 Yingchao Sun
Yingchao Sun Lei Yue1
Lei Yue1 Weiling Hu
Weiling Hu- 1Department of Gastroenterology, Sir Run Run Shaw Hospital, Medical School, Zhejiang University, Hangzhou, China
- 2Department of Gastrointestinal Surgery, Taizhou Hospital, Zhejiang University, Taizhou, China
- 3Institute of Gastroenterology, Zhejiang University (IGZJU), Hangzhou, China
- 4Zhejiang University Cancer Center, Hangzhou, China
Platelet-derived growth factor receptor A (PDGFRA) mutations occur in approximately 10–15% of gastrointestinal stromal tumors (GISTs). These tumors with PDGFRA mutations have a different pathogenesis, clinical characteristics, and treatment response compared to tumors with receptor tyrosine kinase protein (KIT) mutations (60–70%). Many clinical studies have investigated the use of tyrosine kinase inhibitors mainly in patients with KIT mutations; however, there is a lack of attention to the PDGFRA-mutated molecular subtype. The main effective inhibitors of PDGFRA are ripretinib, avapritinib, and crenolanib, and their mechanisms and efficacy in GIST (as confirmed in clinical trials) are described in this review. Some multi-targeted tyrosine kinase inhibitors with inhibitory effects on this molecular subtype are also introduced and summarized in this paper. This review focuses on PDGFRA-mutated GISTs, introduces their clinical characteristics, downstream molecular signaling pathways, and existing resistance mechanisms. We focus on the most recent literature that describes the development of PDGFRA inhibitors and their use in clinical trials, as well as the potential benefits from different combination therapy strategies.
Introduction
Gastrointestinal stromal tumors (GIST) are sarcomas that mostly derive from precursors of the interstitial cells of Cajal (ICC). Although GISTs are the most common sarcoma of the GI tract, they are rare, with an incidence of only 10–15 patients per million per year (1, 2). GISTs are a heterogeneous group of tumors, including multiple molecular subtypes, with various activating oncogene mutations, such as receptor tyrosine kinase protein (KIT; approximately 60%–70%) and platelet-derived growth factor receptor A (PDGFRA; approximately 10%–15%) (3, 4), each of which present with different pathological mechanisms, clinical characteristics, and treatment response (5). PDGFRA is the second most mutated oncogene in GIST, and the annual incidence of PDGFRA-mutated GISTs is < 3 cases per 1 million individuals (6). PDGFRA-mutated GISTs can derive from telocytes and they show an epithelioid pattern (7, 8). PDGFRA-mutated GISTs are mostly located in the stomach (15–18%), followed by the small intestine (5–7%) (9). Around 15% of GISTs are wild-type GIST, which have no mutations in either KIT or PDGFRA, but have other genetic alterations, such as in the succinate dehydrogenase (SDH) gene family, RAS gene family, proto-oncogene B-Raf (BRAF), neurofibromatosis type 1 (NF1), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), gene fusions involving ETS variant transcription factor 6 (ETV6)-neurotrophic tyrosine receptor kinase 3 (NTRK3) or fibroblast growth factor receptor 1 (FGFR1), or other rare driver gene mutations (10–13).
PDGF was first discovered during platelet activation. Its receptor has two similar structures, PDGFRA and PDGFRB, which undergo intracellular activation during transport of the exocytic pathway and are subsequently secreted (14, 15). PDGFRA, similar to KIT, encodes the receptor tyrosine kinase (RTK), is located on chromosome 4q11-q12 (16), and is associated with many physiological processes of human growth and development. PDGFR consists of an extracellular ligand-binding region, a single transmembrane-spanning region, and an intracellular tyrosine kinase domain (17). This dual-switch mechanism carefully regulates cellular kinase activity by control of kinase conformation. Mechanistically, switch control of kinase conformation is mediated by phosphorylation of one or more switch amino acids that turn the kinase “on” or “off” (18). Most primary and secondary resistance mutations in PDGFRA are located within conformation-controlling switch regions embedded in the intracellular kinase domain. Primary resistance is more frequent in the activation loop (6). Secondary resistance is commonly located in the ATP-binding domain (exon 14) or activation loop (exon 18) (7, 19) (Figure 1). Mutations in PDGFRA are mainly found in exons 18 and 12 and rarely occur in exon 14 (4). Exon 18 encodes the activation loop and represents approximately 80% of the PDGFRA-mutated GISTs. A single D842V mutation, substitution of aspartic acid to valine, creates a missense mutation that confers resistance to imatinib, sunitinib and regorafenib. D842V is the most common exon 18 mutation, and it is detected in 62.6% of PDGFRA-mutated tumors (3, 4, 20). Exon 12, encoding the juxta membrane domain, is mutated in approximately 0.6% – 2% of GISTs, and < 1% of PDGFRA mutations will occur in exon 14 (encoding the ATP-binding domain). Studies have shown that novel tyrosine kinase inhibitors (TKIs), avapritinib and ripretinib, target the PDGFRA D842V mutation in GISTs and provide objective responses and long-term tumor control (21). Two clinical trials investigating the use of crenolanib in GISTs (NCT01243346 and NCT02847429), which is also used for targeting the PDGFRA D842V mutation, are still ongoing.
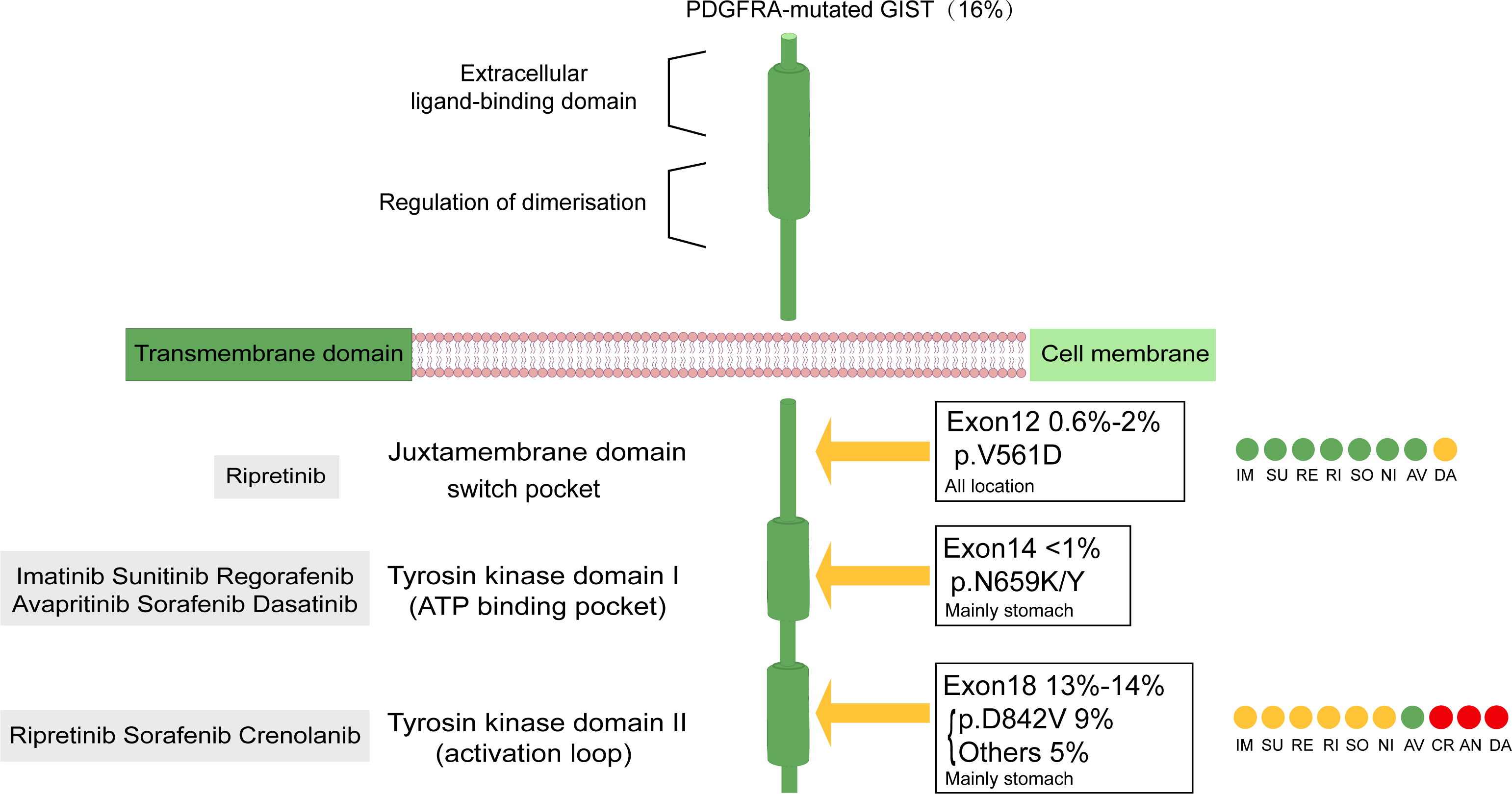
Figure 1 Structure of PDGFRA transmembrane tyrosine kinase receptor. Graphical representation of PDGFRA transmembrane tyrosine kinase receptors with frequency and localization of mutations found in advanced GIST. Gray boxes represent the site of action of the drug. Drug sensitivity of primary and secondary mutations in PDGFRA-mutated GISTs was distinguished by color: green indicates sensitive, yellow indicates mainly sensitive, red indicates in vitro test proved to be valid, but the clinical trial still had no definite result.CR Clinical trials in PDGFRA D842V population still ongoing; DA AN in vitro trial Valid but no clinical trial; NI specifically involving PDGFRA in vitro demonstrated activity against exon 12, diminished activity against D842V. AV, avapritinib; IM, imatinib; RE, regorafenib; RI, ripretinib; SU, sunitinib: SO, sorafenib; CR, crenolanib, DA, dasatinib; Ni, nilotinib; AN, anlotinib.
The PDGFR signaling pathway is an important RTK pathway that is associated with physiological activity in a variety of tumors (22). PDGFRs are transmembrane glycoprotein dimer molecules that initiate dimerization and phosphorylation after binding to the PDGF ligand, thereby activating various downstream signaling pathways, such as phosphatidylinositol 3 kinase (PI3K)/protein kinase B (AKT/PKB) pathway, mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway, and the Notch pathway (23, 24). Inhibition of PDGFR suppresses cancer proliferation, metastasis, invasion, and angiogenesis, and improves the antitumor effects of cancer drugs (25, 26). Some novel therapeutic strategies have emerged based on the PDGFR pathway for cancer treatment (Figure 2).
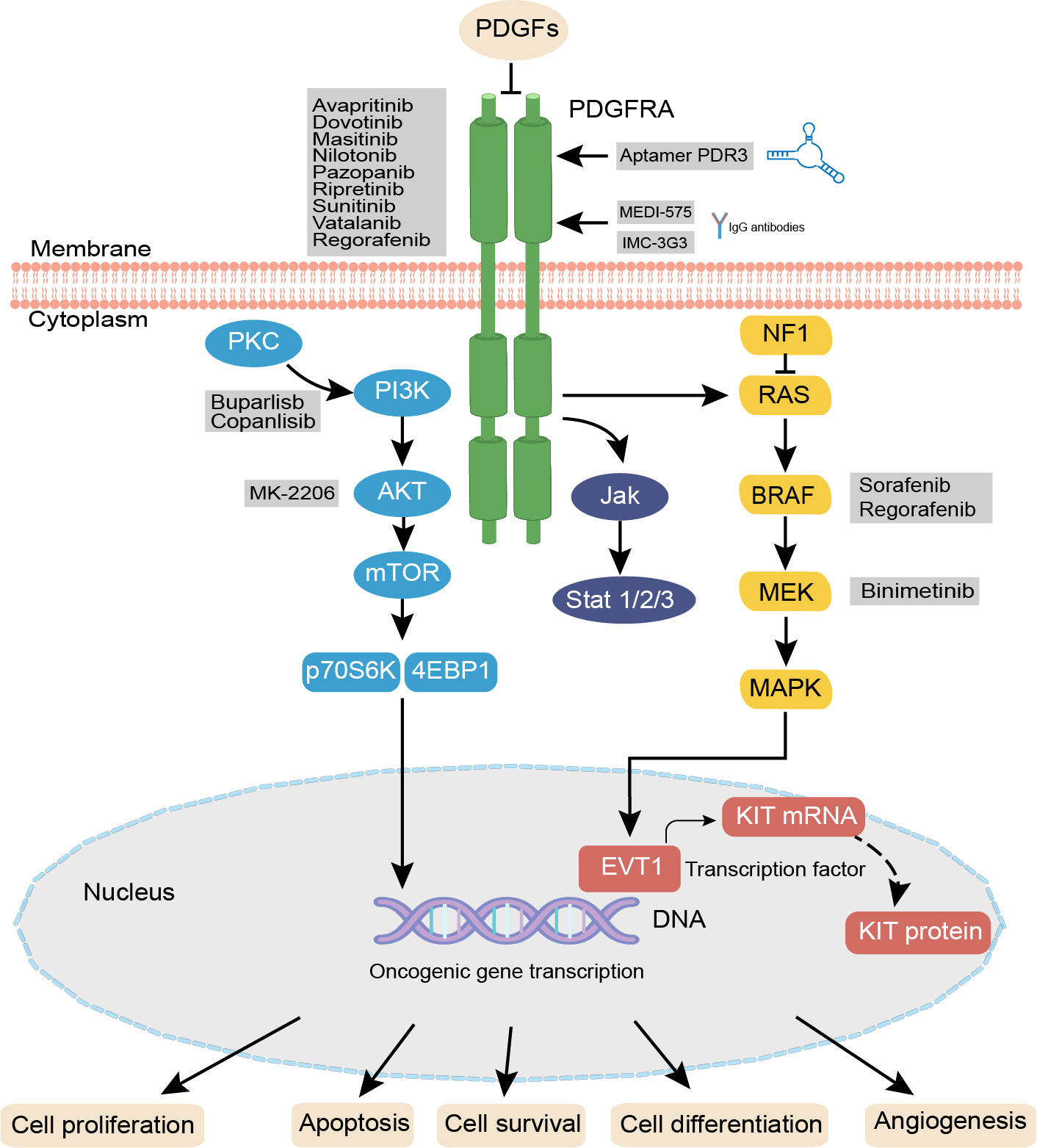
Figure 2 PDGF/PDGFRA signaling pathways and agent treatments. In PDGFRA mutaled GIST. PDGF isoforms bind to the related PDGFRs, initiate distinct receptor dimerization and phosphorylation, thereby activating various signaling pathways including the RAS-MEK-MAPK, PI3K-mTOR, JAK/STAT, and Notch pathway PDGFR antibodies such as INC-3G3, MEDI-575 and the specific RNA aptamer PDR3 can selectively bind to PDGFRA, after binding, these antibodies or aptamers can inhibit the downstream pathways. Activation or inhibition of these pathways influences cellular proliferation, angiogenesis, and apoptosis. PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; PKC, protein kinase C; PI3K, phosphati-dylinositol 3 kinase; AKT/PKB, protein kinase B; JAK, Janus kinase; STAT, signal transducers and activators of transcription; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; PDR3, the specific RNA aptamer.
This review will update the current knowledge on characteristics of PDGFRA-mutated GISTs and further discuss the clinical management of this subtype. After a brief introduction of first-, second-, and third-line TKIs for GIST treatment, we provide an overview of drugs based on other mechanisms, with a focus on new generation TKIs and future combination strategies.
First generation TKIs for treatment of PDGFRA-mutated GISTs: Imatinib, sunitinib, and regorafenib
During the past two decades, GIST has become a successful paradigm for the rational development of molecular targeted agents, based on the poor efficacy of cytotoxic chemotherapy (27) and the satisfactory improvement of prognosis by targeted therapy (28, 29).
Imatinib, an oral small molecule TKI which is a competitive inhibitor of the ATP binding site of KIT and PDGFRA, is the first-line drug for patients receiving initial treatment for advanced GISTs (30). A series of clinical trials have demonstrated the efficacy of imatinib (28, 29, 31). For advanced disease, regardless of mutation type, a daily dose of 400 mg provided a median progression free survival (mPFS) of 18 months. Approximately 45% of patients had a partial response, up to 5% had a complete response, and 32% had stable disease (27). Imatinib is active against non-D842V PDGFRA-mutated GISTs, but lacks efficacy against tumors with the D842V mutation, with significant differences in objective response rates between the two subtypes (32). A total of 58 patients with PDGFRA mutations were included in an international survey study, of whom 55% carried PDGFRA-D842V mutations (33). None of the patients with the D842V mutation achieved a response, and the mPFS was 2.8 months. For patients with other PDGFRA mutations, the mPFS was 28.5 months and overall response rate was 36% (33).
Sunitinib is approved as a second-line treatment for advanced GIST following progression on imatinib, or for patients with intolerance to imatinib. Like imatinib, sunitinib targets KIT and PDGFR, however, sunitinib may be effective in imatinib-resistant GISTs through its broader binding profile and affinity. MPFS was 6.8 months in patients receiving sunitinib and 1.6 months in those on placebo (34). In vitro studies have shown that the efficacy of sunitinib on PDGFRA exon 18 activation loop mutations is poor (35). A non-interventional retrospective analysis (NCT00094029) explored the correlation between PDGFRA mutation status and clinical benefit in patients treated with sunitinib (36). In this study, only 12 patients (5%) had a PDGFRA mutation, thus, the available data is too limited to draw conclusions on the efficacy of sunitinib on survival outcomes in patients. In a Korean cohort, patients with PDGFRA exon 18 mutations treated with sunitinib exhibited stable disease as the best response. Patients with D842V mutations had a 25% probability of stable disease after 24 weeks (37).
Regorafenib has the broadest kinase inhibitory activity among approved agents, which is a competitive inhibitor of the ATP-binding site for PDGFR, vascular endothelial growth factor receptor 1–3 (VEGFR1–3), TEK, KIT, RET, RAF1, BRAF, and FGFR (38). Regorafenib is approved as a third-line treatment for advanced GIST after progression on imatinib and sunitinib (39). In a case report of a patient with PDGFRA D842V mutated GIST, treatment with regorafenib resulted in prolonged response, and 20 months after treatment onset, the patient is still under treatment and maintaining a partial response (40). However, there is limiting evidence relating mutational status to regorafenib response. The mPFS and overall response rate (ORR) of patients with PDGFRA mutations treated with early generation TKIs are summarized in Table 1.

Table 1 Response rates and PFS of approved agents for PDGFRA-mutated GISTs.
There are currently no phase II trials that have specifically included patients with PDGFRA mutations and there is no evidence on the efficacy of sunitinib or regorafenib in PDGFRA mutations available from phase III trials. Imatinib, sunitinib and regorafenib are type II multi-kinase inhibitors, which bind to the ATP-pocket of PDGFRA only in the inactive formation, and they have limited activity against activation loop mutations (45). Recently approved next-generation TKIs, avapritinib and ripretinib, were specifically developed to address this issue.
Next-generation TKIs in GIST: Ripretinib and avapritinib
Ripretinib
Ripretinib was authorized by the FDA in March 2020 as a fourth-line or later-line treatment for GIST patients previously treated with three or more kinase inhibitors, including imatinib. Ripretinib is a novel type II switch-controlled kinase inhibitor that regulates both the kinase switch pocket and the activation loop (45), and the dual mechanism of action maintains KIT and PDGFRA in an inactive conformation independent of the primary and secondary mutation type, thereby inhibiting downstream signaling (46).
The phase III INVICTUS trial (NCT03353753) evaluated ripretinib using a dose of 150 mg daily in a fourth-line or later-line setting compared with placebo in patients who were refractory or intolerant to at least all three approved TKIs (41). In 129 patients, mPFS was 6.3 months in ripretinib-treated patients compared to one month in the placebo group, with a hazard ratio of 0.15 (95% CI 0.09–0.25). Ripretinib and placebo groups showed different response rate and median overall survival (OS) (9% vs 0% and 15.1 months vs 6.6 months, respectively). This trial met the primary end point, and disease stabilization was 47% at 12 weeks.
A second phase III trial, INTRIGUE (NCT03673501), is evaluating the safety and efficacy of ripretinib versus sunitinib as a second-line therapy (47). A total of 453 patients with GIST were enrolled in this study, where 226 patients received continuous dosing of 150 mg of ripretinib and 227 patients received continuous dosing of 50 mg of sunitinib (4-week on and 2-week off). Although the study did not meet its primary endpoint, progression-free survival (PFS) in ripretinib treated patients was not superior those treated with sunitinib, but there were significant advantages in ORR and safety profiles (42). In all population, mPFS was similar in patients treated with ripretinib compared to those treated with sunitinib: 8.0 months vs 8.3 months (HR = 1.05, 95% CI 0.82 to 1.33). The ORR for patients with a KIT exon 11 mutation was higher in those treated with ripretinib compared to those treated with sunitinib (23.9% vs 14.6%, respectively), however, sunitinib treatment improved PFS for patients with KIT exon 9 mutations.
The overall safety profile of ripretinib was favorable, with most side effects being low grade and manageable. In the INVICTUS trial, the most common grade 1/2 non-hematologic adverse events (AEs) occurred in more than 20% of patients and included alopecia (49%), myalgia (27%), nausea (25%), fatigue (24%), hand-foot skin reaction (HFSR) (21%), and diarrhea (20%). The most common grade 3/4 AEs included lipase increase (5%), hypertension (4%), fatigue (2%) and hypophosphatemia (2%) (48). In the INTRIGUE STUDY, the safety profile of ripretinib was improved compared to that of sunitinib, with a lower incidence of grade 3/4 AEs (26.5% vs 55.2%). For grade 3/4 AEs with an incidence of ≥2%, the incidence of these events was also lower in the ripretinib group than in the sunitinib group (42).
Dose escalation can be an alternative treatment option after disease progression, which has been effective in patients receiving imatinib (49). Additionally, there was a study that explored the efficacy of 150 mg of ripretinib twice daily in patients with advanced GIST used as a second, third or later line of therapy (50). In this study, 142 patients were included, and 67 patients received 150 mg of ripretinib twice daily after disease progression, which provided benefit across all lines of therapy. The mPFS was 5.6, 3.3, and 4.6 months in second-, third- and fourth-line therapy, respectively. The rate of partial metabolic response was 35.5%. Similarly, in the Phase III INVICTUS study, 43 patients received a twice daily dose of 150 mg of ripretinib 150 mg after disease progression. The mPFS was 3.7 months and the safety profile was acceptable (51).
Avapritinib
In contrast to early generation TKIs, avapritinib was specifically designed as a potent and highly selective type I inhibitor of mutations affecting the activation loop (encoded by exon 17 in KIT and exon 18 in PDGFRA) (45, 52). Avapritinib was approved by the FDA in 2020 based on the phase I/II trial results (21) for advanced or metastatic PDGFRA-mutated GIST, including the exon 18 D842V mutation.
The safety and efficacy of avapritinib was evaluated in 2 clinical trials, NAVIGATOR (NCT02508532) and VOYAGER (NCT03465722). In the NAVIGATOR study, a first-in-human phase I clinical trial, defined the starting dose as 300 mg and the maximum tolerated dose as 400 mg daily (21). The efficacy results are impressive, as the overall response rate was 91% at a dose of 300 mg daily (51/56 patients), the clinical benefit rate was 98% (55/56 patients), and the mPFS was 34.0 months (43). In this study, avapritinib had an acceptable safety profile, and AEs were generally grade 1 or grade 2. Most AEs that occurred were similar to those observed from other TKIs, such as edema, nausea, vomiting, and diarrhea. Other AEs of special interest were identified, including cognitive effects (48%, including memory impairment (29%), confused mental status (7%), encephalopathy (1%), and other cognitive disorders (11%)) and intracranial hemorrhage (2%) (53). However, the long-term cognitive side effects for avapritinib remain unknown.
The activity of avapritinib was compared to that of regorafenib in GIST as a fourth-line treatment in a phase III trial (VOYAGER), which randomized metastatic GIST patients to either avapritinib (n=240) or regorafenib group (n=236). Early top-line data indicated that avapritinib did not demonstrate an improvement over regorafenib in terms of PFS, the primary end point of the study. The mPFS was reported to be 4.2 months for avapritinib and 5.6 months for regorafenib, disease control rates were 41.7% vs 46.2%, and the overall response rate was 17.1% vs 7.2%, respectively (54).
Although avapritinib remains as the best treatment option for patients with advanced GIST with the PDGFRA D842V mutation, studies have revealed that secondary resistance may still develop in patients. Resistance mutations are observed within PDGFRA exons 13, 14, and 15, and these secondary PDGFRA mutations cause V658A, N659K, Y676C, and G680R substitutions that impair avapritinib binding (55).
Other TKIs beyond standard therapy for GIST
Drug resistance is a typical clinical phenomenon in cancer therapy, therefore, there are new clinical studies being conducted to address this dilemma. Table 2 lists clinical trials of novel next-generation TKIs (ripretinib and avapritinib) and other non-FDA approved agents that target PDGFRA mutations in metastatic or locally advanced GISTs.
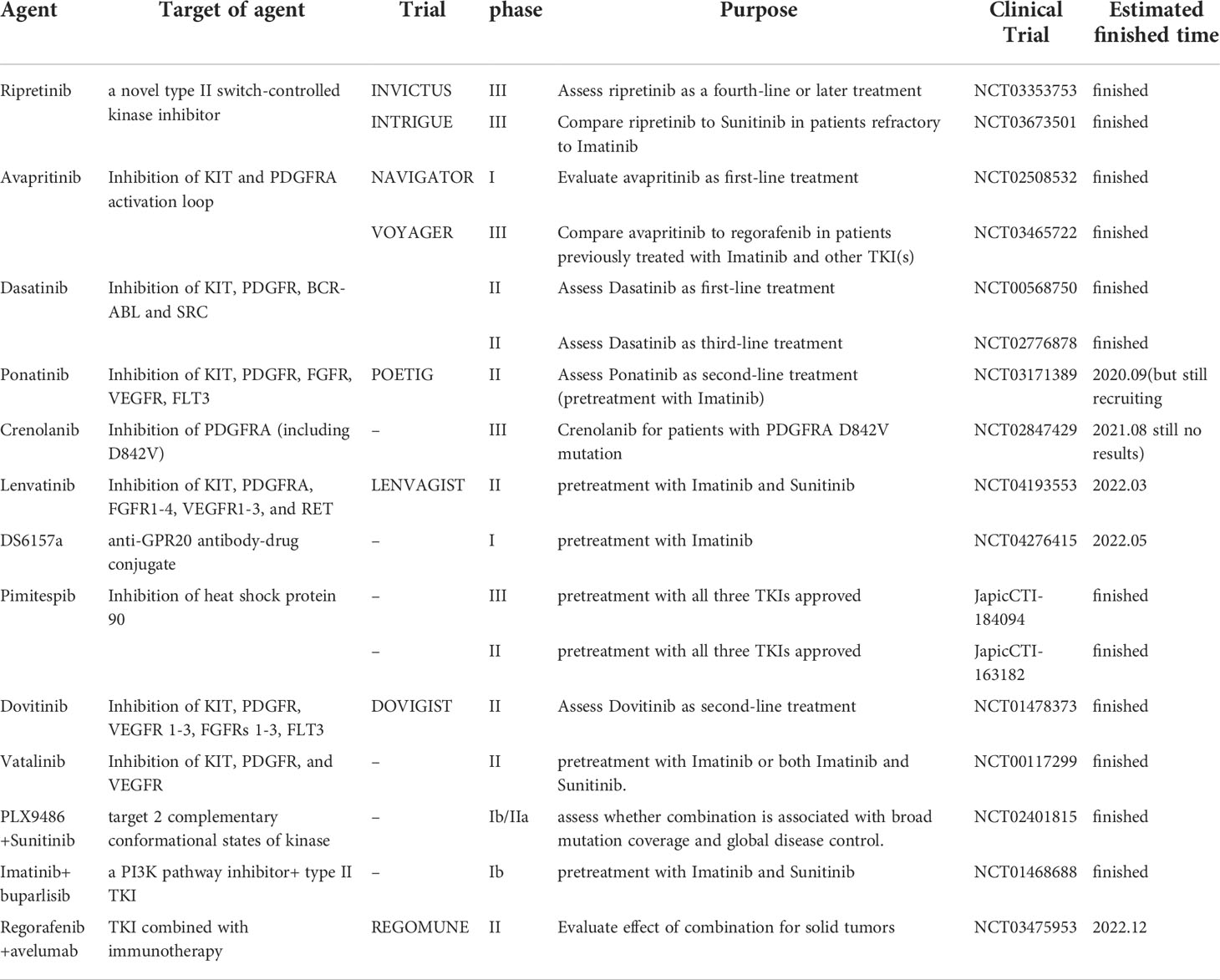
Table 2 Overview of clinical trials for agents targeting PDGFRA mutations in metastatic or locally advanced GIST.
Dasatinib
Dasatinib is a small molecule tyrosine kinase inhibitor with multiple targets including KIT, BCR-ABL, PDGFR, and nonreceptor kinases (SRC family) (56). Structurally, it differs from imatinib and sunitinib as it can bind to the ATP binding pocket regardless of receptor conformation (active and inactive states) (57, 58). A preclinical study has shown that GIST cells harboring the PDGFRA D842V mutation respond to dasatinib, where they exhibited reduced cell proliferation after exposure to dasatinib in vitro (59).
The efficacy and safety of dasatinib as a first-line therapy for GIST was explored in a single-arm phase II trial (60). The FDG-PET/CT response rate at 4 weeks was 74% and mPFS was 13.6 months. Grade 4 AEs occurred in 5% of patients and grade 3 AEs occurred in 48% of patients, most commonly involving gastrointestinal or pulmonary events. These results demonstrate that imatinib offers more benefit and favorable toxicity profile compared to dasatinib. Another single-arm phase II trial investigated the activity of dasatinib as a third-line therapy in 58 GIST patients who failed to respond to imatinib and sunitinib. Preliminary results showed that the 3-month PFS rate was 53.4% and the median OS was 14.0 months (61).
Crenolanib
Crenolanib is an orally available, highly specific inhibitor of PDGFR family members, and has a 25-fold higher affinity for PDGFR than KIT (62). Crenolanib was significantly more potent than imatinib in inhibiting the kinase activity of PDGFRA via the activation loop of exon 18: D842I, D842V, D842Y, DI842-843IM, and deletion I843. In vitro experiments showed that crenolanib was 100 to 150-fold more potent than imatinib against D842V, with an IC50 of approximately 10 nmol/L (63). In KIT-mutant GIST, crenolanib-mediated inhibition of PDGFRA disrupted the KIT-ERK-ETV1-KIT signaling loop by inhibiting ERK activation (64).
Crenolanib has been tested in both phase I and Ib clinical studies and it was found to be well tolerated in patients (65). Currently, there is a phase II trial using 140 mg of crenolanib twice daily in patients with advanced GIST with D842-related mutations and deletions (including D842V mutations) in the PDGFRA gene. Similarly, a randomized phase III trial (NCT02847429) used 100 mg of crenolanib three times daily or matching placebo for patients with advanced or metastatic GIST exhibiting the PDGFRA D842V mutation. The clinical trials are still ongoing and the results have not been disclosed.
Nilotinib
Nilotinib is a selective TKI targeting KIT, PDGFR and BCR-ABL (66). Both in vitro proliferation and in vivo studies indicated that nilotinib exhibited anti-tumor activity against the PDGFRA V561D mutation but significantly reduced efficacy against PDGFRA D842V mutation (67). Nilotinib has been used in 18 clinical trials, including 15 completed trials, two not recruiting, and one active trial (68). Nilotinib was compared with imatinib in a randomized phase III trial as a first-line therapy (69). The trial did not meet the primary endpoint of improvement in PFS. Molecular analysis showed that for patients with other mutations (excluding KIT 11, 9, and wild type), OS rates were comparable in both arms (69). A phase III trial explored the efficacy of nilotinib in patients with advanced GIST after previous imatinib and sunitinib failure (70). The patients were randomized in a 2:1 ratio to either 400 mg of nilotinib twice daily or a best supportive care group (best support only, or with imatinib/sunitinib). Local investigator-based intent-to-treat (ITT) analysis showed a significant increase in mPFS in the nilotinib group (119 days vs 70 days). Median OS was higher in patients with nilotinib (332 days vs 280 days). However, the PFS based on blinded central radiology review (primary endpoint) was not significantly different, therefore, further development of nilotinib as a third-line treatment for GIST was discontinued.
Anlotinib
Anlotinib, a multi-targeted TKI, is characterized as a highly selective and potent c-KIT and PDGFR inhibitor (71). Anlotinib has a broad spectrum of antitumor activity against GIST with D842V, D816H, V560G and V654A mutations both in vitro and in vivo (72). The toxicity profile of anlotinib in the treatment of a variety of advanced tumors was consistent with that reported for sorafenib, sunitinib, and regorafenib (73, 74). In a phase I clinical trial using anlotinib for advanced refractory solid tumors, the most common serious AEs were hand-foot skin reactions, hypertension, fatigue, and lipase elevation (71).
A single-arm, multicenter phase II trial (NCT04106024) is currently exploring the efficacy and safety of anlotinib in patients with advanced GIST refractory to imatinib. The clinical trial plans to enroll 60 participants who will be treated with 12 mg of anlotinib once daily for two weeks, followed by a one week break, and ending with three weeks as a course of treatment. The primary outcome measures is PFS up to 18 months. The trial is currently fully recruited and the results remain undisclosed.
Dovitinib
Dovitinib is a multikinase inhibitor of KIT, PDGFR, VEGFR 1-3, FGFRs 1-3, and FLT3 (75). In a phase I study, dovitinib administration to a patient with GIST resulted in disease control for eight months after failed response to imatinib and sorafenib (76). A phase II trial (DOVIGIST, NCT01478373) evaluated the antitumor activity of dovitinib as a second-line therapy in patients with GIST who were refractory to or intolerant of imatinib, including a total of 38 patients, of which 21 patients had KIT mutations and three patients had PDGFRA mutations. The ORR was 2.6% (1 of 38 patients), all three patients with PDGFRA mutation had stable disease, and the PFS was 4.6 months (77). In a phase II clinical trial of dovitinib as a third-line therapy, dovitinib showed modest anti-tumor activity and manageable toxicity. The study included 30 patients with failed response to imatinib or sunitinib, with a disease control rate of 13% at 24 weeks, a partial response in one patient (3%), stable disease in the other 21 patients, a mPFS of 3.6 months, and a median OS of 9.7 months (78).
Sorafenib
Sorafenib is similar to sunitinib, as it is a multikinase inhibitor with selectivity for KIT, PDGFRA, BRAF and FLT-3 (79, 80), however, it is not approved for treatment of GIST. In vitro studies demonstrated that sorafenib could inhibit imatinib-resistant PDGFRA mutations that occur in exon 14 (encoding the ATP-binding pocket) and exon 18 (encoding the activation loop), except for the substitution of PDGFRA codon 842 (79). A retrospective study has also investigated sorafenib in patients being resistant or intolerant to imatinib or sunitinib and showed a mPFS of 6.4 months after sorafenib treatment (81). Two phase II trials investigated sorafenib as a third-line treatment for patients with advanced GIST. One trial including 31 patients demonstrated a mPFS of 4.9 months and a disease control rate of 36% (82). Another trial performed with 38 patients reported that the disease control rate was 68%, and mPFS was 5.2 months after treatment (83). A case report showed that a patient with deletion of codon p.I843_D846del (located at PDGFRA exon 18), who was highly sensitive to sorafenib, responded in a dose-related manner. This patient had been treated with sorafenib for 12.5 years and still had no signs of recurrence (84). No phase III trials utilizing sorafenib for treatment of GIST have been conducted.
Lenvatinib
Lenvatinib is a type I TKI that also acts as an inhibitor of several RTKs, targets KIT, PDGFRA, FGFR, VEGFR, and RET, and has been approved for various advanced cancers (85, 86). The IC50 of lenvatinib ranged from 29 to 39 nM in in vitro assays (87, 88). It may have profound effects on tumor cell migration and invasion by inhibiting FGFR and PDGFR (89). A prospective, randomized, double-blind, multicenter trial (NCT04193553) aims to evaluate the efficacy and safety of lenvatinib in patients with GISTs who had previously failed imatinib and sunitinib (90). In this trial, 37 patients are treated with a continuous daily oral dose of 24 mg of lenvatinib and they are provided with best supportive care. The expected PFS is 1.5 months in the control group and 3.0 months in the lenvatinib group (HR=0.5), and the study is expected to be completed in March 2023.
Heat shock protein 90 (HSP90) inhibitor pimitespib (TAS116)
HSP90 is a molecular chaperone that assists many proteins, including KIT and PDGFRA, and maintains the structure and activity of certain key signaling proteins by folding and stabilizing proteins (91, 92). In vitro studies have shown that HSP 90 inhibitors can inhibit PDGFRA and attenuate downstream protein phosphorylation (59).
In a single-arm phase II trial (93), the oral HPS90 inhibitor TAS-116 was used in patients who were refractory to imatinib, sunitinib and regorafenib. MPFS was 4.4 months, the progression-free rate at 12 weeks was 74%, and 85% of patients had stable disease after 6 weeks. The most common AEs were gastrointestinal disorders and ocular AEs, and all other grade 3 or higher AEs resolved after dose modification.
In a phase III trial (JapicCTI-184094), pimitespib was compared with placebo in the treatment of patients with advanced GIST refractory to all three approved TKIs (94). The primary endpoint of the study was met and the results showed that the mPFS was 2.8 months vs 1.4 months, respectively, resulting in a 49% reduction in the risk of disease progression or death. This encouraging result suggests that HSP90 inhibitors are a potential novel therapy for patients with advanced GIST.
Azd3229
AZD3229 is a highly potent and selective small molecule inhibitor of KIT/PDGFRA that inhibits a wide range of primary and secondary mutations in GISTs without VEGFR2 inhibition (95, 96). In vitro assays and xenograft models showed that AZD3229 is more potent and selective than other approved agents, including avapritinib, and AZD3229 is 15–60 times more potent than imatinib in inhibiting primary mutations in KIT (96). Therefore, AZD3229 has the potential to be a best-in-class inhibitor for clinically relevant KIT/PDGFRA mutations in GIST.
Anti-GPR20 antibody-drug (DS-6157a)
Previous transcriptional studies have suggested that GIST cells may be rich in a novel gene target called G-protein coupled receptor 20 (GPR20) (97), which is expressed in more than 80% of GIST specimens, although GPR20 expression levels are lower in PDGFRA mutant GISTs (98). DS-6157a is an anti-GPR20 antibody-drug conjugate with a novel tetrapeptide-based linker and a derivative of the DNA topoisomerase I inhibitor exatecan derivative. In GIST xenograft models, including GIST models resistant to imatinib, sunitinib, and regorafenib, anti-GPR20 resulted in anti-tumor activity, and showed a favorable kinetic and safety profile. However, further clinical trials are still needed to verify the safety and efficacy of this novel agent.
Agent combination based on different strategies
Polyclonal resistance is a significant obstacle in GIST treatment, as a single drug is insufficient to target all resistant KIT mutations. Clinical trials have studied TKI therapies with different target inhibitors for tumors with polyclonal secondary mutations to address tumor heterogeneity. Several strategies based on therapeutic combinations aim to overcome the resistance mechanisms. A list of different combination based strategies is provided in Table 3.
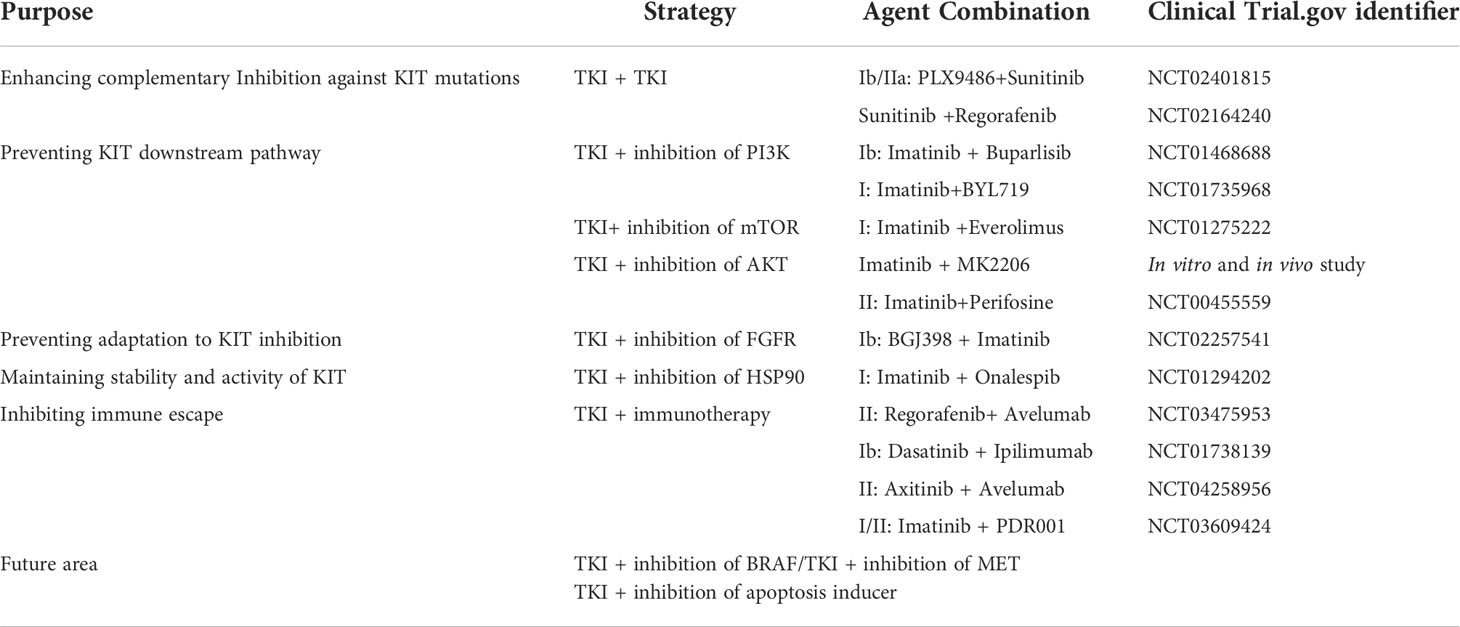
Table 3 Agent combination based on different strategies in the treatment of Imatinib-resistant GIST.
Sunitinib and regorafenib have complementary inhibitory characteristics against KIT mutations, and clinical trials with polyclonal secondary mutant tumors have utilized TKI therapy as a rapid alternating therapy. In the SURE project (NCT02164240), an open-label phase I/II trial, 3 days of sunitinib were followed by 4 days of regorafenib in imatinib-refractory advanced GISTs (99). Four of the 13 patients included had stable illness, and the overall mPFS was 1.9 months (99). Another promising drug combination with complementary efficacy is PLX9486 (a selective type I KIT inhibitor targeting activating loop mutations) and sunitinib. A phase 1b/2a clinical trial (NCT02401815) in 39 patients was conducted to determine whether it is associated with wide mutation coverage, and it demonstrated activity with a mPFS of 12.1 months and an 80% clinical benefit rate (100).
The second strategy is to act on signaling pathways designed to prevent KIT downstream pathway to enhance apoptosis, such as the combination of imatinib with other critical targets (RAS/MAPK or PI3K/mTOR). In GIST patients who had previously failed treatment with imatinib and sunitinib, a Phase 1b study (NCT01468688) using the PI3K inhibitor buparlisib in combination with Imatinib was conducted to assess the clinical profile of the combination (101). However, compared to currently accessible treatments, this combination offered no significant benefit. No partial or full responses were observed, therefore further development of this combination was not pursued. Another Dose-finding Study (NCT01735968) of a Combination of Imatinib and BYL719, PI3K inhibitor in the Treatment of 3rd Line GIST Patients is conducted, but there are no results disclosed (102).
Tumor adaptation to KIT/PDGFRA inhibition leads to apoptosis evasion, and this antiapoptotic response is sustained over time by FGFR- and c-MET–mediated MAPK pathway reactivation (103, 104). It is plausible that GIST follows the same principles of chronic myeloid leukemia, where growth factor receptors are inhibited by a MEK-dependent negative feedback that is released upon BCR-ABL TKI inhibition (105). Binimetinib, a reversible inhibitor of mitogen-activated extracellular signal-regulated kinase 1 (MEK1) and MEK2 activity, is approved for the treatment of unresectable or metastatic melanoma with BRAF V600E or V600K mutations. Binimetinib is currently being investigated in a phase 1 study in patients with advanced GISTs in combination with pexidartinib (NCT03158103) (106). Another phase II trial (NCT0199379) was designed to test the efficacy and safety of binimetinib plus imatinib as a first-line treatment for GIST, which met the primary endpoint and showed good efficacy and manageable toxicity (107). Dual targeting of the GIST lineage-specific master regulators, ETV1 and KIT, by MEK and KIT inhibitors, respectively, may enhance clinical efficacy of these agents, and more studies should be conducted to explore this combination.
The fourth strategy is TKI combined with immunotherapy. REGOMUNE (NCT03475953) is a single arm, multicentric phase II trial investigating the safety and efficacy of regorafenib (160 mg daily for 3 weeks with 1 week rest) in combination with avelumab (once every 2 weeks) in patients with various solid tumors. At present, the results of the GIST subgroup have not been disclosed, but synergistic anti-tumor effect has been shown in biliary tract tumors and colorectal cancer (108, 109).
Future of advanced GIST treatment
Although the combination of imatinib and other targeted inhibitors failed to meet primary clinical outcomes, these studies were conducted in imatinib-resistant GIST, in which case imatinib is unlikely to bind to KIT secondary mutants, and therefore, would have limited efficacy. Future research should consider combinations of other approved TKIs (sunitinib, regorafenib, ripretinib and avapritinib) with other targeted inhibitors. Furthermore, crenolanib is a highly specific inhibitor of PDGFR, including the D842V mutation subtype, thus, further studies should be conducted to explore its combinatorial effects with other agents. Innovative forms of combination therapy such as intermittent or drug rotation regimens can also be explored to achieve effective doses while minimizing overlapping toxicities (99).
Notch signaling is a conserved developmental pathway known to play a critical role in the development of multiple tumors (110, 111). In soft tissue sarcoma, including GIST, multiple studies reported deregulated Notch expression (112). A previous study reported the tumor suppressor effects of the Notch pathway in GISTs via negative feedback with the oncogene KIT and may lead the development of new therapeutic strategies for GISTs patients (113). Only one clinical trial utilizing LY3039478, an oral selective Notch inhibitor, was conducted in patients with GIST to assess its safety, pharmacokinetics, and antitumor efficacy (114). Given the role of Notch inhibitors in the treatment of other cancers, more clinical trials should be done in the future to investigate the effect of notch inhibitors, including gamma-secretase complex inhibitors, as well as anti-Notch2/3 antibody treatments, for the treatment of GIST.
Activation of the PI3K/AKT/mTOR pathway, a key downstream pathway of KIT/PDGFRA signaling, has been shown to be a crucial survival pathway in imatinib-resistant GISTs (115). Many mutations in genes of this signaling pathway, including IGF1R, MTOR, TSC1, FLT4, TSC2, IRS1, INSR, and BRCA1, may mediate resistance to imatinib or other KIT inhibitors (116). Several clinical trials targeting PI3K/AKT/mTOR signaling are currently being investigated as promising targeted therapy strategies for GIST (117). In a phase I/II clinical trial, the oral mTOR inhibitor everolimus demonstrated efficacy in GIST refractory to imatinib and sunitinib, with 37% of patients remaining progression-free for at least 4 months and 36% of patients achieved stable disease (118). Another mTOR inhibitor, sirolimus, has shown promise in a small number of GIST patients with PDGFRA-D842V mutations when combined with TKIs such as imatinib (119). Concurrent inhibition of the two critical KIT-downstream pathways is regarded as a very attractive approach. To investigate the ideal combination dose in the future, additional clinical trials are required.
Studies have shown that adaptive and innate immune cells are present in the GIST tumor microenvironment, which suggests immunotherapy may be a potential future treatment for GIST. Many studies indicate that D842V-mutated tumors exhibited a significant enrichment of immune-related genes and immune cells, such as CD3+, CD8+, and CD68+ cells (120). Compared to KIT mutant GISTs, PDGFRA mutant GISTs express significantly higher levels of chemokines, such as CXCL14, and these tumors could exhibit HLA binding (121). Immunotherapeutic agents used in clinical trials include anti-PD-1 (nivolumab, spartalizumab, pembrolizumab, avelumab, and PDR001)/PD-L1 molecules and ipilimumab (targeting CTLA-4). Nowadays, only one clinical trial each of pembrolizumab and ipilimumab has been completed (122, 123), showing limited activity.
Tumor heterogeneity is another major concern in cancer therapy. Liquid biopsies, combined with sequencing techniques, have the potential to predict TKI treatment sensitivity by capturing circulating tumor DNA (ctDNA) and cells (124). Future efforts in laboratory research should be applied to decipher lineage-specific KIT dependence (125). Parallel efforts should point to studies of high-throughput synthetic lethal screens, for example, the discovery that CDC37 is a critical HSP90 cofactor for oncogenic expression of KIT provides a promising strategy (126). Preclinical and clinical evidence supports the exploration of novel treatment modalities aimed at blocking various mechanisms of resistance or adaptation. Integrating clinical-genomic data and generating robust preclinical models will be the backbone of successful future GIST research.
Conclusion
In conclusion, two decades of active translational and clinical research have demonstrated the paradigm of GISTs as targeted therapies. Multi-kinase inhibitors, such as sunitinib, regorafenib, dasatinib, nilotinib, anlotinib, dovitinib, pazopanib, sorafenib, and lenvatinib, are currently preferred above over other treatment strategies. Efforts were subsequently made to develop highly selective kinase inhibitors, including avapritinib and crenolanib, to improve kinome selectivity. Proteasome inhibitors such as bortezomib and histone deacetylase inhibitors such as SAHA or panobinostat have also been postulated as potential treatments for GISTs. Activation of the PI3K/AKT/mTOR pathway has been shown to be a critical survival pathway, and multiple clinical trials of mTOR inhibitors and PI3K inhibitors in the treatment of GISTs are also being carried out. The majority of current efforts are focused on reducing the hazards brought on by polyclonal heterogeneity in imatinib-refractory GIST. The necessity to research novel agents or, more likely, treatment combinations intended to jointly block multiple resistance mechanisms is being supported by preclinical and clinical evidence more and more.
Author contributions
YS and WH devised the main conceptual ideas and outline. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Platelet-derived growth factor receptor A (PDGFRA), gastrointestinal stromal tumors (GISTs), tyrosine kinase protein (KIT), interstitial cells of Cajal (ICC), succinate dehydrogenase (SDH) gene family, RAS gene family, proto-oncogene B-Raf (BRAF), neurofibromatosis type 1 (NF1), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), gene fusions involving ETS variant transcription factor 6 (ETV6)-neurotrophic tyrosine receptor kinase 3 (NTRK3) or fibroblast growth factor receptor 1 (FGFR1), receptor tyrosine kinase (RTK), phosphatidylinositol 3 kinase (PI3K)/protein kinase B (AKT/PKB) pathway, mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, Janus kinase (JAK)/signal transducers and activators of transcription (STAT), vascular endothelial growth factor receptor 1–3 (VEGFR1–3), median progression free survival (mPFS), overall response rate (ORR), adverse events (AEs), G-protein coupled receptor 20 (GPR20), mitogen-activated extracellular signal-regulated kinase 1 (MEK1).
References
1. Verschoor AJ, Bovee J, Overbeek LIH, group P, Hogendoorn PCW, Gelderblom H. The incidence, mutational status, risk classification and referral pattern of gastro-intestinal stromal tumours in the Netherlands: a nationwide pathology registry (PALGA) study. Virchows Arch (2018) 472(2):221–9. doi: 10.1007/s00428-017-2285-x
2. Soreide K, Sandvik OM, Soreide JA, Giljaca V, Jureckova A, Bulusu VR. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol (2016) 40:39–46. doi: 10.1016/j.canep.2015.10.031
3. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol (2005) 23(23):5357–64. doi: 10.1200/JCO.2005.14.068
4. Martin-Broto J, Martinez-Marin V, Serrano C, Hindi N, Lopez-Guerrero JA, Bisculoa M, et al. Gastrointestinal stromal tumors (GISTs): SEAP-SEOM consensus on pathologic and molecular diagnosis. Clin Transl Oncol (2017) 19(5):536–45. doi: 10.1007/s12094-016-1581-2
5. Szucs Z, Thway K, Fisher C, Bulusu R, Constantinidou A, Benson C, et al. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol (2017) 13(1):93–107. doi: 10.2217/fon-2016-0192
6. Cassier PA, Ducimetiere F, Lurkin A, Ranchere-Vince D, Scoazec JY, Bringuier PP, et al. A prospective epidemiological study of new incident GISTs during two consecutive years in Rhone alpes region: incidence and molecular distribution of GIST in a European region. Br J Cancer (2010) 103(2):165–70. doi: 10.1038/sj.bjc.6605743
7. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer (2011) 11(12):865–78. doi: 10.1038/nrc3143
8. Khalaf S, Shoqeir JH, Scrano L, Karaman R, Bufo SA. Photodegradation using TiO2-activated borosilicate tubes. Environ Sci pollut Res Int (2019) 26(19):19025–34. doi: 10.1007/s11356-018-2858-5
9. Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers (2021) 7(1):22. doi: 10.1038/s41572-021-00254-5
10. Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: A report from the national institutes of health gastrointestinal stromal tumor clinic. JAMA Oncol (2016) 2(7):922–8. doi: 10.1001/jamaoncol.2016.0256
11. Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer (2008) 47(10):853–9. doi: 10.1002/gcc.20589
12. Lasota J, Felisiak-Golabek A, Wasag B, Kowalik A, Zieba S, Chlopek M, et al. Frequency and clinicopathologic profile of PIK3CA mutant GISTs: molecular genetic study of 529 cases. Mod. Pathol (2016) 29(3):275–82. doi: 10.1038/modpathol.2015.160
13. Brcic I, Argyropoulos A, Liegl-Atzwanger B. Update on molecular genetics of gastrointestinal stromal tumors. Diagn (Basel) (2021) 11(2):194. doi: 10.3390/diagnostics11020194
14. Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev (2004) 15(4):197–204. doi: 10.1016/j.cytogfr.2004.03.007
16. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol (2006) 23(2):70–83. doi: 10.1053/j.semdp.2006.09.001
17. Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal (2013) 11:97. doi: 10.1186/1478-811X-11-97
18. Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell> (2002) 109(3):275–82. doi: 10.1016/S0092-8674(02)00741-9
19. Serrano C, Marino-Enriquez A, Tao DL, Ketzer J, Eilers G, Zhu M, et al. Correction: Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer (2019) 121(3):281. doi: 10.1038/s41416-019-0487-5
20. Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod. Pathol (2014) 27 Suppl 1:S1–16. doi: 10.1038/modpathol.2013.173
21. Heinrich MC, Jones RL, von Mehren M, Schoffski P, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol (2020) 21(7):935–46. doi: 10.1016/S1470-2045(20)30269-2
22. Cavalcanti E, Ignazzi A, De Michele F, Caruso ML. PDGFRalpha expression as a novel therapeutic marker in well-differentiated neuroendocrine tumors. Cancer Biol Ther (2019) 20(4):423–30. doi: 10.1080/15384047.2018.1529114
23. Zou X, Tang XY, Qu ZY, Sun ZW, Ji CF, Li YJ, et al. Targeting the PDGF/PDGFR signaling pathway for cancer therapy: A review. Int J Biol Macromol (2022) 202:539–57. doi: 10.1016/j.ijbiomac.2022.01.113
24. Huang XL, Khan MI, Wang J, Ali R, Ali SW, Zahra QU, et al. Role of receptor tyrosine kinases mediated signal transduction pathways in tumor growth and angiogenesis-new insight and futuristic vision. Int J Biol Macromol (2021) 180:739–52. doi: 10.1016/j.ijbiomac.2021.03.075
25. Thies KA, Hammer AM, Hildreth BE 3rd, Steck SA, Spehar JM, Kladney RD, et al. Stromal platelet-derived growth factor receptor-beta signaling promotes breast cancer metastasis in the brain. Cancer Res (2021) 81(3):606–18. doi: 10.1158/0008-5472.CAN-19-3731
26. Balamurugan K, Koehler L, Durig JN, Hempel U, Rademann J, Hintze V, et al. Structural insights into the modulation of PDGF/PDGFR-beta complexation by hyaluronan derivatives. Biol Chem (2021) 402(11):1441–52. doi: 10.1515/hsz-2021-0173
27. Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet (2004) 364(9440):1127–34. doi: 10.1016/S0140-6736(04)17098-0
28. van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrijevic S, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet 2001358(9291):1421–3. doi: 10.1016/S0140-6736(01)06535-7
29. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med (2002) 347(7):472–80. doi: 10.1056/NEJMoa020461
30. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract (2014) 2014:357027. doi: 10.1155/2014/357027
31. Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol (2008) 26(4):620–5. doi: 10.1200/JCO.2007.13.4403
32. Yoo C, Ryu MH, Jo J, Park I, Ryoo BY, Kang YK. Efficacy of imatinib in patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors. Cancer Res Treat (2016) 48(2):546–52. doi: 10.4143/crt.2015.015
33. Cassier PA, Fumagalli E, Rutkowski P, Schoffski P, Van Glabbeke M, Debiec-Rychter M, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res (2012) 18(16):4458–64. doi: 10.1158/1078-0432.CCR-11-3025
34. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet (2006) 368(9544):1329–38. doi: 10.1016/S0140-6736(06)69446-4
35. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol (2008) 26(33):5352–9. doi: 10.1200/JCO.2007.15.7461
36. Reichardt P, Demetri GD, Gelderblom H, Rutkowski P, Im SA, Gupta S, et al. Correlation of KIT and PDGFRA mutational status with clinical benefit in patients with gastrointestinal stromal tumor treated with sunitinib in a worldwide treatment-use trial. BMC Cancer (2016) 16:22. doi: 10.1186/s12885-016-2051-5
37. Yoon DH, Ryu MH, Ryoo BY, Beck M, Choi DR, Cho Y, et al. Sunitinib as a second-line therapy for advanced GISTs after failure of imatinib: relationship between efficacy and tumor genotype in Korean patients. Invest New Drugs (2012) 30(2):819–27. doi: 10.1007/s10637-010-9593-1
38. Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer (2011) 129(1):245–55. doi: 10.1002/ijc.25864
39. George S, Wang Q, Heinrich MC, Corless CL, Zhu M, Butrynski JE, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol (2012) 30(19):2401–7. doi: 10.1200/JCO.2011.39.9394
40. Grellety T, Kind M, Coindre JM, Italiano A. Clinical activity of regorafenib in PDGFRA-mutated gastrointestinal stromal tumor. Future Sci OA (2015) 1(4):FSO33. doi: 10.4155/fso.15.33
41. Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom H, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol (2020) 21(7):923–34. doi: 10.1016/S1470-2045(20)30168-6
42. Heinrich MC, Jones RL, Gelderblom H, George S, Schöffski P, Mehren MV, et al. INTRIGUE: A phase III, randomized, open-label study to evaluate the efficacy and safety of ripretinib versus sunitinib in patients with advanced gastrointestinal stromal tumor previously treated with imatinib. ASCO Plenary Ser (2022). 40(suppl 36):359881. doi: 10.1200/JCO.2022.40.36_suppl.359881
43. Jones RL, Serrano C, von Mehren M, George S, Heinrich MC, Kang YK, et al. Avapritinib in unresectable or metastatic PDGFRA D842V-mutant gastrointestinal stromal tumours: Long-term efficacy and safety data from the NAVIGATOR phase I trial. Eur J Cancer (2021) 145:132–42. doi: 10.1016/j.ejca.2020.12.008
44. George S, Jones RL, Bauer S, Kang YK, Schoffski P, Eskens F, et al. Avapritinib in patients with advanced gastrointestinal stromal tumors following at least three prior lines of therapy. Oncologist (2021) 26(4):e639–e49. doi: 10.1002/onco.13674
45. Evans EK, Gardino AK, Kim JL, Hodous BL, Shutes A, Davis A, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med (2017) 9(414):eaao1690. doi: 10.1126/scitranslmed.aao1690
46. Smith BD, Kaufman MD, Lu WP, Gupta A, Leary CB, Wise SC, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell (2019) 35(5):738–51.e9. doi: 10.1016/j.ccell.2019.04.006
47. Nemunaitis J, Bauer S, Blay JY, Choucair K, Gelderblom H, George S, et al. Intrigue: Phase III study of ripretinib versus sunitinib in advanced gastrointestinal stromal tumor after imatinib. Future Oncol (2020) 16(1):4251–64. doi: 10.2217/fon-2019-0633
48. Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom H, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol (2020) 21(7):923–34. doi: 10.1016/S1470-2045(20)30168-6
49. Zalcberg JR, Verweij J, Casali PG, Le Cesne A, Reichardt P, Blay JY, et al. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer (2005) 41(12):1751–7. doi: 10.1016/j.ejca.2005.04.034
50. George S, Chi P, Heinrich MC, von Mehren M, Jones RL, Ganjoo K, et al. Ripretinib intrapatient dose escalation after disease progression provides clinically meaningful outcomes in advanced gastrointestinal stromal tumour. Eur J Cancer (2021) 155:236–44. doi: 10.1016/j.ejca.2021.07.010
51. Zalcberg JR, Heinrich MC, George S, Bauer S, Schoffski P, Serrano C, et al. Clinical benefit of ripretinib dose escalation after disease progression in advanced gastrointestinal stromal tumor: An analysis of the INVICTUS study. Oncologist (2021) 26(11):e2053–e60. doi: 10.1002/onco.13917
52. Gebreyohannes YK, Wozniak A, Zhai ME, Wellens J, Cornillie J, Vanleeuw U, et al. Robust activity of avapritinib, potent and highly selective inhibitor of mutated KIT, in patient-derived xenograft models of gastrointestinal stromal tumors. Clin Cancer Res (2019) 25(2):609–18. doi: 10.1158/1078-0432.CCR-18-1858
53. Heinrich MC, Jones RL, von Mehren M, Schöffski P, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol (2020) 21(7):935–46. doi: 10.1016/S1470-2045(20)30269-2
54. Kang YK, George S, Jones RL, Rutkowski P, Shen L, Mir O, et al. Avapritinib versus regorafenib in locally advanced unresectable or metastatic GI stromal tumor: A randomized, open-label phase III study. J Clin Oncol (2021) 39(28):3128–39. doi: 10.1200/JCO.21.00217
55. Grunewald S, Klug LR, Muhlenberg T, Lategahn J, Falkenhorst J, Town A, et al. Resistance to avapritinib in PDGFRA-driven GIST is caused by secondary mutations in the PDGFRA kinase domain. Cancer Discovery (2021) 11(1):108–25. doi: 10.1158/2159-8290.CD-20-0487
56. Gnoni A, Marech I, Silvestris N, Vacca A, Lorusso V. Dasatinib: an anti-tumour agent via src inhibition. Curr Drug Targets (2011) 12(4):563–78. doi: 10.2174/138945011794751591
57. O'Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, et al. In vitro activity of bcr-abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant abl kinase domain mutants. Cancer Res (2005) 65(11):4500–5. doi: 10.1158/0008-5472.CAN-05-0259
58. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science (2004) 305(5682):399–401. doi: 10.1126/science.1099480
59. Dewaele B, Wasag B, Cools J, Sciot R, Prenen H, Vandenberghe P, et al. Activity of dasatinib, a dual SRC/ABL kinase inhibitor, and IPI-504, a heat shock protein 90 inhibitor, against gastrointestinal stromal tumor-associated PDGFRAD842V mutation. Clin Cancer Res (2008) 14(18):5749–58. doi: 10.1158/1078-0432.CCR-08-0533
60. Montemurro M, Cioffi A, Domont J, Rutkowski P, Roth AD, von Moos R, et al. Long-term outcome of dasatinib first-line treatment in gastrointestinal stromal tumor: A multicenter, 2-stage phase 2 trial (Swiss group for clinical cancer research 56/07). Cancer (2018) 124(7):1449–54. doi: 10.1002/cncr.31234
61. Zhou Y, Zhang X, Wu X, Zhou Y, Zhang B, Liu X, et al. A prospective multicenter phase II study on the efficacy and safety of dasatinib in the treatment of metastatic gastrointestinal stromal tumors failed by imatinib and sunitinib and analysis of NGS in peripheral blood. Cancer Med (2020) 9(17):6225–33. doi: 10.1002/cam4.3319
62. Ueda Y, Hasegawa M, Yonemitsu Y. Sendai Virus for cancer immunotherapy. Methods Mol Biol (2009) 515:299–308. doi: 10.1007/978-1-59745-559-6_21
63. Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, et al. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res (2012) 18(16):4375–84. doi: 10.1158/1078-0432.CCR-12-0625
64. Hayashi Y, Bardsley MR, Toyomasu Y, Milosavljevic S, Gajdos GB, Choi KM, et al. Platelet-derived growth factor receptor-alpha regulates proliferation of gastrointestinal stromal tumor cells with mutations in KIT by stabilizing ETV1. Gastroenterology (2015) 149(2):420–32.e16. doi: 10.1053/j.gastro.2015.04.006.
65. Lewis NL, Lewis LD, Eder JP, Reddy NJ, Guo F, Pierce KJ, et al. Phase I study of the safety, tolerability, and pharmacokinetics of oral CP-868,596, a highly specific platelet-derived growth factor receptor tyrosine kinase inhibitor in patients with advanced cancers. J Clin Oncol (2009) 27(31):5262–9. doi: 10.1200/JCO.2009.21.8487
66. Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant bcr-abl. Cancer Cell (2005) 7(2):129–41. doi: 10.1016/j.ccr.2005.01.007
67. Weisberg E, Wright RD, Jiang J, Ray A, Moreno D, Manley PW, et al. Effects of PKC412, nilotinib, and imatinib against GIST-associated PDGFRA mutants with differential imatinib sensitivity. Gastroenterology (2006) 131(6):1734–42. doi: 10.1053/j.gastro.2006.09.017
68. Elias B, Starling M, Wilson B, McGreevy P. Influences on infrared thermography of the canine eye in relation to the stress and arousal of racing greyhounds. Anim (Basel) (2021) 11(1):103. doi: 10.3390/ani11010103
69. Blay JY, Shen L, Kang YK, Rutkowski P, Qin S, Nosov D, et al. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): a randomised phase 3 trial. Lancet Oncol (2015) 16(5):550–60. doi: 10.1016/S1470-2045(15)70105-1
70. Reichardt P, Blay JY, Gelderblom H, Schlemmer M, Demetri GD, Bui-Nguyen B, et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann Oncol (2012) 23(7):1680–7. doi: 10.1093/annonc/mdr598
71. Sun Y, Niu W, Du F, Du C, Li S, Wang J, et al. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J Hematol Oncol (2016) 9(1):105. doi: 10.1186/s13045-016-0332-8
72. Ashton S, Song YH, Nolan J, Cadogan E, Murray J, Odedra R, et al. Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo. Sci Transl Med (2016) 8(325):325ra17. doi: 10.1126/scitranslmed.aad2355
73. Porta C, Gore ME, Rini BI, Escudier B, Hariharan S, Charles LP, et al. Long-term safety of sunitinib in metastatic renal cell carcinoma. Eur Urol (2016) 69(2):345–51. doi: 10.1016/j.eururo.2015.07.006
74. Afonso FJ, Anido U, Fernandez-Calvo O, Vazquez-Estevez S, Leon L, Lazaro M, et al. Comprehensive overview of the efficacy and safety of sorafenib in advanced or metastatic renal cell carcinoma after a first tyrosine kinase inhibitor. Clin Transl Oncol (2013) 15(6):425–33. doi: 10.1007/s12094-012-0985-x
75. Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, et al. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood (2005) 105(7):2941–8. doi: 10.1182/blood-2004-10-3913
76. Sarker D, Molife R, Evans TR, Hardie M, Marriott C, Butzberger-Zimmerli P, et al. A phase I pharmacokinetic and pharmacodynamic study of TKI258, an oral, multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clin Cancer Res (2008) 14(7):2075–81. doi: 10.1158/1078-0432.CCR-07-1466
77. Joensuu H, Blay JY, Comandone A, Martin-Broto J, Fumagalli E, Grignani G, et al. Dovitinib in patients with gastrointestinal stromal tumour refractory and/or intolerant to imatinib. Br J Cancer (2017) 117(9):1278–85. doi: 10.1038/bjc.2017.290
78. Kang YK, Yoo C, Ryoo BY, Lee JJ, Tan E, Park I, et al. Phase II study of dovitinib in patients with metastatic and/or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib. Br J Cancer (2013) 109(9):2309–15. doi: 10.1038/bjc.2013.594
79. Heinrich MC, Marino-Enriquez A, Presnell A, Donsky RS, Griffith DJ, McKinley A, et al. Sorafenib inhibits many kinase mutations associated with drug-resistant gastrointestinal stromal tumors. Mol Cancer Ther (2012) 11(8):1770–80. doi: 10.1158/1535-7163.MCT-12-0223
80. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res (2004) 64(19):7099–109. doi: 10.1158/0008-5472.CAN-04-1443
81. Montemurro M, Gelderblom H, Bitz U, Schutte J, Blay JY, Joensuu H, et al. Sorafenib as third- or fourth-line treatment of advanced gastrointestinal stromal tumour and pretreatment including both imatinib and sunitinib, and nilotinib: A retrospective analysis. Eur J Cancer (2013) 49(5):1027–31. doi: 10.1016/j.ejca.2012.10.009
82. Park SH, Ryu MH, Ryoo BY, Im SA, Kwon HC, Lee SS, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs (2012) 30(6):2377–83. doi: 10.1007/s10637-012-9795-9
83. Campbell NP, Wroblewski K, Maki RG, D'Adamo DR, Chow WA, Gandara DR, et al. Sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): final results of a university of Chicago phase II consortium trial. J Clin Oncol (2011) 29(suppl 15):10009. doi: 10.1200/jco.2011.29.15_suppl.10009
84. Brinch C, Dehnfeld M, Hogdall E, Poulsen TS, Toxvaerd A, Al-Farra G, et al. Outstanding response to sorafenib in a patient with metastatic gastrointestinal stromal tumour. Case Rep Oncol (2021) 14(3):1567–73. doi: 10.1159/000519747
85. Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med (2015) 372(7):621–30. doi: 10.1056/NEJMoa1406470
86. Al-Salama ZT, Syed YY, Scott LJ. Lenvatinib: A review in hepatocellular carcinoma. Drugs (2019) 79(6):665–74. doi: 10.1007/s40265-019-01116-x
87. Hussein Z, Mizuo H, Hayato S, Namiki M, Shumaker R. Clinical pharmacokinetic and pharmacodynamic profile of lenvatinib, an orally active, small-molecule, multitargeted tyrosine kinase inhibitor. Eur J Drug Metab Pharmacokinet (2017) 42(6):903–14. doi: 10.1007/s13318-017-0403-4
88. Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer (2008) 122(3):664–71. doi: 10.1002/ijc.23131
89. Glen H, Mason S, Patel H, Macleod K, Brunton VG. E7080, a multi-targeted tyrosine kinase inhibitor suppresses tumor cell migration and invasion. BMC Cancer (2011) 11:309. doi: 10.1186/1471-2407-11-309
90. Le Cesne A, Cropet C, Gautier J, Gagne S, Francourt K, Remir E, et al. LENVAGIST - a multicenter, comparative, placebo-controlled, double-blinded, phase II study of the efficacy of lenvatinib in patients with locally advanced or metastatic GIST after failure of imatinib and sunitinib. J Clin Oncol 2020; (2020) 38(suppl 15):11568. doi: 10.1200/JCO.2020.38.15_suppl.TPS11568
91. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer (2005) 5(10):761–72. doi: 10.1038/nrc1716
92. Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res (2006) 66(18):9153–61. doi: 10.1158/0008-5472.CAN-06-0165
93. Doi T, Kurokawa Y, Sawaki A, Komatsu Y, Ozaka M, Takahashi T, et al. Efficacy and safety of TAS-116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastrointestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: a phase II, single-arm trial. Eur J Cancer (2019) 121:29–39. doi: 10.1016/j.ejca.2019.08.009
94. Honma Y, Kurokawa Y, Sawaki A, Naito Y, Iwagami S, Baba H, et al. Randomized, double-blind, placebo (PL)-controlled, phase III trial of pimitespib (TAS-116), an oral inhibitor of heat shock protein 90 (HSP90), in patients (pts) with advanced gastrointestinal stromal tumor (GIST) refractory to imatinib (IM), sunitinib (SU) and regorafenib (REG. J Clin Oncol (2021) 39(suppl 15):11524. doi: 10.1200/JCO.2021.39.15_suppl.11524
95. Mackay CA, Smit JS, Khan F, Dessai F, Connolly C, Masekela R. Umbilical cord interleukin-6 predicts outcome in very low birthweight infants in a high HIV-burden setting: A prospective cohort study. Arch Dis Child (2020) 105(10):932–7. doi: 10.1136/archdischild-2019-318665
96. Banks E, Grondine M, Bhavsar D, Barry E, Kettle JG, Reddy VP, et al. Discovery and pharmacological characterization of AZD3229, a potent KIT/PDGFRalpha inhibitor for treatment of gastrointestinal stromal tumors. Sci Transl Med (2020) 12(541):eaaz2481. doi: 10.1126/scitranslmed.aaz2481
97. Allander SV, Nupponen NN, Ringner M, Hostetter G, Maher GW, Goldberger N, et al. Gastrointestinal stromal tumors with KIT mutations exhibit a remarkably homogeneous gene expression profile. Cancer Res (2001) 61(24):8624–8.
98. Iida K, Abdelhamid Ahmed AH, Nagatsuma AK, Shibutani T, Yasuda S, Kitamura M, et al. Identification and therapeutic targeting of GPR20, selectively expressed in gastrointestinal stromal tumors, with DS-6157a, a first-in-Class antibody-drug conjugate. Cancer Discovery (2021) 11(6):1508–23. doi: 10.1158/2159-8290.CD-20-1434
99. Serrano C, Leal A, Kuang Y, Morgan JA, Barysauskas CM, Phallen J, et al. Phase I study of rapid alternation of sunitinib and regorafenib for the treatment of tyrosine kinase inhibitor refractory gastrointestinal stromal tumors. Clin Cancer Res (2019) 25(24):7287–93. doi: 10.1158/1078-0432.CCR-19-2150
100. Wagner AJ, Severson PL, Shields AF, Patnaik A, Chugh R, Tinoco G, et al. Association of combination of conformation-specific KIT inhibitors with clinical benefit in patients with refractory gastrointestinal stromal tumors: A phase 1b/2a nonrandomized clinical trial. JAMA Oncol (2021) 7(9):1343–50. doi: 10.1001/jamaoncol.2021.2086
101. Gelderblom H, Jones RL, George S, Valverde Morales C, Benson C, Jean-Yves B, et al. Imatinib in combination with phosphoinositol kinase inhibitor buparlisib in patients with gastrointestinal stromal tumour who failed prior therapy with imatinib and sunitinib: a phase 1b, multicentre study. Br J Cancer (2020) 122(8):1158–65. doi: 10.1038/s41416-020-0769-y
102. A dose-finding study of a combination of imatinib and BYL719 in the treatment of 3rd line GIST patients. NCT01735968.
103. Li F, Huynh H, Li X, Ruddy DA, Wang Y, Ong R, et al. FGFR-mediated reactivation of MAPK signaling attenuates antitumor effects of imatinib in gastrointestinal stromal tumors. Cancer Discov (2015) 5(4):438–51. doi: 10.1158/2159-8290.CD-14-0763
104. Cohen NA, Zeng S, Seifert AM, Kim TS, Sorenson EC, Greer JB, et al. Pharmacological inhibition of KIT activates MET signaling in gastrointestinal stromal tumors. Cancer Res (2015) 75(10):2061–70. doi: 10.1158/0008-5472.CAN-14-2564
105. Asmussen J, Lasater EA, Tajon C, Oses-Prieto J, Jun YW, Taylor BS, et al. MEK-dependent negative feedback underlies BCR-ABL-mediated oncogene addiction. Cancer Discov (2014) 4(2):200–15. doi: 10.1158/2159-8290.CD-13-0235
106. Rosenbaum E, Kelly C, D'Angelo SP, Dickson MA, Gounder M, Keohan ML, et al. A phase I study of binimetinib (MEK162) combined with pexidartinib (PLX3397) in patients with advanced gastrointestinal stromal tumor. Oncologist (2019) 24(10):1309–e983. doi: 10.1634/theoncologist.2019-0418
107. Chi P, Qin LX, Nguyen B, Kelly CM, D'Angelo SP, Dickson MA, et al. Phase II trial of imatinib plus binimetinib in patients with treatment-naive advanced gastrointestinal stromal tumor. J Clin Oncol (2022) 40(9):997–1008. doi: 10.1200/JCO.21.02029
108. Cousin S, Cantarel C, Guegan JP, Mazard T, Gomez-Roca C, Metges JP, et al. Regorafenib-avelumab combination in patients with biliary tract cancer (REGOMUNE): a single-arm, open-label, phase II trial. Eur J Cancer (2022) 162:161–9. doi: 10.1016/j.ejca.2021.11.012
109. Cousin S, Cantarel C, Guegan JP, Gomez-Roca C, Metges JP, Adenis A, et al. Regorafenib-avelumab combination in patients with microsatellite stable colorectal cancer (REGOMUNE): A single-arm, open-label, phase II trial. Clin Cancer Res (2021) 27(8):2139–47. doi: 10.1158/1078-0432.CCR-20-3416
110. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (1999) 284(5415):770–6. doi: 10.1126/science.284.5415.770
111. Koch U, Radtke F. Notch and cancer: a double-edged sword. Cell Mol Life Sci (2007) 64(21):2746–62. doi: 10.1007/s00018-007-7164-1
112. Guo X, Jo VY, Mills AM, Zhu SX, Lee CH, Espinosa I, et al. Clinically relevant molecular subtypes in leiomyosarcoma. Clin Cancer Res (2015) 21(15):3501–11. doi: 10.1158/1078-0432.CCR-14-3141
113. Dumont AG, Yang Y, Reynoso D, Katz D, Trent JC, Hughes DP. Anti-tumor effects of the notch pathway in gastrointestinal stromal tumors. Carcinogenesis (2012) 33(9):1674–83. doi: 10.1093/carcin/bgs221
114. Mir O, Azaro A, Merchan J, Chugh R, Trent J, Rodon J, et al. Notch pathway inhibition with LY3039478 in soft tissue sarcoma and gastrointestinal stromal tumours. Eur J Cancer (2018) 103:88–97. doi: 10.1016/j.ejca.2018.08.012
115. Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene (2007) 26(54):7560–8. doi: 10.1038/sj.onc.1210558
116. Feng Y, Yao S, Pu Z, Cheng H, Fei B, Zou J, et al. Identification of new tumor-related gene mutations in Chinese gastrointestinal stromal tumors. Front Cell Dev Biol (2021) 9:764275. doi: 10.3389/fcell.2021.764275
117. Duan Y, Haybaeck J, Yang Z. Therapeutic potential of PI3K/AKT/mTOR pathway in gastrointestinal stromal tumors: Rationale and progress. Cancers (Basel) (2020) 12(10):2972. doi: 10.3390/cancers12102972
118. Schoffski P, Reichardt P, Blay JY, Dumez H, Morgan JA, Ray-Coquard I, et al. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol (2010) 21(10):1990–8. doi: 10.1093/annonc/mdq076
119. Piovesan C, Fumagalli E, Coco P, Palassini E, Dileo P, Morosi C, et al. Response to sirolimus in combination to tirosine kinase inhibitors (TKI) in three cases of PDGFRA-D842V metastatic gastrointestinal stromal tumor (GIST). J Clin Oncol (2009) 27(1):10565. doi: 10.1200/jco.2009.27.15_suppl.10565
120. Sun X, Sun J, Yuan W, Gao X, Fu M, Xue A, et al. Immune cell infiltration and the expression of PD-1 and PD-L1 in primary PDGFRA-mutant gastrointestinal stromal tumors. J Gastrointest. Surg (2021) 25(8):2091–100. doi: 10.1007/s11605-020-04860-8
121. Vitiello GA, Bowler TG, Liu M, Medina BD, Zhang JQ, Param NJ, et al. Differential immune profiles distinguish the mutational subtypes of gastrointestinal stromal tumor. J Clin Invest (2019) 129(5):1863–77. doi: 10.1172/JCI124108
122. Singh AS, Hecht JR, Rosen L, Wainberg ZA, Wang X, Douek M, et al. A randomized phase II study of nivolumab monotherapy or nivolumab combined with ipilimumab in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res (2022) 28(1):84–94. doi: 10.1158/1078-0432.CCR-21-0878
123. Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: A phase 2 clinical trial. JAMA Oncol (2018) 4(1):93–7. doi: 10.1001/jamaoncol.2017.1617
124. Ravegnini G, Sammarini G, Serrano C, Nannini M, Pantaleo MA, Hrelia P, et al. Clinical relevance of circulating molecules in cancer: focus on gastrointestinal stromal tumors. Ther Adv Med Oncol (2019) 11:1758835919831902. doi: 10.1177/1758835919831902
125. Serrano C, George S. Gastrointestinal stromal tumor: Challenges and opportunities for a new decade. Clin Cancer Res (2020) 26(19):5078–85. doi: 10.1158/1078-0432.CCR-20-1706
Keywords: PDGFRA mutation, targeted therapy, avapritinib, ripretinib, crenolanib, gastrointestinal stromal tumors (GIST)
Citation: Sun Y, Yue L, Xu P and Hu W (2022) An overview of agents and treatments for PDGFRA-mutated gastrointestinal stromal tumors. Front. Oncol. 12:927587. doi: 10.3389/fonc.2022.927587
Received: 24 April 2022; Accepted: 15 August 2022;
Published: 31 August 2022.
Edited by:
Francesca Megiorni, Sapienza University of Rome, ItalyReviewed by:
Judy Sue Crabtree, Louisiana State University, United StatesGiuseppe Badalamenti, University of Palermo, Italy
Copyright © 2022 Sun, Yue, Xu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiling Hu, aHV3ZWlsaW5nQHpqdS5lZHUuY24=