 Alfonso Pompella
Alfonso Pompella Alessandro Corti
Alessandro Corti Athanase Visvikis2
Athanase Visvikis2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Oncol. , 20 May 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.920316
This article is part of the Research Topic Redox Metabolism: a Double Edge Sword Sustaining the Adaptive Resistance to Therapy in Cancer View all 7 articles
Cisplatin (CDDP) is currently employed for the treatment of several solid tumors, but cellular heterogeneity and the onset of drug resistance dictate that suitable biomarkers of CDDP sensitivity are established. Studies on triple-negative breast cancer (TNBC) have recently confirmed the involvement of gamma-glutamyltransferase 1 (GGT1), whose enzyme activity expressed at the cell surface favors the cellular resupply of antioxidant glutathione (GSH) thus offering cancer cells protection against the prooxidant effects of CDDP. However, an additional well-established mechanism depends on GGT1-mediated matabolism of extracellular GSH. It was in fact shown that glycyl-cysteine – the dipeptide originated by GGT1-mediated GSH metabolism at the cell surface – can promptly form adducts with exogenous CDDP, thus hindering its access to the cell, interactions with DNA and overall cytotoxicity. Both mechanisms: mainainance of intracellular GSH levels plus extracellular CDDP detoxication are likely concurring to determine GGT1-dependent CDDP resistance.
The cytotoxic effects of cisplatin (CDDP) are in the first place mediated through its binding to DNA with the formation of intra-strand DNA adducts, leading to the inhibition of DNA synthesis and cell growth as well as to up-regulation of pro-apoptotic/down-regulation of anti-apoptotic genes/proteins, resulting in the induction of both intrinsic and extrinsic pathways of apoptosis. CDDP also induces production of reactive oxygen species (ROS), lipid peroxidation and activation of p53 signaling and other signal transduction pathways (1). Indeed, the molecular and (epi)genetic mechanisms accounting for the onset of CDDP resistance variably involve one or more of such cellular targets, as documented in several solid neoplasias including triple-negative breast cancer (TNBC) (2, 3).
Highly aggressivity, high recurrence rate and poor prognosis make the management of TNBC one of the hardest challenges in oncology. Treatment strategies over the last decades have been largely focused on chemotherapeutics such as taxanes and anthracyclines, but regimens based on CDDP – alone or in combination – have more recently taken the stage as a more versatile and promising tool against TNBC, especially in carriers of BRCA1 mutations or other deficiencies in the DNA repair systems making cells sensitive to clastogenic effects. Major difficulties are however represented by the frequent occurrence of CDDP resistance and the considerable heterogeneity of TNBC, leading to a wide variability in patients’ response to the drug. Several potential biomarkers are under investigation in order to identify patients most likely to benefit from CDDP, including homologous recombination repair deficiency, tumor infiltrating lymphocytes, TP53, cyclin-dependent kinase 2 expression, vascular endothelial growth factor and matrix metalloproteinase-9 (4–6).
An interesting study has recently described a novel cellular/molecular mechanism leading to CDDP resistance of TNBC, highlighting a crucial role for the expression of gamma-glutamyltransferase 1 (GGT1). An interplay of cancer cells with tumor associated macrophages (TAMs) was in fact described, in which TGF-β1 released by TAMs induces TNBC cells to secrete hepatic leukemia factor (HLF), which in turn transactivates GGT1 (7). The expression of this enzyme activity is shown to enable resistance to cisplatin by potentiating TNBC defenses against ferroptosis, the peculiar cell death mode consequent to iron-dependent lipid peroxidation. The finding is interpreted as the result of an improved cellular supply of the antioxidant tripeptide glutathione (GSH), enabling TNBC to protect themselves against the oxidant stress caused by CDDP and prevent the development of cytotoxic lipid peroxidation (7).
However, taking into account the complex set of effects produced by GGT1 enzyme activity (8), such interpretation is likely incomplete. The involvement of GGT1 in drug resistance of cancer cells has been repeatedly reported in quite a number of different neoplasias (reviewed in 9). GGT1 enzyme activity is located at the cell surface and catalyzes the cleavage of gamma-glutamic acid from GSH, thus effecting the first step in a salvage pathway of this critical cellular antioxidant from the extracellular space. In this way GGT1 can favor the maintainance of adequate intracellular GSH levels, and this has been long regarded as the sole mechanism underlying the protection offered by GGT1 against cytotoxicity of prooxidant drugs, including CDDP (10). In addition to its well-established genotoxic effects, the cytotoxicity of CDDP has been in fact variably attributed to production of reactive oxygen species following activation of NADPH oxidase (11) or mitochondrial dysfunction (12, 13). The programmed cell death process termed ferroptosis, in particular, is set in motion when cellular GSH levels are depleted to an extent such that the activity of glutathione peroxidase-4 (GPX4) is compromised. Once this has occurred, lipid peroxidation reactions are no longer regulated and can proceed up to the fragmentation of phospholipids with consequent derangement of cell membranes – a step catalyzed by the presence of free, redox-active iron. These processes are likely taking place in the experimental model used in the mentioned study (7), where the protection afforded by GGT1 expression in TNBC against ROS production, lipid peroxidation and cell death is indeed accompanied by increased levels of intracellular GSH.
A comprehensive interpretation of the role played by GGT1 in CDDP resistance cannot however ignore an additional mechanism documented by previous studies from our own laboratories. We could in fact demonstrate that glycyl-cysteine – the dipeptide originating from the cleavage of GSH effected by GGT1 at the cell surface – accumulates in the extracellular milieu of GGT1-transfected cells (Table 1) and is therefore liable to react with exogenous compounds such as CDDP. GSH itself reacts with CDDP in a 2:1 ratio, forming a complex consisting of a bi-dentate adduct in which both the sulfur and the amide nitrogen of the cysteine residues become bound to the drug (14). Such reaction is however remarkably slow, while formation of a similar adduct of CDDP with GSH metabolite glycyl-cysteine is approximately 10-fold quicker (15). Thus, due to the higher reactivity of its SH group as compared to that of parent tripeptide GSH, glycyl-cysteine can promptly form adducts with exogenous CDDP (Figure 1C) hence hindering it from accessing the cell, resulting in significantly decreased levels of DNA platination (Figure 1B). Overall, the occurrence of such reactions suggests that GSH metabolism effected by GGT1 expressed at the surface of cancer cells can actually mediate sort of an extracellular detoxication of CDDP, which adds to the antioxidant defense offered by intracellular GSH against CDDP-induced oxidative stress, and is likely a major factor concurring to prevent its cytotoxicity (Figures 1A, D) (14–16).

Table 1 Selective accumulation of the reactive GSH metabolite glycyl-cysteine in the extracellular space of GGT1-transfected melanoma c21/GGT cells.
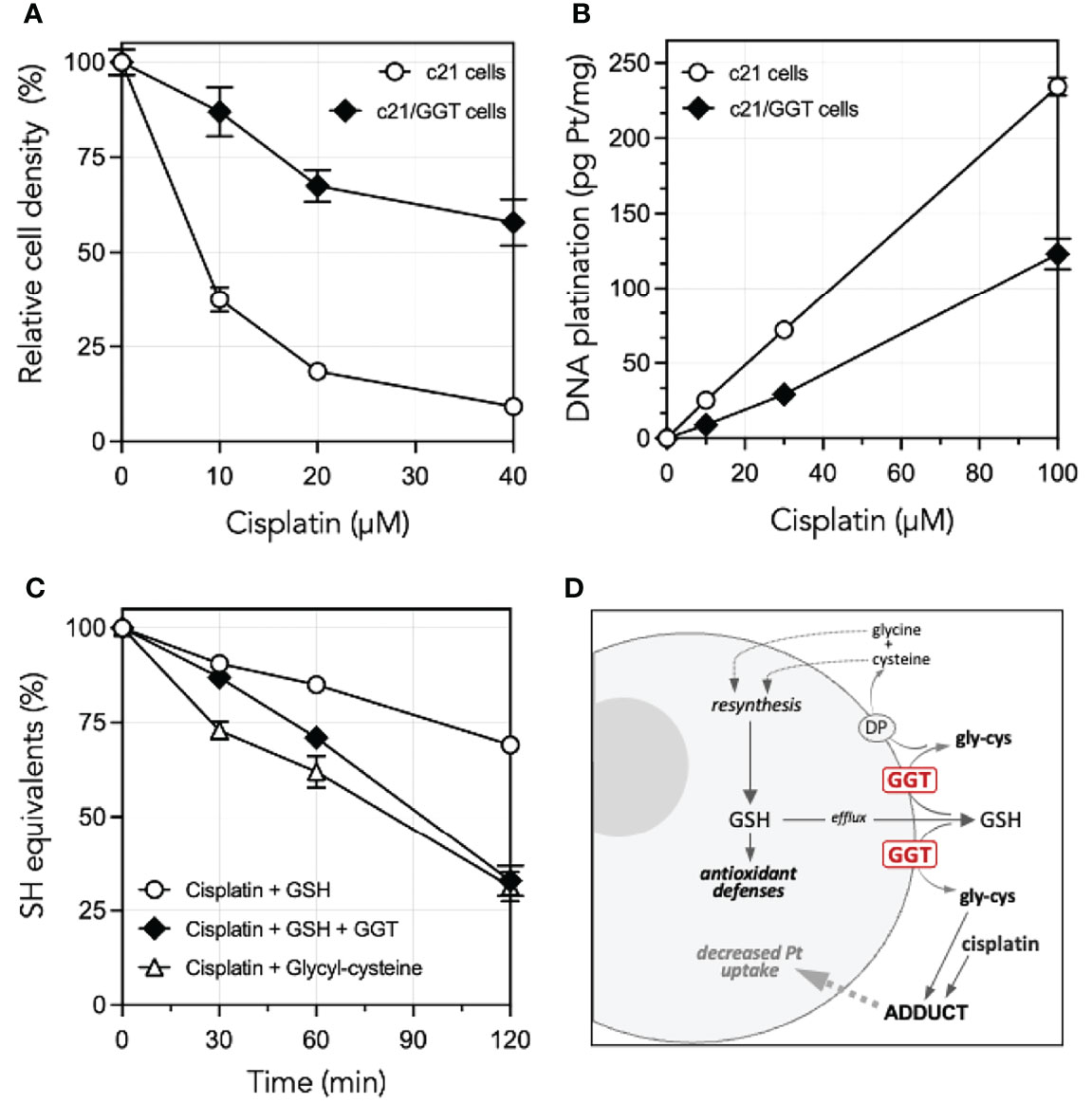
Figure 1 Cisplatin resistance of GGT1-expressing melanoma cells, degrees of DNA platination and differential reactivity of cisplatin with low mol. weight thiols. (A) Cisplatin sensitivity of c21 melanoma cells as compared to their GGT1-transfected (c21/GGT) counterparts. Cells were incubated 24 h with cisplatin and cell number was determined by WST-1 assay. Results are expressed as % of untreated controls, and are means ± SD (N = 12–36). (B) Corresponding levels of DNA platination in the same two clones as in A. Cells were exposed to cisplatin for 1 h. Platination was measured by inductively coupled plasma mass spectroscopy. Values shown are means ± SD (N = 3). (C) Reactivity with cisplatin with GSH in absence (〇) or presence of 200 mU/ml purified GGT1 (♦), or with the product of GGT1-mediated metabolism of GSH, glycyl-cysteine (Δ). SH groups were assessed at time intervals in a mixture containing 1.5 mM cisplatin and 3 mM GSH or glycyl-cysteine. Values shown are means ± SD (N = 3). (D) Proposed mechanisms underlying the GGT1-mediated resistance of cancer cells to cisplatin cytotoxicity. Modified, from Paolicchi et al. (14), Daubeuf et al. (15) and Franzini et al. (13).
In conclusion, the expression of GGT1 represents an important biomarker of CDDP resistance, and studies are warranted in order to investigate its ability to prevent ferroptosis in other malignant neoplasias besides TNBC. The development of CDDP resistance was observed to correlate with the activation of Keap1/Nrf2 transduction pathway in a series of solid tumors (6) and signalling through the Keap1/Nrf2 axis is indeed involved in GGT1 expression (17), raising the possibility that this may be the case in at least some patients. The finding of GGT1 expression should be anyway interpreted as a biomarker of CDDP resistance and an indication towards alternative treatments. In this latter respect, while precluding platinum-based treatments GGT1 expression may conversely offer unique therapeutic opportunities. The specific ability of GGT1 to cleave the gamma-glutamyl residue can in fact be exploited to activate glutathionylated pro-drugs, a pharmacologic strategy whose efficacy was documented in melanoma cells treated with the anti-angiogenic agent GSAO (18).
All authors conceived the article and discussed its content. AP wrote the main text and prepared the figures. All authors contributed to the article and approved the submitted version.
The financial support by University of Pisa (Institutional funds 2021) is gratefully acknowledged.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
HLF, hepatic leukemia factor; GGT1, gamma-glutamyltransferase-1; Gly-cys, glycyl-cysteine; GSAO, 4-(N-(S-glutathionylacetyl)amino) phenylarsonous acid; GSH, glutathione; TAM, tumor associated macrophages; TNBC, triple-negative breast cancer.
1. Tchounwou PB, Dasari S, Noubissi FK, Ray P, Kumar S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J Exp Pharmacol (2021) 13:303–28. doi: 10.2147/JEP.S267383
2. Yang L, Xie HJ, Li YY, Wang X, Liu XX, Mai J. Molecular Mechanisms of Platinum-Based Chemotherapy Resistance in Ovarian Cancer. Oncol Rep (2022) 47:82. doi: 10.3892/or.2022.8293
3. Skowron MA, Oing C, Bremmer F, Ströbel P, Murray MJ, Coleman N, et al. The Developmental Origin of Cancers Defines Basic Principles of Cisplatin Resistance. Cancer Lett (2021) 519:199–210. doi: 10.1016/j.canlet.2021.07.037
4. Zhu Y, Hu Y, Tang C, Guan X, Zhang W. Platinum-Based Systematic Therapy in Triple-Negative Breast Cancer. Biochim Biophys Acta Rev Cancer (2022) 1877:188678. doi: 10.1016/j.bbcan.2022.188678
5. Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New Strategies for Triple-Negative Breast Cancer–Deciphering the Heterogeneity. Clin Cancer Res (2014) 20:782–90. doi: 10.1158/1078-0432.CCR-13-0583
6. Bou Zerdan M, Ghorayeb T, Saliba F, Allam S, Bou Zerdan M, Yaghi M, et al. Triple Negative Breast Cancer: Updates on Classification and Treatment in 2021. Cancers (Basel) (2022) 14:1253. doi: 10.3390/cancers14051253
7. Li H, Yang P, Wang J, Zhang J, Ma Q, Jiang Y, et al. HLF Regulates Ferroptosis, Development and Chemoresistance of Triple-Negative Breast Cancer by Activating Tumor Cell-Macrophage Crosstalk. J Hematol Oncol (2022) 15:2. doi: 10.1186/s13045-021-01223-x
8. Pompella A, Corti A, Paolicchi A, Giommarelli C, Zunino F. Gamma-Glutamyltransferase, Redox Regulation and Cancer Drug Resistance. Curr Opin Pharmacol (2007) 7:360–6. doi: 10.1016/j.coph.2007.04.004
9. Hanigan MH, Gallagher BC, Townsend DM, Gabarra V. Gamma-Glutamyl Transpeptidase Accelerates Tumor Growth and Increases the Resistance of Tumors to Cisplatin In Vivo. Carcinogenesis (1999) 20:553–9. doi: 10.1093/carcin/20.4.553
10. Kim HJ, Lee JH, Kim SJ, Oh GS, Moon HD, Kwon KB, et al. Roles of NADPH Oxidases in Cisplatin-Induced Reactive Oxygen Species Generation and Ototoxicity. J Neurosci (2010) 30:3933–46. doi: 10.1523/JNEUROSCI.6054-09.2010
11. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, et al. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PloS One (2013) 8:e81162. doi: 10.1371/journal.pone.0081162
12. Kleih M, Böpple K, Dong M, Gaißler A, Heine S, Olayioye MA, et al. Direct Impact of Cisplatin on Mitochondria Induces ROS Production That Dictates Cell Fate of Ovarian Cancer Cells. Cell Death Dis (2019) 10:851. doi: 10.1038/s41419-019-2081-4
13. Franzini M, Corti A, Lorenzini E, Paolicchi A, Pompella A, De Cesare M, et al. Modulation of Cell Growth and Cisplatin Sensitivity by Membrane Gamma-Glutamyltransferase in Melanoma Cells. Eur J Cancer (2006) 42:2623–30. doi: 10.1016/j.ejca.2006.04.016
14. Paolicchi A, Sotiropuolou M, Perego P, Daubeuf S, Visvikis A, Lorenzini E, et al. Gamma-Glutamyl Transpeptidase Catalyses the Extracellular Detoxification of Cisplatin in a Human Cell Line Derived From the Proximal Convoluted Tubule of the Kidney. Eur J Cancer (2003) 39:996–1003. doi: 10.1016/s0959-8049(03)00067-4
15. Daubeuf S, Balin D, Leroy P, Visvikis A. Different Mechanisms for Gamma-Glutamyltransferase-Dependent Resistance to Carboplatin and Cisplatin. Biochem Pharmacol (2003) 66:595–604. doi: 10.1016/s0006-2952(03)00343-5
16. Zhang H, Liu H, Iles KE, Liu RM, Postlethwait EM, Laperche Y, et al. 4-Hydroxynonenal Induces Rat Gammaglutamyl Transpeptidase Through Mitogen-Activated Protein Kinase-Mediated Electrophile Response Element/Nuclear Factor Erythroid 2-Related Factor 2 Signaling. Am J Respir Cell Mol Biol (2006) 34:174–81. doi: 10.1165/rcmb.2005-0280OC
17. Dilda PJ, Ramsay EE, Corti A, Pompella A, Hogg PJ. Metabolism of the Tumor Angiogenesis Inhibitor 4-(N-(S-Glutathionylacetyl)amino)phenylarsonous Acid. J Biol Chem (2008) 283:35428–34. doi: 10.1074/jbc.M804470200
Keywords: cisplatin, drug resistance, gamma-glutamyltransferase (GGT), glutathione, biomarkers
Citation: Pompella A, Corti A and Visvikis A (2022) Redox Mechanisms in Cisplatin Resistance of Cancer Cells: The Twofold Role of Gamma-Glutamyltransferase 1 (GGT1). Front. Oncol. 12:920316. doi: 10.3389/fonc.2022.920316
Received: 14 April 2022; Accepted: 26 April 2022;
Published: 20 May 2022.
Edited by:
Cinzia Domenicotti, Università di Genova, ItalyReviewed by:
Barbara Marengo, University of Genoa, ItalyCopyright © 2022 Pompella, Corti and Visvikis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfonso Pompella, YWxmb25zby5wb21wZWxsYUB1bmlwaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.