 Richard P. Tucker
Richard P. Tucker Martin Degen
Martin Degen- 1Department of Cell Biology and Human Anatomy, University of California, Davis, Davis, CA, United States
- 2Laboratory for Oral Molecular Biology, Department of Orthodontics and Dentofacial Orthopedics, University of Bern, Bern, Switzerland
For their full manifestation, tumors require support from the surrounding tumor microenvironment (TME), which includes a specific extracellular matrix (ECM), vasculature, and a variety of non-malignant host cells. Together, these components form a tumor-permissive niche that significantly differs from physiological conditions. While the TME helps to promote tumor progression, its special composition also provides potential targets for anti-cancer therapy. Targeting tumor-specific ECM molecules and stromal cells or disrupting aberrant mesenchyme-cancer communications might normalize the TME and improve cancer treatment outcome. The tenascins are a family of large, multifunctional extracellular glycoproteins consisting of four members. Although each have been described to be expressed in the ECM surrounding cancer cells, tenascin-C and tenascin-W are currently the most promising candidates for exploitability and clinical use as they are highly expressed in various tumor stroma with relatively low abundance in healthy tissues. Here, we review what is known about expression of all four tenascin family members in tumors, followed by a more thorough discussion on tenascin-C and tenascin-W focusing on their oncogenic functions and their potential as diagnostic and/or targetable molecules for anti-cancer treatment purposes.
1 Introduction
The extracellular matrix (ECM) is a complex meshwork of various cell-secreted macromolecules that regulates the development and homeostasis of tissues. In humans, the ECM is broadly composed of a mixture of proteins, polysaccharides, and water that, depending on the specific composition, reflect the physiological requirements for a given tissue (1). The main building blocks of the ECM are (i) collagens, (ii) glycoproteins, (iii) proteoglycans, and (iv) elastin. Collagens include the most abundant fibrous proteins of our body. They maintain the structural integrity of tissues by providing tensile strength. Glycoproteins are responsible for either linking different ECM molecules or mediating cell-ECM interactions, while proteoglycans include gel-like elements within the ECMs that allow a tissue to resist compressive forces. Finally, elastin, as its name indicates, is responsible for tissue elasticity (2). The human genome harbors 274 genes encoding for the proteins of the core matrisome (3). Although typical ECM-like domains were already present in the proteins of unicellular organisms, the complexity and diversity of the matrisome mainly arose during the transition from unicellular (protozoa) to multicellular (metazoa) animals. The emergence of more complex and multicellular organisms required major evolutionary innovations, which were accompanied by the creation of more specialized and sophisticated ECM frameworks. Extensive elaborations of ECM proteins by exon shuffling, multiplications, and gene family expansions provided the rich repertoire of ECM molecules required for building the various structures (4, 5).
Initially, the ECM was mainly perceived as an inert physical scaffold for cells. However, this view has changed considerably during recent decades. Not only does the ECM provide structural and mechanical support for tissue integrity, but it is also responsible for the mobilization and availability of growth factors and cytokines (4). Additionally, the three-dimensional (3D) network provides specific biochemical and biomechanical properties that are sensed by specialized transmembrane receptors, such as integrins, syndecans and discoidin domain receptors, which can bind specific motifs in ECM proteins. Receptor ligation results in its activation, triggering signaling cascades that regulate various vital cellular processes, including cell adhesion, migration, proliferation, survival, and differentiation. Moreover, the ECM is subject to specific modifications and remodeling, which tailors the ECM to the unique needs of tissues. Therefore, each tissue is surrounded by an ECM that is fine-tuned in its composition and arrangement, providing the necessary cues for tissue homeostasis.
Since tissue homeostasis is often disrupted in cancers, the ECM surrounding cancer cells is significantly different from the one found in healthy conditions (6, 7). The role of the tumor microenvironment (TME) on cancer cells has only recently begun to be appreciated. Earlier cancer research was focused on the individual cancer cell, oncogenes and tumor suppressors; novel mutations and hyperactive signaling pathways were then identified and exploited as anti-cancer therapies. While this approach revolutionized cancer treatment, it seems clear today that cancers need to be viewed as complex heterogeneous tissues. Apart from cancer cells, cancers also depend on supporting components present in the TME that are not malignant themselves. These include the vasculature, immune cells, fibroblasts, and the ECM. Cellular sources of this tumor-specific ECM might be tumor cells themselves, but more often this matrix is derived from cancer-associated fibroblasts (CAFs). CAFs are believed to evolve from the activation of quiescent fibroblasts by epithelial-mesenchymal interactions, often involving cancer cell-derived growth factors (8). Hence, while carcinoma growth is initiated by transformed epithelial cells, cancers continuously tailor and orchestrate their microenvironment for their specific needs. Consequently, a unique composition and mix of ECM molecules is created, which provides feedback to malignant cells allowing the modulation of various hallmarks of cancers (9–11).
There is mounting evidence that the tumor ECM plays an active role in driving malignancy (9). Hence, novel anti-cancer drugs might be more powerful if they not only aim to eliminate cancers cells, but also try to normalize the TME. Such an approach requires a better understanding of the aberrant tumor cell-stroma crosstalk as well as a better understanding of the tumor-specific ECM molecules that are present. At least two members of the tenascin family of ECM glycoproteins, tenascin-C (TN-C) and tenascin-W (TN-W), are highly expressed in most solid cancers, but are expressed at much lower levels in normal tissues. Both of these tenascins provide multifunctional pro-tumorigenic activities (12). Therefore, tenascins, as specific components of tumor-permissive ECMs, are interesting candidates for potential exploitation for cancer therapies. Herein, we will briefly revisit the tenascin family with a focus on their structure and patterns of expression in physiological as well as in cancerous conditions. We will further focus on their cancer-related functions providing the basis for their exploitability and value for translational applications and clinical benefit.
2 A Brief Encounter With the Tenascins
Tenascins are a family of modular ECM glycoproteins (Figure 1). The family members share both a basic domain architecture and a common evolutionary ancestor. Near their amino-termini most tenascins have a coiled-coil domain that supports trimerization. This is followed by one or more EGF-like domains, numerous fibronectin-type III (FNIII) repeats, and a fibrinogen-like globe (FBG) at their carboxy-termini. The founding member of the tenascin family is TN-C, with the C designating “cytotactin”, which was one of its original monikers. This was followed by descriptions of tenascin-R (TN-R), tenascin-X (TN-X) and TN-W (31, 32). While the roles of the EGF domains remain to be clarified, one or more of a tenascin’s FNIII repeats typically contains integrin-binding motifs (33, 34), and tenascin FBGs bind to and activate toll-like receptor 4 (TLR4) (35). Tenascins are only found in chordates, with all four family members present in bony fishes and tetrapods (36).
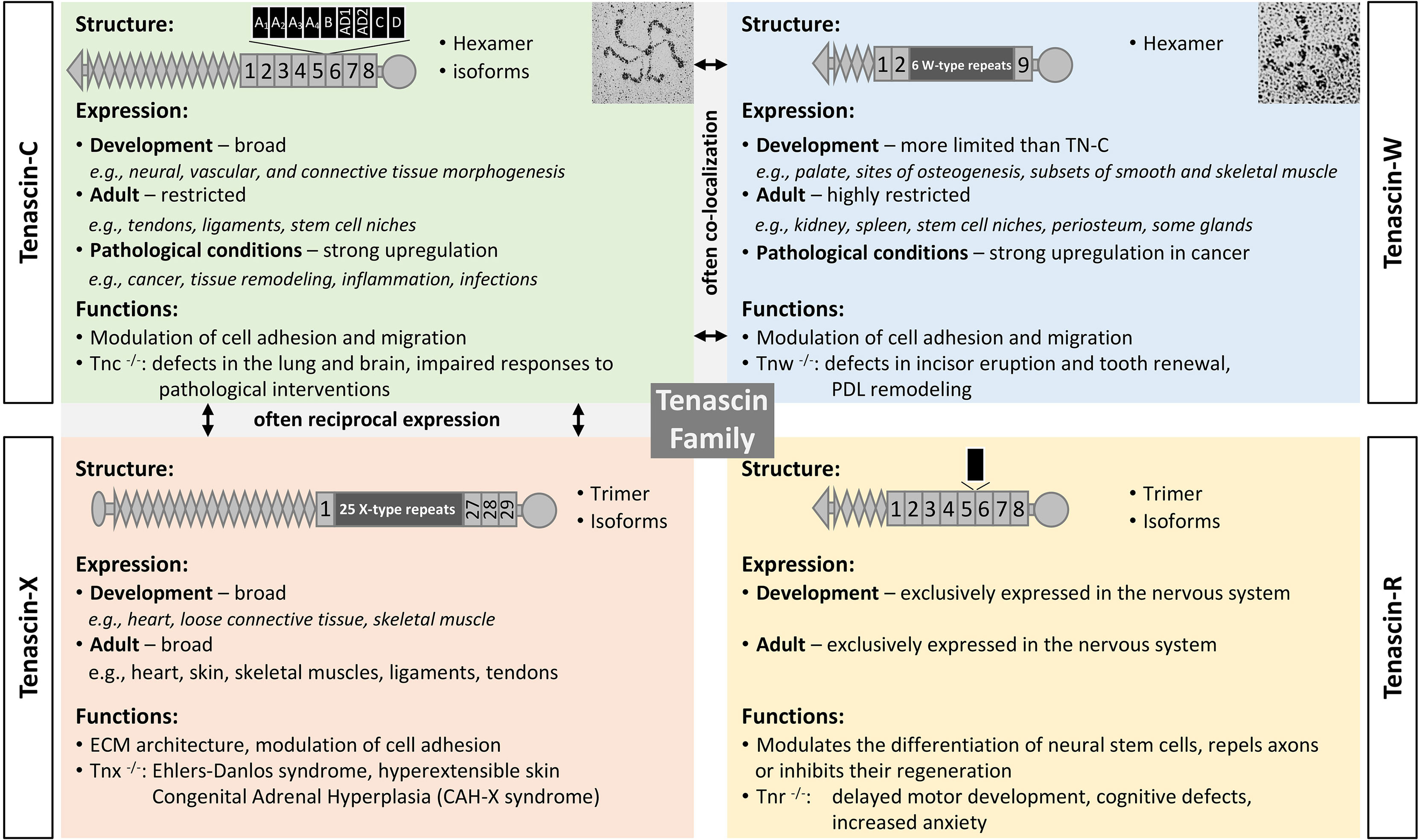
Figure 1 The tenascin family. A brief description of the four tenascin members, including their structure and alternative splicing, major sites of expression, and functions. Original rotary shadowing micrograph for TN-C is from Chiquet-Ehrismann et al. (13) and for TN-W from Scherberich et al. (14) both with permission. The following symbols have been used to describe the structural domains: EGF-like domains (diamonds), FNIII domains (boxes), fibrinogen globe (circle). FNIII repeats shown above the structure are subject to alternative splicing (black boxes). Selected references: for TN-C (15–17), TN-X (18–21), TN-R (22–24), TN-W (25–27), and for tenascins in general (28–30).
After considering each member of the tenascin family, this review emphasizes what we have learned about the two tenascins that are most abundant in the TME: TN-C and TN-W. In humans, TN-C has 14 EGF-like domains and 8 constant FNIII repeats. Between the 5th and 6th constant FNIII repeats alternative splicing can add up to 9 additional FNIII repeats, resulting in hundreds of TN-C variants with significantly different molecular weights (32, 37). The potential to exploit differential expression patterns of TN-C isoforms in normal tissues and in cancer is described below. Human TN-W has just three EGF-like domains and 9 FNIII repeats. There is no evidence of alternative splicing in TN-W (32). In addition to trimerization via the coiled-coil domain, the amino-termini of TN-C and TN-W have free cysteine residues that support the formation of hexamers from two trimers (i.e., hexabrachions). As well as being integrin ligands, both TN-C and TN-W can bind to Wnt3a (38).
TN-C and TN-W are both widely expressed during embryonic development. For example, TN-C is found around migrating neural crest cells, at sites of branching morphogenesis and epithelial-mesenchymal interactions, and throughout the central nervous system in zones where glial precursors are proliferating and migrating. TN-C and TN-W are both expressed at sites of smooth muscle morphogenesis as well as chondrogenesis and osteogenesis, where their expression patterns often partially overlap (32, 37). In the adult, both remain highly expressed in dense connective tissues (28) and in certain stem cell niches (39), but they are largely absent from most other tissues (Figure 2).
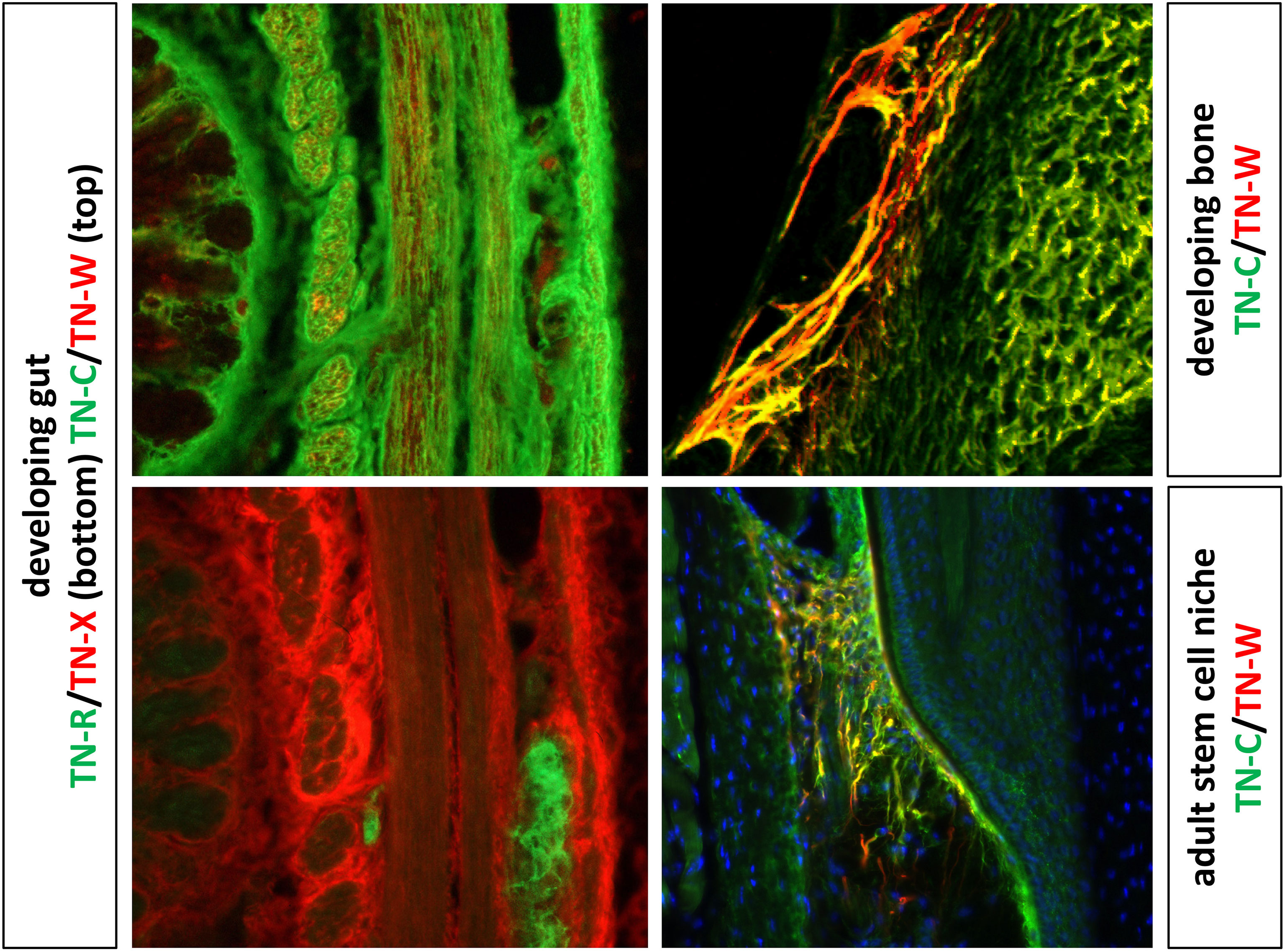
Figure 2 The images to the left show double label immunohistochemistry of the developing gut with antibodies to TN-C, TN-W, TN-R and TN-X. TN-C expression is widespread, while TN-W is found in small patches in a subset of the smooth muscle. TN-R is limited to the autonomic ganglia of the gut, while TN-X is concentrated in the epimysium surrounding bundles of smooth muscle. On the top right is a section of a developing ulna immunostained with antibodies to TN-C and TN-W. Both are strongly expressed in developing bone, with partially overlapping patterns (yellow). On the bottom right is a section though the whisker follicle stem cell niche of an adult mouse. The patterns of TN-C and TN-W expression are more limited in the adult, but they are found in partly overlapping patterns in this stem cell niche. Original images of the developing gut and bone from Meloty-Kapella et al. (40) and adult stem cell niches from Tucker et al. (41) with permission.
3 Expression of the Tenascin Members in Cancer
The TME plays an active role in cancer progression. Therefore, full awareness of the composition and expression patterns of tumor-specific ECM, compared to the matrix of corresponding healthy tissues, is of high clinical importance. Tumor-specific ECM molecules might be harnessed and targeted in combination with more classical anti-cancer treatment approaches for the benefit of patients. Almost 40 years ago, TN-C was identified as an ECM protein enriched in the stroma of glioma (42). As other members of the tenascin family were identified, each was studied in turn to see if it was also expressed in tumor stroma. The next section will focus on the expression patterns of all four members of the tenascin family in solid cancers.
3.1 TN-C: “The Founding Member”
As described above, TN-C is transiently expressed in a highly spatio-temporal manner in various tissues during development (Figure 2). In the adult, its expression is only preserved in a few tissues that mainly have to withstand tensile stress (e.g., tendons, ligaments) (28). A robust re-expression in solid cancers is one of the most intriguing features of TN-C. One of the earliest studies analyzing tumor-specific TN-C expression was performed in mouse models of mammary breast cancer (43). These data described TN-C as an ECM molecule present in malignant breast cancer, but absent in the normal healthy mammary parenchyma as well as in benign tumors of the breast. This initial discovery fueled subsequent studies analyzing various cancers for the presence of TN-C, which resulted in an extremely rich literature on its expression in human tumors. TN-C has been identified in the tumor stroma of basically all cancer types analyzed, including cancers derived from breast, brain, uterus, ovary, prostate, pancreas, colorectal, kidney, biliary tract, stomach, gastric, osteosarcoma, oral cavity, urinary tract, skin tissue and many more [for reviews see (44–47)]. This abundant tumor-specific TN-C expression is compelling as TN-C is only mildly expressed in normal adult tissues, although most of them are not completely deprived of it. In certain cancers, such as renal cell carcinoma, brain and lung cancer, TN-C is expressed adjacent to CD31-positive vascular endothelial cells (48). TN-C is not only abundantly expressed in the tissues of cancer patients, but also detectable in their sera. Several studies reported increased TN-C levels in serum of cancer patients compared to healthy controls in non-small cell lung cancer, squamous cell carcinoma of the head and neck, colorectal cancer, breast cancer, and ovarian cancer (49–52).
The TNC gene is subject to extensive alternative splicing within its FNIII repeats (Figure 1). Hence, many different TN-C isoforms can be produced, with the larger isoforms with additional FNIII repeats typically present during development and in tumors. Indeed, the TN-C isoform spectrum present in the ECM surrounding cancer cells significantly differs from the one in normal ECM (53–56). Consequently, not only general detection of TN-C in tumor stroma is important, but also knowledge about the precise expression of tumor-specific TN-C isoforms is relevant for translational use, as different isoforms in the stroma might affect cancer cell behavior in a distinct way (57). While alternative splicing adds another level of complexity to the multifunctional activities of TN-C, tumor-specific TN-C isoforms also represent promising exploitable targets for anti-cancer therapies (58, 59).
The observation of a tight regulation of TN-C expression during development, in the adult, and in cancers prompted numerous studies trying to elucidate the regulatory mechanisms for this multifunctional ECM molecule. Many factors have been identified, both in vivo and in vitro, that lead to induction of TN-C. Inducing stimuli include patterning genes during development such as paired-related homeobox 1, as well as growth factors (e.g., PDGF, TGFβ1, FGF2), inflammatory cytokines (e.g., IL1α, IL1β), biophysical properties of the microenvironment (e.g., hypoxia, ECM stiffness), mechanical stress, activated signaling pathways, and micro RNAs (miRNAs). In contrast, glucocorticoids are known to repress TN-C, though the exact molecular mechanism underlying this repression is not clear (60, 61). While some of these regulatory mechanisms might also be relevant for TN-C expression in tumors (e.g., TGFβ1 in breast cancer), the transcription factor SOX4 has been shown to induce TN-C in many malignancies (62). In addition, a crosstalk between TN-C and Notch signaling has also been described in glioma (63). The proximal TNC promoter contains a crucial RBPJκ binding element and in glioblastoma, TN-C was shown to be trans-activated by Notch2 signaling in an RBPJκ-dependent manner. How alternative splicing of the TNC is gene is regulated is not well understood. Apart from some specific splice factors (e.g., Sam68), certain growth factors/cytokines as well as biophysical properties of the microenvironment might also be involved in the splicing regulation of TNC (64–67). Clearly, regulation of TNC is a very complex process that reflects its highly specific expression pattern both in physiological conditions as well as in cancers.
TNC mRNA has been detected both in stromal as well as in cancer cells. Epithelial-mesenchymal interactions between cancer cells and stromal cells results in the differentiation of quiescent fibroblasts into CAFs, which show a unique gene expression profile. TN-C is one of the specific proteins that is secreted and incorporated into a tumor-permissive ECM by CAFs (8). Furthermore, hyperplastic endothelial cells are also a source for TN-C, which showed strong TNC expression in astrocytoma but not in vessels of normal brains (68). Similarly, TNC was also identified as a gene highly enriched in endothelial cells isolated from a panel of malignant compared to normal tissues (69). However, TN-C expression has also been found in many cancer cells, including cancers of the breast, brain, skin, colon, pharynx, and larynx as well as in oral squamous cell carcinoma (46). The strongest TN-C positivity is often observed in the cancer cells at the periphery of malignant nests suggesting a role for this tenascin in driving invasion and migration (70). The importance of epithelial-mesenchymal interaction for TN-C expression is illustrated by the fact that in co-culture experiments cancer cells are able to induce TN-C expression in otherwise TN-C-negative fibroblasts (71), while TN-C-negative cancer cells start to express this tenascin when co-cultured with embryonic mesenchyme (72).
3.2 TN-R: “The Brain-Specific Member”
TN-R shows a very tissue-specific expression pattern during development and in the adult as it is only detectable in the nervous system (Figure 2) (22, 73). Relatively few studies focused on the expression and function of TN-R in cancers, and its expression was only revealed in brain cancer. As often is the case with a low number of studies and lack of functional analyses, the expression as well as the role of TN-R in brain cancer remains controversial. Although TN-R was found to be expressed in fetal cerebellum, TN-R was not detectable in the most common malignant brain tumor in children, medulloblastoma, as assessed by a cDNA microarray as well as immunohistochemistry (74). In contrast, analysis by high resolution northern blot displayed TNR in both astrocytoma and meningioma (75). Ayachi et al. show by quantitative real-time PCR (qPCR) and in situ hybridization (ISH) that TNR was over-expressed in pilocytic astrocytoma and ganglioglioma, two non-invasive tumors, while in low-grade infiltrative glioma and glioblastoma, TNR expression was moderate and negative, respectively (76). These results suggest that TN-R could act as a suppressor of glioma invasion. But clearly, more studies are required to allow a conclusive verdict to be given about the role of TN-R in brain and in other cancers.
Regulation of TNR remains not well-understood at the moment. Some growth factors and cytokines, including PDGF and NGF, have been shown to induce TN-R in vitro. TN-R is mainly produced by oligodendrocytes. Co-culturing these cells with either astrocytes, neurons, or conditioned medium from activated microglia modulates TN-R expression (60). Whether any of these or related mechanisms play a role in aberrant TN-R expression in cancer is not known.
3.3 TN-X – “The Controversial Member”
TN-X is found in epimysium and loose connective tissues (Figure 2) (32). As with TN-R, only a few studies have addressed TN-X expression in tumor-specific ECMs, and the results of these studies have proven to be controversial. TN-X levels were found to be elevated in the tumor stroma of malignant mesothelioma, ovarian cancer, and low-grade astrocytoma. In the latter, TN-X was expressed in the adventita and the perivascular niche (77–80). In mesothelioma, high TN-X expression allowed the differential diagnosis between malignant mesothelioma and lung adenocarcinoma (78) as well as between malignant mesothelioma and ovarian carcinoma/peritoneal serous carcinoma (77). TN-X is also expressed by Hodgkin lymphoma-derived Reed Sternberg cells (81). Additionally, elevated TN-X concentrations were detectable in serum of breast cancer and ovarian cancer patients compared to control serum (79). Contradictory results were observed in high-grade astrocytoma, malignant nerve sheath tumors, melanoma, and cancers of the uterus (leiomyoma) (80, 82–84). In these malignancies, TN-X was markedly reduced compared to their corresponding normal tissues or to their benign forms. The most elaborate study on TN-X expression in cancer, which used in silico analyses of expression data as well as immunohistochemical screening of tissue microarrays, was recently published (85). In this study, TN-X was found to be downregulated in the six cancers with highest incidence and mortality in the world (lung, breast, colon, prostate, stomach, and liver) as well as in all other cancers tested. Notably, diminished TN-X levels were found in the cancers reported to have upregulated TN-X expression by others (see above). Only in brain cancers (astrocytoma, glioblastoma multiforme and oligodendroglioma) was TN-X found to be enriched compared to healthy tissue. For lung adenocarcinoma and breast cancer, clinical data matching the in silico datasets were also available, which allowed to establish a negative correlation between TN-X expression and tumor stage as well as patient survival. These data suggest that TN-X levels are reduced in most cancers and that TN-X might represent a promising prognostic biomarker (85). Tumor-specific TN-X downregulation was also shown in another study examining The Cancer Genome Atlas for ECM molecule dysregulation in a large panel of malignancies (86). While 58 of the 249 ECM molecules analyzed were dysregulated in cancers, TNXB was found to be the most significantly downregulated gene overall. However, all these data have to be interpreted carefully since (i) TN-X RNA and protein levels did not always correlate; (ii), TN-X downregulation was not observed in every clinical specimen analyzed from the same cancer type; (iii) TN-X was still readily detectable in cancer tissues. However, these observations definitely warrant additional studies looking at TN-X expression and function in cancers.
Very limited data are available regarding TNXB gene regulation. So far, there is no evidence that TNXB responds to any growth factors or cytokines, which is in contrast to the other tenascin members. This is somewhat surprising since several putative binding sites for Sp1/Sp3 transcription factors were identified close to the transcription start site and found to be functional and required for driving TNXB expression. However, the upstream signaling pathways remain elusive. As with other tenascin family members, it is known that glucocorticoids have a negative effect on TNXB levels. However, details remain unresolved (60).
3.4 TN-W: “The Most Tumor-Specific Member”
TN-W is the newest member of the tenascin family, which was often found co-expressed or at least in the vicinity of TN-C during development (Figure 2) (25, 26). Since TN-C has been known for its high expression in the tumor stroma of breast cancer, initial studies on TN-W in tumors focused on this tissue as well. Using different breast cancer mouse models, TN-W was found to be enriched in the tumor stroma surrounding breast cancer cells. However, tumor stroma was only positive for TN-W in the models with a high likelihood to metastasize and not in the non-metastatic ones (87). Encouraged by these findings in mice, the human orthologue was cloned, expressed, and antibodies raised against it. The first tissues analyzed resulted in intriguing observations. In healthy normal breast and colorectal tissues, TN-W was not detectable at all, while their corresponding cancerous tissues displayed a robust and highly significant de novo expression of TN-W (52, 88). Screening of additional human tumor types confirmed these initial observations: TN-W was strongly enriched in the stroma of brain (glioblastoma, oligodendroglioma, astrocytoma), prostate, kidney (clear cell carcinoma, papillary carcinoma, chromophobe renal carcinoma, and oncocytoma), ovarian, prostate, pancreas, biliary tract (liver carcinoma, gallbladder carcinoma), and lung cancers as well as in melanoma (48, 89, 90). Most significantly, TN-W was not detectable in the corresponding healthy tissues. However, TN-W expression in tumors was heterogenous, varying from low to very high levels, but was significantly increased compared to the normal tissues in most of the cases tested. For some metastatic melanoma patients, lymph node metastases as well as metastases from diverse locations (spleen, lung and skin) were found to be TN-W positive (87.5% of lymph node metastases, and 85% of other metastases) (48). Similar to TN-C, TN-W was often found to be expressed in close proximity to the vasculature as assessed by co-staining of TN-W with endothelial cell markers CD31/Pecam-1, von-Willebrand-factor, and Desmin in at least kidney, lung, breast, colon, ovary, and brain cancers (48, 90). Elevated concentrations of TN-W could also be measured in the serum of breast and colon cancer patients compared to healthy controls using a sensitive TN-W sandwich ELISA (52).
Although the list of solid cancers with prominent upregulation of TN-W is constantly growing, some very elementary questions about TN-W in tumor stroma remain. For instance, what is the cellular source of its expression? It is believed that TN-W is mainly expressed by stromal cells mediated by epithelial-mesenchymal interactions. This statement is supported by immunohistochemical analyses of TN-W that never revealed any TN-W expression in cancer cells as well as by in vitro experiments unsuccessfully trying to detect endogenous TN-W in numerous cancer cell lines of various origin (26). The concept of stromal cell origin of TN-W was reinforced by observations that bone marrow-derived stromal cells exclusively expressed TN-W when co-cultured with malignant cells in a xenograft model of breast cancer cell displaying metastatic propensity toward bone (91). However, a recent study screening for TNW transcripts in 20 biliary tract cancer cell lines is challenging the idea about pure stromal cell origin of TN-W (89). While most of the cells displayed very modest or negligible TNW levels, Huh-28 cells, an intrahepatic cholangiocarcinoma cell line, showed high levels of TNW. These results were further confirmed by immunoblotting and immunocytochemistry. Notably, when cultured alone, TN-W in Huh-28 cells was mainly observed to be intracellular without being organized and incorporated into the ECM. However, when co-cultured with bone marrow-derived stromal cells, Huh28-derived TN-W organized into ECM fibrils surrounding nests of tumor cells, a situation that closely mimics the situation of tumors in vivo (89). Therefore, there is strong evidence that proper epithelial-mesenchymal crosstalk is required for formation of TN-W-positive fibrils, but also that cancer cells themselves might represent a cellular source for TN-W, which is similar to the situation for TN-C. Additional experiments, such as ISH on tumor samples, are required to fully elucidate the cell types responsible for TN-W expression and to know whether the cellular source might differ among distinct tumors.
Another open question about TN-W expression in tumor stroma concerns the regulatory mechanisms of its de novo expression. Most of the available knowledge on this topic has been gained in in vitro studies of mouse and chicken cells. Growth factors, such as BMP2, BMP7, and TNFα induce TN-W in mouse embryonic fibroblasts, mouse cranial fibroblasts, mouse C2C12 myoblasts, HC11 normal mouse mammary gland epithelial cells, mouse bone marrow-derived Kusa-A1 cells as well as in primary chicken osteoblasts (14, 92, 93). In addition, Wnt5a seems also indirectly involved in triggering TN-W expression through p38/MAPK signaling (94). However, the molecule that acts as the upstream inducer has not been identified. In contrast, none of these factors have been shown to be able to activate TNW in human cells. Today, we only know that TGFβ1 signaling has an inducing effect on TN-W. Breast cancer cells secrete TGFβ1 into the tumor stroma, where the growth factor is sensed by bone marrow-derived stromal cells, which leads to a Smad4-dependent induction of TNW in the stromal cells (91). Negative impact on TNW transcripts has been attributed to glucocorticoids, which is similar to their effect on TNC and TNXB (60). More studies and research are required to better understand cancer-specific expression of TN-W.
There is not much evidence available for the presence of specific TN-W isoforms or TN-W modifications. The only observation in this regard was gained in the Huh-28 cells, which showed a TN-W protein with a higher molecular weight than expected (89). Whether this result hints at a tumor-specific isoform, modification, or multimerization of TN-W in this cell line and whether it has any relevance in vivo remains to be seen.
4 Potential Cancer-Promoting Activities of Tenascins
As mentioned before, tenascins are multifunctional matricellular proteins with a highly specific pattern of expression during development and in cancer. Although each family member might be involved in cancers to some extent (see above), currently there is only robust and consistent data about expression and function in tumors for two of the family members: TN-C and TN-W. Therefore, we put the focus in the following chapters on TN-C and TN-W and try to emphasize their role in regulating various cellular processes required for tumor progression (Figure 3).
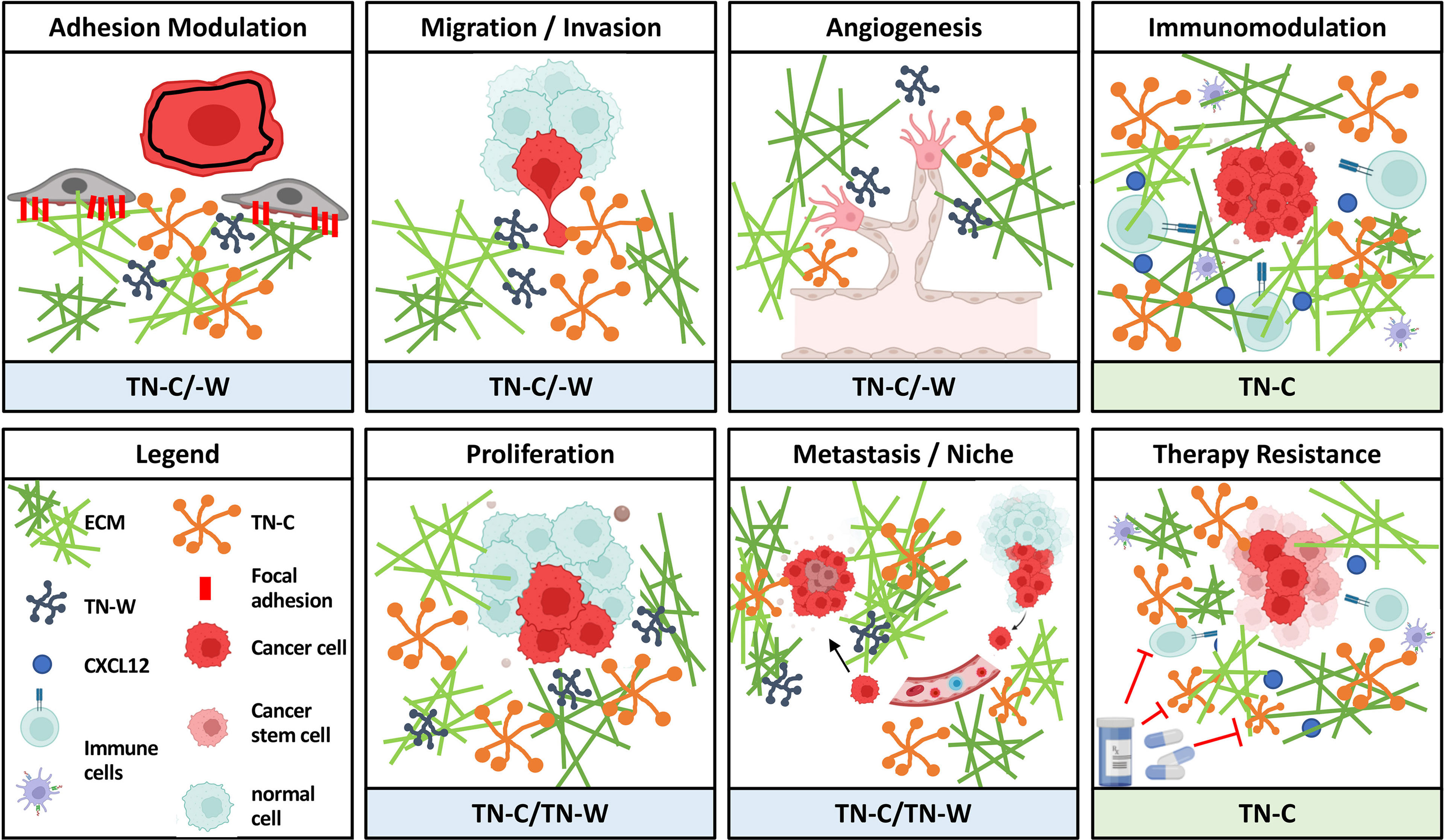
Figure 3 Overview of oncogenic activities of TN-C and TN-W. As matricellular proteins, TN-C and TN-W can influence various cellular processes involved in tumor progression by modulating the interplay between stromal and epithelial cells or other cells. Schemes were created using www.biorender.com.
4.1 Modulation of Cell Adhesion
The potential to modulate cell adhesion is probably one of the most described and studied characteristics of TN-C. TN-C is an anti-adhesive molecule, which, as substratum, does not promote attachment and spreading of either epithelial cells (e.g., breast cancer cells) or fibroblasts (95). The most prominent feature of TN-C is its inhibitory effect on fibronectin (FN)-mediated cell adhesion (13). Attachment and spreading on FN requires cells to engage synergistically both the heparan sulfate proteoglycan receptor syndecan-4 and integrin α5β1. However, TN-C can bind to FN at a heparin binding motif located around the 13th FNIII repeat, which leads to a competitive hindrance of syndecan-4 binding to FN. Consequently, the presence of TN-C (such as in tumors) inhibits cell spreading and increases cell proliferation (96). Similar, but less consistent and less understood properties, have been found for TN-W in regulating cell adhesion. Initially, a TN-W substratum was found to be anti-adhesive for human breast cancer cells, while it promoted human fibroblast attachment, but not proper spreading. However, for both cell types, the presence of TN-W did not interfere with FN-mediated attachment (88). Different results were observed in primary chicken osteoblasts, mouse C2C12 myoblasts, embryonic fibroblasts, and whisker follicle stem cells, for which TN-W was able to interfere with FN-mediated cell spreading when soluble or used as substratum (41, 97). These results suggest that the activity of TN-W to modulate cell adhesion and spreading is cell-type specific. For cancer cells, TN-W seems to be anti-adhesive and not able to interfere with FN-mediated cell attachment, while for normal cells, TN-W shares the anti-adhesive properties of TN-C towards FN-mediated cell spreading. However, the mechanistical details underlying the latter observation remain unknown, as direct TN-W/FN interactions have yet to be reported.
4.2 Cell Proliferation
In vitro, cancer cell lines can be stimulated to become hyperproliferative by the addition of recombinant TN-C (45). This is most likely the consequence of TN-C impairing cell adhesion to FN by blocking the binding site of syndecan-4 (96). As a result, tumor cell proliferation was enhanced in the presence of TN-C. Gene expression profiling revealed the mechanistical details for this effect: TN-C disrupts the actin cytoskeleton by downregulation of tropomyosin 1, while it was able to de-repress and activate Wnt and MAPK signaling, respectively (98). Besides having a pro-proliferative effect on cancer cells, TN-C is also able to stimulate proliferation of endothelial and smooth muscle cells (45). The mitogenic activity of TN-C is supported in vivo by immunohistochemistry showing TN-C co-expression with the proliferation marker Ki-67/Mib-1 in many invasive carcinomas (99), and the observation that TN-C is associated with proliferative epithelial cells in regenerating epithelia (100). A study by Swindle et al. also revelaed that select EGF-like repeats of TN-C might promote proliferation in an EGFR-dependent way by activating downstream MAPK signaling (101). In contrast, there are not many studies available on the effect of TN-W on cell proliferation. While an anti-proliferative activity was reported for TN-W in the murine osteoblastic cells MC3T3 (92), treatment of MDA-MB231-1833 breast cancer cells with different concentrations of recombinant TN-W resulted in a significant increase of BrdU incorporation, suggesting that TN-W is able to stimulate cancer cell proliferation (91).
4.3 Cell Migration/Invasion
In clinical specimens, TN-C is often expressed in the intratumoral stroma but also at the invasive front of the tumor nests (46). TN-C expression at the invasive front is correlated with aggressive tumor behavior and poor prognosis and is suggestive of a role for TN-C in promoting migration and invasion of cancer cells (45, 46). These observations could be recapitulated in vitro by showing increased motility of various cell types, including cancer cells, fibroblasts, endothelia cells, and smooth muscle cells, in the presence of soluble TN-C or TN-C as substratum (102–104). Again, the TN-C-mediated cell adhesion modulating effect might explain the mechanisms underlying the pro-migratory outcome. In the presence of TN-C, cancer cells cannot properly spread, favoring an intermediate state of adhesions with suppressed Rho activation and actin-rich filipodia, which enhances their motile behavior (105). In contrast, lack of TN-C allows cell spreading and the formation of stable focal adhesions with stress fibers. Similar pro-migratory properties can be attributed to TN-W, although the molecular mechanisms have not been elucidated yet. First evidence for this finding has been gained in the highly metastatic murine mammary cancer cell line 4T1, which showed a faster migratory behavior across a transwell filter coated with TN-W (87). A follow-up study using human T47D breast cancer cells lines confirmed this result and showed that treatment of the cells with soluble TN-W increased their migratory phenotype (88). Accordingly, the migration of chicken osteoblasts across filters coated with TN-W was stimulated and TN-W added to the medium increased osteoblast migration when cultured on FN-coated filters when compared to controls (106).
Another prominent feature that is linked to altered motile behavior is the process of epithelial-mesenchymal-transition (EMT). While EMT is crucial during development, cancers often use this process for invasion and their dissemination into distant organs (107). TN-C has been found to be able to promote EMT. In MCF7 breast cancer cells, TN-C induced EMT-like morphological changes, which, on a molecular level, included the delocalization of E-cadherin and β-catenin correlating with tyrosine-protein kinase SRC activation and the SRC-mediated phosphrorylation of FAK (108). Subsequently, these changes were accompanied by cell detachment from the substratum and an enhanced migratory cell phenotype. In clinical breast cancer specimens, a frequent upregulation and co-localization of TN-C with the mesenchymal marker vimentin was observed at the invasive fronts of cancers in those cells having a more scattered and mesenchymal-like morphology. Such a cancer fingerprint could be correlated to increased malignancy in breast cancer (109). These data suggest an active role for TN-C in promoting EMT-like features. In regard to this, a recent in vitro study identified the highly conserved C-terminal FBG of TN-C and TN-W as activators of latent TGFβ (110). Subsequently, the mature TGFβ is presented to cells, which elicits an intracellular smad-dependent signaling cascade. Since TGFβ is a potent inducer of EMT (111), these data indicate that the FBGs of tenascins might be able to promote a TGFβ-mediated EMT. Whether this process plays an active role in TN-C- and/or TN-W-rich tumor stroma remains to be elucidated. However, EMT-promotion by tenascins represent intriguing options for them to drive cancer progression.
4.4 Metastasis and the “Cancer Stem Cell” Niche
Metastasis is a complex multistep process by which cancer cells gain the power to escape from the primary tumor mass, disseminate into the vasculature and throughout the body, and finally home and colonize distant organ sites to form secondary cancers (112). On this journey, cancer cells must overcome several hurdles and challenges. Successfully disseminated cancer cells often are able to produce a specific microenvironment, a so-called niche, that protects them and enables them longer survival in a hostile environment during their metastatic journey (70, 113). The invasive front of tumors has been suggested to be rich in cancer-initiating cells (“cancer stem cells”) and the same region is known to express high levels of TN-C, indicating that TN-C might be a component of the metastatic niche (70). Disseminating breast cancer cells that express their own TN-C have survival advantages early on until the CAFs take over and produce TN-C, creating a niche that supports their fitness by modulating stemness-like signaling pathways such as Wnt and Notch (70, 114). A more recent study promotes TN-C as a specific and highly-expressed molecule of the lymph node pre-metastatic niche in muscle invasive bladder cancer (115). In this niche, TN-C induction by CAFs is believed to be triggered by cytokines present within tumor cell-derived extracellular vesicles (EV). Hence, infiltrating tumor cells secrete EVs with signaling cues that prepare a TN-C-rich niche, which provides survival advantages (115). Similar findings have recently emerged for TN-W (91). Breast cancer cells on their way to metastasize to bone are surrounded by a specific niche, established by epithelial-mesenchymal interactions. Cancer cell-derived TGFβ1 induces de novo expression of TN-W in the stromal cells, providing an important component for the metastatic niche. Accordingly, aberrant presence of TN-W in the metastatic niche, shaped and tailored by the cancer cells, provides the stimuli for increased cancer cell proliferation and migration, thereby increasing the risk for bone metastases (91). In vitro studies in biliary tract cancers revealed that TN-W fibrils are only formed in the presence of stromal cells (89). This observation might reflect such a metastatic niche as well. In there, TN-W can carry out its potential oncogenic activities leading to the successful dissemination of the cancer cells.
TN-C is also believed to influence metastasis by its very specific organization in the tumor matrix networks. Together with additional ECM proteins, TN-C remodels the ECM to form channel-forming, migratory tracks, which provides signaling cues for the survival of cancer cells and supports dissemination of cancer cells during the metastatic process (116, 117). Notably, periostin (POSTN), another matricellular ECM molecule associated with the metastatic niche (118), interacts with TN-C and is responsible for TN-C incorporation into this specialized ECM organizational units. While doing so, POSTN might enhance collagen cross-linking, which leads to the formation of migratory tracks during metastasis (119). Functionally, both POSTN and TN-C are known to be able to activate Wnt signaling in cancer cells by recruiting Wnt ligands and the downregulation of the Wnt signaling pathway inhibitor DKK1, respectively (98, 118). The education of stromal cells to produce POSTN and TN-C by infiltrating cancer cells represents a crucial step during the metastatic process, which might be exploitable for anti-cancer therapies.
4.5 Angiogenesis
TN-C as well as TN-W are found associated with endothelial cells in various cancers suggesting that both tenascins might play a role in promoting tumor angiogenesis. In vitro, both tenascins trigger an elongated morphology and motility of human umbilical vein endothelial cells (HUVEC), properties related to angiogenic endothelial cells (90). Presence of either TN-W or TN-C in a collagen gel induces endothelial cell sprouts in gel-embedded HUVEC spheroids compared to collagen alone (90). Similarly, injection of malignant melanoma cells into TN-C-deficient mice revealed significantly less vascularization within the tumor mass (120). All these data point towards an active role for TN-C and TN-W in promoting angiogenesis in cancers. Indeed, TN-C in the TME contributes to the angiogenic switch in neuroendocrine tumors (121) and belongs to the AngioMatrix signature (a panel of 110 ECM genes) that correlates with poor prognosis in glioma patients (122). In addition, a higher vessel density, but less functional vessels, are also observations attributed to high TN-C levels in tumors (121). How TN-W triggers angiogenesis mechanistically has yet to be addressed. For TN-C, it is known that its presence in the mesenchyme results in increased levels of the pro-angiogenic factor VEGF in A375 melanoma cell transplant experiments (120) and TN-C is able to repress Wnt signaling by DKK1 in tumor and endothelial cells creating a proangiogenic TME (121). Increased complexity was revealed by a more recent study showing that TN-C can regulate both pro- and anti-angiogenic factors (123). Direct contact of TN-C with endothelial cells inhibits YAP signaling by disrupting actin polymerization. Since YAP signaling is required for the induction of pro-angiogenic genes (e.g., CTGF and CYR61), endothelial cell survival, proliferation, and vessel formation is impaired. In contrast, brain cancer cells exposed to TN-C respond by secretion of pro-angiogenic signals promoting tubulogenesis of endothelial cells. Hence, TN-C can play multiple and opposing roles during tumor angiogenesis depending on the cells with which it interacts.
4.6 Immunomodulation
Although it has been known that TN-C is able to shape innate and adaptive immunity (124, 125), its impact on tumor immunity is not fully elucidated yet. Studies have revealed that the C-terminal FBG of TN-C as well as of TN-W is able to activate TLR4 on either macrophages or fibroblasts (35, 126). TLR4 is a key component of the innate immune system and when activated stimulates the secretion of pro-inflammatory cytokines such as IL6, IL8, or TNFα (127). In vivo relevance for TLR4 activation by the FBG of TN-C was established in an orthotopic grafting model of breast cancer (128). Application of a therapeutic monoclonal antibody raised against the FBG of TN-C blocked TLR4 activation and reduced primary tumor growth and metastasis (128). The same authors reported a dual role for TN-C on tumor immunity depending on its cellular source: while tumor cell-derived TN-C polarized macrophages towards a pathogenic, immune-suppressive phenotype, host stromal cell-derived TN-C promoted immunity by recruiting anti-tumoral macrophages. Additional roles for TN-C in modulating the immune system in cancers are only recently emerging. In a mouse model, prostate cancer-initiating cells were protected from immune surveillance by over-expressing TN-C, which in turn was able to inhibit T-cell proliferation and effector functions (129). Similar results were obtained in a study in brain cancer. There, TN-C is packaged into exosomes derived from brain tumor-initiating cells and released into the microenvironment, where it can suppress systemic T-cell responses through an α5β1 or αvβ6 integrin-dependent inhibition of T-cell activation and proliferation (130). Both studies promote TN-C as a molecule providing protection for cancer-initiating cells from host-derived immune surveillance. Moreover, TN-C was recently shown to contribute to an immune-suppressive microenvironment in oral squamous cell carcinoma by mobilizing dendritic cells in the tumor stroma through binding CCL21 (131). Similarly, TN-C was responsible for the induction of CXCL12, which in turn immobilized CD8+ tumor infiltrating T lymphocytes (CD8 TIL) in the matrix. Trapped in the TN-C-rich matrix, CD8 TIL could not combat and kill breast cancer cells, suggesting a role for TN-C in anti-tumor immunity escape (132). The newest discovery that the FBGs of both TN-C and TN-W can activate latent TGFβ renews speculation about a potential involvement of tenascins in tumor immunomodulation (110) since TGFβ is a potent suppressor of immune responses (133). Data on the effect of TN-W in immunomodulation remain absent apart from the potential of its FBG to activate TLR4 and TGFβ. Whether such activities might play a role in tumor progression has to be addressed in the future.
4.7 Therapy-Related Activities
Cancer therapy resistance represents a major hurdle and some evidence suggests that TN-C might be involved in this process. In a melanoma model, knockdown of TN-C not only significantly diminished the stem cell-like cells in melanoma spheres, but also lowered their resistance to doxorubicin treatment (134). Therefore, it can be assumed that TN-C is a driver of melanoma progression as it provides protective cues for therapy-resistant melanoma-initiating cells. A tumor-specific large TN-C isoform was shown to confer gemcitabine resistance in pancreatic cancer cells by the canonical phosphatidylinositol 3-kinase/AKT/NF-κB signaling pathway by its interaction with Annexin2 (135). In a study aiming to determine changes of stromal proteins in breast cancer following neoadjuvant chemotherapy, TN-C was identified as a molecule being important for conferring cancer cell resistance to doxorubicin- and docetaxel-based neoadjuvant chemotherapy by activating the integrinβ1/mTOR pathway (136). Moreover, a correlation of high TNC expression with tamoxifen resistance was identified in a clinical study analyzing 1286 primary breast tumors by qPCR (137).
Having reviewed the functions of TN-C in tumor immunity, TN-C might also affect the success rate of anti-cancer immunotherapies. Regarding this, a recent study identified a molecular mechanism explaining the observation that triple-negative breast cancers (TNBC) often do not respond to T-cell-mediated immunotherapies (138). In TNBC, defective autophagy-mediated TN-C degradation results in TN-C accumulation, which impairs T-cell-mediated tumor cytotoxicity. This phenotype could be reversed by inhibition of TN-C suggesting an important role for TN-C in autophagy deficiency-mediated immunosuppression. Consequently, blocking TN-C by specific anti-TN-C antibodies could boost the efficiency of immunotherapies in TNBC.
In addition to chemo- and immunotherapies, radiotherapy is a frequent cancer treatment option. Radiotherapy triggers DNA damage leading to the death of rapidly dividing cancer cells. However, tissues adjacent to the tumor and other normal cells are also affected. Ionization can lead to the modulation of the ECM and indeed, TN-C has been found to be induced upon this treatment (47). Moreover, it is known that ionization also stimulates an inflammatory reaction, fibrosis, and hypoxia. These conditions are known to mediate high TN-C expression (28). With all the knowledge we have gained about the tumor-promoting activities of TN-C, radiation, while killing cancer cells, might also create a pro-tumorigenic stroma. Whether or not this radiation-induced TN-C plays a role in potential tumor relapses, or secondary tumor formations after radio-treatment, remains to be investigated. Nothing is known yet about TN-W function in therapy resistance.
5 Can We Make Use of the Prominent Expression of TNC and TNW in Tumor Stroma?
Having summarized the expression patterns of TN-C and TN-W in tumors as well as their potential tumor-promoting activities, we plan to shed light on the possibilities they offer to be harnessed for clinical use and for the benefit of cancer patients.
5.1 Diagnostic and/or Prognostic Tumor Biomarker
Cancers are most dangerous when they are able to form distant metastases. Unfortunately, some cancers are only detected when such metastases have already formed rendering therapy even more challenging. In order to prevent this, there is an urgent need for diagnostic tumor markers that recognize premalignant lesion with high specificity and sensitivity. The best-case scenario would be that in adult healthy tissues a potential biomarker is not detectable, while its levels are sharply elevated in early premalignant dysplasia. This would allow an early detection of dysplastic and pre-malignant tissues. Both TN-C and TN-W seem to be interesting cancer biomarker candidates as they are often over-expressed in the tumor stroma (Figure 4A). However, the two tenascins differ significantly in their expression in healthy and noncancerous tissues. A recent screening of a healthy tissue microarray revealed limited basal TN-W expression. Apart for some expression in spleen, kidney, and some adult stem cell niches (e.g., hair follicles), as has been already observed in the initial mouse studies, TN-W was also weakly present in the female reproductive system and in certain glands (89). In contrast, TN-C is readily detectable in a larger array of healthy tissues (Figure 4A). Moreover, it is well established that TN-C upregulation is not restricted to cancers, but that abundant TN-C expression is a prominent feature of many other pathological conditions. These include inflammatory diseases (e.g. arthritis and asthma), infections (e.g. viral infections), and tissue remodeling processes such as wound healing and fibrosis (28). This is in stark contrast to TN-W, for which there is no evidence for de novo expression in pathological conditions other than cancers. Even in conditions with strong TN-C expression such as healing wounds and inflammatory bowel diseases, TN-W is not detectable (Figure 4B). These observations promote TN-W as a better and more tumor-specific tissue biomarker than TN-C (48). However, we have mentioned that TN-C is subject to alternative splicing generating very tissue- and condition-specific isoforms. Therefore, it might be also worth studying tumor-specific expression of distinct TN-C isoforms. It is known that certain splice variants, especially the variants with additional FNIII domains, are expressed in cancerous conditions. Hence, even if normal healthy tissue is expressing TN-C, detection of tumor-specific TN-C isoforms might still be of clinical value as a diagnostic readout.
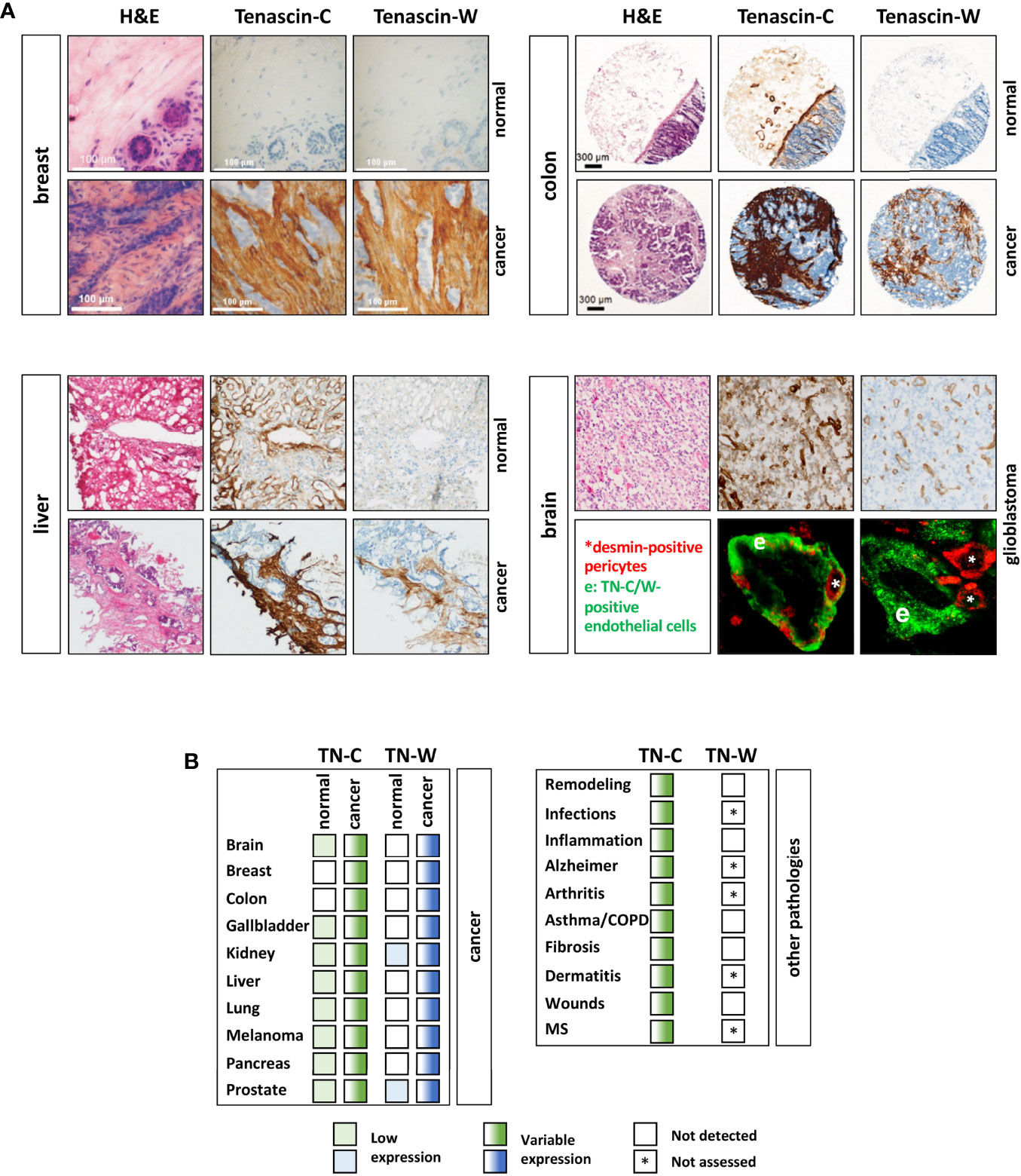
Figure 4 TN-C and TN-W expression in human cancers. (A) Examples of immunohistochemical analyses of TN-C and TN-W in human breast, liver, and colorectal cancer as well as in adjacent normal tissues. Note that while TN-W is not expressed in healthy tissues, TN-C is detectable in colon and liver. In glioblastoma TN-C and TN-W show strong expression around vessel-like structures (top). Double immunofluorescence staining of TN-W or TN-C (green) with desmin (red) shows that TN-C encloses all pericytes, while TN-W staining is adjacent to them. Original images of breast tissue from Degen et al. (88), of liver tissue from Hendaoui et al. (89), of colon tissue from Degen et al. (52), and of brain tissue from Martina et al. (90) with permission. (B) Table of all cancer/normal tissues that have been analyzed so far for TN-W and TN-C by immunohistochemistry and/or immunoblotting (left). Right table shows non-cancerous conditions with TN-C upregulation and non-detectable (white box) TN-W expression. Fully colored boxes: expressed; gradient-colored boxes: variable expression ranging from low (light) to high (dark). * not assessed yet; MS, multiple sclerosis.
TN-C and TN-W can also be detected in elevated concentrations in serum of cancer patients. But similar to the situation in tissues, increased TN-C levels in serum are also found in additional pathologies. Some of the conditions known to have increased serum TN-C levels include spondylitis, sepsis, myocardial defects, traumatic brain injuries, rheumatic diseases, pulmonary hypertension, Alzheimer’s disease, systemic lupus, idiopathic inflammatory myositis, muscular dystrophy, chronic hepatitis C infection, and chronic renal diseases (139–146). Common to most of these conditions is the fact that they involve an inflammatory reaction. Therefore, it is not surprising that TN-C in serum correlates with C- reactive protein, a common inflammation marker, and represents a questionable tumor biomarker (147, 148). As mentioned above, the presence of certain TN-C splice variants in serum may still have some value as a marker of cancer. In contrast, to date elevated TN-W has only been detected in the serum of cancer patients (52).
Numerous studies also assessed the prognostic value of TN-C. Although these results are often ambiguous, there seems to be a clear trend towards a positive correlation between TN-C levels and local tumor recurrence, the metastatic potential, and poor prognosis in many aggressive human cancers. This correlation was observed in clinical specimens of brain, breast, colon, head and neck, and lung cancer (45). Why this observation is not valid for other cancers is not understood, but it might reflect the multifunctional roles of TN-C and tumor heterogeneity. There are also two studies that report a correlation of serum TN-C with prognostic factors of cancers: In non-small cell lung cancer patients TN-C serum levels correlated with larger tumor size, lymph node metastases and patient’s overall survival (51), while serum TN-C levels in grade 3 breast cancer were significantly higher than in grade 1-2 tumors (50). Moreover, TN-C is known to be a substrate for various matrix metalloproteinases and serine proteases, which can lead to TN-C fragmentation (149). Significantly more small TN-C fragments were found in lung cancer patients who had a higher risk to develop lymph node as well as poor overall survival suggesting that TN-C degradation might be a marker for the metastatic potential of lung tumors (150, 151). Unfortunately, little has been done to date to study the prognostic value of TN-W. Initially, TN-W was found to be mainly enriched in mouse models of mammary cancer that metastasize (87). However, such a correlation could not be confirmed in human breast cancer where TN-W was more strongly expressed in low-grade tumors than in high-grade tumors (88). In lung cancer, TN-W showed a tendency towards higher expression in high-grade compared to low-grade cancers. Similarly, the more severe clear cell carcinoma expressed more TN-W (and TN-C) than the less severe oncocytoma (48). In colorectal carcinoma patients with tumor recurrence, serum TN-W levels were higher than in those without recurrence (52). Although these data suggest some prognostic value of TN-W in certain malignancies, the number of cases analyzed for TN-W are too small to draw any definite conclusions yet.
5.2 Modulating Tumor-Specific Tenascin Expression
TN-C and TN-W are enriched in most solid human tumors. Both molecules also share a broad spectrum of tumor-promoting activities (Figure 3). Reversing their high expression in tumors could be a promising concept to normalize the TME thereby impairing its effect on promoting tumor progression. This approach should be attempted in combination with more classical, cancer cell-targeting therapies with the ultimate goal to limit the metastasis of, and to kill, cancer cells. To target tumor-expression of tenascins and prevent TN-C and TN-W from joining the TME, there are broadly two options: pharmacological inhibition and gene-based approaches.
5.2.1 Pharmacological and Natural Inhibitors
A simple concept for reducing the expression levels of tenascins includes the inhibition of the signaling pathways required for to their activation. However, this approach requires full knowledge about how tenascins are regulated in tumors. While for TN-C several pathways have been reported to lead to its induction (e.g. TGFβ signaling), knowledge of TN-W regulation in tumors remains sparse. In addition, inhibiting such important and canonical pathways will likely have unwanted side effects and therefore, does not represent a feasible approach to target tenascins in tumor stroma.
The use of miRNAs might represent an alternative approach to target tenascins. miRNAs are regulators of post-transcriptional gene regulation, which are also known to play a critical role in different cancer types as their dysregulation has been shown to affect the hallmarks of cancer (152). Therefore, tenascin-specific miRNA could be utilized in combination with additional treatment strategies to increase treatment success against cancer. A major challenge for this approach is the efficient delivery of these agents to target cells, which might require antibodies or peptides. Several miRNAs have been identified to suppress TN-C levels in cancers: miRNA-198 in colorectal cancer (153), miRNA-335 in breast cancer (154), miRNA-150 in head and neck squamous cell carcinoma (155), and miRNA-218 in glioma (156). In brain cancer, miRNA-107 indirectly diminishes TN-C levels by targeting Notch2, a direct inducer of TN-C (157). So far, no data exist about a possible regulation of TN-W by miRNAs.
Glucocorticoids are known repressors of both TN-C and TN-W (60). Therefore, the use of corticoids, which dampen tenascin expression, might represent a promising addition to the treatment cocktail of specific cancer types. Such an approach is applied in the treatment of castration-resistant prostate cancer patients, who receive dexamethasone as monotherapy or as part of their chemotherapeutic cocktail (158, 159). However, the effects of a widespread use of dexamethasone either to attenuate the side effects of chemotherapy or to combat malignancies might be double-edged as it has been shown that the glucocorticoid can enhance lung metastasis of breast cancer cells (160).
Recently, an approach to reduce TN-C in the tumor stroma of pancreatic cancer involved interfering with the aberrant cancer cell-mesenchymal crosstalk, which is required for stellate cell activation (161). These activated stellate cells are responsible for TN-C secretion into the stroma, in which TN-C can activate oncogenic signaling cascades (Wnt/β-catenin via inhibition of DKK1 and YAP/TAZ signaling) leading to tumor progression. The metastasis suppressor, N-myc downstream-regulated gene-1 (NDRG1) is able to dampen tumorigenesis by inhibiting these two pathways as well as by decreasing TGFβ production by cancer cells. The latter factor is required for stellate cell activation, which enables TN-C synthesis. Consequently, upregulation of NDRG1 might have the power to normalize the TME by impairing stellate cell activation thereby preventing induction of TN-C. Interestingly, the anti-cancer compounds thiosemicarbazone potently upregulate NDRG1, which both prevents aberrant epithelial-stellate cell crosstalk and synthesis of TN-C.
5.2.2 Gene-Based Approaches
Another strategy for anti-cancer treatment includes the use of RNA interference (RNAi) technologies. With RNAi, it is possible to specifically target a gene that is known to play a key role in tumor progression. As such it is not surprising that several attempts used RNAi to target TN-C in cancers (Table 1). Double-stranded RNAs specific for the TN-C sequence (ATN-RNA) were used to inhibit its expression in glioma, which are rich in TN-C. ATN-RNA was injected into the brain of 11 patients after initial tumor surgery, and treatment specificity and effects were assessed (162). Thereafter, the study was extended to 46 patients with grade II, III, and IV glioma (163, 164). Both the well-being as well as progression-free and overall survival of the patients were significantly increased in the ATN-RNA treated patients compared to the ones only obtaining brachytherapy. Although promising, this treatment only provided small advantages in survival prolongation. Whether the foreign dsRNA elicited an immune response affecting the patient’s outcome was not assessed. A recent study used the same approach in vitro to show proof of concept for ATN-RNA application in breast cancer (165). ATN-RNA treatment significantly downregulated TN-C in MDA-MB-231 cells and reduced many TN-C-mediated activities for tumor progression, such as cancer cell proliferation and migration. Although, the ATN-RNA approach shows some apparent benefits, systemic delivery of RNAi to the patients remains one of the most difficult challenges in the clinic. We are not aware of any RNAi approaches targeting TN-W in the tumor stroma.
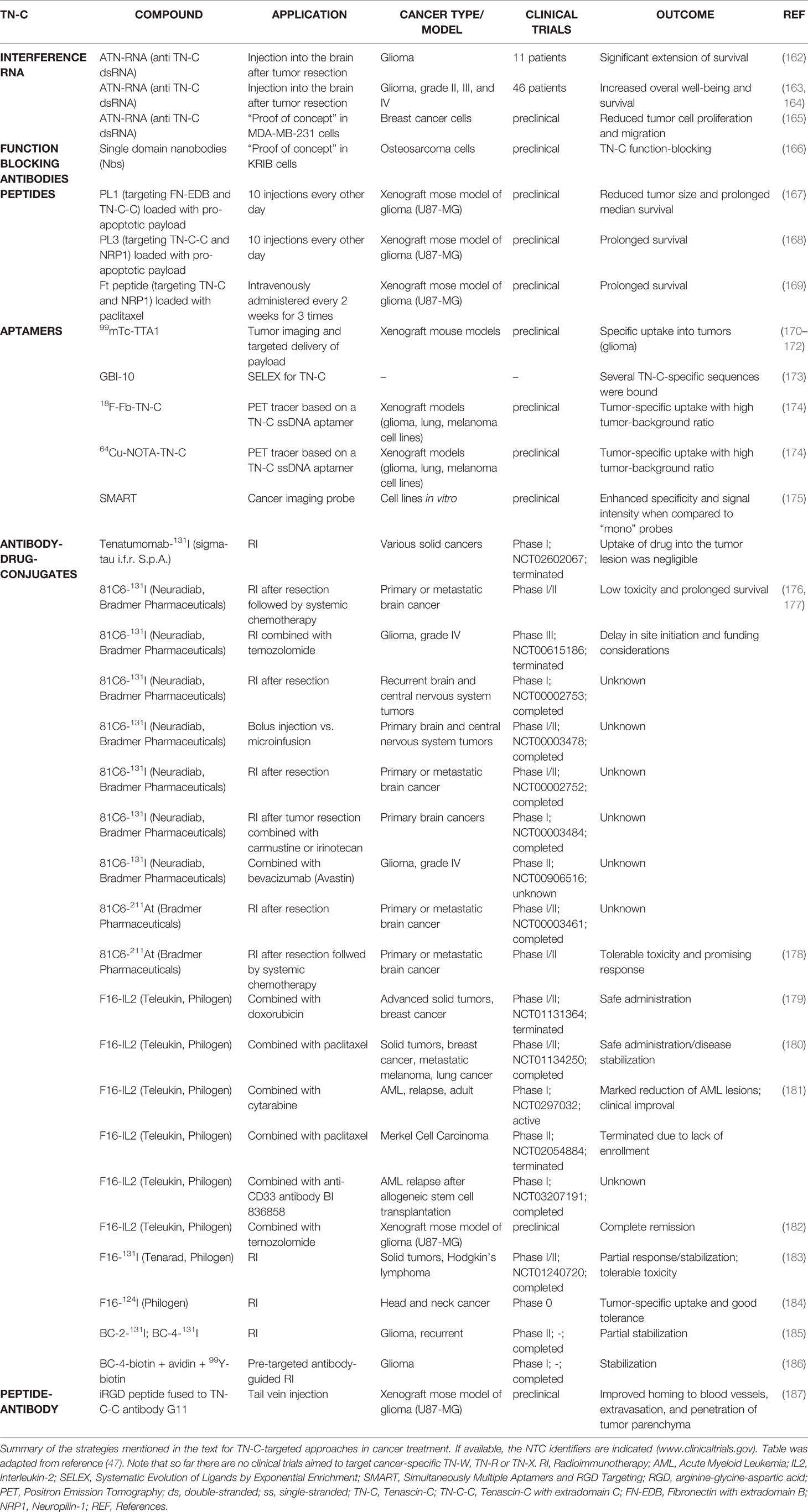
Table 1 Strategies for targeting TN-C in cancer treatment.
5.2.2.1 Function Blocking Antibodies
In an effort to develop novel and better tools to detect human TN-C, recombinant single-domain nanobodies (Nbs) have been recently raised in the dromedary (166). Nbs possess favorable properties over regular antibodies as they present a high stability, solubility, specificity as well as low immunogenicity. These Nbs recognized the 5th FNIII repeat of TN-C and were able to specifically detect TN-C in formalin-fixed and paraffin-embedded tumor sections. The Nbs also abolished certain pro-tumorigenic functions of TN-C, such as cell adhesion modulation on a FN substratum as well as stromal retention of dendritic cells DC2.4 by CCL21/TN-C. These results promote the newly developed TN-C-specific Nbs as novel molecular tools for detecting TN-C in tumors (diagnostic biomarker) as well as for their potential use as anti-cancer treatment options by blocking the oncogenic functions of TN-C (Table 1). Although TN-W-specific single-domain antibodies have been developed in the dromedary as well (188), so far they only have been tested for the specific detection and imaging of TN-W and not for blocking its functions.
5.2.2.2 Affinity Ligands to TN-C and TN-W as Anti-Cancer Strategy
Well-known challenges faced by conventional cytotoxic anti-cancer drugs are their side effects in normal tissues and the barriers in the organism that the drugs need to cross to reach the target site. Specificity is lacking and high drug doses are required for reaching the required local compound concentration. For these reasons, drug-targeting strategies represent promising new avenues for anti-cancer treatment. High-affinity targeting ligands, such as antibodies, peptides, and aptamers can be used that specifically recognize a molecule overexpressed at the target site. By coupling active pharmacological molecules, it should be possible to maximize the therapeutic effects by specific drug delivery. The idea of targeted therapy was initially developed by Ehrlich more than 100 years ago, who proposed to kill pathogens without harming the body [reviewed by (189)]. This simple concept can be and is currently applied to cancer therapy where malignancies must be specifically targeted and eliminated. TN-C, including tumor-specific isoforms, and TN-W represent very promising targets for high-affinity ligands as they are both explicitly enriched in various tumor stroma and as ECM proteins represent quite stable antigens. The benefits in doing so are twofold: TN-C and TN-W are expressed and accessible in the TME (target site), and their levels represent excellent biomarkers for treatment success. As indicated before, available data so far speak in favor of TN-W being superior to TN-C as a tumor biomarker. This holds true both in regard to its tumor-specific expression and to its more significant threshold defining the stage of transformation from normal to malignant tissues as compared to TN-C. However, as it was identified almost 40 years ago, there is much more known about TN-C biology, function, and expression and more TN-C-specific tools have been developed for studying and targeting TN-C in animal models, preclinical, and clinical trials than for TN-W. Indeed, TN-C targeting approaches are actively exploited in in vivo models as well as in clinical trials for delivering drugs (e.g., cytokines, radionuclides) specifically to distinct cancer tissues (Table 1). Several different tumor-homing agents have been used to target TN-C, including antibody fragments, monoclonal antibodies, peptides, and aptamers. Optimally, the ligand should possess high-binding affinity for TN-C and low immunogenicity. We present a short overview about the most promising approaches, tools and studies, which utilize TN-C as delivery address for anti-cancer payloads.
5.2.2.2.1 Peptides
During the last two years several novel peptide-based approaches for TN-C-targeted anti-cancer therapies have been developed. A peptide PL1 that targets bi-specifically the oncofetal FN isoform (FN-EDB) and a large TN-C isoform (TN-C-C) was identified and shown to home exclusively to the TME as well as to glioblastoma and prostate carcinoma cells in xenografts models (167). When loaded with a pro-apoptotic peptide, the PL1-targeted model nanocarrier was able to shrink glioblastoma in mice and significantly increase their survival. The same authors also developed a specific, octameric PL3 peptide targeting TN-C and cell- and tissue-penetration receptor NRP1. Promisingly, nanoparticles containing PL3-targeted anticancer payloads proved to be very useful in detection and systemic therapy in prostate carcinoma, glioma, and melanoma cells and in the corresponding xenograft mouse models, as the animal significantly survived for longer periods (168). A similar approach was applied by Kang et al. They also used the combination of TN-C and NRP1 as homing addresses for targeting the tumor ECM and neovasculature, respectively. When loaded with paclitaxel, the synergistic dual-targeting nanocarrier showed higher cytotoxic effects and apoptosis rate compared with mono-targeting approaches in glioma models (169).
5.2.2.2.2 Aptamers
Aptamers are short single-stranded oligonucleotides (DNA or RNA) and represent an alternative approach for targeting approaches in tumors. They possess high affinity and selectivity for a specific target, while there is low immunogenicity, amenability to modifications and conjugations, as well as favorable pharmacokinetics. Several TN-C-specific aptamers have been developed that can be used as imaging tools [99mTc-TTA1 (170–172) and GBI-10 aptamers (173)] and PET tracers [18F-Fb-TN-C and 64Cu-NOTA-TN-C aptamers (174)] to localize and detect tumor-specific TN-C expression allowing the planning of personalized treatment strategies as well as tumor monitoring. Aptamers are usually taken up by a variety of solid tumors and are rapidly cleared from the blood and other nontarget tissues, which add to their beneficial properties. A multimodal nanoparticle-based Simultaneously Multiple Aptamers and RGD Targeting (SMART) cancer imaging probe, which is able to simultaneously target nucleolin (AS1411 aptamer), integrin avβ3 (RGD), and TN-C (TTA1 aptamer) hes been described by Ko et al. (175). This probe showed enhanced tumor targeting efficacy and a better signal-to-noise ratio compared to mono-biomarker targeting approaches in various cancers (175). Moreover, TN-C targeting aptamers can also be used as vehicles for delivering radioisotopes or chemical agents to cancerous tissues and present novel potential therapeutic applications. So far, clinical trials with aptamers targeting TN-W have not been reported.
5.2.2.2.3 Antibodies
Antibody-drug-conjugate (ADC) technology aims to home a toxic agent specifically into the tumor using target-specific antibodies. Several ADCs have shown impressive results in treating cancers, which resulted in the approval of 11 different ADCs by the FDA that target hematologic as well as solid malignancies (190). Four crucial features might decide the success or failure of ADC applications: (i) specific tumor targeting; (ii) the property of the antibody itself; (iii) the nature of the cytotoxic payload; and (iv) the method used for linking the payload to the antibody. As described, TN-C and especially tumor-specific large TN-C isoforms (as well as TN-W) represent interesting tumor marker candidates for ADCs. Indeed, several monoclonal antibodies recognizing isoforms of TN-C are in preclinical and clinical stages. For instance, 131I- and 211At-labeled chimeric 81C6 monoclonal antibody (Neuradiab recognizing TN-C-C/D) as monotreatment or in combination with other compounds for the treatment of patients with non-Hodgkin lymphoma, with recurrent glioma, with primary or metastatic brain cancer showed encouraging initial results with relatively low toxicity (176–178). Additional TN-C-specific antibodies have been tested in recurrent malignant glioma patients (antibodies BC-2-131I, BC-4-131I; clinical phase II) and in anaplastic astrocytoma and glioblastoma patients (BC4-biotin + avidin + 99Y-biotin; clinical phase I) and revealed disease stabilization with partial remission (185, 186). Another promising tool consisting of a fully humanized antibody F16 recognizing the variable FNIII domain A1 of TN-C has been developed by Philogen (191). In the small immunoprotein format (SIP), F16 was able to recognize TN-C in several different cancers such as glioblastoma, lymphoma, melanoma, head and neck, and renal cell carcinoma (182, 192–196). The promising biodistribution of F16 prompted its testing for radionuclide as well as cytokine/chemokine combination therapy. F16-124I has been applied in four head and neck cancer patients resulting in a tumor-specific signal in all patients within 24 hours (184). Phase I/II clinical dose finding and efficacy trials (www.clinicaltrials.gov; NCT01240720) have been performed with F16-131I (Tenarad) in eight Hodgkin lymphoma patients (183). Treatment of the patients with F16-131I did not cause any severe toxicity problems and was effective in providing clinical benefit in the majority of the patients. Apart from radiotherapies, F16SIP is currently also evaluated for its potential as combinatory treatment option when coupled to IL2, which elicits an immune response that is able to kill cancer cells. So far, F16-IL2 (Teleukin, Philogen) has been applied in combination with paclitaxel for breast cancer, melanoma, non-small lung cancer [NCT01134250 (180)] as well as in metastatic Merkel cell carcinoma patients (NCT02054884), together with low doses of cytarabine in AML patients with advanced disease (NCT02957032 (181), and simultaneously with doxorubicin in advanced solid tumor and metastatic breast cancer patients (NCT01131364). Initial results for the studies using F16-IL2 are promising as it can be safely and repeatedly administered to patients with lung cancer in combination with paclitaxel (180) and to patients with different solid and metastatic breast cancer together with doxorubicin (179). The strong safety profile and the appearance of early signs of clinical activity of F16-IL2 warrant further studies.
Tumor-homing, extravasation, and penetration of cancer drugs can be improved by conjugation to tumor penetrating iRGD peptides, which is a widely used approach (197–199). Several clinical trials are ongoing in pancreas, colon, and digestive cancers testing the effect of iRGD alone [NCT05052567 (not yet recruiting)] in combination with anti-cancer drugs nabpaclitaxel and gemcitabine [NCT03517176 (completed), NCT05042128 (not yet recruiting)], or with panitumumab [NCT05121038 (recruiting)]. Recently, a study analyzed the effect of genetic fusion of the iRGD peptide to recombinant anti-TN-C-C single-chain antibody clone G11 (58) in U87-MG glioma tumor models compared to the parental antibody (187). Comparative biodistribution studies revealed improved homing to blood vessels, extravasation, and tumor penetration in the presence of the iRGD peptide (187). Such an approach using a tumor-penetrating peptide-functionalized TN-C antibody could be more efficient in delivering therapeutic agents into solid tumor lesions and may develop into a useful strategy for affinity targeting of solid tumors.
6 Concluding Remarks
Tenascins are a family of matricellular proteins that show intriguing expression patterns. During development they are expressed widely in various tissues reflecting their important role in organ and tissue development. In most healthy adult tissues their levels of expression are greatly diminished. Upon disturbance of tissue homeostasis, which often accompanies the initiation of a tumor (200), TN-C and TN-W are sharply upregulated in the TME. There, they make use of their multifunctionality and interact with several other ECM proteins and cell surface receptors, eliciting signaling pathways that drive tumor progression (201).
In the embryo and in the TME tenascins are part of a complex microenvironment with many other ECM molecules and growth factors. With these many potential interaction partners available, tenascins have multiple possibilities to influence cell fate. This enormous complexity makes the study of the real in vivo effects and functions of tenascins in physiological as well in pathological conditions challenging. Can we really study tenascin function in a 2D tissue culture dish? Is the use of 3D conditions or Matrigel of any advantage to determine the in vivo function of these molecules? What all these study models have in common is the limitation that none can fully recapitulate the microenvironment as it exists in vivo. Still, the proper understanding and a thorough characterization of the tenascin members is of utmost importance. Even if the study conditions do not fully reflect the complexity of the in vivo situation, much has been and still can be learned from these experimental approaches. This is especially true for TN-C and TN-W, which are both prominently over-expressed in tumor stroma. The many pre-clinical and clinical studies trying to harness their tumor-specific expression for diagnostic and therapeutic use could not have been done without understanding the basics about TN-C and TN-W biology.
Fundamental to the study of TN-C and TN-W in clinical settings is the availability of excellent antibodies for their detection. For both proteins, outstanding antibodies have been raised (88, 147, 188), and more recent efforts generated TN-C-specific Nbs (166), which might be highly useful for future studies and clinical trials. Unfortunately, research on TN-W lags behind the research focused on its older brother, TN-C. In contrast to TN-C, TN-W function has not been thoroughly elucidated yet and there still remains a lot to be learned about the role of TN-W in tumors, which might clarify why it is expressed at a high level in many cancers. A better understanding of TN-W function and regulation, as well as its interactions with cellular receptors in tumors, might also provide the basis for more attempts in using it as a cancer biomarker or therapeutic target. This, of course, raises the questions about the importance of TN-C and TN-W in tumor ECM. Is it feasible to expect better cancer management or a normalization of the TME by targeting one specific aberrantly expressed ECM protein present in the tumor stroma, such as TN-C, while we know that the TME consists of a 3D meshwork with seemingly countless components? Perhaps after targeting TN-C in tumors stroma, TN-W can compensate for its absence and, therefore, a positive treatment effect is only short-lived or only achieved if both tenascins are targeted. The clinical trials with drug-delivery vehicles targeting TN-C produced some encouraging results in their initial phases (see above). Certainly, it is worth continuing this path of research and making use of their highly regulated expression in tumors. In this regard, TN-W might also represent an interesting and promising tumor-specific homing address for drug delivery: (i) TN-W is often expressed around blood vessels in the tumor stroma, which are often leaky making antibody administration feasible; (ii) TN-W is not known to be upregulated in pathologies other than cancers; and (iii) TN-W is generally not expressed in normal tissues with some few exceptions. However, we are currently not aware of such attempts. It also remains to be understood why among patients suffering from the same type of tumor, TN-W and TN-C expression is very heterogenous. Some patients display very high levels of the tenascins, while other patients only show modest expression [see for example (88)]. Which biochemical, biophysical or clinical properties dictate their regulation of expression in cancer?
Herein, we attempted to summarize some of the knowledge about the tenascin family of ECM proteins in health and cancer. Although we have learned much over the last 40 years, there is still much to be discovered. We are still far from being able to depict the full picture of the specific roles of each of the tenascin members and how they are incorporated into complex ECM networks, and how they are precisely regulated in health and disease. This is true for all four members. As of today, TN-C and TN-W are the most promising family members to be exploited for anti-cancer treatment. However, this does not diminish the potential roles that TN-R and TN-X might play in tumorigenesis. Clearly, more work is required to address and recognize the pleiotropic roles of all tenascin members in physiological as well as in pathological conditions. Only a broader understanding of tenascin biology might give us the chance to use them as exploitable matricellular proteins for cancer diagnosis and monitoring as well as for anti-cancer therapies.
Author Contributions
RT wrote the first draft of the abstract and section 2, MD wrote the first draft of the remaining sections. Both authors contributed to the figures and the final draft. All authors contributed to the article and approved the submitted version.
Funding
Open access publication fees have been paid by the University of Bern, Switzerland.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We apologize to all colleagues whose work could not be mentioned due to space restrictions.
References
1. Frantz C, Stewart KM, Weaver VM. The Extracellular Matrix at a Glance. J Cell Sci (2010) 123(Pt 24):4195–200. doi: 10.1242/jcs.023820
2. Karamanos NK, Theocharis AD, Piperigkou Z, Manou D, Passi A, Skandalis SS, et al. A Guide to the Composition and Functions of the Extracellular Matrix. FEBS J (2021) 288(24):6850–912. doi: 10.1111/febs.15776
3. Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The Extracellular Matrix: Tools and Insights for the “Omics” Era. Matrix Biol (2016) 49:10–24. doi: 10.1016/j.matbio.2015.06.003
4. Hynes RO. The Extracellular Matrix: Not Just Pretty Fibrils. Science (2009) 326(5957):1216–9. doi: 10.1126/science.1176009
5. Ozbek S, Balasubramanian PG, Chiquet-Ehrismann R, Tucker RP, Adams JC. The Evolution of Extracellular Matrix. Mol Biol Cell (2010) 21(24):4300–5. doi: 10.1091/mbc.E10-03-0251
6. Mueller MM, Fusenig NE. Friends or Foes - Bipolar Effects of the Tumour Stroma in Cancer. Nat Rev Cancer (2004) 4(11):839–49. doi: 10.1038/nrc1477
7. Shekhar MP, Pauley R, Heppner G. Host Microenvironment in Breast Cancer Development: Extracellular Matrix-Stromal Cell Contribution to Neoplastic Phenotype of Epithelial Cells in the Breast. Breast Cancer Res (2003) 5(3):130–5. doi: 10.1186/bcr580
8. Kalluri R, Zeisberg M. Fibroblasts in Cancer. Nat Rev Cancer (2006) 6(5):392–401. doi: 10.1038/nrc1877
9. Bonnans C, Chou J, Werb Z. Remodelling the Extracellular Matrix in Development and Disease. Nat Rev Mol Cell Biol (2014) 15(12):786–801. doi: 10.1038/nrm3904
10. Lu P, Weaver VM, Werb Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J Cell Biol (2012) 196(4):395–406. doi: 10.1083/jcb.201102147
11. Pickup MW, Mouw JK, Weaver VM. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep (2014) 15(12):1243–53. doi: 10.15252/embr.201439246
12. Brellier F, Chiquet-Ehrismann R. How Do Tenascins Influence the Birth and Life of a Malignant Cell? J Cell Mol Med (2012) 16(1):32–40. doi: 10.1111/j.1582-4934.2011.01360.x
13. Chiquet-Ehrismann R, Kalla P, Pearson CA, Beck K, Chiquet M. Tenascin Interferes With Fibronectin Action. Cell (1988) 53(3):383–90. doi: 10.1016/0092-8674(88)90158-4
14. Scherberich A, Tucker RP, Samandari E, Brown-Luedi M, Martin D, Chiquet-Ehrismann R. Murine Tenascin-W: A Novel Mammalian Tenascin Expressed in Kidney and at Sites of Bone and Smooth Muscle Development. J Cell Sci (2004) 117(Pt 4):571–81. doi: 10.1242/jcs.00867
15. Saga Y, Yagi T, Ikawa Y, Sakakura T, Aizawa S. Mice Develop Normally Without Tenascin. Genes Dev (1992) 6(10):1821–31. doi: 10.1101/gad.6.10.1821
16. Mackie EJ, Tucker RP. The Tenascin-C Knockout Revisited. J Cell Sci (1999) 112(Pt 22):3847–53. doi: 10.1242/jcs.112.22.3847
17. Chiquet M. Tenascin-C: From Discovery to Structure-Function Relationships. Front Immunol (2020) 11:611789. doi: 10.3389/fimmu.2020.611789
18. Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, et al. Tenascin-X Deficiency Is Associated With Ehlers-Danlos Syndrome. Nat Genet (1997) 17(1):104–8. doi: 10.1038/ng0997-104
19. Mao JR, Taylor G, Dean WB, Wagner DR, Afzal V, Lotz JC, et al. Tenascin-X Deficiency Mimics Ehlers-Danlos Syndrome in Mice Through Alteration of Collagen Deposition. Nat Genet (2002) 30(4):421–5. doi: 10.1038/ng850
20. Tucker RP, Chiquet-Ehrismann R. The Regulation of Tenascin Expression by Tissue Microenvironments. Biochim Biophys Acta (2009) 1793(5):888–92. doi: 10.1016/j.bbamcr.2008.12.012
21. Valcourt U, Alcaraz LB, Exposito JY, Lethias C, Bartholin L. Tenascin-X: Beyond the Architectural Function. Cell Adh Migr (2015) 9(1-2):154–65. doi: 10.4161/19336918.2014.994893
22. Anlar B, Gunel-Ozcan A. Tenascin-R: Role in the Central Nervous System. Int J Biochem Cell Biol (2012) 44(9):1385–9. doi: 10.1016/j.biocel.2012.05.009
23. Weber P, Bartsch U, Rasband MN, Czaniera R, Lang Y, Bluethmann H, et al. Mice Deficient for Tenascin-R Display Alterations of the Extracellular Matrix and Decreased Axonal Conduction Velocities in the Cns. J Neurosci (1999) 19(11):4245–62. doi: 10.1523/JNEUROSCI.19-11-04245.1999
24. Pesheva P, Probstmeier R, Skubitz AP, McCarthy JB, Furcht LT, Schachner M. Tenascin-R (J1 160/180 Inhibits Fibronectin-Mediated Cell Adhesion–Functional Relatedness to Tenascin-C. J Cell Sci (1994) 107(Pt 8):2323–33. doi: 10.1242/jcs.107.8.2323
25. Degen M, Scherberich A, Tucker RP. Tenascin-W: Discovery, Evolution, and Future Prospects. Front Immunol (2020) 11:623305. doi: 10.3389/fimmu.2020.623305
26. Tucker RP, Degen M. The Expression and Possible Functions of Tenascin-W During Development and Disease. Front Cell Dev Biol (2019) 7:53. doi: 10.3389/fcell.2019.00053
27. Imhof T, Balic A, Heilig J, Chiquet-Ehrismann R, Chiquet M, Niehoff A, et al. Pivotal Role of Tenascin-W (-N) in Postnatal Incisor Growth and Periodontal Ligament Remodeling. Front Immunol (2020) 11:608223. doi: 10.3389/fimmu.2020.608223
28. Chiquet-Ehrismann R, Chiquet M. Tenascins: Regulation and Putative Functions During Pathological Stress. J Pathol (2003) 200(4):488–99. doi: 10.1002/path.1415
29. Hsia HC, Schwarzbauer JE. Meet the Tenascins: Multifunctional and Mysterious. J Biol Chem (2005) 280(29):26641–4. doi: 10.1074/jbc.R500005200
30. Jones FS, Jones PL. The Tenascin Family of Ecm Glycoproteins: Structure, Function, and Regulation During Embryonic Development and Tissue Remodeling. Dev Dyn (2000) 218(2):235–59. doi: 10.1002/(SICI)1097-0177(200006)218:2<235::AID-DVDY2>3.0.CO;2-G
31. Tucker RP, Drabikowski K, Hess JF, Ferralli J, Chiquet-Ehrismann R, Adams JC. Phylogenetic Analysis of the Tenascin Gene Family: Evidence of Origin Early in the Chordate Lineage. BMC Evol Biol (2006) 6:60. doi: 10.1186/1471-2148-6-60
32. Chiquet-Ehrismann R, Tucker RP. Tenascins and the Importance of Adhesion Modulation. Cold Spring Harb Perspect Biol (2011) 3(5):a004960. doi: 10.1101/cshperspect.a004960
33. Tucker RP, Chiquet-Ehrismann R. Tenascin-C: Its Functions as an Integrin Ligand. Int J Biochem Cell Biol (2015) 65:165–8. doi: 10.1016/j.biocel.2015.06.003
34. Yoshida T, Akatsuka T, Imanaka-Yoshida K. Tenascin-C and Integrins in Cancer. Cell Adh Migr (2015) 9(1-2):96–104. doi: 10.1080/19336918.2015.1008332
35. Zuliani-Alvarez L, Marzeda AM, Deligne C, Schwenzer A, McCann FE, Marsden BD, et al. Mapping Tenascin-C Interaction With Toll-Like Receptor 4 Reveals a New Subset of Endogenous Inflammatory Triggers. Nat Commun (2017) 8(1):1595. doi: 10.1038/s41467-017-01718-7
36. Adams JC, Chiquet-Ehrismann R, Tucker RP. The Evolution of Tenascins and Fibronectin. Cell Adh Migr (2015) 9(1-2):22–33. doi: 10.4161/19336918.2014.970030
37. Midwood KS, Chiquet M, Tucker RP, Orend G. Tenascin-C at a Glance. J Cell Sci (2016) 129(23):4321–7. doi: 10.1242/jcs.190546
38. Hendaoui I, Tucker RP, Zingg D, Bichet S, Schittny J, Chiquet-Ehrismann R. Tenascin-C Is Required for Normal Wnt/Beta-Catenin Signaling in the Whisker Follicle Stem Cell Niche. Matrix Biol (2014) 40:46–53. doi: 10.1016/j.matbio.2014.08.017
39. Chiquet-Ehrismann R, Orend G, Chiquet M, Tucker RP, Midwood KS. Tenascins in Stem Cell Niches. Matrix Biol (2014) 37:112–23. doi: 10.1016/j.matbio.2014.01.007
40. Meloty-Kapella CV, Degen M, Chiquet-Ehrismann R, Tucker RP. Avian Tenascin-W: Expression in Smooth Muscle and Bone, and Effects on Calvarial Cell Spreading and Adhesion in Vitro. Dev Dyn (2006) 235(6):1532–42. doi: 10.1002/dvdy.20731
41. Tucker RP, Ferralli J, Schittny JC, Chiquet-Ehrismann R. Tenascin-C and Tenascin-W in Whisker Follicle Stem Cell Niches: Possible Roles in Regulating Stem Cell Proliferation and Migration. J Cell Sci (2013) 126(Pt 22):5111–5. doi: 10.1242/jcs.134650
42. Bourdon MA, Wikstrand CJ, Furthmayr H, Matthews TJ, Bigner DD. Human Glioma-Mesenchymal Extracellular Matrix Antigen Defined by Monoclonal Antibody. Cancer Res (1983) 43(6):2796–805.
43. Mackie EJ, Chiquet-Ehrismann R, Pearson CA, Inaguma Y, Taya K, Kawarada Y, et al. Tenascin Is a Stromal Marker for Epithelial Malignancy in the Mammary Gland. Proc Natl Acad Sci U S A (1987) 84(13):4621–5. doi: 10.1073/pnas.84.13.4621
44. Midwood KS, Hussenet T, Langlois B, Orend G. Advances in Tenascin-C Biology. Cell Mol Life Sci (2011) 68(19):3175–99. doi: 10.1007/s00018-011-0783-6
45. Orend G, Chiquet-Ehrismann R. Tenascin-C Induced Signaling in Cancer. Cancer Lett (2006) 244(2):143–63. doi: 10.1016/j.canlet.2006.02.017
46. Lowy CM, Oskarsson T. Tenascin C in Metastasis: A View From the Invasive Front. Cell Adh Migr (2015) 9(1-2):112–24. doi: 10.1080/19336918.2015.1008331
47. Spenle C, Saupe F, Midwood K, Burckel H, Noel G, Orend G. Tenascin-C: Exploitation and Collateral Damage in Cancer Management. Cell Adh Migr (2015) 9(1-2):141–53. doi: 10.1080/19336918.2014.1000074
48. Brellier F, Martina E, Degen M, Heuze-Vourc’h N, Petit A, Kryza T, et al. Tenascin-W Is a Better Cancer Biomarker Than Tenascin-C for Most Human Solid Tumors. BMC Clin Pathol (2012) 12:14. doi: 10.1186/1472-6890-12-14
49. Pauli C, Stieber P, Schmitt UM, Andratschke M, Hoffmann K, Wollenberg B. The Significance of Tenascin-C Serum Level as Tumor Marker in Squamous Cell Carcinoma of the Head and Neck. Anticancer Res (2002) 22(5):3093–7.
50. Didem T, Faruk T, Senem K, Derya D, Murat S, Murat G, et al. Clinical Significance of Serum Tenascin-C Levels in Epithelial Ovarian Cancer. Tumour Biol (2014) 35(7):6777–82. doi: 10.1007/s13277-014-1923-z
51. Gebauer F, Gelis S, Zander H, Meyer KF, Wolters-Eisfeld G, Izbicki JR, et al. Tenascin-C Serum Levels and Its Prognostic Power in Non-Small Cell Lung Cancer. Oncotarget (2016) 7(15):20945–52. doi: 10.18632/oncotarget.7976
52. Degen M, Brellier F, Schenk S, Driscoll R, Zaman K, Stupp R, et al. Tenascin-W, a New Marker of Cancer Stroma, Is Elevated in Sera of Colon and Breast Cancer Patients. Int J Cancer (2008) 122(11):2454–61. doi: 10.1002/ijc.23417
53. Adams M, Jones JL, Walker RA, Pringle JH, Bell SC. Changes in Tenascin-C Isoform Expression in Invasive and Preinvasive Breast Disease. Cancer Res (2002) 62(11):3289–97.
54. Hindermann W, Berndt A, Borsi L, Luo X, Hyckel P, Katenkamp D, et al. Synthesis and Protein Distribution of the Unspliced Large Tenascin-C Isoform in Oral Squamous Cell Carcinoma. J Pathol (1999) 189(4):475–80. doi: 10.1002/(SICI)1096-9896(199912)189:4<475::AID-PATH462>3.0.CO;2-V
55. Dueck M, Riedl S, Hinz U, Tandara A, Moller P, Herfarth C, et al. Detection of Tenascin-C Isoforms in Colorectal Mucosa, Ulcerative Colitis, Carcinomas and Liver Metastases. Int J Cancer (1999) 82(4):477–83. doi: 10.1002/(SICI)1097-0215(19990812)82:4<477::AID-IJC2>3.0.CO;2-5
56. Borsi L, Carnemolla B, Nicolo G, Spina B, Tanara G, Zardi L. Expression of Different Tenascin Isoforms in Normal, Hyperplastic and Neoplastic Human Breast Tissues. Int J Cancer (1992) 52(5):688–92. doi: 10.1002/ijc.2910520504
57. Hancox RA, Allen MD, Holliday DL, Edwards DR, Pennington CJ, Guttery DS, et al. Tumour-Associated Tenascin-C Isoforms Promote Breast Cancer Cell Invasion and Growth by Matrix Metalloproteinase-Dependent and Independent Mechanisms. Breast Cancer Res (2009) 11(2):R24. doi: 10.1186/bcr2251
58. Silacci M, Brack SS, Spath N, Buck A, Hillinger S, Arni S, et al. Human Monoclonal Antibodies to Domain C of Tenascin-C Selectively Target Solid Tumors in Vivo. Protein Eng Des Sel (2006) 19(10):471–8. doi: 10.1093/protein/gzl033
59. Galler K, Junker K, Franz M, Hentschel J, Richter P, Gajda M, et al. Differential Vascular Expression and Regulation of Oncofetal Tenascin-C and Fibronectin Variants in Renal Cell Carcinoma (Rcc): Implications for an Individualized Angiogenesis-Related Targeted Drug Delivery. Histochem Cell Biol (2012) 137(2):195–204. doi: 10.1007/s00418-011-0886-z
60. Chiovaro F, Chiquet-Ehrismann R, Chiquet M. Transcriptional Regulation of Tenascin Genes. Cell Adh Migr (2015) 9(1-2):34–47. doi: 10.1080/19336918.2015.1008333
61. Degen M, Goulet S, Ferralli J, Roth M, Tamm M, Chiquet-Ehrismann R. Opposite Effect of Fluticasone and Salmeterol on Fibronectin and Tenascin-C Expression in Primary Human Lung Fibroblasts. Clin Exp Allergy (2009) 39(5):688–99. doi: 10.1111/j.1365-2222.2009.03220.x
62. Scharer CD, McCabe CD, Ali-Seyed M, Berger MF, Bulyk ML, Moreno CS. Genome-Wide Promoter Analysis of the Sox4 Transcriptional Network in Prostate Cancer Cells. Cancer Res (2009) 69(2):709–17. doi: 10.1158/0008-5472.CAN-08-3415
63. Sivasankaran B, Degen M, Ghaffari A, Hegi ME, Hamou MF, Ionescu MC, et al. Tenascin-C Is a Novel Rbpjkappa-Induced Target Gene for Notch Signaling in Gliomas. Cancer Res (2009) 69(2):458–65. doi: 10.1158/0008-5472.CAN-08-2610
64. Borsi L, Allemanni G, Gaggero B, Zardi L. Extracellular Ph Controls Pre-Mrna Alternative Splicing of Tenascin-C in Normal, But Not in Malignantly Transformed, Cells. Int J Cancer (1996) 66(5):632–5. doi: 10.1002/(SICI)1097-0215(19960529)66:5<632::AID-IJC9>3.0.CO;2-U
65. Borsi L, Balza E, Castellani P, Carnemolla B, Ponassi M, Querze G, et al. Cell-Cycle Dependent Alternative Splicing of the Tenascin Primary Transcript. Cell Adhes Commun (1994) 1(4):307–17. doi: 10.3109/15419069409097262
66. Moritz S, Lehmann S, Faissner A, von Holst A. An Induction Gene Trap Screen in Neural Stem Cells Reveals an Instructive Function of the Niche and Identifies the Splicing Regulator Sam68 as a Tenascin-C-Regulated Target Gene. Stem Cells (2008) 26(9):2321–31. doi: 10.1634/stemcells.2007-1095
67. Zhao Y, Young SL. Tgf-Beta Regulates Expression of Tenascin Alternative-Splicing Isoforms in Fetal Rat Lung. Am J Physiol (1995) 268(2 Pt 1):L173–80. doi: 10.1152/ajplung.1995.268.2.L173
68. Zagzag D, Friedlander DR, Dosik J, Chikramane S, Chan W, Greco MA, et al. Tenascin-C Expression by Angiogenic Vessels in Human Astrocytomas and by Human Brain Endothelial Cells in Vitro. Cancer Res (1996) 56(1):182–9.
69. Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes That Distinguish Physiological and Pathological Angiogenesis. Cancer Cell (2007) 11(6):539–54. doi: 10.1016/j.ccr.2007.04.017
70. Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast Cancer Cells Produce Tenascin C as a Metastatic Niche Component to Colonize the Lungs. Nat Med (2011) 17(7):867–74. doi: 10.1038/nm.2379
71. Chiquet-Ehrismann R, Kalla P. Pearson CA. Participation of Tenascin and Transforming Growth Factor-Beta in Reciprocal Epithelial-Mesenchymal Interactions of Mcf7 Cells and Fibroblasts. Cancer Res (1989) 49(15):4322–5.
72. Hiraiwa N, Kida H, Sakakura T, Kusakabe M. Induction of Tenascin in Cancer Cells by Interactions With Embryonic Mesenchyme Mediated by a Diffusible Factor. J Cell Sci (1993) 104(Pt 2):289–96. doi: 10.1242/jcs.104.2.289
73. Rathjen FG, Hodge R. Early Days of Tenascin-R Research: Two Approaches Discovered and Shed Light on Tenascin-R. Front Immunol (2020) 11:612482. doi: 10.3389/fimmu.2020.612482
74. Park PC, Taylor MD, Mainprize TG, Becker LE, Ho M, Dura WT, et al. Transcriptional Profiling of Medulloblastoma in Children. J Neurosurg (2003) 99(3):534–41. doi: 10.3171/jns.2003.99.3.0534
75. Carnemolla B, Leprini A, Borsi L, Querze G, Urbini S, Zardi L. Human Tenascin-R. Complete Primary Structure, Pre-Mrna Alternative Splicing and Gene Localization on Chromosome 1q23-Q24. J Biol Chem (1996) 271(14):8157–60. doi: 10.1074/jbc.271.14.8157
76. El Ayachi I, Baeza N, Fernandez C, Colin C, Scavarda D, Pesheva P, et al. Kiaa0510, the 3’-Untranslated Region of the Tenascin-R Gene, and Tenascin-R Are Overexpressed in Pilocytic Astrocytomas. Neuropathol Appl Neurobiol (2010) 36(5):399–410. doi: 10.1111/j.1365-2990.2010.01074.x
77. Yuan Y, Nymoen DA, Stavnes HT, Rosnes AK, Bjorang O, Wu C, et al. Tenascin-X Is a Novel Diagnostic Marker of Malignant Mesothelioma. Am J Surg Pathol (2009) 33(11):1673–82. doi: 10.1097/PAS.0b013e3181b6bde3
78. Nakayama K, Seike M, Noro R, Takeuchi S, Matsuda K, Kunugi S, et al. Tenascin Xb Is a Novel Diagnostic Marker for Malignant Mesothelioma. Anticancer Res (2019) 39(2):627–33. doi: 10.21873/anticanres.13156
79. Kramer M, Pierredon S, Ribaux P, Tille JC, Petignat P, Cohen M. Secretome Identifies Tenascin-X as a Potent Marker of Ovarian Cancer. BioMed Res Int (2015) 2015:208017. doi: 10.1155/2015/208017
80. Hasegawa K, Yoshida T, Matsumoto K, Katsuta K, Waga S, Sakakura T. Differential Expression of Tenascin-C and Tenascin-X in Human Astrocytomas. Acta Neuropathol (1997) 93(5):431–7. doi: 10.1007/s004010050636
81. Wallentine JC, Kim KK, Seiler CE 3rd, Vaughn CP, Crockett DK, Tripp SR, et al. Comprehensive Identification of Proteins in Hodgkin Lymphoma-Derived Reed-Sternberg Cells by Lc-Ms/Ms. Lab Invest (2007) 87(11):1113–24. doi: 10.1038/labinvest.3700672
82. Geffrotin C, Horak V, Crechet F, Tricaud Y, Lethias C, Vincent-Naulleau S, et al. Opposite Regulation of Tenascin-C and Tenascin-X in Melim Swine Heritable Cutaneous Malignant Melanoma. Biochim Biophys Acta (2000) 1524(2-3):196–202. doi: 10.1016/s0304-4165(00)00158-6
83. Levy P, Ripoche H, Laurendeau I, Lazar V, Ortonne N, Parfait B, et al. Microarray-Based Identification of Tenascin C and Tenascin Xb, Genes Possibly Involved in Tumorigenesis Associated With Neurofibromatosis Type 1. Clin Cancer Res (2007) 13(2 Pt 1):398–407. doi: 10.1158/1078-0432.CCR-06-0182
84. Lee SO, Lee SY, Lee SR, Ju W, Kim SC. Tenascin-X and Leukemia Inhibitory Factor Receptor Are Down-Regulated in Leiomyoma Compared With Normal Myometrium. J Gynecol Oncol (2008) 19(2):139–44. doi: 10.3802/jgo.2008.19.2.139
85. Liot S, Aubert A, Hervieu V, Kholti NE, Schalkwijk J, Verrier B, et al. Loss of Tenascin-X Expression During Tumor Progression: A New Pan-Cancer Marker. Matrix Biol Plus (2020) 6-7:100021. doi: 10.1016/j.mbplus.2020.100021
86. Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. Tgf-Beta-Associated Extracellular Matrix Genes Link Cancer-Associated Fibroblasts to Immune Evasion and Immunotherapy Failure. Nat Commun (2018) 9(1):4692. doi: 10.1038/s41467-018-06654-8
87. Scherberich A, Tucker RP, Degen M, Brown-Luedi M, Andres AC, Chiquet-Ehrismann R. Tenascin-W Is Found in Malignant Mammary Tumors, Promotes Alpha8 Integrin-Dependent Motility and Requires P38mapk Activity for Bmp-2 and Tnf-Alpha Induced Expression in Vitro. Oncogene (2005) 24(9):1525–32. doi: 10.1038/sj.onc.1208342
88. Degen M, Brellier F, Kain R, Ruiz C, Terracciano L, Orend G, et al. Tenascin-W Is a Novel Marker for Activated Tumor Stroma in Low-Grade Human Breast Cancer and Influences Cell Behavior. Cancer Res (2007) 67(19):9169–79. doi: 10.1158/0008-5472.CAN-07-0666
89. Hendaoui I, Lahmar A, Campo L, Mebarki S, Bichet S, Hess D, et al. Tenascin-W Is a Novel Stromal Marker in Biliary Tract Cancers. Front Immunol (2020) 11:630139. doi: 10.3389/fimmu.2020.630139
90. Martina E, Degen M, Ruegg C, Merlo A, Lino MM, Chiquet-Ehrismann R, et al. Tenascin-W Is a Specific Marker of Glioma-Associated Blood Vessels and Stimulates Angiogenesis in Vitro. FASEB J (2010) 24(3):778–87. doi: 10.1096/fj.09-140491
91. Chiovaro F, Martina E, Bottos A, Scherberich A, Hynes NE, Chiquet-Ehrismann R. Transcriptional Regulation of Tenascin-W by Tgf-Beta Signaling in the Bone Metastatic Niche of Breast Cancer Cells. Int J Cancer (2015) 137(8):1842–54. doi: 10.1002/ijc.29565
92. Kimura H, Akiyama H, Nakamura T, de Crombrugghe B. Tenascin-W Inhibits Proliferation and Differentiation of Preosteoblasts During Endochondral Bone Formation. Biochem Biophys Res Commun (2007) 356(4):935–41. doi: 10.1016/j.bbrc.2007.03.071
93. d’Amaro R, Scheidegger R, Blumer S, Pazera P, Katsaros C, Graf D, et al. Putative Functions of Extracellular Matrix Glycoproteins in Secondary Palate Morphogenesis. Front Physiol (2012) 3:377. doi: 10.3389/fphys.2012.00377
94. Morgan JM, Wong A, Yellowley CE, Genetos DC. Regulation of Tenascin Expression in Bone. J Cell Biochem (2011) 112(11):3354–63. doi: 10.1002/jcb.23265
95. Midwood KS, Orend G. The Role of Tenascin-C in Tissue Injury and Tumorigenesis. J Cell Commun Signal (2009) 3(3-4):287–310. doi: 10.1007/s12079-009-0075-1
96. Huang W, Chiquet-Ehrismann R, Moyano JV, Garcia-Pardo A, Orend G. Interference of Tenascin-C With Syndecan-4 Binding to Fibronectin Blocks Cell Adhesion and Stimulates Tumor Cell Proliferation. Cancer Res (2001) 61(23):8586–94.
97. Brellier F, Martina E, Chiquet M, Ferralli J, van der Heyden M, Orend G, et al. The Adhesion Modulating Properties of Tenascin-W. Int J Biol Sci (2012) 8(2):187–94. doi: 10.7150/ijbs.8.187
98. Ruiz C, Huang W, Hegi ME, Lange K, Hamou MF, Fluri E, et al. Growth Promoting Signaling by Tenascin-C [Corrected]. Cancer Res (2004) 64(20):7377–85. doi: 10.1158/0008-5472.CAN-04-1234
99. Vollmer G. Biologic and Oncologic Implications of Tenascin-C/Hexabrachion Proteins. Crit Rev Oncol Hematol (1997) 25(3):187–210. doi: 10.1016/s1040-8428(97)00004-8
100. Yoshimura E, Majima A, Sakakura Y, Sakakura T, Yoshida T. Expression of Tenascin-C and the Integrin Alpha 9 Subunit in Regeneration of Rat Nasal Mucosa After Chemical Injury: Involvement in Migration and Proliferation of Epithelial Cells. Histochem Cell Biol (1999) 111(4):259–64. doi: 10.1007/s004180050356
101. Swindle CS, Tran KT, Johnson TD, Banerjee P, Mayes AM, Griffith L, et al. Epidermal Growth Factor (Egf)-Like Repeats of Human Tenascin-C as Ligands for Egf Receptor. J Cell Biol (2001) 154(2):459–68. doi: 10.1083/jcb.200103103
102. Schenk S, Chiquet-Ehrismann R, Battegay EJ. The Fibrinogen Globe of Tenascin-C Promotes Basic Fibroblast Growth Factor-Induced Endothelial Cell Elongation. Mol Biol Cell (1999) 10(9):2933–43. doi: 10.1091/mbc.10.9.2933
103. McKean DM, Sisbarro L, Ilic D, Kaplan-Alburquerque N, Nemenoff R, Weiser-Evans M, et al. Fak Induces Expression of Prx1 to Promote Tenascin-C-Dependent Fibroblast Migration. J Cell Biol (2003) 161(2):393–402. doi: 10.1083/jcb.jcb.200302126
104. Ishigaki T, Imanaka-Yoshida K, Shimojo N, Matsushima S, Taki W, Yoshida T. Tenascin-C Enhances Crosstalk Signaling of Integrin Alphavbeta3/Pdgfr-Beta Complex by Src Recruitment Promoting Pdgf-Induced Proliferation and Migration in Smooth Muscle Cells. J Cell Physiol (2011) 226(10):2617–24. doi: 10.1002/jcp.22614
105. Wenk MB, Midwood KS, Schwarzbauer JE. Tenascin-C Suppresses Rho Activation. J Cell Biol (2000) 150(4):913–20. doi: 10.1083/jcb.150.4.913
106. Meloty-Kapella CV, Degen M, Chiquet-Ehrismann R, Tucker RP. Effects of Tenascin-W on Osteoblasts in Vitro. Cell Tissue Res (2008) 334(3):445–55. doi: 10.1007/s00441-008-0715-4
107. Derynck R, Weinberg RA. Emt and Cancer: More Than Meets the Eye. Dev Cell (2019) 49(3):313–6. doi: 10.1016/j.devcel.2019.04.026
108. Nagaharu K, Zhang X, Yoshida T, Katoh D, Hanamura N, Kozuka Y, et al. Tenascin C Induces Epithelial-Mesenchymal Transition-Like Change Accompanied by Src Activation and Focal Adhesion Kinase Phosphorylation in Human Breast Cancer Cells. Am J Pathol (2011) 178(2):754–63. doi: 10.1016/j.ajpath.2010.10.015
109. Dandachi N, Hauser-Kronberger C, More E, Wiesener B, Hacker GW, Dietze O, et al. Co-Expression of Tenascin-C and Vimentin in Human Breast Cancer Cells Indicates Phenotypic Transdifferentiation During Tumour Progression: Correlation With Histopathological Parameters, Hormone Receptors, and Oncoproteins. J Pathol (2001) 193(2):181–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH752>3.0.CO;2-V
110. Aubert A, Mercier-Gouy P, Aguero S, Berthier L, Liot S, Prigent L, et al. Latent Tgf-Beta Activation Is a Hallmark of the Tenascin Family. Front Immunol (2021) 12:613438. doi: 10.3389/fimmu.2021.613438
111. Xu J, Lamouille S, Derynck R. Tgf-Beta-Induced Epithelial to Mesenchymal Transition. Cell Res (2009) 19(2):156–72. doi: 10.1038/cr.2009.5
112. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell (2017) 168(4):670–91. doi: 10.1016/j.cell.2016.11.037
113. Descot A, Oskarsson T. The Molecular Composition of the Metastatic Niche. Exp Cell Res (2013) 319(11):1679–86. doi: 10.1016/j.yexcr.2013.04.017
114. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat Commun (2020) 11(1):5120. doi: 10.1038/s41467-020-18794-x
115. Silvers CR, Messing EM, Miyamoto H, Lee YF. Tenascin-C Expression in the Lymph Node Pre-Metastatic Niche in Muscle-Invasive Bladder Cancer. Br J Cancer (2021) 125(10):1399–407. doi: 10.1038/s41416-021-01554-z
116. Spenle C, Gasser I, Saupe F, Janssen KP, Arnold C, Klein A, et al. Spatial Organization of the Tenascin-C Microenvironment in Experimental and Human Cancer. Cell Adh Migr (2015) 9(1-2):4–13. doi: 10.1080/19336918.2015.1005452
117. Tomko LA, Hill RC, Barrett A, Szulczewski JM, Conklin MW, Eliceiri KW, et al. Targeted Matrisome Analysis Identifies Thrombospondin-2 and Tenascin-C in Aligned Collagen Stroma From Invasive Breast Carcinoma. Sci Rep (2018) 8(1):12941. doi: 10.1038/s41598-018-31126-w
118. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions Between Cancer Stem Cells and Their Niche Govern Metastatic Colonization. Nature (2011) 481(7379):85–9. doi: 10.1038/nature10694
119. Kii I, Nishiyama T, Li M, Matsumoto K, Saito M, Amizuka N, et al. Incorporation of Tenascin-C Into the Extracellular Matrix by Periostin Underlies an Extracellular Meshwork Architecture. J Biol Chem (2010) 285(3):2028–39. doi: 10.1074/jbc.M109.051961
120. Tanaka K, Hiraiwa N, Hashimoto H, Yamazaki Y, Kusakabe M. Tenascin-C Regulates Angiogenesis in Tumor Through the Regulation of Vascular Endothelial Growth Factor Expression. Int J Cancer (2004) 108(1):31–40. doi: 10.1002/ijc.11509
121. Saupe F, Schwenzer A, Jia Y, Gasser I, Spenle C, Langlois B, et al. Tenascin-C Downregulates Wnt Inhibitor Dickkopf-1, Promoting Tumorigenesis in a Neuroendocrine Tumor Model. Cell Rep (2013) 5(2):482–92. doi: 10.1016/j.celrep.2013.09.014
122. Langlois B, Saupe F, Rupp T, Arnold C, van der Heyden M, Orend G, et al. Angiomatrix, a Signature of the Tumor Angiogenic Switch-Specific Matrisome, Correlates With Poor Prognosis for Glioma and Colorectal Cancer Patients. Oncotarget (2014) 5(21):10529–45. doi: 10.18632/oncotarget.2470
123. Rupp T, Langlois B, Koczorowska MM, Radwanska A, Sun Z, Hussenet T, et al. Tenascin-C Orchestrates Glioblastoma Angiogenesis by Modulation of Pro- and Anti-Angiogenic Signaling. Cell Rep (2016) 17(10):2607–19. doi: 10.1016/j.celrep.2016.11.012
124. Marzeda AM, Midwood KS. Internal Affairs: Tenascin-C as a Clinically Relevant, Endogenous Driver of Innate Immunity. J Histochem Cytochem (2018) 66(4):289–304. doi: 10.1369/0022155418757443
125. Udalova IA, Ruhmann M, Thomson SJ, Midwood KS. Expression and Immune Function of Tenascin-C. Crit Rev Immunol (2011) 31(2):115–45. doi: 10.1615/critrevimmunol.v31.i2.30
126. Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C Is an Endogenous Activator of Toll-Like Receptor 4 That Is Essential for Maintaining Inflammation in Arthritic Joint Disease. Nat Med (2009) 15(7):774–80. doi: 10.1038/nm.1987
127. Medzhitov R, Janeway CA Jr. Decoding the Patterns of Self and Nonself by the Innate Immune System. Science (2002) 296(5566):298–300. doi: 10.1126/science.1068883
128. Deligne C, Murdamoothoo D, Gammage AN, Gschwandtner M, Erne W, Loustau T, et al. Matrix-Targeting Immunotherapy Controls Tumor Growth and Spread by Switching Macrophage Phenotype. Cancer Immunol Res (2020) 8(3):368–82. doi: 10.1158/2326-6066.CIR-19-0276
129. Jachetti E, Caputo S, Mazzoleni S, Brambillasca CS, Parigi SM, Grioni M, et al. Tenascin-C Protects Cancer Stem-Like Cells From Immune Surveillance by Arresting T-Cell Activation. Cancer Res (2015) 75(10):2095–108. doi: 10.1158/0008-5472.CAN-14-2346
130. Mirzaei R, Sarkar S, Dzikowski L, Rawji KS, Khan L, Faissner A, et al. Brain Tumor-Initiating Cells Export Tenascin-C Associated With Exosomes to Suppress T Cell Activity. Oncoimmunology (2018) 7(10):e1478647. doi: 10.1080/2162402X.2018.1478647
131. Spenle C, Loustau T, Murdamoothoo D, Erne W, Beghelli-de la Forest Divonne S, Veber R, et al. Tenascin-C Orchestrates an Immune-Suppressive Tumor Microenvironment in Oral Squamous Cell Carcinoma. Cancer Immunol Res (2020) 8(9):1122–38. doi: 10.1158/2326-6066.CIR-20-0074
132. Murdamoothoo D, Sun Z, Yilmaz A, Riegel G, Abou-Faycal C, Deligne C, et al. Tenascin-C Immobilizes Infiltrating T Lymphocytes Through Cxcl12 Promoting Breast Cancer Progression. EMBO Mol Med (2021) 13(6):e13270. doi: 10.15252/emmm.202013270
133. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming Growth Factor-Beta Regulation of Immune Responses. Annu Rev Immunol (2006) 24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737
134. Fukunaga-Kalabis M, Martinez G, Nguyen TK, Kim D, Santiago-Walker A, Roesch A, et al. Tenascin-C Promotes Melanoma Progression by Maintaining the Abcb5-Positive Side Population. Oncogene (2010) 29(46):6115–24. doi: 10.1038/onc.2010.350
135. Gong XG, Lv YF, Li XQ, Xu FG, Ma QY. Gemcitabine Resistance Induced by Interaction Between Alternatively Spliced Segment of Tenascin-C and Annexin A2 in Pancreatic Cancer Cells. Biol Pharm Bull (2010) 33(8):1261–7. doi: 10.1248/bpb.33.1261
136. Wang T, Srivastava S, Hartman M, Buhari SA, Chan CW, Iau P, et al. High Expression of Intratumoral Stromal Proteins Is Associated With Chemotherapy Resistance in Breast Cancer. Oncotarget (2016) 7(34):55155–68. doi: 10.18632/oncotarget.10894
137. Helleman J, Jansen MP, Ruigrok-Ritstier K, van Staveren IL, Look MP, Meijer-van Gelder ME, et al. Association of an Extracellular Matrix Gene Cluster With Breast Cancer Prognosis and Endocrine Therapy Response. Clin Cancer Res (2008) 14(17):5555–64. doi: 10.1158/1078-0432.CCR-08-0555
138. Li ZL, Zhang HL, Huang Y, Huang JH, Sun P, Zhou NN, et al. Autophagy Deficiency Promotes Triple-Negative Breast Cancer Resistance to T Cell-Mediated Cytotoxicity by Blocking Tenascin-C Degradation. Nat Commun (2020) 11(1):3806. doi: 10.1038/s41467-020-17395-y
139. Bubova K, Prajzlerova K, Hulejova H, Gregova M, Mintalova K, Husakova M, et al. Elevated Tenascin-C Serum Levels in Patients With Axial Spondyloarthritis. Physiol Res (2020) 69(4):653–60. doi: 10.33549/physiolres.934414
140. Yuan W, Zhang W, Yang X, Zhou L, Hanghua Z, Xu K. Clinical Significance and Prognosis of Serum Tenascin-C in Patients With Sepsis. BMC Anesthesiol (2018) 18(1):170. doi: 10.1186/s12871-018-0634-1
141. Hasanzadeh Z, Nourazarian A, Nikanfar M, Laghousi D, Vatankhah AM, Sadrirad S. Evaluation of the Serum Dkk-1, Tenascin-C, Oxidative Stress Markers Levels and Wnt Signaling Pathway Genes Expression in Patients With Alzheimer’s Disease. J Mol Neurosci (2021) 71(4):879–87. doi: 10.1007/s12031-020-01710-9
142. Zavada J, Uher M, Svobodova R, Olejarova M, Husakova M, Ciferska H, et al. Serum Tenascin-C Discriminates Patients With Active Sle From Inactive Patients and Healthy Controls and Predicts the Need to Escalate Immunosuppressive Therapy: A Cohort Study. Arthritis Res Ther (2015) 17:341. doi: 10.1186/s13075-015-0862-4
143. Page TH, Charles PJ, Piccinini AM, Nicolaidou V, Taylor PC, Midwood KS. Raised Circulating Tenascin-C in Rheumatoid Arthritis. Arthritis Res Ther (2012) 14(6):R260. doi: 10.1186/ar4105
144. Niebroj-Dobosz I, Madej-Pilarczyk A, Marchel M, Sokolowska B, Hausmanowa-Petrusewicz I. Circulating Tenascin-C Levels in Patients With Dilated Cardiomyopathy in the Course of Emery-Dreifuss Muscular Dystrophy. Clin Chim Acta (2011) 412(17-18):1533–8. doi: 10.1016/j.cca.2011.04.033
145. Tanaka H, El-Karef A, Kaito M, Kinoshita N, Fujita N, Horiike S, et al. Circulating Level of Large Splice Variants of Tenascin-C Is a Marker of Piecemeal Necrosis Activity in Patients With Chronic Hepatitis C. Liver Int (2006) 26(3):311–8. doi: 10.1111/j.1478-3231.2005.01229.x
146. Horstrup JH, Gehrmann M, Schneider B, Ploger A, Froese P, Schirop T, et al. Elevation of Serum and Urine Levels of Timp-1 and Tenascin in Patients With Renal Disease. Nephrol Dial Transplant (2002) 17(6):1005–13. doi: 10.1093/ndt/17.6.1005
147. Schenk S, Muser J, Vollmer G, Chiquet-Ehrismann R. Tenascin-C in Serum: A Questionable Tumor Marker. Int J Cancer (1995) 61(4):443–9. doi: 10.1002/ijc.2910610402
148. Schenk S, Muser J, Vollmer G, Chiquet-Ehrismann R. Tenascin-C in Serum: An Acute-Phase Protein or a Carcinoma Marker? Int J Cancer (1995) 60(1):145. doi: 10.1002/ijc.2910600121
149. Imai K, Kusakabe M, Sakakura T, Nakanishi I, Okada Y. Susceptibility of Tenascin to Degradation by Matrix Metalloproteinases and Serine Proteinases. FEBS Lett (1994) 352(2):216–8. doi: 10.1016/0014-5793(94)00960-0
150. Kusagawa H, Onoda K, Namikawa S, Yada I, Okada A, Yoshida T, et al. Expression and Degeneration of Tenascin-C in Human Lung Cancers. Br J Cancer (1998) 77(1):98–102. doi: 10.1038/bjc.1998.15
151. Cai M, Onoda K, Takao M, Kyoko IY, Shimpo H, Yoshida T, et al. Degradation of Tenascin-C and Activity of Matrix Metalloproteinase-2 Are Associated With Tumor Recurrence in Early Stage Non-Small Cell Lung Cancer. Clin Cancer Res (2002) 8(4):1152–6.
152. Peng Y, Croce CM. The Role of Micrornas in Human Cancer. Signal Transduct Target Ther (2016) 1:15004. doi: 10.1038/sigtrans.2015.4
153. Murakami T, Kikuchi H, Ishimatsu H, Iino I, Hirotsu A, Matsumoto T, et al. Tenascin C in Colorectal Cancer Stroma Is a Predictive Marker for Liver Metastasis and Is a Potent Target of Mir-198 as Identified by Microrna Analysis. Br J Cancer (2017) 117(9):1360–70. doi: 10.1038/bjc.2017.291
154. Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, et al. Endogenous Human Micrornas That Suppress Breast Cancer Metastasis. Nature (2008) 451(7175):147–52. doi: 10.1038/nature06487
155. Koshizuka K, Nohata N, Hanazawa T, Kikkawa N, Arai T, Okato A, et al. Deep Sequencing-Based Microrna Expression Signatures in Head and Neck Squamous Cell Carcinoma: Dual Strands of Pre-Mir-150 as Antitumor Mirnas. Oncotarget (2017) 8(18):30288–304. doi: 10.18632/oncotarget.16327
156. Dang S, Zhang R, Tian S, Hou P, Li G, Ji M. Microrna218 Inhibits the Malignant Phenotypes of Glioma by Modulating the Tnc/Akt/Ap1/Tgfbeta1 Feedback Signaling Loop. Int J Mol Med (2021) 48(5):205. doi: 10.3892/ijmm.2021.5038
157. Chen L, Zhang R, Li P, Liu Y, Qin K, Fa ZQ, et al. P53-Induced Microrna-107 Inhibits Proliferation of Glioma Cells and Down-Regulates the Expression of Cdk6 and Notch-2. Neurosci Lett (2013) 534:327–32. doi: 10.1016/j.neulet.2012.11.047
158. Venkitaraman R, Lorente D, Murthy V, Thomas K, Parker L, Ahiabor R, et al. A Randomised Phase 2 Trial of Dexamethasone Versus Prednisolone in Castration-Resistant Prostate Cancer. Eur Urol (2015) 67(4):673–9. doi: 10.1016/j.eururo.2014.10.004
159. Holder SL, Drabick J, Zhu J, Joshi M. Dexamethasone May Be the Most Efficacious Corticosteroid for Use as Monotherapy in Castration-Resistant Prostate Cancer. Cancer Biol Ther (2015) 16(2):207–9. doi: 10.1080/15384047.2014.1002687
160. Zhang Y, Shi G, Zhang H, Xiong Q, Cheng F, Wang H, et al. Dexamethasone Enhances the Lung Metastasis of Breast Cancer Via a Pi3k-Sgk1-Ctgf Pathway. Oncogene (2021) 40(35):5367–78. doi: 10.1038/s41388-021-01944-w
161. Geleta B, Tout FS, Lim SC, Sahni S, Jansson PJ, Apte MV, et al. Targeting Wnt/Tenascin C-Mediated Cross Talk Between Pancreatic Cancer Cells and Stellate Cells Via Activation of the Metastasis Suppressor Ndrg1. J Biol Chem (2022) 298(3):101608. doi: 10.1016/j.jbc.2022.101608
162. Zukiel R, Nowak S, Wyszko E, Rolle K, Gawronska I, Barciszewska MZ, et al. Suppression of Human Brain Tumor With Interference Rna Specific for Tenascin-C. Cancer Biol Ther (2006) 5(8):1002–7. doi: 10.4161/cbt.5.8.2886
163. Wyszko E, Rolle K, Nowak S, Zukiel R, Nowak M, Piestrzeniewicz R, et al. A Multivariate Analysis of Patients With Brain Tumors Treated With Atn-Rna. Acta Pol Pharm (2008) 65(6):677–84.
164. Rolle K, Nowak S, Wyszko E, Nowak M, Zukiel R, Piestrzeniewicz R, et al. Promising Human Brain Tumors Therapy With Interference Rna Intervention (Irnai). Cancer Biol Ther (2010) 9(5):396–406. doi: 10.4161/cbt.9.5.10958
165. Wawrzyniak D, Grabowska M, Glodowicz P, Kuczynski K, Kuczynska B, Fedoruk-Wyszomirska A, et al. Down-Regulation of Tenascin-C Inhibits Breast Cancer Cells Development by Cell Growth, Migration, and Adhesion Impairment. PloS One (2020) 15(8):e0237889. doi: 10.1371/journal.pone.0237889
166. Dhaouadi S, Ben Abderrazek R, Loustau T, Abou-Faycal C, Ksouri A, Erne W, et al. Novel Human Tenascin-C Function-Blocking Camel Single Domain Nanobodies. Front Immunol (2021) 12:635166. doi: 10.3389/fimmu.2021.635166
167. Lingasamy P, Tobi A, Haugas M, Hunt H, Paiste P, Asser T, et al. Bi-Specific Tenascin-C and Fibronectin Targeted Peptide for Solid Tumor Delivery. Biomaterials (2019) 219:119373. doi: 10.1016/j.biomaterials.2019.119373
168. Lingasamy P, Tobi A, Kurm K, Kopanchuk S, Sudakov A, Salumae M, et al. Tumor-Penetrating Peptide for Systemic Targeting of Tenascin-C. Sci Rep (2020) 10(1):5809. doi: 10.1038/s41598-020-62760-y
169. Kang T, Zhu Q, Jiang D, Feng X, Feng J, Jiang T, et al. Synergistic Targeting Tenascin C and Neuropilin-1 for Specific Penetration of Nanoparticles for Anti-Glioblastoma Treatment. Biomaterials (2016) 101:60–75. doi: 10.1016/j.biomaterials.2016.05.037
170. Hicke BJ, Marion C, Chang YF, Gould T, Lynott CK, Parma D, et al. Tenascin-C Aptamers Are Generated Using Tumor Cells and Purified Protein. J Biol Chem (2001) 276(52):48644–54. doi: 10.1074/jbc.M104651200
171. Schmidt KS, Borkowski S, Kurreck J, Stephens AW, Bald R, Hecht M, et al. Application of Locked Nucleic Acids to Improve Aptamer in Vivo Stability and Targeting Function. Nucleic Acids Res (2004) 32(19):5757–65. doi: 10.1093/nar/gkh862
172. Hicke BJ, Stephens AW, Gould T, Chang YF, Lynott CK, Heil J, et al. Tumor Targeting by an Aptamer. J Nucl Med (2006) 47(4):668–78.
173. Daniels DA, Chen H, Hicke BJ, Swiderek KM, Gold L. A Tenascin-C Aptamer Identified by Tumor Cell Selex: Systematic Evolution of Ligands by Exponential Enrichment. Proc Natl Acad Sci U S A (2003) 100(26):15416–21. doi: 10.1073/pnas.2136683100
174. Jacobson O, Yan X, Niu G, Weiss ID, Ma Y, Szajek LP, et al. Pet Imaging of Tenascin-C With a Radiolabeled Single-Stranded DNA Aptamer. J Nucl Med (2015) 56(4):616–21. doi: 10.2967/jnumed.114.149484
175. Ko HY, Choi KJ, Lee CH, Kim S. A Multimodal Nanoparticle-Based Cancer Imaging Probe Simultaneously Targeting Nucleolin, Integrin Alphavbeta3 and Tenascin-C Proteins. Biomaterials (2011) 32(4):1130–8. doi: 10.1016/j.biomaterials.2010.10.034
176. Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, Herndon JE 2nd, et al. Salvage Radioimmunotherapy With Murine Iodine-131-Labeled Antitenascin Monoclonal Antibody 81c6 for Patients With Recurrent Primary and Metastatic Malignant Brain Tumors: Phase Ii Study Results. J Clin Oncol (2006) 24(1):115–22. doi: 10.1200/JCO.2005.03.4082
177. Reardon DA, Zalutsky MR, Akabani G, Coleman RE, Friedman AH, Herndon JE 2nd, et al. A Pilot Study: 131i-Antitenascin Monoclonal Antibody 81c6 to Deliver a 44-Gy Resection Cavity Boost. Neuro Oncol (2008) 10(2):182–9. doi: 10.1215/15228517-2007-053
178. Zalutsky MR, Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, et al. Clinical Experience With Alpha-Particle Emitting 211at: Treatment of Recurrent Brain Tumor Patients With 211at-Labeled Chimeric Antitenascin Monoclonal Antibody 81c6. J Nucl Med (2008) 49(1):30–8. doi: 10.2967/jnumed.107.046938
179. Catania C, Maur M, Berardi R, Rocca A, Giacomo AM, Spitaleri G, et al. The Tumor-Targeting Immunocytokine F16-Il2 in Combination With Doxorubicin: Dose Escalation in Patients With Advanced Solid Tumors and Expansion Into Patients With Metastatic Breast Cancer. Cell Adh Migr (2015) 9(1-2):14–21. doi: 10.4161/19336918.2014.983785
180. De Braud FG, Catania C, Onofri A, Pierantoni C, Cascinu S, Maur M, et al. Combination of the Immunocytokine F16-Il2 With Doxorubicin or Paclitaxel in Patients With Solid Tumors: Results From Two Phase Ib Trials. J Clin Oncol (2011) 29(15):Strategies for targeting TN-C in cancer treatment. doi: 10.1200/jco.2011.29.15_suppl.2595
181. Gutbrodt KL, Schliemann C, Giovannoni L, Frey K, Pabst T, Klapper W, et al. Antibody-Based Delivery of Interleukin-2 to Neovasculature Has Potent Activity Against Acute Myeloid Leukemia. Sci Transl Med (2013) 5(201):201ra118. doi: 10.1126/scitranslmed.3006221
182. Pedretti M, Verpelli C, Marlind J, Bertani G, Sala C, Neri D, et al. Combination of Temozolomide With Immunocytokine F16-Il2 for the Treatment of Glioblastoma. Br J Cancer (2010) 103(6):827–36. doi: 10.1038/sj.bjc.6605832
183. Aloj L, D’Ambrosio L, Aurilio M, Morisco A, Frigeri F, Caraco C, et al. Radioimmunotherapy With Tenarad, a 131i-Labelled Antibody Fragment Targeting the Extra-Domain A1 of Tenascin-C, in Patients With Refractory Hodgkin’s Lymphoma. Eur J Nucl Med Mol Imaging (2014) 41(5):867–77. doi: 10.1007/s00259-013-2658-6
184. Heuveling DA, de Bree R, Vugts DJ, Huisman MC, Giovannoni L, Hoekstra OS, et al. Phase 0 Microdosing Pet Study Using the Human Mini Antibody F16sip in Head and Neck Cancer Patients. J Nucl Med (2013) 54(3):397–401. doi: 10.2967/jnumed.112.111310
185. Riva P, Arista A, Tison V, Sturiale C, Franceschi G, Spinelli A, et al. Intralesional Radioimmunotherapy of Malignant Gliomas. Effective Treat Recurrent Tumors Cancer (1994) 73(3 Suppl):1076–82. doi: 10.1002/1097-0142(19940201)73:3+<1076::aid-cnr2820731347>3.0.co;2-z
186. Paganelli G, Bartolomei M, Ferrari M, Cremonesi M, Broggi G, Maira G, et al. Pre-Targeted Locoregional Radioimmunotherapy With 90y-Biotin in Glioma Patients: Phase I Study and Preliminary Therapeutic Results. Cancer Biother Radiopharm (2001) 16(3):227–35. doi: 10.1089/10849780152389410
187. Lingasamy P, Laarmann AH, Teesalu T. Tumor Penetrating Peptide-Functionalized Tenascin-C Antibody for Glioblastoma Targeting. Curr Cancer Drug Targets (2021) 21(1):70–9. doi: 10.2174/1568009620666201001112749
188. Dhaouadi S, Murdamoothoo D, Tounsi A, Erne W, Benabderrazek R, Benlasfar Z, et al. Generation and Characterization of Dromedary Tenascin-C and Tenascin-W Specific Antibodies. Biochem Biophys Res Commun (2020) 530(2):471–8. doi: 10.1016/j.bbrc.2020.05.077
189. Strebhardt K, Ullrich A. Paul Ehrlich’s Magic Bullet Concept: 100 Years of Progress. Nat Rev Cancer (2008) 8(6):473–80. doi: 10.1038/nrc2394
190. Tong JTW, Harris PWR, Brimble MA, Kavianinia I. An Insight Into Fda Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules (2021) 26(19). doi: 10.3390/molecules26195847
191. Brack SS, Silacci M, Birchler M, Neri D. Tumor-Targeting Properties of Novel Antibodies Specific to the Large Isoform of Tenascin-C. Clin Cancer Res (2006) 12(10):3200–8. doi: 10.1158/1078-0432.CCR-05-2804
192. Pedretti M, Soltermann A, Arni S, Weder W, Neri D, Hillinger S. Comparative Immunohistochemistry of L19 and F16 in Non-Small Cell Lung Cancer and Mesothelioma: Two Human Antibodies Investigated in Clinical Trials in Patients With Cancer. Lung Cancer (2009) 64(1):28–33. doi: 10.1016/j.lungcan.2008.07.013
193. Schliemann C, Wiedmer A, Pedretti M, Szczepanowski M, Klapper W, Neri D. Three Clinical-Stage Tumor Targeting Antibodies Reveal Differential Expression of Oncofetal Fibronectin and Tenascin-C Isoforms in Human Lymphoma. Leuk Res (2009) 33(12):1718–22. doi: 10.1016/j.leukres.2009.06.025
194. Berndt A, Kollner R, Richter P, Franz M, Voigt A, Berndt A, et al. A Comparative Analysis of Oncofetal Fibronectin and Tenascin-C Incorporation in Tumour Vessels Using Human Recombinant Sip Format Antibodies. Histochem Cell Biol (2010) 133(4):467–75. doi: 10.1007/s00418-010-0685-y
195. Frey K, Fiechter M, Schwager K, Belloni B, Barysch MJ, Neri D, et al. Different Patterns of Fibronectin and Tenascin-C Splice Variants Expression in Primary and Metastatic Melanoma Lesions. Exp Dermatol (2011) 20(8):685–8. doi: 10.1111/j.1600-0625.2011.01314.x
196. Schwager K, Villa A, Rosli C, Neri D, Rosli-Khabas M, Moser G. A Comparative Immunofluorescence Analysis of Three Clinical-Stage Antibodies in Head and Neck Cancer. Head Neck Oncol (2011) 3:25. doi: 10.1186/1758-3284-3-25
197. Sha H, Li R, Bian X, Liu Q, Xie C, Xin X, et al. A Tumor-Penetrating Recombinant Protein Anti-Egfr-Irgd Enhance Efficacy of Paclitaxel in 3d Multicellular Spheroids and Gastric Cancer in Vivo. Eur J Pharm Sci (2015) 77:60–72. doi: 10.1016/j.ejps.2015.05.020
198. Sha H, Zou Z, Xin K, Bian X, Cai X, Lu W, et al. Tumor-Penetrating Peptide Fused Egfr Single-Domain Antibody Enhances Cancer Drug Penetration Into 3d Multicellular Spheroids and Facilitates Effective Gastric Cancer Therapy. J Control Release (2015) 200:188–200. doi: 10.1016/j.jconrel.2014.12.039
199. Zhu A, Sha H, Su S, Chen F, Wei J, Meng F, et al. Bispecific Tumor-Penetrating Protein Anti-Egfr-Irgd Efficiently Enhances the Infiltration of Lymphocytes in Gastric Cancer. Am J Cancer Res (2018) 8(1):91–105.
200. Henke E, Nandigama R, Ergun S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci (2019) 6:160. doi: 10.3389/fmolb.2019.00160
Keywords: tenascins, extracellular matrix, tumor stroma, biomarker, anti-cancer therapy
Citation: Tucker RP and Degen M (2022) Revisiting the Tenascins: Exploitable as Cancer Targets? Front. Oncol. 12:908247. doi: 10.3389/fonc.2022.908247
Received: 30 March 2022; Accepted: 16 May 2022;
Published: 17 June 2022.
Edited by:
Alberto Passi, University of Insubria, ItalyReviewed by:
Sherif Abdelaziz Ibrahim, Cairo University, EgyptNikolaos A. Afratis, National and Kapodistrian University of Athens, Greece
Copyright © 2022 Tucker and Degen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Degen, bWFydGluLmRlZ2VuQHptay51bmliZS5jaA==