 Wiktoria Strasenburg1*†
Wiktoria Strasenburg1*† Jakub Jóźwicki1†
Jakub Jóźwicki1† Justyna Durślewicz1
Justyna Durślewicz1 Błażej Kuffel2Martyna Parol Kulczyk1Adam Kowalewski1Dariusz Grzanka1Tomasz Drewa2
Błażej Kuffel2Martyna Parol Kulczyk1Adam Kowalewski1Dariusz Grzanka1Tomasz Drewa2 Jan Adamowicz2
Jan Adamowicz2- 1Department of Clinical Pathomorphology, Faculty of Medicine, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University, Toruń, Poland
- 2Department of General and Oncological Urology, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University in Toruń, Toruń, Poland
Tumor cells have the ability to induce platelet activation and aggregation. This has been documented to be involved in tumor progression in several types of cancers, such as lung, colon, breast, pancreatic, ovarian, and brain. During the process, platelets protect circulating tumor cells from the deleterious effects of shear forces, shield tumor cells from the immune system, and provide growth factors, facilitating metastatic spread and tumor growth at the original site as well as at the site of metastasis. Herein, we present a wider view on the induction of platelet aggregation by specific factors primarily developed by cancer, including coagulation factors, adhesion receptors, growth factors, cysteine proteases, matrix metalloproteinases, glycoproteins, soluble mediators, and selectins. These factors may be presented on the surface of tumor cells as well as in their microenvironment, and some may trigger more than just one simple receptor–ligand mechanism. For a better understanding, we briefly discuss the physiological role of the factors in the platelet activation process, and subsequently, we provide scientific evidence and discuss their potential role in the progression of specific cancers. Targeting tumor cell-induced platelet aggregation (TCIPA) by antiplatelet drugs may open ways to develop new treatment modalities. On the one hand, it may affect patients’ prognosis by enhancing known therapies in advanced-stage tumors. On the other hand, the use of drugs that are mostly easily accessible and widely used in general practice may be an opportunity to propose an unparalleled antitumor prophylaxis. In this review, we present the recent discoveries of mechanisms by which cancer cells activate platelets, and discuss new platelet-targeted therapeutic strategies.
Introduction
The primary hemostatic function of platelets is well known; however, increasing evidence supports the crucial role of platelets in cancer biology (1, 2). In 1865 Armand Trousseau first described cases of thrombophlebitis in patients with cancer. He emphasized the association of malignancies with the creation of venous and arterial platelet–rich microthrombi in the vasculature (3). Further studies demonstrated that tumor cells can induce platelet activation and aggregation. This mechanism is now called tumor cell–induced platelet aggregation (TCIPA) (4). TCIPA has been documented to be involved in tumorigenesis in several types of cancers including breast (5)lung (6)and pancreatic (7). During TCIPAplatelets protect circulating tumor cells from the deleterious effects of shear forces and also preserve the tumor cells from the immune system by creating a physical barrier around cancer cells (8). These actions may contribute to metastatic spread and tumor growth (2). One of the reasons why TCIPA is in the spotlight of current research is the explored possibility to involve antiplatelet drugs in cancer therapy. Targeting TCIPA with antiplatelet drugs may open new ways to affect cancer environment and develop new treatment modalities. In this reviewwe present the recent discoveries of mechanisms explaining platelet activation by cancer cells. Moreoverwe discuss new platelet–targeted therapeutic strategies as potential inhibitors of TCIPA (Figure 1).
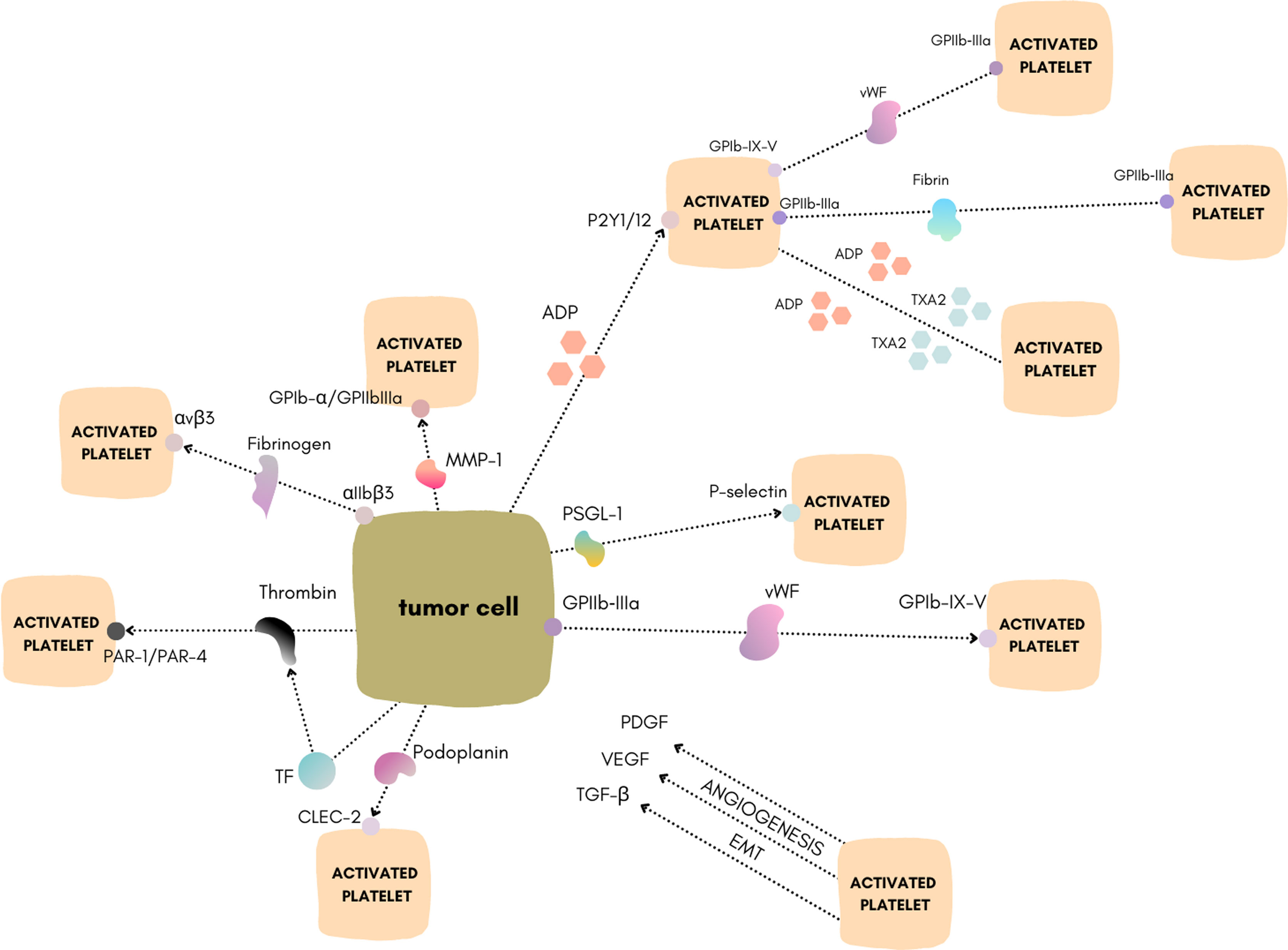
Figure 1 Schematic of the different mechanisms of TCIPA. Figure created using https://www.canva.com/.
Clotting Factors
Thrombin
Thrombin is a serine proteaseplaying a pivotal role in blood coagulation (9). It converts fibrinogen into fibrin and activates various coagulation factorsincluding VVIIIXIand XIIIand the protease–activated receptors (PAR) on plateletsendothelial cellsmyocytesand neurons (2). In this processreceptorssuch as PAR–1PAR–3and PAR–4are activated (1). PAR–1 is the most effective receptor for thrombin (10). There is increasing evidence suggesting a crucial role of thrombin in cancer biology (11). Thrombin is proven to be generated by lung cancer cells (6). Thrombin–activated platelets express factors that facilitate contact with tumor cells andin turnenhance TCIPA through full activation of specific membrane receptors on platelets (12). Similarlyoverexpression of the PAR–1 receptor was associated with cancer progression and development. As demonstrated by Cisowski et al.silencing or pharmacologic blocking of PAR–1 results in a significant decrease in motility of the lung cancer cell lines A549 and HOP62 (13). PAR–4 is overexpressed in colorectal and prostate cancer and PAR–3 in kidney and liver cancer (14). Thrombin also increases the surface exposure of GPIIb–IIIa on platelets and tumor cellsthereby enhancing the interactions between tumor cells and platelets (15). Another potential mechanism linking thrombin with tumorigenesis is its major function during fibrinogen activation and conversion to fibrin. Thrombin may promote abnormal and upregulated fibrin deposition within the tumor matrix. Fibrin itself has also binding motifs for numerous integrins like GPIIb–IIIa and αVβ3making it capable of influencing numerous cell types including platelets and tumor cells (12). Interestinglythrombin may also contribute to breast cancer metastasis via other factorsunrelated to the TCIPA mechanism. It cleaves osteopontin (OPN) andthusincreases its biological activity. Schulze et al. reported that inhibition of thrombin in breast cancer cells overexpressing OPN leads to its more indolent behavior (16).
Tissue Factor
Tissue factor (TF) is a membrane glycoprotein that is crucial to initiate the extrinsic coagulation cascade (17). Expression of TF has been detected in several types of cancersincluding breast cancer (18)prostate cancer (19)and lung cancer (20). TF is expressed on the cell membrane to activate the plasmatic coagulation cascade (21) that causes the generation of thrombinwhich in turn induces platelet activation (2). Furthermoreit has been discovered that TF plays an important role in tumor angiogenesis and progression as well as in metastasis (22). TF expression is under the control of E–cadherinPTENK–rasand p53. The activation of E–cadherin and K–ras or the loss–of–function of PTEN and p53 results in the implication of the mitogen–activated protein kinase (MAPK)/phosphoinositide–3 kinase (PI3K) signaling pathway and the subsequent increase of TF expression (23–25). High TF expression is correlated with the histological grade and poor prognosis in some tumor typesincluding non–small–cell lung carcinoma (26) and breast cancer (27). In bladder cancer patientshigh TF serum levels were previously shown to be associated with rapid disease progression (28). A study by John et al. has shown that despite the high expression of TF in bladder cancer cellsthe plasmatic coagulation was not induced. The authors explained this phenomenon by the comparably high levels of thrombomodulin that binds and inactivates thrombin on the cell surface (29). The TF–bearing extracellular vesicles (EVs) can be secreted by cancer cells andthusmay trigger TCIPA (30). For instanceSasano et al. demonstrated that TF–expressing EVs from ovarian cancer cells impact platelet aggregation and thrombosis (31). TF+ EVs from two human pancreatic adenocarcinoma cell lines affect resting platelets and activate them via thrombin generation (30). Geddings and colleagues presented that patients with advanced breast cancer had elevated levels of TF–bearing EVs compared with healthy controls (30).
von Willebrand Factor
von Willebrand factor (vWF) is an adhesive and multimeric glycoprotein present in plasmasynthesized by endothelial cells and megakaryocytes. vWF has a central role in primary hemostasis where it mediates platelet adhesion to the exposed extracellular matrix at the site of vascular damage (32, 33). vWF promotes platelet accumulation in the classical first wave of hemostasis by binding the platelet glycoprotein Ib–IX–V (GPIb–IX–V) complex (32). Monomers of pro–vWF undergo dimerization in the endoplasmic reticulum through C–terminal disulfide bonds and then undergo multimerization in the Golgi apparatus through N–terminal disulfide bonds (34). The newly synthesized vWF multimers are stored in the Weibel–Palade bodies (WPB) of endothelial cells and in the α–granules of megakaryocytes and platelets. vWF occurs in a range of sizesreferred to as vWF multimersincluding ultra–largehighintermediateand low molecular weight forms. In addition to endothelial and platelet–derived vWFthere is also another pool of circulating large heterogeneous multimers composed of repeating monomeric unitsup to 40,000 kDa in lengththat are the most biologically active form of vWF. This pool is reported to be released upon endothelial cell activation in response to inflammatory and ischemic injuries (35) and in response to a variety of factors such as thrombinhistamineadenosine diphosphate (ADP)collagenand other immune or tumor cell–secreted factors (32). The circulating multimers act dynamicallydepending on shear conditions or the presence of vessel wall damage. In low–shear conditionsplatelet adhesion is not permissible (36). On the other handplatelet–derived vWF exists as a hyposialylated glycoformrendering it less susceptible to ADAMTS13–mediated proteolysis (37). Intriguinglytumors may also sequester circulating vWF from plasma into the tumor stroma. It is reported that the main role in this process is played by the collagen–binding motif within the A3 domain of vWF (38). Taking into consideration these argumentsthe endothelial cell–derived pool of vWF seems to be more relevant in investigating TCIPA. It is considered that vWF is one of the major platelet adhesion ligands that may also mediate cancer progression and metastasis (39). Upon tumor–induced endothelial cell activationthe vWF within WPB is secreted into the lumen of the blood vessel as well as basolaterally into the subendothelium (40). In the tumor microenvironmentit can contribute to increased angiogenesisblood vessel permeabilityand epithelial–mesenchymal transitioningwhich was reported in osteosarcoma cells (41). Intraluminal accumulation of the vWF can result in the deposition of platelet–rich thrombin within the vasculature and serve to increase the number of metastatic focias it was found in the murine melanoma cell lines Ret and B16F10 (42). Yang et al. reported that patients with late–stage gastric cancer had higher serum levels of vWFand suggested that the expression of vWF in gastric cancer cells may contribute to its progression in vivo. The authors also found that this may be regulated by the vascular endothelial growth factor (VEGF)–VEGFR2 signaling pathway (43). Endothelial cell activation followed by vWF fiber formation was found to be the main culprit of platelet aggregation in malignant melanoma vasculature (44). On the other handTerraube et al. found that the presence of vWF plays a protective role against murine melanoma and lung cancer metastasis in vivo (45). Studies reported that gastric cancer cells express vWFsecrete it into the circulationand thus mediate TCIPA. vWF potentiates TCIPAwhile inhibition of this factor reduces platelet–cancer cell interactions (46). vWF–cleaving protease (ADAMTS13) serum levels are associated with poor prognosis and metastasis (47, 48). Jurasz et al. have shown that vWF potentiates the platelet–aggregatory activity of human fibrosarcoma HT1080 cells. This effect appears to be mediated via upregulation of platelet GPIIb/IIIa (49). vWFin addition to promoting pro‐inflammatory signalingcan also regulate angiogenesis and vascular permeability andthereforecan facilitate tumor cell growth and extravasation (50).
Adhesion Receptors
Glycoprotein Ib–IX–V
GPIb–IX–V is a membrane receptor complex originating in megakaryocytes and belonging to the leucine–rich repeat family of proteins. The complex consists of four distinct transmembrane proteinsnamelyGPIbαGPIbβGPIXand GPVand is expressed on platelets. The most important component of the complex is the glycoprotein component GPIbα which contains the binding sites for vWFP–selectinthrombinthrombospondinfactor XIIfactor Xkininogenand integrin αMβ2 (Mac–1). GPIb–IX–V plays a critical role in thrombosisinflammationmetastasisand the life cycle of platelets (51–55). One of its functions is the interaction with vWF on sites of vascular injury. The GPIb–IX–V complex binds to vWF and initiates signaling that results in GPIIb–IIIa activation and platelet aggregation (56, 57). Several lines of evidence implicated the role of GPIb–IX–V in TCIPA. GPIb was recently discovered to be expressed in breast cancer cells (22, 58). It plays an important role in tumor cell–host cell interactions. Deregulated expression of GPIbα is associated with cell transformation and global genomic destabilization (59). Interestinglyinhibition of GPIb–IX–V or vWF function reduced platelet–cancer cell interactions suggesting that these receptors play a role in tumor–induced platelet aggregation (58). GPIb–IX–V has also been shown to contribute to tumor malignancy and metastasis in lung cancer (60).
Glycoprotein IIb‐IIIa
Glycoprotein IIb‐IIIa (GPIIb‐IIIa) is an important platelet membrane receptor for fibrinogenfibronectinand vWF. It provides adhesive properties andhenceis necessary for platelet aggregation (58). The function of the GPIIb‐IIIa receptor in TCIPA has been established for decades (61). Platelets recruited in TCIPA can attach to the surface of tumor cells by a GPIIb–IIIa–fibrinogen bridge to secure tumor cells from immune surveillance (62). It also contributes to tumor progression and metastasis (63). Studies on breast cancer cell lines show that expression of the GPIIb–IIIa subunit occurs on the surface of MCF–7 cells and plays an important role in tumor metastasis (58). As demonstrated by Zhang et al.platelet GPIIb–IIIa is involved in the formation of the human melanoma A375 cell complex with platelets. Evidence showed that blocking the function of platelet GPIIa–IIIb by antagonists or antibodies could prevent hematogenous cancer metastasis (64). This mechanism was also observed in a study on lung and prostate cancer cases (64, 65). These results suggest that GPIIa–IIIb plays a key role in tumor progression and metastasismaking it an interesting target for anticancer therapy (64–66). Apart from therapyadvances in understanding the TCIPA mechanism may result in developing new highly specific diagnostic methods targeting cancer cells at the molecular level. Yap et al. introduced a specifically designed antibody that binds an activated form of the integrin receptor GPIIb–IIIa. The authors claimed that this method could be a new approach for enhancing ultrasound and PET imaging of tumors (67).
Integrin αvβ3
Integrin αvβ3 is a transmembrane heterodimer which belongs to the family of cell adhesion receptors (68). Platelets express αvβ3 integrin at their surfacewhich binds several adhesive proteins including fibrinogenfibronectinvWFand vitronectin. It is possible that the role of αvβ3 is triggering platelet adhesion and aggregation at sites of vascular injury (69). Moreoverthe αvβ3 integrin is expressed in breast cancer cells and may influence TCIPA by binding tumor cells to platelets using plasma proteins such as fibrinogen (70). The platelet GPIIb–IIIa can link fibrin with tumor αvβ3 and mediate tumor cell–platelet aggregation (71). In malignant melanoma cellsαvβ3 mediates platelet aggregation cell arrest during flow (72). This interaction creates a physical shield around cancer cells by protecting them from the deleterious effects of shear forces and immune cells (71, 72). Interestinglythe combined blockade of platelet GPIIb–IIIa and tumor cell–expressed αvβ3 is more effective at inhibiting tumor growth when compared with the single blockade of integrin receptors (73). The role of αvβ3 in tumor biology is even more complex since it plays an important role in tumor angiogenesisprogressionand metastasis (73). In recent yearsmany studies have demonstrated that the increased expression of αvβ3 integrin is related to a metastatic phenotype in many types of cancers such as ovarian (74) prostate (75)and breast (76).
Growth Factors
Vascular Endothelial Growth Factor
VEGF is the basic regulator of vascular growth. The actions of VEGF include the regulation of proliferationmigrationand permeability of endothelial cells. VEGF increases the expression of adhesion molecules and coagulation factors. It is stored in high quantities in platelet alpha granules (77). Elevated serum VEGF levels in cancer patients correlate with a poorer prognosis (78, 78). The molecules accumulated in platelets entrapped within the tumor matrix are gradually released depending on the protease activity such as the matrix metalloproteinase (MMP) family (79, 80). Extracellular proteases act on VEGF in several ways: on the one handthey can release matrix–bound VEGFon the other handthey can suppress VEGF’s proangiogenic activity. As a result of platelet interaction with cancer cellsVEGF is released into the tumor microenvironment and stimulates neoangiogenesiswhich ultimately enhances tumor growth (77). The overexpression of VEGF is one of the main factors leading to the occurrence and progression of cancerincluding renal cancerbreast cancernon–small cell lung cancerand pancreatic cancer (81). Another source is cellular hypoxiaoccurring in tumors rapidly overgrowing their blood supply. It induces the production of VEGF through factor–1α. The released VEGF binds to the VEGFR of endothelial cellsfavoring the formation of tumor–associated microvesselsand thusincreases tumor oxygen deliverydecreases hypoxiaand contributes to its further growth through positive feedback (82).
Transforming Growth Factor Beta
Platelets are a potent reservoir of transforming growth factor–beta (TGF–β) carrying a higher proportion of this molecule in the blood (up to 40%). Its release from TCIPA is widely described in the literature (83, 84). Howeverthere is no available research on the TGF–β mechanism primarily triggering TCIPAleaving a promising field for further studies. TGF–β is one of the most pleiotropic cytokines belonging to the transforming growth factor superfamily that includes three different mammalian isoforms (1–3) and many other signaling proteins. TGF–β proteins are produced by all white blood cell lineages. They are secreted in a latent form in which they bind with two other polypeptides: latent TGF–β–binding protein (LTBP) and latency–associated peptide (LAP). Thereforethe regulation of TGF–β levels is unique among other cytokines as they are not dependent on transcription factors but rather on proteases (such as plasmin)catalyzing the release of its active form (85). In this situationTGF–β may be upregulatedbypassing the transcription factors’ alterations. Activated TGF–β complexes form a serine/threonine kinase complex that binds to TGF–β receptors. These receptors are composed of both type 1 and type 2 receptor subunits. After binding of TGF–βthe type 2 receptor kinase phosphorylates and activates the type 1 receptor kinase that activates different downstream substrates and regulatory proteinsinducing the transcription of several target genes that promote differentiationchemotaxisproliferationand activation of immune cells (86). Recentlysome authors have focused on the role of TGF–β in the downstream activation of VEGF in tumorsthus leading to settling in an environment that is both nutritious through angiogenesis (87) and immunotolerant (88). Howeverthe initial clinical experience with drugs selectively targeting the tumor neovasculaturesuch as bevacizumabsunitiniband sorafenibhas been sobering: the major clinical responses to these drugs are rare and have minimal effects on overall survival after long–term follow–up (89–91). Some authors have explained that these effects were due to the promotion of compensatory angiogenic pathways (92) as well as to NK attenuation or activation of other intracellular pathways. Another mechanism of TGF–β–related tumorigenesis is through the promotion of epithelial–mesenchymal transition (EMT) (93).
Platelet–Derived Growth Factor
Platelet–derived growth factor (PDGF) is a two–chain polypeptidewhich belongs to the family of growth factors (94) and is a potent cell–cycle regulator acting on multiple levels and affecting numerous tissues and structures. Originallythe PDGF was discovered in plateletshoweverPDGF and PDGF–like peptides have been recognized in various normal and malignant cellsencompassing the bone matrix and osteosarcoma cell (95, 96). The mechanism of action of PDGF is mediated by a specific membranous receptor—the platelet–derived growth factor receptor (PDGFR). The receptor belongs to a large family of tyrosine kinases regulating cellular function and proliferation. PDGF is stored in the α–granules of platelets (97). Platelet–tumor cell crosstalk in the tumor microenvironment leads to platelet activation and secretion of stored growth factors. Platelet aggregation induced through the thrombin pathway leads to the release of the whole content from α–granules of plateletsincluding PDGFTGF–βand VEGF (94, 98–100). In addition to its main role of inducing tumor growthit can also promote angiogenesis and neovascularization (101). On the other handPDGF stimulation triggers the repression of platelet aggregation (102). Niitsu et al. demonstrated that the human cell line of fibrosarcoma proliferated more rapidly in a medium containing platelet lysatewith PDGF alone substantially promoting growth activity (103). Tsuruo et al. proved that stimulation with PDGF can affect the growth of metastatic clones of mouse colon adenocarcinoma in a concentration–dependent way. They hypothesized that the PDGF pathway might be engaged in the promotion of metastasis. Accordinglymigrating tumor cells that get “arrested” in microvessels may attract platelet adherence and activationultimately leading to their aggregation and formation of a “safe cuff” (104). It was proven that PDGF can play an important role in the EMT of prostate cancer cells. Overexpression of PDGF–D (a variant of PDGF) in prostate cancer cells was related to enhanced adhesive and invasive behaviors and increased tumor growth (105). An additional example of the impact of PDGF on EMT was demonstrated in hepatocellular carcinoma and was linked with TGF–β–mediated progression (106). Activation of PDGFR with PDGF was proven to stimulate cellular proliferation in autocrine and paracrine ways (107). A couple of studies have shown that intraplatelet PDGF concentrations were significantly elevated in patients with colorectal cancer when compared to healthy individualswhich may indicate an even more important in–vivo interaction between platelet–produced PDGF and the tumor microenvironment (108109). Interestinglyit was shown that PDGF–producing platelets are expressing PDGFR on their own surface allowing autocrine feedback regulation of PDGF release. A study has shown the inhibitory influence of the activated PDGFR–alpha variant on platelet activation (108). The findings presented lead us to the hypothesis that PDGF plays a substantial role in tumor progression and metastasis.
Cysteine Proteases
Cathepsins
Cathepsins are a family of globular proteases that primarily were discovered as intracellularly functioning peptide hydrolaseshowevermultiple cathepsins have extracellular activity (109). The cathepsin family consists of a number of proteases named from “A” to “X” (110). Cathepsins play different physiological rolessuch as bone remodeling and activation of granzymes and mast cell proteases triggered by cathepsin K and Crespectively. In tumorscathepsins contribute to the maintenance of inflammatory processes. Neverthelesstheir main function is associated with tumor progression and metastasis. Cysteine cathepsins function in concert with serine proteases and matrix metalloproteinases (111). The expression of human cysteine cathepsins is highly upregulated in numerous cancerssuch as melanomacolorectal cancerglioblastomaprostate carcinomabreast carcinomalung cancerbladder cancerand gastric cancer (111–113). Cathepsin G seems to be involved in platelet activation in Trousseau syndrome (114): circulating mucins trigger granulocyte activationand in turngranulocytes release cathepsin Gwhich splits the PAR–4 and stimulates G proteins (Gq and G12/13) to prompt the shape change and activation of platelets (114, 115). Cathepsins K and B are secreted to an extracellular matrix as soluble enzymes where they remain in the active form (116–119). It has been found that overactivity of cathepsin K stimulates the initiation of the mTOR signal transduction pathway andthusthe proliferationmigrationand invasion of NSCLC cells (120). Under physiological conditionscathepsin B participates in the maintenance of cellular metabolism (106). Cathepsin B acts as a cysteine cathepsin often associated with tumor progression (121). Overexpressed cathepsin B level was associated with the notably shorter overall survival of colon cancer patients (122). A strong correlation between cathepsin B expression and tumor angiogenesisinvasionand metastasis has been widely described in the literature (123). Cathepsin B and cancer procoagulant factor (9) were widely described to participate in TCIPA (124–126).
Matrix Metalloproteinases
MMPs are structurally similarzinc–dependent endopeptidases. The major function of the MMP family is controlled degeneration of the extracellular matrix (127). Their influence extends from embryonic developmentmorphogenesisand tissue remodeling to the regulation of vascular reactions and leukocyte and platelet activity (128). MMPs are involved in all steps of cancer progression: from primary tumor development to distant metastasis (129). The expression of MMP–2 on the surface of cancer cells was described in studies conducted on fibrosarcoma and colorectal and breast cancerwhere the authors have revealed that platelet and MMP–2 manifested by cancer cells contribute to TCIPA (130). Other MMPs of similar function include the membrane type I–matrix metalloproteinase (MT1–MMP)MMP–1and MMP–9also involved in platelet aggregation and TCIPA (131–134). MMP–1 expressed on breast cancer cells interacts with both GPIb–α and GPIIb–IIIaleading to their upregulation and providing ADP release and thus promoting TCIPA (2). The role of MMP–2 in the TCIPA pathway has been confirmed in HT1080 human fibrosarcoma cells and MCF–7 breast carcinoma cells (49, 134) as well as in human prostate cancer (130). The effect of MMP–2–induced platelet aggregation depends on the activation of proMMP–2 to MMP–2 through MMP–14 (126). It is considered that the communication between MMPs and glycoprotein receptors such as GPIIb–IIIaGPIband integrin αvβ3 is responsible for the MMP–mediated stimulation of platelets and tumor cells (135–137)howeverthe mechanism of action is still not completely understood (134). MMPs have presented the ability to stimulate TCIPA in vitrowhich is similar to cathepsin B (133). MMPs can be released from both platelets and tumor cells in vivo (138).
Sialomucin Glycoproteins
Podoplanin
Podoplanin (PDPN) is a mucin–type protein that mediates effects on cell migration and adhesion through its multiple partners. During embryonic developmentit plays a role in blood and lymphatic vessel separation by binding platelet C–type lectin–like receptor 1B (CLEC1B)triggering CLEC1B activation in platelets and leading to platelet activation and/or aggregation (139, 140). PDPN directly interacts with the CLEC2which promotes platelet aggregation and activation. Although elevated levels of PDPN have been correlated with increased malignancy in different tumorsits relevance for tumor progression is still unclear. Recentlya hypothesis of two different mechanisms of PDPN–related TCIPA in brain tumor patients has emerged:
1. PDPN may be released into the circulationeither in soluble form or on the surface of tumor–derived microvesicles.
2. Circulating tumor cells may be a source of circulating PDPN. Trapping of tumor cells in the venous system might lead to further local platelet activation and aggregation (141).
PDPN is known to contribute to tumor progression by inducing cancer cell migration and tumor invasion connected to the EMT mechanism (145146) and in the absence of EMT markers (142). High PDPN expression in primary brain tumors is associated with an increased risk of venous thromboembolism (VTE) (141) cancer progressionand overall poor prognosis (143, 144). Thereforethe PDPN–CLEC2 axis is a potential drug target for both reducing the risk of VTE and improving prognosis (141). Anti–PDPN therapies are expected to be of robust potential for future treatment strategies (145). In recent preclinical studiesa few anti–PDPN factors were targetedincluding types of recombinant immunotoxin NZ (146–148) and CD9an inhibitor of Aggrus/PDPN–induced platelet aggregation recognized to reduce the metastatic potential of HT1080 cells (149).
Soluble Mediators
Adenosine Diphosphate
ADP is a strong proaggregatory factoraccumulated in platelet–dense granulesand constitutes a secondary mediator of platelet aggregation (124). ADP has the capacity to communicate with platelet receptors P2Y12 and P2Y1resulting in the activation of platelet aggregation and changes of shape as well as the release of thromboxane A2 (TXA2) by platelets (150) and other multiple growth factors (151–153). ADP has been found to be expressed on cancer cells and involved in TCIPA. The P2Y12 receptor plays the main role in the process (134, 154). ADP–induced platelet activation is connected normally to VEGF release. Bambace et al. proved this by the termination of platelet activation by selective repression of the P2Y12 receptor (155). Interestinglyderegulation of ADP molecules may influence the reduction of metastases. Uluçkan et al. examined the mouse models of breast cancer and melanoma metastases treating them with acetylsalicylic acid and APT102 (a soluble apyrase/ADPase). Their results stand in favor of anti–ADP therapy in cancer (156).
Thromboxane A2
TXA2 is considered a powerful modulator of platelet activation and aggregation as well as a stimulator of vascular constrictionwhich acts via binding to the thromboxane prostanoid receptor (TP) (157). TXA2 is considered a crucial molecule associated with tumor metastasis. Some evidence supported this hypothesis: 1) TXA2 is a strong platelet–aggregatory eicosanoidfacilitating the binding of tumor cell–platelet aggregates to the surface of endothelial cells (158). 2) TXA2 enhanced the migration and angiogenesis of endothelial cells in both in–vitro and in–vivo models (164165). FurthermoreTXA2 and ADP are recognized as “soluble stimulators” of platelet aggregation (159–161). TXA2similar to ADPis secreted in an autocrine/paracrine manner and triggers platelet activation through positive feedback (162). The release of both TXA2 and ADP factors stimulates the conversion of the GPIIb/IIIa receptor into an active form mediating platelet aggregation. Lian et al. have noticed that both TXA2 and ADP signaling pathways are prompted during the MCF–7 cell–initiated TCIPA (162). TXA2 synthesis is catalyzed by cyclooxygenase 1 (COX–1). COX–1 in platelets enzymatically converts arachidonic acid into PGG2 and then into PGH2 and generates prothrombotic TXA2. Lucotti and colleagues have provided evidence that aspirin reduces the metastasis of different murine tumors (melanomabreast cancercolorectal cancer) by inhibition of platelet COX–1 and its product TXA2. High and medium doses of aspirin reduced the number of metastatic lung nodules by more than 50%. Howeverthe authors concluded that the use of more specific TXA2 inhibitorssuch as picotamidecould be more beneficial since they do not affect gastroprotective COX–1 products (163). Recent meta–analyses of 88 cohort trials have revealed that routine aspirin administration correlates with diminished risk of several types of cancersincluding colorectalgastricbreastand prostate. Unexpectedlythere was no correlation with the risk of lung cancer (164).
Selectins
P–selectin
P–selectin is a Ca2+–dependent receptor for myeloid cells that binds to carbohydrates on neutrophils and monocytes (165). It mediates the interaction of activated endothelial cells or platelets with leukocytes. The ligands recognized are sialyl–Lewis X (sLeX) and P–selectin glycoprotein ligand 1 (PSGL–1) (166). P–selectin functions as a cell adhesion molecule (CAM) on the surfaces of activated endothelial cells and activated platelets. In inactive endothelium and plateletsit is stored within the Weibel–Palade bodies and α–granulesrespectively. It is responsible for rapid leukocyte rolling over vascular surfaces during the initial steps of inflammation. It is widely known that P–selectin induces TCIPA andthuspromotes tumor growth (167). The rapid mobilization of P–selectin primarily to TCIPA was observed in tumor blood vessels in different speciessuch as oil miceC57BL6 miceand nude miceand in different tumor types like lung carcinomacolon carcinomabreast carcinomaand gliomas in response to radiotherapy. In contrastnormal tissue did not reveal increased post–treatment expression (168). This may suggest that the tumor and its environment can stimulate P–selectin to be in a closely preactivated stateready to externalize when a non–specific trigger occurs (such as radiation therapy). Other authors noticed P–selectin upregulation to be triggered by contact with the tumor cell–surface mucin (169) and non–mucin ligands (170). Mucins associated with cancer progression are MUC1MUC2MUC4and MUC16. As demonstrated by Kim et al.large mucin molecules on the surface of tumor cells bearing multiple P–selectin–binding sites could bridge tumor cells and P–selectin–expressing platelets (167). These interactions protect tumor cells within the bloodstreamhiding them from NK cells (1)which could influence metastatic spread and may also contribute to tumor progression (171). Studies on mice show that platelet–tumor cell interactions are significantly reduced in P–selectin–deficient miceand consequentlyattenuation of metastasis is observed. Furthermoreenzymatic removal of carcinoma mucins results in attenuated metastasis comparable to the absence of P–selectin (172).
An abbreviated description of all aforementioned factors is gathered in Supplementary Table 1.
TCIPA Targeting in Cancer Management
In recent yearsthe successful adoption and implementation of selective cancer therapies in clinical practice has increased research efforts aimed to identify and target various anticancer mechanisms. TCIPA is one of the pathways explored in the search for new options for cancer treatment. The crosstalk between plateletstheir receptorsreleased moleculesand clotting factors is subject to extensive and long–time research from different medical fields. These extensive studies resulted in the development of blockbuster therapies in cardiology and vascular medicine. The major principle of those protocols is to affect aggregation and clotting in a safe and controlled manner on different regulatory levels. The rich experience gained in the design of antiplatelet and antithrombotic treatment modalities may be transferred to oncology. For instanceTCIPA is a mechanism worthy of further studies as it demonstrates the involvement of platelets in carcinogenesis. It is highly probable that TCIPA inhibition would be beneficial for patients due to reduced risk of cancer–related thrombosis and associated clinical conditions such as strokepulmonary embolismand deep vein thrombosis. Howeverin this articlewe focused on exposing antitumor effects mediated by influencing TCIPA. The proof of concept is several studies reporting the potential benefit of targeting TCIPA in various tumors.
Thrombin and Factor X
In pancreatic cancerthe expression of the PAR–1 receptor in the tumor microenvironment was proven to drive progression and induce chemoresistance (173). Thereforethe next step was to use a thrombin inhibitor in cancer therapy. In a study on micea direct thrombin inhibitor dabigatranwidely used as an anticoagulant drug in numerous indicationswas employed. The study showed that it significantly potentiated gemcitabine–induced growth inhibition of pancreatic cancer (174).
Dabigatran and a direct factor Xa inhibitor rivaroxaban are known as novel oral anticoagulants (NOACs) and have gained widespread use in medicine. The latter was found to inhibit cancer stem cell (CSC) activity in the in–vitro functional CSC assay of mammosphere formation (175). Thenin 2020it was included in a clinical trial to evaluate the impact of rivaroxaban on tumor progression in ER–negative stage I–III early breast cancer patients. Up to datethe results of the trial are still concealed. Another indirect antitumor mechanism of antithrombotic drugs is the reduction of angiogenic potential by limiting the VEGF platelet release (176).
Adhesion Receptors
The inhibition of adhesion receptors using monoclonal antibodies has been already the subject of several studies. Qi et al. revealed that inhibition of GPIba leads to reduced interaction between platelets and tumor cellswhich results in the diminished metastatic potential of lung cancer cells. The study was performed in vitro and in vivo on animal models (177). Another study by Zhang et al. has demonstrated the promising effect of the anti–GPIIIa antibody on lung carcinoma cells in rat models. The mechanism of action was based on the fragmentation of activated platelets (178). The inhibition of GPIIb/IIIa in breast cancer cells was studied by Kononczuk et al. They have used specific antagonists of GPIIb/IIIa—abciximab and eptifibatide—to observe their proapoptotic effect on human breast cancer cells (179). Their promising results encouraged further trials. Another experimental anti–GPIIb/IIIa antagonist is the newly synthesized XV454 tested for its anticancer properties against lung cancer in rats. The results indicated a significant influence of this drug on tumor cell–platelet interaction and metastasis (65).
PDPN and CLEC2
The PDPN–CLEC2 axis might provide a potential drug target for both reducing the risk of VTE and improving prognosis. Anti–PDPN therapies are expected to be of robust potential for future treatment strategies. In recent preclinical studiesseveral molecules were evaluated to interfere with the PDPN–CLEC2 pathway. CD9an inhibitor of PDPN–induced platelet aggregationwas recognized to reduce the metastatic potential of human fibrosarcoma cells (149). The recombinant immunotoxin NZ–1–(scdsFv)–PE38KDEL was recognized to delay the growth of glioblastoma and medulloblastoma tumor cells (144). The first synthesized selective inhibitor of PDPN–CLEC2 interaction is the 5–nitrobenzoate compound 2CP. Chang et al. proved its selective TCIPA inhibition on osteosarcoma and glioma cells and cisplatin therapy efficacy augmentation (180).
Soluble Mediators
Another potential target is the ADP receptor P2Y12. The study of Cho et al. has shown that the growth of ovarian cancer in murine models was reduced with the specific P2Y12 inhibitor ticagrelor (181). Aspirin’s effect on platelet–mediated tumor progression is another potential therapeutic target: the study of Guillem–Llobat et al. has proven that COX–1 inhibition by aspirin could lower the metastatic potential of human colon adenocarcinoma cells (182). Ifetrobana potent selective TXA receptor antagonistpresumed to be decreasing cancer metastatic potentialhas been recently included in a second phase clinical trial involving patients with malignant solid tumors at high risk of metastatic recurrence. The results of that trial are planned to be available after 2025 (183).
P–selectin
Studies reported that heparin is an outstanding inhibitor of P–selectinwhich binds to its natural ligandsthus inhibiting the initial platelet–tumor cell interactions. Even a single heparin dose that transiently blocks this interaction is sufficient to prevent long–term organ colonization. These discoveries indicate that P–selectin and its ligands could be a potential therapeutic target (169).
The therapeutic targets among the TCIPA mechanisms are shown in Supplementary Table 2.
Conclusion and Future Perspectives
Among the unorthodox mechanisms of tumor progressionTCIPA has established by far a genuine target for potential therapies. The discussed studies demonstrated the encouraging influence of newly manufactured as well as widely used antiplatelet drugs on the inhibition of tumorigenesisprogressionand metastasis. These revelations should lay the groundwork for next–level clinical trials to optimize and determine the oncological efficiency of antiplatelet treatment. Howeverbecause of the simultaneous influence on blood coagulation and the wide variety of individual sensitivity to antiplatelet drugsit is still difficult to specify the optimal criteria for such studies. Hopefullyfurther research in the field of TCIPA will soon give us another instrument for aiding antitumor therapies.
Author Contributions
The first draft of the manuscript was written by WS, JJ, JD, BK, MK, and AK. Supervision and review were exercised by DG, TD, and JA. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by Nicolaus Copernicus University in ToruńFaculty of MedicineCollegium Medicum in Bydgoszcz (Research Task No. 141 within the framework of the Basic Research Activity).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.909767/full#supplementary-material
References
1. Palacios-Acedo AL, Mège D, Crescence L, Dignat-George F, Dubois C, Panicot-Dubois L. Platelets, Thrombo-Inflammation, and Cancer: Collaborating With the Enemy. Front Immunol (2019) 31:1805. doi: 10.3389/fimmu.2019.01805
2. Mezouar S, Frère C, Darbousset R, Mege D, Crescence L, Dignat-George F, et al. Role of Platelets in Cancer and Cancer-Associated Thrombosis: Experimental and Clinical Evidences. Thromb Res (2016) 139:65–76. doi: 10.1016/j.thromres.2016.01.006
3. Contursi A, Sacco A, Grande R, Dovizio M, Patrignani P. Platelets as Crucial Partners for Tumor Metastasis: From Mechanistic Aspects to Pharmacological Targeting. Cell Mol Life Sci (2017) 74(19):3491–507. doi: 10.1007/s00018-017-2536-7
4. Jurasz P, Alonso-Escolano D, Radomski MW. Platelet-Cancer Interactions: Mechanisms and Pharmacology of Tumour Cell-Induced Platelet Aggregation. Br J Pharmacol (2004) 143(7):819–26. doi: 10.1038/sj.bjp.0706013
5. Zarà M, Canobbio I, Visconte C, Canino J, Torti M, Guidetti GF. Molecular Mechanisms of Platelet Activation and Aggregation Induced by Breast Cancer Cells. Cell Signal (2018) 48:45–53. doi: 10.1016/j.cellsig.2018.04.008
6. Heinmöller E, Weinel RJ, Heidtmann HH, Salge U, Seitz R, Schmitz I, et al. Studies on Tumor-Cell-Induced Platelet Aggregation in Human Lung Cancer Lines. J Cancer Res Clin Oncol (1996) 122(12):735–44. doi: 10.1007/BF01209121
7. Heinmöller E, Schropp T, Kisker O, Simon B, Seitz R, Weinel RJ. Tumor Cell-Induced Platelet Aggregation In Vitro By Human Pancreatic Cancer Cell Lines. Encycl Cancer. (1995) 30(10):1008–16. doi: 10.3109/00365529509096346
8. Schlesinger M. Role of Platelets and Platelet Receptors in Cancer Metastasis. J Hematol Oncol (2018) 11:125. doi: 10.1186/s13045-018-0669-2
9. Danckwardt S, Hentze MW, Kulozik AE. Pathologies at the Nexus of Blood Coagulation and Inflammation: Thrombin in Hemostasis, Cancer, and Beyond. J Mol Med (2013) 91(11):1257–71. doi: 10.1007/s00109-013-1074-5
10. Tantry US, Liu F, Chen G, Gurbel PA. Vorapaxar in the Secondary Prevention of Atherothrombosis. Expert Rev Cardiovasc Ther (2015) 13(12):1293–305. doi: 10.1586/14779072.2015.1109447
11. Mitrugno A, Yunga ST, Sylman JL, Zilberman- Rudenko J, Shirai T, Hebert JF, et al. The Role of Coagulation and Platelets in Colon Cancer Associated Thrombosis. Am J Physiol Cell Physiol (2019) 316(2):264–73. doi: 10.1152/ajpcell.00367.2018
12. Adams GN, Rosenfeldt L, Frederick M, Miller W, Waltz D, Kombrinck K, et al. Colon Cancer Growth and Dissemination Relies Upon Thrombin Stromal PAR1 and Fibrinogen. Cancer Res (2015) 75(19):4235–43. doi: 10.1158/0008-5472.CAN-15-0964
13. Cisowski J, O’Callaghan K, Kuliopulos A, Yang J, Nguyen N, Deng Q, et al. Targeting Protease-Activated Receptor-1 With Cell-Penetrating Pepducins in Lung Cancer. Am J Pathol (2011) 179(1):513–23. doi: 10.1016/j.ajpath.2011.03.025
14. Zhang H, Jiang P, Zhang C, Lee S, Wang W, Zou H. PAR4 Overexpression Promotes Colorectal Cancer Cell Proliferation and Migration. Oncol Lett (2018) 16(5):5745–52. doi: 10.3892/ol.2018.9407
15. Wojtukiewicz MZ, Tang DG, Nelson KK, Walz DA, Diglio CA, Honn KV. Thrombin Enhances Tumor Cell Adhesive and Metastatic Properties via Increased αiibβ3 Expression on the Cell Surface. Thromb Res (1992) 68(3):233–45. doi: 10.1016/0049-3848(92)90081-k
16. Schulze EB, Hedley BD, Goodale D, Postenka CO, Al-Katib W, Tuck AB, et al. The Thrombin Inhibitor Argatroban Reduces Breast Cancer Malignancy and Metastasis via Osteopontin-Dependent and Osteopontin-Independent Mechanisms. Breast Cancer Res Treat (2008) 112(2):243–54. doi: 10.1007/s10549-007-9865-4
17. Cole M, Bromberg M. Tissue Factor as a Novel Target for Treatment of Breast Cancer. Oncologist. (2013) 18:14–8:1. doi: 10.1634/theoncologist.2012-0322
18. Contrino J, Hair G, Kreutzer DL, Rickles FR. In Situ Detection of Tissue Factor in Vascular Endothelial Cells: Correlation With the Malignant Phenotype of Human Breast Disease. Nat Med (1996) 2(2):209–15. doi: 10.1038/nm0296-209
19. Lwaleed BA, Lam L, Lasebai M, Cooper AJ. Expression of Tissue Factor and Tissue Factor Pathway Inhibitor in Microparticles and Subcellular Fractions of Normal and Malignant Prostate Cell Lines. Blood Coagul Fibrinolysis (2013) 24(3):339–43. doi: 10.1097/MBC.0b013e32835e98a6
20. Eisenreich A, Zakrzewicz A, Huber K, Thierbach H, Pepke W, Goldin-Lang P, et al. Regulation of Pro-Angiogenic Tissue Factor Expression in Hypoxia-Induced Human Lung Cancer Cells. Oncol Rep (2013) 30(1):462–70. doi: 10.3892/or.2013.2413
21. Auwerda JJA, Yuana Y, Osanto S, De Maat MPM, Sonneveld P, Bertina RM, et al. Microparticle-Associated Tissue Factor Activity and Venous Thrombosis in Multiple Myeloma. Thromb Haemost. (2011) 105(1):14–20. doi: 10.1160/TH10-03-0187
22. Suter CM, Hogg PJ, Price JT, Chong BH, Ward RL. Identification and Characterisation of a Platelet GPIb/V/IX-Like Complex on Human Breast Cancers: Implications for the Metastatic Process. Japanese J Cancer Res (2001) 92(10):1082–92. doi: 10.1111/j.1349-7006.2001.tb01063.x
23. Rao B, Gao Y, Huang J, Gao X, Fu X, Huang M, et al. Mutations of P53 and K-Ras Correlate TF Expression in Human Colorectal Carcinomas: TF Downregulation as a Marker of Poor Prognosis. Int J Colorectal Dis (2011) 26(5):593–601. doi: 10.1007/s00384-011-1164-1
24. Lau MT, Klausen C, Leung PCK. E-Cadherin Inhibits Tumor Cell Growth by Suppressing PI3K/Akt Signaling via B-Catenin-Egr1-Mediated PTEN Expression. Oncogene (2011) 30(24):2753–66. doi: 10.1038/onc.2011.6
25. Rong Y, Belozerov VE, Tucker-burden C, Chen G, Donald L, Olson JJ, et al. Epidermal Growth Factor Receptor and PTEN Modulate Tissue Factor Expression in Glioblastoma Through JunD/Activator Protein-1 Transcriptional Activity. Cancer Res (2009) 69(6):2540–9. doi: 10.1158/0008-5472.CAN-08-1547
26. Koomägi R, Volm M. Tissue-Factor Expression in Human non-Small-Cell Lung Carcinoma Measured by Immunohistochemistry: Correlation Between Tissue Factor and Angiogenesis. Int J Cancer (1998) 79(1):19–22. doi: 10.1002/(sici)1097-0215(19980220)79:1<19::aid-ijc4>3.0.co;2-z
27. Ueno T, Toi M, Koike M, Nakamura S, Tominaga T. Tissue Factor Expression in Breast Cancer Tissues: Its Correlation With Prognosis and Plasma Concentration. Br J Cancer (2000) 83(2):164–70. doi: 10.1054/bjoc.2000.1272
28. Förster Y, Meye A, Albrecht S, Kotzsch M, Füssel S, Wirth MP, et al. Tissue Specific Expression and Serum Levels of Human Tissue Factor in Patients With Urological Cancer. Cancer Lett (2003) 193(1):65–73. doi: 10.1016/s0304-3835(02)00687-0
29. John A, Robador JR, Vidal-Y-Sy S, Houdek P, Wladykowski E, Gunes C, et al. Urothelial Carcinoma of the Bladder Induces Endothelial Cell Activation and Hypercoagulation. Mol Cancer Res (2020) 18(7):1099–109. doi: 10.1158/1541-7786.MCR-19-1041
30. Geddings JE, Hisada Y, Boulaftali Y, Getz TM, Wheliha M, Fuentes R, et al. Tissue Factor-Positive Tumor Microvesicles Activate Platelets and Enhance Thrombosis in Mice. J Thromb Haemost. (2016) 14(1):153–66. doi: 10.1111/jth.13181
31. Sasano T, Cho MS, Rodriguez-Aguayo C, Bayraktar E, Taki M, Afshar-Kharghan V, et al. Role of Tissue-Factor Bearing Extracellular Vesicles Released From Ovarian Cancer Cells in Platelet Aggregation In Vitro and Venous Thrombosis in Mice. Thromb Updat. (2021) 2:1–8. doi: 10.1016/j.tru.2020.100020
32. Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Culleré M, Hynes RO, et al. A Mouse Model of Severe Von Willebrand Disease: Defects in Hemostasis and Thrombosis. Proc Natl Acad Sci U.S.A. (1998) 95(16):9524–9. doi: 10.1073/pnas.95.16.9524
33. Schick PK, Walker J, Profeta B, Denisova L, Bennett V. Synthesis and Secretion of Von Willebrand Factor and Fibronectin in Megakaryocytes at Different Phases of Maturation. Arterioscler Thromb Vasc Biol (1997) 17(2):797–801. doi: 10.1161/01.atv.17.4.797
34. Katsumi A, Tuley EA, Bodo I, Sadler JE. Localization of Disulfide Bonds in the Cystine Knot Domain of Human Von Willebrand Factor. J Biol Chem (2000) 275(33):25585–94. doi: 10.1074/jbc.M002654200
35. Gragnano F, Sperlongano S, Golia E, Natale F, Bianchi R, Crisci M, et al. The Role of Von Willebrand Factor in Vascular Inflammation: From Pathogenesis to Targeted Therapy. Mediators Inflamm (2017) 2017:1–13. doi: 10.1155/2017/5620314
36. De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the Von Willebrand Factor Strings Puzzle. Blood (2013) 121(2):270–7. doi: 10.1182/blood-2012-07-442285
37. McGrath RT, Van Den Biggelaar M, Byrne B, O’Sullivan JM, Rawley O, O’Kennedy R, et al. Altered Glycosylation of Platelet-Derived Von Willebrand Factor Confers Resistance to ADAMTS13 Proteolysis. Blood (2013) 122(25):4107–10. doi: 10.1182/blood-2013-04-496851
38. Ishihara J, Ishihara A, Sasaki K, Lee SSY, Williford JM, Yasui M, et al. Targeted Antibody and Cytokine Cancer Immunotherapies Through Collagen Affinity. Sci Transl Med (2019) 11:487. doi: 10.1126/scitranslmed.aau3259
39. Bazo IG, González VC, Gutiérrez ÁA, Rodríguez JR, Fernández JAP, de la Cámara Gómez J, et al. Impact of Surgery and Chemotherapy on Von Willebrand Factor and Vascular Endothelial Growth Factor Levels in Colorectal Cancer. Clin Transl Oncol (2005) 7(4):150–5. doi: 10.1007/BF02708752
40. Da Silva ML, Cutler DF. Von Willebrand Factor Multimerization and the Polarity of Secretory Pathways in Endothelial Cells. Blood (2016) 128(2):277–85. doi: 10.1182/blood-2015-10-677054
41. Ling J, Sun Y, Pan J, Wang H, Ma Z, Yin J, et al. Feedback Modulation of Endothelial Cells Promotes Epithelial-Mesenchymal Transition and Metastasis of Osteosarcoma Cells by Von Willebrand Factor Release. J Cell Biochem (2019) 120(9):15971–9. doi: 10.1002/jcb.28875
42. Goertz L, Schneider SW, Desch A, Mayer FT, Koett J, Nowak K, et al. Heparins That Block VEGF-A-Mediated Von Willebrand Factor Fiber Generation are Potent Inhibitors of Hematogenous But Not Lymphatic Metastasis. Oncotarget (2016) 7(42):68527–45. doi: 10.18632/oncotarget.11832
43. Yang X, Sun HJ, Li ZR, Zhang H, Yang WJ, Ni B, et al. Gastric Cancer-Associated Enhancement of Von Willebrand Factor is Regulated by Vascular Endothelial Growth Factor and Related to Disease Severity. BMC Cancer (2015) 15(1):1–11. doi: 10.1186/s12885-015-1083-6
44. Bauer AT, Suckau J, Frank K, Desch A, Goertz L, Wagner AH, et al. Von Willebrand Factor Fibers Promote Cancer-Associated Platelet Aggregation in Malignant Melanoma of Mice and Humans. Blood (2015) 125(20):3153–63. doi: 10.1182/blood-2014-08-595686
45. Terraube V, Pendu R, Baruch D, Gebbink MFBG, Meyer D, Lenting PJ, et al. Increased Metastatic Potential of Tumor Cells in Von Willebrand Factor-Deficient Mice. J Thromb Haemost (2006) 4(3):519–26. doi: 10.1111/j.1538-7836.2005.01770.x
46. Yang AJ, Wang M, Wang Y, Cai W, Li Q, Zhao TT, et al. Cancer Cell-Derived Von Willebrand Factor Enhanced Metastasis of Gastric Adenocarcinoma. Oncogenesis (2018) 7:1. doi: 10.1038/s41389-017-0023-5
47. Wang WS, Lin JK, Lin TC, Chiou TJ, Liu JH, Yen CC, et al. Plasma Von Willebrand Factor Level as a Prognostic Indicator of Patients With Metastatic Colorectal Carcinoma. World J Gastroenterol (2005) 11(14):2166–70. doi: 10.3748/wjg.v11.i14.2166
48. Pépin M, Kleinjan A, Hajage D, Büller HR, Di Nisio M, Kamphuisen PW, et al. ADAMTS-13 and Von Willebrand Factor Predict Venous Thromboembolism in Patients With Cancer. J Thromb Haemost. (2016) 14(2):306–15. doi: 10.1111/jth.13205
49. Jurasz P, Stewart MW, Radomski A, Khadour F, Duszyk M, Radomski MW. Role of Von Willebrand Factor in Tumour Cell-Induced Platelet Aggregation: Differential Regulation by NO and Prostacyclin. Br J Pharmacol (2001) 134(5):1104–12. doi: 10.1038/sj.bjp.0704343
50. Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TAJ, Sutton RE, et al. Endothelial Von Willebrand Factor Regulates Angiogenesis. Blood (2011) 117(3):1071–80. doi: 10.1182/blood-2010-01-264507
51. Nierodzik ML, Plotkin A, Kajumo F, Karpatkin S. Thrombin Stimulates Tumor-Platelet Adhesion In Vitro and Metastasis In Vivo. J Clin Invest (1991) 87:229–36. doi: 10.1172/JCI114976
52. Ware J, Russell S, Ruggeri ZM. Generation and Rescue of a Murine Model of Platelet Dysfunction: The Bernard-Soulier Syndrome. Proc Natl Acad Sci U.S.A. (2000) 97(6):2803–8. doi: 10.1073/pnas.050582097
53. Ravanat C, Strassel C, Hechler B, Schuhler S, Chicanne G, Payrastre B, et al. A Central Role of GPIb-IX in the Procoagulant Function of Platelets That is Independent of the 45-kDa Gpibα N-Terminal Extracellular Domain. Blood (2010) 116(7):1157–64. doi: 10.1182/blood-2010-01-266080
54. Kato K, Martinez C, Russell S, Nurden P, Nurden A, Fiering S, et al. Genetic Deletion of Mouse Platelet Glycoprotein Ibβ Produces a Bernard-Soulier Phenotype With Increased α-Granule Size. Blood (2004) 104(8):2339–44. doi: 10.1182/blood-2004-03-1127
55. Chen M, Geng JG. P-Selectin Mediates Adhesion of Leukocytes, Platelets, and Cancer Cells in Inflammation, Thrombosis, and Cancer Growth and Metastasis. Arch Immunol Ther Exp (Warsz) (2006) 54(2):75–84. doi: 10.1007/s00005-006-0010-6
56. Sakariassen KS, Nievelstein PF, Coller BS, Sixma JJ. The Role of Platelet Membrane Glycoproteins Ib and IIb-IIIa in Platelet Adherence to Human Artery Subendothelium. Br J Haematol (1986) 63(4):681–91. doi: 10.1111/j.1365-2141.1986.tb07552.x
57. Nesbitt WS, Giuliano S, Kulkarni S, Dopheide SM, Harper IS, Jackson SP. Intercellular Calcium Communication Regulates Platelet Aggregation and Thrombus Growth. J Cell Biol (2003) 160:7 1151–1161. doi: 10.1083/jcb.200207119
58. Oleksowicz L, Mrowiec Z, Schwartz E, Khorshidi M, Dutcher JP, Puszkin E. Characterization of Tumor-Induced Platelet Aggregation. The Role of Immunorelated GPIb and GPIIb IIIa Expression by MCF-7 Breast Cancer Cells. Thromb Res (1995) 79(3):261–74. doi: 10.1016/0049-3848(95)00113-6
59. Li Y, Lu J, Cohen D, Prochownik EV. Transformation, Genomic Instability and Senescence Mediated by Platelet/Megakaryocyte Glycoprotein Ibα. Oncogene (2008) 27(11):1599–609. doi: 10.1038/sj.onc.1210794
60. Jain S, Zuka M, Liu J, Russell S, Dent J, Guerrero JA, et al. Platelet Glycoprotein Ibα Supports Experimental Lung Metastasis. Proc Natl Acad Sci U.S.A. (2007) 104(21):9024–8. doi: 10.1073/pnas.0700625104
61. Grossi IM, Taylor JD, Honn KV, Kendall A, Sloane BF, Fitzgerald LA. Inhibition of Human Tumor Cell Induced Platelet Aggregation by Antibodies to Platelet Glycoproteins Lb and Llb/Llla. Proc Soc Exp Biol Med (1987) 186(3):378–83. doi: 10.3181/00379727-186-3-rc1
62. Lou XL, Sun J, Gong SQ, Yu XF, Gong R, Deng H. Interaction Between Circulating Cancer Cells and Platelets: Clinical Implication. Chin J Cancer Res (2015) 27(5):450–60. doi: 10.3978/j.issn.1000-9604.2015.04.10
63. Honn Kv, Chen YQ, Timar J, Onoda JM, Hatfield JS, Fligiel SEG, et al. αiibβ3 Integrin Expression and Function in Subpopulations of Murine Tumors. Exp Cell Res (1992) 201(1):23–32. doi: 10.1016/0014-4827(92)90344-8
64. Zhang L, Shan X, Meng X, Gu T, Lu Q, Zhang J, et al. The First Integrins β3-Mediated Cellular and Nuclear Targeting Therapeutics for Prostate Cancer. Biomaterials (2019) 223:1–10. doi: 10.1016/j.biomaterials.2019.119471
65. Amirkhosravi A, Mousa SA, Amaya M, Blaydes S, Desai H, Meyer T, et al. Inhibition of Tumor Cell-Induced Platelet Aggregation and Lung Metastasis by the Oral GpIIb/IIIa Antagonist XV454. Thromb Haemost. (2003) 90:549–54:3. doi: 10.1160/TH03-02-0102
66. Zhang CM, Liu Y, Gao YG, Shen J, Zheng S, Wei M, et al. Modified Heparins Inhibit Integrin αiibβ3 Mediated Adhesion of Melanoma Cells to Platelets In Vitro and In Vivo. Int J Cancer (2009) 125(9):2058–65. doi: 10.1002/ijc.24561
67. Yap ML, McFadyen JD, Wang X, Zia NA, Hohmann JD, Ziegler M, et al. Targeting Activated Platelets: A Unique and Potentially Universal Approach for Cancer Imaging. Theranostics (2017) 7(10):2565–74. doi: 10.7150/thno.19900
68. Stucci S, Tucci M, Passarelli A, Silvestris F. Avβ3 Integrin: Pathogenetic Role in Osteotropic Tumors. Crit Rev Oncol Hematol (2015) 96(1):183–93. doi: 10.1016/j.critrevonc.2015.05.018
69. Lavergne M, Janus-Bell E, Schaff M, Gachet C, Mangin PH. Platelet Integrins in Tumor Metastasis: Do They Represent a Therapeutic Target? Cancers (Basel) (2017) 9:10. doi: 10.3390/cancers9100133
70. Felding-Habermann B, O’Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, et al. Integrin Activation Controls Metastasis in Human Breast Cancer. Proc Natl Acad Sci U.S.A. (2001) 98(4):1853–68. doi: 10.1073/pnas.98.4.1853
71. Felding-Habermann B, Habermann R, Saldívar E, Ruggeri ZM. Role of β3 Integrins in Melanoma Cell Adhesion to Activated Platelets Under Flow. J Biol Chem (1996) 271(10):5892–900. doi: 10.1074/jbc.271.10.5892
72. Pilch J, Habermann R, Felding-Habermann B. Unique Ability of Integrin Alpha(V)Beta 3 to Support Tumor Cell Arrest Under Dynamic Flow Conditions. J Biol Chem (2002) 277:24. doi: 10.1074/jbc.M201630200
73. Trikha M, Zhou Z, Timar J, Raso E, Kennel M, Emmell E, et al. Multiple Roles for Platelet GPIIb/IIIa and αvβ3 Integrins in Tumor Growth, Angiogenesis, and Metastasis. Cancer Res (2002) 62(10):2824–33.
74. Landen CN, Kim TJ, Lin YG, Merritt WM, Kamat AA, Han LY, et al. Tumor-Selective Response to Antibody-Mediated Targeting of αvβ3 Integrin in Ovarian Cancer. Neoplasia (2008) 10(11):1259–67. doi: 10.1593/neo.08740
75. McCabe NP, De S, Vasanji A, Brainard J, Byzova TV. Prostate Cancer Specific Integrin αvβ3 Modulates Bone Metastatic Growth and Tissue Remodeling. Oncogene (2007) 26(42):6238–43. doi: 10.1038/sj.onc.1210429
76. Takayama S, Ishii S, Ikeda T, Masamura S, Doi M, Kitajima M. The Relationship Between Bone Metastasis From Human Breast Cancer and Integrin αvβ3 Expression. Anticancer Res (2005) 25(1A):79–83.
77. Battinelli EM, Markens BA, Italiano JE. Release of Angiogenesis Regulatory Proteins From Platelet Alpha Granules: Modulation of Physiologic and Pathologic Angiogenesis. Blood (2011) 118(5):1359–69. doi: 10.1182/blood-2011-02-334524
78. Ikeda M, Furukawa H, Imamura H, Shimizu J, Ishida H, Masutani S, et al. Poor Prognosis Associated With Thrombocytosis in Patients With Gastric Cancer. Ann Surg Oncol (2002) 9(3):287–91. doi: 10.1007/BF02573067
79. Quintero-Fabián S, Arreola R, Becerril-Villanueva E, Torres-Romero JC, Arana-Argáez V, Lara-Riegos J, et al. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front Oncol (2019) 6:1370(9). doi: 10.3389/fonc.2019.01370
80. Vizovisek M, Ristanovic D, Menghini S, Christiansen MG, Schuerle S. The Tumor Proteolytic Landscape: A Challenging Frontier in Cancer Diagnosis and Therapy. Int J Mol Sci (2021) 22(5):1–30. doi: 10.3390/ijms22052514
81. Costache MI, Ioana M, Iordache S, Ene D, Costache CA, Săftoiu A. VEGF Expression in Pancreatic Cancer and Other Malignancies: A Review of the Literature. Rom J Intern Med (2015) 53(3):199–208. doi: 10.1515/rjim-2015-0027
82. Chouaib S, Messai Y, Couve S, Escudier B, Hasmim M, Noman MZ. Hypoxia Promotes Tumor Growth in Linking Angiogenesis to Immune Escape. Front Immunol (2012) 3:21(21). doi: 10.3389/fimmu.2012.00021
83. Takemoto A, Okitaka M, Takagi S, Takami M, Sato S, Nishio M, et al. A Critical Role of Platelet TGF-β Release in Podoplanin-Mediated Tumour Invasion and Metastasis. Sci Rep (2017) 7:1–12. doi: 10.1038/srep42186
84. Labelle M, Begum S, Hynes RO. Direct Signaling Between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell (2011) 20(5):576–90. doi: 10.1016/j.ccr.2011.09.009
85. Letterio JJ, Roberts AB. Regulation of Immune Responses by TGF- β. Annu Rev Immunol (1998) 16:137–61. doi: 10.1146/annurev.immunol.16.1.137
87. Ten Dijke P, Arthur HM. Extracellular Control of Tgfβ Signalling in Vascular Development and Disease. Nat Rev Mol Cell Biol (2007) 8(11):857–69. doi: 10.1038/nrm2262
88. Courau T, Nehar-Belaid D, Florez L, Levacher B, Vazquez T, Brimaud F, et al. TGF-β and VEGF Cooperatively Control the Immunotolerant Tumor Environment and the Efficacy of Cancer Immunotherapies. JCI Insight (2016) 1(9):1–16. doi: 10.1172/jci.insight.85974
89. Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, et al. Bevacizumab Plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N Engl J Med (2004) 350(23):2335–42. doi: 10.1056/NEJMoa032691
90. Szczylik C, Oudard S, Siebels M, Hutson TE, Pharm D, Gore M, et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. N Engl J Med (2007) 356(2):125–34. doi: 10.1056/NEJMoa060655
91. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib Versus Interferon Alfa in Metastatic Renal Cell Carcinoma. N Engl J Med (2015) 356(2):687–96. doi: 10.1056/NEJMoa065044
92. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug Resistance by Evasion of Antiangiogenic Targeting of VEGF Signaling in Late-Stage Pancreatic Islet Tumors. Cancer Cell (2005) 8(4):299–309. doi: 10.1016/j.ccr.2005.09.005
93. Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-β Induced Transdifferentiation of Mammary Epithelial Cells to Mesenchymal Cells: Involvement of Type I Receptors. J Cell Biol (1994) 127(6 II):2021–36. doi: 10.1083/jcb.127.6.2021
94. Shah P, Keppler L, Rutkowski J. A Review of Platelet Derived Growth Factor Playing Pivotal Role in Bone Regeneration. J Oral Implantol. (2014) 40(3):330–40. doi: 10.1563/AAID-JOI-D-11-00173
95. Canalis E, MacCarthy T, Centrella M. Growth Factors and the Regulation of Bone Remodeling. J Clin Invest. (1988) 81:277–81. doi: 10.1172/JCI113318
96. Ross R, Glomset J, Kariya B, Harker L. A Platelet Dependent Serum Factor That Stimulates the Proliferation of Arterial Smooth Muscle Cells In Vitro. Proc Natl Acad Sci U S A. (1974) 71(4):1207–10. doi: 10.1073/pnas.71.4.1207
97. Pagel O, Walter E, Jurk K, Zahedi RP. Taking the Stock of Granule Cargo: Platelet Releasate Proteomics. Platelets (2017) 28(2):119–28. doi: 10.1080/09537104.2016.1254762
98. Karpatkin S, Ambrogio CPE. The Role of Tumor-Induced Platelet Aggregation, Platelet Adhesion and Adhesive Proteins in Tumor Metastasis. Prog Clin Biol Res (1988) 283:585–606.
99. Wang S, Li Z, Xu R. Human Cancer and Platelet Interaction, a Potential Therapeutic Target. Int J Mol Sci (2018) 19(4):1–15. doi: 10.3390/ijms19041246
100. Lee S, Margolin K. Cytokines in Cancer Immunotherapy. Cancers (Basel) (2011) 3(4):3856–93. doi: 10.3390/cancers3043856
101. Wojtukiewicz MZ, Sierko E, Hempel D, Tucker SC, Honn KV. Platelets and Cancer Angiogenesis Nexus. Cancer Metastasis Rev (2017) 36(2):249–62. doi: 10.1007/s10555-017-9673-1
102. Yang M, Khachigian LM, Hicks C, Chesterman CN, Chong BH. Identification of PDGF Receptors on Human Megakaryocytes and Megakaryocytic Cell Lines. Thromb Haemost. (1997) 78(2):892–6. doi: 10.1055/s-0038-1657648
103. Niitsu Y, Koshida Y, Mahara K, Ishigaki S, Kogawa K, Watanabe N, et al. Growth Promoting Activity of PDGF, EGF and TGF-β on Highly Metastatic Subline of Meth a Cells. Immunopharmacol Immunotoxicol. (1968) 10(1):67–78. doi: 10.3109/08923978809014402
104. Tsuruo T, Watanabe M, Oh-hara T. Stimulation of the Growth of Metastatic Clones of Mouse Colon Adenocarcinoma 26 In Vitro by Platelet-Derived Growth Factor. Japanese J Cancer Res (1989) 80(2):136–40. doi: 10.1111/j.1349-7006.1989.tb02281.x
105. Kong D, Wang Z, Sarkar HS, Li Y, Banerjee S, Saliganan A, et al. Platelet-Derived Growth Factor-D Overexpression Contributes to Epithelial-Mesenchymal Transition of PC3 Prostate Cancer Cells. Stem Cells (2008) 26(6):1425–35. doi: 10.1634/stemcells.2007-1076
106. Podgorski I, Sloane BF. Cathepsin B and its Role(s) in Cancer Progression. Biochem Soc Symp (2003) 276(70):263–76. doi: 10.1042/bss0700263
107. Williams LT, Escobedo JA, Keating MT, Coughlin SR. The Stimulation of Paracrine and Autocrine Mitogenic Pathways by the Platelet-Derived Growth Factor Receptor. J Cell Physiol (1987) 5:27–30. doi: 10.1002/jcp.1041330406
108. Vassbotn FS, Havnen OK, Heldin CH, Holmsen H. Negative Feedback Regulation of Human Platelets via Autocrine Activation of the Platelet-Derived Growth Factor α-Receptor. J Biol Chem (1994) 269(19):13874–9. doi: 10.1016/S0021-9258(17)36728-5
109. Tan GJ, Peng ZK, Lu JP, Tang FQ. Cathepsins Mediate Tumor Metastasis. World J Biol Chem (2013) 4(4):91–101. doi: 10.4331/wjbc.v4.i4.91
110. Kuester D, Lippert H, Roessner A, Krueger S. The Cathepsin Family and Their Role in Colorectal Cancer. Pathol - Res Pract (2008) 204(7):491–500. doi: 10.1016/j.prp.2008.04.010
111. Jedeszko C, Sloane BF. Cysteine Cathepsins in Human Cancer. Biol Chem (2004) 385(11):1017–27. doi: 10.1515/BC.2004.132
112. Andrade SS, Gouvea IE, Silva MCC, Castro ED, de Paula CAA, Okamoto D, et al. Cathepsin K Induces Platelet Dysfunction and Affects Cell Signaling in Breast Cancer - Molecularly Distinct Behavior of Cathepsin K in Breast Cancer. BMC Cancer (2016) 16(1):1–19. doi: 10.1186/s12885-016-2203-7
113. Berdowska I. Cysteine Proteases as Disease Markers. Clin Chim Acta (2004) 342(1–2):41–69. doi: 10.1016/j.cccn.2003.12.016
114. Shao B, Wahrenbrock MG, Yao L, David T, Coughlin SR, Xia L, et al. Carcinoma Mucins Trigger Reciprocal Activation of Platelets and Neutrophils in a Murine Model of Trousseau Syndrome. Blood (2011) 118(15):4015–23. doi: 10.1182/blood-2011-07-368514
115. Faraday N, Schunke K, Saleem S, Fu J, Wang B, Zhang J, et al. Cathepsin G-Dependent Modulation of Platelet Thrombus Formation In Vivo by Blood Neutrophils. PloS One (2013) 8:8. doi: 10.1371/journal.pone.0071447
116. Brix K, Dunkhorst A, Mayer K, Jordans S. Cysteine Cathepsins: Cellular Roadmap to Different Functions. Biochimie (2008) 90(2):194–207. doi: 10.1016/j.biochi.2007.07.024
117. Brömme D, Lecaille F. Cathepsin K Inhibitors for Osteoporosis and Potential Off-Target Effects. Expert Opin Investig Drugs (2009) 18(5):585–600. doi: 10.1517/13543780902832661
118. Reiser J, Adair B, Reinheckel T. Specialized Roles for Cysteine Cathepsins in Health and Disease. J Clin Invest. (2010) 120(10):3421–31. doi: 10.1172/JCI42918
119. Cattaruzza F, Lyo V, Jones E, Pham D, Hawkins J, Kirkwood K, et al. Cathepsin S is Activated During Colitis and Causes Visceral Hyperalgesia by a PAR2-Dependent Mechanism in Mice. Gastroenterology (2011) 141(5):1864–74. doi: 10.1053/j.gastro.2011.07.035
121. Aggarwal N, Sloane BF. Cathepsin B: Multiple Roles in Cancer. Proteomics Clin Appl (2014) 8(5-6):427–37. doi: 10.1002/prca.201300105
122. Koblinski JE, Ahram M, Sloane BF. Unraveling the Role of Proteases in Cancer. Clin Chim Acta (2000) 291(2):113–35. doi: 10.1016/s0009-8981(99)00224-7
123. Ruan H, Hao S, Young P, Zhang H. Targeting Cathepsin B for Cancer Therapies. Horizons Cancer Res (2015) 56:23–39.
124. Bambace NM, Holmes CE. The Platelet Contribution to Cancer Progression. J Thromb Haemost. (2011) 9(2):237–49. doi: 10.1111/j.1538-7836.2010.04131.x
125. Olas B, Mielicki WP, Wachowicz B, Krajewski T. Cancer Procoagulant Stimulates Platelet Adhesion. Thromb Res (1999) 94(3):199–203. doi: 10.1016/s0049-3848(98)00214-x
126. Honn KV, Cavanaugh P, Evens C, Taylor JD, Sloane BF. Tumor Cell-Platelet Aggregation: Induced by Cathepsin B-Like Proteinase and Inhibited by Prostacyclin. Science (1982) 217(4559):540–2. doi: 10.1126/science.7046053
127. Sternlicht MD, Werb Z. How Matrix Metalloproteinases Regulate Cell Behavior. Annu Rev Cell Dev Biol (2001) 17:463–516. doi: 10.1146/annurev.cellbio.17.1.463
128. Jurasz P, Chung AWY, Radomski A, Radomski MW. Nonremodeling Properties of Matrix Metalloproteinases: The Platelet Connection. Circ Res (2002) 90(10):1041–3. doi: 10.1161/01.res.0000021398.28936.1d
129. Plantureux L, Mège D, Crescence L, Dignat-George F, Dubois C, Panicot-Dubois L. Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis. Cancers (Basel) (2018) 10(11):1–23. doi: 10.3390/cancers10110441
130. Jurasz P, North S, Venner P, Radomski MW. Matrix Metalloproteinase-2 Contributes to Increased Platelet Reactivity in Patients With Metastatic Prostate Cancer: A Preliminary Study. Thromb Res (2003) 112(1–2):59–64. doi: 10.1016/j.thromres.2003.10.012
131. Kazes I, Elalamy I, Sraer JD, Hatmi M, Nguyen G. Platelet Release of Trimolecular Complex Components MT1-MMP/TIMP2/MMP2: Involvement in MMP2 Activation and Platelet Aggregation. Blood (2000) 96(9):3064–9. doi: 10.1182/blood.V96.9.3064
132. Chung AWY, Jurasz P, Hollenberg MD, Radomski MW. Mechanisms of Action of Proteinase-Activated Receptor Agonists on Human Platelets. Br J Pharmacol (2002) 135(5):1123–32. doi: 10.1038/sj.bjp.0704559
133. Jurasz P, Sawicki G, Duszyk M, Sawicka J, Carlos Miranda IM, Radomski MW. Matrix Metalloproteinase 2 in Tumor Cell-Induced Platelet Aggregation: Regulation by Nitric Oxide. Cancer Res (2001) 7(4):646–51.
134. Alonso-Escolano D, Strongin AY, Chung AW, Deryugina EI, Radomski MW. Membrane Type-1 Matrix Metalloproteinase Stimulates Tumour Cell-Induced Platelet Aggregation: Role of Receptor Glycoproteins. Br J Pharmacol (2004) 141(2):241–52. doi: 10.1038/sj.bjp.0705606
135. Deryugina EI, Bourdon MA, Jungwirth K, Smith JW, Strongin AY. Functional Activation of Integrin αvβ3 in Tumor Cells Expressing Membrane-Type 1 Matrix Metalloproteinase. Int J Cancer. (2000) 86(1):15–23. doi: 10.1002/(sici)1097-0215(20000401)86:1<15::aid-ijc3>3.0.co;2-b
136. Martinez A, Salas E, Radomski MW. Matrix Metalloproteinase-2 in Platelet Adhesion to Fibrinogen: Interactions With Nitric Oxide. Med Sci Monit (2001) 7(4):646–51.
137. Galt SW, Lindemann S, Allen L, Medd DJ, Falk JM, McIntyre TM, et al. Outside-In Signals Delivered by Matrix Metalloproteinase-1 Regulate Platelet Function. Circ Res (2002) 90(10):1093–9. doi: 10.1161/01.res.0000019241.12929.eb
138. Deryugina EI, Quigley JP. Matrix Metalloproteinases and Tumor Metastasis. Cancer Metastasis Rev (2006) 25(1):9–34. doi: 10.1007/s10555-006-7886-9
139. Christou CM, Pearce AC, Watson AA, Mistry AR, Pollitt AY, Fenton-May AE, et al. Renal Cells Activate the Platelet Receptor CLEC-2 Through Podoplanin. Biochem J (2008) 411(1):133–40. doi: 10.1042/BJ20071216
140. Kaneko MK, Kato Y, Kameyama A, Ito H, Kuno A, Hirabayashi J, et al. Functional Glycosylation of Human Podoplanin: Glycan Structure of Platelet Aggregation-Inducing Factor. FEBS Lett (2007) 581(2):331–6. doi: 10.1016/j.febslet.2006.12.044
141. Riedl J, Preusser M, Nazari PMS, Posch F, Panzer S, Marosi C, et al. Podoplanin Expression in Primary Brain Tumors Riedl J, Preusser M, Seyed Nazari PM, Posch F, Panzer S, Marosi Ch, Birner P, Thaler J, Brostjan Ch, Lötsch D, Berger W, Hainfellner JA, Pabinger I, Ay C. Podoplanin Expression in Primary Brain Tumors Induces Platelet Aggregation and Increases Risk of Venous Thromboembolism. Blood (2017) 129(13):1831–9. doi: 10.1182/blood-2016-06-720714
142. Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor Invasion in the Absence of Epithelial-Mesenchymal Transition: Podoplanin-Mediated Remodeling of the Actin Cytoskeleton. Cancer Cell (2006) 9(4):261–72. doi: 10.1016/j.ccr.2006.03.010
143. Birner P, Pusch S, Christov C, Mihaylova S, Toumangelova-Uzeir K, Natchev S, et al. Mutant IDH1 Inhibits PI3K/Akt Signaling in Human Glioma. Cancer (2014) 120(16):2440–7. doi: 10.1002/cncr.28732
144. Chandramohan V, Bao X, Kaneko MK, Kato Y, Keir ST, Szafranski SE, et al. Recombinant Anti-Podoplanin (NZ-1) Immunotoxin for the Treatment of Malignant Brain Tumors. Int J Cancer (2013) 132(10):1–20. doi: 10.1002/ijc.27919
145. Xie L, Lin C, Zhang Q, Piao H, Bigner DD, Zhang Z, et al. Elevated Expression of Podoplanin and its Clinicopathological, Prognostic, and Therapeutic Values in Squamous non-Small Cell Lung Cancer. Cancer Manag Res (2018) 10:1329–40. doi: 10.2147/CMAR.S163510
146. Kaneko MK, Yamada S, Nakamura T, Abe S, Nishioka Y, Kunita A, et al. Antitumor Activity of Chlpmab-2, a Human–Mouse Chimeric Cancer-Specific Antihuman Podoplanin Antibody, via Antibody-Dependent Cellular Cytotoxicity. Cancer Med (2017) 6(4):768–77. doi: 10.1002/cam4.1049
147. Kaneko MK, Abe S, Ogasawara S, Fujii Y, Yamada S, Murata T, et al. Chimeric Anti-Human Podoplanin Antibody NZ-12 of Lambda Light Chain Exerts Higher Antibody-Dependent Cellular Cytotoxicity and Complement-Dependent Cytotoxicity Compared With NZ-8 of Kappa Light Chain. Monoclon Antib Immunodiagn Immunother. (2017) 36(1):25–9. doi: 10.1089/mab.2016.0047
148. Abe S, Kaneko MK, Tsuchihashi Y, Izumi T, Ogasawara S, Okada N, et al. Antitumor Effect of Novel Anti-Podoplanin Antibody NZ-12 Against Malignant Pleural Mesothelioma in an Orthotopic Xenograft Model. Cancer Sci (2016) 107:9 1198–1205. doi: 10.1111/cas.12985
149. Nakazawa Y, Sato S, Naito M, Kato Y, Mishima K, Arai H, et al. Tetraspanin Family Member CD9 Inhibits Aggrus/podoplanin-Induced Platelet Aggregation and Suppresses Pulmonary Metastasis. Blood (2008) 112:5 1730–1739. doi: 10.1182/blood-2007-11-124693
150. Fabre JE, Nguyen M, Latour A, Keifer JA, Audoly LP, Coffman TM, et al. Decreased Platelet Aggregation, Increased Bleeding Time and Resistance to Thromboembolism in P2Y1-Deficient Mice. Nat Med (1999) 5:10 1199–1202. doi: 10.1038/13522
151. Hechler B, Léon C, Vial C, Vigne P, Frelin C, Cazenave JP, et al. The P2Y1 Receptor is Necessary for Adenosine 5’-Diphosphate-Induced Platelet Aggregation. Blood (1998) 92(1):152–9. doi: 10.1182/blood.V92.1.152.413k27_152_159
152. Communi D, Janssens R, Suarez-Huerta N, Robaye B, Boeynaems JM. Advances in Signalling by Extracellular Nucleotides the Role and Transduction Mechanisms of P2Y Receptors. Cell Signal (2000) 12(6):351–60. doi: 10.1016/s0898-6568(00)00083-8
153. Jin J, Kunapuli SP. Coactivation of Two Different G Protein-Coupled Receptors is Essential for ADP-Induced Platelet Aggregation. Proc Natl Acad Sci U.S.A. (1998) 95(14):8070–4. doi: 10.1073/pnas.95.14.8070
154. Dorsam RT, Kunapuli SP. Central Role of the P2Y12 Receptor in Platelet Activation. J Clin Invest. (2004) 113(3):340–5. doi: 10.1172/JCI20986
155. Bambace NM, Levis JE, Holmes CE. The Effect of P2Y-Mediated Platelet Activation on the Release of VEGF and Endostatin From Platelets. Platelets (2010) 21(2):85–93. doi: 10.3109/09537100903470298
156. Uluçkan O, Eagleton MC, Floyd DH, Morgan EA, Hirbe AC, Kramer M, et al. APT102, a Novel ADPase, Cooperates With Aspirin to Disrupt Bone Metastasis in Mice. J Cell Biochem (2008) 104(4):1311–23. doi: 10.1002/jcb.21709
157. Matsui Y, Amano H, Ito Y, Eshima K, Suzuki T, Ogawa F, et al. Thromboxane a 2 Receptor Signaling Facilitates Tumor Colonization Through P-Selectin-Mediated Interaction of Tumor Cells With Platelets and Endothelial Cells. Cancer Sci (2012) 103(4):700–7. doi: 10.1111/j.1349-7006.2012.02200.x
158. Honk KV. Inhibition of Tumor Cell Metastasis by Modulation of the Vascular Prostacyclin/Thromboxane A2 System. Clin Exp Metastasis (1983) 1(2):103–14. doi: 10.1007/BF00121490
159. De Leval X, Benoit V, Delarge J, Julémont F, Masereel B, Pirotte B, et al. Pharmacological Evaluation of the Novel Thromboxane Modulator BM-567 (II/II). Effects of BM-567 on Osteogenic Sarcoma-Cell-Induced Platelet Aggregation. Prostaglandins Leukot Essent Fatty Acids (2003) 68(1):55–9. doi: 10.1016/s0952-3278(02)00235-1
160. Boukerche H, Berthier-Vergnes O, Penin F, Tabone E, Lizard G, Bailly M, et al. Human Melanoma Cell Lines Differ in Their Capacity to Release ADP and Aggregate Platelets. Br J Haematol (1994) 87(4):763–72. doi: 10.1111/j.1365-2141.1994.tb06736.x
161. Bastida E, Escolar G, Almirall L, Ordinas A. Platelet Activation Induced by a Human Neuroblastoma Tumor Cell Line is Reduced by Prior Administration of Ticlopidine. Thromb Haemost. (1986) 55(3):333–7. doi: 10.1055/s-0038-1661558
162. Lian L, Li W, Li ZY, Mao YX, Zhang YT, Zhao YM, et al. Inhibition of MCF-7 Breast Cancer Cell-Induced Platelet Aggregation Using a Combination of Antiplatelet Drugs. Oncol Lett (2013) 5(2):675–80. doi: 10.3892/ol.2012.1074
163. Lucotti S, Cerutti C, Soyer M, Gil-Bernabé AM, Gomes AL, Allen PD, et al. Aspirin Blocks Formation of Metastatic Intravascular Niches by Inhibiting Platelet-Derived COX-1/ Thromboxane A2. J Clin Invest. (2019) 129(5):1845–62. doi: 10.1172/JCI121985
164. Wang L, Zhang R, Yu L, Xiao J, Zhou X, Li X, et al. Aspirin Use and Common Cancer Risk: A Meta-Analysis of Cohort Studies and Randomized Controlled Trials. Front Oncol (2021) 11:690219. doi: 10.3389/fonc.2021.690219
165. Mehta-D’souza P, Klopocki AG, Oganesyan V, Terzyan S, Mather T, Li Z, et al. Glycan Bound to the Selectin Low Affinity State Engages Glu-88 to Stabilize the High Affinity State Under Force. J Biol Chem (2017) 292(6):2510–8. doi: 10.1074/jbc.M116.767186
166. Pouyani T, Seed B. PSGL-1 Recognition of P-Selectin is Controlled by a Tyrosine Sulfation Consensus at the PSGL-1 Amino Terminus. Cell (1995) 83(2):333–43. doi: 10.1016/0092-8674(95)90174-4
167. Kim YJ, Borsig L, Varki NM, Varki A. P-Selectin Deficiency Attenuates Tumor Growth and Metastasis. Proc Natl Acad Sci U.S.A. (1998) 95(16):9325–30. doi: 10.1073/pnas.95.16.9325
168. Hallahan DE, Staba-Hogan MJ, Virudachalam S, Kolchinsky A. X-Ray-Induced P-Selectin Localization to the Lumen of Tumor Blood Vessels. Cancer Res (1998) 58(22):5216–20.
169. Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A. Heparin and Cancer Revisited: Mechanistic Connections Involving Platelets, P-Selectin, Carcinoma Mucins, and Tumor Metastasis. Proc Natl Acad Sci U.S.A. (2001) 98(6):3352–7. doi: 10.1073/pnas.061615598
170. Borsig L, Wong R, Hynes RO, Varki NM, Varki A. Synergistic Effects of L- and P-Selectin in Facilitating Tumor Metastasis can Involve non-Mucin Ligands and Implicate Leukocytes as Enhancers of Metastasis. Proc Natl Acad Sci U.S.A. (2002) 99(4):2193–8. doi: 10.1073/pnas.261704098
171. Mannori G, Crottet P, Cecconi O, Hanasaki K, Aruffo A, Nelson RM, et al. Differential Colon Cancer Cell Adhesion to E-, P-, and L-Selectin: Role of Mucin-Type Glycoproteins. Cancer Res (1995) 55(19):4425–31.
172. Häuselmann I, Borsig L. Altered Tumor-Cell Glycosylation Promotes Metastasis. Front Oncol (2014) 4:28(28). doi: 10.3389/fonc.2014.00028
173. Queiroz KCS, Shi K, Duitman JW, Aberson HL, Wilmink JW, Van Noesel CJM, et al. Protease-Activated Receptor-1 Drives Pancreatic Cancer Progression and Chemoresistance. Int J Cancer (2014) 135(10):2294–304. doi: 10.1002/ijc.28726
174. Shi K, Damhofer H, Daalhuisen J, Brink Mt, Richel DJ, Spek CA. Dabigatran Potentiates Gemcitabine-Induced Growth Inhibition of Pancreatic Cancer in Mice. Mol Med (2017) 23:13–23. doi: 10.2119/molmed.2016.00214
175. Castle J, Farnie G, Kirwan CC. PO-11 - Thrombin and Cancer Stem-Like Cells: In Vitro Support for Breast Cancer Anticoagulation. Thromb Res (2016) 140:S180. doi: 10.1016/S0049-3848(16)30144-X
176. Battinelli EM, Markens BA, Kulenthirarajan RA, Machlus KR, Flaumenhaft R, Italiano JE. Anticoagulation Inhibits Tumor Cell-Mediated Release of Platelet Angiogenic Proteins and Diminishes Platelet Angiogenic Response. Blood (2014) 123(1):101–12. doi: 10.1182/blood-2013-02-485011
177. Qi Y, Chen W, Liang X, Xu K, Gu X, Wu F, et al. Novel Antibodies Against Gpibα Inhibit Pulmonary Metastasis by Affecting vWF-Gpibα Interaction. J Hematol Oncol (2018) 11:1. doi: 10.1186/s13045-018-0659-4
178. Zhang W, Dang S, Hong T, Tang J, Fan J, Bu D, et al. A Humanized Single-Chain Antibody Against Beta 3 Integrin Inhibits Pulmonary Metastasis by Preferentially Fragmenting Activated Platelets in the Tumor Microenvironment. Blood (2012) 120(14):2889–98. doi: 10.1182/blood-2012-04-425207
179. Kononczuk J, Surazynski A, Czyzewska U, Prokop I, Tomczyk M, Palka J, et al. αiibβ3-Integrin Ligands: Abciximab and Eptifibatide as Proapoptotic Factors in MCF-7 Human Breast Cancer Cells. Curr Drug Targets (2015) 16(13):1429–37. doi: 10.2174/1389450115666140804220441
180. Chang YW, Hsieh PW, Chang YT, Lu MH, Huang TF, Chong KY, et al. Identification of a Novel Platelet Antagonist That Binds to CLEC-2 and Suppresses Podoplanin-Induced Platelet Aggregation and Cancer Metastasis. Oncotarget (2015) 6(40):42733–48. doi: 10.18632/oncotarget.5811
181. Cho MS, Noh K, Haemmerle M, Li D, Park H, Hu Q, et al. Role of ADP Receptors on Platelets in the Growth of Ovarian Cancer. Blood (2017) 130(10):1235–42. doi: 10.1182/blood-2017-02-769893
182. Guillem-Llobat P, Dovizio M, Bruno A, Ricciotti E, Cufino V, Sacco A, et al. Aspirin Prevents Colorectal Cancer Metastasis in Mice by Splitting the Crosstalk Between Platelets and Tumor Cells. Oncotarget (2016) 7(22):32462–77. doi: 10.18632/oncotarget.8655
Keywords: TCIPA, platelets, aggregation, activation, cancer
Citation: Strasenburg W, Jóźwicki J, Durślewicz J, Kuffel B, Kulczyk MP, Kowalewski A, Grzanka D, Drewa T and Adamowicz J (2022) Tumor Cell-Induced Platelet Aggregation as an Emerging Therapeutic Target for Cancer Therapy. Front. Oncol. 12:909767. doi: 10.3389/fonc.2022.909767
Received: 31 March 2022; Accepted: 16 May 2022;
Published: 23 June 2022.
Edited by:
Abdel-Majid Khatib, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Fabrizio Martelli, National Institute of Health (ISS), ItalyPavel Davizon-Castillo, University of Colorado, United States
Copyright © 2022 Strasenburg, Jóźwicki, Durślewicz, Kuffel, Kulczyk, Kowalewski, Grzanka, Drewa and Adamowicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wiktoria Strasenburg, d2lrdG9yaWEuc3RyYXNlbmJ1cmc5NUBnbWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship