 Kairav Shah
Kairav Shah Sanchari Basu Mallik
Sanchari Basu Mallik Praveer Gupta
Praveer Gupta Abishek Iyer
Abishek Iyer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 22 June 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.908156
Tumours develop within complex tissue environments consisting of aberrant oncogenic cancer cells, diverse innate and adaptive immune cells, along with structural stromal cells, extracellular matrix and vascular networks, and many other cellular and non-cellular soluble constituents. Understanding the heterogeneity and the complex interplay between these cells remains a key barrier in treating tumours and cancers. The immune status of the pre-tumour and tumour milieu can dictate if the tumour microenvironment (TME) supports either a pro-malignancy or an anti-malignancy phenotype. Identification of the factors and cell types that regulate the dysfunction of the TME is crucial in order to understand and modulate the immune status of tumours. Among these cell types, tumour-associated fibroblasts are emerging as a major component of the TME that is often correlated with poor prognosis and therapy resistance, including immunotherapies. Thus, a deeper understanding of the complex roles of tumour-associated fibroblasts in regulating tumour immunity and cancer therapy could provide new insight into targeting the TME in various human cancers. In this review, we summarize recent studies investigating the role of immune and key stromal cells in regulating the immune status of the TME and discuss the therapeutic potential of targeting stromal cells, especially tumour-associated fibroblasts, within the TME as an adjuvant therapy to sensitize immunosuppressive tumours and prevent cancer progression, chemo-resistance and metastasis.
The tumour and its microenvironment (TME) not just consists of aberrant oncogenic cancer cells but are additionally composed of recruited and resident host cells such as various immune cells and other structural stromal cells (1). The tumour stroma contains components of extracellular matrix (ECM) and a host of different cell types such as immune cells, fibroblasts and vascular endothelial cells. For a long time, a cancer cell centric view of tumours led us to believe that mutations in oncogenes and tumour suppressor genes were sufficient to determine tumour survival, growth and progression. However, the current paradigm is that cancer cell derived signals delivered to immune and stromal cells reprogram the phenotype of these cells and determine, to a large extent, the survival and proliferation of oncogenic cancer cells. The exact phenotype and polarisation state of these stromal and immune cells will determine the immune status of the TME and further dictate tumour initiation, progression and metastasis (2). This change in paradigm from a cancer cell centric view to a TME centric view has opened new avenues for cancer therapy. Indeed, even conventional, tumour targeted therapies such as cytotoxic chemotherapies derive significant clinical benefit from their ability to stimulate anticancer immunity upon inducing cancer cell death, by increasing the tumour’s antigenicity and adjuvanticity (Figure 1). Further, the understanding that the TME is capable of reprograming and normalising tumour cells has led to development of novel approaches such as re-educating and reprograming immune and other stromal cells to prevent tumour progression and induce effective cancer remission (3).
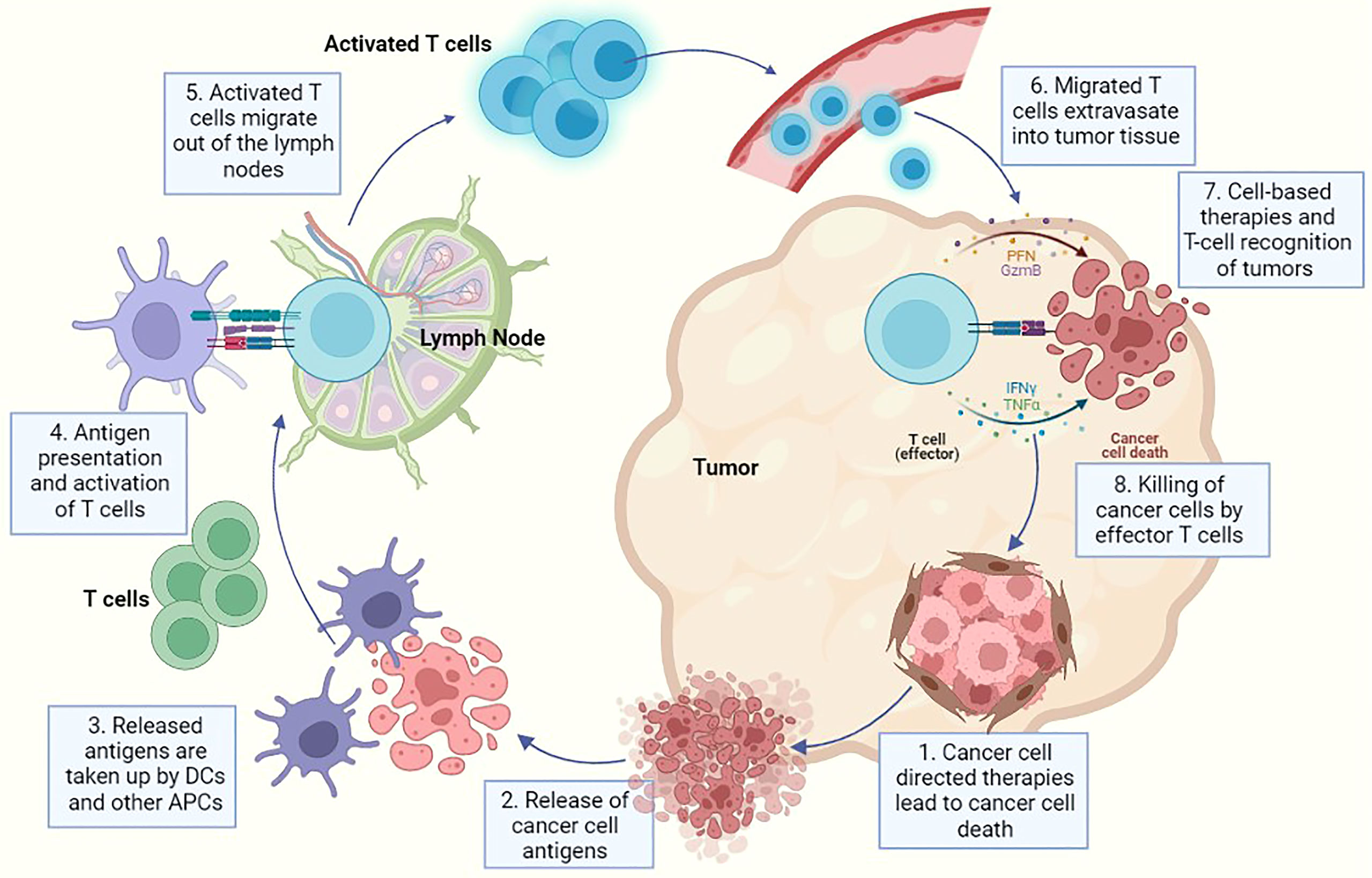
Figure 1 A representative diagram for cancer progression and associated immune reaction where each step indicates a potential method of therapeutic intervention using either cancer-cell directed therapies or immune cell directed therapies. In this image - 1. Indicates killing of tumour cells using cancer-cell directed therapies like chemotherapy, radiation etc., 2. Represents the release of cancer cell specific antigens after cancer cell death has been induced, 3. Represents the cancer antigen presentation to dendritic cells and antigen presenting cells, 4. Indicates priming of APCs and T cell activation, 5. Demonstrates the migration of activated T cells from the lymph node into the circulation, 6. Represents the migration of T cells into tumour tissue via extravasation through endothelial cells in blood vessels, 7. Demonstrates the recognition of cancer cells by effector T cells and also represents the point of action of multiple cell based therapies and (8). Finally represents the killing of cancer cells after recognition by effector T cells. Multiple immunotherapies have been developed capitalizing on this particular step e.g. checkpoint inhibitors, metabolic reprogramming therapies etc.
Apart from malignant tumour cells, various types of resident sentinel and infiltrated innate and adaptive immune cells, and stromal cells such as vascular endothelial cells, tumour-associated fibroblasts (TAFs), together with extracellular matrix (ECM) as well as multiple extracellular soluble factors such as cytokines, proteases, chemotactic factor, growth factors and others, are now recognized as part of the TME (4). The recent evolution of single cell technologies has provided significant insights into the heterogenous nature of the TME. Studies focused on single cell RNA sequencing (scRNA-seq) have yielded data on the diversity of cellular phenotypes in the TME, revealing a wealth of new targets as well as unknown cell types that can potentially play a pivotal role in cancer progression (5). Developed tumours that have a pro-inflammatory milieu and increased ratio of tumour fighting effector cells compared to tumour promoting immunosuppressive cells are often referred to as ‘Hot’ tumours. On the other hand, when the balance tilts towards tumour promoting immunosuppressive cells, these tumours are referred to as ‘Cold’ tumours (4). Understanding and identifying the key factors and cell types that regulate cold to hot transition of the TME, and vice versa, is an intense area of investigation. Key innate immune cells such as macrophages, dendritic cells and neutrophils have been prime suspects, as these cells are notorious for their functional plasticity and have been found in both hot and cold TMEs. Among these cells, tumour macrophages are the most studied to date and the key mechanisms and approaches to re-educate these cells in the TME have been extensively reviewed elsewhere (6). However, apart from macrophages and other immune cells, stromal cells represent a large, often under-appreciated cell population present in the TME that could be re-educated to prevent tumour progression and induce effective cancer remission. Activated fibroblasts trained by cancer cells, called tumour associated fibroblasts (TAFs or CAFs), consist of a large proportion of this stromal component (7). TAFs have largely been characterized as master controllers of matrix remodelling in tumours as they are the major producers of collagen, other matrix proteins and enzymes. Emerging evidence suggests that cells of the fibroblast lineage play a central role in tumour-related inflammation by engaging in complex interactions with cancer cells, as well as other stromal and tumour resident and infiltrating immune cells. These cells promote cancer cell proliferation and survival, angiogenesis and suppress anti-tumour T-cell responses. Several factors, including hypoxia, chemokines, cytokines and metabolic products of cancer cells (for example, lactic acid) are involved in TAF activation and functional polarization. Recent evidence indicates that TAFs also bear significant responsibility in influencing tumour metabolism. They may also play an important role in the hot to cold tumour immune phenotype transition in the TME, that represents one of the biggest challenges in cancer immunotherapy (8). In this review, we will describe known fibroblast biology and summarise the multifaceted roles that TAFs play in cancer progression and in sculpting an immunosuppressive tumour-microenvironment. We will also evaluate the rationale for targeting TAFs in combination with current immunotherapies, and explore current strategies employed in the field to re-educate and reprogram TAFs.
TAFs are defined as fibroblast cells that surround both primary and metastatic cancers and represent one of the most abundant heterogeneous cell population in the tumour stroma. In general, fibroblasts as a cell type are difficult to identify since they lack specific and unique markers. Hence, identification of fibroblasts relies primarily on morphology and tissue location (9). Under resting conditions, fibroblasts appear as spindle shaped cells and are normally located in the interstitial space (7). Understanding the roles of fibroblasts in normal tissues can provide crucial insight into how cancers may hijack their functions for their own proliferation (Figure 2). Fibroblasts are otherwise quiescent cells that demonstrate functional phenotypes only after activation. Broadly, fibroblasts are key producers and modulators of ECM, regulators of tissue interstitial pressure, and are intimately involved in wound healing (7, 10). Underlying most fibroblast functions is the production of collagens and other ECM proteins along with metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) which together modulate ECM degradation (10). In wound healing, activated fibroblasts become stellate shaped and can gain pro-angiogenic functions to promote a pro-M2 secretome by producing factors such as VEGFA, TGF-β, IL-6 and CXCL-10. Tumours can capitalise on the release of these cytokines and growth factors to survive and grow (7).
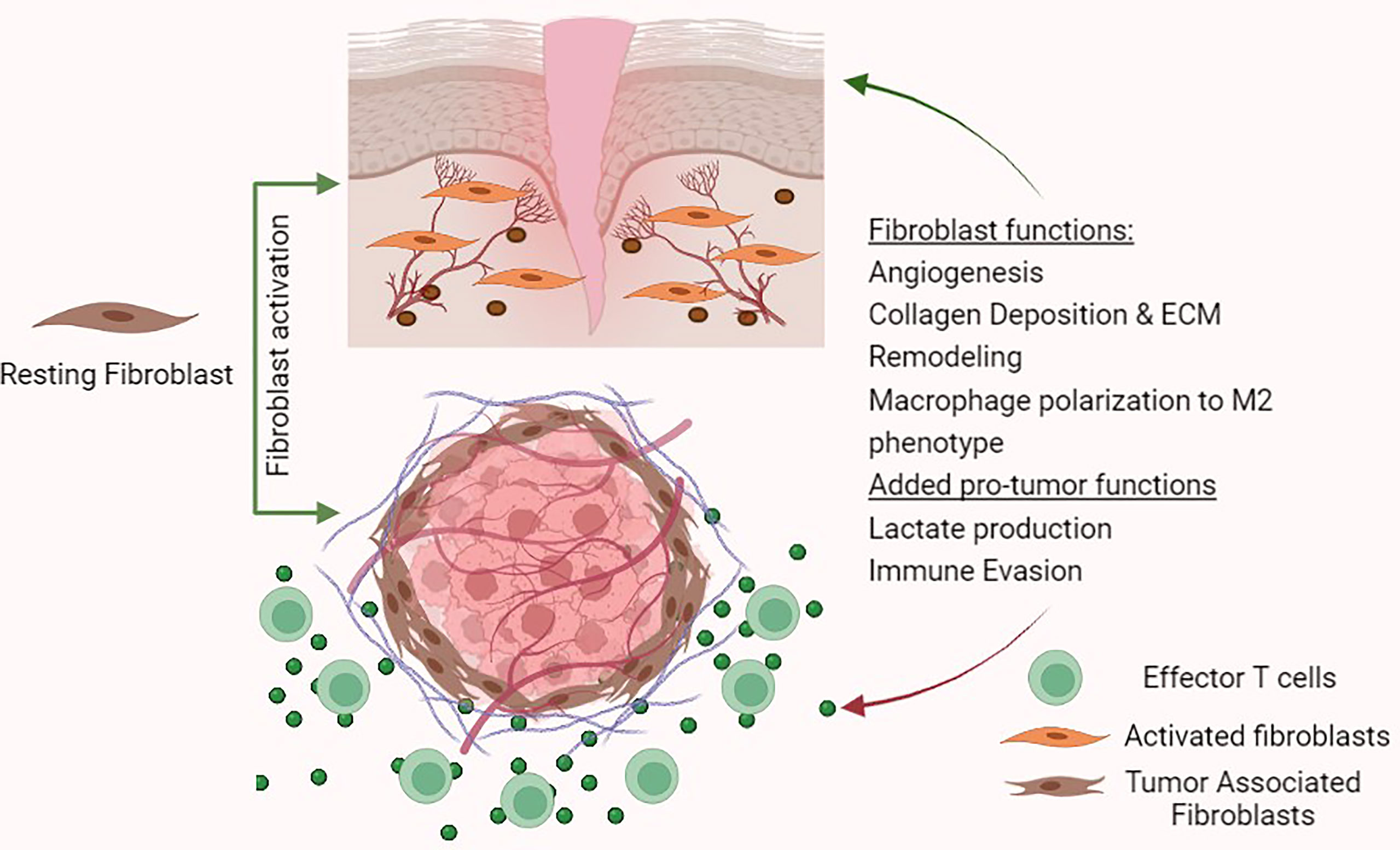
Figure 2 Fibroblast functions in wound healing and tumour- promoting functions in the tumour micro-environment.
Under the influence of the TME or in response to tissue injury caused by the growing tumour, fibroblasts can become activated to a distinct phenotype, and are termed tumour associated fibroblasts (TAFs) (also known as cancer-associated fibroblasts or CAFs) (9, 11, 12). TAFs can arise from quiescent tissue resident fibroblasts and from the activation of myofibroblasts. Alternatively, other cell types such as bone marrow derived mesenchymal stem cells (BM MSCs), pericytes, adipose, endothelial and epithelial cells can also differentiate into TAFs (9, 11, 13). Accurate tracing of TAF origin is important for differentiating between TAF subsets, which may have distinct phenotypes, and consequently allow specific targeting of pro-tumourigenic subsets.
The heterogeneity observed even in normal fibroblast populations gives us a fair idea of the complex fibroblast subtypes involved in cancer. Recent advances in single cell technologies have led to greater understanding of the heterogeneity in fibroblast subpopulations in the dermis (14, 15) and also in TAF populations along with the diversity of markers which are expressed by these subsets. Heterogeneity in TAF populations was also elucidated by the scRNA-seq of 52698 stromal cells isolated from lung tumours (with matching non-malignant controls). The study found five distinct TAF populations, each identifiable by its distinct set of associated collagen and ECM proteins (16). In other studies myofibroblasts and TAFs were identified to be present in tumour samples from head and neck squamous cancer patients through scRNA seq (17). Similar observations were made in colorectal cancer patients where normal fibroblasts, myofibroblasts and TAFs were identified (18). Other studies in cancer populations such as breast cancer have also noted the presence of multiple TAF subgroups, one of which had immune-regulatory function (14, 19). Largely, these studies explain the vast heterogeneity in TAF populations in different subsets of cancer and the need to identify specific markers for targeting TAF-related functions.
However, a common theme emerges from the wealth of information brought forward by single cell technologies, and based on these studies, TAFs can be broadly classified into two key subpopulations: a matrix-producing and contractile phenotype termed myTAFs and an inflammatory phenotype termed iTAFs. For a long time, myTAFs with an α-SMA positive signature were considered to be the only TAF population and these played a predominant role in the expression of ECM components, myofibroblast markers and cell contraction. With the advancement in single cell RNA sequencing technologies, inflammatory TAFs or iTAFs were identified as another distinct population characterized by an inflammatory secretome (20). TAFs have been predominantly implicated to be pro-tumour. However, some contradictory studies have also highlighted anti-tumour roles of TAFs. For example, myTAFs expressing α-SMA, have been observed to have anti-tumour properties in the TME in some studies (9, 21).
Understanding the multiple pathways that could lead to activation of the TAF phenotype is crucial to developing strategies that can prevent or even reverse this activation. Of the many factors released into the TME by cancer cells, TGF-β is one of the most well-known inducers of TAF activation, leading to the αSMA+ myofibroblasts (myTAFs) which have a pro-invasive phenotype (22). These activated TAFs engage in TGF-β and SDF-1 autocrine signalling loops that can amplify and maintain their activated phenotype (23). Additionally, TGF-β and SDF-1 produced by TAFs can subsequently lead to immune suppression (24) and angiogenesis (25) respectively. Although these changes may be a result of transcription by the downstream SMAD proteins, evidence suggests that reactive oxygen species (ROS) could play a prominent role in this pathway (26, 27). Importantly, TGF-β can increase the production of mitochondrial ROS, which in turn activates latent TGF-β and upregulates TGF-β gene expression, in a feed-forward loop (26). NADPH oxidases (NOXs) such as NOX4 are key producers of ROS downstream of TGF-β1 signalling (26). Other factors like chemotherapy can also indirectly contribute to the activation of fibroblasts and cause an increase in levels of TGF-β and IL-17A secretion (28) as a result of ROS production. In spite of its immune suppressive role, targeted inhibition of TGF-β may lack pathway specificity. Thus, inhibition of downstream players such as Nox4 could represent alternative targets for modulating TAF activation and polarisation. Other factors released in the TME like Platelet Derived Growth Factor (PDGF) signalling can also drive TAF activation (29, 30). Extracellular matrix (ECM) stiffness and organisation can also induce a pro-invasive phenotype via focal adhesion kinase (FAK) signalling (31). Activation of NF-κB, by IL-1β (32) or tumour derived exosomes (33), can also lead to a pro-inflammatory TAF phenotype in the TME. Overall, the different factors that could contribute to TAF activation suggest that differences in the resulting TAF populations may not be solely dependent on the cellular origin of the TAF subset, but also result from the combination of activating signals it receives in the TME. This could explain some of the phenotypic heterogeneity of TAFs seen within the TME. Recently, single cell profiling of stromal cells has revealed largely heterogenous populations of TAFs in pan cancer studies and along with specific TAF populations in conditions such as breast cancer, pancreatic ductal carcinoma and cholangiocarcinoma, that could possibly promote tumour cell growth through cellular crosstalk (5). Targeting TAF activation should thus take into consideration the dominant phenotype in the specific TME as well as alternative pathways to activation.
The ECM is composed of various collagens, proteoglycans and glycoproteins (34) and undergoes dynamic remodelling which is essential for development, wound healing and normal organ homeostasis. The ECM can change in composition and organisation to regulate both biochemical and biomechanical pathways (35). Life-threatening pathological conditions arise when ECM remodeling becomes excessive or uncontrolled (36). Cells can interact with the ECM via surface receptors, such as integrins, which affect cell adhesion and signalling (37) and through mechanical cues which can direct cell fate (38). The ECM can be divided into two forms: the basement membrane, which is a sheet-like structure surrounding cells and tissues that separates them from one another, and the interstitial matrix. Under conditions of wound healing, fibroblasts coordinate homeostatic ECM production and wound contraction (39). After completion of the repair process, they either undergo apoptosis or return to their quiescent state. However, this protective physiological process is dysregulated in cancer where TAFs retain their activated state and produce excessive amounts of collagen along with the collagen crosslinking enzymes like lysyl oxidase (LOX) and lysyl oxidase-like 1 (LOXL1), which increase ECM stiffness and direct tumour progression. On the other hand, upregulation of ECM degrading enzymes like MMPs can create spaces for migration (40) and release ECM bound factors such as TGF-β, fibroblast growth factor 2 (FGF-2) and vascular endothelial growth factor A (VEGFA). These factors can further increase angiogenesis and promote fibroblast activation along with ECM deposition in the TME (41). Additionally, the protein fibronectin is assembled into fibrils by TAFs, which allows directional migration of cancer cells (42), that is supportive of the pro-tumourigenic role of TAFs by modulating ECM remodelling.
The production of the ECM is closely linked to the process of angiogenesis. Angiogenesis is the formation of new capillaries and blood vessels that are required to provide oxygen and nutrients to cells. Tumours cannot grow or metastasize without access to new blood vessels (43) and often even utilize abnormal vasculature to evade immune attack. The tumour vasculature consists of immature vessels that are hyper-permeable in nature. Abnormal tumour vasculature can result in decreased perfusion, hypoxia and can even promote glycolysis resulting in an acidic environment. This can potentially deter immune cell function. Recent evidence from RNA sequencing studies from 11,069 patients, have revealed that low angiogenic immune tumour types are more responsive to immune checkpoint blockade (44, 45). The balance of pro-angiogenic versus anti-angiogenic factors is crucial in characterizing the ‘angiogenic switch’, which can be triggered by hypoxia, mechanical stress, inflammatory cells or by stromal derived factors. In the normal wound healing process, fibroblasts promote angiogenesis to replace damaged vessels and restore nutrient supply to regenerating tissue. This is accomplished by VEGF production, which can be enhanced by the release of hypoxia-inducible factor-1alpha (HIF-1α), along with FGF-2 and TNFα (46). Other proteins such as MYCT1, expressed almost exclusively in tumour associated endothelial cells, have been identified to be regulators of angiogenesis. Targeting MYCT1 has been demonstrated to be synergistic with checkpoint blockade in inhibiting tumour growth in pre-clinical models (47). Tumour cells can capitalise on this new vascularization to maintain and promote their own growth. For example, TAF derived IL-6 has been shown to be critical in promoting angiogenesis in colon cancer through increased production of VEGFA in other TAFs and fibroblasts. This effect could be abrogated by the addition of an anti-IL-6 receptor antibody (48). In another study, it was observed that endothelial progenitor cells are recruited via TAF mediated SDF-1/CXCL12 release and signalling that can also lead to angiogenesis in growing tumours (49). Other cytokines such as CXCL8 (IL-8), a known pro-angiogenic factor, along with CCL2 have been implicated in increasing tumour angiogenesis in a mouse model of PDAC (50) These pre-clinical studies have therefore aided in establishing a link between cancer cells and their ability to direct TAFs to produce a variety of factors that support tumour growth via angiogenesis. However, in-spite of this growing body of evidence, the exact molecular mechanisms by which fibroblasts educate vascular cells in the TME remain to be completely elucidated.
A key hallmark of cancer cells is their ability to reprogram metabolism of glucose to produce lactic acid (“Warburg metabolism”) instead of oxidative phosphorylation. In the nutrient starved TME, cells need to adapt their metabolic pathways to survive and function efficiently. Some cells have the capacity to proliferate faster by having access to energy at a much higher rate through glycolysis. Cancer cells have long been thought to utilize glycolysis for energy supply, particularly in the (hypoxic) interior of tumours where oxygen cannot be supplied due to interrupted blood flow (51). More recently, certain types of immune cells under infectious stress have also been reported to similarly undergo glycolysis, converting glucose to lactic acid (52–54). This phenomenon is believed to provide uncertain advantages to cancer cell survival and influence functions of surrounding cells in the TME. Lactate is considered a key mediator in the metabolic crosstalk between cancer cells and the TME. For example, tumour-derived lactate contributes to tumour-associated macrophage (TAM) polarization towards the pro-tumour/anti-inflammatory phenotype (M2 type) (55). Angiogenesis can also be stimulated by increased extracellular lactate which can stabilise HIF-1α and increase NF-kB as well VEGF production (56). Cancer cells can induce myofibroblasts to perform Warburg metabolism for further generation of energy. Here, cancer cells induce Caveolin-1 deficient TAFs to conduct aerobic glycolysis and produce metabolites like lactate and pyruvate that cancer cells could then take up and process to derive energy for proliferation (57). This switch to aerobic glycolysis in fibroblasts can be induced by TGF-β1 or PDGF and is reflected in a decrease in the levels of isocitrate dehydrogenase 3α (IDH3α) (58). In an alternative pathway, cancer cells also secrete TGF-β1 to induce p38 MAPK signalling in TAFs, which can in turn release factors that promote the metabolism of glycogen to glucose in cancer cells, representing another source of ATP (59).
At a cellular level, an immune response requires substantial energy, necessitating metabolic changes to rapidly meet an urgent need for greater ATP production (60). Besides production of ATP for proliferation, the Warburg effect can also produce intermediate glucose metabolites that act as signalling molecules in regulating expression and activity of genes and proteins involved metabolic pathways and immunity (52, 54, 61, 62). We now know that immune and stromal cells including TAFs also share this functional phenotype. TAFs release pro-inflammatory cytokines including pro-type II cytokines such as thymic stromal lymphopoietin (TSLP) under the influence of unknown stimuli. TSLP-activated resident dendritic cells migrate to draining lymph nodes where they prime naïve T cells into CD4+ Th2 cells. Further, Th2 cells are recruited to the TME by chemo-attractants such as TARC and MDC that are released by activated DCs and tumour cells. Th2 cells also release cytokines IL-5 and IL-13 that foster fibrosis by increasing extracellular matrix deposition and development of M2-like TAMs. However, the specific triggers and receptors, and the signalling mechanisms, that effect metabolism and the immune response in TAFs and the TME are still obscure.
Apart from supporting cancer growth, angiogenesis, tumour metabolism and metastasis, recent evidence suggests that TAFs can also cripple the immune response to cancer and promote tumour immune escape by creating an immunosuppressive TME. For example, two TAF derived cytokines that have been implicated in multiple pathways of immune suppression are TGF-β and SDF-1/CXCL12. TAFs can therefore modulate the function of various immune cells in the TME, which have been discussed in the following sections.
Among other antigen presenting cells, conventional dendritic cells (cDCs) are vital in initiating a T-cell response in cancer and represent the start of the cancer immunity cycle. DCs can endocytose dead cells and debris within the TME. Specifically, a small subset of cDC1s are required to cross-present antigens to CD8+ T cells (63). TAFs are major producers of growth factors like VEGF, which are known to inhibit the maturation of DCs (64). The relevance and mechanisms of this suppression in the TME are not completely understood. However anti-VEGF therapy has been shown to improve DC function and number (65, 66). Tumour metabolism can also influence DC differentiation, with lactate promoting tolerogenic, IL-10 producing DCs (67).
TGF-β functions as a key inhibitor of DC migration and their ability to prime T cells. As discussed earlier, TAFs are major producers of TGF-β in many tumour types and also contribute significantly to the release of latent ECM bound TGF-β (24). TGF-β was shown to immobilise DCs in skin tumours, hence preventing their migration to lymph nodes for antigen presentation (68). TGF-β can also stimulate the downregulation of MHC class II expression and that of co-stimulatory molecules like CD40, CD80 and CD86. This results in immature DCs ultimately leading to the generation of immunosuppressive Tregs and tolerogenic T cells (69). Such T cell suppressive DCs are also characterized by the expression of the tryptophan degrading enzyme indoleamine 2,3-dioxygenase (IDO) (70). In TME, TAF derived IL-6 can stimulate the production of IDO in a STAT3 dependent manner and thereby contribute in decreased T cell immune response towards tumours (71).
T-cell infiltration into the tumour is an essential step that precludes T cell mediated killing of tumour cells, and thus, also represents a mode of resistance or lack of response to immunotherapies that aim to enhance T-cell activity. A subset of TAFs expressing fibroblast associated protein (FAP) have been documented to cause T-cell exclusion from cancer cell nests in multiple cancer models (72). Studies indicate that FAP+ TAFs produce CXCL12/SDF-1 that may be responsible for coating cancer cells and causing T-cell exclusion. Further evidence for the role of CXCL12 in T-cell infiltration came from other preclinical studies, where inhibition of CXCR4 (the receptor for CXCL12) enhanced the response to checkpoint inhibition in a pancreatic cancer model (73). However, it is important to note that in spite of these discoveries, the complete mechanism of T-cell exclusion propagated by TAF-mediated CXCL12/CXCR4 axis remains unclear.
TAFs can restrict cytotoxic T-cell mediated cancer cell killing by direct or indirect upregulation of checkpoint inhibition molecules in the TME. TAFs themselves exhibit immune checkpoint ligands. However, the impact this has on T-cell function is not clear (74). TAFs can also boost PD-L1 expression on other cells in the TME. For example, TAF derived CXCL5 was shown to upregulate PD-L1 expression on cancer cells in murine melanoma and colorectal cancer models (75). In animal models of hepatocellular carcinoma, TAFs were shown to induce PD-L1 expression in neutrophils through a IL-6 STAT signalling pathway (76). In other murine tumour models, TAFs were also identified to be the main source of CD73 which is another key immune checkpoint molecule (77). Collagen in the tumour ECM which is primarily secreted by TAFs, may also lead to CD8+ T-cell exhaustion and cause resistance to anti-PD-L1 therapy. In models of lung tumours, expression of the receptor LAIR1 on CD8+ T-cells caused by CD18 interaction with collagen was shown to prompt T-cell exhaustion through SHP-1 (78). Interestingly, studies have suggested that TAFs may protect tumours from T-cell responses by direct elimination of tumour specific CD8+ T-cells. These studies revealed an enhanced ability of TAFs to cross-present antigen from exogenous sources by complexing with MHC-I, which lead to T-cell death via interaction with immune checkpoints FAS and PD-1 in an antigen specific manner. On the contrary, normal fibroblasts did not exhibit this protection to tumours. Furthermore, non-deleted T-cells also showed increased expression of LAG3, a marker for exhaustion (79).
TAFs can also influence the cancer immunity cycle indirectly by modulating recruitment and differentiation of immunosuppressive cell types in the TME. Macrophages recruited to the TME from circulating monocytes are called TAMs. These cells are particularly abundant, and in general promote tumour growth (80). In a rodent model of breast cancer, TAFs were observed to promote monocyte migration into the TME by secreting pro-inflammatory cytokines such as MCP-1 and CXCL12/SDF-1. Increased expression of CXCL12 chemokine receptor, CXCR4, was seen to be induced on monocytes by TGF-β (81). Unlike normal fibroblasts, TAFs were shown to coerce these monocytes to a more M2-like immunosuppressive phenotype, showing increased expression of markers associated with M2 macrophages such as CD163 and CD206, along with increased expression of PD-1 (82). A similar phenomenon of promoting M2 differentiation in macrophages was observed in other models of prostate cancer, PDAC, colorectal cancer and oral squamous cell carcinoma (83–86). Myeloid derived suppressor cells (MDSCs) are another immunosuppressive and pro-tumour immune cell population present in the TME that can aid in angiogenesis, metastasis and resistance to cancer immunotherapies (65). TAFs have been identified as recruiters of MDSCs in multiple rodent models of cancer. TAF derived SDF-1 was again implicated in the recruitment of MDSCs in murine breast and hepatocellular carcinoma models (87, 88). In a rodent model of lung squamous cell cancer, TAFs were found to recruit MDSCs via CCL2 (89). Most interestingly, contrary to TGF-β pro-tumour role in the TME, depletion of TGF-β resulted in increased MDSC infiltration in multiple studies (24).
Another population of adaptive immune cells that TAFs influence is that of the Foxp3+ regulatory T cell (Treg). Treg cells guard against autoimmunity and are well known to suppress anti-tumour responses. Infiltration of Treg cells into the TME is correlated with a worse prognosis in most cancers (90). In various cancers such as breast cancer, high grade serous ovarian cancer, and murine ovarian and pancreatic cancer models (91), a subset of TAFs were shown to associate with the recruitment and differentiation of Tregs in the TME, again via the secretion of SDF-1 (CXCL12) and its interaction with its receptor CXCR4. This effect was also seen in lung cancer models (92), however the study did not identify the exact mechanisms involved. Another cytokine that has been shown to induce upregulation of FOXP3 on CD4+ T cells to promote their differentiation into Tregs is TGF-β. This upregulation of FOXP3 on CD4+ T cells abrogated by TGF-β inhibition or by deletion of downstream transcription factors (24).
Stemming from the diverse roles that TAFs play in cancer survival and growth as well as immunosuppression, targeting TAFs for slowing disease progression in cancer could be potentially beneficial and enhance the effect immuno/chemo-therapy. In the following section, we describe current efforts and approaches targeting TAFs to sensitise the immunosuppressive TME and prevent tumour progression, chemoresistance and metastasis.
TAFs regulate an array of functions in the TME. TAFs secrete various cytokines, chemokines, and growth factors and play an essential role in promoting tumourigenesis, angiogenesis and chemo resistance, etc. Due to their phenotypic and functional heterogeneity, they have essential and sometimes contradictory roles in both eradication and progression of tumours (21). Although no ubiquitous marker exists for studying the pathology of these fibroblasts, certain TAF-specific proteins such as alpha smooth muscle actin (α-SMA), fibroblast activation protein (FAP), fibroblast specific protein -1 (FSP-1), podoplannin (PDPN), and vimentin, insulin-like growth factor-binding protein-7 (IGFBP7) etc. have been utilized to identify these cells. These markers also provide an opportunity to specifically target various types of tumours by modulating TAF mediated cancer progression. Lately, the pro-tumourigenic role of TAFs has garnered a lot of attention, making them a potential therapeutic target for TME modulating cancer interventions (21). Additionally, TAFs are present in various cancer types and unlike tumour cells, they are genetically stable, thus ensuring that they do not acquire resistance to therapies. A number of promising therapies (small molecules, peptides, antibodies and nano-delivery systems) targeting TAFs for potentiating innate immune responses in the immunosuppressive TME have been developed (Figure 3) and are briefly presented in the following sections. We have categorized the representative therapeutic strategies, which are in either preclinical or clinical development stages into- (a) targeting cell surface markers leading to TAF depletion, (b) targeting TAF activation, signaling pathways and ECM remodeling, and (c) reprogramming strategies to de-differentiate and re-educate activated TAFs to normalized fibroblasts (Figure 3).
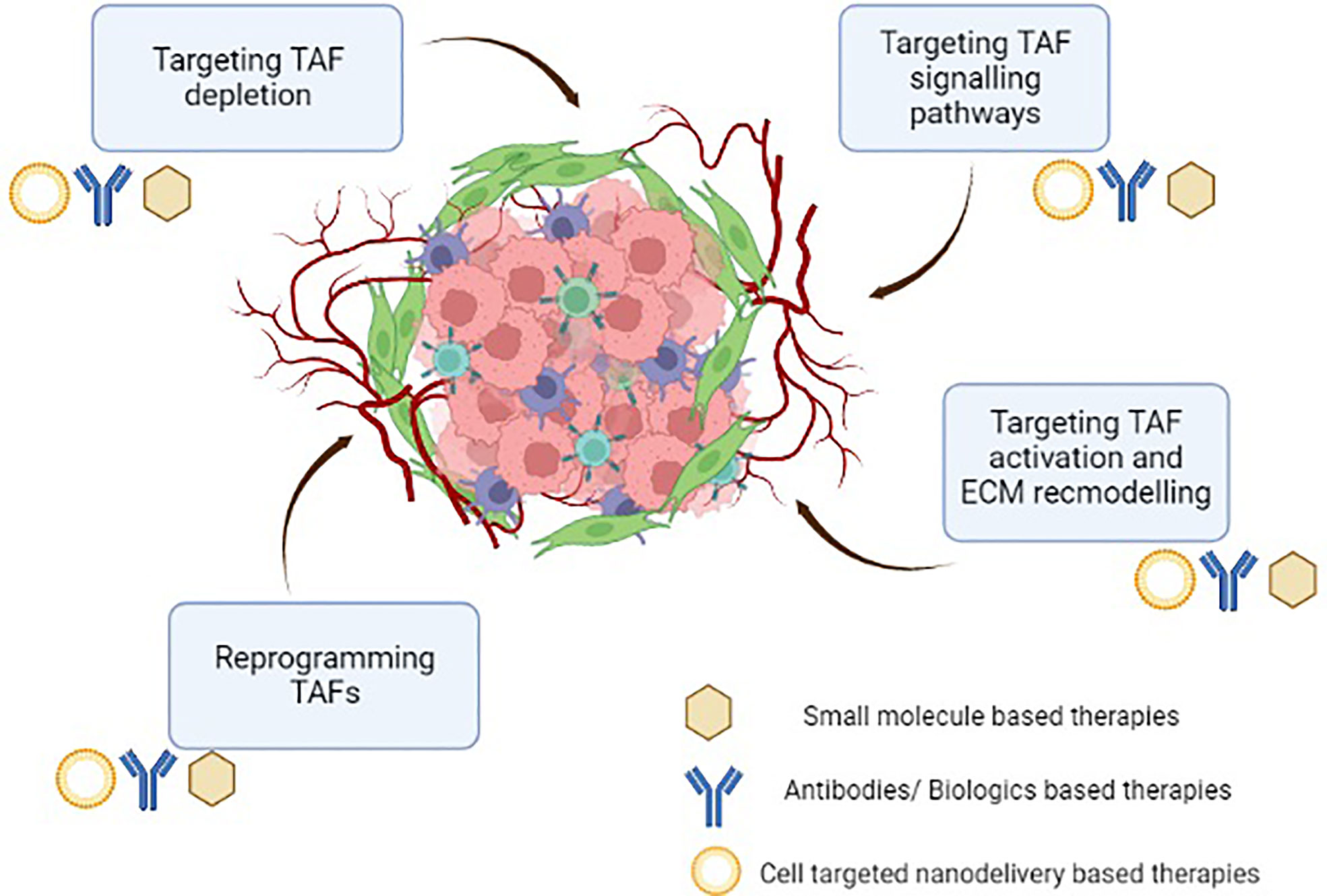
Figure 3 Therapeutic intervention targeting tumour-associated fibroblasts in the immunosuppressive tumour microenvironment.
Given the diverse role of TAFs in cancer progression, one probable way to target TAF function is to simply deplete their population, which can potentially alleviate TAF-mediated immunosuppression in the TME (93). Various strategies to target TAF depletion have been evaluated in preclinical models and clinical studies (Figures 3, 4; Table 1). From these studies, targeting TAF population depletion through cell surface markers has emerged as the most promising approach.
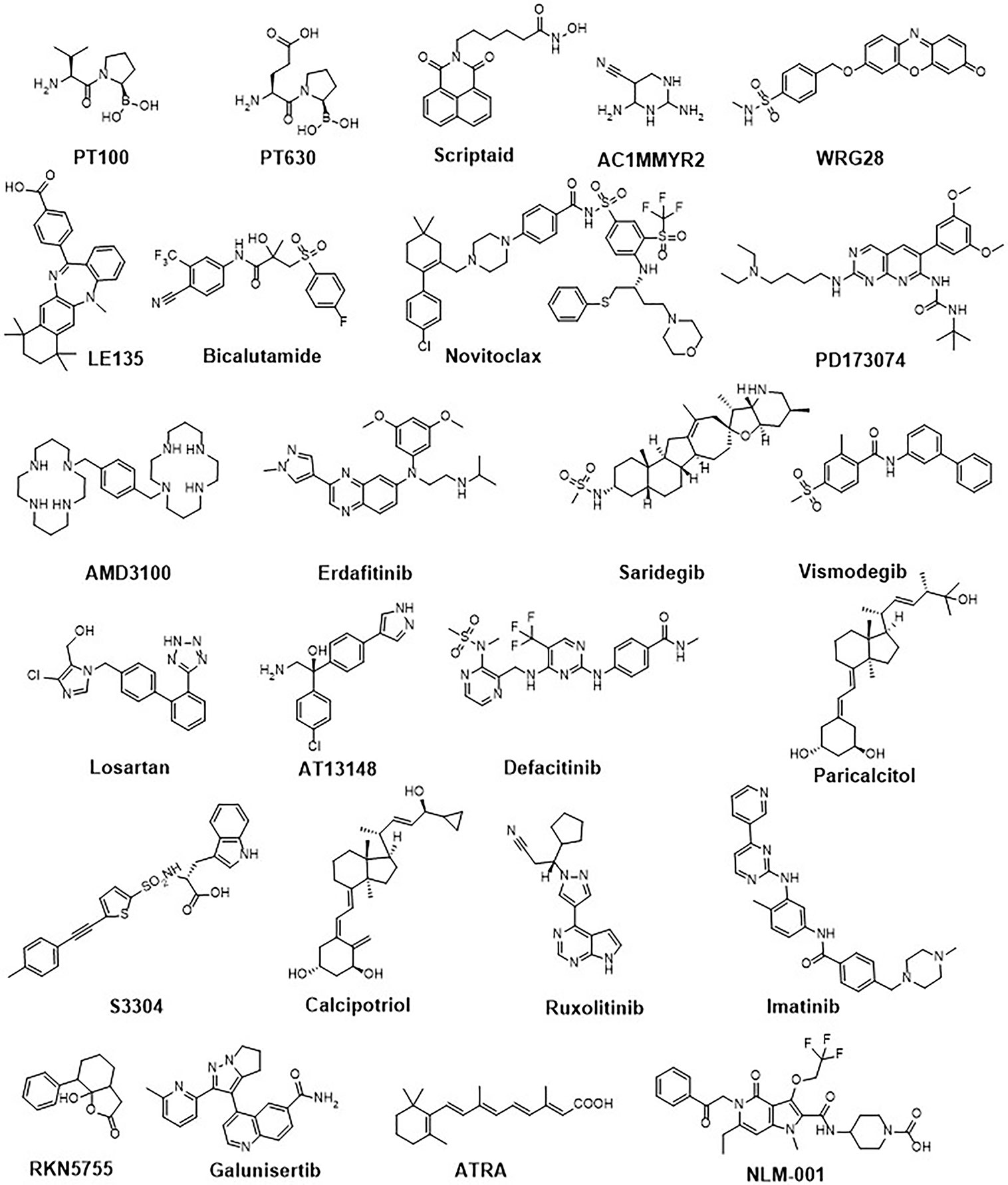
Figure 4 Chemical structures of a representative panel of small molecule drugs targeting tumour-associated fibroblasts.

Table 1 A representative panel of pharmacological interventions targeting tumour-associated fibroblasts.
One such important cell surface marker in activated stromal fibroblasts is Fibroblast Associated Protein (FAP) that has garnered a lot of attention lately. FAP is a type-II membrane bound serine protease which has dipeptidyl peptidase (DPP) and collagenase-like activity and is important in remodelling the ECM. Various small molecules targeting the enzymatic activity of FAP have been explored in the past decade. Talabostat (also known as PT100), a boronic acid-based peptide inhibitor (from amino boronic dipeptide class) of FAP DPP activity, is the only FAP inhibitor to be tested in clinical studies. A combination of PT100 with Oxaliplatin reduced tumour growth and decreased recruitment of tumour associated macrophages in pre-clinical models of cancers (94). However, it is interesting to note that the compound did not elicit any clinically meaningful efficacy in Phase II clinical trials. Further, combination treatment of Talabostat (PT100) with docetaxel or cisplatin also did not demonstrate any effect on disease progression and survival in patients. Similar to PT-100, another amino boronic dipeptide inhibitor of FAP activity, PT-630 (GluBoroPro dipeptide), demonstrated retardation of tumour growth in preclinical animal models (95). Clinical efficacy of this compound is not known at present. Depletion of FAP+ TAF population using humanized antibodies targeting has also been evaluated in a number of studies. Pre-clinical efficacy studies were conducted by Kraman et al. (2010), where stromal cells expressing FAP were conditionally deleted. In animal models of lewis lung carcinoma and pancreatic ductal carcinoma, this selective ablation led to control of tumour growth (145). A FAP targeting strategy was also employed by Gottschalk et al. (2013) who developed a compound DC vaccine with three targets: silencing of the zinc finger A20 to improve DC activation, targeting of TAFs through FAP, and targeting of the tumour antigen tyrosine related protein 2 (TRP2). The authors observed T-cell responses towards FAP and TRP2, enhanced CD8+ T-cell infiltration and antigen spreading, leading to potent anti-tumour activity (135). Similar CD8+ T cell mediated effects were seen in another DC vaccine targeting FAP in a CT26 mouse colon cancer model (96). A few of these FAP targeting antibodies have also been taken up in clinical trials with varying success. One such example is Sibrotuzumab which is the humanized form of the murine anti-FAP mAb F19. Sibrotuzumab was evaluated in a Phase I clinical trial involving patients with metastatic colorectal cancer, but showed little or no clinical benefit (97). It is worth mentioning here about the FAP targeting antibody Simlukafusp alfa (RO6874281; developed by Roche) which is a novel monomeric bispecific IL-2v immunocytokine having picomolar range binding affinity to FAP. A key advantage of this concept is that apart from binding to IL-2R expressed on immune effector cells, binding to FAP allows for the retention of IL-2v in the tumour, which can further affect immune cells in the TME. A number of phase I trials of Simlukafusp alfa as a single agent or in combinations are currently underway (146) (ClinicalTrials.gov identifiers: NCT03875079, NCT03063762, NCT02627274, NCT03386721 & NCT03193190). Next, another approach to target FAP+ TAFs was utilized by Ostermann et al. (2008) where an antibody-maytanisoid conjugate FAP5-DM1 containing anti-FAP-mAb was covalently linked to a tubulin binding maytansinoid with anti-mitotic activity (DM1). Treatment with FAP5-DM1 lead to increased apoptosis of malignant epithelial cells and also induced long lasting tumour inhibition in mice xenograft models of lung, pancreas, head and neck cancers (132). Other studies targeted FAP+ TAFs using a FAP-targeted immunotoxin αFAP-PE38. In a mouse model of metastatic breast cancer (4T1), treatment with αFAP-PE38 led to significant reduction in the recruitment of tumour infiltrating immune cells and tumour growth as monotherapy and in combination with paclitaxel (133).
Alternatively, the adoptive transfer of FAP targeted CAR T-cells is also an exciting approach for targeting TAFs in TME. In a study by Wang et al. (2014) and Lo et al. (2015), the use of FAP targeted CAR T cells produced inhibition of cancer growth both by improving CD8+ T-cell function (147), and by reducing ECM proteins and vascular density without eliciting severe toxicity in their models. However, targeting FAP+ cells can be non-selective towards cancer due to the wide expression of FAP in different cells. It was further observed that FAP targeted CAR T-cells elicited lethal toxicity and cachexia due to reactions against bone marrow stromal cells, which also express FAP on the cell surface (148). In support of this, studies using bioluminescent imaging in mice revealed that FAP+ cells were found in most tissues and their depletion led to cachexia and anemia. In the face of such potential systemic toxicity concerns, nanoparticle-aided targeted delivery of FAP targeting therapies are currently being explored to minimize toxicity and improve tumour specificity (149). For example, Zhen et al. (2017) have described the use of ferritin (a protein nanocage) conjugated with a FAP targeted single chain variable fragment that can attach to FAP+ cells selectively in the TME. Further, localized photo-irradiation enabled killing of FAP+ TAFs in TME are being evaluated to provide efficacy without causing significant toxicity (101). In another study, PNP-D-mAb based nano-delivery system, was evaluated to selectively target TAF in the TME. In this system, a cell penetrating peptide (CPP) was synthesized with nine arginine residues and cholesterol was embedded in it to provide as a hydrophobic tail. These modifications resulted into the self-assembly of this peptide to form peptide nanoparticles (PNP) and doxorubicin was encapsulated into this construct to form PNP-D. Finally, these particles were modified to construct PNP-D-mAb (102) that demonstrated preclinical efficacy by targeting FAP in both in vitro and in vivo studies. In another study, bispecific targeted liposomes combining single chain antibody fragments specific for FAP along with either variable antibody fragments or Trastuzumab for targeting HER2 were developed. Treatment with these bispecific liposomes showed greater depletion of HER2 expressing tumour cells and aided in tumour specific distribution in pre-clinical models (137). Aside from directly targeting FAP receptors, use of a novel cleavable amphiphilic peptide (CAP) as a substrate for FAP has also been explored in preclinical studies. These self-assembling peptides can form nano-fiber like structures in aqueous solution and enable further loading of cytotoxic drugs, which can be delivered upon FAP mediated cleavage of the particle (138). On similar lines, Hou and colleagues (2019) designed a nanoparticle (HA@DSP-pep-DSP) based on the FAP cleavable peptide DATGPA, from which dox loaded poly (amidoamine) particles were released (mediated by glutathione) and accumulated in the tumour stroma (139).
Several other cell surface markers which can help define TAF subsets have also been evaluated for selectively targeting TAFs in the TME. Among small molecules, Navitoclax, an inhibitor of the programmed cell death regulator BCL-2 that functions as a pro-apoptotic factor, has been shown to induce cell death in activated fibroblasts. Navitoclax is currently under investigation as an anti-fibrotic and anti-cancer agent, with phase II and III clinical trials underway (150). Similarly, antibodies targeting other TAF specific cell surface receptors have also been explored. Su et al. (2018) described a subset of TAFs expressing the surface markers CD10 and GPR77, which was found to promote survival of cancer stem cells in breast cancers. In CD10+GPR77+ -TAFs, NF-κB signalling via p65 phosphorylation leads to IL-6 and IL-8 secretion, that aids in the development of chemoresistance. However, this effect could be reversed by blocking GPR77 with a neutralizing antibody (151). Another approach that has been evaluated is targeting connective tissue growth factor (CTGF) using monoclonal antibodies. The monoclonal antibody FG-3019 targeting CTGF was investigated in a murine model of PDAC. Treatment with FG-3019 in combination with gemcitabine, led to increased killing of cancer cells through the modulation of the TME and not solely due to the increase in delivery of chemotherapy to the tumour site (130). Other studies have evaluated the role of fibroblast growth factor (FGFR) in conditions such as head and neck squamous cell cancers (HNSCC). The FGFR inhibitor PD173074 restrained fibroblast and endothelial cell proliferation and led to suppressed growth of tumour cells both in vitro and in a murine xenograft model of HNSCC (110).
Multiple studies have employed nanoparticles for targeted delivery of drugs to the TME and the same has been extended to targeting TAFs. For example, TAF targeted micelles incorporating Vismodegib, an abnormal sonic hedgehog (SHH) signaling inhibitor and SN38 (active metabolite of an anticancer drug Irinotecan) encapsulated in a polymeric PEG5k-P (HEMASN 38)x nanoparticle, have been studied (99). Other SHH inhibitors like cyclopamine were similarly co-delivered with Paclitaxel and this resulted in reduction αSMA+ (28%) and FAP-α+ (56%) TAF populations (152). Chen and colleagues reported the use of a novel TAF targeted nanovesicles (FH-SSL-NV) to completely deplete TAF and enhance the antitumour activity of Doxorubicin. The authors used the FH-peptide which targets tenascin C protein that is highly expressed in TAFs. This peptide was conjugated to prepare TAF targeted Navitoclax loaded liposomes (103). In addition, there are other receptors overexpressed on TAFs, for example SPARC which is an albumin binding protein, highly overexpressed in some desmoplastic tumours (prostate and breast cancers). Li and colleagues have utilised this approach of targeting SPARC by using Cellax. Cellax is a polymeric nanoparticle that is synthesised by conjugating docetaxel and PEG. Cellax was found to be more effective in depleting stroma in the TME as compared to docetaxel and nab-paclitaxel (104).
However, it is pertinent to re-emphasize that the heterogeneity in TAF populations and their specific context roles in tumour progression needs to be taken into consideration before translating TAF targeted therapies to the clinic. Indiscriminate TAF depletion may in fact lead to worse outcomes. This was observed in a murine model for pancreatic cancer, where depletion of αSMA+ myofibroblasts unexpectedly resulted in reduced animal survival. The resulting highly invasive tumours were characterized with higher levels of hypoxia, epithelial to mesenchymal transition (EMT) and cancer stem cell-like phenotype, along with increased recruitment of immunosuppressive Foxp3+ Tregs (93). Another study using a murine model of PDAC showed that SHH deficient mice with lower stromal content displayed more aggressive and proliferative tumours with increased vascularity (113). Therefore, enhanced specificity in targeting TAFs in the TME is warranted and other potential approaches to achieve this are discussed in the next two sections.
An alternative to depleting TAF populations can be to neutralize their effects on the TME by hampering TAF activation, related signaling, or their ability to regulate the ECM. This approach was utilized by Shen et al. (2018) to inhibit TAF activation related signaling. The authors used engineered lipid-protamine-DNA (LPD) nanoparticles carrying trap genes for IL-10 and CXCL12 to alleviate T cell inhibition. In pre-clinical studies, this approach led to changes in ECM remodeling with decreased levels of collagen and α-SMA as well as fewer immunosuppressive cells in a murine PDAC model (73, 141). Inhibition of CXCL12 released from FAP expressing TAFs can be another potential method to alleviate immune suppression. For example, the pre-clinical efficacy of a clinically approved inhibitor of CXCL12 receptor CXCR4, AMD3100, has been evaluated in various animal models of cancer. However, due to its limited efficacy as monotherapy, a combination therapy regimen has been employed with anti-PD-L1 and anti-CTLA4 immunotherapy (73, 153), docetaxel chemotherapy (154), radiation therapy (155) and a siRNA based lipid nanoparticle system (156) in preclinical studies. AMD3100 was synergistic with these various combinations and suppressed tumour growth in these studies. Lang et al. developed a cell-penetrant peptide based nanoparticle which targets TAFs through FAP-α antibody and delivers siRNA targeting CXCL12 into the TME. In an animal model of prostate cancer, this nanoparticle decreased tumour cell invasion, migration and angiogenesis accompanied by a reduced potential for metastasis (142). In order to target matrix metalloproteinases (MMPs), a key mechanism by which fibroblasts orchestrate ECM deposition, Chiappori et al. (2007) conducted a phase I clinical trial of the MMP inhibitor S-3304. In this study, S-3304 was found to be safe and well tolerated (124). The detailed efficacy of S-3304 is yet to be evaluated.
Targeting intracellular signaling in TAFs to modulate their phenotype is another potential therapeutic approach to modulate the TME. Suvarna et al. (2018) have targeted fibroblast migration using RKN5755, a ligand for beta-arrestin1 to interfere with downstream cofilin signaling pathways and restrict the migratory phenotype of activated fibroblasts (121). In another study, platelet derived growth factor (PDGF) receptor signaling was inhibited using imatinib in a mouse model of cervical carcinogenesis. This inhibition led to the reduced expression of fibroblast growth factor 2 (FGF-2) and FGF-7 leading to suppression tumour growth (30). A number of other agents targeting TAFs are also under investigation in clinical trials. Losartan, an angiotensin II blocker, capable of inhibiting the TGFβ pathway in TAFs to prevent their activation was evaluated in the clinic. Losartan treatment led to reduced levels of collagen and hyaluronan, consequently causing hypoxia in the TME. A phase II clinical study combining losartan with chemotherapy and radiotherapy reported increased R0 resection rates (157). Another phase II clinical study with losartan in combination with immunotherapy is also underway (ClinicalTrials.gov Identifier: NCT03563248). Similarly, using Galunisertib, a TGF-β receptor type I inhibitor, in combination with sorafenib showed an acceptable safety profile and an improved overall survival in patients with advanced hepatocellular carcinoma enrolled in a phase II human clinical trial (123). Other compounds like Erdafitinib, which is a pan fibroblast growth factor receptor (FGFR) inhibitor, is already approved for use in patients with advanced urothelial cancer having specific FGFR mutations (111). Angiogenesis in cancer is critical for its survival, growth and also metastasis. This is one of the key functions mediated by TAFs that can be a potential therapeutic strategy. In a Phase III clinical trial aimed to evaluate a combination of bevacizumab (VEGF inhibitor) and cisplatin plus pemetrexed which is the standard of care regimen for advanced malignant pleural mesothelioma, an increase in the OS was observed. However, this combination therapy did lead to increased toxic adverse effects (128).
In summary, Targeting TAF derived cytokines and other upstream signaling modulators remains challenging in spite of the promise it holds as a potential therapeutic approach. Given the various different mechanisms by which TAFs can promote tumour progression— multiple cytokines, metabolites, exosomes, force-mediated contraction, and even direct contact with cancer cells and transcytosis (158)— the scope of effects meditated by these single inhibitors, even in combinations remain unclear and needs further investigation.
Another approach to deter TAF function in the TME is to target fibroblasts which have already differentiated into a tumour promoting phenotype and to reprogram them to their native quiescent state. This reprogramming of TAFs has been attempted at the transcriptional level by several researchers. For example, Kim et al. identified the small molecule Scriptaid, an HDAC 1/3/8 inhibitor, as a factor that can reduce TGF-β-induced TAF differentiation. This in turn led to lower ECM secretion, cell invasiveness and stiffness in preclinical animal models (105). Use of DNA methyl transferase (DNMT) inhibitors along with JAK inhibition has also demonstrated preclinical efficacy in reverting TAFs to a wild-type phenotype by targeting the proinflammatory cytokine leukemia inhibitory factor (LIF) (159). Another study identified the methyltransferase nicotinamide N-methyltransferase (NNMT) enzyme as a central regulator of TAF activation in the TME. Here, treatment with an NNMT inhibitor alone decreased tumour burden in a mouse model of ovarian cancer (160). Chan et al. targeted two nuclear receptors (ligand responsive transcription factors), retinoid acid receptor β and androgen receptor, with their respective antagonists LE135 and bicalutamide in combination with cisplatin therapy to abrogate chemotherapy resistance in a mouse xenograft model of squamous cell carcinoma (SCC) (108). Ren et al. (2016) combined AC1MMTYR2, a small molecule inhibitor of miR-21, with taxol and were able to observe reduced tumour migration and invasion in rodent models of cancers. The authors also reported lower levels of FAP and α-SMA in a mouse model of breast cancer (106). In a mouse model of metastatic breast cancer, the small molecule WRG-28 was shown to inhibit the collagen receptor discoidin domain receptor 2 (DDR2) and block communication pathways between the tumour, stromal cells and the ECM. Following treatment with WRG-28, a reduction in tumour migration and invasiveness was also observed in these studies (107). Other reports involve inhibition of Rho-associated protein kinase, a small GTPase involved in cell contractility, that is upregulated in PDAC. Treatment with inhibitors of Rho-associated protein kinase, Fasudil and AT13148, resulted in reduced tumour growth and invasiveness in animal models of cancers (115). Paricalcitol, a vitamin D analog, has been shown to revert pancreatic stellate cells (PSCs) to a non-activated phenotype (161). Another vitamin D receptor ligand, Calcipotriol, yielded similar results (118). Since the deficiency of vitamin A has been observed in PDAC and it is known to play a role in PSC activation, ATRA treatment was initiated in a murine disease model and in human samples to reprogram PSCs to their quiescent state (162). Similarly, Han et al. (2018) also employed ATRA along with siRNA targeting heat shock protein 47 (HSP47), using a gold nanoparticle construct, to induce PSC quiescence in a PDAC xenograft model (163).
Aside from these, kinases involved in the complex signalling network in TAFs can also be potential targets for inhibition of TAF function. A multi-kinase receptor inhibitor Nintedanib was shown to downregulate the induction of collagens and α-SMA in TGF-β1 stimulated fibroblasts (164). Borriello et al. investigated the effect of inhibiting JAK2/STAT3 and MEK/ERK/1/2 using ruxolitinib and trametinib in a murine model of neuroblastoma to reverse the activation of a subset of pro-tumourigenic TAFs. They observed an increase in response to etoposide and overall survival (119). Ford et al. focused on inhibition of NOX4, a ROS-producing enzyme which is a downstream target of TGF-β1, and can regulate TAF phenotype. Interestingly, TGF-β inhibition could prevent myofibroblast differentiation, but not revert activated TAF phenotype to normal fibroblasts. On the contrary, NOX4 inhibition using Setanaxib (GKT137831) could revert TAFs to their normal phenotype. Treatment with Setanaxib also improved CD8+ T-cell infiltration which was initially limited to the periphery in TAF-rich tumours but increased to the center of tumours upon normalization. Consequently, tumours rich in TAFs were resensitized to anticancer vaccination and PD-1 checkpoint inhibition (165). Miao et al. made clever use of the off target uptake of anticancer nanoparticles by fibroblasts by creating and delivering plasmids encoding sTRAIL— a secretable form of TNF-related apoptosis-inducing ligand, which can induce apoptosis selectively in cancer cells. Thus, a majority of TAFs were reprogrammed to factories of sTRAIL secretion, which led potent tumour suppression in these preclinical studies. They also observed a normalization of residual TAFs to a quiescent state, which further inhibited tumour growth (166). Overall, multiple approaches have been utilized in preclinical and clinical studies to target the pro-tumourigenic role of TAFs including their effective depletion from the TME, inhibition of signaling pathways and their reprogramming to varying success. The heterogeneity of TAF population in the TME poses a big challenge to specifically target the pro-tumourigenic populations while not effectively deleting their anti-tumour properties. With the identification of TAF specific roles and markers along with the advent of newer targeting approaches, we anticipate that a greater success at therapy can potentially be achieved in the coming years.
Overall, fibroblasts are a dominant stromal cell type present in many tumours, which can be activated within the TME and ‘educated’ by cancer cells into overall tumour-promoting TAFs. Usually, quiescent fibroblasts become activated in tissue repair where they manipulate the ECM, regulate new vessel formation, and generate a skew towards an immunosuppressive, wound healing immune phenotype. Once the repair is complete, activated fibroblasts revert back to a normal phenotype. The continued activation of fibroblasts is thus characteristic of pathological responses such as organ remodelling and fibrosis, as well as tumours and cancers. Within the TME, cancer cells can program fibroblasts to pro-tumourigenic TAFs, which take on unique, heterogenous phenotypes, and are activated by different factors, including TGF-β. Further research is warranted to completely understand TAF differentiation and distinctions need to be made between the various subsets of TAFs that may influence tumour progression and metastasis in unique ways. The major hurdle for specifically targeting pro-tumourigenic TAF subsets is identifying reliable and specific markers that have largely remained elusive to date.
TAFs are capable of promoting tumour progression in many ways, which include ECM modulation, promoting angiogenesis, modulating the metabolic environment and creating an immunosuppressive TME. However, some mechanisms involved in these processes remain yet to be elucidated. For example, a more concrete understanding of how TAFs affect metabolism of tumour cells and other stromal cells, such as immune cells, is vital. Similarly, their impact on different stages of the cancer immunity cycle should be carefully examined to uncover opportunities for combination treatments. Despite the bulk of evidence that suggests that key TAF subsets support tumour progression, it is interesting that non-specific TAF depletion leads to pro-tumour responses, while re-educating TAFs and reversal to a normalized phenotype is protective. This warrants closer study of phenotypes and signalling mechanisms involved in the anti-tumour functions of certain TAF subsets. Various preclinical studies indicate that TAF content in common syngeneic murine models is lower than in human tumours. Examining TAF content and developing TAF rich preclinical tumour models that mimic human tumours may make them more accurate and reveal further details of TAF influence on tumour progression, metastasis and chemo-resistance. It is likely that characterizing and targeting the tumour stromal component may become essential in future cancer treatment, and reprogramming TAFs could unlock the potential of many existing therapies. Additionally, positive outcomes from the various ongoing clinical trials compiled in Table 2 will ultimately help unlock the true clinical potential of targeting TAFs in regulating the TME in various human cancers.
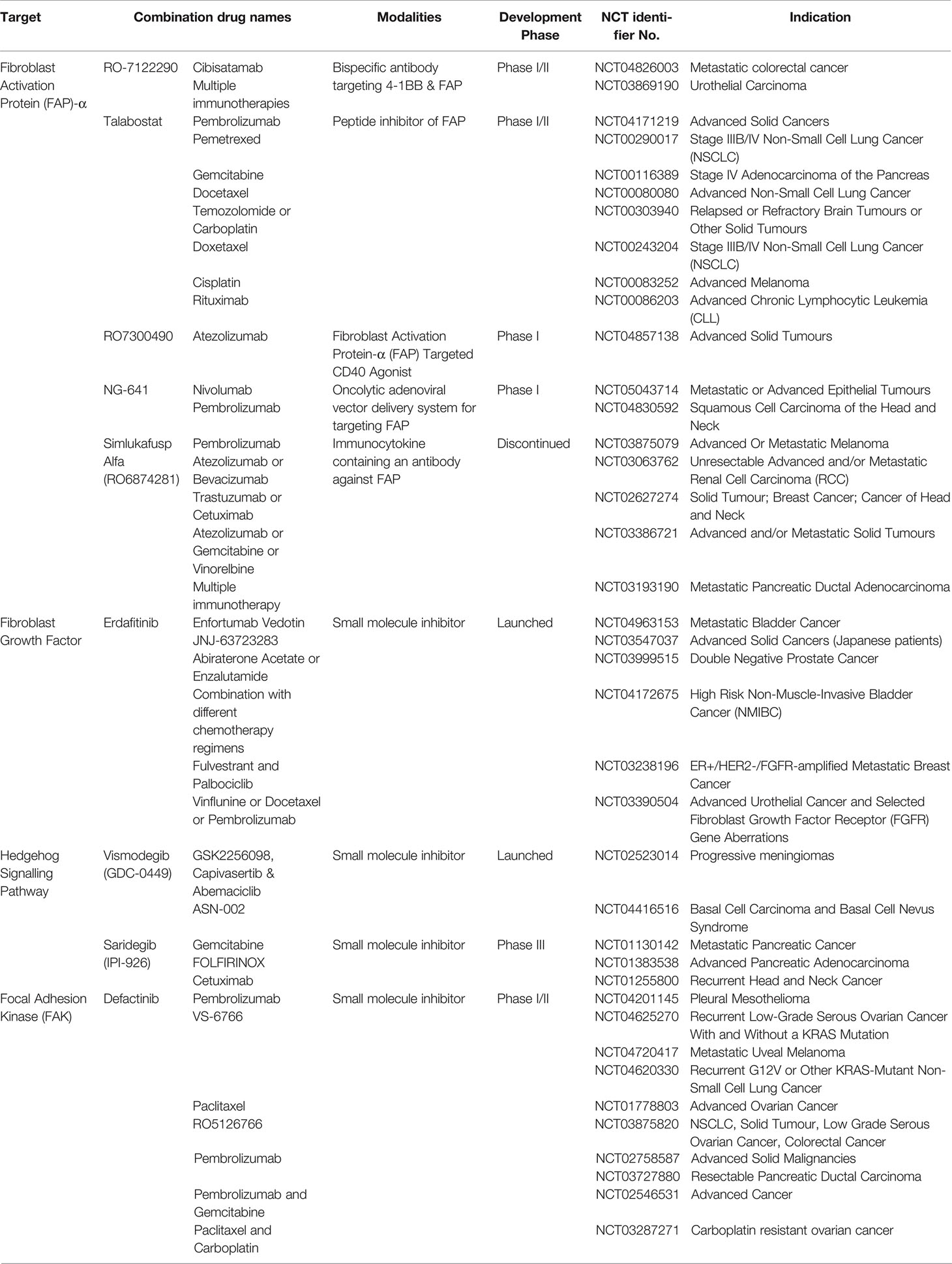
Table 2 A representative panel of current clinical trials with either solid tumour microenvironment modifying or tumour-associated fibroblasts targeting agents in combination with FDA-approved immune-oncology therapies.
KS initiated and wrote the manuscript and was the major content provider. KS, SB, PG and AI contributed to determining the content of each section. KS, SB, PG and AI collected data and contributed to writing each section of the manuscript. All authors contributed to editing and have read and approved the final manuscript.
This research was supported by Alembic Pharmaceuticals, India including salary support to SB, PG and AI. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Authors KS, SB, PG and AI were employed by the company Alembic Pharmaceuticals Ltd., India.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun Signal (2020) 18(1):1–19. doi: 10.1186/s12964-020-0530-4
2. Greten FR, Grivennikov SIJI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
3. Dimberg J, Shamoun L, Landerholm K, Andersson RE, Kolodziej B, Wågsäter D. Genetic Variants of the Il2 Gene Related to Risk and Survival in Patients With Colorectal Cancer. Anticancer Res (2019) 39(9):4933–40. doi: 10.21873/anticanres.13681
4. Duan Q, Zhang H, Zheng J, Zhang L. Turning Cold Into Hot: Firing Up the Tumor Microenvironment. Trends Cancer(2020) 6(7):605–18. doi: 10.1016/j.trecan.2020.02.022
5. Pan D, Jia D. Application of Single-Cell Multi-Omics in Dissecting Cancer Cell Plasticity and Tumor Heterogeneity. Front Mol Biosci. (2021) 8:757024. doi: 10.3389/fmolb.2021.757024
6. Xiang X, Wang J, Lu D, Xu XJST, Therapy T. Targeting Tumor-Associated Macrophages to Synergize Tumor Immunotherapy. Signal Transduct Target Ther (2021) 6(1):1–12. doi: 10.1038/s41392-021-00484-9
7. Kalluri R. The Biology and Function of Fibroblasts in Cancer. Nat Rev Cancer (2016) 16(9):582–98. doi: 10.1038/nrc.2016.73
8. Dobosz P, Dzieciątkowski T. The Intriguing History of Cancer Immunotherapy. Front Immunol (2019) 10:2965. doi: 10.3389/fimmu.2019.02965
9. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
10. McAnulty RJ. Fibroblasts and Myofibroblasts: Their Source, Function and Role in Disease. Int J Biochem Cell Biol (2007) 39(4):666–71. doi: 10.1016/j.biocel.2006.11.005
11. LeBleu VS, Kalluri R. A Peek Into Cancer-Associated Fibroblasts: Origins, Functions and Translational Impact. Dis Model Mech (2018) 11(4):dmm029447. doi: 10.1242/dmm.029447
12. Chen X, Song E. Turning Foes to Friends: Targeting Cancer-Associated Fibroblasts. Nat Rev Drug Discov (2019) 18(2):99–115. doi: 10.1038/s41573-018-0004-1
13. Raz Y, Cohen N, Shani O, Bell RE, Novitskiy SV, Abramovitz L, et al. Bone Marrow–Derived Fibroblasts Are a Functionally Distinct Stromal Cell Population in Breast Cancer. J Exp Med (2018) 215(12):3075–93. doi: 10.1084/jem.20180818
14. Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front Cell Dev Biol. (2019) 7:60. doi: 10.3389/fcell.2019.00060
15. Philippeos C, Telerman SB, Oulès B, Pisco AO, Shaw TJ, Elgueta R, et al. Spatial and Single-Cell Transcriptional Profiling Identifies Functionally Distinct Human Dermal Fibroblast Subpopulations. J Invest Dermatol (2018) 138(4):811–25. doi: 10.1016/j.jid.2018.01.016
16. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype Molding of Stromal Cells in the Lung Tumor Microenvironment. Nat Med (2018) 24(8):1277–89. doi: 10.1038/s41591-018-0096-5
17. Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell (2017) 171(7):1611–24.e24. doi: 10.1016/j.cell.2017.10.044
18. Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, et al. Reference Component Analysis of Single-Cell Transcriptomes Elucidates Cellular Heterogeneity in Human Colorectal Tumors. Nat Genet (2017) 49(5):708–18. doi: 10.1038/ng.3818
19. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell (2018) 33(3):463–79.e10. doi: 10.1016/j.ccell.2018.01.011
20. Huang H, Brekken RA. Recent Advances in Understanding Cancer-Associated Fibroblasts in Pancreatic Cancer. Am J Physiol Cell Physiol (2020) 319(2):C233–C43. doi: 10.1152/ajpcell.00079.2020
21. Barrett RL, Puré E. Cancer-Associated Fibroblasts and Their Influence on Tumor Immunity and Immunotherapy. Elife (2020) 9:. doi: 10.7554/eLife.57243
22. Casey TM, Eneman J, Crocker A, White J, Tessitore J, Stanley M, et al. Cancer Associated Fibroblasts Stimulated by Transforming Growth Factor Beta1 (Tgf-β1) Increase Invasion Rate of Tumor Cells: A Population Study. Breast Cancer Res Treat (2008) 110(1):39–49. doi: 10.1007/s10549-007-9684-7
23. Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine Tgf-β and Stromal Cell-Derived Factor-1 (Sdf-1) Signaling Drives the Evolution of Tumor-Promoting Mammary Stromal Myofibroblasts. Proc Natl Acad Sci (2010) 107(46):20009–14. doi: 10.1073/pnas.1013805107
24. Batlle E, Massagué J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
25. Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S, et al. Stromal Cell–Derived Factor 1 Promotes Angiogenesis Via a Heme Oxygenase 1–Dependent Mechanism. J Exp Med (2007) 204(3):605–18. doi: 10.1084/jem.20061609
26. Liu R-M, Desai LP. Reciprocal Regulation of Tgf-β and Reactive Oxygen Species: A Perverse Cycle for Fibrosis. Redox Biol (2015) 6:565–77. doi: 10.1016/j.redox.2015.09.009
27. Sampson N, Brunner E, Weber A, Puhr M, Schäfer G, Szyndralewiez C, et al. Inhibition of Nox4-Dependent Ros Signaling Attenuates Prostate Fibroblast Activation and Abrogates Stromal-Mediated Protumorigenic Interactions. Int J Cancer (2018) 143(2):383–95. doi: 10.1002/ijc.31316
28. Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A, et al. Chemotherapy Activates Cancer-Associated Fibroblasts to Maintain Colorectal Cancer-Initiating Cells by Il-17a. J Exp Med (2013) 210(13):2851–72. doi: 10.1084/jem.20131195
29. Herrera A, Herrera M, Guerra-Perez N, Galindo-Pumariño C, Larriba MJ, García-Barberán V, et al. Endothelial Cell Activation on 3d-Matrices Derived From Pdgf-Bb-Stimulated Fibroblasts Is Mediated by Snail1. Oncogenesis (2018) 7(9):1–15. doi: 10.1038/s41389-018-0085-z
30. Pietras K, Pahler J, Bergers G, Hanahan D. Functions of Paracrine Pdgf Signaling in the Proangiogenic Tumor Stroma Revealed by Pharmacological Targeting. PLoS Med (2008) 5(1):. doi: 10.1371/journal.pmed.0050019
31. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat Med (2016) 22(8):851–60. doi: 10.1038/nm.4123
32. Erez N, Truitt M, Olson P, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an Nf-κb-Dependent Manner. Cancer cell (2010) 17(2):135–47. doi: 10.1016/j.ccr.2009.12.041
33. Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor-Derived Exosomal Mir-1247-3p Induces Cancer-Associated Fibroblast Activation to Foster Lung Metastasis of Liver Cancer. Nat Commun (2018) 9(1):1–13. doi: 10.1038/s41467-017-02583-0
34. Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The Extracellular Matrix: Tools and Insights for the “Omics” Era. Matrix Biology (2016) 49:10–24. doi: 10.1016/j.matbio.2015.06.003
35. Kai F, Drain AP, Weaver VM. The Extracellular Matrix Modulates the Metastatic Journey. Dev Cell (2019) 49(3):332–46. doi: 10.1016/j.devcel.2019.03.026
36. Cox TR, JTJDm E. Remodeling and Homeostasis of the Extracellular Matrix: Implications for Fibrotic Diseases and Cancer. Dis Model Mech (2011) 4(2):165–78. doi: 10.1242/dmm.004077
37. Heino J, Kapyla J. Cellular Receptors of Extracellular Matrix Molecules. Curr Pharm Des (2009) 15(12):1309–17. doi: 10.2174/138161209787846720
38. Romani P, Brian I, Santinon G, Pocaterra A, Audano M, Pedretti S, et al. Extracellular Matrix Mechanical Cues Regulate Lipid Metabolism Through Lipin-1 and Srebp. Nat Cell Biol (2019) 21(3):338–47. doi: 10.1038/s41556-018-0270-5
39. Bainbridge PJ. Wound Healing and the Role of Fibroblasts. J Wound Care (2013) 22(8):407–8, 410–12. doi: 10.12968/jowc.2013.22.8.407
40. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell (2009) 139(5):891–906. doi: 10.1016/j.cell.2009.10.027
41. Yamauchi M, Gibbons DL, Zong C, Fradette JJ, Bota-Rabassedas N, Kurie JM. Fibroblast Heterogeneity and Its Impact on Extracellular Matrix and Immune Landscape Remodeling in Cancer. Matrix Biol (2020) 91:8–18. doi: 10.1016/j.matbio.2020.05.001
42. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-Associated Fibroblasts Promote Directional Cancer Cell Migration by Aligning Fibronectin. J Cell Biol (2017) 216(11):3799–816. doi: 10.1083/jcb.201704053
43. Carmeliet P, Jain RK. Angiogenesis in Cancer and Other Diseases. Nature (2000) 407(6801):249–57. doi: 10.1038/35025220
44. Fischbeck AJ, Ruehland S, Ettinger A, Paetzold K, Masouris I, Noessner E, et al. Tumor Lactic Acidosis: Protecting Tumor by Inhibiting Cytotoxic Activity Through Motility Arrest and Bioenergetic Silencing. Front Oncol (2020) 10:589434. doi: 10.3389/fonc.2020.589434
45. Subramanian M, Kabir AU, Barisas DA, Krchma K, Choi K. Conserved Angio-Immune Subtypes of the Cancer Microenvironment Predict Response to Immune Checkpoint Blockade Therapy. BioRxiv (2021). doi: 10.1101/2021.12.23.470799
46. Greaves NS, Ashcroft KJ, Baguneid M, Bayat AJ. Current Understanding of Molecular and Cellular Mechanisms in Fibroplasia and Angiogenesis During Acute Wound Healing. J Dermatol Sci (2013) 72(3):206–17. doi: 10.1016/j.jdermsci.2013.07.008
47. Kabir AU, Subramanian M, Lee DH, Wang X, Krchma K, Wu J, et al. Dual Role of Endothelial Myct1 in Tumor Angiogenesis and Tumor Immunity. Sci Transl Med (2021) 13(583). doi: 10.1126/scitranslmed.abb6731
48. Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 Released by Colon Cancer-Associated Fibroblasts Is Critical for Tumour Angiogenesis: Anti-Interleukin-6 Receptor Antibody Suppressed Angiogenesis and Inhibited Tumour–Stroma Interaction. Br J Cancer (2014) 110(2):469–78. doi: 10.1038/bjc.2013.748
49. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis Through Elevated Sdf-1/Cxcl12 Secretion. Cell (2005) 121(3):335–48. doi: 10.1016/j.cell.2005.02.034
50. Pausch TM, Aue E, Wirsik NM, Valls AF, Shen Y, Radhakrishnan P, et al. Metastasis-Associated Fibroblasts Promote Angiogenesis in Metastasized Pancreatic Cancer Via the Cxcl8 and the Ccl2 Axes. Sci Rep (2020) 10(1):1–12. doi: 10.1038/s41598-020-62416-x
51. Warburg O. On Respiratory Impairment in Cancer Cells. Science (1956) 124(3215):269–70. doi: 10.1126/science.124.3215.269
52. Cairns RA, Harris IS, Mak TW. Regulation of Cancer Cell Metabolism. Nat Rev Cancer (2011) 11(2):85–95. doi: 10.1038/nrc2981
53. Mills E, O’Neill LA. Succinate: A Metabolic Signal in Inflammation. Trends Cell Biol (2014) 24(5):313–20. doi: 10.1016/j.tcb.2013.11.008
54. Kelly B, O'neill LA. Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res (2015) 25(7):771–84. doi: 10.1038/cr.2015.68
55. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-Derived Lactate Induces M2 Macrophage Polarization Via the Activation of the Erk/Stat3 Signaling Pathway in Breast Cancer. Cell Cycle (2018) 17(4):428–38. doi: 10.1080/15384101.2018.1444305
56. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
57. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell cycle (2009) 8(23):3984–4001. doi: 10.4161/cc.8.23.10238
58. Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, et al. Metabolic Reprogramming of Cancer-Associated Fibroblasts by Idh3α Downregulation. Cell Rep (2015) 10(8):1335–48. doi: 10.1016/j.celrep.2015.02.006
59. Curtis M, Kenny HA, Ashcroft B, Mukherjee A, Johnson A, Zhang Y, et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab (2019) 29(1):141–55. doi: 10.1016/j.cmet.2018.08.007
60. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-Associated Idh1 Mutations Produce 2-Hydroxyglutarate. Nature (2009) 462(7274):739–44. doi: 10.1038/nature08617
61. Palsson-McDermott EM, O'neill LA. The Warburg Effect Then and Now: From Cancer to Inflammatory Diseases. Bioessays (2013) 35(11):965–73. doi: 10.1002/bies.201300084
62. Mills E, O’Neill LA. Succinate: A Metabolic Signal in Inflammation. Trends Cell Biol (2014) 24(5):313–20. doi: 10.1016/j.tcb.2013.11.008
63. Gardner A, Ruffell B. Dendritic Cells and Cancer Immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
64. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular Endothelial Growth Factor Affects Dendritic Cell Maturation Through the Inhibition of Nuclear Factor-κb Activation in Hemopoietic Progenitor Cells. J. Immunol. (1998) 160(3):1224–32.
65. Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to Vascular Endothelial Growth Factor Enhance the Efficacy of Cancer Immunotherapy by Improving Endogenous Dendritic Cell Function. Clin Cancer Res (1999) 5(10):2963–70.
66. Terme M, Colussi O, Marcheteau E, Tanchot C, Tartour E, Taieb JJC, et al. Modulation of Immunity by Antiangiogenic Molecules in Cancer. Clin Dev Immunol. (2012) 2012:492920. doi: 10.1155/2012/492920
67. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-Derived Lactic Acid Modulates Dendritic Cell Activation and Antigen Expression. Blood (2006) 107(5):2013–21. doi: 10.1182/blood-2005-05-1795
68. Weber F, Byrne SN, Le S, Brown DA, Breit SN, Scolyer RA, et al. Transforming Growth Factor-β 1 Immobilises Dendritic Cells Within Skin Tumours and Facilitates Tumour Escape From the Immune System. Cancer Immunol Immunother (2005) 54(9):898–906. doi: 10.1007/s00262-004-0652-3
69. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The Polarization of Immune Cells in the Tumour Environment by Tgfβ. Nat Rev Immunol (2010) 10(8):554–67. doi: 10.1038/nri2808
70. Mellor AL, Munn DH. Ido Expression by Dendritic Cells: Tolerance and Tryptophan Catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi: 10.1038/nri1457
71. Cheng J, Deng Y, Yi H, Wang G, Fu B, Chen W, et al. Hepatic Carcinoma-Associated Fibroblasts Induce Ido-Producing Regulatory Dendritic Cells Through Il-6-Mediated Stat3 Activation. Oncogenesis. (2016) 5(2):e198–e. doi: 10.1038/oncsis.2016.7
72. Fearon DT. The Carcinoma-Associated Fibroblast Expressing Fibroblast Activation Protein and Escape From Immune Surveillance. Cancer Immunol Res. (2014) 2(3):187–93. doi: 10.1158/2326-6066.CIR-14-0002
73. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting Cxcl12 From Fap-Expressing Carcinoma-Associated Fibroblasts Synergizes With Anti–Pd-L1 Immunotherapy in Pancreatic Cancer. Proc Natl Acad Sci (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
74. Harryvan TJ, Verdegaal EM, Hardwick JC, Hawinkels LJ, van der Burg SH. Targeting of the Cancer-Associated Fibroblast—T-Cell Axis in Solid Malignancies. J Clin Med. (2019) 8(11):1989. doi: 10.3390/jcm8111989
75. Li Z, Zhou J, Zhang J, Li S, Wang H, Du J. Cancer-Associated Fibroblasts Promote Pd-L1 Expression in Mice Cancer Cells Via Secreting Cxcl5. Int J Cancer. (2019) 145(7):1946–57. doi: 10.1002/ijc.32278
76. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-Associated Fibroblasts Induce Pdl1+ Neutrophils Through the Il6-Stat3 Pathway That Foster Immune Suppression in Hepatocellular Carcinoma. Cell Death Dis. (2018) 9(4):1–11. doi: 10.1038/s41419-018-0458-4
77. Yu M, Guo G, Huang L, Deng L, Chang C-S, Achyut BR, et al. Cd73 on Cancer-Associated Fibroblasts Enhanced by the a 2b-Mediated Feedforward Circuit Enforces an Immune Checkpoint. Nat Commun. (2020) 11(1):1–17. doi: 10.1038/s41467-019-14060-x
78. Peng DH, Rodriguez BL, Diao L, Chen L, Wang J, Byers LA, et al. Collagen Promotes Anti-Pd-1/Pd-L1 Resistance in Cancer Through Lair1-Dependent Cd8+ T Cell Exhaustion. Nat Commun (2020) 11(1):1–18. doi: 10.1038/s41467-020-18298-8
79. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-Associated Fibroblasts Induce Antigen-Specific Deletion of Cd8+ T Cells to Protect Tumour Cells. Nat Commun (2018) 9(1):1–9. doi: 10.1038/s41467-018-03347-0
80. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity. (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
81. Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, et al. A Unidirectional Transition From Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep (2018) 23(5):1239–48. doi: 10.1016/j.celrep.2018.04.007
82. Yavuz BG, Gunaydin G, Gedik ME, Kosemehmetoglu K, Karakoc D, Ozgur F, et al. Cancer Associated Fibroblasts Sculpt Tumour Microenvironment by Recruiting Monocytes and Inducing Immunosuppressive Pd-1+ Tams. Sci Rep. (2019) 9(1):1–15. doi: 10.1038/s41598-019-39553-z
83. Comito G, Giannoni E, Segura C, Barcellos-de-Souza P, Raspollini M, Baroni G, et al. Cancer-Associated Fibroblasts and M2-Polarized Macrophages Synergize During Prostate Carcinoma Progression. Oncogene (2014) 33(19):2423–31. doi: 10.1038/onc.2013.191
84. Zhang A, Qian Y, Ye Z, Chen H, Xie H, Zhou L, et al. Cancer-Associated Fibroblasts Promote M2 Polarization of Macrophages in Pancreatic Ductal Adenocarcinoma. Cancer Med (2017) 6(2):463–70. doi: 10.1002/cam4.993
85. Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-Associated Fibroblasts Enhance Tumor-Associated Macrophages Enrichment and Suppress Nk Cells Function in Colorectal Cancer. Cell Death Dis (2019) 10(4):1–14. doi: 10.1038/s41419-019-1435-2
86. Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-Associated Fibroblasts Promote an Immunosuppressive Microenvironment Through the Induction and Accumulation of Protumoral Macrophages. Oncotarget (2017) 8(5):8633. doi: 10.18632/oncotarget.14374
87. Ouyang L, Chang W, Fang B, Qin J, Qu X, Cheng F. Estrogen-Induced Sdf-1α Production Promotes the Progression of Er-Negative Breast Cancer Via the Accumulation of Mdscs in the Tumor Microenvironment. Sci Rep (2016) 6(1):1–10. doi: 10.1038/srep39541
88. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic Carcinoma-Associated Fibroblasts Enhance Immune Suppression by Facilitating the Generation of Myeloid-Derived Suppressor Cells. Oncogene (2017) 36(8):1090–101. doi: 10.1038/onc.2016.273
89. Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, et al. Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing Ros-Generating Monocytic Mdscs in Lung Squamous Cell Carcinoma. Cancer Immunol Res. (2020) 8(4):436–50. doi: 10.1158/2326-6066.CIR-19-0507
90. Tanaka A, Sakaguchi S. Regulatory T Cells in Cancer Immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
91. Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol (2019) 10:1835. doi: 10.3389/fimmu.2019.01835
92. Kinoshita T, Ishii G, Hiraoka N, Hirayama S, Yamauchi C, Aokage K, et al. Forkhead Box P3 Regulatory T Cells Coexisting With Cancer Associated Fibroblasts Are Correlated With a Poor Outcome in Lung Adenocarcinoma. Cancer Sci (2013) 104(4):409–15. doi: 10.1111/cas.12099
93. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer With Reduced Survival. Cancer cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
94. Li M, Li M, Yin T, Shi H, Wen Y, Zhang B, et al. Targeting of Cancer−Associated Fibroblasts Enhances the Efficacy of Cancer Chemotherapy by Regulating the Tumor Microenvironment. Mol Med Rep (2016) 13(3):2476–84. doi: 10.3892/mmr.2016.4868
95. Santos AM, Jung J, Aziz N, Kissil JL, Puré E. Targeting Fibroblast Activation Protein Inhibits Tumor Stromagenesis and Growth in Mice. J Clin Investig (2009) 119(12):3613–25. doi: 10.1172/JCI38988
96. Wen Y, Wang CT, Ma TT, Li ZY, Zhou LN, Mu B, et al. Immunotherapy Targeting Fibroblast Activation Protein Inhibits Tumor Growth and Increases Survival in a Murine Colon Cancer Model. J Clin Investig (2010) 101(11):2325–32. doi: 10.1111/j.1349-7006.2010.01695.x
97. Scott AM, Wiseman G, Welt S, Adjei A, Lee F-T, Hopkins W, et al. A Phase I Dose-Escalation Study of Sibrotuzumab in Patients With Advanced or Metastatic Fibroblast Activation Protein-Positive Cancer. Clin Cancer Res (2003) 9(5):1639–47.
98. Lo A, Wang L-CS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting Fap-Expressing Stromal Cells. Cancer Res (2015) 75(14):2800–10. doi: 10.1158/0008-5472.CAN-14-3041
99. Wang L, Liu X, Zhou Q, Sui M, Lu Z, Zhou Z, et al. Terminating the Criminal Collaboration in Pancreatic Cancer: Nanoparticle-Based Synergistic Therapy for Overcoming Fibroblast-Induced Drug Resistance. Biomaterials (2017) 144:105–18. doi: 10.1016/j.biomaterials.2017.08.002
100. Smits VA, Cabrera E, Freire R, Gillespie DA. Claspin–Checkpoint Adaptor and DNA Replication Factor. FEBS J (2019) 286(3):441–55. doi: 10.1111/febs.14594
101. Zhen Z, Tang W, Wang M, Zhou S, Wang H, Wu Z, et al. Protein Nanocage Mediated Fibroblast-Activation Protein Targeted Photoimmunotherapy to Enhance Cytotoxic T Cell Infiltration and Tumor Control. Nano Letters (2017) 17(2):862–9. doi: 10.1021/acs.nanolett.6b04150
102. Ji T, Ding Y, Zhao Y, Wang J, Qin H, Liu X, et al. Peptide Assembly Integration of Fibroblast-Targeting and Cell-Penetration Features for Enhanced Antitumor Drug Delivery. Adv Mater (2015) 27(11):1865–73. doi: 10.1002/adma.201404715
103. Chen B, Dai W, Mei D, Liu T, Li S, He B, et al. Comprehensively Priming the Tumor Microenvironment by Cancer-Associated Fibroblast-Targeted Liposomes for Combined Therapy With Cancer Cell-Targeted Chemotherapeutic Drug Delivery System. J Control Release (2016) 241:68–80. doi: 10.1016/j.jconrel.2016.09.014
104. Murakami M, Ernsting MJ, Undzys E, Holwell N, Foltz WD, Li S-D. Docetaxel Conjugate Nanoparticles That Target α-Smooth Muscle Actin–Expressing Stromal Cells Suppress Breast Cancer Metastasis. Cancer Res. (2013) 73(15):4862–71. doi: 10.1158/0008-5472.CAN-13-0062
105. Kim DJ, Dunleavey JM, Xiao L, Ollila DW, Troester MA, Otey CA, et al. Suppression of Tgfβ-Mediated Conversion of Endothelial Cells and Fibroblasts Into Cancer Associated (Myo) Fibroblasts Via Hdac Inhibition. Br J Cancer (2018) 118(10):1359–68. doi: 10.1038/s41416-018-0072-3
106. Ren Y, Zhou X, Liu X, Jia H-h, Zhao X-h, Wang Q-x, et al. Reprogramming Carcinoma Associated Fibroblasts by Ac1mmyr2 Impedes Tumor Metastasis and Improves Chemotherapy Efficacy. Cancer Lett (2016) 374(1):96–106. doi: 10.1016/j.canlet.2016.02.003
107. Grither WR, Longmore GD. Inhibition of Tumor–Microenvironment Interaction and Tumor Invasion by Small-Molecule Allosteric Inhibitor of Ddr2 Extracellular Domain. Proc Natl Acad Sci (2018) 115(33):E7786–E94. doi: 10.1073/pnas.1805020115
108. Chan JSK, Sng MK, Teo ZQ, Chong HC, Twang J, Tan N. Targeting Nuclear Receptors in Cancer-Associated Fibroblasts as Concurrent Therapy to Inhibit Development of Chemoresistant Tumors. Oncogene (2018) 37(2):160–73. doi: 10.1038/onc.2017.319
109. Mertens JC, Fingas CD, Christensen JD, Smoot RL, Bronk SF, Werneburg NW, et al. Therapeutic Effects of Deleting Cancer-Associated Fibroblasts in Cholangiocarcinoma. Cancer Res (2013) 73(2):897–907. doi: 10.1158/0008-5472.CAN-12-2130
110. Sweeny L, Liu Z, Lancaster W, Hart J, Hartman YE, Rosenthal EL. Inhibition of Fibroblasts Reduced Head and Neck Cancer Growth by Targeting Fibroblast Growth Factor Receptor. Laryngoscope (2012) 122(7):1539–44. doi: 10.1002/lary.23266
111. Roubal K, Myint ZW, Kolesar JM. Erdafitinib: A Novel Therapy for Fgfr-Mutated Urothelial Cancer. Am J Health Syst Pharm (2020) 77(5):346–51. doi: 10.1093/ajhp/zxz329
112. Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/Ii Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol (2015) 33(36):4284. doi: 10.1200/JCO.2015.62.8719
113. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
114. Hauge A, Rofstad EK. Antifibrotic Therapy to Normalize the Tumor Microenvironment. J Transl Med (2020) 18:1–11. doi: 10.1186/s12967-020-02376-y
115. Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP, et al. Caf Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer (2019) 5(11):724–41. doi: 10.1016/j.trecan.2019.09.010
116. Demircioglu F, Wang J, Candido J, Costa AS, Casado P, de Luxan Delgado B, et al. Cancer Associated Fibroblast Fak Regulates Malignant Cell Metabolism. Nat Commun (2020) 11(1):1–18. doi: 10.1038/s41467-020-15104-3
117. Abyaneh HS, Regenold M, McKee TD, Allen C, Gauthier MA. Towards Extracellular Matrix Normalization for Improved Treatment of Solid Tumors. Theranostics (2020) 10(4):1960. doi: 10.7150/thno.39995
118. Sherman MH, Ruth TY, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell (2014) 159(1):80–93. doi: 10.1016/j.cell.2014.08.007
119. Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity With Mesenchymal Stromal Cells. Cancer Res (2017) 77(18):5142–57. doi: 10.1158/0008-5472.CAN-16-2586
120. Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM III, et al. Randomized, Double-Blind, Phase Ii Study of Ruxolitinib or Placebo in Combination With Capecitabine in Patients With Metastatic Pancreatic Cancer for Whom Therapy With Gemcitabine Has Failed. J Clin Oncol (2015) 33(34):4039. doi: 10.1200/JCO.2015.61.4578
121. Suvarna K, Honda K, Kondoh Y, Osada H, Watanabe N. Identification of a Small-Molecule Ligand of β-Arrestin1 as an Inhibitor of Stromal Fibroblast Cell Migration Accelerated by Cancer Cells. Cancer Med. (2018) 7(3):883–93. doi: 10.1002/cam4.1339
122. Edelman MJ, Wang X, Hodgson L, Cheney RT, Baggstrom MQ, Thomas SP, et al. Phase Iii Randomized, Placebo-Controlled, Double-Blind Trial of Celecoxib in Addition to Standard Chemotherapy for Advanced Non–Small-Cell Lung Cancer With Cyclooxygenase-2 Overexpression: Calgb 30801 (Alliance). J Clin Oncol. (2017) 35(19):2184. doi: 10.1200/JCO.2016.71.3743
123. Kelley R, Gane E, Assenat E, Siebler J, Galle P, Merle P, et al. A Phase 2 Study of Galunisertib (Tgf-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin Transl Gastroenterol. (2019) 10(7):e00056. doi: 10.14309/ctg.0000000000000056
124. Chiappori AA, Eckhardt SG, Bukowski R, Sullivan DM, Ikeda M, Yano Y, et al. A Phase I Pharmacokinetic and Pharmacodynamic Study of S-3304, a Novel Matrix Metalloproteinase Inhibitor, in Patients With Advanced and Refractory Solid Tumors. Clin Cancer Res. (2007) 13(7):2091–9. doi: 10.1158/1078-0432.CCR-06-1586
125. Ohashi T, Oguro Y, Tanaka T, Shiokawa Z, Tanaka Y, Shibata S, et al. Discovery of the Investigational Drug Tak-441, a Pyrrolo [3, 2-C] Pyridine Derivative, as a Highly Potent and Orally Active Hedgehog Signaling Inhibitor: Modification of the Core Skeleton for Improved Solubility. Bioorg Med Chem. (2012) 20(18):5507–17. doi: 10.1016/j.bmc.2012.07.034
126. Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, Herndon JE, et al. Salvage Radioimmunotherapy With Murine Iodine-131–Labeled Antitenascin Monoclonal Antibody 81c6 for Patients With Recurrent Primary and Metastatic Malignant Brain Tumors: Phase Ii Study Results. J Clin Oncol. (2006) 24(1):115–22. doi: 10.1200/JCO.2005.03.4082
127. Wang Z, Tang Y, Tan Y, Wei Q, Yu W. Cancer-Associated Fibroblasts in Radiotherapy: Challenges and New Opportunities. Cell Commun Signal (2019) 17(1):1–12. doi: 10.1186/s12964-019-0362-2
128. Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, et al. Bevacizumab for Newly Diagnosed Pleural Mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (Maps): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet (2016) 387(10026):1405–14. doi: 10.1016/S0140-6736(15)01238-6
129. Wang FT, Sun W, Zhang JT, Fan YZ. Cancer−Associated Fibroblast Regulation of Tumor Neo−Angiogenesis as a Therapeutic Target in Cancer. Oncol Lett (2019) 17(3):3055–65. doi: 10.3892/ol.2019.9973
130. Neesse A, Frese KK, Bapiro TE, Nakagawa T, Sternlicht MD, Seeley TW, et al. Ctgf Antagonism With Mab Fg-3019 Enhances Chemotherapy Response Without Increasing Drug Delivery in Murine Ductal Pancreas Cancer. Proc Natl Acad Sci (2013) 110(30):12325–30. doi: 10.1073/pnas.1300415110
131. Benson AB III, Wainberg ZA, Hecht JR, Vyushkov D, Dong H, Bendell J, et al. A Phase Ii Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination With Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. oncologist (2017) 22(3):241–e15. doi: 10.1634/theoncologist.2017-0024
132. Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective Immunoconjugate Therapy in Cancer Models Targeting a Serine Protease of Tumor Fibroblasts. Clin Cancer Res (2008) 14(14):4584–92. doi: 10.1158/1078-0432.CCR-07-5211
133. Fang J, Xiao L, Joo KI, Liu Y, Zhang C, Liu S, et al. A Potent Immunotoxin Targeting Fibroblast Activation Protein for Treatment of Breast Cancer in Mice. Int J Cancer (2016) 138(4):1013–23. doi: 10.1002/ijc.29831
134. Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR. Targeting Carcinoma-Associated Fibroblasts Within the Tumor Stroma With a Fibroblast Activation Protein-Activated Prodrug. J Natl Cancer Inst (2012) 104(17):1320–34. doi: 10.1093/jnci/djs336
135. Gottschalk S, Yu F, Ji M, Kakarla S, Song X-T. A Vaccine That Co-Targets Tumor Cells and Cancer Associated Fibroblasts Results in Enhanced Antitumor Activity by Inducing Antigen Spreading. PLoS One (2013) 8(12):. doi: 10.1371/journal.pone.0082658
136. Sunshine J, Taube JM. Pd-1/Pd-L1 Inhibitors. Curr Opin Pharmacol (2015) 23:32–8. doi: 10.1016/j.coph.2015.05.011
137. Tansi FL, Rüger R, Böhm C, Steiniger F, Kontermann RE, Teichgraeber UK, et al. Activatable Bispecific Liposomes Bearing Fibroblast Activation Protein Directed Single Chain Fragment/Trastuzumab Deliver Encapsulated Cargo Into the Nuclei of Tumor Cells and the Tumor Microenvironment Simultaneously. Acta Biomater (2017) 54:281–93. doi: 10.1016/j.actbio.2017.03.033
138. Ji T, Zhao Y, Ding Y, Wang J, Zhao R, Lang J, et al. Transformable Peptide Nanocarriers for Expeditious Drug Release and Effective Cancer Therapy Via Cancer-Associated Fibroblast Activation. Angew Chem Int Ed Engl (2016) 55(3):1050–5. doi: 10.1002/anie.201506262
139. Hou L, Chen D, Hao L, Tian C, Yan Y, Zhu L, et al. Transformable Nanoparticles Triggered by Cancer-Associated Fibroblasts for Improving Drug Permeability and Efficacy in Desmoplastic Tumors. Nanoscale (2019) 11(42):20030–44. doi: 10.1039/C9NR06438A
140. Kim M-G, Shon Y, Kim J, Oh Y-K. Selective Activation of Anticancer Chemotherapy by Cancer-Associated Fibroblasts in the Tumor Microenvironment. J Natl Cancer Inst (2017) 109(1):djw186. doi: 10.1093/jnci/djw186
141. Shen L, Li J, Liu Q, Song W, Zhang X, Tiruthani K, et al. Local Blockade of Interleukin 10 and Cxc Motif Chemokine Ligand 12 With Nano-Delivery Promotes Antitumor Response in Murine Cancers. ACS Nano (2018) 12(10):9830–41. doi: 10.1021/acsnano.8b00967
142. Lang J, Zhao X, Qi Y, Zhang Y, Han X, Ding Y, et al. Reshaping Prostate Tumor Microenvironment to Suppress Metastasis Via Cancer-Associated Fibroblast Inactivation With Peptide-Assembly-Based Nanosystem. ACS Nano (2019) 13(11):12357–71. doi: 10.1021/acsnano.9b04857
143. Von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The Role of Stromal Cancer-Associated Fibroblasts in Pancreatic Cancer. J Hematol Oncol (2017) 10(1):1–8. doi: 10.1186/s13045-017-0448-5
144. Schnittert J, Kuninty PR, Bystry TF, Brock R, Storm G, Prakash J. Anti-Microrna Targeting Using Peptide-Based Nanocomplexes to Inhibit Differentiation of Human Pancreatic Stellate Cells. Nanomedicine (2017) 12(12):1369–84. doi: 10.2217/nnm-2017-0054
145. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein–α. Science (2010) 330(6005):827–30. doi: 10.1126/science.1195300
146. Waldhauer I, Gonzalez-Nicolini V, Freimoser-Grundschober A, Nayak TK, Fahrni L, Hosse RJ, et al. Simlukafusp Alfa (Fap-Il2v) Immunocytokine Is a Versatile Combination Partner for Cancer Immunotherapy. Mabs (2021) 13(1):1913791. doi: 10.1080/19420862.2021.1913791
147. Wang L-CS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting Fibroblast Activation Protein in Tumor Stroma With Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity Without Severe Toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi: 10.1158/2326-6066.CIR-13-0027
148. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee C-CR, Restifo NP, et al. Immune Targeting of Fibroblast Activation Protein Triggers Recognition of Multipotent Bone Marrow Stromal Cells and Cachexia. J Exp Med (2013) 210(6):1125–35. doi: 10.1084/jem.20130110
149. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of Stromal Cells Expressing Fibroblast Activation Protein-α From Skeletal Muscle and Bone Marrow Results in Cachexia and Anemia. J Exp Med (2013) 210(6):1137–51. doi: 10.1084/jem.20122344
150. Mohamad Anuar NN, Nor Hisam NS, Liew SL, Ugusman A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front Pharmacol (2020) 11:1817. doi: 10.3389/fphar.2020.564108
151. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. Cd10+ Gpr77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell (2018) 172(4):841–56.e16. doi: 10.1016/j.cell.2018.01.009
152. Zhao J, Wang H, Hsiao C-H, Chow DS-L, Koay EJ, Kang Y, et al. Simultaneous Inhibition of Hedgehog Signaling and Tumor Proliferation Remodels Stroma and Enhances Pancreatic Cancer Therapy. Biomaterials (2018) 159:215–28. doi: 10.1016/j.biomaterials.2018.01.014
153. Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking Cxcr4 Alleviates Desmoplasia, Increases T-Lymphocyte Infiltration, and Improves Immunotherapy in Metastatic Breast Cancer. Proc Natl Acad Sci (2019) 116(10):4558–66. doi: 10.1073/pnas.1815515116
154. Domanska UM, Timmer-Bosscha H, Nagengast WB, Munnink THO, Kruizinga RC, Ananias HJ, et al. Cxcr4 Inhibition With Amd3100 Sensitizes Prostate Cancer to Docetaxel Chemotherapy. Neoplasia (2012) 14(8):709–18. doi: 10.1593/neo.12324
155. Zhou K, Xie L, Peng X, Guo Q, Wu Q, Wang W, et al. CXCR4 Antagonist AMD3100 Enhances the Response of MDA-MB-231 Triple-Negative Breast Cancer Cells to Ionizing Radiation. Cancer Lett (2018) 418:196–203. doi: 10.1016/j.canlet.2018.01.009
156. Muralidharan R, Panneerselvam J, Chen A, Zhao YD, Munshi A, Ramesh R. Hur-Targeted Nanotherapy in Combination With Amd3100 Suppresses Cxcr4 Expression, Cell Growth, Migration and Invasion in Lung Cancer. Cancer Gene Ther (2015) 22(12):581–90. doi: 10.1038/cgt.2015.55
157. Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY, et al. Total Neoadjuvant Therapy With Folfirinox in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol (2019) 5(7):1020–7. doi: 10.1001/jamaoncol.2019.0892
158. Arwert EN, Milford EL, Rullan A, Derzsi S, Hooper S, Kato T, et al. Sting and Irf3 in Stromal Fibroblasts Enable Sensing of Genomic Stress in Cancer Cells to Undermine Oncolytic Viral Therapy. Nat Cell Biol (2020) 22(7):758–66. doi: 10.1038/s41556-020-0527-7
159. Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic Switch Drives the Conversion of Fibroblasts Into Proinvasive Cancer-Associated Fibroblasts. Nat Commun (2015) 6(1):1–15. doi: 10.1038/ncomms10204
160. Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, et al. Proteomics Reveals Nnmt as a Master Metabolic Regulator of Cancer-Associated Fibroblasts. Nature (2019) 569(7758):723–8. doi: 10.1038/s41586-019-1173-8
161. Abyaneh HS, Regenold M, McKee TD, Allen C, Gauthier MA. Towards Extracellular Matrix Normalization for Improved Treatment of Solid Tumors. Theranostics (2020) 10(4):1960. doi: 10.7150/thno.39995
162. Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, et al. Retinoic Acid–Induced Pancreatic Stellate Cell Quiescence Reduces Paracrine Wnt–β-Catenin Signaling to Slow Tumor Progression. Gastroenterology (2011) 141(4):1486–97.e14. doi: 10.1053/j.gastro.2011.06.047
163. Han X, Li Y, Xu Y, Zhao X, Zhang Y, Yang X, et al. Reversal of Pancreatic Desmoplasia by Re-Educating Stellate Cells With a Tumour Microenvironment-Activated Nanosystem. Nat Commun (2018) 9(1):1–18. doi: 10.1038/s41467-018-05906-x
164. Gabasa M, Ikemori R, Hilberg F, Reguart N, Alcaraz J. Nintedanib Selectively Inhibits the Activation and Tumour-Promoting Effects of Fibroblasts From Lung Adenocarcinoma Patients. Br J Cancer (2017) 117(8):1128–38. doi: 10.1038/bjc.2017.270
165. Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, et al. Nox4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated Cd8 T-Cell Exclusion From Tumors. Cancer Res (2020) 80(9):1846–60. doi: 10.1158/0008-5472.CAN-19-3158
Keywords: cancer-associated fibroblasts, tumour microenvironment, inflammation, cancer, immunotherapy
Citation: Shah K, Mallik SB, Gupta P and Iyer A (2022) Targeting Tumour-Associated Fibroblasts in Cancers. Front. Oncol. 12:908156. doi: 10.3389/fonc.2022.908156
Received: 30 March 2022; Accepted: 23 May 2022;
Published: 22 June 2022.
Edited by:
Yun Dai, First Affiliated Hospital of Jilin University, ChinaReviewed by:
Christian Stockmann, University of Zurich, SwitzerlandCopyright © 2022 Shah, Mallik, Gupta and Iyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Praveer Gupta, cHJhdmVlci5ndXB0YUBhbGVtYmljLmNvLmlu; Abishek Iyer, YWJpc2hlay5peWVyQGFsZW1iaWMuY28uaW4=
†ORCID: Abishek Iyer, orcid.org/0000-0001-9533-5265
‡Present address: Kairav Shah, School of Medicine, Washington University, St. Louis, MO, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.