 Rosaria Chilà1†‡
Rosaria Chilà1†‡ Michela Chiappa1‡
Michela Chiappa1‡ Federica Guffanti1Nicolò Panini2
Federica Guffanti1Nicolò Panini2 Donatella Conconi3Andrea Rinaldi4
Donatella Conconi3Andrea Rinaldi4 Luciano Cascione4,5
Luciano Cascione4,5 Francesco Bertoni4,6
Francesco Bertoni4,6 Maddalena Fratelli7
Maddalena Fratelli7 Giovanna Damia1*
Giovanna Damia1*- 1Laboratory of Experimental Oncology, Department of Oncology, Istituto di Ricerche Farmacologiche Mario Negri Istituito di Ricovero e Cura a Carattere Scientifico (IRCCS), Milan, Italy
- 2Laboratory of Cancer Pharmacology, Department of Oncology, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milan, Italy
- 3School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
- 4Institute of Oncology Research, Faculty of Biomedical Sciences, USI, Bellinzona, Switzerland
- 5SIB Swiss Institute of Bioinformatics, Lausanne, Switzerland
- 6Oncology Institute of Southern Switzerland (IOSI), Bellinzona, Switzerland
- 7Department of Biochemistry, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milan, Italy
Cyclin-dependent kinase 12 (CDK12) is a serine/threonine kinase involved in the regulation of RNA polymerase II and in the transcription of a subset of genes involved in the DNA damage response. CDK12 is one of the most mutated genes in ovarian carcinoma. These mutations result in loss-of-function and can predict the responses to PARP1/2 inhibitor and platinum. To investigate the role of CDK12 in ovarian cancer, CRISPR/Cas9 technology was used to generate a stable CDK12 knockout (KO) clone in A2780 ovarian carcinoma cells. This is the first report on a CDK12 null cell line. The clone had slower cell growth and was less clonogenic than parental cells. These data were confirmed in vivo, where CDK12 KO transplanted cells had a much longer time lag and slightly slower growth rate than CDK12-expressing cells. The slower growth was associated with a higher basal level of apoptosis, but there were no differences in the basal level of autophagy and senescence. While cell cycle distribution was similar in parental and knockout cells, there was a doubling in DNA content, with an almost double modal number of chromosomes in the CDK12 KO clone which, however did not display any increase in γH2AX, a marker of DNA damage. We found partial down-regulation of the expression of DNA repair genes at the mRNA level and, among the down-regulated genes, an enrichment in the G2/M checkpoint genes. Although the biological features of CDK12 KO cells are compatible with the function of CDK12, contrary to some reports, we could not find any difference in the sensitivity to cisplatin and olaparib between wild-type and CDK12 KO cells.
Introduction
Cyclin-dependent kinase 12 (CDK12) is a serine/threonine kinase involved in the regulation of RNA polymerase II and mRNA processing (1–3). CDK12 is important for the maintenance of genomic stability as it regulates the transcription of a subset of genes involved in the DNA damage response (DDR) (4, 5). In murine embryonic cells, CDK12 sustains self-renewal and its knock-down reduces the expression of self-renewal genes and increases the expression of differentiation markers (6). CDK12 knockout (KO) mice are not viable, as CDK12 deletion is fatal during the peri-implantation stage, when CDK12-/- blastocysts fail to undergo inner cell mass outgrowth and do not survive in vitro due to the induction of apoptosis (5).
CDK12 mutations and amplifications have been reported in different tumor types (7, 8). Not only it is one of the most mutated genes in ovarian carcinoma (9), but its mutations have been correlated with alterations of its catalytic activity, leading to genomic instability, downregulation of genes in the homologous recombination (HR) repair pathway (10, 11) and increased sensitivity to platinum agents and PARP inhibitors (PARPi) (12, 13). HR genes have multiple intronic to poly-adenylation sites that are susceptible to CDK12 inhibition, leading to decreases in their expression (14, 15). In addition, CDK12 inhibition induces an RNA Pol II elongation defect with the subsequent use of proximal poly(A) sites that leads to premature cleavage and polyadenylation especially in DDR gene, known to have longer lengths (16).
High-grade serous ovarian carcinomas (HGSOC) with mutated CDK12 present a unique signature of genomic instability characterized by tens to hundreds of large tandem duplications scattered throughout the genome, probably due to defects in DNA repair (17). In breast cancer CDK12 amplification co-occurs with ERBB2 amplification (18, 19) and its over-expression has been associated with aggressive disease (18). The absence of CDK12 protein, however, correlates with the triple negative phenotype and with reduced expression of some DDR proteins, but not with lower DDR mRNA levels (20). CDK12 mutations in primary and castration-resistant prostate cancer have also been reported, mutually exclusive with other mutations in DNA repair genes (21–23).
With the aim of understanding the role of CDK12 in tumor growth and response to therapy better, we used CRISPR/Cas9 technology to generate a CDK12 knock-out system in A2780 ovarian cancer cell lines.
Materials and Methods
CDK12 KO Clone
The ovarian carcinoma cell line A2780 was obtained from the American Type Culture Collection (ATCC) and was authenticated by the authors within the last six months. Cell lines were maintained in RPMI culture medium supplemented with 10% FBS and 1% glutamine, at 37°C with 5% CO2. A2780 cells were initially transfected with CRISPR/Cas9 plasmid directed against exon 1 of CDK12 (U6gRNA-Cas9-2A-GFP, Sigma-Aldrich). After 48 hours, cells were seeded at very low density in order to isolate, expand and analyze single clones by western blot and DNA sequencing. We obtained only a CDK12 heterozygous KO clone, out of about 150 clones screened. On this clone, we ran a second round of transfection and selection and obtained an A2780 homozygous KO clone (A2780 KO).
DNA Sequencing
Total DNA was purified from cells (Maxwell Total DNA Purification Kit, Promega) and the selected CDK12 locus was amplified with a polymerase chain reaction (PCR) (Supplementary Table 1). Amplified DNAs were separated through electrophoresis, and Sanger-sequenced.
Cell Growth
Growth curves were obtained seeding the cells at 15000 cells/mL in six-well plates and counting them at different time points with a cell counter (Multisizer 3, Beckman Coulter). For clonogenic assay in six-well plates, cells were seeded at 125 cells/mL; colonies were left to grow for about ten days then stained with Gram’s Crystal Violet solution (Merck). For the limiting dilution assay, cells were seeded at 0.5 cells/well in 96-well plates and colonies were left to grow for about 30 days.
Flow Cytometry Analysis of DNA Content
Exponentially growing cells were washed twice in ice-cold PBS, fixed in ice-cold 70% ethanol, washed in PBS, re-suspended in 2 mL of a solution containing 25 μg/mL of propidium iodide in PBS and 25 μL of RNase 1 mg/mL in water, and stained overnight at 4°C in the dark. We used the FACS Calibur (Becton Dickinson) for cell cycle analysis.
Chromosome Analysis
Exponentially growing A2780, and A2780 CDK12 KO cells were treated for 6 hours with colcemid (Roche) 0.2 μg/mL in order to block them in metaphase, then treated with hypotonic solution (75 mM KCl) and fixed with 3:1 (vol/vol) methanol:acetic acid. We karyotyped 19 metaphases for A2780 and fifteen for A2780 CDK12 KO cells lines. Chromosome preparations were analyzed by QFQ banding (Q-Bands by Fluorescence using Quinacrine) according to routine procedures and the karyotype was described using the International System for Chromosome Nomenclature 2016.
Cell Death Analysis
To investigate apoptosis in the cells, we used a caspase 3/7 luminescence-based assay (Promega) and the β-galactosidase staining assay (Cell Signaling Technology) for the detection of senescent cells.
Western Blotting
Proteins were extracted and processed as already described (24). The primary antibodies used are listed in Supplementary Table 2. All the protein blots for western analysis have been cropped before antibody hybridization to be able to detect in the same filter different proteins.
Real-Time (RT)-PCR
Total RNA from cell lines was purified with the Maxwell 16 Total RNA Purification Kit (Promega) and reverse transcribed with the cDNA Archive Kit (Applied Biosystems). mRNA expression of the genes was detected using Sybr Green assays (Applied Biosystems). Reactions were run in a total volume of 20 μL containing 10 ng of cDNA with Sybr Green and the forward and reverse primers of the gene, in triplicate (Supplementary Table 1). All the data were normalized to the levels of the cyclophillin gene and expressed as the -fold increase over the wild type CDK12 cells.
Gene Expression
As already described (25), NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England BioLabs Inc., Ipswich, MA, USA) was used for RNA-seq experiments and the NEBNext Multiplex Oligos for Illumina (New England BioLabs Inc.) for cDNA synthesis. The pre-pool sequencing was done using the NextSeq 500 (Illumina, San Diego, CA, USA) with the NextSeq 500/550 High Output Kit v2.5 (75 cycles; Illumina) and for all the samples we used stranded, single-ended 75bp-long sequencing reads.
Drugs and Treatments
Cisplatin was purchased from Sigma Aldrich; paclitaxel from ChemieTek; olaparib from LC Laboratories; VE822, MK1775 and KU55933 from Axon Medchem; THZ1 and THZ1 hydrochloride were from Insight Biotechnology Limited; ET-743 from PharmaMar and PF-00477736 from Pfizer. For cytotoxicity experiments, cell lines were seeded in 96-well plates and were treated after 48 hours with different concentrations of the drugs. After 72 hours cell viability was examined with the MTS assay (Promega) and absorbance was acquired using a plate reader (Infinite M200, TECAN). Drug concentrations inhibiting growth in 50% of the cells (IC50s) were calculated for each cell line, with the interpolation method.
Chemotaxis and Chemo-Invasion Tests
The cell lines were tested for their ability to migrate through a Nucleopore Track-Etch Membrane (pore size 8 μm, Whatman International), coated with Matrigel Basement Membrane Matrix for chemo-invasion, using Boyden chambers. We added 05 to 2 x 104 cells, re-suspended in DMEM culture medium with 0.1% FBS, to each well of the upper chamber for chemotaxis and chemo-invasion tests; DMEM+0.1% FBS (control wells) or 3T3 cell line supernatant (chemo-attractive agent) were added to the lower chamber. The chambers were incubated at 37°C overnight, then the cells from the upper side of the filter were removed and filters were fixed in methanol and stained for nuclear and cytoplasmic detection. Cells that had migrated the lower side of the filters were quantified by brightfield microscopy using a 40× objective, and the average number of cells per field was calculated.
In Vivo Studies
NCr-nu/nu mice (five-week-old females) were from Harlan S.p.a Italy and maintained under standard pathogen-free conditions. The Istituto di Ricerche Farmacologiche Mario Negri IRCCS adheres to national and international laws, regulations, and policies on the maintenance, care and use of laboratory animals, as already reported (26). The in vivo experiments were approved by an institutional review board and the Italian Ministry of Health (Authorization no. 705/2016-PR). Exponentially growing A2780 and A2780 CDK12 KO cells (approximately 7.5 x 106 cells per mouse) were injected subcutaneously in the flank of four mice per group. A Vernier caliper was used to measure tumor diameters, and tumor volumes were calculated following the formula: [(smallest diameter)2 x biggest diameter]/2. Tumor and body weights were recorded at different time points from implant and mice were euthanized by carbon dioxide (CO2) overdose when tumors reached 10% of the animal’s body weight.
Statistical Analyses
Statistical analyses were done with GraphPad Prism software. We used a t-test to compare cell growth, colony formation, IC50s of cytotoxic experiments and chemo-attraction and chemo-invasion data between the CDK12 wild type (WT) and CDK12 KO cell lines. To compare the activation of apoptosis between CDK12 WT and CDK12 KO cells we used two-way ANOVA followed by a Bonferroni test.
RNA-Seq was analyzed as previously described (27). Briefly, after quality control using fastqc and mapping against the human GRCh38 genome build, alignment was done with STAR, reads counted with HTSeq-Count, and differently expressed transcripts defined with the voom/limma R package. Genes were considered up-regulated or down-regulated if the adjusted p-value was <0.05 and log2 (KO vs WT) was respectively >1 or <-1. Genes deregulated in both cell lines were analyzed for enrichment in cancer hallmarks using the web-based tool of the Molecular Signaling Database (MsigDB, http://software.broadinstitute.org/gsea/msigdb), filtering for a false discovery rate (FDR) <0.05.
Results
CDK12 KO Ovarian Cancer Cell Lines
A2780 cells were selected to generate KO clones for CDK12 with the CRISPR/Cas9 gene editing tool. As already reported (28), this was not very successful as we obtained only one clone from A2780 (A2780 KO). Sequencing of genomic DNA showed the A2780 KO clone was homozygous in exon 1 of CDK12, carrying one allele harboring a deletion of 30 base pairs and one containing an insertion of 270 base pairs (Figure 1A). These data were confirmed by the cDNA sequencing of the CDK12 KO cells (data not shown).
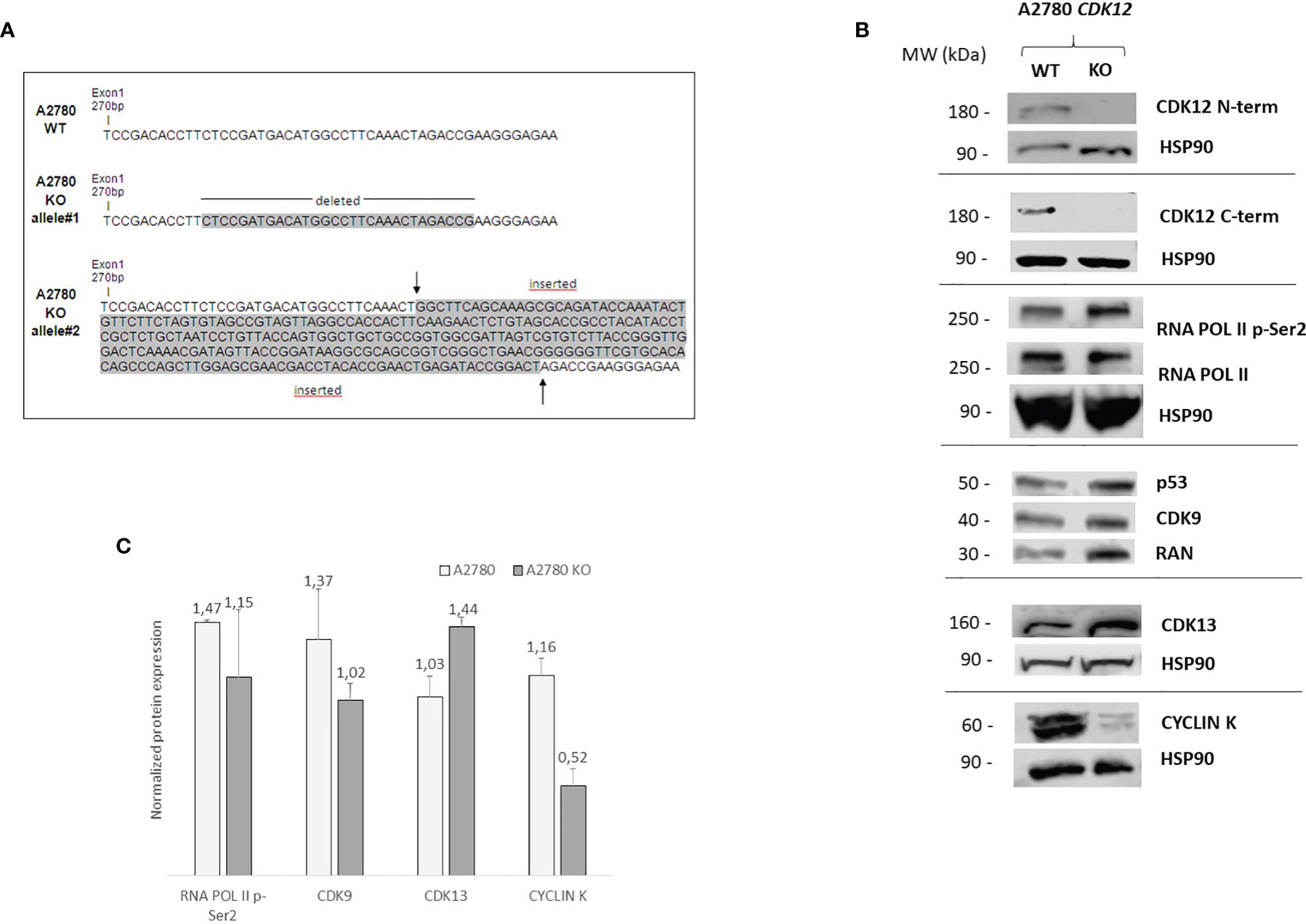
Figure 1 Molecular characterization of A2780 and A2780 CDK12 KO cells. (A) Nucleotide sequences of the loci targeted by CRISPR/Cas9 in exon 1 of CDK12, generating CDK12 KO clones. Upper panel: nucleotide sequences of A2780 CDK12 WT and KO; shaded nucleotides were deleted and inserted in KO clone allele #1 and #2, respectively. (B) Western blot analysis showing CDK12, HSP90, RNA pol II p-Ser2, RNA pol II, p53, CDK9, cyclin K, CDK13, RAN protein levels in A2780 and A2780 CDK12 KO cell lines. All the protein blots for western analysis have been cropped before antibody hybridization to be able to detect in the same filter different proteins. (C) Quantification of the western blot data by densitometric analyses. Values are normalized by actin and are the mean+SD of three replicates.
We tested whether our guide RNAs could be directed against other genes besides CDK12 (off-target genes). We used the online CRISPR Design tool by Zhang laboratory (http://crispr.mit.edu/) and found no off-target sequence in other genes.
Alterations in CDK12 sequence are predicted to code for probably non-functional proteins. In fact, deletion in one allele (30 bp deletion) would produce a protein lacking 10 AA; the other allele has a 270 bp insertion that causes a number of stop codons, implying that no full protein should be formed. No expression of CDK12 protein in A2780 KO cells was observed, using both a C-terminal and a N-terminal epitope directed CDK12 antibody (Figure 1B). We carefully checked for extra bands (possibly CDK12) with different sizes, but we never found any. Considering that one allele is predicted to produce a protein lacking 10 AA, the fact that we could not find any CDK12 protein by western blot suggests that the encoded protein is very unstable.
We checked the levels of a number of proteins that have been associated and regulated by CDK12 (7, 29). We found that the basal level of RNA polymerase II phosphorylation was similar in WT and KO CDK12, while slightly lower levels of the CDK12 cyclin partner cyclin K (Figures 1B, C). The levels of CDK9, the other transcriptional CDK involved in Ser2 RNA polymerase II phosphorylation, were not changed, nor were those of CDK13 (Figure 1B). p53 protein levels were similar in A2780 and the corresponding CDK12 KO clone (Figure 1B).
Biological Characterization of the CDK12 KO Ovarian Cancer Cells
CDK12 KO cells were morphologically indistinguishable from their parental counterparts (data not shown). We characterized the clone for its proliferation rate and clonogenicity. KO clones had slower growth than their parental cells (Figure 2A). Colony and limiting dilution assays suggested that A2780 CDK12 KO cells had lower clonogenic ability than parental cells. CDK12 KO cells did have less colony formation ability (Figure 2B) and less single cell growth in the limiting dilution assay (Figure 2C). We wondered whether the absence of CDK12 led to an increase in cell death that could partly explain the differences in cell growth. As shown in Figure 2D, the percentage of cells undergoing apoptosis was higher in CDK12 KO cells than in their parental cell line at different times. There was no activation of autophagy, as demonstrated by the absence of a cleaved form of LC3 protein and the increase in p62 (Supplementary Figure 1A), or senescence, as indicated by the absence of β-galactosidase staining (Supplementary Figure 1B) in CDK12 KO cells.
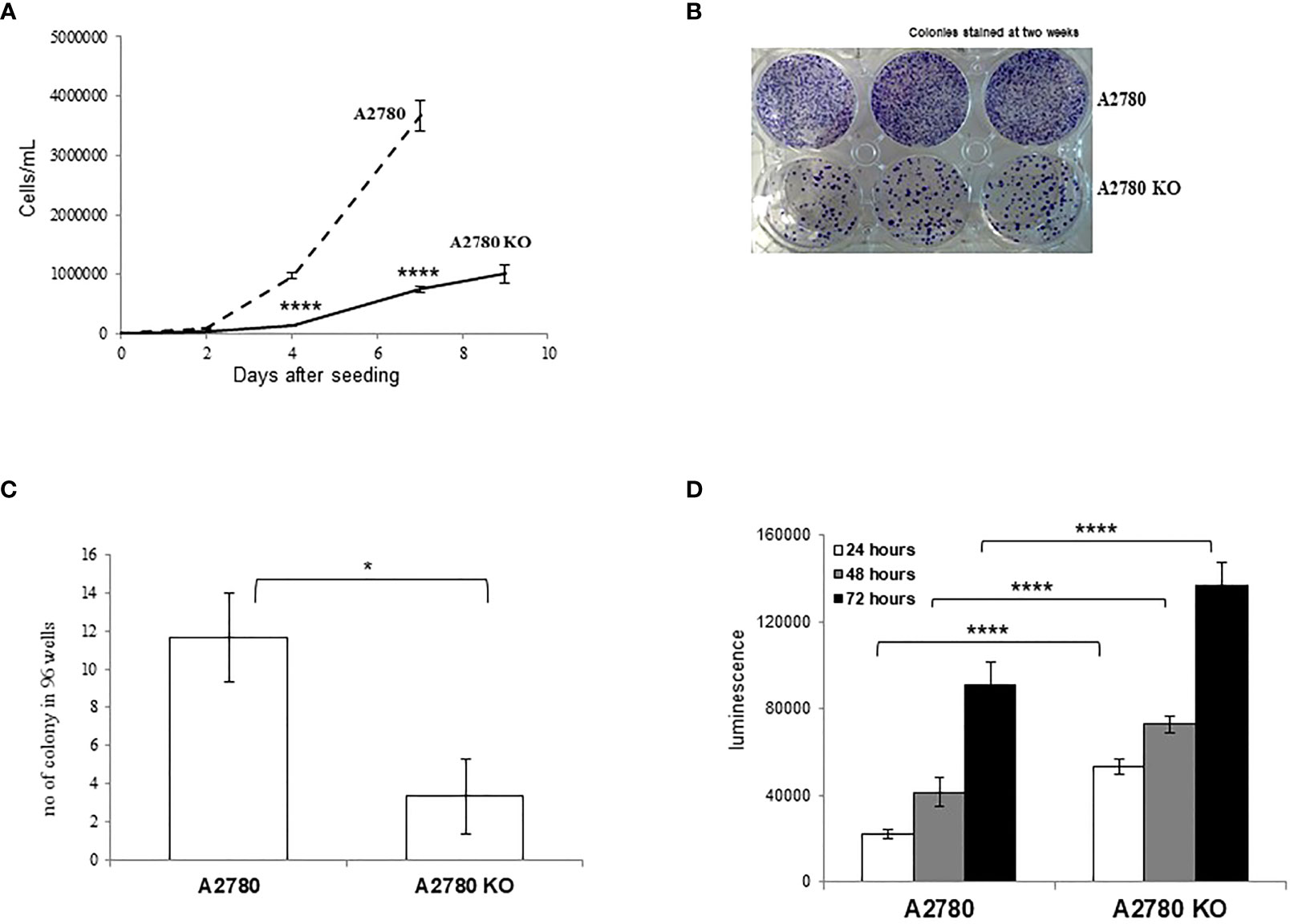
Figure 2 Biological characterization of A2780 and A2780 CDK12 KO cells. (A) Growth curves for cells/mL (— A2780 cells; ---- A2780 CDK12 KO cells) at different times from seeding. Data are the mean ± SD of three replicates and each experiment was performed three times. t-test was used for statistical analysis (****p < 0.0001). (B) Colony assays performed in six-wells plates of CDK12 WT (upper wells) and KO (lower wells) cell lines. Cells were stained two weeks after seeding. The results shown are representative of three independent experiments. (C) Number of colonies in 96-wells plates. Data are the mean ± SD of three replicates done three times (*= p<0.0082). (D) Apoptotic signals 24 (white), 48 (grey) and 72 (black) hours from the seeding. Data are the mean ± SD of six replicates done three times. For statistical analysis, two-way ANOVA, followed by Tukey’s test was used to compare CDK12 KO with the WT cell line at each time (****p < 0.0001).
A2780 and CDK12 KO cells were analyzed for their DNA content and distribution in the different phases of the cell cycle. A2780 and A2780 CDK12 KO cells were regularly distributed in the different phases of the cell cycle (Figure 3A). However, A2780 CDK12 KO cells had a twice DNA content as compared to parental cells.
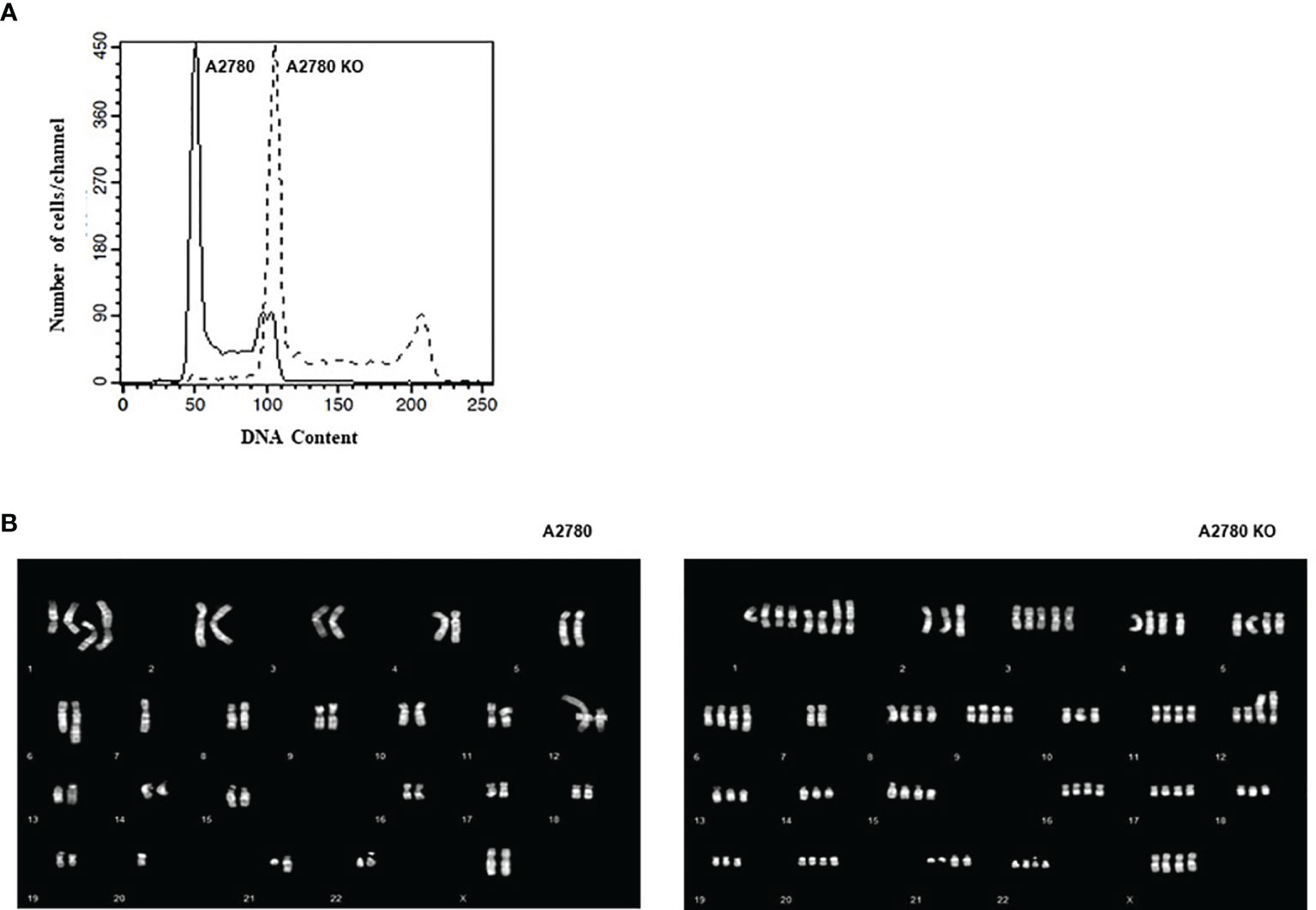
Figure 3 (A) Flow cytometric analysis of DNA content of the cell lines. –: CDK12 WT cell lines, - - : CDK12 KO cell lines. (B) Composite karyotypes of A2780 cell line (left panel) and of A2780 CDK12 KO (right panel).
Considering the recent report of an association of bi-allelic inactivation of CDK12 with tandem duplication (17), we analyzed the karyotype of the cells to investigate the number and structure of the chromosomes. The composite reconstructions of WT and KO karyotypes (Figure 3B) showed that structural rearrangements of A2780 were maintained in A2780 CDK12 KO cells, but duplicated. The A2780 cell line (upper panel) showed a modal number of 46 chromosomes, ranging from 44 to 47 corresponding to a diploid DNA content. Its composite karyotype is 44~47,XX,-1x2,+der (1) dup (1q),+del(1)(p36.3p32),+der(1)t(1);?(q11);?x2,-4,der(6)t(1;6)(q21.2;q22),7, der(7)add(7)(q32), der (12)t(?;12),del(14)(q23),der(21)add(21)(q22). The A2780 CDK12 KO cells line had a modal number of 90 chromosomes, ranging from 88 to 95 (lower panel), in line with the doubled DNA content shown by FACS analysis. Its composite karyotype is 88~95,XXXX,-1x4, +der(1)dup(1q)x3, +del(1)(p36.3p32)x2,+der(1)t(1);?(q11);?x4,+3,-4,+4,+5,der(6)t(1;6)(q21.2;q22)x2,-7x2,der(7) add (7) (q32) x 2, -10, der (12) t (?;12) x 2, -13x2, der (13;14)(q10;q10),-14,del(14)(q23)x2,-17,-18,-19,-21, der (21)add(21)(q22)x2.
CDK12 KO Effect on Tumor Growth In Vivo
We transplanted parental and CDK12 KO cells in nude mice and examined their tumor take and tumor growth. The A2780 KO tumor lag was much longer than A2780, all the mice having a palpable tumor by day 135, while all the animals transplanted with A2780 cells were dead with tumors by day 25 (Table 1). Tumor growth rates of A2780 CDK12 KO cells were slightly lower than A2780 cells (Supplementary Figure 2).

Table 1 No of mice tumor/no of transplanted mice with A2780 and CDK12 KO A2780 cells.
CDK12 KO Effect on Invasiveness
CDK12 was reported to modulate the alternative splicing of specific genes, increasing the tumorigenicity and aggressiveness of breast cancer cells (30). We therefore investigated whether CDK12 KO cells showed a difference in migrating from their parental cells. In chemotaxis experiments (Supplementary Figure 3) there was no difference in migration between parental and CDK12 KO clone. In wound healing assay, we found no differences in the ability to repair the wound in CDK12 KO cells compared to the parental cells (data not shown).
CDK12 KO Effect on DNA Damage and Repair
CDK12 down-regulation has been reported to be associated with lower mRNA and protein levels of DNA repair genes (4). We investigated the levels of a number of DNA damage genes in CDK12 KO cells compared to those in parental cells. Figure 4A shows the relative mRNA levels of some of the genes acting in DNA damage and repair, expressed as the fold of increase over the parental cells. There was a partial downregulation of BRCA1, CHK1, and WEE1 in A2780 KO cells, while PARP1 was downregulated. Protein levels of the same genes (Figures 4B, D) assessed by western blot, confirmed the downregulation of PARP1, while no significant change of BRCA1, CHK1, and WEE1 protein levels. We then checked for the level of endogenous DNA damage evaluating the activation of H2AX, a bone fide marker of this. No increase in p-Ser139 H2AX was detected in CDK12 KO cells compared to the WT cells, suggesting no greater DNA damage in CDK12 KO cells (Figure 4C).
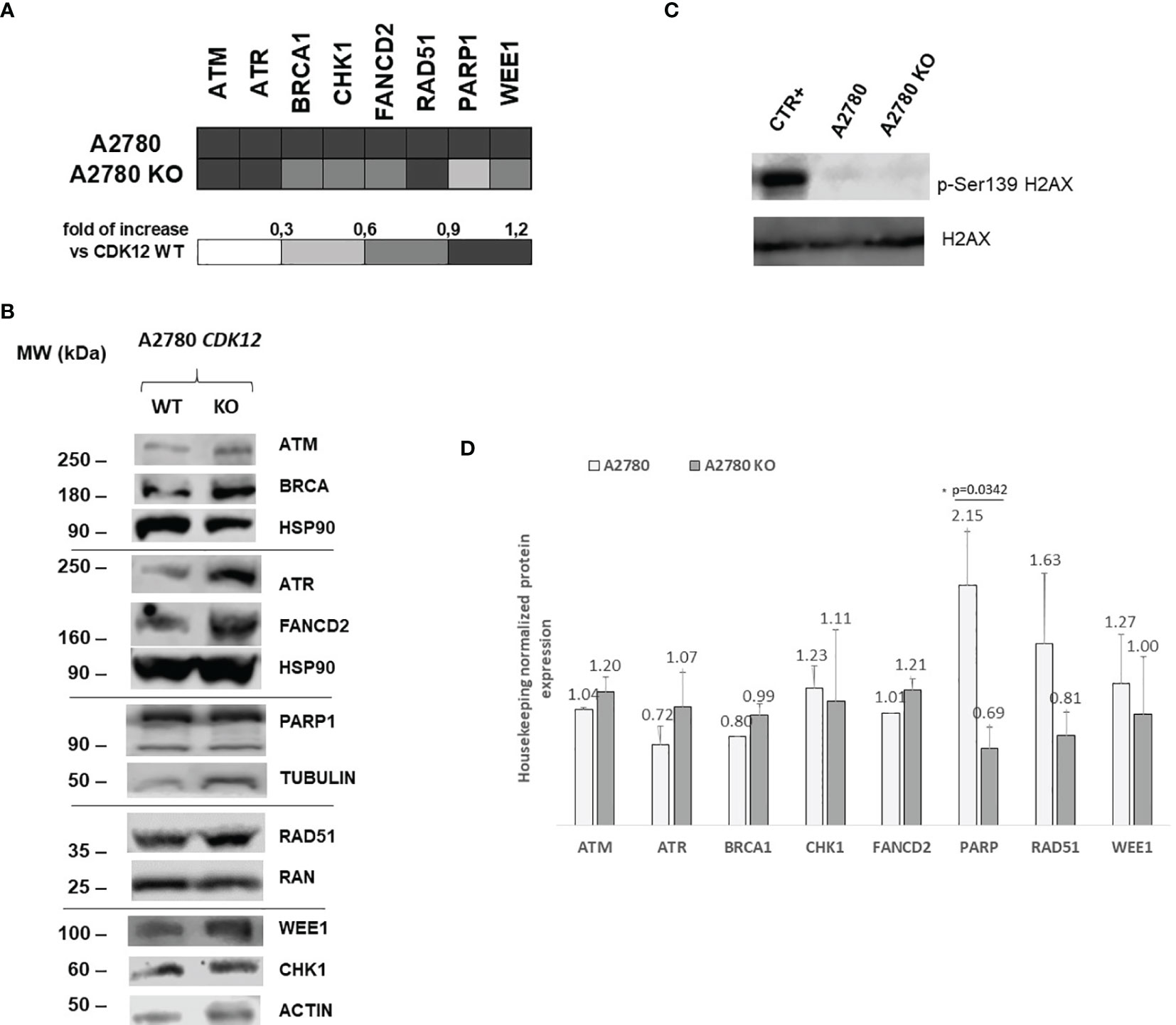
Figure 4 DNA repair gene expression in A2780 and A2780 CDK12 KO cells. (A) mRNA levels of ATM, ATR, BRCA1, CHK1, FANCD2, RAD51, PARP1, WEE1 in A2780 and A2780 CDK12 KO cell lines obtained by RT-PCR. Values are the mean+SD of three independent experiments, done in triplicates and are expressed as the fold of change over the CDK12 WT cell line. (B) Western blot analysis showing ATM, HSP90, ATR, FANCD2, BRCA1, PARP1, RAD51, β tubulin, CHK1, WEE1, ACTIN, p-Ser139 H2AX, H2AX protein levels in A2780 and A2780 CDK12 KO cells lines. All the protein blots for western analysis have been cropped before antibody hybridization to be able to detect in the same filter different proteins. (C) Western blot analysis showing p-Ser139 H2AX, H2AX protein levels in A2780 and A2780 CDK12 KO cells. All the protein blots for western analysis have been cropped before antibody hybridization to be able to detect in the same filter different proteins. (D) Quantification of the western blot data by densitometric analyses. Values are normalized by actin and are the mean+SD of three replicates.*p = 0.0342.
Considering the reported role of the cyclin K/CDK12 complex in eukaryotic gene expression and to gain a broader picture of changes in gene expression, we compared the baseline gene expression profiles in KO versus WT cells, as described in Materials and Methods. In CDK12 KO cells, 6.8% of genes were up-regulated in A2780 KO and 7.6% were down-regulated (Supplementary Table 3). We did not find the reported (4) enrichment in DNA repair genes in A2780 CDK12 KO cells. However, among the up-regulated genes there was significant enrichment of genes involved in apoptosis, in agreement with the experimental data (Figure 2D) (Supplementary Table 4).
CDK12 KO Effect on Sensitivity to Anticancer Drugs
Recent reports suggest that cells with CDK12 mutations and/or transient downregulation with small interference RNA are more sensitive to alkylating agents, PARP inhibitors and irinotecan (11, 12, 31). We pharmacologically characterized A2780 CDK12 KO cells compared to parental cells using agents with different mechanisms of action; DNA interfering agents: cisplatin (DDP), ET-743, olaparib (OLA); antitubulin agents: paclitaxel (PTX), and agents targeting the DNA damage response pathway: ATR inhibitor (VE822), ATM inhibitor (KU55933), CHK1 inhibitor (PF477736), WEE1 inhibitor (MK1775) and CDK7/CDK12 inhibitors (THZ1 and THZ1 HYDRO). Contrary to some reports, KO clones were not more sensitive to DDP and OLA; the patterns of sensitivity were also similar with ET-743, PTX, KU55933, PF477736 and MK1775. CDK12 KO cells showed the same sensitivity to THZ1 and THZ1 HYDRO as WT cells (Figure 5). Interestingly, CDK12 KO cells were five fold more resistant to the ATR inhibitor VE822 with an IC50 value of 1.99 + 0.81μM versus 0.39 + 0.07 μM (Figure 5; Supplementary Table 5).
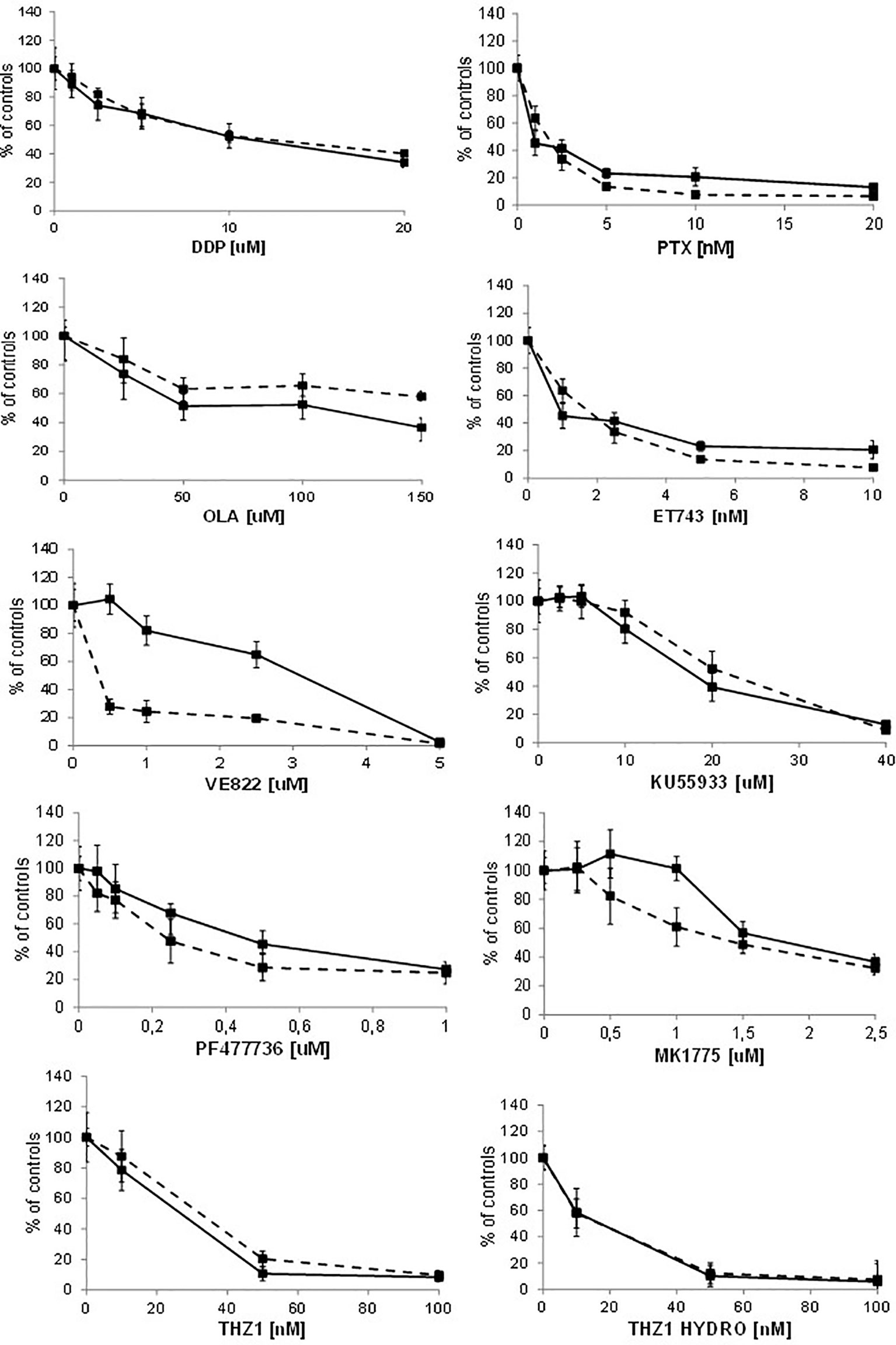
Figure 5 Drug dose-response curves. Dose-response curves of A2780 - - -■- - - and A2780 KO ▬▬■▬▬ to cisplatin, paclitaxel, olaparib, ET-743, VE822, KU55933, PF477736, MK1775, THZ1, THZ1 hydrocloride. Data are expressed as survival percentages over controls (+ standard error) of least three experiments done in quintuplicate.
To see whether the cells’ sensitivities to DDP and VE822 were due to some changes in apoptosis, we ran an apoptosis test 24, 48 and 72 hours after treatment with different drug concentrations. Supplementary Figure 4 shows the apoptotic levels of treated and untreated A2780 and A2780 CDK12 KO cells. There were no differences in DDP-induced apoptosis between the WT and CDK12 KO cells, while VE822 caused weaker induction of apoptosis in KO than in WT cells. These data suggest that the lower sensitivity of A2780 CDK12 KO to VE822 than A2780 may be partly due to a lower induction of apoptosis.
Discussion
Cyclin-dependent kinase 12 (CDK12) has been reported to favor the maintenance of genomic stability as it promotes the transcription of a subset of genes involved in the DNA damage response (4, 5). CDK12 inactivation causes downregulation of genes involved in DNA repair (10, 11) and increases sensitivity to platinum agents and PARP inhibitors (12, 13). Mutations in CDK12 have been reported in different tumor types (7, 8) and it is one of the most mutated genes in ovarian carcinoma (9). Recently, we reported that CDK12 mRNA levels were predictive of platinum response in a xenobank of patient-derived ovarian carcinomas (32). To understand CDK12’s role in tumor growth and response to therapy better, we here generated ovarian cancer cells knocked out for CDK12.
The very low rate of success in obtaining CDK12 KO clones using the effective CRISPR/Cas9 tool suggests CDK12 is important for cell viability (5, 28). To the best of our knowledge, no stable CDK12 KO cells have been described; only an analog sensitive CDK12 cellular system has been published (28), a single HeLa cell clone in which the only functional copy of CDK12 was selectively inhibited by a cell-permeable adenine analog. In this system, CDK12 inhibition resulted in an arrest in cell proliferation and perturbation of the phosphorylation pattern of RNA polymerase II CTD. The cell growth of CDK12 KO cells confirmed this, as the lack of CDK12 delayed cell growth and reduced the clonogenic ability of the CDK12 KO cells. The data were also partially confirmed in vivo, where A2780 KO tumors were as tumorigenic as A2780 cells. However, their time lags were almost 9-13 times those of A2780 cells, and their growth rates were slightly lower than the parental cells. These data seem to contrast with the reported evidence of CDK12 as tumor suppressor. However, as recently suggested (33), if important events in oncogenesis depend on the loss of CDK12 catalytic activity leading, among others to deregulation of DDR, we did not observed in our experimental setting an impairment of response to damage in CDK12 KO cells. As regards CDK12’s implication in tumor invasiveness and aggressiveness (30), we did not find any differences in migration capacity between WT and KO clones.
The absence of the CDK12 protein is not associated with any clear decrease in RNA pol II p-Ser2 phosphorylation, probably because CDK9 and CDK13 levels-which induce the same phosphorylation- were unchanged (34). Of note, CDK13 is evolutionarily and structurally related to CDK12 kinase, sharing the same cognate cyclin K and biochemical assays reported similar kinase activity and phosphorylation of Ser2 of the CTD heptad sequence (33). Even some studies support gene selective modulation and non-overlapping function for CDK12 and CDK13, evidence have recently been put forward on a significant redundancy between these two kinases and on the fact that both are key regulators of global POLII processivity (35). There was a slight decrease in cyclin K levels, never reaching statistical significance in CDK12 KO cells. The pre-replicative assembly complex (licensing) is a requisite for DNA replication and sustains cell proliferation (36). Cyclin K is essential to promote efficient licensing in G1 phase and CDK12 knockdown prevents the pre-replicative complex assembly in G1 (37). This might explain a DNA replication defect in CDK12 KO cells, leading to slower growth.
Contributing to the slower growth in CDK12 KO cells is the significant downregulation of genes involved in the G2/M checkpoint observed with gene expression analysis, and the higher basal level of apoptosis in CDK12 KO than WT cells. In A2780 CDK12 KO cells we did not find any increase in DNA damage, as demonstrated by the comparable levels of γH2AX phosphorylation in contrast to the reports in reduced licensing conditions (38) and in CDK12-null embryos (4, 5), suggesting a more complex picture.
CDK12 KO cells have double the DNA content of WT cells, even with regular distribution in the cell cycle, with doubling of the chromosomes and retention of the structural rearrangements seen in the parental WT cells. It has been recently demonstrated that ovarian cancers with inactivating mutations of CDK12 present genomic instability characterized by hundreds of tandem duplications of up to ten megabases (MB) in size (CDK12 TD-plus phenotype) (17). In prostate cancer, the bi-allelic inactivation of CDK12 is associated with a large number of focal copy-number gains dispersed across the genome (39). This phenotype was mutually exclusive with BRCA1/2 mutations (germline and/or somatic) and with BRCA1 promoter methylation and was not associated with genomic homologous recombination deficiency, partially contrasting with the data suggesting a decrease in the expression of genes involved in homologous recombination repair (4, 40). It was also reported that large genes (including the DNA repair genes) were not markedly abundant among the downregulated ones.
Given the role of CDK12 in regulating the expression of genes involved in DNA repair, we investigated the levels of some of these genes in our CDK12 KO cells. There was a slight tendency to mRNA downregulation of some genes involved in DNA repair (i.e. BRCA1, CHK1, WEE1 and PARP1) not always supported by a downregulation at protein level. This apparently inconsistency could be due to other level of regulation at protein level, such as post-translation modifications interfering with their stability (41). As we focused on a limited number of DNA repair genes (those whose mRNAs were downregulated in CDK12 transient KO conditions), we might not have captured the whole spectrum of altered transcription. However, a wide gene expression analysis indicated a certain proportion of deregulated genes (both up- and down-regulated), but we did not find any clear enrichment in DNA repair genes among the downregulated transcripts as reported elsewhere (4, 40). However, the fact that CDK12 KO cells have double DNA content suggests that the two-fold increase in DNA repair genes copy number could potentially lead to an increase in their transcripts, even in the absence of CDK12. A detailed gene copy number analysis coupled with transcriptome of the clones would help clarify this issue.
Although the biological and molecular characterization of our CDK12 KO clones is compatible with the known function of CDK12 in cells, we could not find any differences in the sensitivity of CDK12 KO cells to cisplatin, PARP inhibitor olaparib, ET-743, KU55933 (an ATM inhibitor) and paclitaxel compared to WT cells. The ATR inhibitor VE822 was less active in CDK12 KO clones than in WT cells.
These results partially contrast with reports of increased sensitivity to platinum-based drugs and olaparib in cells transiently transfected with shRNA and siRNA against CDK12 (11, 31, 42). Possible explanations can be offered: i) the experimental settings are quite different, as our CDK12 KO cells had a stable, not transient, loss of CDK12, and may have developed some mechanisms of adaptation to survive cells; ii) given the functional redundancy between CDK12 and CDK13 (35), it could be that CDK13 can compensate for CDK12 loss; iii) our CDK12 KO cells have double the DNA content and no significant decrease in DDR proteins. The lower sensitivity to ATR inhibitor in CDK12 KO cells - partly explained by the less induction of apoptosis - would be due to the fact that these cells are less dependent on the ATR-CHK1 axis. Finally, the similar sensitivity to the reported CDK12 inhibitors in A2780 CDK12 KO cells would suggest that these inhibitors are not specific for CDK12.
In conclusion, here we report for the first time a CDK12 null cell line. We realize that all the data here rely on only one clone, but nevertheless all are compatible with the known function of CDK12 and support its role for survival in tumor cells too. CDK12 KO cells showed a duplication of DNA, with doubling of the chromosomal number, corroborating the role of CDK12 in genomic maintenance. CDK12 KO cells had reduced cell growth, no increase in endogenous DNA damage levels and no major downregulation of DNA repair genes, reported to be downregulated in transient CDK12 KO cells. The lack of DDR gene downregulation might be partially explained by the doubling of DNA content and could justify the lack of increase sensitivity to platinum and PARPi. Detailed genome molecular study coupled with a global gene expression profile will help us understand better what kind of selection this KO clone has undergone and explain the differences from the transient CDK12 downregulation experimental setting.
Data Availability Statement
All data will be available at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) database; GSE183041.
Ethics Statement
The animal study was reviewed and approved by the Italian Ministry of Health Institutional Review Board. The IRB also approved all the in vivo experiments performed with PDXs (Authorization n.705/2016-PR).
Author Contributions
RC performed research, collected data, analysed and interpreted data, performed statistical analyses and wrote the manuscript; MC and FG performed the western blots, interpret the data and performed statistical analyses; NP collected data and analysed and interpreted data; DC performed research, analysed and interpreted data; AR and LC performed research, analysed and interpreted data, performed statistical analysis; FB and MF analysed and interpreted data, performed statistical analysis and wrote the manuscript; GD designed experiments, analysed and interpreted data; wrote and reviewed the manuscript. All authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The research was supported by a grant from the The Italian Association for Cancer Research (GD IG 19797).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The generous contribution of AIRC (The Italian Association for Cancer Research) is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.903536/full#supplementary-material
References
1. Lim S, Kaldis P. Cdks, Cyclins and CKIs: Roles Beyond Cell Cycle Regulation. Development (2013) 140(15):3079–93. doi: 10.1242/dev.091744
2. Cheng SW, Kuzyk MA, Moradian A, Ichu TA, Chang VC, Tien JF, et al. Interaction of Cyclin-Dependent Kinase 12/CrkRS With Cyclin K1 Is Required for the Phosphorylation of the C-Terminal Domain of RNA Polymerase II. Mol Cell Biol (2012) 32(22):4691–704. doi: 10.1128/MCB.06267-11
3. Liang K, Gao X, Gilmore JM, Florens L, Washburn MP, Smith E, et al. Characterization of Human Cyclin-Dependent Kinase 12 (CDK12) and CDK13 Complexes in C-Terminal Domain Phosphorylation, Gene Transcription, and RNA Processing. Mol Cell Biol (2015) 35(6):928–38. doi: 10.1128/MCB.01426-14
4. Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, et al. The Cyclin K/Cdk12 Complex Maintains Genomic Stability via Regulation of Expression of DNA Damage Response Genes. Genes Dev (2011) 25(20):2158–72. doi: 10.1101/gad.16962311
5. Juan HC, Lin Y, Chen HR, Fann MJ. Cdk12 is Essential for Embryonic Development and the Maintenance of Genomic Stability. Cell Death Differ (2016) 23(6):1038–48. doi: 10.1038/cdd.2015.157
6. Dai Q, Lei T, Zhao C, Zhong J, Tang YZ, Chen B, et al. Cyclin K-Containing Kinase Complexes Maintain Self-Renewal in Murine Embryonic Stem Cells. J Biol Chem (2012) 287(30):25344–52. doi: 10.1074/jbc.M111.321760
7. Chila R, Guffanti F, Damia G. Role and Therapeutic Potential of CDK12 in Human Cancers. Cancer Treat Rev (2016) 50:83–8. doi: 10.1016/j.ctrv.2016.09.003
8. Paculova H, Kramara J, Simeckova S, Fedr R, Soucek K, Hylse O, et al. BRCA1 or CDK12 Loss Sensitizes Cells to CHK1 Inhibitors. Tumour Biol (2017) 39(10):1010428317727479. doi: 10.1177/1010428317727479
9. Cancer Genome Atlas Research N. Integrated Genomic Analyses of Ovarian Carcinoma. Nature (2011) 474(7353):609–15. doi: 10.1038/nature10166
10. Ekumi KM, Paculova H, Lenasi T, Pospichalova V, Bosken CA, Rybarikova J, et al. Ovarian Carcinoma CDK12 Mutations Misregulate Expression of DNA Repair Genes via Deficient Formation and Function of the Cdk12/CycK Complex. Nucleic Acids Res (2015) 43(5):2575–89. doi: 10.1093/nar/gkv101
11. Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian Cancer-Associated Mutations Disable Catalytic Activity of CDK12, A Kinase That Promotes Homologous Recombination Repair and Resistance to Cisplatin and Poly(ADP-Ribose) Polymerase Inhibitors. J Biol Chem (2014) 289(13):9247–53. doi: 10.1074/jbc.M114.551143
12. Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res (2014) 74(1):287–97. doi: 10.1158/0008-5472.CAN-13-2541
13. Natrajan R, Wilkerson PM, Marchio C, Piscuoglio S, Ng CK, Wai P, et al. Characterization of the Genomic Features and Expressed Fusion Genes in Micropapillary Carcinomas of the Breast. J Pathol (2014) 232(5):553–65. doi: 10.1002/path.4325
14. Dubbury SJ, Boutz PL, Sharp PA. CDK12 Regulates DNA Repair Genes by Suppressing Intronic Polyadenylation. Nature (2018) 564(7734):141–5. doi: 10.1038/s41586-018-0758-y
15. Krajewska M, Dries R, Grassetti AV, Dust S, Gao Y, Huang H, et al. CDK12 Loss in Cancer Cells Affects DNA Damage Response Genes Through Premature Cleavage and Polyadenylation. Nat Commun (2019) 10(1):1757. doi: 10.1038/s41467-019-09703-y
16. Bayles I, Krajewska M, Pontius WD, Saiakhova A, Morrow JJ, Bartels C, et al. Ex Vivo Screen Identifies CDK12 as a Metastatic Vulnerability in Osteosarcoma. J Clin Invest (2019) 129(10):4377–92. doi: 10.1172/JCI127718
17. Popova T, Manie E, Boeva V, Battistella A, Goundiam O, Smith NK, et al. Ovarian Cancers Harboring Inactivating Mutations in CDK12 Display a Distinct Genomic Instability Pattern Characterized by Large Tandem Duplications. Cancer Res (2016) 76(7):1882–91. doi: 10.1158/0008-5472.CAN-15-2128
18. Capra M, Nuciforo PG, Confalonieri S, Quarto M, Bianchi M, Nebuloni M, et al. Frequent Alterations in the Expression of Serine/Threonine Kinases in Human Cancers. Cancer Res (2006) 66(16):8147–54. doi: 10.1158/0008-5472.CAN-05-3489
19. Kauraniemi P, Kuukasjarvi T, Sauter G, Kallioniemi A. Amplification of a 280-Kilobase Core Region at the ERBB2 Locus Leads to Activation of Two Hypothetical Proteins in Breast Cancer. Am J Pathol (2003) 163(5):1979–84. doi: 10.1016/S0002-9440(10)63556-0
20. Naidoo K, Wai PT, Maguire SL, Daley F, Haider S, Kriplani D, et al. Evaluation of CDK12 Protein Expression as a Potential Novel Biomarker for DNA Damage Response-Targeted Therapies in Breast Cancer. Mol Cancer Ther (2018) 17(1):306–15. doi: 10.1158/1535-7163.MCT-17-0760
21. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature (2012) 487(7406):239–43. doi: 10.1038/nature11125
22. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell (2015) 161(5):1215–28. doi: 10.1016/j.cell.2015.06.053
23. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational Landscape of Metastatic Cancer Revealed From Prospective Clinical Sequencing of 10,000 Patients. Nat Med (2017) 23(6):703–13. doi: 10.1038/nm.4333
24. Restelli V, Vagni M, Arribas AJ, Bertoni F, Damia G, Carrassa L. Inhibition of CHK1 and WEE1 as a New Therapeutic Approach in Diffuse Large B Cell Lymphomas With MYC Deregulation. Br J Haematol (2018) 181(1):129–33. doi: 10.1111/bjh.14506
25. Mensah AA, Spriano F, Sartori G, Priebe V, Cascione L, Gaudio E, et al. Study of the Antilymphoma Activity of Pracinostat Reveals Different Sensitivities of DLBCL Cells to HDAC Inhibitors. Blood Adv (2021) 5(10):2467–80. doi: 10.1182/bloodadvances.2020003566
26. Guffanti F, Alvisi MF, Caiola E, Ricci F, De Maglie M, Soldati S, et al. Impact of ERCC1, XPF and DNA Polymerase Beta Expression on Platinum Response in Patient-Derived Ovarian Cancer Xenografts. Cancers (Basel) (2020) 12(9):2398–3411. doi: 10.3390/cancers12092398
27. Cascione L, Rinaldi A, Bruscaggin A, Tarantelli C, Arribas AJ, Kwee I, et al. Novel Insights Into the Genetics and Epigenetics of MALT Lymphoma Unveiled by Next Generation Sequencing Analyses. Haematologica (2019) 12, e558–61. doi: 10.3324/haematol.2018.214957
28. Bartkowiak B, Yan C, Greenleaf AL. Engineering an Analog-Sensitive CDK12 Cell Line Using CRISPR/Cas. Biochim Biophys Acta (2015) 1849(9):1179–87. doi: 10.1016/j.bbagrm.2015.07.010
29. Choi SH, Kim S, Jones KA. Gene Expression Regulation by CDK12: A Versatile Kinase in Cancer With Functions Beyond CTD Phosphorylation. Exp Mol Med (2020) 52(5):762–71. doi: 10.1038/s12276-020-0442-9
30. Tien JF, Mazloomian A, Cheng SG, Hughes CS, Chow CCT, Canapi LT, et al. CDK12 Regulates Alternative Last Exon mRNA Splicing and Promotes Breast Cancer Cell Invasion. Nucleic Acids Res (2017) 45(11):6698–716. doi: 10.1093/nar/gkx187
31. Quereda V, Bayle S, Vena F, Frydman SM, Monastyrskyi A, Roush WR, et al. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell (2019) 36(5):545–58.e7. doi: 10.1016/j.ccell.2019.09.004
32. Guffanti F, Fratelli M, Ganzinelli M, Bolis M, Ricci F, Bizzaro F, et al. Platinum Sensitivity and DNA Repair in a Recently Established Panel of Patient-Derived Ovarian Carcinoma Xenografts. Oncotarget (2018) 9(37):24707–17. doi: 10.18632/oncotarget.25185
33. Greenleaf AL. Human CDK12 and CDK13, Multi-Tasking CTD Kinases for the New Millenium. Transcription (2019) 10(2):91–110. doi: 10.1080/21541264.2018.1535211
34. Franco LC, Morales F, Boffo S, Giordano A. CDK9: A Key Player in Cancer and Other Diseases. J Cell Biochem (2018) 119(2):1273–84. doi: 10.1002/jcb.26293
35. Fan Z, Devlin JR, Hogg SJ, Doyle MA, Harrison PF, Todorovski I, et al. CDK13 Cooperates With CDK12 to Control Global RNA Polymerase II Processivity. Sci Adv (2020) 6(18):eaaz5041. doi: 10.1126/sciadv.aaz5041
36. Falaschi A, Abdurashidova G, Biamonti G. DNA Replication, Development and Cancer: A Homeotic Connection? Crit Rev Biochem Mol Biol (2010) 45(1):14–22. doi: 10.3109/10409230903365608
37. Lei T, Zhang P, Zhang X, Xiao X, Zhang J, Qiu T, et al. Cyclin K Regulates Prereplicative Complex Assembly to Promote Mammalian Cell Proliferation. Nat Commun (2018) 9(1):1876. doi: 10.1038/s41467-018-04258-w
38. McIntosh D, Blow JJ. Dormant Origins, the Licensing Checkpoint, and the Response to Replicative Stresses. Cold Spring Harb Perspect Biol (2012) 4(10):a012955. doi: 10.1101/cshperspect.a012955
39. Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell (2018) 173(7):1770–82.e14. doi: 10.1016/j.cell.2018.04.034
40. Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, et al. Covalent Targeting of Remote Cysteine Residues to Develop CDK12 and CDK13 Inhibitors. Nat Chem Biol (2016) 12(10):876–84. doi: 10.1038/nchembio.2166
41. Keenan EK, Zachman DK, Hirschey MD. Discovering the Landscape of Protein Modifications. Mol Cell (2021) 81(9):1868–78. doi: 10.1016/j.molcel.2021.03.015
Keywords: CDK12, ovarian carcinoma, PARP inhibitor, cisplatin, DNA damage
Citation: Chilà R, Chiappa M, Guffanti F, Panini N, Conconi D, Rinaldi A, Cascione L, Bertoni F, Fratelli M and Damia G (2022) Stable CDK12 Knock-Out Ovarian Cancer Cells Do Not Show Increased Sensitivity to Cisplatin and PARP Inhibitor Treatment. Front. Oncol. 12:903536. doi: 10.3389/fonc.2022.903536
Received: 25 March 2022; Accepted: 24 May 2022;
Published: 13 July 2022.
Edited by:
Arianna Palladini, University of Pavia, ItalyReviewed by:
Kamal Eltayeb, University of Parma, ItalyCarmen Blanco Aparicio, Spanish National Cancer Research Center (CNIO), Spain
Copyright © 2022 Chilà, Chiappa, Guffanti, Panini, Conconi, Rinaldi, Cascione, Bertoni, Fratelli and Damia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanna Damia, Z2lvdmFubmEuZGFtaWFAbWFyaW9uZWdyaS5pdA==
†Present address: Rosaria Chilà, IFOM-the FIRC Institute of Molecular Oncology, Milan, Italy
‡These authors have contributed equally to this work