 Siddharth Mehra
Siddharth Mehra Samara Singh
Samara Singh Nagaraj Nagathihalli
Nagaraj Nagathihalli- 1Division of Surgical Oncology, Department of Surgery, University of Miami Miller School of Medicine, Miami, FL, United States
- 2Sylvester Comprehensive Cancer Center, University of Miami, Miami, FL, United States
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive solid malignancy with a high rate of metastasis and therapeutic resistance as its major hallmarks. Although a defining mutational event in pancreatic cancer initiation is the presence of oncogenic KRAS, more advanced PDAC lesions accumulate additional genomic alterations, including loss of tumor suppressor gene TP53. Co-occurrence of mutant KRAS and TP53 in PDAC promotes hyperactivation of cancer cell signaling pathways driving epithelial to mesenchymal plasticity (EMP). The cellular process of EMP influences the biological behavior of cancer cells by increasing their migratory and invasive properties, thus promoting metastasis. Our previous work has demonstrated that oncogenic KRAS-mediated activation of cyclic AMP response element-binding protein 1 (CREB) is one of the critical drivers of PDAC aggressiveness. The therapeutic approach of targeting this key transcription factor attenuates tumor burden in genetically engineered mouse models (GEMMs) of this disease. Herein, we discuss the significant role of CREB in perpetuating disease aggressiveness and therapeutic resistance through the EMP process. Furthermore, this review updates the therapeutic implications of targeting CREB, highlighting the challenges and emerging approaches in PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive solid malignancy associated with significant mortality and is projected to be the second leading cause of cancer-related deaths by 2030 (1, 2). Delayed diagnosis, lack of effective treatments, and high metastatic propensity put this disease in the category of cancers with an extremely poor 5-year survival. PDAC originates from normal pancreatic epithelium transitioning to a neoplastic precursor state known as a pancreatic intraepithelial neoplasm (PanIN), instigating the oncogenic transformation into a ductal adenocarcinoma (3). This gradual progression towards invasive cancer is supported by a unique dependency on the mutated KRAS oncogene, prevalent in more than 90% of PDAC patients. Other than the presence of KRAS, inactivating mutation in TP53 also co-occur in more than 70% of PDAC patients. The underlying cooperativity between these two key dominant oncogenic drivers promotes PDAC progression and contributes to metastatic dissemination of this disease (4–7). Therefore, a major unmet need is to understand the cellular mechanisms responsible for promoting disease aggressiveness and to further identify actionable therapeutic strategies to improve the prognosis of this malignancy (8).
The presence of dominant genomic alterations modulates several oncogenic cellular signaling events in PDAC, which enables tumor cells to exhibit a distinctive cellular plasticity, enabling them to transform into an invasive and migratory mesenchymal phenotype, in a process known as epithelial to mesenchymal transition (EMT) and subsequent reversal from mesenchymal to epithelial transition (MET). These two biological phenomena give rise to the concept of epithelial-mesenchymal plasticity (EMP) (Figure 1). This cellular transformation of EMP in cancer cells leads to the disruption of tissue homeostasis and facilitates crosstalk between different stromal components within the tumor microenvironment (TME), contributing to intratumoral heterogeneity (9).
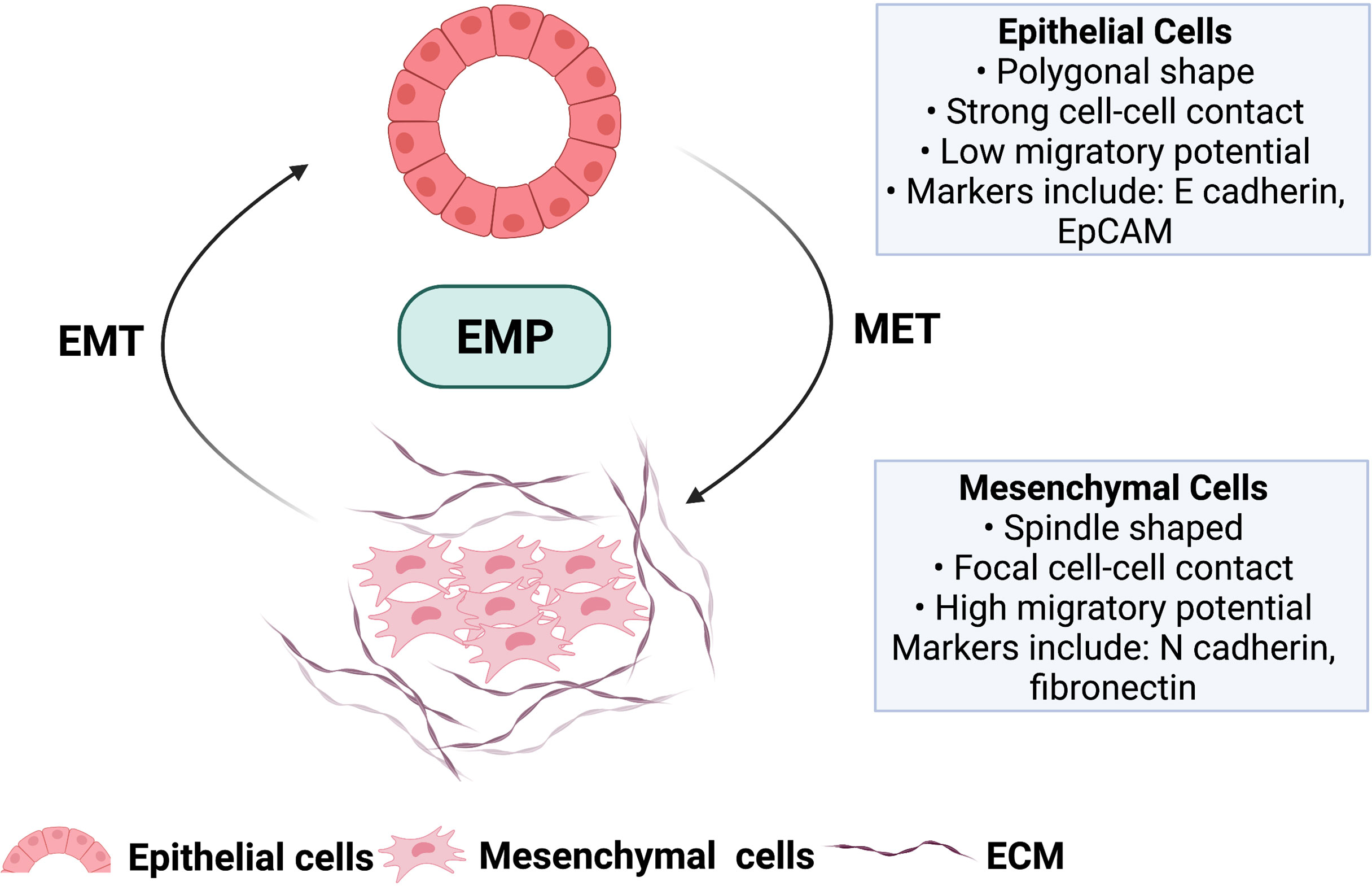
Figure 1 Hallmark features of malignant epithelial cells undergoing forward differentiation towards mesenchymal cell type (EMT) and the reversal mesenchymal to epithelial transition (MET). These two biological processes give rise to the concept of epithelial to mesenchymal plasticity (EMP) involved in tumor progression and metastasis. ECM; Extracellular matrix. Image created with BioRender.com (Agreement number TL23SM1HJO).
Cyclic AMP response element-binding protein 1 (CREB) is a transcriptional coactivator that has been shown to be activated downstream of Ras-dependent oncogenic signaling pathways (10, 11). Once activated through phosphorylation at Ser133, CREB binds to its coactivator, the CREB-binding protein (CBP), enabling the recruitment of additional transcriptional machinery elements necessary to drive transcriptional programs of malignant progression (12). The present review will focus on the various mechanisms of CREB-dependent EMP, downstream of mutant KRAS in PDAC and other cancer types. Additionally, the therapeutic potential and challenges of targeting CREB in the attenuation of cellular plasticity and overcoming drug resistance will be examined. Overall, identifying novel molecular targets to attenuate the cellular process of EMP may improve clinical outcomes in patients with PDAC.
EMP and Metastasis in PDAC
Mortalities associated with PDAC do not occur due to the primary tumor but are often found to be associated with the metastatic dissemination of tumor cells, which begins early in most patients (2, 13). More than 90% of the patients diagnosed display local or distant metastatic disease. This rapid progression associated with this malignancy warrants new studies on the key cellular processes driving this metastatic behavior (8). Mechanistically, PDAC cells exhibit the unique hallmark of EMT, displaying higher invasive characteristics, cancer stem cell-like behavior, and greater resistance to therapies (14). During this forward differentiation process, they lose the expression of epithelial markers (including E cadherin, occludin, claudin, and laminin) and gain mesenchymal phenotypic plasticity with elevated expression of markers such as N-cadherin, vimentin, and fibronectin (13, 15, 16). These changes are often associated with the activation of EMT-driven transcriptional programs led by transcription factors such as TWIST, SNAIL, and ZEB, which coordinatively repress E-cadherin levels while promoting expression of mesenchymal differentiation markers. Once these invading cancer cells colonize the metastatic sites, they undergo the reverse EMT process known as MET, which helps them to adopt a high proliferation rate in the invaded TME (17–19). Taken together, these two distinct but related cellular programs- EMT, and the reversal mesenchymal-epithelial transition MET, play a significant role in PDAC tumorigenesis. EMP mediated cellular plasticity allows PDAC tumor cells to detach and migrate from their site of origin (invasion) and gain access to lymphatic blood vasculature and distant sites (extravasation), to form metastases (20). Therefore, understanding the molecular regulation of these steps can help to elucidate therapeutic options to restrict EMP-mediated tumor metastases in PDAC.
RAS/TP53 and EMP in PDAC
In mutant KRAS/TP53-driven tumors, including PDAC, these cellular plasticity programs and tumorigenesis are interconnected; this was established using genetically engineered mouse models (GEMMs) of PDAC, where tumor cells harboring these oncogenic mutation displayed EMT like features at an early stage after tumor initiation (13). Additionally, the presence of soluble ligands, including growth factors mediating activation of receptor tyrosine kinases (RTKs), also drives EMP programs in PDAC in a RAS-dependent manner (21, 22). Shedding of EGFR ligands, including amphiregulin from PDAC cells, results in an autocrine feedback loop to further promote the KRAS hyperactivation (21–23). Once activated, signaling networks downstream of KRAS, including MEK/ERK and PI3K/AKT, can promote EMP. Targeted inhibition of these downstream effector kinases has been shown to reverse KRAS-mediated epithelial plasticity (24). Transcriptomics-based gene set enrichment analysis (GSEA) of highly metastatic cell lines derived from KPC (LSL-KrasG12D/+;Trp53R273H/+;Pdx1Cre/+) GEMM of PDAC identified significant enrichment of KRAS-dependent gene signatures compared to cell clones with low metastatic potential (25). Several studies over recent years have established the vast contribution of various molecular regulators and downstream KRAS effectors to perpetuate PDAC aggressiveness. Direct targeting of KRAS has largely been unsuccessful; therefore, efforts have now shifted to targeting its downstream effectors (26–28), providing opportunities for novel therapeutic interventions in PDAC.
CREB Activation in PDAC
Targeting individual KRAS-mediated factors may provide therapeutic insights and understanding the cooperative interactions between different transcriptional and signaling networks remains an essential need to target the heterogenous tumor cell population of KRAS-driven PDAC effectively. With long-standing efforts of our laboratory in deciphering the molecular underpinnings of KRAS-mediated signaling pathways in the pathogenesis of PDAC, our work has demonstrated that oncogenic KRAS activates a master transcription regulator CREB (11). We have uncovered its role as a critical driver of PDAC aggressiveness, and its overexpression is associated with poor prognosis in the patients. Activation of CREB is mediated through MEK and AKT-dependent signaling pathways in KRAS mutant PDAC (Figure 2) (11). Additional work from our lab by Srinivasan et al. further illuminated GM-CSF-mediated CREB activation as a critical driver of the development and progression of smoking-associated PDAC tumorigenesis to promote disease aggressiveness. Therapeutic targeting of CREB significantly attenuated tumor burden in our PDAC disease model (29). Taken together, it is vital to understand CREB’s function and its role in EMP regulation in this disease.
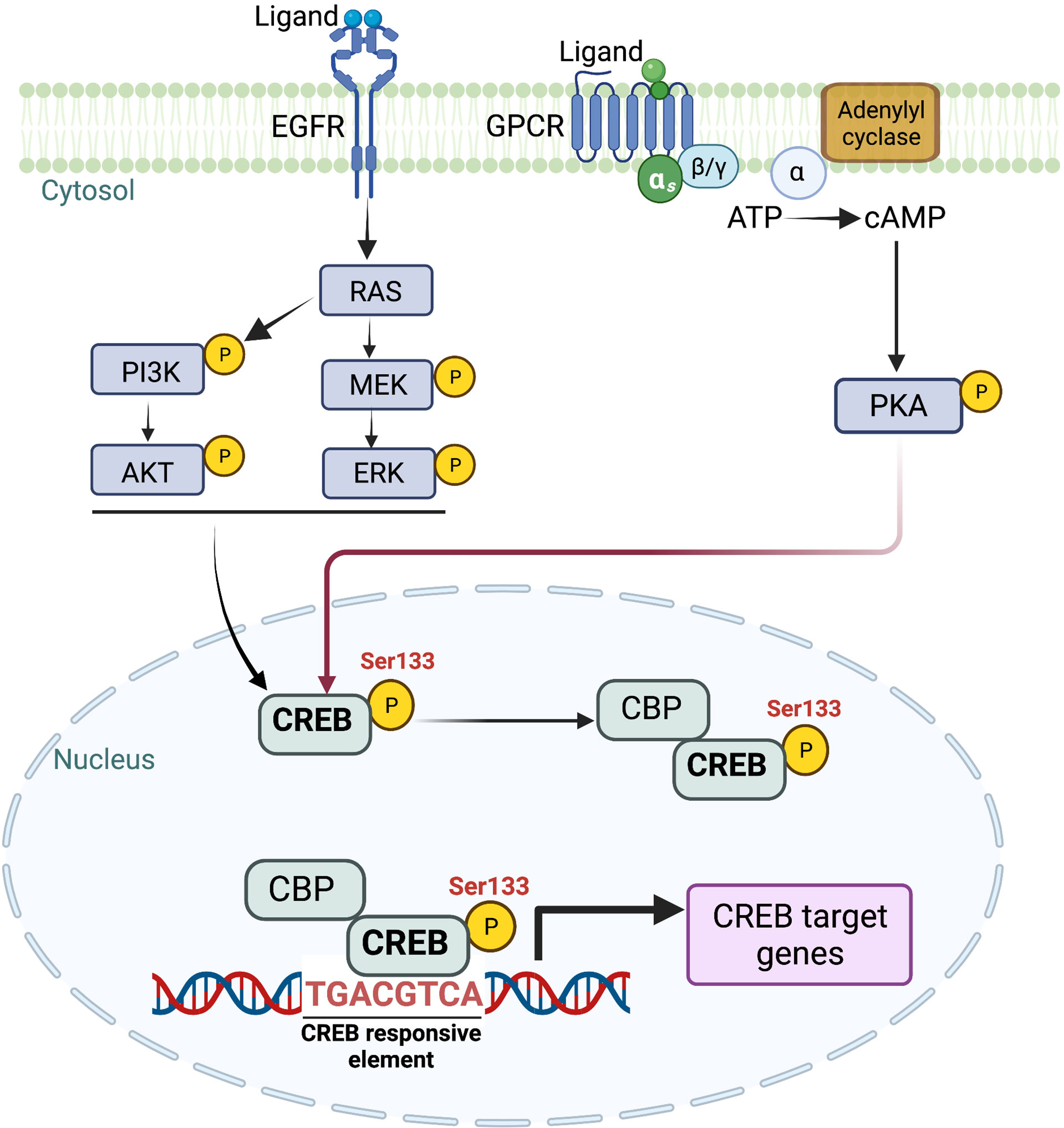
Figure 2 Schematic representation of multiple upstream signaling cascades involved in intracellular CREB activation. CREB, once phosphorylated at Ser 133, translocates to the nucleus. Once translocated, CREB binds to CRE (cAMP response element) and provides transcriptional ability to activate multiple downstream targets. Image created with BioRender.com (Agreement number ZW23SM1LR3).
CREB Structure and Function
CREB1 was the first reported basic leucine zipper family transcription factor whose activity was shown to be regulated by auto-phosphorylation. This was found to be a shared feature among its family members, including activating transcription factor (ATF1) and cAMP response element modulator (CREM) (30). CREB, once translated into protein, constitutes 341 amino acids in length, forming a 37 kDa transcription factor (31). The protein structure of CREB consists of an NH2-terminal activation domain, COOH-terminal basic region/leucine zipper domain (bZIP), DNA-binding, and dimerization domain (32). The primary structure (Figure 3) of the CREB has a centrally located 60-amino-acid kinase-inducible domain (KID), which comprises multiple phosphorylation sites. Hydrophobic glutamine-rich domains (Q1 and Q2) are present on either side of the KID. Q1 is a basal transcriptional activation domain involved in the interaction of CREB with TATA-binding protein to regulate gene expression. In contrast, the other Q2 domain is responsible for interaction with RNA polymerase II transcriptional initiation complex leading to recognition and binding of CREB to its canonical CREB responsible element (CRE) sequence, 5’- TGACGTCA-3’ (32, 33). Furthermore, the bZIP dimerization domain at the carboxy-terminal is involved in its dimerization and is required for CREB binding to DNA regulatory sequences (34).
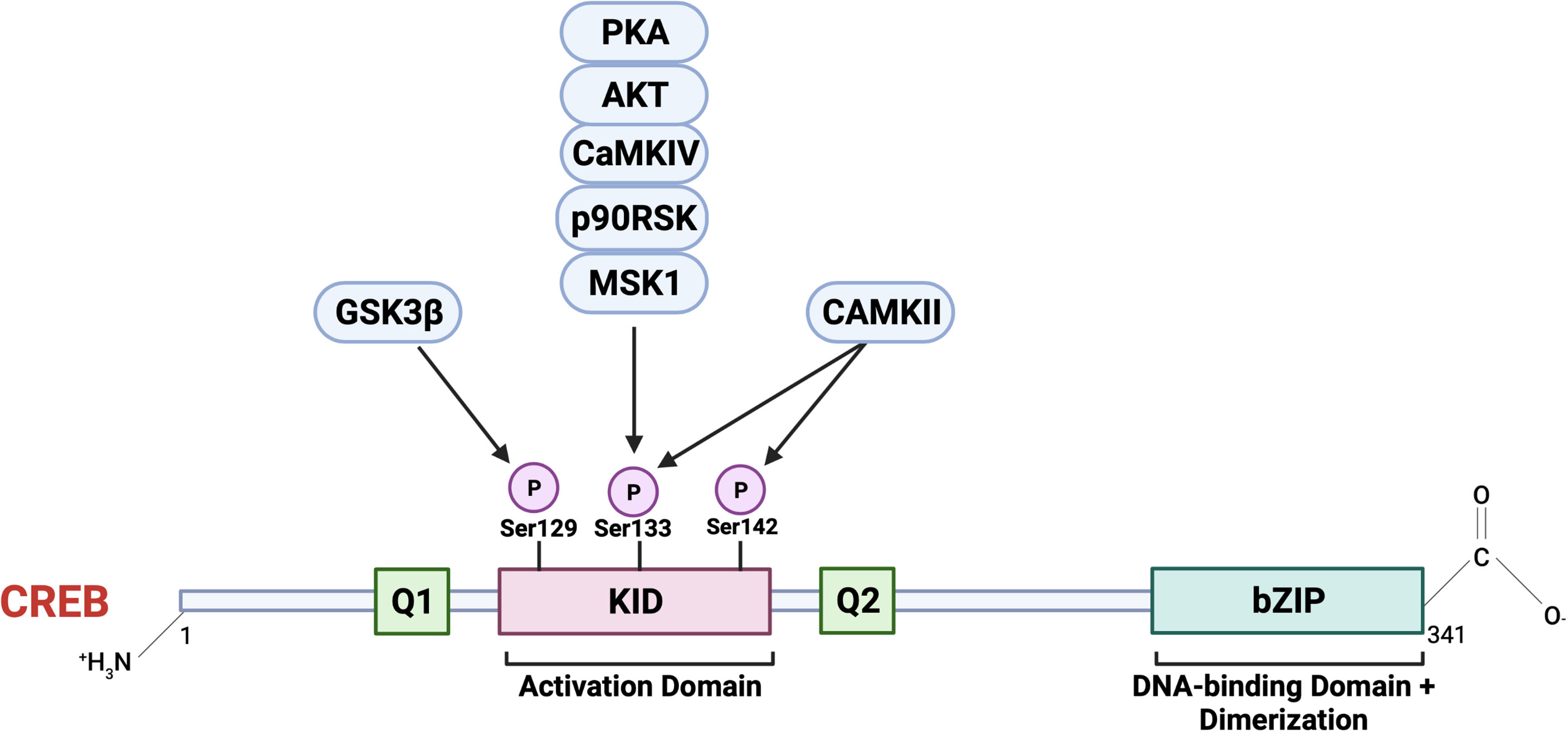
Figure 3 The primary structure of CREB consists of an N terminal containing an activation domain and a smaller C terminal with a basic region/leucine zipper (bZIP) DNA-binding and dimerization domain. CREB also contains kinase inducible domain (KID) and hydrophobic glutamate-rich domains (Q1 and Q2). A critical event involving CREB activation is the phosphorylation at Ser 133 in KID by multiple upstream cellular effector kinases. CREB, once activated, acts as a master transcriptional regulator of multiple downstream cellular targets. Image created with BioRender.com (Agreement number NZ23SM1ZAY).
Activation of CREB involves reversible phosphorylation at numerous serine residues positioned at 129 (S129), 133 (S133), and 142 (S142). This phosphorylation event is triggered via multiple cellular effector kinases in response to growth factors or extracellular stress stimuli (35, 36). Some of the critical upstream activators involved in CREB phosphorylation, and its activation include protein kinase A (PKA), protein kinase B (PKB/AKT), the mitogen-activated kinase (MAPK), 90 kDa ribosomal S6 kinase (RSK), AKT, protein kinase C (PKC), calcium/calmodulin-dependent protein kinase II (CaMKII) (10, 35–37). The significance of phosphorylation at the position Ser 133 is extensively studied within the context of CREB activation and mediating its downstream effects on target genes. In response to intracellular signals, including cAMP, induces recruitment of CBP, a transcriptional co-activator, and its paralogue p300 (37), together this complex is active and promotes nuclear gene expression via targeting various promoter regions containing the highly conserved cAMP-responsive element (CRE) TGACGTCA (38–40). Some CREB target genes contain CRE and half-CRE elements (TGACG/CGTCA), and binding is dependent on the TATA box in their promoters. Out of the 4000 identified promoter regions with a predicted solid CREB binding affinity, only around 339 genes contain complete CRE and TATA boxes, and less than 100 genes have been validated as direct CREB targets in a cAMP-responsive manner (41). This suggests the role of CREB as a transcriptional regulator independent of the presence of CRE.
With the advancements of high throughput technologies in the field of cancer signaling and therapeutics, compelling scientific evidence demonstrates that CREB and CREB-regulated gene targets play an essential role in promoting tumor initiation, progression, and aggressiveness, implicating CREB as a critical transcription factor with proto-oncogene characteristics across multiple cancer types (42–47). In addition the role of CREB in the regulating protein-coding genes involved in tumorigenesis, several genomic-wide studies have also identified CREB-dependent regulation of non-coding genes (microRNAs), specifically within the context of EMP mediated tumor growth and aggressiveness.
CREB-microRNA(miRNAs) and EMP
The cellular process of EMP during carcinogenesis is regulated by activating multiple transcription factors, including TWIST, SNAIL, and ZEB1, which are involved in modulating the expression of several cell adhesion and tight junction proteins (48). Studies highlighting the direct involvement of CREB in regulating the expression of genes involved in epithelial cell plasticity are limited. However, it still plays a pivotal role in regulating an EMP program by interacting with multiple microRNAs (miRNAs). miRNAs are small non-coding RNAs constituted of 18-22 nucleotides and are involved in regulating gene expression by binding to the 3’UTR of their target mRNA transcript (49–51). Mounting scientific evidence recently suggests that CREB transcriptionally regulates the expression of multiple miRNAs; additionally, CREB expression itself can also be modulated by miRNAs, thus forming a feedback loop.
The CREB-miRNA axis has been found to play a crucial role in influencing the tumorigenic potential of cancer cells as well as in EMP-mediated metastasis. Within the context of PDAC, Zhang et al. demonstrated that intracellular activation of zinc-dependent transcription factor (ZIP-4) promotes CREB activation, which upregulates transcription of miRNA-373 to influence PDAC tumor growth both in vitro and in vivo (52). Additionally, in bladder cancer, CREB regulation by miRNA-433 alters the EMT potential of tumor cells by targeting the c-Met/AKT/GSK-3β/SNAIL signaling pathway (53). The strong association of the CREB transcription factor promoting miRNAs in the development and progression of gastric cancer metastasis was reported by Liu et al., where overexpression of CREB was associated with the loss of the tumor-suppressive mir-520b/GATA6 signaling axis, thereby promoting migration and metastasis of gastric cancer cells (54). Additionally, in a recent study describing the regulation of gene expression by profiling miRNAs expression and transcription factors CREB emerged as a master regulator in multiple cancer types (55). Given the critical role of the CREB-miRNA network in influencing tumor progression by inducing transcriptional and post-transcriptional changes through multiple cellular mechanisms. It becomes imperative to comprehensively understand and utilize regulatory network information involving CREB-miRNA to design novel therapeutic strategies across several human malignancies.
Therapeutic Targeting of CREB With Possible Implications in EMP
Given the critical role of CREB as a proto-oncogene involved in tumor initiation, progression, and metastasis, therapeutic targeting of this key transcription factor has achieved success in preclinical studies of PDAC (29). The recent findings by Kim et al. describe that activation of CREB downstream to KRAS signaling led to physical interaction with oncogenic mutant p53 (56). This interaction, in turn, activated multiple pro-tumor transcriptional programs, including FOXA1, promoting PDAC metastasis via the activation of the downstream WNT/β-catenin signaling axis (56). Additionally, pharmacologic inhibition using a CREB inhibitor (666–15) significantly attenuated PDAC metastasis in vivo (56), highlighting the essential role of CREB in the pathogenesis of this disease
Additional studies have established the involvement of the Wnt/β-Catenin signaling pathway as a major culprit of pancreatic tumorigenesis and therapeutic resistance. Wnt ligands act through autocrine or paracrine manners to bind to cognate receptors, thereby initiating a phosphorylation cascade. This permits dissociation of β-catenin degradation complex, allowing for translocation of β-catenin across the nuclear membrane (57, 58). Once inside the nucleus, β-catenin further regulates the expression of target genes, including the cAMP response element-binding protein (CBP, CREB binding protein). Similarly, Arensman et al. demonstrated that the small molecule ICG-001 binds cAMP-responsive element-binding (CREB)-binding protein (CBP) to disrupt its interaction with β-catenin and inhibit CBP function as a coactivator of Wnt/β-catenin–mediated transcription. Treatment with this inhibitor significantly improved overall survival in an in-vivo orthotopic xenograft model of PDAC, further establishing that disruption of CBP activity impacts PDAC tumor burden (59).
EMP in PDAC is mediated through multiple signaling pathways, among which TGF-β signaling has been shown to be the most prominent cellular pathway (60, 61). Previous studies have shown the correlation of TGF-β overexpression with poor prognosis in PDAC patients and directly associated with promoting tumor cell proliferation and invasion (62). Although it is currently unclear how CREB signaling regulates EMP in PDAC, previous studies have suggested that CREB signaling influences TGF-β signaling in pancreatic cancer cells and fibroblasts. Along with E1A binding protein, EP300, CREB can influence the EMP by regulating the E-cadherin expression (63). There are studies that established the association of CREB activity as an essential driver of tumor aggressiveness; however, there is a lack of molecular and cellular evidence exploring the regulation and activation of CREB as well as its role in influencing the EMP transcriptional program, warranting future investigations of these mechanisms in PDAC. Therefore, there are many remaining questions about the mechanisms through which CREB functions, and elucidation of this critical transcriptional regulator may have significant implications for the success of targeting strategies.
Conclusions and Future Perspectives
The prognosis of patients with pancreatic cancer has not improved notably despite considerable research efforts. The complex interplay of signaling pathways driving EMP programs promoting therapeutic resistance and local and distal recurrence presents a significant obstacle to the current treatment regimen for this disease. Therefore, identifying new candidate molecules, such as CREB, responsible for these aberrant cellular processes, is critical for targeted therapies against the tumor heterogeneity in PDAC. Future investigations using in vivo manipulation of CREB expression in PDAC to better recapitulate the spontaneity and heterogeneity of human tumors may provide more robust scientific evidence of its role in regulating EMP, metastasis, and therapeutic resistance in PDAC. Importantly, understanding how transcription factors regulate EMP is still an area of intense study with emerging therapeutically relevant insights.
Author Contributions
SM, SS, and NN were involved in the initial review design and composition. SM, SS, and NN contributed to the manuscript content and format. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the American Cancer Society IRG 98-277-13, NIH NCI R03 CA249401, and James Esther and King Biomedical Research Program by Florida Department of Health grant 22K06 to NN.
Author Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to acknowledge Dr. Christine Isabelle Rafie and Dr. Chandrashekar Joshi for providing their critical insights and stimulating discussions during the preparation of this manuscript.
References
1. Miller KD, Goding Sauer A, Ortiz AP, Fedewa SA, Pinheiro PS, Tortolero-Luna G, et al. Cancer Statistics for Hispanics/Latinos, 2018. CA: A Cancer J Clin (2018) 68(6):425–45. doi: 10.3322/caac.21494
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res (2014) 74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155
3. Aichler M, Seiler C, Tost M, Siveke J, Mazur PK, Da Silva-Buttkus P, et al. Origin of Pancreatic Ductal Adenocarcinoma From Atypical Flat Lesions: A Comparative Study in Transgenic Mice and Human Tissues. J Pathol (2012) 226(5):723–34. doi: 10.1002/path.3017
4. di Magliano MP, Logsdon CD. Roles for KRAS in Pancreatic Tumor Development and Progression. Gastroenterology (2013) 144(6):1220–9. doi: 10.1053/j.gastro.2013.01.071
5. Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. Selective Requirement of PI3K/PDK1 Signaling for Kras Oncogene-Driven Pancreatic Cell Plasticity and Cancer. Cancer Cell (2013) 23(3):406–20. doi: 10.1016/j.ccr.2013.01.023
6. Connor AA, Denroche RE, Jang GH, Lemire M, Zhang A, Chan-Seng-Yue M, et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell (2019) 35(2):267–82.e7. doi: 10.1016/j.ccell.2018.12.010
7. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell (2005) 7(5):469–83. doi: 10.1016/j.ccr.2005.04.023
8. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA: A Cancer J Clin (2018) 68(1):7–30. doi: 10.3322/caac.21442
9. Arner EN, Du W, Brekken RA. Behind the Wheel of Epithelial Plasticity in KRAS-Driven Cancers. Front Oncol (2019) 9. doi: 10.3389/fonc.2019.01049
10. Johannessen M, Delghandi MP, Moens U. What Turns CREB on? Cell Signalling (2004) 16(11):1211–27. doi: 10.1016/j.cellsig.2004.05.001
11. Castellanos J, Srinivasan S, Honnenahally K, Shi C, VanSaun M, Robbins D, et al. Abstract 2618: Oncogenic Mutant KRAS Modulates CREB Activation Through MEK-ERK and AKT Signaling in Pancreatic Cancer. Cancer Res (2016) 76(14 Supplement):2618. doi: 10.1158/1538-7445.AM2016-2618
12. Shaywitz AJ, Greenberg ME. CREB: A Stimulus-Induced Transcription Factor Activated by a Diverse Array of Extracellular Signals. Annu Rev Biochem (1999) 68:821–61. doi: 10.1146/annurev.biochem.68.1.821
13. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell (2012) 148(1-2):349–61. doi: 10.1016/j.cell.2011.11.025
14. Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell (2019) 49(3):361–74. doi: 10.1016/j.devcel.2019.04.010
15. Lamouille S, Xu J, Derynck R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi: 10.1038/nrm3758
16. Kalluri R, Weinberg RA. The Basics of Epithelial-Mesenchymal Transition. J Clin Invest (2009) 119(6):1420–8. doi: 10.1172/JCI39104
17. Pitarresi JR, Rustgi AK. Mechanisms Underlying Metastatic Pancreatic Cancer. Adv Exp Med Biol (2019) 1164:3–10. doi: 10.1007/978-3-030-22254-3_1
18. Lawlor RT, Veronese N, Nottegar A, Malleo G, Smith L, Demurtas J, et al. Prognostic Role of High-Grade Tumor Budding in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis With a Focus on Epithelial to Mesenchymal Transition. Cancers (Basel) (2019) 11(1):113. doi: 10.3390/cancers11010113
19. Pelosi E, Castelli G, Testa U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines (2017) 5(4):65. doi: 10.3390/biomedicines5040065
20. Tsai JH, Yang J. Epithelial-Mesenchymal Plasticity in Carcinoma Metastasis. Genes Dev (2013) 27(20):2192–206. doi: 10.1101/gad.225334.113
21. Chen SJ, Chen YT, Zeng LJ, Zhang QB, Lian GD, Li JJ, et al. Bmi1 Combines With Oncogenic KRAS to Induce Malignant Transformation of Human Pancreatic Duct Cells In Vitro. Tumour Biol (2016) 37(8):11299–309. doi: 10.1007/s13277-016-4840-5
22. Tripathi K, Garg M. Mechanistic Regulation of Epithelial-to-Mesenchymal Transition Through RAS Signaling Pathway and Therapeutic Implications in Human Cancer. J Cell Commun Signal (2018) 12(3):513–27. doi: 10.1007/s12079-017-0441-3
23. Nagathihalli NS, Beesetty Y, Lee W, Washington MK, Chen X, Lockhart AC, et al. Novel Mechanistic Insights Into Ectodomain Shedding of EGFR Ligands Amphiregulin and TGF-α: Impact on Gastrointestinal Cancers Driven by Secondary Bile Acids. Cancer Res (2014) 74(7):2062–72. doi: 10.1158/0008-5472.CAN-13-2329
24. Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, et al. Ras and TGF[beta] Cooperatively Regulate Epithelial Cell Plasticity and Metastasis: Dissection of Ras Signaling Pathways. J Cell Biol (2002) 156(2):299–313. doi: 10.1083/jcb.200109037
25. Genovese G, Carugo A, Tepper J, Robinson FS, Li L, Svelto M, et al. Synthetic Vulnerabilities of Mesenchymal Subpopulations in Pancreatic Cancer. Nature (2017) 542(7641):362–6. doi: 10.1038/nature21064
26. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment of Cancer. Oncogene (2007) 26(22):3291–310. doi: 10.1038/sj.onc.1210422
27. Wicki A, Mandalà M, Massi D, Taverna D, Tang H, Hemmings BA, et al. Acquired Resistance to Clinical Cancer Therapy: A Twist in Physiological Signaling. Physiol Rev (2016) 96(3):805–29. doi: 10.1152/physrev.00024.2015
28. Nagathihalli NS, Castellanos JA, Lamichhane P, Messaggio F, Shi C, Dai X, et al. Inverse Correlation of STAT3 and MEK Signaling Mediates Resistance to RAS Pathway Inhibition in Pancreatic Cancer. Cancer Res (2018) 78(21):6235–46. doi: 10.1158/0008-5472.CAN-18-0634
29. Srinivasan S, Totiger T, Shi C, Castellanos J, Lamichhane P, Dosch AR, et al. Tobacco Carcinogen-Induced Production of GM-CSF Activates CREB to Promote Pancreatic Cancer. Cancer Res (2018) 78(21):6146–58. doi: 10.1158/0008-5472.CAN-18-0579
30. Mayr B, Montminy M. Transcriptional Regulation by the Phosphorylation-Dependent Factor CREB. Nat Rev Mol Cell Biol (2001) 2(8):599–609. doi: 10.1038/35085068
31. Steven A, Seliger B. Control of CREB Expression in Tumors: From Molecular Mechanisms and Signal Transduction Pathways to Therapeutic Target. Oncotarget (2016) 7(23):35454–65. doi: 10.18632/oncotarget.7721
32. Mayr BM, Guzman E, Montminy M. Glutamine Rich and Basic Region/Leucine Zipper (bZIP) Domains Stabilize cAMP-Response Element-Binding Protein (CREB) Binding to Chromatin. J Biol Chem (2005) 280(15):15103–10. doi: 10.1074/jbc.M414144200
33. Schumacher MA, Goodman RH, Brennan RG. The Structure of a CREB bZIP.Somatostatin CRE Complex Reveals the Basis for Selective Dimerization and Divalent Cation-Enhanced DNA Binding. J Biol Chem (2000) 275(45):35242–7. doi: 10.1074/jbc.M007293200
34. Thakur JK, Yadav A, Yadav G. Molecular Recognition by the KIX Domain and Its Role in Gene Regulation. Nucleic Acids Res (2013) 42(4):2112–25. doi: 10.1093/nar/gkt1147
35. Johannessen M, Moens U. Multisite Phosphorylation of the cAMP Response Element-Binding Protein (CREB) by a Diversity of Protein Kinases. Front Biosci (2007) 12:1814–32. doi: 10.2741/2190
36. Naqvi S, Martin KJ, Arthur JS. CREB Phosphorylation at Ser133 Regulates Transcription via Distinct Mechanisms Downstream of cAMP and MAPK Signalling. Biochem J (2014) 458(3):469–79. doi: 10.1042/BJ20131115
37. Wheaton KL, Hansen KF, Aten S, Sullivan KA, Yoon H, Hoyt KR, et al. The Phosphorylation of CREB at Serine 133 Is a Key Event for Circadian Clock Timing and Entrainment in the Suprachiasmatic Nucleus. J Biol Rhythms (2018) 33(5):497–514. doi: 10.1177/0748730418791713
38. Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH. Identification of a Cyclic-AMP-Responsive Element Within the Rat Somatostatin Gene. Proc Natl Acad Sci (1986) 83(18):6682–6. doi: 10.1073/pnas.83.18.6682
39. Comb M, Birnberg NC, Seasholtz A, Herbert E, Goodman HM. A Cyclic AMP- and Phorbol Ester-Inducible DNA Element. Nature (1986) 323(6086):353–6. doi: 10.1038/323353a0
40. Short JM, Wynshaw-Boris A, Short HP, Hanson RW. Characterization of the Phosphoenolpyruvate Carboxykinase (GTP) Promoter-Regulatory Region. II. Identification of cAMP and Glucocorticoid Regulatory Domains. J Biol Chem (1986) 261(21):9721–6.
41. Conkright MD, Guzmán E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-Wide Analysis of CREB Target Genes Reveals A Core Promoter Requirement for cAMP Responsiveness. Mol Cell (2003) 11(4):1101–8. doi: 10.1016/S1097-2765(03)00134-5
42. Sapio L, Salzillo A, Ragone A, Illiano M, Spina A, Naviglio S. Targeting CREB in Cancer Therapy: A Key Candidate or One of Many? An Update. Cancers (Basel) (2020) 12(11):3166. doi: 10.3390/cancers12113166
43. Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A, et al. The Role of CREB as a Proto-Oncogene in Hematopoiesis and in Acute Myeloid Leukemia. Cancer Cell (2005) 7(4):351–62. doi: 10.1016/j.ccr.2005.02.018
44. Xie S, Price JE, Luca M, Jean D, Ronai Z, Bar-Eli M. Dominant-Negative CREB Inhibits Tumor Growth and Metastasis of Human Melanoma Cells. Oncogene (1997) 15(17):2069–75. doi: 10.1038/sj.onc.1201358
45. Kovach SJ, Price JA, Shaw CM, Theodorakis NG, McKillop IH. Role of Cyclic-AMP Responsive Element Binding (CREB) Proteins in Cell Proliferation in a Rat Model of Hepatocellular Carcinoma. J Cell Physiol (2006) 206(2):411–9. doi: 10.1002/jcp.20474
46. Linnerth NM, Greenaway JB, Petrik JJ, Moorehead RA. cAMP Response Element-Binding Protein Is Expressed at High Levels in Human Ovarian Adenocarcinoma and Regulates Ovarian Tumor Cell Proliferation. Int J Gynecol Cancer (2008) 18(6):1248–57. doi: 10.1111/j.1525-1438.2007.01177.x
47. Zheng T, Huang J, Xiang X, Li S, Yu J, Qu K, et al. Systematical Analysis Reveals a Strong Cancer Relevance of CREB1-Regulated Genes. Cancer Cell Int (2021) 21(1):530. doi: 10.1186/s12935-021-02224-z
48. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell (2009) 139(5):871–90. doi: 10.1016/j.cell.2009.11.007
49. Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell (2004) 116(2):281–97. doi: 10.1016/S0092-8674(04)00045-5
50. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray Analysis Shows That Some microRNAs Downregulate Large Numbers of Target mRNAs. Nature (2005) 433(7027):769–73. doi: 10.1038/nature03315
51. Kröger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, et al. Acquisition of a Hybrid E/M State is Essential for Tumorigenicity of Basal Breast Cancer Cells. Proc Natl Acad Sci (2019) 116(15):7353–62. doi: 10.1073/pnas.1812876116
52. Zhang Y, Yang J, Cui X, Chen Y, Zhu VF, Hagan JP, et al. A Novel Epigenetic CREB-miR-373 Axis Mediates ZIP4-Induced Pancreatic Cancer Growth. EMBO Mol Med (2013) 5(9):1322–34. doi: 10.1002/emmm.201302507
53. Xu X, Zhu Y, Liang Z, Li S, Xu X, Wang X, et al. C-Met and CREB1 are Involved in miR-433-Mediated Inhibition of the Epithelial–Mesenchymal Transition in Bladder Cancer by Regulating Akt/GSK-3β/Snail Signaling. Cell Death Dis (2016) 7(2):e2088. doi: 10.1038/cddis.2015.274
54. Liu H, Du F, Sun L, Wu Q, Wu J, Tong M, et al. GATA6 Suppresses Migration and Metastasis by Regulating the miR-520b/CREB1 Axis in Gastric Cancer. Cell Death Disease (2019) 10(2):35. doi: 10.1038/s41419-018-1270-x
55. Yan Z, Shah PK, Amin SB, Samur MK, Huang N, Wang X, et al. Integrative Analysis of Gene and miRNA Expression Profiles With Transcription Factor–miRNA Feed-Forward Loops Identifies Regulators in Human Cancers. Nucleic Acids Res (2012) 40(17):e135. doi: 10.1093/nar/gks395
56. Kim MP, Li X, Deng J, Zhang Y, Dai B, Allton KL, et al. Oncogenic KRAS Recruits an Expansive Transcriptional Network Through Mutant P53 to Drive Pancreatic Cancer Metastasis. Cancer Discov (2021) 11(8):2094–111. doi: 10.1158/2159-8290.CD-20-1228
57. Ram Makena M, Gatla H, Verlekar D, Sukhavasi S, KP M. And K CP. Wnt/β-Catenin Signaling: The Culprit in Pancreatic Carcinogenesis and Therapeutic Resistance. Int J Mol Sci (2019) 20(17):4242. doi: 10.3390/ijms20174242
58. Jeong WJ, Ro EJ, Choi KY. Interaction Between Wnt/β-Catenin and RAS-ERK Pathways and an Anti-Cancer Strategy via Degradations of β-Catenin and RAS by Targeting the Wnt/β-Catenin Pathway. NPJ Precis Oncol (2018) 2(1):5. doi: 10.1038/s41698-018-0049-y
59. Arensman MD, Telesca D, Lay AR, Kershaw KM, Wu N, Donahue TR, et al. The CREB-Binding Protein Inhibitor ICG-001 Suppresses Pancreatic Cancer Growth. Mol Cancer Ther (2014) 13(10):2303–14. doi: 10.1158/1535-7163.MCT-13-1005
60. Ellenrieder V, Hendler SF, Boeck W, Seufferlein T, Menke A, Ruhland C, et al. Transforming Growth Factor Beta1 Treatment Leads to an Epithelial-Mesenchymal Transdifferentiation of Pancreatic Cancer Cells Requiring Extracellular Signal-Regulated Kinase 2 Activation. Cancer Res (2001) 61(10):4222–8.
61. Jungert K, Buck A, Buchholz M, Wagner M, Adler G, Gress TM, et al. Smad-Sp1 Complexes Mediate TGFbeta-Induced Early Transcription of Oncogenic Smad7 in Pancreatic Cancer Cells. Carcinogenesis (2006) 27(12):2392–401. doi: 10.1093/carcin/bgl078
62. Ostapoff KT, Cenik BK, Wang M, Ye R, Xu X, Nugent D, et al. Neutralizing Murine Tgfβr2 Promotes a Differentiated Tumor Cell Phenotype and Inhibits Pancreatic Cancer Metastasis. Cancer Res (2014) 74(18):4996–5007. doi: 10.1158/0008-5472.CAN-13-1807
63. Schiller M, Dennler S, Anderegg U, Kokot A, Simon JC, Luger TA, et al. Increased cAMP Levels Modulate Transforming Growth Factor-Beta/Smad-Induced Expression of Extracellular Matrix Components and Other Key Fibroblast Effector Functions. J Biol Chem (2010) 285(1):409–21. doi: 10.1074/jbc.M109.038620
Keywords: pancreatic cancer, CREB, RAS, epithelial to mesenchymal plasticity, MicroRNAs, metastasis, therapeutic resistance
Citation: Mehra S, Singh S and Nagathihalli N (2022) Emerging Role of CREB in Epithelial to Mesenchymal Plasticity of Pancreatic Cancer. Front. Oncol. 12:925687. doi: 10.3389/fonc.2022.925687
Received: 21 April 2022; Accepted: 12 May 2022;
Published: 21 June 2022.
Edited by:
Aamir Ahmad, University of Alabama at Birmingham, United StatesReviewed by:
Chethan Ashokkumar, University of Pittsburgh, United StatesManish Tripathi, The University of Texas Rio Grande Valley, United States
Copyright © 2022 Mehra, Singh and Nagathihalli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nagaraj Nagathihalli, bm5hZ2F0aGloYWxsaUBtZWQubWlhbWkuZWR1