 Kun Ke
Kun Ke Li Li2,3†
Li Li2,3† Chao Lu
Chao Lu Yuanyu Wang
Yuanyu Wang Huiju Wang
Huiju Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 12 August 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.916082
This article is part of the Research Topic Ion Channels & Homeostasis of Ions in Cancer Cell Fate View all 7 articles
Ferroptosis is an iron-dependent cell death process characterized by excessive accumulation of reactive oxygen species and lipid peroxidation. The elucidation of ferroptosis pathways may lead to novel cancer therapies. Current evidence suggests that the mechanism of ferroptosis can be summarized as oxidative stress and antioxidant defense mechanisms. During this process, ferrous ions play a crucial role in cellular oxidation, plasma membrane damage, reactive oxygen species removal imbalance and lipid peroxidation. Although, disregulation of intracellular cations (Fe2+, Ca2+, Zn2+, etc.) and anions (Cl-, etc.) have been widely reported to be involved in ferroptosis, their specific regulatory mechanisms have not been established. To further understand the crosstalk effect between ferrous and other ions in ferroptosis, we reviewed the ferroptosis process from the perspective of ions metabolism. In addition, the role of ferrous and other ions in tumor therapy is briefly summarized.
Ferroptosis is an iron-dependent cell death characterized by reactive oxygen species (ROS) accumulation and lipid peroxidation. Different from cuprotosis, apoptosis and necrosis, it does not depend on classical death pathways such as caspase activation (1–3). The process of ferroptosis is a dynamic equilibrium that includes oxidation and antioxidation mechanisms. On the one hand, ROS accumulation and lipid peroxidation are caused by ferrous metabolism imbalance culminating in plasma membrane damage and cell death. On the other hand, a series of antioxidant systems have been established in humans and other organisms, including cystine-GSH-GPX4, CoQ10-FSP1 and GCH1-BH4-DHFR, which can effectively inhibit lipid peroxidation and thus prevent ferroptosis. Accordingly, it is widely thought that targeting the process of iron metabolism and ferroptosis may provide a potential clinical application for anti-tumor therapy (4).
Iron metabolism plays a central role in ferroptosis. Thus, any imbalance regarding its uptake, distribution and output can initiate ferroptosis. It is well-established that complex mechanisms affect iron absorption, storage, and outflow to prevent or promote the occurrence of ferroptosis, and iron has a bidirectional regulatory effect on the occurrence and progression of tumors (5, 6). In addition to ferrous ions, other ions (Ca2+, Zn2+, Cl-, etc.) have also been reported to participate in ferroptosis, but the regulatory network between ferrous and other ions has not been cleared. Calcium (Ca2+) influx in extracellular space is closely related to the ferroptosis process, characterized by depletion of the intracellular antioxidant glutathione, increased reactive oxygen species in the cytoplasm and mitochondrial dysfunction (7). Zinc (Zn2+) is widely acknowledged as the second most abundant trace element in humans after iron. Zn2+ mainly acts on mitochondria, and its mobilization is the prerequisite for triggering irreversible mitochondrial dysfunction (8, 9). Zinc overload can destroy intracellular calcium homeostasis by damaging mitochondrial function and promoting organelle death in coordination with Ca2+-induced damage, eventually leading to cell death such as ferroptosis. Moreover, selenium is an important regulator of ferroptosis, and the utilization of selenium by GPX4 is a necessary condition to prevent ferroptosis induced by hydrogen peroxide (10). GPX4 harbors selenocysteine making cells resistant to irreversible excessive oxidation and ferroptosis. Chloride ions (Cl-) are well-recognized as the most abundant anion in the body. Interestingly, it has been shown that an imbalance of chloride ions and channels is associated with ferroptosis (11).
In this review, we discuss the core mechanisms of ferroptosis and introduce the regulatory roles of cations (Fe2+, Ca2+, Zn2+, etc.) and anions (Cl-, etc.) in ferroptosis. Furthermore, the potential application value of iron and other ions in treating ferroptosis-associated tumors is also discussed.
Ferroptosis is a passive cell death process caused by the accumulation of iron-dependent lipid peroxidation and plasma membrane damage. Ever since ferroptosis was defined as a distinct cell death different from necrosis and apoptosis in 2012 by Dixon, vast strides have been made in revealing its main signaling pathways (2), which are mainly categorized into the classical or non-classical signaling pathways. Specifically, the process of ferroptosis is induced by ROS, LOX, and the Fenton reaction, and suppressed by the Cystine-GSH-GPX4 axis and FSP1-COQ10 (Figure 1).
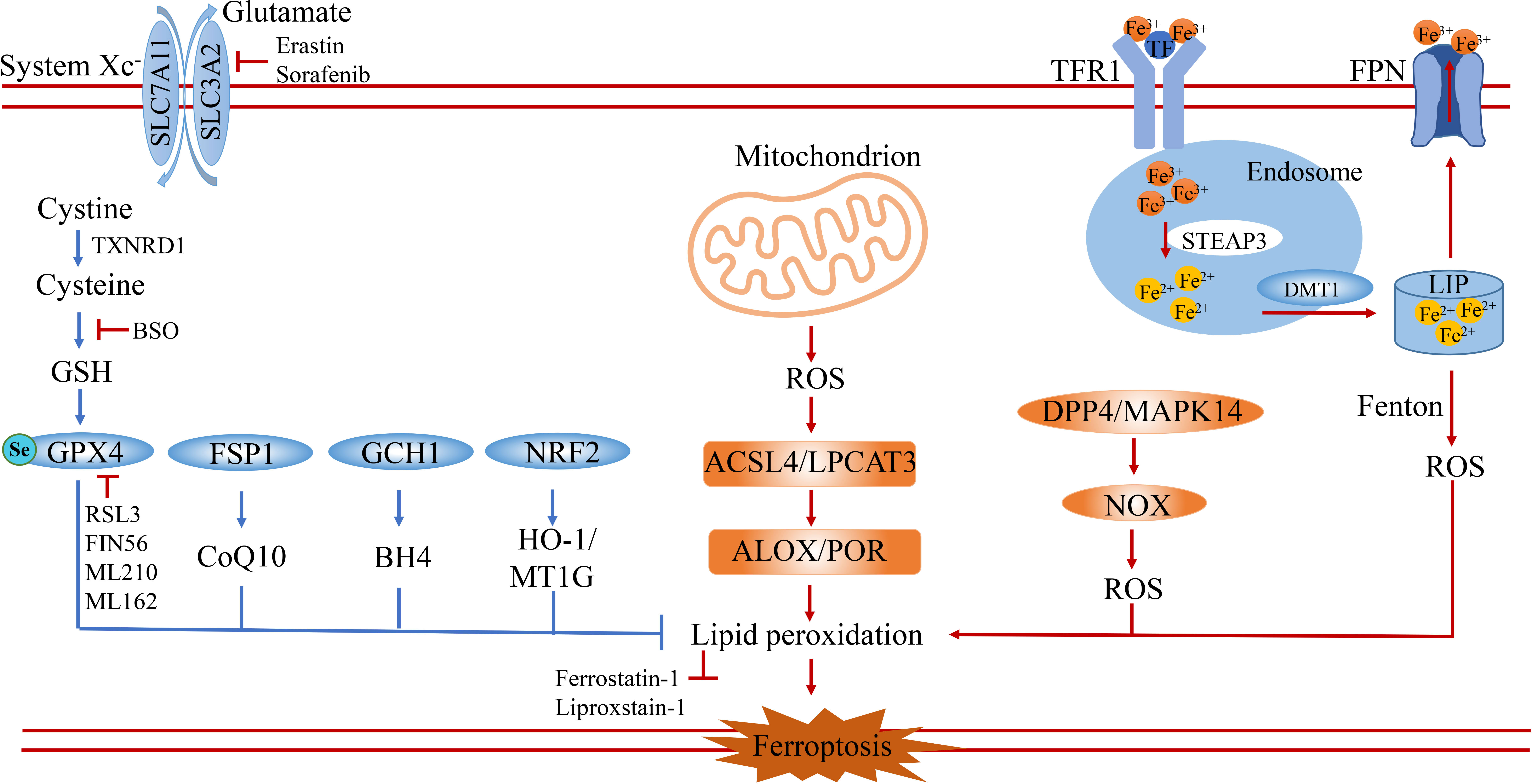
Figure 1 The core signaling pathway of ferroptosis. Ferroptosis is a special type of cell death characterized by excessive accumulation of reactive oxygen species and lipid peroxidation. It is induced by the imbalance of oxidation and anti-oxidation. On the one hand, ferroptosis is induced by the accumulation of ROS from iron-induced Fenton reaction, NOX family and mitochondrion dysfunction. On the other hand, ferroptosis is prevented by anti-oxidation systems including the GSH-GPX4 pathway, FSP1-CoQ10 pathway, GCH1-BH4 pathway and NRF2-HMOX1/MT1G pathway. SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; TXNRD1, thioredoxin reductase 1; GSH, glutathione; GPX4, glutathione peroxidase 4; FSP1, ferroptosis suppressor protein 1; GCH1, GTP Cyclohydrolase 1; NRF2(NFE2L2), nuclear factor erythroid 2-related factor 2; HO-1, also known as HMOX1, heme oxygenase-1; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; DPP4, dipeptidyl peptidase 4; MAPK14, mitogen-activated protein kinase 14; ALOX, arachidonate lipoxygenase; POR, cytochrome P450 oxidoreductase; TFR1, transferrin receptor 1; TF, transferrin; FPN, ferroportin; DMT1, divalent metal transporter 1; LIP, labile iron pool; BSO, buthionine sulfoximine.
ROS. ROS are produced by human and other mammalian cells during cell metabolism, mainly including superoxide (O2-), hydrogen peroxide (H2O2), and hydroxyl radical (OH•), which are associated with ferroptosis in redox reactions. The inability to scavenge ROS in time, leads to excessive accumulation of reactive oxygen species, resulting in cell membrane damage and triggering cell ferroptosis. Ferrous ions-mediated Fenton response, mitochondrial ROS, and membrane-associated ROS driven by the NOX protein family are the main sources of intracellular ROS (Figure 1). The reaction of Fe2+ with H2O2 is called the Fenton reaction (12). In the Fenton reaction, Fe2+ is oxidized to Fe3+, and electrons are transferred to H2O2 to form HO•. The lipid radical (L-OH) binds to O2 to form the lipid peroxide radical (L-OO•), which then grabs hydrogen from the adjacent PUFA to produce L-OOH and a new lipid radical. In addition, mitochondria are important sources of intracellular ROS and organelles that regulate ferroptosis (13). Mitochondrial ROS are derived from oxidative respiration in the inner mitochondrial membrane. During electron transfer to O2 by mitochondrial electron transport chain complex, part of O2 is reduced to O2- and H2O2. Subsequently, O2- is rapidly decomposed to hydrogen peroxide (H2O2) by the superoxide dismutase 2 (SOD2) in the mitochondrial matrix and superoxide dismutase 1 (SOD1) in the membrane gap (14). Another important source of ROS is the membrane-associated ROS driven by the NOX protein family. NADPH oxidase 2 (NOX2) is expressed on the cell membrane and transports electrons through NOX to produce O2· or H2O2 (15). In addition, studies have shown that toxic lipid metabolites produced by lipid peroxidation can trigger NOX expression and form positive feedback to induce ferroptosis in cells (16). The excessive accumulation of ROS is a key factor in lipid peroxidation, leading to ferroptosis in tumor cells.
Interestingly, similar to the mechanism by which tumor cells evade immune cell attack, tumor cells with increased iron and ROS also maintain intracellular ROS balance through complex anti-ferroptosis mechanisms (17, 18).
System xc-. The cysteine-glutamate antiporter (also known as the System xc-) is a complex composed of SLC7A11 and SLC3A2 dimers embedded on the surface of the cell membrane (19). SLC7A11 is the main functional subunit and inhibition of SLC7A11 expression can induce ferroptosis, which has been confirmed in vivo by SLC7A11 depletion leading to ferroptosis in pancreatic cancer (20). Erastin, the first identified ferroptosis inducer, acts by inhibiting the activity of the cysteine-glutamate anti-transporter (System xc-) (2, 21). In addition to Erastin, drugs such as Salazosulfapyridine (SAS) and Sorafenib have also been shown to be inhibitors of system xc- mediated cystine importation. The system xc- mediates the uptake of cystine during ferroptosis, which is reduced to cysteine by glutathione and/or thioredoxin reductase 1 (TXNRD1) for glutathione biosynthesis. NRF2 (E2-related factor 2) is an important regulator of SLC7A11. Treatment with the drugs such as Erastin, Sorafenib, and Buthionine sulfoximine (BSO) enhances NRF2 activity and prevents ferroptosis (Figure 1) (22, 23).
Cystine/GSH/GPX4 axis. Glutathione (GSH) is a tripeptide containing an R-amide bond and sulfhydryl. It is composed of glutamic acid, cysteine, and glycine, and exists in two states: reduction (GSH) and oxidation (GSSG). Glutathione peroxidase 4 (GPX4) belongs to the glutathione peroxidase (GPXs) family and is a peroxidase containing selenium. The active center of GPX4 is selenocysteine, which catalyzes GSH to form GSSG. In this process, GSH acts as a reducing agent to reduce toxic lipid peroxides to non-toxic hydroxyl compounds (lipid alcohols), while promoting the decomposition of H2O2. Inhibition of GPX4 activity (RSL3, FIN56, etc.) leads to the accumulation of lipid peroxides in cells and induces ferroptosis. Current evidence suggests that the Cystine/GSH/GPX4 axis based on the System xc-, produces cysteine via the sulfur-conversion pathway and selenocysteine through the mevalonate pathway. Therefore, limited cystine input is the initiating factor of ferroptosis. GSH is an essential molecule in GPX4-catalyzed reactions because GSH acts as an electron donor to reduce toxic lipid peroxides (such as P-OOH) to non-toxic lipid alcohols (such as P-OH). In this process, the reduced NADPH acts as an electron donor. Hence, GSH is a key factor in maintaining GPX4 activity and inhibits ferroptosis. On the contrary, treatment with GPX4 active inhibitors such as RSL3 and FIN56 can induce ferroptosis. Indeed, cystine/GSH/GPX4 axis regulation plays an important role in tumor formation. It has been found that SLC7A11 is extensively expressed in various tumors and prevents ferroptosis (2, 24–27). These studies suggest that ferroptosis can modulate the tolerance of tumor cell death.
Coenzyme Q10 (CoQ10) is widely distributed on the cell membrane of mammalian cells and consists of a benzoquinone ring and a polyisoprene tail. Non-mitochondrial CoQ10 reportedly plays an important role as a reversible redox carrier in the electron transport of plasma membrane and Golgi body membrane, which can directly remove lipid peroxy radicals. In addition, oral CoQ10 has been reported to treat various human diseases, such as cardiomyopathy, Parkinson’s disease, and diabetes. In contrast, low CoQ10 levels caused by mutations in CoQ10 biosynthase or related enzymes have been associated with a variety of diseases. The CoQ10 biosynthetic pathway is tightly regulated at both transcription and translation levels. Moreover, CoQ10 can be synthesized by acetyl-CoA through the MVA pathway.
An increasing body of evidence suggests that the sensitivity of GPX4 inhibitors varies greatly between different cell lines. Screening of ferroptosis-related genes using CRISPR/Cas9 revealed that FSP1 is a previously unrecognized ferroptosis suppressor gene (28, 29).FSP1 was initially identified as a p53 response gene (PRG), named PRG3. Due to its homology to human apoptosis-inducing factor (AIF), it is also named mitochondrial-associated apoptosis-inducing factor 2 (AIFM2) (28, 29). Unlike AIF family proteins, FSP1 is predominantly cytoplasmic and may have an affinity for the cytoplasmic surface of the mitochondrial outer membrane. FSP1 is modified by myristoylation and is associated with various cell membrane structures including cytoplasmic membrane, Golgi apparatus, and perinuclear structures. Mutations of the memorial modification site of FSP1 decrease its resistance to ferroptosis. During ferroptosis, FSP1 mainly catalyzes non-mitochondrial CoQ10 to form ubiquinol, and reduced CoQ10H2 or ubiquitin acts as lipophilic free radical to capture antioxidants to prevent lipid peroxides and ultimately block ferroptosis (Figure 1) (28, 30). Due to its NADH activity, FSP1 directly reduces lipid free radicals and prevents lipid peroxidation by reducing ubiquinone, or indirectly promotes free radical regeneration of oxidative alpha-tocopherol (a natural antioxidant) to inhibit lipid peroxidation and ferroptosis (31). FIN56, a novel ferroptosis inducer, consumes CoQ10 by modulating the activity of squalene synthase (an enzyme in the mevalonic acid pathway) and enhancing the sensitivity of cells to iron (2, 32).
GTP cyclic hydrolase 1 (GCH1) has been reported to resist ferroptosis through its metabolites tetrahydrobiopterin (BH4) and dihydrobiopterin (BH2) (Figure 1) (33). BH4 can reportedly protect phospholipids containing PUFA tails from oxidative degradation through a dual mechanism. Although the role of GCH1 in protecting tissues and organs from ferroptosis remains to be clarified, deletion of GCH1 in mice has been shown to cause bradycardia and embryonic death in the second trimester of pregnancy. BH4 is an effective free radical capturing antioxidant that reduces lipid peroxidation and inhibits ferroptosis through GPX4. It has been shown that the GTP-dependent cyclic hydrolase 1 (GCH1)/tetrahydrobiopterin (BH4)/phospholipid axis inhibits ferroptosis by synthesizing lipophilic and antioxidant BH4, which enhances cell membrane resistance to oxidative damage and selectively prevents the depletion of phospholipid (34).
GSH-GPX4, FSP1-CoQ10 and GCH1-BH4 are the three main parallel pathways that inhibit ferroptosis, while the NRF2 axis and other antioxidants (such as prominin 2) also play a significant role in suppressing ferroptosis (Figure 1). Another endogenous antioxidant defense system is the nuclear factor erythroid 2-associated factor 2 (NRF2), which is usually maintained at low levels by ubiquitination mediated by the tumor suppressor Kelch ECH-associated protein 1 (KEAP1) (35–37). Under oxidative stress, NRF2 is separated from KEAP1 and activated to inhibit ferroptosis through NRF2/SLC7A11/heme oxygenase-1 (HO-1) and NRF2-FTH1 signaling pathways (36, 38). What’s more, NRF2, a transcription factor can regulate ferroptosis in response to oxidative stress through HO-1 and FTH1 and via transcription-induced expression of GPX4, SLC7A11, and p53 (35, 39). In terms of tumor drug resistance, NRF2 is one of the core factors leading to drug insensitivity or drug resistance of cancer cells in response to oxidative stress (40). P450 oxidoreductase (POR) and CYB5R1 generate H2O2 by transferring electrons from NAD(P)H to O2, which then reacts with Fe2+ to drive lipid peroxidation and promote ferroptosis (41). Haploid cell screening has helped identify acyl-CoA synthase long-chain isomer 4 (ACSL4) as a key factor in sensitizing cells to RSL3-induced ferroptosis. Moreover, Prominin 2 reduces iron levels by promoting iron output and is considered another negative regulator of ferroptosis (42).
Iron is an important component of complex proteins such as hemoglobin, myoglobin, heme enzyme and non-heme compounds. It facilitates the transport and exchange of oxygen in the blood and participates in electron transfer and redox reactions (43). Iron homeostasis is achieved by dynamic coordination of iron absorption, utilization, circulation, storage and output. The imbalance of iron content or distribution leads to the accumulation of iron in cells, which leads to the occurrence of iron-dependent cell death (2).
Iron is mostly derived from food absorption and iron recycling in the body. Non-heme iron in food mainly exists as insoluble Fe3+ and needs to be reduced to Fe2+ until it is absorbed. Iron is transported mainly by transferrin (TF), which is mainly synthesized by the liver and then released into serum globulin, which has a good chelating ability for Fe3+ ions (44). Fe3+ binds to TF in serum and is then recognized by Transferrin receptor 1(TFR1) on the cell membrane, which delivers the transferrin binding Fe3+ into the cell and is located in the endosome (Figure 2) (45). In addition to transferrin, lactotransferrin (LTF) participates in the transport of Fe3+. Subsequently, Fe3+ is released from the transferrin-1 complex in the low-pH acidic endosomes and reduced to Fe2+ by six-transmembrane epithelial antigen of the prostate 3 (STEAP3) (6, 46). Finally, the release of Fe2+ from endosomes to the labile iron pool (LIP) of the cytoplasm is mediated by divalent metal ion transporter 1 (DMT1, also known as solute carrier family 11 member 2, SLC11A2) (47). Thus, LIP and ferritin represent the two main forms of intracellular iron storage. LIP causes active oxidative stress-related toxicity and is responsible for regulating cellular iron homeostasis through the iron regulatory protein-iron response element system. Ferritin is an iron sequester protein consisting of ferritin light chain (FTL) and Ferritin Heavy Chain 1 (FTH1), containing up to 4500 irons, which can be dissolved by lysosomes or ferritinophagy to release iron ions (48). Ferritin exhibits a variety of functions in iron transport, cell proliferation, angiogenesis and immune suppression, but it is not directly involved in ROS production. Evidence suggests that increased iron in LIP is the main cause of triggering the Fenton reaction and promoting ferroptosis (5).
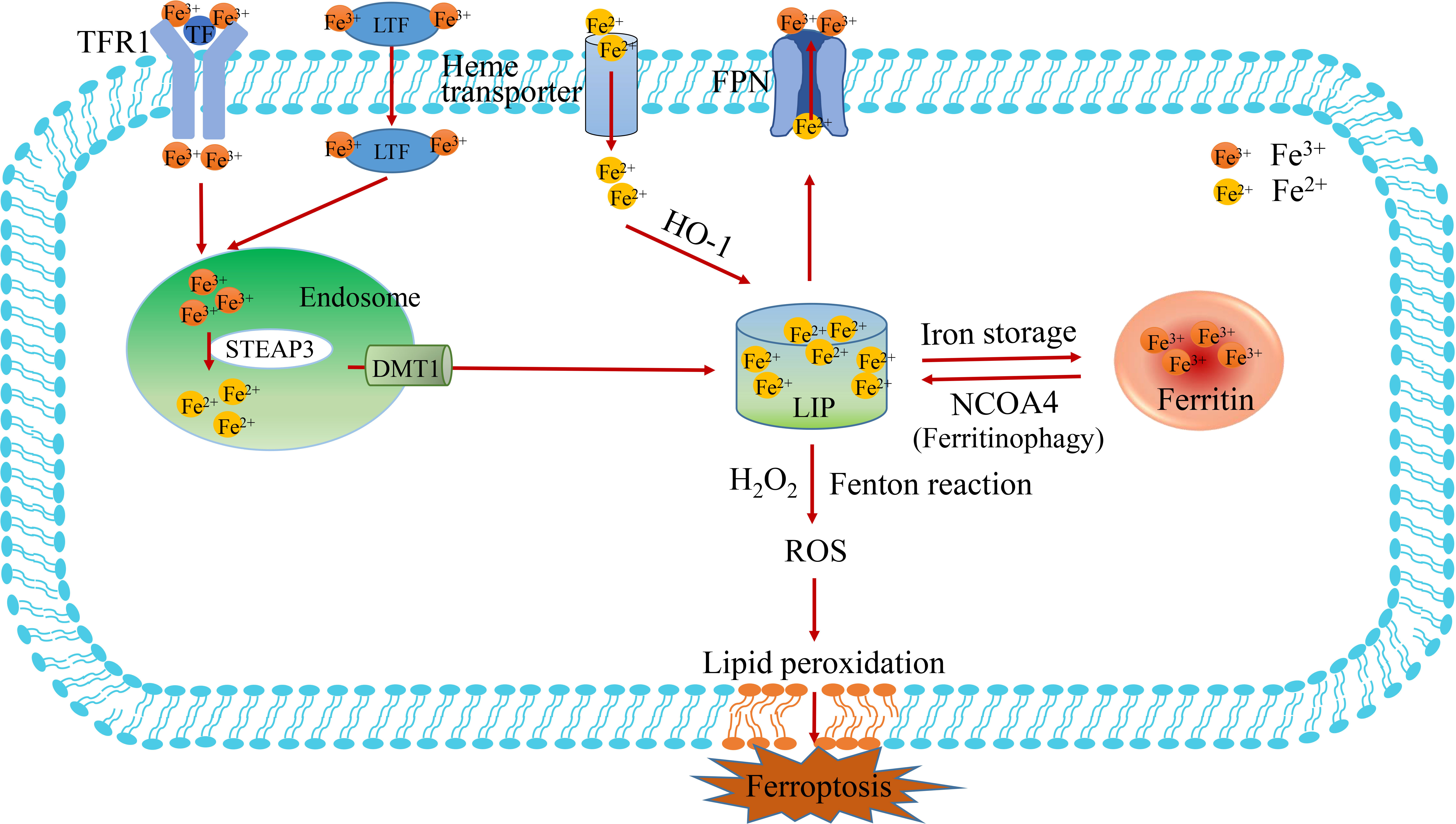
Figure 2 The metabolism of iron. The process of iron import, transduction, storage and export is described in figure 2. The Fe3+-loaded transferrin (TF) binds to transferrin receptors1 (TFR1) on the cell membrane to enter the cell. Then, Fe3+ is deoxidized to Fe2+ by iron oxidoreductase STEAP3 in the endosome, which subsequently Fe2+ is released into the labile iron pool (LIP) by DMT1. The pathways of Fe3+deoxidization by STEAP3-DMT1, ferritin degradation by NCOA4 (also known as ferritinophagy) and Heme transport are the main sources of Fe2+. LIP is the primary storage site for unstable iron, whereas ferritin is the key storage site for stable iron. TF, transferrin; TFR1, transferrin receptor 1; FPN, ferroportin; LIP, labile iron pool; NCOA4, nuclear receptor coactivator 4.
In terms of utilization, mitoferritin 1 and 2 (MFRN1 and MFRN2) transport iron to the mitochondria to assist cellular respiration and heme synthesis. Ferritin can also release free iron by degradation of nuclear receptor coactivator 4 (NCOA4), a process known as “ferritinophagy”(Figure 2). In terms of iron output, ferroportin (FPN, also known as Solute carrier family 40 Member 1, SLC40A1) mediates iron efflux and is the only known protein in mammals that synergistically exports intracellular iron with ceruloplasmin or heparin (49). Ceruloplasmin inhibits ferroptosis by regulating iron homeostasis in HepG2 and Hep3B cells. It has been showed that the consumption of ceruloplasmin leads to intracellular accumulation of iron ions and lipid ROS and promotes ferroptosis induced by erastin or RSL3. Iron regulatory proteins (IRP1/2) are also involved in intracellular iron homeostasis, regulating the expression of genes related to intracellular iron uptake, storage or excretion through post-transcriptional regulation (50). In addition, prominin protein 2 promotes the formation of ferritin poly-vesicles and exosomes that transport iron out of cells, thereby promoting breast cancer resistance to ferroptosis (42).
Excessive iron can produce ROS through the Fenton reaction and activate iron-containing enzymes (such as lipoxygenase) to promote lipid peroxidation and lead to ferroptosis. It is widely thought that complex mechanisms affecting iron absorption, iron storage and iron outflow in tumor cells, to prevent or promote iron death and have a bidirectional effect. A study demonstrated higher iron content in tumor cells than in normal cells, and found that tumor cells promote cell proliferation by increasing iron content (51). Ferroportin (FPN) is the most important iron exporting protein, and FPN exhibits significantly lower expression in tumor cells (49, 52). Overwhelming evidence substantiates that excessive dietary iron or direct iron injection can produce excessive reactive oxygen species (ROS), increasing the risk of solid tumors (53, 54). Moreover, iron imbalance in cells can lead to the development of cancer or the initiation of cell death processes. In addition to causing ferroptosis, iron-mediated ROS may also lead to different types of cell death, such as apoptosis and necrosis. Unfortunately, there is no clinical evidence that supplementing exogenous iron or regulating the intracellular distribution abundance of iron can be used to treat tumors.
Calcium (Ca2+) is an important second messenger in eukaryotic cells, which affects the changes in intracellular signaling pathways and regulates many different cellular processes (55). Cells generally maintain low cytoplasmic free Ca2+ levels under physiological conditions. In this respect, calcium is strictly regulated intracellularly, and the disruption of calcium homeostasis can lead to cell damage and even cell death (56). The distribution of calcium is not uniform, of which 50% is stored in the nucleus, 30% in the mitochondria, 14% in the endoplasmic reticulum, and the rest in the plasma membrane and solute. Calcium is usually transported by calcium ion channels, calcium pumps and other calcium transporters. Ca2+ entry into the cytoplasm depends on external calcium influx (Ca2+ channels, calcium pumps, Na+/Ca2+ exchangers) and internal calcium release (IP3 and Ryonadine pathways) (31, 57). The endoplasmic reticulum (ER) is an intracellular Ca2+ storage organelle that plays an important role in many cellular metabolic processes. When calcium ions stored in the ER are released via IP3R, it can induce extracellular calcium ions to rapidly enter the cell, a process called store-operated Ca2+ entry (SOCE) (58). SOCE is mediated by the calcium receptor stromal interaction molecule (STIM) on ER and Orai on the cell membrane (Figure 3) (59). The calcium receptor STIM1 on ER moves to the plasma membrane and then binds to Orai1, resulting in the entry of Ca2+ from extracellular to intracellular, which regulates various death pathways (60–62). In addition, mitochondria can participate in cellular Ca2+ signaling through their transporters, such as mitochondrial calcium uniporter (MCU), which transport cytoplasmic calcium to mitochondria, and mitochondrial sodium-calcium exchange (NCX), which transfer calcium out of mitochondria (63, 64). The regulation of NCX activity can also inhibit Ca2+ efflux, promote intracellular Ca2+ accumulation by Na+ influx, and activate nicotinamide adenine dinucleotide phosphate (NADPH) oxidase to produce superoxide. On the contrary, the outflow of Ca2+ mainly depends on the cytoplasmic membrane, the endoplasmic reticulum calcium pump and Na+/Ca2+ exchangers. Moreover, SARCO/ER calcium ATPase (Ca-ATPase, SERCA) has been proved to be a regulator that restores ER calcium homeostasis by transporting Ca2+ from the cytoplasm back to SR/ER.
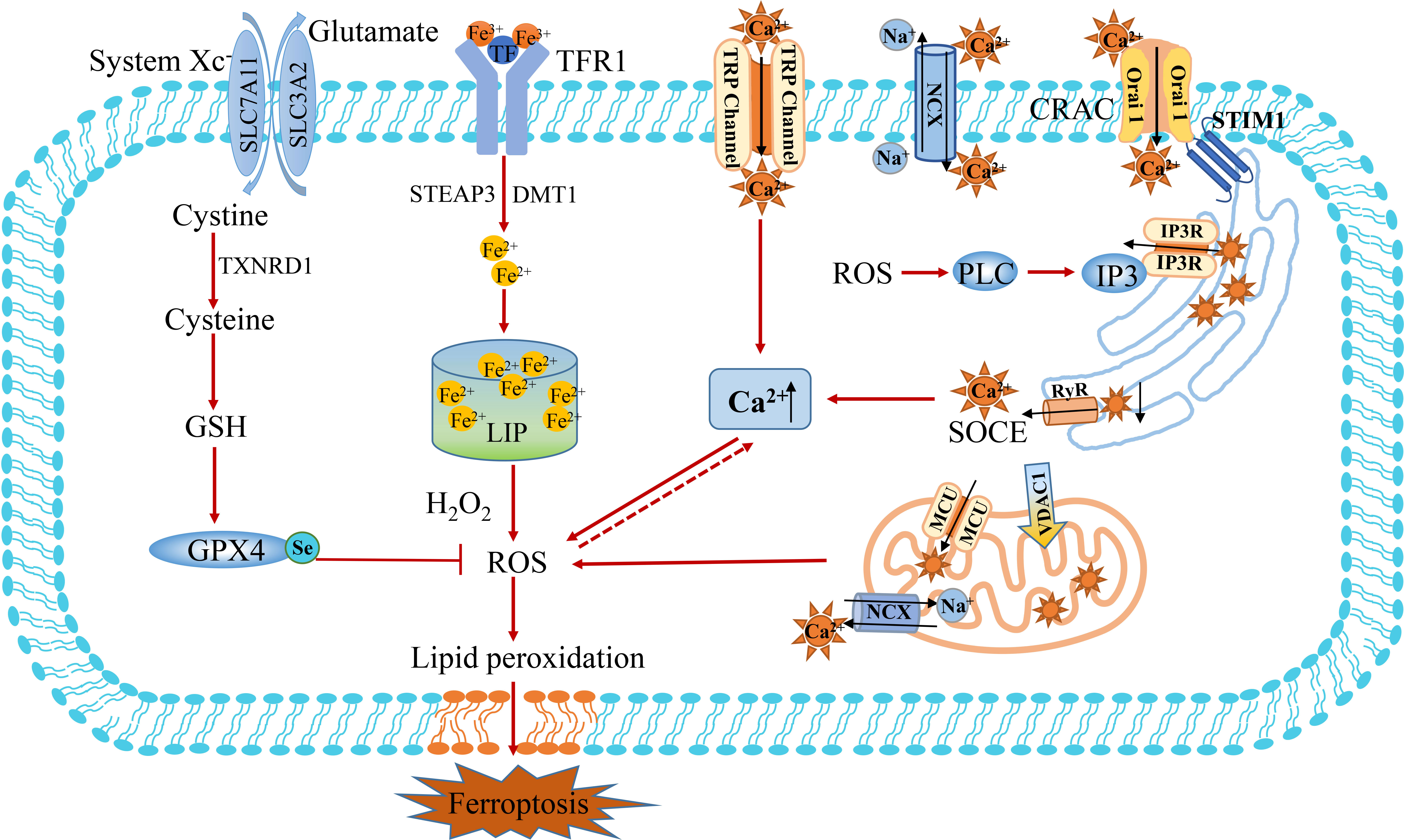
Figure 3 The crosstalk between Ca2+ and Fe2+ metabolism. Ca2+ entry into the cytoplasm depends on the influx of external calcium (Ca2+ channels of TRP, calcium pumps, Na+/Ca2+ exchanger of MCX) and release of internal calcium (IP3 and Ryonadine pathways). The endoplasmic reticulum (ER) binds to Orai1 through calcium receptor STIM1 and mediates the SOCE mechanism to promote calcium influx. Mitochondria participate in cellular Ca2+ signaling through their transporters, such as mitochondrial calcium uniporter (MCU) transport cytoplasmic calcium to mitochondria, while mitochondrial sodium-calcium exchangers (NCX) transfer calcium out of mitochondria. The influx of extracellular Ca2+can induces the opening of the permeability pore of the mitochondrial bilayer membrane, resulting in mitochondrial swelling and rupture. The release of Ca2+ from the mitochondrial intermembrane calcium pool can overload the cytoplasmic Ca2+ and induce ferroptosis resulting from the accumulation of ROS. MCU, mitochondrial calcium uniporter; NCX, sodium-calcium exchanger; SOCE, store-operated Ca2+ entry; STIM, stromal interaction molecule; TRP, transient receptor potential; VDAC1, voltage-dependent anionic channel 1; CRAC, calcium released activated Ca2+ channel.
Oxidative glutamate toxicity of Ca2+. Disrupted calcium homeostasis can lead to cell death, such as ferroptosis. Ca2+ influx in the extracellular space occurs during the late stages of the cascade characterized by depletion of the intracellular antioxidant glutathione, production of reactive oxygen species in the cytoplasm and mitochondrial dysfunction (65). As early as 1989, Murphy et al. reported that inhibition of cystine input through the cystine/glutamate anti-transporter system xc¯ in neuroblastoma X retinal cell line would lead to consumption of antioxidant glutathione (GSH) and Ca2+ dependent cell death (66). This type of cytotoxicity has been named oxidative glutamate toxicity or oxytosis, and recently this mode of death has been defined by most researchers as ferroptosis (67). Two reasons account for the conclusion that the process is similar to ferroptosis and that oxidative glutamate toxicity can be inhibited by iron chelating agents. In addition, it has been shown that the ferroptosis inhibitor ferrostatin-1 could inhibit oxidative glutamate toxicity (68). Maher et al. found that compounds can inhibit oxidative glutamate toxicity by blocking mitochondrial ROS production or reducing Ca2+ influx to protect cells from erastin or sulfapyridine-induced cell death (2, 67). Mouse hippocampal cell HT22 represents a good model for studying the effects of endogenous oxidative stress. After the extracellular addition of glutamate to inhibit the system xc-, glutathione levels decreased in a time-dependent manner (69). Levels of reactive oxygen species (ROS) increased exponentially, triggering surge activation of the signaling pathway, leading to an influx of cGMP-dependent Ca2+ and cell death (70). Indeed, it is widely acknowledged that Ca2+ influx is essential for cell death. Glutamate-treated HT22 cells did not die when cells were cultured in Ca2+-free medium or calcium antagonists blocking Ca2+ influx (66, 71). To elucidate the mechanism of Ca2+-induced glutamate oxidative toxicity, the researcher analyzed the calcium-sensitive status in glutamate-resistant HT22 cells and found that the difference in sensitivity was due to the downregulation of Ca2+ channel ORAI1 rather than the downregulation of Ca2+ sensor STIM1 or STIM2. This regulation resulted in a significant reduction in store-operated Ca2+ entry (SOCE) (72).
Zinc (Zn2+) is the second-largest trace element in organisms after iron, playing an important role in regulating cell metabolism, signal transduction, gene expression and cell death (73). Zinc ions can exist in the form of structural stability and instability. Structural Zn2+ is an important cofactor in more than 300 cellular enzymes. It affects organelle functions by binding to enzymes and regulatory proteins as catalytic subunits, such as zinc finger proteins and copper-zinc superoxide dismutase. Zn2+-Importing proteins (ZIPs) promote influx of Zn2+ into the cytoplasm through activated voltaic gated Ca2+ channels (VGCCs) or into neurons via Ca2+ and Zn2+ permeable GluR2-lacking AMPA receptors (Figure 4) (74). In contrast, ZnT transporters are homodimers that specifically exchange Zn2+/H+, removing Zn2+ from the cytoplasm (75). Proton-coupled divalent metal ion transporter (DMT1, also known as NRAMP2 and SLC11A2) may be a candidate for Zn2+ efflux from lysosomes. DMT1 is an important iron ion transporter, which has been described previously (76). Part of the labile Zn2+ in cells is stored in organelles, such as glutamate neurons, synaptic vesicles, mitochondria, lysosomes, endoplasmic reticulum (ER) and Golgi apparatus, and the other part binds to metallothionein (MTs) to form a stable structure (77). MT is a small family of cysteine-rich proteins capable of binding up to seven zinc atoms. Similar to iron, Zn2+ can be released from the unstable pool and affect the function of mitochondrial and lysosomes, which participated in the ferroptosis banquet (78). As signaling molecules, Zn2+ and Ca2+ share many transport and signaling pathways (79). Therefore, Ca2+ and Zn2+ are synergistically regulated oxidative stress to participate in Fe2+-induced oxidative stress and ferroptosis signal.
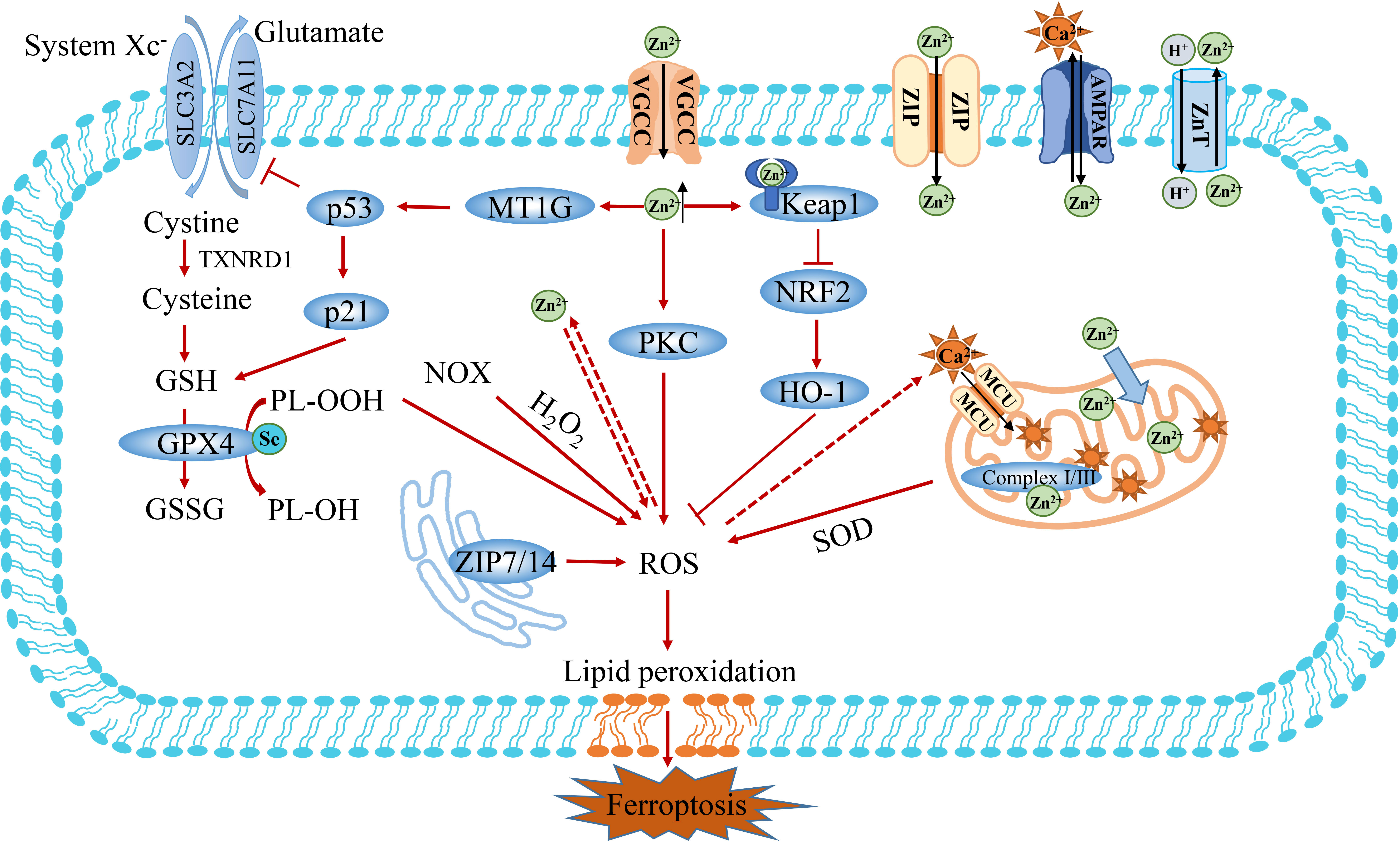
Figure 4 The crosstalk between Zn2+ and Fe2+ metabolism. Zn2+ is transported into cells via Zn2+-importing proteins (ZIPs), Volage-gated Ca2+ channels (VGCCs) and Ca2+ and Zn2+ permeable GluR2-deficient AMPA channels. Zn2+ exposure has been shown to increase the production of ROS and inhibit the activity of GSH. Furthermore, Zn2+ can indirectly promote the accumulation of ROS through PKC or Keap1/NFR2 pathways. In addition, the overload of Zn2+ induces ferroptosis by activating MT1G/p53 to regulate the expression of SLC7A11 and GSH. Finally, the mitochondrion is an important organelle for the action of Zn2+, which affects ROS production by regulating Complex I/III. Keap1, Kelch-like ECH-associated protein 1; FTH1, ferritin heavy chain 1; MT1G, metallothionein 1G; PKC, protein kinase C.
Overwhelming evidence substantiates that zinc is not only an antioxidant and an oxidative stress sensor, but also a redox inert metal that does not react directly with oxidants (80–82). Zn2+ exposure has been shown to increase intracellular ROS production levels. Nonetheless, it remains unclear how Zn2+ induces ROS production. It has been established that NADPH oxidoreductase (NOX) and mitochondria are key targets of oxidative stress induced by Zn2+(Figure 4) (83). Zn2+ promotes oxidative stress by activating NADPH oxidase and protein kinase C (PKC) (84, 85). Moreover, Zn2+ also acts on mitochondria, and its mobilization is the prerequisite for triggering irreversible mitochondrial dysfunction. The overload of Zinc can disrupt intracellular calcium homeostasis by destabilizing mitochondrial function and promoting organelle death in coordination with Ca2+-induced injury, eventually leading to cell death (86–88). Zn2+ and Ca2+ share many transport and signaling pathways. However, the specific molecular mechanisms underlying the regulatory effect of zinc on calcium are poorly understood.
Cu/Zn superoxide dismutase (SOD1) is an important reactive oxygen scavenging protein, that fights against reactive oxygen-mediated oxidative stress by catalyzing the conversion of superoxide anion to hydrogen peroxide (89). Excess H2O2 is usually reduced to water by glutathione peroxidase or catalase, preventing the production of hydroxyl radicals. Growing evidence suggests that Zn2+ can alter intracellular glutathione content and GSH/GSSG ratio in many cell types (90, 91). Importantly, zinc has been shown to inhibit the activity of enzymes related to glutathione metabolism, especially glutathione reductase (GR), and affect the clearance of endogenous peroxides. Studies have shown that zinc can reduce GSH in astrocytes and increase the output of GSSG to slow down the clearance of exogenous H2O2, thus promoting the production of intracellular ROS (90). A study showed that zinc could decrease GSSG reductase activity in lung fibroblasts and alveolar cell lines to promote cell death (92). Studies in humans and animals have shown that iron and zinc are negatively correlated and complement each other in intestinal absorption (93–95). Zinc may prevent iron-induced oxidative damage in rats by regulating intracellular iron signaling pathways (96). Zn2+ can enhance the expression of Metallothionein 1G (MT1G) in colon cancer cells and enhance cytotoxicity by activating the p53 pathway to promote the sensitivity of cancer cells to oxaliplatin (OXA) and 5-Fu (Figure 4) (97, 98). What’s more, Zn2+ can increase the expression of heme oxygenase-1 (HO-1) mRNA and protein in colorectal cancer cell lines, which can be inhibited by the iron-binding agent deferoxamine (DFO) (99). Considering the effect of Zn2+ exposure on HO-1 expression, the recently documented presence of a conserved Zn2+ binding site on the NRF2 inhibitor Keap1 seems to validate the association with Fe2+ metabolism (100). Moreover, zinc overload may lead to morphological changes induced by mitochondrial stress, leading to Hela cell death, which was inhibited by the zinc chelating agent TPEN (101). ZIP14 is a transport protein that can mediate cellular uptake of iron, and zinc (102). Increased zinc levers and ZIP14 were observed in the muscle of patients with pancreatic cancer cachexia, it suggested that zinc chelating agents might potentially improve quality of life and prolong treatment survival in patients with PDAC (103). Interestingly, ZnO NPs nanomaterials based on Zn2+ can interfere with iron metabolism and induce ferroptosis by regulating iron intake, storage and export. This finding confirms that both ZnO nanoparticles and Zn2+ can induce ferroptosis (8). Therefore, zinc supplementation provides a potential therapeutic strategy to sensitize tumor therapy and induce tumor cell death.
The trace element selenium (Se) was discovered by Jons Jacob Berzelius in 1817 (104). Later, it was found that selenium in the human body mainly exists in the form of selenoproteins, with 25 kinds divided into two categories. There are 16 kinds of free selenoproteins in cells, the rest are located on the cell membrane, called membrane selenoproteins. Selenoproteins are rare proteins among all kingdoms of life and contain the 21st amino acid, selenocysteine. Selenocysteine is similar to cysteine, except that selenium is substituted for sulfur (105). Albeit the synthesis of selenoprotein is complicated, but it is of great significance to the life activities of humans and other mammals (106).
Studies on selenium-deficient mouse models demonstrated a close association with glutathione peroxidase 4 (GPX4) (107). GPX4 is one of 25 selenoproteins in the human body, and its expression is regulated by selenium content in cells. A study demonstrated that GPX4-deficient mice exhibited embryonic lethality and died at 7.5 days of the embryonic stage. GPX4 harbors selenocysteine making cells resistant to irreversible excessive oxidation and preventing cell ferroptosis (10). GPX4 contains sulfoxide form (GPX-SeOH) and reduction form (GPX-SeH). Under physiological conditions, oxidized GPX-SeOH can be reduced by 2 molecules of GSH to form GPX4-SHE. When GSH depletion or peroxidation occurs in cells, the accumulation of PLOOH leads to ferroptosis (Figure 5) (108). Selenium supplementation effectively inhibits GPX4-dependent ferroptosis and inhibits cell death induced by neuroexcitatory toxicity or ER stress in a GPX4-independent manner (109).
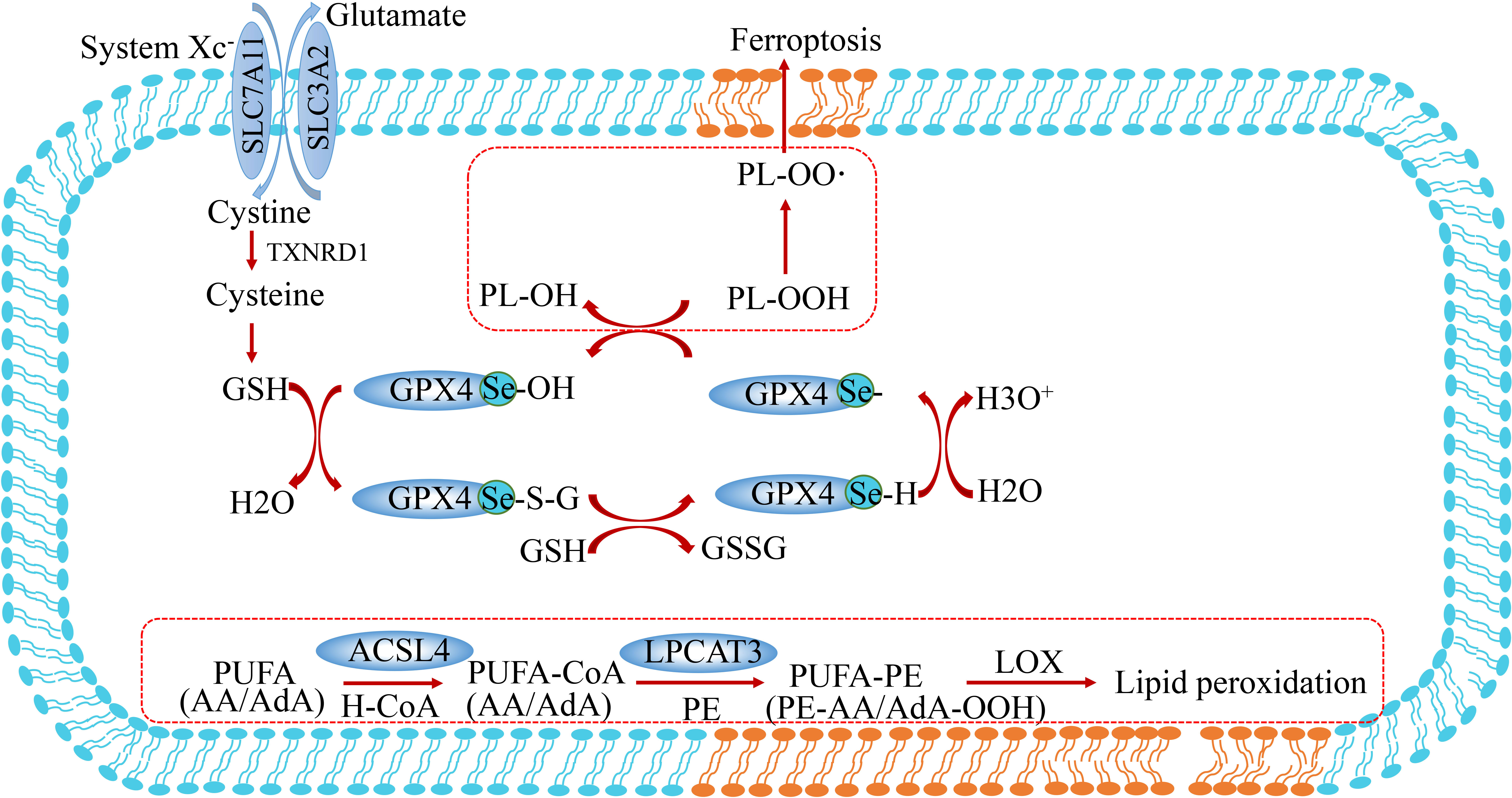
Figure 5 Lipid peroxidation and selenium metabolism in ferroptosis. These peroxides are produced when polyunsaturated fatty acids (PUFA) form PUFA-OOH catalyzed by Acyl-coenzyme A(CoA) synthetase-4 (ASCL4) and LPCAT3. GPX4 converts L-OOH into lipid alcohol (L-OH) by consuming two glutathione (GSH) molecules. PUFA, polyunsaturated fatty acid; PUFA-CoA, PUFA-coenzyme A; FSP1, ferroptosis suppressor protein 1; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3.
The catalytic action of GPX4 is indispensable for normal embryogenesis in mammals, while constitutive deletion of GPX4 has a similar effect to Trsp knockout (110). Moreover, selenium is an important regulator of ferroptosis. The utilization of selenium by GPX4 is essential to prevent ferroptosis induced by hydrogen peroxide. The effect of GPX4 on selenocysteine makes cells resistant to excessive oxidation and ferroptosis (Figure 5). SEPHS2 enzymes in the selenocysteine biosynthesis pathway participate in selenium detoxification in cancer cells, which is essential for the survival of cancer cells, highlighting the role of selenium metabolism in cancer. This research suggests that toxic metabolite produced in this process may be a potential direction for cancer treatment (111). In contrast, selenium deficiency has been proved to increase the incidence of many cancers (112). In several clinical trials, oral administration of 200 ug Se per day significantly reduced the incidence of lung, colorectal, and prostate cancers. In this respect, combining sulforaphane and selenium in food leads to synergistic upregulation of TrxR-1, which contributes to enhanced protection against free radical-mediated oxidative damage in human hepatocytes (113). The established role of selenium and GPX4 in anti-oxidation and prevention of ferroptosis suggests they may have potential value for application against the occurrence and prevention of tumors.
Chloride ions are the most abundant anions in the human body. Apart from participating in general physiological functions (regulating cell membrane potential, PH and cell volume, etc.), chloride ions can also act as intracellular signal effectors or second messengers to widely participate in the occurrence and development of diseases and tumors (114–116). There are four types of chloride channels in eukaryotes, including voltage-gated chloride channels (CLCs), ligand-gated chloride channels, and cystic fibrosis transmembrane conductance regulatory channels (CFTR) and calcium-activated chloride channels (CaCCs). Under normal circumstances, immune cells such as neutrophils maintain high levels of cellular chloride ion concentration, which is conducive to the phagocytosis of phagocytes. For example, Cl-/H+ antiporter and CFTR transporter were found on neutrophil phagosomes (117). What’s more, chloride intracellular channel 1 (CLIC1) also contributes to the acidification and ROS production of macrophage phagosomes. CLIC5 on mitochondrial intima inhibits mitochondrial ROS production by acting on mitochondrial Complex II/III (118). Under pathological conditions, the dysfunction of chloride ion transport can lead to diseases. For instance, the cystic fibrosis transmembrane conductance regulator (CFTR) mutation has been widely reported to be an important cause of pulmonary cystic fibrosis (119).
In recent years, studies have reported that an imbalance of chloride ions and channels is associated with iron-dependent cell death (Figure 6) (11, 120). TMEM16A-K (also known as Anoctamin 1-10) is a TMEM protein family consisting of 10 homologous proteins, belonging to calcium-activated chloride channels (121). TMEM16F (also known as ANO6) exposes phosphatidylserine to the cell surface by disrupting the phospholipids of the cell membrane, thereby activating a non-selective ionic current that destabilizes the plasma membrane and leads to cell death (122, 123). Further studies showed that TMEM16F is activated during ferroptosis, and the use of ferroptosis inhibitors Ferrostatin-1 or TMEM16F inhibitors can block chloride ion currents and inhibit cell death (Figure 6) (11). The ionic current generated by TMEM16F depolarizes the cell membrane, causing the cell swelling. Due to cell swelling and phospholipid disorder, the plasma membrane is unstable lysis, which further explains the process of cell ferroptosis. Therefore, activation of TMEM16F may be a new strategy against cancer growth, although there is no small molecule activator targeting TMEM16F at present (11). In addition, increased intracellular Cl- concentration exerts a pro-inflammatory effector, inducing the expression and secretion of IL-1β, induces the production of ROS, reduces the activity of mitochondrial Complex I, and indirectly regulates the process of ferroptosis (Figure 6) (116).
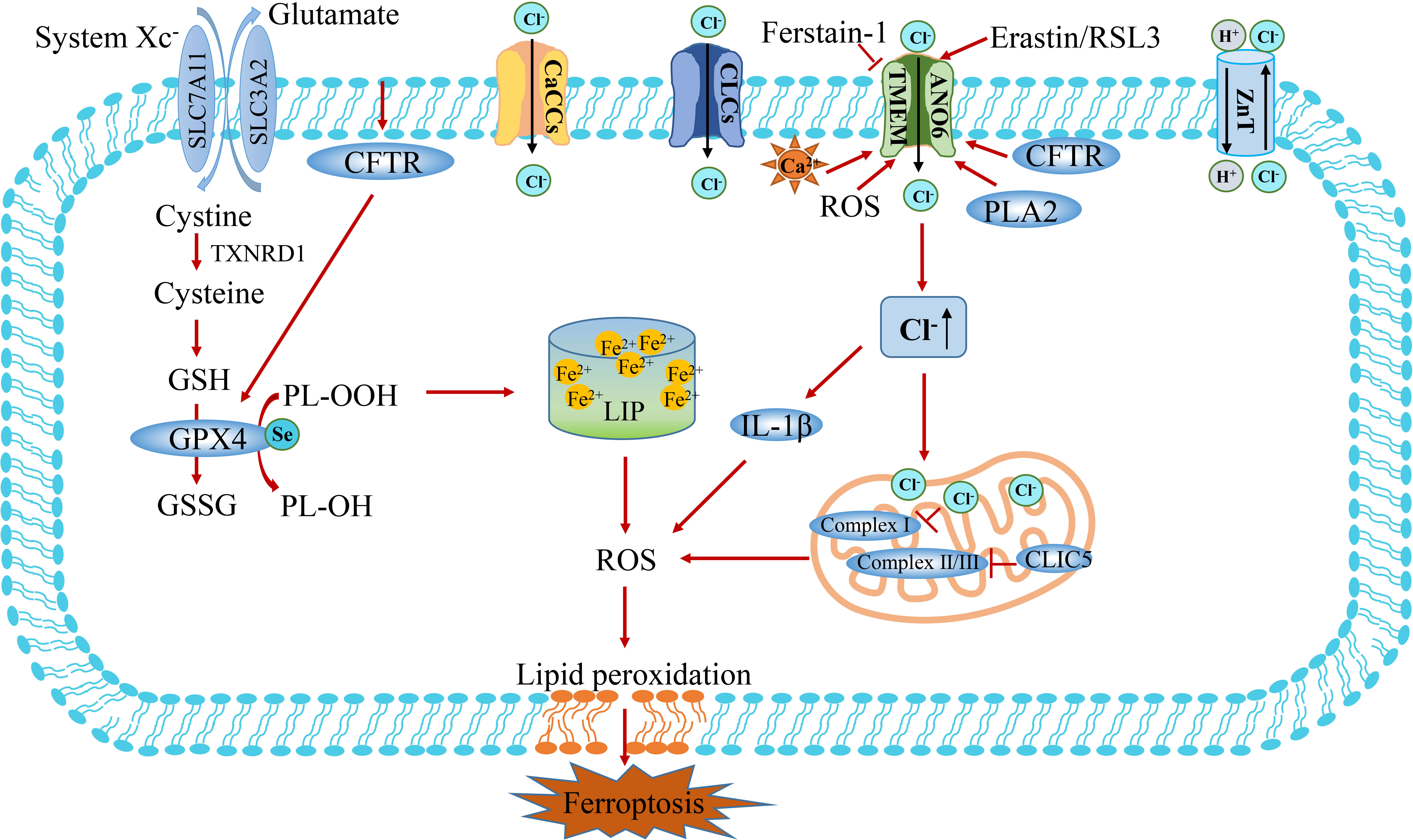
Figure 6 The crosstalk between Cl- and Fe2+ metabolism. There are four types of chloride channels in eukaryotes, including voltage-gated chloride channels (CLCs), ligand-gated chloride channels, and cystic fibrosis transmembrane conductance regulatory channels (CFTR) and calcium-activated chloride channels (CaCCs). Calcium-activated chloride channel TMEM and intracellular chloride channel protein play a vital role in bridging Cl- and ferroptosis. CLIC5 in mitochondrial intima inhibits the production of ROS by acting on mitochondrial complex II/III. TMEM16F (also known as ANO6) induces cell ferroptosis by regulating the flow of Cl-. Increased intracellular Cl- concentration can act as a pro-inflammatory effector, induce the expression of IL-1β and production of ROS, reduce the activity of mitochondrial Complex I, and indirectly regulate the process of ferroptosis. CLIC5, chloride intracellular channel protein 5; TMEM16F, transmembrane protein 16F; PLA2, phospholipase A2.
In summary, it has been established that apart from iron ions participating in the ferroptosis process, other ions (such as Ca2+, Zn2+ and Cl-) also play an important role in ferroptosis. The crosstalk between ferrous and other ions in ferroptosis is summarized in Table 1. In particular, Ca2+-induced cell death is similar to Fe2+-dependent ferroptosis, which ferroptosis inhibitors can inhibit to a certain extent (55, 65). Zn2+ and Ca2+ share many transport and signaling pathways and can be used as novel intracellular signaling molecules (87, 88, 94). Therefore, we found that Ca2+ and Zn2+ may synergistically participate in Fe2+-induced oxidative stress and ferroptosis signal. Selenium is an important component of selenoproteins (such as GPX4). Selenoproteins exert biological functions in redox signaling and antioxidant defense, playing a regulatory role in ferroptosis. Cl- is widely acknowledged as the most important anion. In recent years, it has been found that chlorine ion channel proteins are closely related to ferroptosis (11).
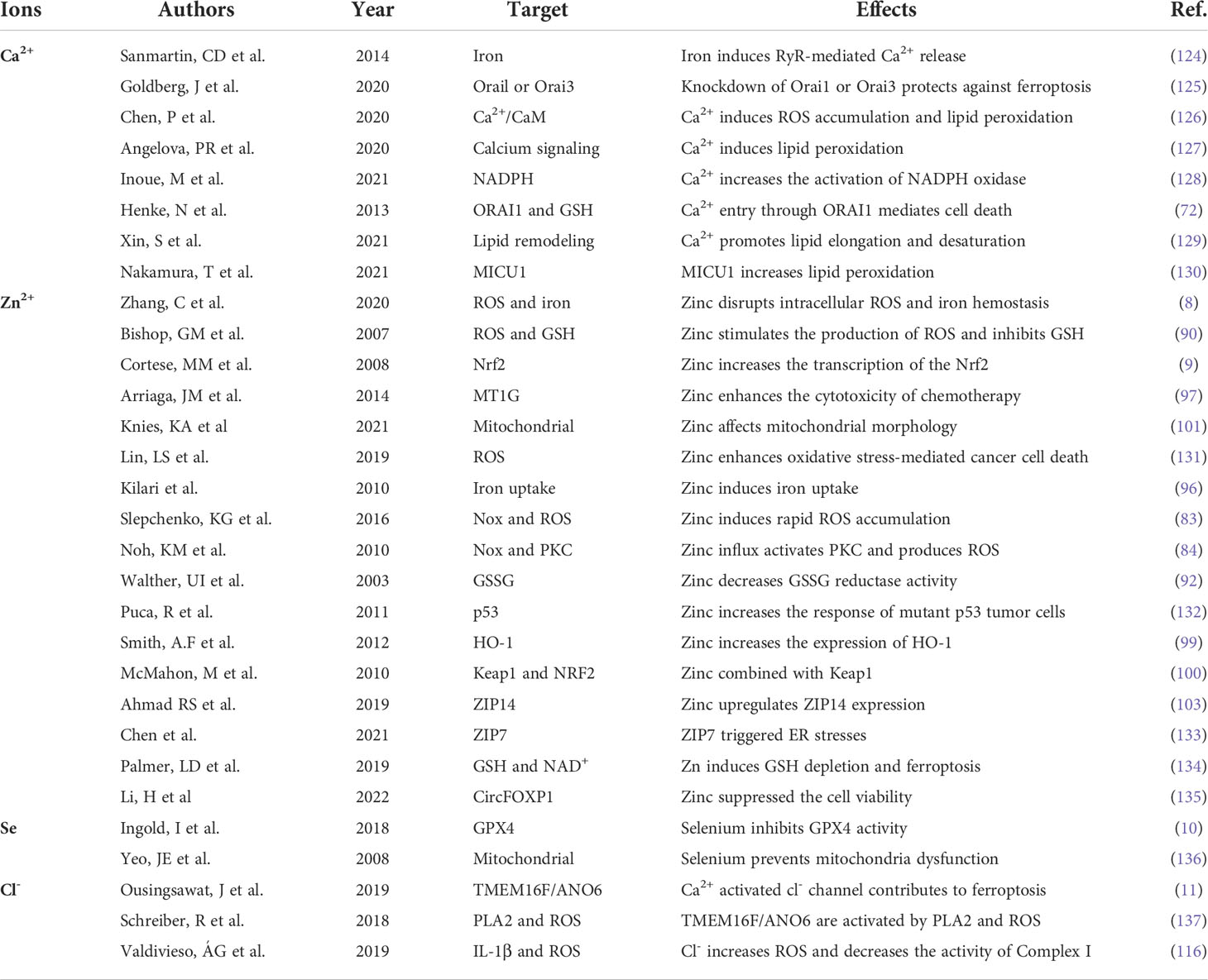
Table 1 The crosstalk between iron and other ions in ferroptosis.
Ferroptosis involves regulating cell death based on iron metabolism, which opens up new ideas and therapeutic strategies for tumor treatment. In addition, the crosstalk between ferrous ions and other ions is closely related, which may be gradually confirmed in future studies and provide a supplement for the treatment of ferroptosis-mediated tumors.
Induction of ferroptosis can sensitize anti-tumor drugs and provide novel insights for improving the therapeutic effect of tumor treatment. Studies have substantiated that several drugs commonly used clinically, such as sorafenib, cisplatin, sulfadiazine, and artesunate, can induce ferrous-induced ferroptosis in various types of cancer (138–141). Moreover, it was found that changes in oncogene and tumor suppressor gene activity, such as Ras, Kras, p53 and BAP1, can regulate the expression of ferroptosis pathway proteins and mediate ferroptosis in tumor cells (20, 142–145). What’s more, immune checkpoint inhibitors have broadened the therapeutic landscape for patients with advanced tumors. Traditional immune-induced death methods include necrosis and apoptosis, but efficiency is observed in less than 1/3 of cases. Altering the mode of tumor cell death (e.g., ferroptosis) may increase the sensitivity of tumor immune killing (4, 146, 147). Similar studies have reported that combining immunotherapy and ferroptosis may have good stacking effects on tumor treatment sensitization and overcoming drug resistance, representing important perspectives for future tumor research. Finally, tumor cells often metastasize through the lymphatic system and the blood system. Interestingly, researchers found that intravenous injection of melanoma cells produced more metastatic tumors, and lymph node melanoma cells were more resistant to ferroptosis, suggesting that ferroptosis has an anti-tumor effect on lymph node metastasis (148).
It is widely thought that ferroptosis acts as a double-edged sword for tumor treatment, with the ability to promote or inhibit tumor death. Therefore, it is necessary to further explore therapeutic strategies for inducing ferroptosis in future studies, including screening the most optimal drugs, dosage, and interval of drug use, and clarifying indications for targeting ferroptosis to regulate tumor therapy.
Iron-induced cell death plays a central role in ferroptosis, but the mechanism of how the plasma membrane ruptured leads to the final cell death remains poorly understood. It has been reported that a sustained increase of cytoplasmic Ca2+ before cell rupture and death is another characteristic of ferroptosis, suggesting that Ca2+ plays an important role in the terminal event of ferroptosis (149). Chen et al. found that a natural product isolated from Dendrobium Chrysostom Lindl, can exert an anti-lu by inducing Ca2+/CaM-dependent ferroptosis (126). These results provide substantial evidence for the synergistic involvement of Fe2+ and Ca2+ in inducing ferroptosis. In a study by Plamena R et al. excessive α-synuclein in neurons could induce abnormal calcium influx signal, further inducing lipid peroxidation and ferroptosis (127). Xin et al. found that the MS4A15 protein located in the endoplasmic reticulum inhibits lipid elongation and desaturation by consuming Ca2+ stored in the endoplasmic reticulum lumen, thus protecting phospholipids from ferroptosis. Increasing intracavitary Ca2+ levels can be used to enhance the therapeutic sensitivity of refractory tumor cells, and manipulating Ca2+ homeostasis can balance lipid and cell death pathways, providing potential clinical applications for tumor therapy (129). Cell death induced by zinc ion toxicity is well confirmed, although recent studies reported that different zinc ion concentrations might mediate different death mechanisms. Palmer and colleagues found that nanogram zinc ion exposure can induce tumor ferroptosis, providing a theoretical basis for zinc as an anticancer drug in the treatment of non-small cell lung cancer (134). Chen demonstrated that although ferroptosis must depend on ferrous ion death, zinc is also an essential element for inducing breast cancer and kidney cancer cell death. Genome-wide RNAi screening identified SLC39A7 (also named ZIP7) as a novel determinant of ferroptosis by regulating zinc from the endoplasmic reticulum to the cytoplasm (133). In addition, zinc has been found to induce iron death in lung cancer by regulating circFOXP1 expression (135). Further studies substantiated that various zinc nanomaterials can destroy intracellular ROS and iron homeostasis, and thus induce ferroptosis, suggesting their potential for tumor treatment (8).
Ferroptosis is an iron-dependent cell death process characterized by excessive accumulation of reactive oxygen species and lipid peroxidation. As the mechanism of ferroptosis has been clarified, the comprehensive regulation based on iron and other ions metabolism brings potential clinical application value for anti-tumor therapy (4). Up to now, some researchers have tried to develop artificial ion transporters with anticancer activity for tumor treatment. Targeting ion channels and oxidative stress processes offers potential applications for diagnosing and treating of liver cancer and other tumors (150–152). Iron ions and the related mechanisms of ferroptosis have been established. However, there remain some unsolved problems in the mechanism of ferroptosis between iron and other ions, such as the essential difference and relationship between Ca2+ induced oxidative glutamate toxicity and cell death process and ferroptosis. What’s more, it remains unclear whether Fe2+ changes are accompanied by changes of concentration of other ions. Interestingly, Tsvetkov and Golub recently reported a novel type of cell death caused by Cu2+, which is called cuprotosis (3). They found that cells that rely on mitochondria for energy production are more sensitive to copper ionophores than cells treated with glucose. Whether there is a connection between cuprotosis and ferroptosis is worth of further investigation. Indeed, future studies should also investigate the specific role of ferroptosis involving different ions in diseases or tumors and develop new approaches to regulate ferroptosis.
The review was designed and revised by WJ and HW. KK and LL conducted the literature collection and wrote the manuscript, which contributed equally to the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from scientific research fund of national health commission of China, key health science and technology program of Zhejiang Province (Grant No.WKJ-ZJ-2201); the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (Grant No.2022C03099); Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (Grant No.2022502425).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chen X, Kang R, Kroemer G, Tand D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
2. Dixon Scott J, Lemberg Kathryn M, Lamprecht Michael R, Skouta R, Zaitsev Eleina M, Gleason Caroline E, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
3. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Sci (New York N.Y.) (2022) 375(6586):1254–61. doi: 10.1126/science.abf0529
4. Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol (2020) 13(1):110. doi: 10.1186/s13045-020-00946-7
5. Zhou L, Zhao B, Zhang L, Wang S, Dong D, Lv H, et al. Alterations in cellular iron metabolism provide more therapeutic opportunities for cancer. Int J Mol Sci (2018) 19(5):1545–. doi: 10.3390/ijms19051545
6. Crielaard B, Lammers T, Rivella S. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discovery (2017) 16(6):400–23. doi: 10.1038/nrd.2016.248
7. Maher P, van Leyen K, Dey PN, Honrath B, Dolga A, Methner A. The role of Ca(2+) in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium (2018) 70:47–55. doi: 10.1016/j.ceca.2017.05.007
8. Zhang C, Liu Z, Zhang Y, Liu Z, Zhang Y, Song Y. "Iron free" zinc oxide nanoparticles with ion-leaking properties disrupt intracellular ROS and iron homeostasis to induce ferroptosis. Cell Death Dis (2020) 11(3):183. doi: 10.1038/s41419-020-2384-5
9. Cortese MM, Suschek CV, Wetzel W, Kroncke KD, Kolb-Bachofen V. Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free Radic Biol Med (2008) 44(12):2002–12. doi: 10.1016/j.freeradbiomed.2008.02.013
10. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell (2018) 172(3):409–22.e21. doi: 10.1016/j.cell.2017.11.048
11. Ousingsawat J, Schreiber R, Kunzelmann K. TMEM16F/Anoctamin 6 in ferroptotic cell death. Cancers (2019) 11(5):625. doi: 10.3390/cancers11050625
12. Fenton HJH. Oxidation of tartaric acid in presence of iron. J Chem Soc (1894) 65:899–0. doi: 10.1039/CT8946500899
13. Bock FJ, Tait S. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol (2019) 21(2):85–100. doi: 10.1038/s41580-019-0173-8
14. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94(3):909. doi: 10.1152/physrev.00026.2013
15. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev (2007) 87(1):245–313. doi: 10.1152/physrev.00044.2005
16. Taylor-Fishwick DA. NOX, NOX who is there? the contribution of NADPH oxidase one to beta cell dysfunction. Front Endocrinol (2013) 4:40. doi: 10.3389/fendo.2013.00040
17. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol (2014) 10(1):9. doi: 10.1038/nchembio.1416
18. Galluzzi L, Vitale I, Aaronson SA, Kroemer G. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
19. Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids (2012) 42(1):231–46. doi: 10.1007/s00726-011-0867-5
20. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science (2020) 368(6486):85–9. doi: 10.1126/science.aaw9872
21. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, et al. Pharmacological inhibition of cystine- glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
22. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology (2016) 63(1):173–84. doi: 10.1002/hep.28251
23. Fan Z, Wirth AK, Chen D, Wruck C J, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis (2017) 6(8):e371. doi: 10.1038/oncsis.2017.65
24. Kan X, Yin Y, Song C, Tan L, Ding C. Newcastle Disease virus induced ferroptosis through p53-SLC7A11-GPX4 axis mediated nutrient deprivation in tumor cells. SSRN (2021) 24(8). doi: 10.1101/2021.01.03.424919
25. Huang Y, Dai Z, Barbacioru C, Sadée W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res (2005) 65(16):7446–54. doi: 10.1158/0008-5472.CAN-04-4267
26. Liu X, Li X, Zhang B, Liang Y, Zhou C, Cao D, et al. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett (2011) 585(9):1363–7. doi: 10.1016/j.febslet.2011.04.018
27. Roh J, Kim E, Jang H, Park J, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett (2016) 381(1):96–103. doi: 10.1016/j.canlet.2016.07.035
28. Bersuker K, Hendricks J, Li Z, Magtanong L, Ford B, Tang P, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
29. Doll S, Freitas F, Shah R, Aldrovandi M, da Silva M, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
30. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell (2021) 81(2):355–69.e10. doi: 10.1016/j.molcel.2020.11.024
31. Fill M, Copello J. Ryanodine receptor calcium release channels. Physiol Rev (2002) 82(4):893–922. doi: 10.1152/physrev.00013.2002
32. Shimada K, Skouta R, Kaplan A, Yang W, Hayano M, Dixon S, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol (2016) 12(7):497–503. doi: 10.1038/nchembio.2079
33. Kraft V, Bezjian C, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci (2020) 6(1):41–53. doi: 10.1021/acscentsci.9b01063
34. Jiang X, Stockwell B, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
35. Anandhan A, Dodson M, Schmidlin C, Liu P, Zhang D. Breakdown of an ironclad defense system: The critical role of NRF2 in mediating ferroptosis. Cell Chem Biol (2020) 27(4):436–47. doi: 10.1016/j.chembiol.2020.03.011
36. Rojo de la Vega M, Chapman E, Zhang D. NRF2 and the hallmarks of cancer. Cancer Cell (2018) 34(1):21–43. doi: 10.1016/j.ccell.2018.03.022
37. Jaramillo M, Zhang D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev (2013) 27(20):2179–91. doi: 10.1101/gad.225680.113
38. Zhang L, Zhang J, Jin Y, Yao G, Zhao H, Qiao P, et al. Nrf2 is a potential modulator for orchestrating iron homeostasis and redox balance in cancer cells. Front Cell Dev Biol (2021) 9:728172. doi: 10.3389/fcell.2021.728172
39. Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell (2017) 68(1):224–32.e4. doi: 10.1016/j.molcel.2017.09.009
40. Wu Y, Yu C, Luo M, Cen C, Qiu J, Zhang S, et al. Ferroptosis in cancer treatment: Another way to Rome. Front Oncol (2020) 10:571127. doi: 10.3389/fonc.2020.571127
41. Koppula P, Zhuang L, Gan B. Cytochrome P450 reductase (POR) as a ferroptosis fuel. Protein Cell (2021) 12:675–9. doi: 10.1007/s13238-021-00823-0
42. Brown C, Amante J, Chhoy P, Elaimy A, Liu H, Zhu L, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell (2019) 51(5):575–86.e4. doi: 10.1016/j.devcel.2019.10.007
43. Singh M, Sanderson P, Hurrell RF, Fa Irweather-Tait SJ, Geissler C, Prentice A, et al. Iron bioavailability. Br J Nutr (2006) 96(05):985–90. doi: 10.1017/BJN20061894
44. Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol (2007) 69(1):69–85. doi: 10.1146/annurev.physiol.69.031905.164337
45. Feng H, Schorpp K, Jin J, Yozwiak CE, Stockwell BR. Transferrin receptor is a specific ferroptosis marker. Cell Rep (2020) 30(10):3411–23.e7. doi: 10.1016/j.celrep.2020.02.049
46. Dautry-Varsat A, Ciechanover A, Lodish H. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc Natl Acad Sci United States America (1983) 80(8):2258–62. doi: 10.1073/pnas.80.8.2258
47. Hubert N, Hentze M. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci United States America (2002) 99(19):12345–50. doi: 10.1073/pnas.192423399
48. Bou-Abdallah and F. The iron redox and hydrolysis chemistry of the ferritins. Biochim Et Biophys Acta (2010) 1800:719–31. doi: 10.1016/j.bbagen.2010.03.021
49. Ganz T. Cellular iron: ferroportin is the only way out. Cell Metab (2005) 1(3):155–7. doi: 10.1016/j.cmet.2005.02.005
50. Terzi E, Sviderskiy V, Alvarez S, Sviderskiy V, Alvarez S, Whiten G, Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv (2021) 7(22):1–11. doi: 10.1126/sciadv.abg4302
51. Torti S, Manz D, Paul B, Blanchette-Farra N, Torti F. Iron and cancer. Annu Rev Nutr (2018) 38:97–125. doi: 10.1146/annurev-nutr-082117-051732
52. Chen Y, Zhang S, Wang X, Guo W, Wang L, Zhang D, et al. Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell signalling (2015) 27(1):168–76. doi: 10.1016/j.cellsig.2014.11.002
53. Choi J, Neuhouser M, Barnett M, Hong C, Kristal A, Thornquist M, et al. Iron intake, oxidative stress-related genes (MnSOD and MPO) and prostate cancer risk in CARET cohort. Carcinogenesis (2008) 29(5):964–70. doi: 10.1093/carcin/bgn056
54. Quintana Pacheco D, Sookthai D, Graf M, Schübel R, Johnson T, Katzke V, et al. Iron status in relation to cancer risk and mortality: Findings from a population-based prospective study. Int J Cancer (2018) 143(3):561–9. doi: 10.1002/ijc.31384
55. Berridge M, Lipp P, Bootman M. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol (2000) 1(1):11–21. doi: 10.1038/35036035
56. Bettaieb L, Brulé M, Chomy A, Diedro M, Fruit M, Happernegg E, et al. Ca Signaling and its potential targeting in pancreatic ductal carcinoma. Cancers (2021) 13(12):3085–105. doi: 10.3390/cancers13123085
57. Woll K, Van Petegem F. Calcium release channels: Structure and function of IP3 receptors and ryanodine receptors. Physiol Rev (2022) 102:209–68. doi: 10.1152/physrev.00033.2020
58. Maus M, Cuk M, Patel B, Lian J, Ouimet M, Kaufmann U, et al. Store-operated Ca entry controls induction of lipolysis and the transcriptional reprogramming to lipid metabolism. Cell Metab (2017) 25(3):698–712. doi: 10.1016/j.cmet.2016.12.021
59. Stathopulos P, Zheng L, Li G, Plevin M, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell (2008) 135(1):110–22. doi: 10.1016/j.cell.2008.08.006
60. Zhang S, Yu Y, Roos J, Kozak J, Deerinck T, Ellisman M, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature (2005) 437(7060):902–5. doi: 10.1038/nature04147
61. Yeromin A, Zhang S, Jiang W, Yu Y, Safrina O, Cahalan M, et al. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of orai. Nature (2006) 443(7108):226–9. doi: 10.1038/nature05108
62. Zhivotovsky B, Orrenius S. Calcium and cell death mechanisms: a perspective from the cell death community. Cell calcium (2011) 50(3):211–21. doi: 10.1016/j.ceca.2011.03.003
63. Kamer K, Mootha V. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol (2015) 16(9):545–53. doi: 10.1038/nrm4039
64. Nita L, Hershfinkel M, Sekler I. Life after the birth of the mitochondrial Na+/Ca2+ exchanger, NCLX. Sci China Life Sci (2015) 58(1):59–65. doi: 10.1007/s11427-014-4789-9
65. Maher P, van Leyen K, Dey P, Honrath B, Dolga A, Methner A, et al. The role of Ca in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell calcium (2018) 70:47–55. doi: 10.1016/j.ceca.2017.05.007
66. Murphy TH, Miyamoto M, Sastre A, Schnaar R L, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron (1989) 2(6):1547–58. doi: 10.1016/0896-6273(89)90043-3
67. Shirlee Tan DS, Maher P. Oxytosis: A novel form of programmed cell death. Curr Topics Med Chem (2001) 1(6):497–506. doi: 10.2174/1568026013394741
68. Kang Y, Tiziani S, Park G, Kaul M, Paternostro G. Cellular protection using Flt3 and PI3Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat Commun (2014) 5:3672. doi: 10.1038/ncomms4672
69. CHOI DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron (1988) 1(8):623–34. doi: 10.1016/0896-6273(88)90162-6
70. Li and Y. Requirement for cGMP in nerve cell death caused by glutathione depletion. J Cell Biol (1997) 139(5):1317–24. doi: 10.1083/jcb.139.5.1317
71. Davis JB, Maher P. Protein kinase c activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res (1994) 652(1):169–73. doi: 10.1016/0006-8993(94)90334-4
72. Henke N, Albrecht P, Bouchachia I, Ryazantseva M, Knoll K, Lewerenz J, et al. The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis (2013) 4(1):e470. doi: 10.1038/cddis.2012.216
73. Mccall KA, Huang C, Fierke CA. Function and mechanism of zinc metalloenzymes. J Nutr (2000) 130(5S Suppl):1437S. doi: 10.1093/jn/130.5.1437S
74. Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci (2009) 10(11):780–91. doi: 10.1038/nrn2734
75. Kambe T. An overview of a wide range of functions of ZnT and zip zinc transporters in the secretory pathway. Biosci Biotechnol Biochem (2011) 75(6):1036–43. doi: 10.1271/bbb.110056
76. Fleming M, Romano MA, Su MA, Garrick LM, Garrick M, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal irontransport. Proc Natl Acad Sci United States America (1998) 95(3):1148–53. doi: 10.1073/pnas.95.3.1148
77. Maret W. Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc Natl Acad Sci (1994) 91(1):237–41. doi: 10.1073/pnas.91.1.237
78. Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal (2006) 8(9-10):1419–41. doi: 10.1089/ars.2006.8.1419
79. Yamasaki S, Sakata-Sogawa K, Hasegawa A, Suzuki T, Hirano T. Zinc is a novel intracellular second messenger. J Cell Biol (2007) 177(4):637–45. doi: 10.1083/jcb.200702081
80. Powell SR. The antioxidant properties of zinc. J Nutr (2000) 130(5):1447S–54S. doi: 10.1093/jn/130.5.1447S
81. Bray TM, Bettger WJ. The physiological role of zinc as an antioxidant - ScienceDirect. Free Radical Biol Med (1990) 8(3):281–91. doi: 10.1016/0891-5849(90)90076-U
82. Girotti AW, Thomas JP, Jordan JE. Inhibitory effect of zinc (II) on free radical lipid peroxidation in erythrocyte membranes. J Free Radicals Biol Med (1985) 1(5):395–401. doi: 10.1016/0748-5514(85)90152-7
83. Slepchenko KG, Lu Q, Yang VL. Zinc wave during the treatment of hypoxia is required for initial reactive oxygen species activation in mitochondria. Int J Physiol Pathophysiol Pharmacol (2016) 8(1):44–51.
84. Noh KM, Yang HK, Koh JY. Mediation by membrane protein kinase c of zinc-induced oxidative neuronal injury in mouse cortical cultures. J Neurochem (2010) 72(4):1609–16. doi: 10.1046/j.1471-4159.1999.721609.x
85. Kim YH, Koh JY. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced Poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol (2002) 177(2):407–18. doi: 10.1006/exnr.2002.7990
86. Bozym RA, Chimienti F, Giblin LJ, Gross G W, Korichneva I, Li Y, et al. Original research free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Exp Biol Med (2010) 235(6):741–50. doi: 10.1258/ebm.2010.009258
87. Sensi SL, Yin HZ, Carriedo SG. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad USA (1999) 96(5):2414–9. doi: 10.1073/pnas.96.5.2414
88. Sensi SL, Yin HZ, Weiss JH. Glutamate triggers preferential Zn2+ flux through Ca2+ permeable AMPA channels and consequent ROS production. Neuroreport (1999) 10(8):1723–7. doi: 10.1097/00001756-199906030-00018
89. Fridovich I. Superoxide radical and superoxide dismutase. Accounts Chem Res (1972) 5(10):321–26. doi: 10.1021/ar50058a001
90. Bishop GM, Dringen R, Robinson SR. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radical Biol Med (2007) 42(8):1222–30. doi: 10.1016/j.freeradbiomed.2007.01.022
91. Dineley KE, Richards LL, Votyakova TV, Reynolds IJ. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion (2005) 5(1):55–65. doi: 10.1016/j.mito.2004.11.001
92. Walther UI, Czermak A, Mückter H, Walther SC, Fichtl B. Decreased GSSG reductase activity enhances cellular zinc toxicity in three human lung cell lines. Arch Toxicol (2003) 77(3):131–7. doi: 10.1007/s00204-002-0421-z
93. Sre Ed Har B, Subramaniyan R, Nair KM. A protective role for zinc on intestinal peroxidative damage during oral iron repletion. Biochem Biophys Res Commun (2004) 318(4):992–7. doi: 10.1016/j.bbrc.2004.04.132
94. Olivares M, Pizarro F, Ruz M. Zinc inhibits nonheme iron bioavailability in humans. Biol Trace Elem Res (2007) 117(1-3):7–14. doi: 10.1007/BF02698079
95. Arredondo M, MARTÍNEZ R, NÚÑEZ MT, Ruz M, Olivares M. Inhibition of iron and copper uptake by iron, copper and zinc. Biol Res (2006) 39(1):95–102. doi: 10.4067/S0716-97602006000100011
96. Kilari S, Pullakhandam R, Nair KM. Zinc inhibits oxidative stress-induced iron signaling and apoptosis in caco-2 cells. Free Radical Biol Med (2010) 48(7):961–8. doi: 10.1016/j.freeradbiomed.2010.01.019
97. Arriaga JM, Greco A, Mordoh J, Bianchini M. Metallothionein 1G and zinc sensitize human colorectal cancer cells to chemotherapy. Mol Cancer Ther (2014) 13(5):1369–81. doi: 10.1158/1535-7163.MCT-13-0944
98. Margalit O, Simon AJ, Yakubov E, Puca R, Yosepovich A, Avivi C, et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int J Cancer (2012) 131(4):E562–8. doi: 10.1002/ijc.26441
99. Smith AF, Loo G. Upregulation of haeme oxygenase-1 by zinc in HCT-116 cells. Free Radical Res (2012) 46(9):1099–107. doi: 10.3109/10715762.2012.690872
100. McMahon M, Lamont D, Beattie K, Hayes J. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci United States America (2010) 107(44):18838–43. doi: 10.1073/pnas.1007387107
101. Knies KA, Li YV. Zinc cytotoxicity induces mitochondrial morphology changes in hela cell line. Int J Physiol Pathophysiol Pharmacol (2021) 13(2):43–51.
102. Wu K, Fei L, Wang X, Lei Y, Liu Y, Xu W, et al. ZIP14 is involved in iron deposition and triggers ferroptosis in diabetic nephropathy. Metallomics integrated biometal Sci (2022) 14(7) mfac034. doi: 10.1093/mtomcs/mfac034
103. Shakri AR, Zhong TJ, Ma W, Coker C, Acharyya S. Upregulation of ZIP14 and altered zinc homeostasis in muscles in pancreatic cancer cachexia. Cancers (2019) 12(3):1–17. doi: 10.3390/cancers12010003
104. Reich H, Hondal R. Why nature chose selenium. ACS Chem Biol (2016) 11(4):821–41. doi: 10.1021/acschembio.6b00031
105. Hatfield D, Tsuji P, Carlson B, Gladyshev V. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci (2014) 39(3):112–20. doi: 10.1016/j.tibs.2013.12.007
107. Bösl MR, Takaku K, Oshima M, Taketo NMM. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene(Trsp). Proc Natl Acad Sci United States America (1997) 94(11):5531–4. doi: 10.1073/pnas.94.11.5531
108. Conrad M, Proneth B. Selenium: Tracing another essential element of ferroptotic cell death. Cell Chem Biol (2020) 27(4):409–19. doi: 10.1016/j.chembiol.2020.03.012
109. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell (2019) 177(5):1262–1279 e25. doi: 10.1016/j.cell.2019.03.032
110. Yant L, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter J, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radical Biol Med (2003) 34(4):496–502. doi: 10.1016/S0891-5849(02)01360-6
111. Carlisle A, Lee N, Matthew-Onabanjo A, Spears M, Park S, Youkana D, et al. Selenium detoxification is required for cancer-cell survival. Nat Metab (2020) 2(7):603–11. doi: 10.1038/s42255-020-0224-7
112. Clark LC, Combs GF, Turnbull BW, Slate EH, Dan KC, Chow J, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin: A randomized controlled trial. JAMA (1996) 276(24):1957–63. doi: 10.1001/jama.1996.03540240035027
113. Li D, Wang W, Shan Y, Barrera LN, Howie AF, Beckett GJ, et al. Synergy between sulforaphane and selenium in the up-regulation of thioredoxin reductase and protection against hydrogen peroxide-induced cell death in human hepatocytes. Food Chem (2012) 133(2):300–7. doi: 10.1016/j.foodchem.2012.01.026
114. Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol (2010) 72(1):95–121. doi: 10.1146/annurev-physiol-021909-135811
115. Graves AR, Curran PK, et al. The cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature (2008) 2:309. doi: 10.1038/nature06907
116. Valdivieso AG, Santa-Coloma TA. The chloride anion as a signalling effector. Biol Rev Camb Philos Soc (2019) 94(5):1839–56. doi: 10.1111/brv.12536
117. Painter RG, Wang G. Direct measurement of free chloride concentrations in the phagolysosomes of human neutrophils. Analytical Chem (2006) 78(9):3133–7. doi: 10.1021/ac0521706
118. Ponnalagu D, Gururaja Rao S, Farber J, Xin W, Hussain AT, Shah K, et al. Molecular identity of cardiac mitochondrial chloride intracellular channel proteins. Mitochondrion (2016) 27:6–14. doi: 10.1016/j.mito.2016.01.001
119. Paulino C, Kalienkova V, Lam A, Neldner Y, Dutzler R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature (2017) 552:421–5. doi: 10.1038/nature24652
120. Simões F, Ousingsawat J, Wanitchakool P, Fonseca A, Cabrita I, Benedetto R, et al. CFTR supports cell death through ROS-dependent activation of TMEM16F (anoctamin 6). Pflügers Archiv - Eur J Physiol (2017) 470:305–14. doi: 10.1007/s00424-017-2065-0
121. Tian Y, Schreiber R, Kunzelmann K. Anoctamins are a family of Ca2+-activated cl channels. J Cell Sci (2012) 125(21):4991–8. doi: 10.1242/jcs.109553
122. Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature (2010) 468(7325):834. doi: 10.1038/nature09583
123. Grubb S, Poulsen KA, et al. TMEM16F (Anoctamin 6), an anion channel of delayed Ca2+activation. J Gen Physiol (2013) 141(5):585–600. doi: 10.1085/jgp.201210861
124. Sanmartín C, Paula-Lima A, García A, Barattini P, Hartel S, Núñez M, et al. Ryanodine receptor-mediated Ca(2+) release underlies iron-induced mitochondrial fission and stimulates mitochondrial Ca(2+) uptake in primary hippocampal neurons. Front Mol Neurosci (2014) 7:13. doi: 10.3389/fnmol.2014.00013
125. Goldberg J, Currais A, Ates G, Huang L, Shokhirev M, Maher P, et al. Targeting of intracellular Ca stores as a therapeutic strategy against age-related neurotoxicities. NPJ Aging Mech Dis (2020) 6:10. doi: 10.1038/s41514-020-00048-1
126. Chen P, Wu Q, Feng J, Yan L, Sun Y, Liu S. Erianin, a novel dibenzyl compound in dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal transduct targeted Ther (2020) 5(1):51. doi: 10.1038/s41392-020-0149-3
127. Angelova P, Choi M, Berezhnov A, Horrocks M, Hughes C, De S, et al. Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death differ (2020) 27(10):2781–96. doi: 10.1038/s41418-020-0542-z
128. Inoue M, Enomoto M, Yoshimura M, Mizowaki T. Pharmacological inhibition of sodium-calcium exchange activates NADPH oxidase and induces infection-independent NETotic cell death. Redox Biol (2021) 43:101983. doi: 10.1016/j.redox.2021.101983
129. Xin S, Mueller C, Pfeiffer S, Kraft V, Merl-Pham J, Bao X, et al. MS4A15 drives ferroptosis resistance through calcium-restricted lipid remodeling. Cell Death differ (2022) 29(3):670–86. doi: 10.1038/s41418-021-00883-z
130. Nakamura T, Ogawa M, Kojima K, Takayanagi S, Ishihara S, Hattori K, et al. The mitochondrial Ca uptake regulator, MICU1, is involved in cold stress-induced ferroptosis. EMBO Rep (2021) 22(5):e51532. doi: 10.15252/embr.202051532
131. Lin L, Wang J, Song J, Liu Y, Zhu G, Dai Y, et al. Cooperation of endogenous and exogenous reactive oxygen species induced by zinc peroxide nanoparticles to enhance oxidative stress-based cancer therapy. Theranostics (2019) 9(24):7200–9. doi: 10.7150/thno.39831
132. Puca R, Nardinocchi L, Porru M, Simon A, Rechavi G, Leonetti C, et al. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle (Georgetown Tex.) (2011) 10(10):1679–89. doi: 10.4161/cc.10.10.15642
133. Chen P, Wu J, Xu Y, Ding C, Mestre A, Lin C, et al. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis (2021) 12(2):198. doi: 10.1038/s41419-021-03482-5
134. Palmer L, Jordan A, Maloney K, Farrow M, Gutierrez D. Zinc intoxication induces ferroptosis in A549 human lung cells. Metallomics integrated biometal Sci (2019) 11(5):982–93. doi: 10.1039/c8mt00360b
135. Li H, Liu L. Zinc moderates circular RNA CircFOXP1 expression in order to regulate ferroptosis during lung adenocarcinoma. Chemico-biol Interact (2022) 352:109760. doi: 10.1016/j.cbi.2021.109760
136. Yeo J, Kim J, Kang S. Selenium attenuates ROS-mediated apoptotic cell death of injured spinal cord through prevention of mitochondria dysfunction; in vitro and in vivo study. Cell Physiol Biochem Int J Exp Cell physiol biochem Pharmacol (2008) 21:225–38. doi: 10.1159/000113764
137. Schreiber R, Ousingsawat J, Wanitchakool P, Sirianant L, Benedetto R, Reiss K, et al. Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca and plasma membrane lipid. J Physiol (2018) 596(2):217–29. doi: 10.1113/JP275175
138. Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res (2014) 34(11):6417–22. doi: 10.1007/978-3-642-27841-9_7139-1
139. Zhang X, Sui S, Wang L, Li H, Zhang L, Xu S, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol (2020) 235(4):3425–37. doi: 10.1002/jcp.29232
140. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res Treat (2018) 50(2):445–60. doi: 10.4143/crt.2016.572
141. Chen G, Benthani F, Wu J, Liang D, Bian Z, Jiang X, et al. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death differ (2020) 27(1):242–54. doi: 10.1038/s41418-019-0352-3
142. Yang W, Stockwell B. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol (2008) 15(3):234–45. doi: 10.1016/j.chembiol.2008.02.010
143. Jiang H, Muir RK, Gonciarz RL, Olshen AB, Renslo AR. Exploiting KRAS-driven ferroaddiction in cancer through ferrous iron-activatable drug conjugates (FeADC). Cold Spring Harbor Lab (2020). doi: 10.1101/2020.05.12.088971
144. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radical Biol Med (2019) 133:162–8. doi: 10.1016/j.freeradbiomed.2018.05.074
145. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell (2021) 12(8):599–620. doi: 10.1007/s13238-020-00789-5
146. Wang W, Green M, Choi J, Gijón M, Kennedy P, Johnson J, et al. CD8 T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
147. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain treg cell activation and suppression of antitumor immunity. Cell Rep (2021) 35(11):109235. doi: 10.1016/j.celrep.2021.109235
148. Ubellacker J, Tasdogan A, Ramesh V, Shen B, Mitchell E, Martin-Sandoval M, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature (2020) 585(7823):113–8. doi: 10.1038/s41586-020-2623-z
149. Pedrera L, Espiritu R, Ros U, Weber J, Schmitt A, Stroh J, et al. Ferroptotic pores induce Ca fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death differ (2021) 28(5):1644–57. doi: 10.1038/s41418-020-00691-x
150. Park SH, Park SH, Howe ENW, Hyun JY, Chen LJ, Hwang I, et al. Determinants of ion-transporter cancer cell death. Chem (2019) 5(8):2079–98. doi: 10.1016/j.chempr.2019.05.001
151. Busschaert N, Park SH, Baek KH, Choi YP, Park J, Howe ENW, et al. A synthetic ion transporter that disrupts autophagy and induces apoptosis by perturbing cellular chloride concentrations. Nat Chem (2017) 9(7):667–75. doi: 10.1038/nchem.2706
Keywords: ion metabolism, ferroptosis, glutathione metabolism, lipid peroxidation, cancer therapy
Citation: Ke K, Li L, Lu C, Zhu Q, Wang Y, Mou Y, Wang H and Jin W (2022) The crosstalk effect between ferrous and other ions metabolism in ferroptosis for therapy of cancer. Front. Oncol. 12:916082. doi: 10.3389/fonc.2022.916082
Received: 08 April 2022; Accepted: 19 July 2022;
Published: 12 August 2022.
Edited by:
Wei Cheng, Dalian Medical University, ChinaReviewed by:
Devanjan Sinha, Banaras Hindu University, IndiaCopyright © 2022 Ke, Li, Lu, Zhu, Wang, Mou, Wang and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiwei Jin, amlud3dAemp1LmVkdS5jbg==; Huiju Wang, d2hqMzM0NEAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.