gy (57%) relative to the control group (in blue). To confirm and further elucidate these results, we performed cell cycle analysis using propidium iodide and Annexin V apoptosis detection assay. Our cell cycle analysis (Figure 3B) showed no remarkable changes in cell cycle distribution of NOK cells exposed to 1 and 2 Gy. However, 4 and 8 Gy treatments resulted in a dose-dependent increase in G2/M phase (32.9% and 47.5%, respectively), providing an explanation for the decrease in cell density at these doses. A minimum dose-dependent increase in subG0 was also obtained in NOK cells exposed to 1-8 Gy, indicating a minor induction of cell death. On the other hand, our Annexin V/7AAD profile did not show any significant increase in the levels of apoptotic and necrotic cells in any of the treated cells, except for a slight increase in the number of Annexin V+/7AAD- pre-apoptotic cells in 4 Gy (9.48%) and 8 Gy (12.9%) (Figure 3C), validating our trypan blue assay and cell cycle analysis results.
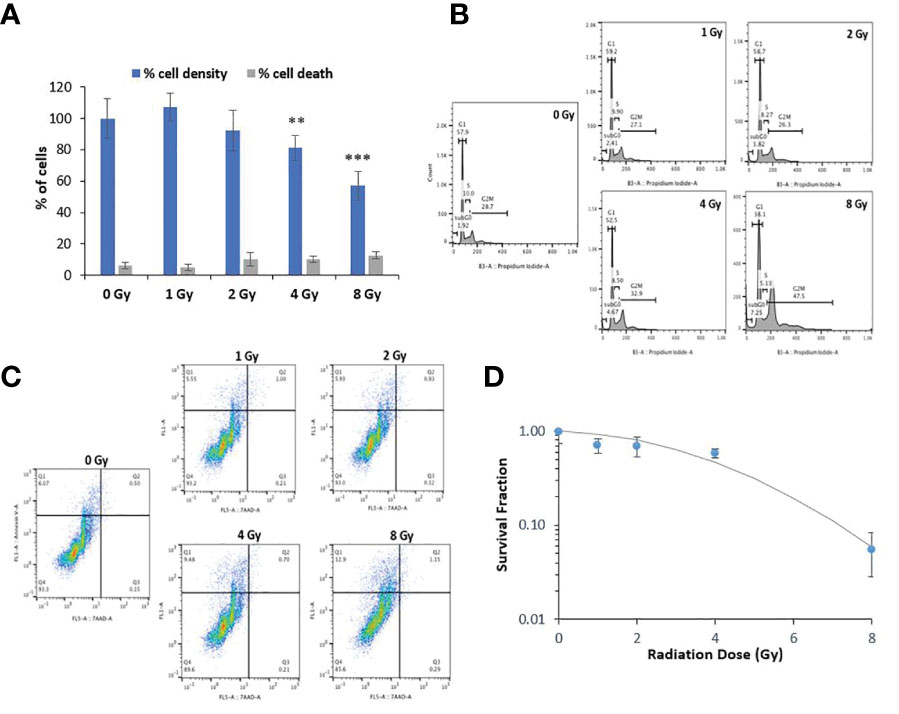
Figure 3 Proton radiation at 1-2 Gy does not cause significant toxicity but causes G2/M cell cycle arrest at higher doses. (A) Acute cytotoxic effect of proton exposure on NOK cells was assessed using the trypan blue exclusion test at 48-72 hr. The ratio of stained cells to the total number of cells was used to determine the percentage of cell death in each treated group (in gray), while the total cell density relative to the control group as 100% (in blue) was used to estimate the rate of cell proliferation. Asterisks on bars represent statistical significance in total cell density relative to control (** for p<0.01; *** for p<0.001). (B) Representative image of one of three experiments showing the cell cycle phases of NOK cells exposed to 0-8 Gy of protons at 48-72 hr. (C) Representative image of one of three experiments showing the Annexin V/7AAD profile in NOK cells exposed to 0-8 Gy of protons at 48-72 hr. Annexin V-/7AAD− cell population is considered healthy, Annexin V-/7AAD+ is indicative of necrotic cells, whereas the Annexin V+/7AAD− and Annexin V+/7AAD+ are representative of early and late apoptotic cells, respectively. (D) Analysis of cell survival fraction by colony formation assay in NOK cells treated with 0-8 Gy of protons 1-2 weeks after exposure.
Delayed toxicity (1-2 weeks) was also examined in irradiated NOK cells using the standard colony formation assay (Figure 3D). Consistent with the other cytotoxicity assays, 1 and 2 Gy did not cause any significant effect on the survival and ability of the cells to undergo “unlimited” division; 4 Gy showed some significant cytotoxicity, whereas 8 Gy was found to be highly lethal.
Proton exposure induces integration of plasmid DNA into NOK cells
To investigate whether exposure to proton radiation increases the rate at which circular plasmid DNA integrates into the human genome, we performed a clonogenic assay. NOK cells were irradiated with the selected doses (0, 1, 2, 4 Gy), following which they were transfected with pMetLuc plasmid encoding for puromycin resistance. To normalize for transfection efficiency, the expression of Metridia luciferase was monitored in the media. After puromycin selection for 2-3 weeks, the resistant colonies were stained with crystal violet and counted (Figure 4A). The number of colonies normalized to luciferase activity is presented in Figure 4B. As anticipated, no antibiotic resistant clones were observed in the untreated wells following selection, while numerous stable clones were noted in wells treated with 1 and 2 Gy, demonstrating that DNA damage induced by protons promotes the integration of plasmid DNA in NOK cells. On the other hand, 4 Gy-treated wells had relatively fewer number of clones likely due to the observed toxicity.
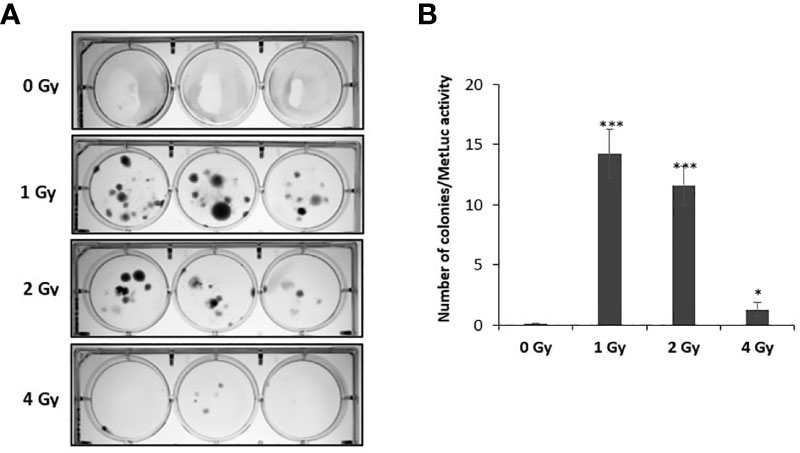
Figure 4 Exposing NOK cells to proton radiation results in increased frequency of pMetLuc plasmid integration. 5 × 105 cells/well of NOK cells were exposed to proton radiation (0, 1, 2, 4 Gy), following which they were transfected with pMetluc puro plasmid. To normalize for transfection efficiency, the media was collected 48 hr post-transfection, and the expression of secreted luciferase was measured. Selection of cells resistant to puromycin (5 μg/ml) was performed daily for 2-3 weeks, following which the colonies produced were stained with crystal violet (A), and normalized for transfection efficiency (B). (*), and (***) correspond to p < 0.05, and 0.001, respectively.
Discussion
The ability of radiation to damage DNA, thereby leading to single or double-stranded breaks, larger-scale damage and cancer, is well established (53). In the study reported here, we hypothesize that the impact of proton-induced DNA damage is magnified in the case of oncogenic DNA viruses such as HPV, because such damage has the additional effect of increasing integration frequency and inducing carcinogenesis. Therefore, our overall goal was to explore the effect of DNA damage induced by ionizing radiation such as protons on the integration of foreign DNA into the human genome.
DNA is one of the critical targets of ionizing radiation. The most dangerous type of DNA lesion caused by ionizing radiation is the complete break of the DNA double helix, and one of the well-characterized markers of double stranded DNA breaks is the phosphorylation of the histone H2AX (γ-H2AX) by the ATM and/or DNA-PK kinases at dsDNA break sites. Therefore, formation of γ-H2AX foci indicates the presence of dsDNA breaks, while foci disappearance is associated with the repair of the damaged regions of the DNA. In this study, we first determined the kinetics of direct DNA damage caused by proton exposure in irradiated normal oral keratinocytes at different time-points. Our results show that histone H2AX is rapidly phosphorylated following radiation exposure, with a peak of H2AX phosphorylation at 30 minutes after irradiation, after which it continuously decreases with time to be almost undetectable by 12 hr (Figure 1A). These results are consistent with a study by Mariotti et al. (62), in which cells exposed to 1 and 2 Gy of X-ray reached the maximum number of γ-H2AX foci formation at 30 min after irradiation, after which the resulting foci kinetics followed a clear negative exponential response with very little residual damage detectable by 24 hr post exposure. After determining the kinetics of γ-H2AX foci formation in normal oral keratinocytes following proton exposure, we assessed the dose-response of proton exposure on direct DNA damage 30 min after exposure. As expected, foci formation was detected to be dose-dependently increased following exposure to 0-4 Gy of protons (Figure 1B), similar to previous reports in other cell lines exposed to X-ray and alpha-particles (63, 64).
When cells, which are composed of more than 70% of water, are exposed to ionizing radiation, radiolysis of water molecules occurs, resulting in the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Persistent and excessive generation of ROS causes oxidative insults to cellular components including nucleic acids, proteins and lipids (65, 66). These oxidizing events induced by ionizing radiation can persist for days and months due to the continuous generation of ROS species. Furthermore, radiation-induced oxidative stress may spread from targeted cells to non-targeted bystander cells through intercellular communication mechanisms (51). Therefore, the molecular and biochemical events that promote oxidative stress in irradiated cells play crucial roles in mediating the harmful effects of ionizing radiation. From the many types of oxidative DNA damage products, 8-hydroxy-2’-deoxyguanosine, derived from hydroxyl radical attack of deoxyguanosine residues, is the most used and representative biomarker of oxidative DNA damage (67, 68). We therefore analyzed the levels of ROS and the resulting 8-oxoG hydroxylation product in irradiated cells to assess the effect of proton radiation on indirect ROS-induced DNA damage. As anticipated, indirect DNA damage was delayed for hours in NOK cells exposed to protons, due to the time required for ROS particles to build up and cause oxidative DNA damage (Figure 2A). An increase in ROS levels was detected as early as 1 hr post-exposure and was continuously increased to reach a plateau at 11 hr. It then decreased to be non-significant at 21 hr, likely due to the activation of antioxidant defensive mechanisms of the cells. These results are consistent with a study by Yamamori et al. (69) showing that X-ray irradiation induces a time-dependent increase in ROS levels, peaking at 12 hr, and declining by 24 hr. On the other hand, our results demonstrate that oxidative DNA damage is induced at 3 hr, reaches a peak at 6-8 hrs, and becomes completely repaired at 11 hr and 21 hr. Importantly, our oxidative DNA damage results are concomitant with those of ROS levels in irradiated NOK cells, in that ROS accumulation occurs first, which later leads to oxidative damage to the DNA. Finally, similar to our observations with direct DNA damage, we also demonstrated that indirect ROS-induced DNA damage increases dose-dependently following exposure of NOK cells to 1-4 Gy of protons for 6-8 hr (Figure 2B).
In response to DNA damage induced by ionizing radiation, numerous cellular signaling pathways are anticipated to be activated in irradiated cells, which have the potential to result in cell cycle checkpoint activation/DNA repair, apoptosis, and cellular senescence (70). To ensure that the proton doses used in our experiments do not cause major cytotoxic effects which could interfere with our clonogenic integration study, we carried out cytotoxicity assays. Acute cytotoxicity assessed by the trypan blue exclusion test showed a dose-dependent decrease in total cell density in NOK cells exposed to 4 and 8 Gy of protons compared to the untreated group, though no significant increase in the percentage of dead cells was noted at these doses (Figure 3A). The decrease in the cell density at 4 and 8 Gy is explained by the observed G2/M cell cycle arrest at these doses (Figure 3B). On the other hand, the Annexin-V/7AAD profile did not display an increase in the number of apoptotic or necrotic cells at any of the doses, except for a slight increase in the levels of pre-apoptotic cells at 4-8 Gy (Figure 3C), validating the limited number of dead cells noted in our trypan blue results. Together, the remarkable halt in cell cycle progression and the relative absence of apoptotic/necrotic cells point toward an effect of proton exposure on cell proliferation rather than on cell viability. In response to DNA damage, cell cycle progression often becomes blocked by the activation of cell cycle checkpoints, allowing the cells to repair the damage prior to replication. In cases where the damage is irreversible or the cell cycle checkpoint is dysfunctional, apoptosis may be triggered to eliminate the damaged cells (71). While the cell cycle arrest detected at 4 Gy seems to be temporary, the cells exposed to 8 Gy appears to be more permanent, as the results of colony formation assay show 4 Gy to be slightly cytotoxic, while 8 Gy to be highly lethal (Figure 3D). Interestingly, previous studies have reported that the mitochondrial content, mitochondrial electron transport chain function, and mitochondrial ROS production peak in the G2/M phase of the cell cycle (72, 73), and that accumulation of irradiated cells in G2/M phase under the control of G2/M checkpoint result in a corresponding increase in mitochondrial ROS level (69). Hence, it has been suggested that ROS can also be released from biological sources such as mitochondria in irradiated cells, in addition to its byproduct generation from radiolysis of water molecules. Our results showing proton exposure inducing excessive ROS generation may therefore be at least partly due to the ionizing radiation-induced cell cycle arrest at the G2/M phase.
Following our establishment of an experimental system in which proton exposure induces both types of DNA damage without causing major cytotoxicity, we explored the impact of high doses of proton radiation on the integration of foreign DNA, the pMetLuc plasmid, into the host genome using a clonogenic assay. This integration model is well established in our lab and has been previously employed to examine the effect of chronic oxidative stress on HPV integration frequency (35. The genetic material used in this system works as a good model to study HPV integration, since it shares many critical features with the HPV genome. They are both small in size (<8kb), circular, double stranded DNA, and most importantly, they rarely integrate into the host genome spontaneously. Consistent with our working model, we demonstrated that exposure to proton radiation induces the integration of HPV-like foreign DNA into the human genome (Figures 4A, B). These findings are in line with previous studies, in which various doses of γ- and X-ray ionizing radiation have been shown to stimulate integration of different DNA vectors into the host cell genome (54, 57, 74, 75). However, to the best of our knowledge, we are the first to explore the effect of high doses of protons on DNA integration, and particularly in the context of HPV. Therefore, our results add to the broad data on the deleterious effects of ionizing radiation and allow us to predict that proton-induced damage to the host DNA puts the exposed population at higher risk of developing HPV-related cancers by stimulating HPV integration. The concept of ionizing radiation inducing DNA integration has also been explored as a tool to enhance DNA-mediated gene transfer in mammalian cells (55, 56, 76, 77). Administering radiation prior to adenovirus-mediated gene therapy holds promise to greatly improve the adenoviral transduction efficiency and overcome the low rate of stable gene transfer. However, this combined modality of radiation-guided gene therapy should be considered with extra caution since it could result in unintentional integrations of oncogenic viruses such as HPV, putting infected individuals at higher risks for oncovirus-mediated carcinogenesis.
It is well established that exposure to elevated doses of ionizing radiation contribute to carcinogenesis. The carcinogenic potential of ionizing radiation was recognized shortly after Roentgen’s discovery of X-rays, when the first radiation-induced skin cancer was reported in 1902. Ever since, several experimental studies have identified the general characteristics of radiation carcinogenesis, and various epidemiological studies in human populations exposed to occupational, medical, and accidental sources of radiation have supported the emerging findings (78). Radiation can induce a broad spectrum of DNA lesions including damage to nucleotide bases, cross-linking and DNA single- and double-strand breaks, with the latter being most important type of biological lesion (79, 80). Most of these dsDNA breaks are repaired by the error prone non-homologous end joining (NHEJ) or microhomology-mediated end joining (MHEJ) mechanisms, and thereby facilitate the generation of gene mutations, chromosomal abnormalities and other large-scale changes that are frequently detected in irradiated cells (81–85). While radiation is considered to be a relatively weaker carcinogen and mutagen compared to certain chemicals such as the polycyclic hydrocarbons, various secondary factors can modulate its hazardous effects and contribute to cancer development (78). In this study, we report for the first time that proton-induced damage to DNA stimulates integration of foreign DNA into the human genome, amplifying the carcinogenic potential of oncogenic viruses such as HPV.
Integration of the viral genome into the host genome is a pivotal step in the process of malignant transformation by several oncogenic viruses, including Hepatitis B virus (HBV), Merkel cell polyomavirus (MCV) and HPV [reviewed in (30)]. These viruses do not encode genes that produce integrase enzymatic activity protein; therefore, the integrative process of these oncogenic viruses is likely mediated by cellular genes involved in DNA replication and repair mechanisms (86–88). Since viral integration requires breakage of both the viral and the host DNA, the likelihood of integration depends on the levels of DNA damage. In support of this idea, several pieces of evidence have linked oxidative DNA damage and dsDNA breaks to a higher integration frequency of HBV (89–91). Furthermore, following dsDNA breaks, the recruitment of DNA damage repair complexes ensures the accessibility of ligases that can reconnect the recombined host and viral sequences, creating the perfect microenvironment for viral integration. In the case of HPV, the homologous recombination process is unlikely to contribute to HPV integration, as there is insufficient homology between HPV sequences and the human genome. On the other hand, microhomology-mediated DNA repair pathways have been found to be involved in the ligation of linearized HPV16 and host DNA, as evidenced by enriched micro-homologous sequences between HPV and human genomes at the integration breakpoints in both cervical and oropharyngeal cancers (25, 92–94). This integration model of HPV is consistent with our working model that radiation-induced dsDNA breaks activate DNA repair pathways mediated by end joining which would in its turn facilitate the integration of HPV into the host genome.
Conclusions
We determined the kinetics of DNA damage in normal oral keratinocytes exposed to proton radiation by assessing γ-H2AX foci formation using immunofluorescence (direct damage) and measuring ROS and 8-oxoG levels via DCFDA and Avidin-FITC (indirect damage). As anticipated, direct DNA damage was observed promptly, within 30 min, whereas indirect DNA damage was delayed due to the time required for ROS to accumulate and cause oxidative damage. Although protons were toxic at high doses (4-8 Gy), we were able to establish an experimental system where exposure to proton radiation induced DNA damage dose-dependently without causing major cytotoxic effects as assessed by several cytotoxicity assays. Most importantly, we explored the impact of proton exposure on integration frequency using a clonogenic assay, and showed that as predicted, DNA damage induced by proton exposure promotes the integration of foreign DNA in oral keratinocytes, increasing the risks of HPV malignancies. Our results demonstrating the amplified impact of proton-induced DNA damage in the case of oncogenic DNA viruses such as HPV accentuate the deleterious effects of ionizing radiation and shed light on the extra precautions that need to be taken particularly in HPV-infected populations. Overall, the insights gained from this work enable us to better understand the contribution of proton exposure and DNA damage to HPV integration and risk of subsequent carcinogenesis and direct us toward strategies aimed at preventing malignancies in HPV-infected individuals.
Data availability statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
Author contributions
MK performed the experimental work and data analysis and drafted this manuscript. AB and MV carried out the ionizing radiation exposures of the cells and contributed to the overall study design. VF participated in developing the overall concepts. XC helped with insightful discussion regarding DNA integration. PD-H was responsible for the overall study design and manuscript finalization. All authors contributed to the article and approved the submitted version.
Funding
Funding for this study was provided by Loma Linda University (LLU). Other than the clear contributions by the listed LLU-affiliated authors, the funding body had no role in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
Acknowledgments
We thank Dr. Charles Wang and Dr. Isaac Kremsky for helpful and critical discussions of this work. We also thank to Dr. Jerry D. Slater for providing access to the proton facilities and supporting laboratories.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee SL, Tameru AM. A mathematical model of human papillomavirus (HPV) in the united states and its impact on cervical cancer. J Cancer (2012) 3:262–8. doi: 10.7150/jca.4161
2. Franco EL, Villa LL, Sobrinho JP, Prado JM, Rousseau MC, Desy M, et al. Epidemiology of acquisition and clearance of cervical human papillomavirus infection in women from a high-risk area for cervical cancer. J Infect Dis (1999) 180(5):1415–23. doi: 10.1086/315086
3. Molano M, van den Brule A, Plummer M, Weiderpass E, Posso H, Arslan A, et al. Determinants of clearance of human papillomavirus infections in Colombian women with normal cytology: A population-based, 5-year follow-up study. Am J Epidemiol (2003) 158(5):486–94. doi: 10.1093/aje/kwg171
4. Braaten KP, Laufer MR. Human papillomavirus (HPV), HPV-related disease, and the HPV vaccine. Rev Obstetrics Gynecol (2008) 1(1):2–10.
5. Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine (2006) 24:S11–25. doi: 10.1016/j.vaccine.2006.05.111
6. Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer (2006) 118(12):3030–44. doi: 10.1002/ijc.21731
7. Colón-López V, Ortiz AP, Palefsky J. Burden of human papillomavirus infection and related comorbidities in men: implications for research, disease prevention and health promotion among Hispanic men. Puerto Rico Health Sci J (2010) 29(3):232–40.
8. Yim E-K, Park J-S. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res Treat (2005) 37(6):319–24. doi: 10.4143/crt.2005.37.6.319
9. Yeo-Teh NSL, Ito Y, Jha S. High-risk human papillomaviral oncogenes E6 and E7 target key cellular pathways to achieve oncogenesis. Int J Mol Sci (2018) 19(6):1706. doi: 10.3390/ijms19061706
10. Xue Y, Lim D, Zhi L, He P, Abastado J-P, Thierry F. Loss of HPV16 E2 protein expression without disruption of the E2 ORF correlates with carcinogenic progression. Open Virol J (2012) 6:163–72. doi: 10.2174/1874357901206010163
11. Tomaić V. Functional roles of E6 and E7 oncoproteins in HPV-induced malignancies at diverse anatomical sites. Cancers (2016) 8(10):95. doi: 10.3390/cancers8100095
12. Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol (1987) 61(4):962–71. doi: 10.1128/jvi.61.4.962-971.1987
13. Peitsaro P, Johansson B, Syrjänen S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J Clin Microbiol (2002) 40(3):886–91. doi: 10.1128/JCM.40.3.886-891.2002
14. Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer (2002) 2:342. doi: 10.1038/nrc798
15. Oyervides-Muñoz MA, Pérez-Maya AA, Rodríguez-Gutiérrez HF, Gómez-Macias GS, Fajardo-Ramírez OR, Treviño V, et al. Understanding the HPV integration and its progression to cervical cancer. Infection Genet Evol (2018) 61:34–144. doi: 10.1016/j.meegid.2018.03.003
16. Rosty C, Sheffer M, Tsafrir D, Stransky N, Tsafrir I, Peter M, et al. Identification of a proliferation gene cluster associated with HPV E6/E7 expression level and viral DNA load in invasive cervical carcinoma. Oncogene (2005) 24(47):7094–104. doi: 10.1038/sj.onc.1208854
17. Szalmás A, Kónya J. Epigenetic alterations in cervical carcinogenesis. Semin Cancer Biol (2009) 19(3):144–52. doi: 10.1016/j.semcancer.2009.02.011
18. Gray E, Pett MR, Ward D, Winder DM, Stanley MA, Roberts I, et al. In vitro progression of human papillomavirus 16 episome-associated cervical neoplasia displays fundamental similarities to integrant-associated carcinogenesis. Cancer Res (2010) 70(10):4081–91. doi: 10.1158/0008-5472.CAN-09-3335
19. Durzynska J, Lesniewicz K, Poreba E. Human papillomaviruses in epigenetic regulations. Mutat Research/Reviews Mutat Res (2017) 772:36–50. doi: 10.1016/j.mrrev.2016.09.006
20. Shukla S, Mahata S, Shishodia G, Pande S, Verma G, Hedau S, et al. Physical state & copy number of high risk human papillomavirus type 16 DNA in progression of cervical cancer. Indian J Med Res (2014) 139(4):531–43.
21. Cullen AP, Reid R, Campion M, Lörincz AT. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J Virol (1991) 65(2):606–12. doi: 10.1128/jvi.65.2.606-612.1991
22. Burk RD, Chen Z, Saller C, Tarvin K, Carvalho AL, Scapulatempo-Neto C, et al. Integrated genomic and molecular characterization of cervical cancer. Nature (2017) 543(7645):378–84. doi: 10.1038/nature21386
23. Valmary-Degano S, Jacquin E, Prétet J-L, Monnien F, Girardo B, Arbez-Gindre F, et al. Signature patterns of human papillomavirus type 16 in invasive anal carcinoma. Hum Pathol (2013) 44(6):992–1002. doi: 10.1016/j.humpath.2012.08.019
24. Olthof NC, Speel E-JM, Kolligs J, Haesevoets A, Henfling M, Ramaekers FCS, et al. Comprehensive analysis of HPV16 integration in OSCC reveals no significant impact of physical status on viral oncogene and virally disrupted human gene expression. PloS One (2014) 9(2):e88718. doi: 10.1371/journal.pone.0088718
25. Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci (2014) 111(43):15544–9. doi: 10.1073/pnas.1416074111
26. Vojtechova Z, Sabol I, Salakova M, Turek L, Grega M, Smahelova J, et al. Analysis of the integration of human papillomaviruses in head and neck tumours in relation to patients' prognosis. Int J Cancer (2016) 138(2):386–95. doi: 10.1002/ijc.29712
27. Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene (2003) 22:1225. doi: 10.1038/sj.onc.1206170
28. Yu T, Ferber MJ, Cheung TH, Chung TKH, Wong YF, Smith DI. The role of viral integration in the development of cervical cancer. Cancer Genet Cytogenetics (2005) 158(1):27–34. doi: 10.1016/j.cancergencyto.2004.08.021
29. Pett M, Coleman N. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? J Pathol (2007) 212(4):356–67. doi: 10.1002/path.2192
30. Chen Y, Williams V, Filippova M, Filippov V, Duerksen-Hughes P. Viral carcinogenesis: factors inducing DNA damage and virus integration. Cancers (Basel) (2014) 6(4):2155–86. doi: 10.3390/cancers6042155
31. Barzilai A, Yamamoto K-I. DNA Damage responses to oxidative stress. DNA Repair (2004) 3(8):1109–15. doi: 10.1016/j.dnarep.2004.03.002
32. Alfadda AA, Sallam RM. Reactive oxygen species in health and disease. J biomed. Biotechnol (2012) 2012:936486–6. doi: 10.1155/2012/936486
33. Maluf SW, Marroni NP, Heuser VD, Prá D. DNA Damage and oxidative stress in human disease. BioMed Res Int (2013) 2013:696104–4. doi: 10.1155/2013/696104
34. Katerji M, Filippova M, Wongworawat YC, Siddighi S, Bashkirova S, Duerksen-Hughes PJ. Oxidative stress markers in patient-derived non-cancerous cervical tissues and cells. Sci Rep (2020) 10(1):19044. doi: 10.1038/s41598-020-76159-2
35. Chen Wongworawat Y, Filippova M, Williams VM, Filippov V, Duerksen-Hughes PJ. Chronic oxidative stress increases the integration frequency of foreign DNA and human papillomavirus 16 in human keratinocytes. Am J Cancer Res (2016) 6(4):764–80.
36. Hawes SE, Kiviat NB. Are genital infections and inflammation cofactors in the pathogenesis of invasive cervical cancer? J Natl Cancer Inst (2002) 94(21):1592–3. doi: 10.1093/jnci/94.21.1592
37. Finan RR, Musharrafieh U, Almawi WY. Detection of chlamydia trachomatis and herpes simplex virus type 1 or 2 in cervical samples in human papilloma virus (HPV)-positive and HPV-negative women. Clin Microbiol Infection (2006) 12(9):927–30. doi: 10.1111/j.1469-0691.2006.01479.x
38. Almonte M, Albero G, Molano M, Carcamo C, García PJ, Pérez G. Risk factors for human papillomavirus exposure and Co-factors for cervical cancer in Latin America and the Caribbean. Vaccine (2008) 26:L16–36. doi: 10.1016/j.vaccine.2008.06.008
39. Nagata C, Shimizu H, Yoshikawa H, Noda K, Nozawa S, Yajima A, et al. Serum carotenoids and vitamins and risk of cervical dysplasia from a case-control study in Japan. Br J Cancer (1999) 81(7):1234–7. doi: 10.1038/sj.bjc.6690834
40. Manju V, Balasubramanian V, Nalini N. Oxidative stress and tumor markers in cervical cancer patients. J Biochem Mol Biol Biophys (2002) 6(6):387–90. doi: 10.1080/1025814021000036115
41. Naidu MSK, Suryakar AN, Swami SC, Katkam RV, Kumbar KM. Oxidative stress and antioxidant status in cervical cancer patients. Indian J Clin Biochem IJCB (2007) 22(2):140–4. doi: 10.1007/BF02913333
42. De Marco F. Oxidative stress and HPV carcinogenesis. Viruses (2013) 5(2):708–31. doi: 10.3390/v5020708
43. Schauer DA, Linton OW. National council on radiation protection and measurements report shows substantial medical exposure increase. Radiology (2009) 253(2):293–6. doi: 10.1148/radiol.2532090494
44. Badhwar GD, Atwell W, Badavi FF, Yang TC, Cleghorn TF. Space radiation absorbed dose distribution in a human phantom. Radiat Res (2002) 157(1):76–91. doi: 10.1667/0033-7587(2002)157[0076:SRADDI]2.0.CO;2
45. Cucinotta FA, Kim MH, Willingham V, George KA. Physical and biological organ dosimetry analysis for international space station astronauts. Radiat Res (2008) 170(1):127–38. doi: 10.1667/RR1330.1
46. Hu S, Kim MH, McClellan GE, Cucinotta FA. Modeling the acute health effects of astronauts from exposure to large solar particle events. Health Phys (2009) 96(4):465–76. doi: 10.1097/01.HP.0000339020.92837.61
47. Newhauser WD, Zhang R. The physics of proton therapy. Phys Med Biol (2015) 60(8):R155–209. doi: 10.1088/0031-9155/60/8/R155
48. Kandula S, Zhu X, Garden AS, Gillin M, Rosenthal DI, Ang KK, et al. Spot-scanning beam proton therapy vs intensity-modulated radiation therapy for ipsilateral head and neck malignancies: a treatment planning comparison. Med Dosim (2013) 38(4):390–4. doi: 10.1016/j.meddos.2013.05.001
49. Stuschke M, Kaiser A, Abu-Jawad J, Pöttgen C, Levegrün S, Farr J. Re-irradiation of recurrent head and neck carcinomas: comparison of robust intensity modulated proton therapy treatment plans with helical tomotherapy. Radiat Oncol (2013) 8(1):93. doi: 10.1186/1748-717X-8-93
50. Kooy HM, Grassberger C. Intensity modulated proton therapy. Br J Radiol (2015) 88(1051):20150195. doi: 10.1259/bjr.20150195
51. Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett (2012) 327(1-2):48–60. doi: 10.1016/j.canlet.2011.12.012
52. Desouky O, Ding N, Zhou G. Targeted and non-targeted effects of ionizing radiation. J Radiat Res Appl Sci (2015) 8(2):247–54. doi: 10.1016/j.jrras.2015.03.003
53. Chandel V, Seth G, Shukla P, Kumar D. Role of radiation in DNA damage and radiation induced cancer. In: Kesari KK, editor. Networking of mutagens in environmental toxicology. Cham: Springer International Publishing (2019). p. 1–23.
54. Stoker M. Effect of X-irradiation on susceptibility of cells to transformation by polyoma virus. Nature (1963) 200(4908):756–8. doi: 10.1038/200756a0
55. Debenham PG, Webb MB. The effect of X-rays and ultraviolet light on DNA-mediated gene transfer in mammalian cells. Int J Radiat Biol Relat Stud Phys Chem Med (1984) 46(5):555–68. doi: 10.1080/09553008414551761
56. Zeng M, Cerniglia GJ, Eck SL, Stevens CW. High-efficiency stable gene transfer of adenovirus into mammalian cells using ionizing radiation. Hum Gene Ther (1997) 8(9):1025–32. doi: 10.1089/hum.1997.8.9-1025
57. Zelensky AN, Schoonakker M, Brandsma I, Tijsterman M, van Gent DC, Essers J, et al. Low dose ionizing radiation strongly stimulates insertional mutagenesis in a γH2AX dependent manner. PloS Genet (2020) 16(1):e1008550. doi: 10.1371/journal.pgen.1008550
58. Piboonniyom S-O, Duensing S, Swilling NW, Hasskarl J, Hinds PW, Münger K. Abrogation of the retinoblastoma tumor suppressor checkpoint during keratinocyte immortalization is not sufficient for induction of centrosome-mediated genomic instability. Cancer Res (2003) 63(2):476–83.
59. Coutrakon G, Bauman M, Lesyna D, Miller D, Nusbaum J, Slater J, et al. A prototype beam delivery system for the proton medical accelerator at loma Linda. Med Phys (1991) 18(6):1093–9. doi: 10.1118/1.596617
60. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc (2006) 1(5):2315–9. doi: 10.1038/nprot.2006.339
61. Braselmann H, Michna A, Heß J, Unger K. CFAssay: statistical analysis of the colony formation assay. Radiat Oncol (London England) (2015) 10:223–3. doi: 10.1186/s13014-015-0529-y
62. Mariotti LG, Pirovano G, Savage KI, Ghita M, Ottolenghi A, Prise KM, et al. Use of the γ-H2AX assay to investigate DNA repair dynamics following multiple radiation exposures. PloS One (2013) 8(11):e79541. doi: 10.1371/journal.pone.0079541
63. Macphail SH, BanÁth JP, Yu TY, Chu EHM, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol (2003) 79(5):351–9. doi: 10.1080/0955300032000093128
64. Staaf E, Brehwens K, Haghdoost S, Czub J, Wojcik A. Gamma-H2AX foci in cells exposed to a mixed beam of X-rays and alpha particles. Genome integrity (2012) 3:8. doi: 10.1186/2041-9414-3-8
65. Le Caër S. Water radiolysis: influence of oxide surfaces on H2 production under ionizing radiation. Water (2011) 3(1):235–53. doi: 10.3390/w3010235
66. Wei J, Wang B, Wang H, Meng L, Zhao Q, Li X, et al. Radiation-induced normal tissue damage: Oxidative stress and epigenetic mechanisms. Oxid Med Cell Longev (2019) 2019:3010342. doi: 10.1155/2019/3010342
67. Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-hydroxyguanine, an abundant form of oxidative DNA damage, causes G–-T and a–-C substitutions. J Biol Chem (1992) 267(1):166–72. doi: 10.1016/S0021-9258(18)48474-8
68. Guo C, Li X, Wang R, Yu J, Ye M, Mao L, et al. Association between oxidative DNA damage and risk of colorectal cancer: Sensitive determination of urinary 8-Hydroxy-2′-deoxyguanosine by UPLC-MS/MS analysis. Sci Rep (2016) 6(1):32581. doi: 10.1038/srep32581
69. Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, et al. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radical Biol Med (2012) 53(2):260–70. doi: 10.1016/j.freeradbiomed.2012.04.033
70. Hein AL, Ouellette MM, Yan Y. Radiation-induced signaling pathways that promote cancer cell survival (review). Int J Oncol (2014) 45(5):1813–9. doi: 10.3892/ijo.2014.2614
71. Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem (2004) 73(1):39–85. doi: 10.1146/annurev.biochem.73.011303.073723
72. Martínez-Diez M, Santamaría G, Ortega Á.D, Cuezva JM. Biogenesis and dynamics of mitochondria during the cell cycle: Significance of 3′UTRs. PloS One (2006) 1(1):e107. doi: 10.1371/journal.pone.0000107
73. Jahnke VE, Sabido O, Freyssenet D. Control of mitochondrial biogenesis, ROS level, and cytosolic Ca2+ concentration during the cell cycle and the onset of differentiation in L6E9 myoblasts. Am J Physiol Cell Physiol (2009) 296(5):C1185–1194. doi: 10.1152/ajpcell.00377.2008
74. Coggin JH Jr. Enhanced virus transformation of hamster embryo cells in vitro. J Virol (1969) 3(5):458–62. doi: 10.1128/jvi.3.5.458-462.1969
75. Su ZZ, Zhang P, Geard CR, Fisher PB. Enhancement of adenovirus transformation of cloned rat embryo fibroblast cells by gamma irradiation. Mol Carcinogenesis (1990) 3(3):141–9. doi: 10.1002/mc.2940030307
76. Stevens CW, Zeng M, Cerniglia GJ. Ionizing radiation greatly improves gene transfer efficiency in mammalian cells. Hum Gene Ther (1996) 7(14):1727–34. doi: 10.1089/hum.1996.7.14-1727
77. Hillman GG, Xu M, Wang Y, Wright JL, Lu X, Kallinteris NL, et al. Radiation improves intratumoral gene therapy for induction of cancer vaccine in murine prostate carcinoma. Hum Gene Ther (2003) 14(8):763–75. doi: 10.1089/104303403765255156
78. Little JB. Radiation carcinogenesis. Carcinogenesis (2000) 21(3):397–404. doi: 10.1093/carcin/21.3.397
79. Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol (1994) 65(1):7–17. doi: 10.1080/09553009414550021
80. Ward JF. Radiation mutagenesis: The initial DNA lesions responsible. Radiat Res (1995) 142(3):362–8. doi: 10.2307/3579145
81. Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J (2009) 417(3):639–50. doi: 10.1042/BJ20080413
82. Scuric Z, Chan CY, Hafer K, Schiestl RH. Ionizing radiation induces microhomology-mediated end joining in trans in yeast and mammalian cells. Radiat Res (2009) 171(4):454–63. doi: 10.1667/RR1329.1
83. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem (2010) 79:181–211. doi: 10.1146/annurev.biochem.052308.093131
84. Rodgers K, McVey M. Error-prone repair of DNA double-strand breaks. J Cell Physiol (2016) 231(1):15–24. doi: 10.1002/jcp.25053
85. Dutta A, Eckelmann B, Adhikari S, Ahmed KM, Sengupta S, Pandey A, et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res (2017) 45(5):2585–99. doi: 10.1093/nar/gkw1262
86. Archambault J, Melendy T. Targeting human papillomavirus genome replication for antiviral drug discovery. Antiviral Ther (2013) 18(3):271–83. doi: 10.3851/IMP2612
87. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology (2013) 435(1):118–30. doi: 10.1016/j.virol.2012.09.029
88. Hai H, Tamori A, Kawada N. Role of hepatitis b virus DNA integration in human hepatocarcinogenesis. World J Gastroenterol (2014) 20(20):6236–43. doi: 10.3748/wjg.v20.i20.6236
89. Petersen J, Dandri M, Burkle A, Zhang L, Rogler CE. Increase in the frequency of hepadnavirus DNA integrations by oxidative DNA damage and inhibition of DNA repair. J Virol (1997) 71(7):5455–63. doi: 10.1128/jvi.71.7.5455-5463.1997
90. Cougot D, Neuveut C, Buendia MA. HBV induced carcinogenesis. J Clin Virol (2005) 34 Suppl 1:S75–78. doi: 10.1016/S1386-6532(05)80014-9
91. Hu X, Lin J, Xie Q, Ren J, Chang Y, Wu W, et al. DNA Double-strand breaks, potential targets for HBV integration. J Huazhong Univ. Sci Technolog. Med Sci (2010) 30(3):265–70. doi: 10.1007/s11596-010-0341-8
92. Winder DM, Pett MR, Foster N, Shivji MKK, Herdman MT, Stanley MA, et al. An increase in DNA double-strand breaks, induced by Ku70 depletion, is associated with human papillomavirus 16 episome loss and de novo viral integration events. J Pathol (2007) 213(1):27–34. doi: 10.1002/path.2206
93. Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet (2015) 47(2):158–63. doi: 10.1038/ng.3178
Keywords: ionizing radiation, DNA damage, DNA integration, carcinogenesis, human papillomaviruses, reactive oxygen species
Citation: Katerji M, Bertucci A, Filippov V, Vazquez M, Chen X and Duerksen-Hughes PJ (2022) Proton-induced DNA damage promotes integration of foreign plasmid DNA into human genome. Front. Oncol. 12:928545. doi: 10.3389/fonc.2022.928545
Received: 25 April 2022; Accepted: 16 August 2022;
Published: 02 September 2022.
Edited by:
Laure Marignol, Trinity College Dublin, IrelandReviewed by:
Harikrishna Reddy Rallabandi, Oklahoma Medical Research Foundation, United StatesMartin Vlk, Czech Technical University in Prague, Czechia
Copyright © 2022 Katerji, Bertucci, Filippov, Vazquez, Chen and Duerksen-Hughes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Penelope J. Duerksen-Hughes, cGRodWdoZXNAbGx1LmVkdQ==