 Yihui Gu1†
Yihui Gu1† Wei Wang1†
Wei Wang1† Yuehua Li1†
Yuehua Li1† Haibo Li2†
Haibo Li2† Zizhen Guo1
Zizhen Guo1 Chengjiang Wei1
Chengjiang Wei1 Manmei Long3
Manmei Long3 Manhon Chung1
Manhon Chung1 Rehanguli Aimaier1
Rehanguli Aimaier1 Qingfeng Li1*
Qingfeng Li1* Zhichao Wang1*
Zhichao Wang1*- 1Department of Plastic and Reconstructive Surgery, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Plastic Surgery, The Third Xiangya Hospital, Central South University, Changsha, China
- 3Department of Pathology, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Malignant peripheral nerve sheath tumors (MPNSTs) are rare soft-tissue sarcomas refractory to standard therapies. Inactivation of NF1 and subsequent upregulation of RAS/RAF/MEK/ERK signaling exist in the majority of MPNSTs. However, the lack of preclinical assessment of MEK inhibitors in MPNSTs hinders the clinical application as well as the development of combination therapy. To guide further clinical studies, we evaluated different MEK inhibitors in terms of efficacy, safety, and mechanism of adaptive response in treating MPNSTs. Using a MPNST tissue microarray, we found that p-ERK could serve as a biomarker for predicting the prognosis of MPNST patients as well as an effective therapeutic target. Through in vitro and in vivo experiments, we identified trametinib as the most potent MEK inhibitor for the treatment of MPNSTs. Mechanistically, reduced reactivation of the MAPK pathway and compensatory activation of the parallel pathways contributed to better efficacy. Our results provide a basis for the further clinical application of MEK inhibitors as single agents or combinational therapies.
Introduction
Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive soft tissue sarcomas arising in patients with neurofibromatosis type 1(NF1) or sporadically. MPNSTs are refractory to standard therapies (1, 2), and extended resection is currently the only potential curative management for MPNSTs, which is not suitable for advanced MPNST patients. In addition, the local recurrence rate remains high after surgical resection and traditional chemo/radio therapies show mild benefits (3–6). The overall 5-year survival rate has remained poor at only 16-52% for decades, mainly owing to the lack of effective treatments. Therefore, there is an unmet clinical demand to discover novel therapeutic strategies, particularly targeted therapies, to treat advanced MPNSTs.
Genomic profiling of MPNST cohorts revealed that mutation in Neurofibromin 1(NF1) occurred in 100% of NF1-associated MPNSTs and 82% of other types of MPNSTs. As a RAS GTPase–activating protein, inactivation of NF1 leads to uncontrolled activation of RAS and subsequent upregulation of downstream signaling pathways including mitogen-activated protein kinase (MAPK), PI3K/Akt, and Ral-GEF pathways (7–12). Among these dysregulated signaling contributing to the occurrence and progression of MPNSTs, MAPK is the most well-studied signaling pathway which is critical for cell proliferation, differentiation, and survival, suggesting that this pathway is a promising therapeutic target in MPNST treatment. In addition, MAPK signaling pathway is also an effective therapeutic target for a variety of human cancers, including melanoma and NSCLC (13–15).
MEK inhibitors, such as trametinib and cobimetinib, are highly specific, non-ATP competitive, small-molecule inhibitors of MEK1/2. MEK inhibitors have shown promising therapeutic efficacy in RAS-driven tumors including plexiform neurofibroma (pNF), the precancerous lesion of MPNSTs (13, 16–20). In clinical trials, the MEK inhibitor selumetinib led to clinical responses in up to 70% of children with inoperable pNF and subsequently gained FDA approval for this indication (17, 21). However, the efficacy of MEK inhibitors in the treatment of MPNSTs remains controversial. Currently, the research related to MEK inhibitors mainly focused on the development of effective combinational strategies. Several combinational therapies including MEK inhibitors in combination with mTOR inhibitors, MET inhibitors, and others were evaluated in preclinical studies and clinical trials (22–24). However, clinically sufficient combinational therapies still do not exist.
We found that the efficacy of different MEK inhibitors as single agents and the mechanism underlying the difference in treatment response are poorly investigated in preclinical studies. In addition, the low incidence of MPNSTs makes the recruitment of clinical trials extremely difficult. These deficiencies and challenges hinder the implications of MEK inhibitors in MPNST therapy. Therefore, a comprehensive preclinical evaluation of MEK inhibitors is necessary to guide further clinical studies.
In this study, we evaluated different MEK inhibitors in terms of efficacy, safety, and mechanism of adaptive response in treating MPNSTs. We assessed the expression and clinical relevance of MAPK pathway in MPNST patients using a tissue microarray. The efficacy and safety of different MEK inhibitors were evaluated in vivo and in vitro. In addition, the mechanism of adaptive response in 8 MEK inhibitors was also explored. This study provided a basis for the further clinical application of MEK inhibitors as single agents or combinational therapies.
Materials & Methods
Cell Lines and Reagents
The NF1+/- Schwann cell line (sNF02.3 2λ) was obtained from the American Type Culture Collection (ATCC). NF1-associated (ST88-14, T265, S462, and S462TY) and sporadic (STS-26T) MPNST lines were kindly granted by Prof. Vincent Keng and Prof. Jilong Yang (Table S1). All MPNST cell lines were maintained in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a 37°C, 5% CO2 incubator, sNF02.3 2λ cells were cultured under the same conditions with 1% L-glutamine added.
Reagents and antibodies used in this article are described in Supplementary Table S2.
Immunohistochemical Staining
A tissue microarray (TMA) was constructed using 57 paraffin-embedded MPNST specimens obtained from 49 patients who underwent surgical treatment at Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine in Shanghai, China. IHC staining was performed as previously described (25). The assessment of TMA IHC staining was accomplished by two qualified pathologists independently. The score was assigned according to the proportion of positive cells within carcinomatous areas so that no staining, <25% of malignant cells staining, 25%-50% of malignant cells staining, >50% of malignant cells staining were recorded as 0, 1, 2, 3. Clinical information was collected through electronic medical records and telephone follow-ups. Twenty-eight of 49 patients were lost to follow-up and were not able to be contacted through multiple methods.
Cell Line-Based Assays
A Cell Counting Kit-8 (CCK-8) assay was implemented to assess cytotoxicity. A total of 1*103 or 3*103 cells per well were seeded into 96-well culture plates and treated with 0.1% DMSO or the indicated drug. After 72 h or 96 h, 10 µL CCK-8 solution (Dojindo, Japan) dispersed in 90 µL DMEM was added per well to measure the 450 nm OD value after a 2 h incubation. Percentage cell viability was calculated as 100% × (OD of drug-treated cells - OD of background control)/(OD of untreated cells - OD of background control). The IC50 of indicated drugs was calculated by Prism 8.4.0 using [inhibitor] vs. normalized response – Variable slope analysis.
Annexin V–FITC and propidium iodide (PI) assays were implemented to detect cell apoptosis. MPNST cells were seeded into 6-well culture plates and treated with DMSO or the indicated MEKi for 24 h or 48 h. Afterwards, MPNST cells were stained with annexin V–FITC and PI at room temperature in the dark for 15 minutes, followed by analysis using a flow cytometer (CytoFLEX LX, Beckman Coulter, Shanghai) equipped with CytExpert software (Beckman Coulter, Shanghai).
A cell cycle assay was implemented to monitor the cell cycle. MPNST cells treated as described above were fixed with cold 70% ethanol, stained with PI/RNase solution, and analyzed on a flow cytometer as described above.
Western Blot Analysis
Cells were lysed in RIPA buffer (Beyotime, China) with protease and phosphatase inhibitor cocktails (Beyotime, China). Proteins separated by SDS-PAGE were transferred onto PVDF membranes and blocked in 3% bovine serum. Afterwards, membranes were incubated overnight in primary antibody solution at 4°C, followed by incubation with an HRP-conjugated secondary antibody for 1 h at room temperature. Band signals were detected using an Amersham Imager 600 (General Electric Company, Boston, MA, United States), and quantification was performed using ImageJ software after normalization to GAPDH.
Xenografts
Four-week-old male NOD-SCID IL-2 receptor gamma-null mice were purchased from Shanghai Model Organisms Co., Ltd. (Shanghai, China). A total of 5×106 S462TY cells suspended in 100 µL phosphate-buffered saline was engrafted subcutaneously into the armpit of each mouse. Tumor volume (based on caliper measurements) was calculated by the modified ellipsoidal formula: tumor volume = 1/2 length* width2. When tumors reached a volume of approximately 100 mm3, mice were randomly assigned to treatment with vehicle (n=4), trametinib (0.3 mg/kg, daily gavage, n=4), TAK-733 (2.4 mg/kg, daily gavage, n=4) or selumetinib (15.2 mg/kg, 2 times daily gavage, n=4). After 3 weeks of treatment, mice were sacrificed, and grafts were removed, weighed, and photographed. Grafts were stained with p-ERK, Ki-67, c-MYC, and cleaved Caspase 3 for histological analysis according to standard protocols.
Statistics
Data are presented as the mean ± standard error of the mean (SEM) or standard deviation (SD). Statistical analysis was conducted using Prism 8.4 (GraphPad Software, San Diego, CA). Statistical analyses were performed using the student’s t-test, the log-rank test, and the Pearson correlation analysis. P-values < 0.05 were considered to indicate statistical significance, and asterisks (*) are used to indicate significant differences between two specified groups. * indicates a P-value < 0.05, ** indicates a P-value < 0.01, *** indicates a P-value < 0.001.
Ethics Approval and Consent to Participate
The study was approved by the Ethics Committee of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, and informed consent was achieved from patients under institutional reviewer board protocols.
Results
High Expression of p-ERK Correlates With Dismal Prognosis in MPNSTs
To evaluate the expression of MAPK pathway in MPNSTs, we performed IHC staining of p-ERK using a TMA containing 57 MPNST specimens (Table S3). The levels of p-ERK in tumor tissues were classified as score 0 (negative, 20.8%), 1 (low positive, 28.3%), 2 (medium positive, 32.1%) or 3 (high positive, 18.9%) (Figure 1A). The expression of p-ERK was positively correlated with that of p-MEK, Ki-67, and CD34 (Table 1; Figures S1A–C). Analysis of clinicopathological correlations indicated that MPNST patients with high p-ERK expression had a shorter overall survival duration than the p-ERK low expression patients (p = 0.007, Figure 1B).
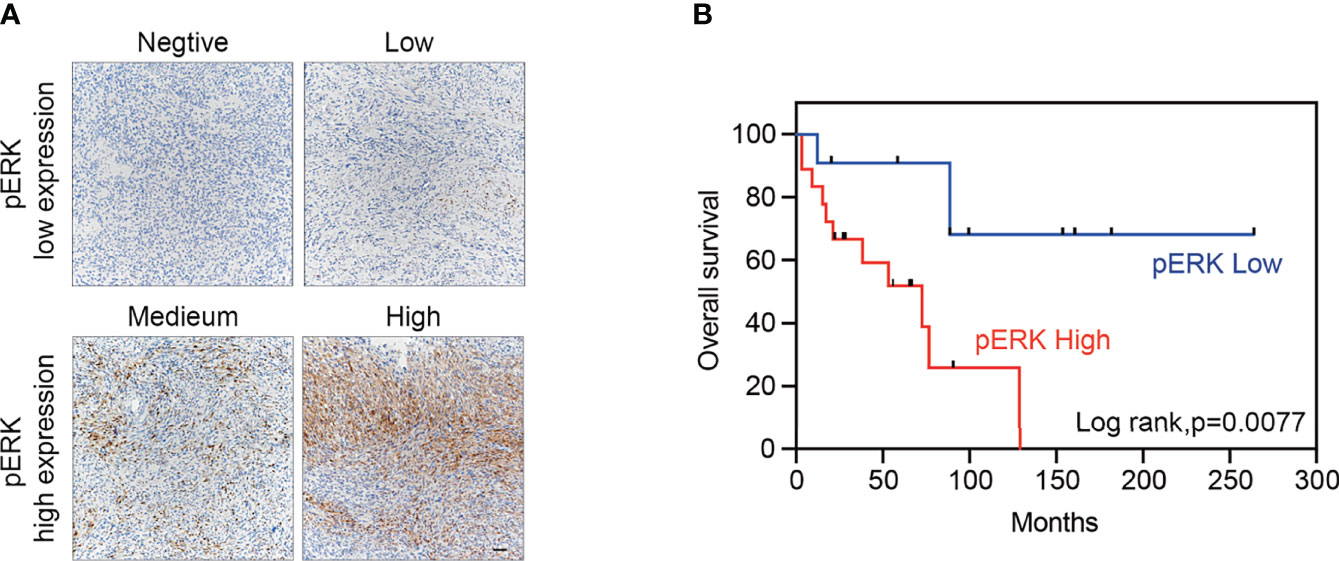
Figure 1 High expression of p-ERK correlates with dismal prognosis in MPNSTs. (A) Typical images of p-ERK in MPNST tissues following immunohistochemical staining, scored as 0 (negative), 1 (low positive), 2 (medium positive), and 3 (high positive). Scale bar = 50 μm (B) Kaplan–Meier curves for cumulative survival according to the p-ERK level in the MPNST cohort with available follow-up data (n = 29, p = 0.007 by the log-rank test). Negative and low positive were defined as low expression, while medium and high positive were defined as high expression.

Table 1 Pathological parameter with p-ERK expression.
Screening 8 MEK Inhibitors in MPNST Cells Revealed Differences in Efficiency and the Adaptive Response
After identifying p-ERK as a potential therapeutic target for MPNSTs, we screened 8 different MEK inhibitors, trametinib, TAK-733, PD0325901, cobimetinib, pimasertib, refametinib, binimetinib and selumetinib, to investigate their efficacy in MPNSTs. We performed cell viability assays in S462 cells with loss of heterozygosity (LOH) at the NF1 locus. While STS26T with the oncogenic V600E mutation in BRAF served as a control since the BRAF V600E mutation has been shown to be associated with resistance to MEK inhibitors (26). The cell viability curves and corresponding IC50 values of these two cell lines are presented in Figure 2. Trametinib and TAK-733 were the most efficient due to having the lowest IC50 values. At the same time, the IC50 values of Trametinib and TAK-733 in S462 were significantly lower than those in STS26T.
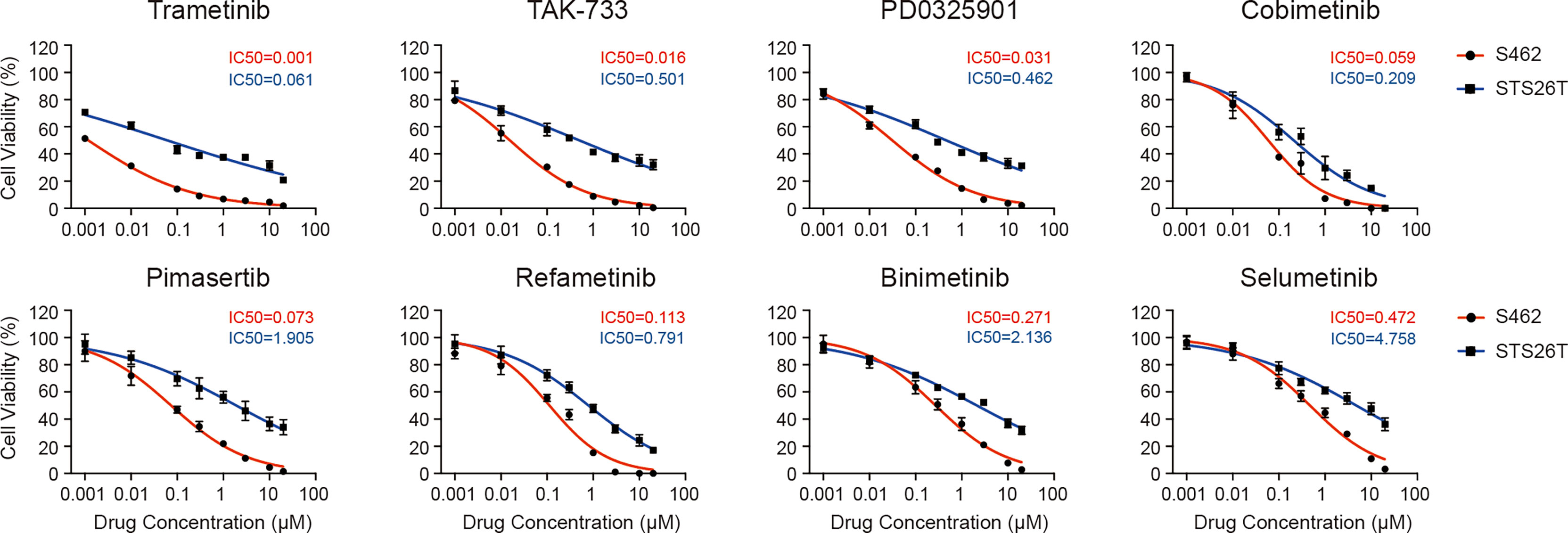
Figure 2 Different efficacy of MEK inhibitors in reducing the viability of MPNST cells. Cell viability of MPNST cell line S462 (the NF1-deficient human MPNST cell line) and STS26T (the NF1-wild-type human MPNST cell line with BRAF V600E and PTEN loss) after treated with MEK inhibitors for 96 h.
To explore the mechanism underlying the difference in efficacy, we next assessed the alterations in protein expression, including p-ERK and p-MEK in the MAPK signaling pathway, the downstream MEK/ERK effector cyclin D1, and p-AKT in the parallel PI3K/AKT pathway. P-ERK levels decreased with increasing concentrations of MEK inhibitors. Trametinib and TAK-733 were the most potent inhibitors, as p-ERK was not detected in S462 cells following 24 h of treatment with 0.01 μM. Selumetinib was the least effective in reducing p-ERK levels. A similar variation tendency in cyclin D1 expression was observed. As demonstrated in previous studies, MEK inhibition could reactivate MAPK signaling or upregulate parallel signaling pathways, particularly, the PI3K pathway, resulting in reduced efficacy of MEK inhibitors (27). It was also observed in S462 cells that compensatory increases in p-MEK and p-AKT occurred after 24 h of MEK inhibitor treatment. Trametinib and TAK-733 induced upregulation of p-MEK less obvious than other MEK inhibitors (Figure 3, Figure S2).
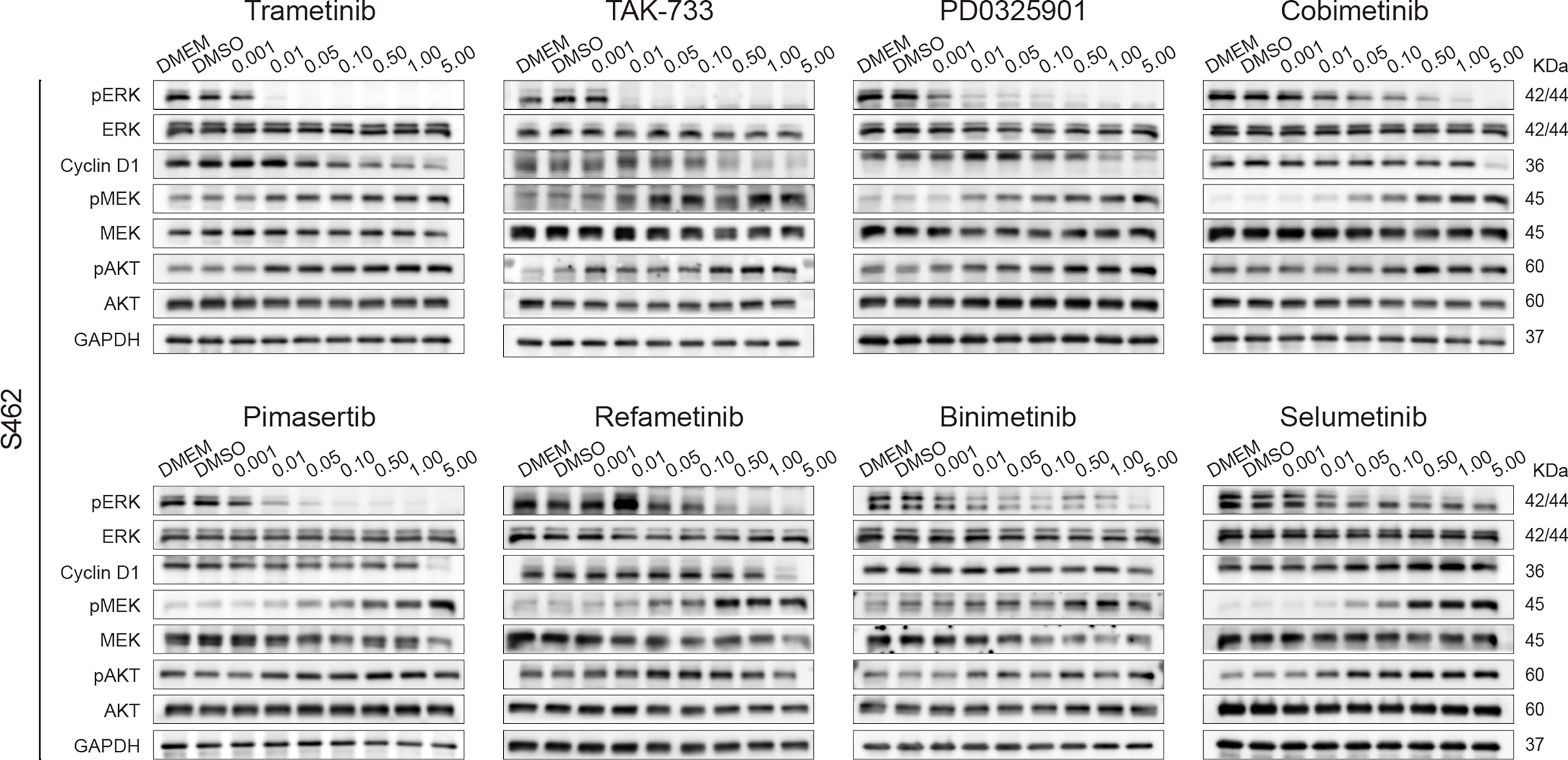
Figure 3 Screening 8 MEK inhibitors in MPNST cells revealed differences in adaptive response. Expression of p-ERK, ERK, cyclin D1, p-MEK, MEK, p-AKT, AKT, and GAPDH in S462 cells treated with different MEK inhibitors at the indicated concentrations for 24h.
Inhibition of p-ERK Induced Cell Cycle Arrest and Apoptosis in MPNST Cells
We next explored the efficiency and safety of MEK inhibitors in a variety of MPNST cell models. We selected the most potent MEK inhibitors trametinib and TAK-733, as well as selumetinib, the first FDA-approved MEK inhibitor for the treatment of pediatric PNFs, for further investigations. Cell viability assays were conducted in 4 NF1-MPNST cell lines S462, T265, ST88-14, S462TY, and schwann cell line 02.3 2λ. As shown in Figure 4A, trametinib inhibited all the MPNST cells most efficiently with submicromolar IC50 values. In addition, MEK inhibitors trametinib, TAK-733, and selumetinib were more efficient in MPNST cell lines compared with healthy Schwann cell line 02.3 2λ (Figure S3).
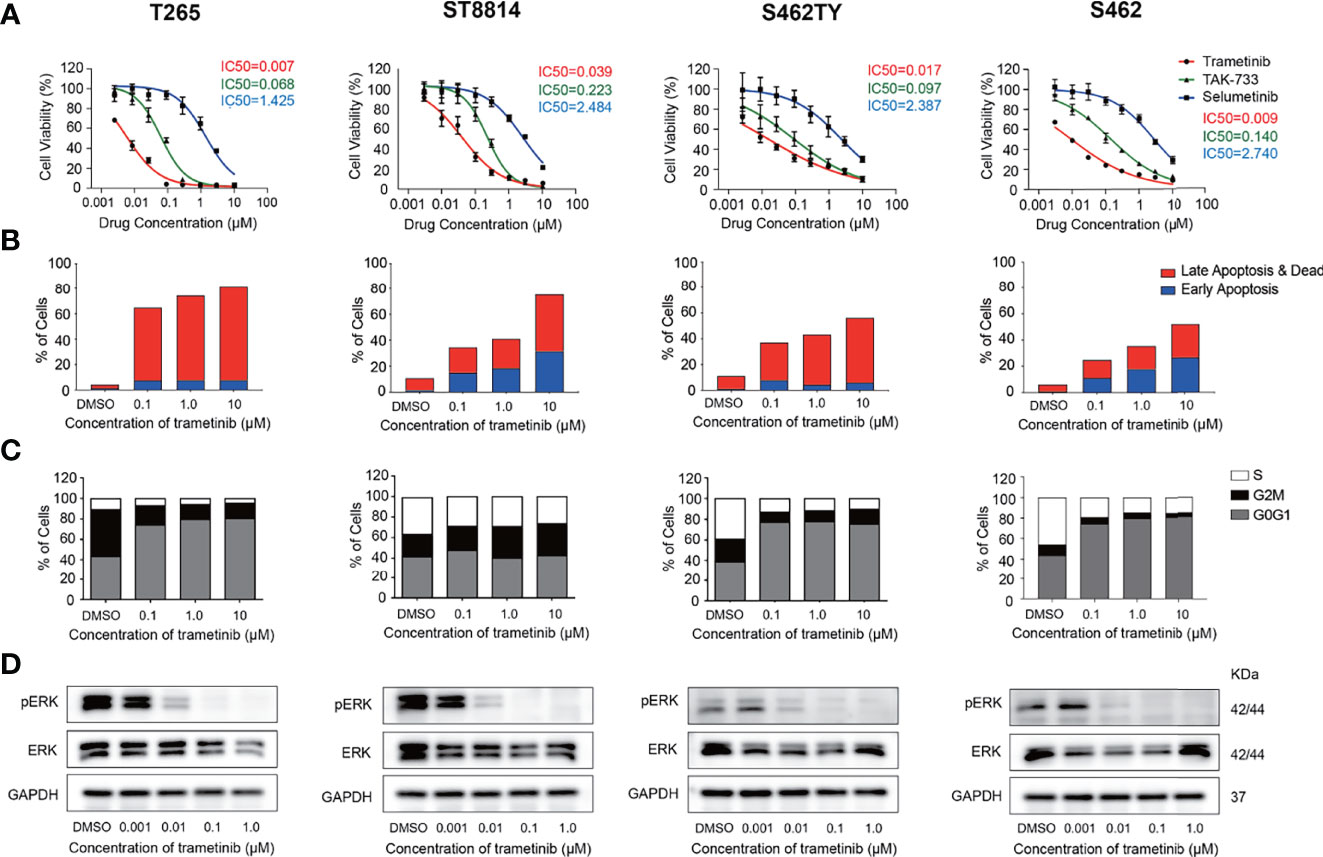
Figure 4 Inhibition of p-ERK induced cell cycle arrest and apoptosis in MPNST cells. (A) Cell viability of MPNST cell lines T265, ST8814, S462, S462TY, and Schwann cell line 02.3 2λ treated with MEK inhibitors for 72h. (B) apoptosis ratios of MPNST cell lines treated with MEK inhibitor trametinib for 48h. (C) Cell cycle and (D) p-ERK/ERK expression levels of MPNST cell lines treated with MEK inhibitor trametinib for 24h.
Furthermore, alterations in cell proliferation, apoptosis, and p-ERK expression following trametinib treatment were also explored in these MPNST cells. All the MPNST cell lines showed a high apoptosis ratio after trametinib treatment in a time-dependent manner (Figure 4B, S4). Significant blockage of the G0/G1 phase was observed in S462, T265, and S462TY cells while blockage of the G2/M phase was observed in ST88-14 cells (Figure 4C). As shown in Figure 4D and Figure S5, MEK inhibitor trametinib induced a significant reduction of p-ERK expression in S462, T265, ST88-14, and S462TY at a concentration of 0.01μM. The p-ERK expression in 02.3 2λ cells was significantly inhibited only at a higher concentration of 0.1μM (Figure S6).
MEK Inhibitors Significantly Inhibited the Growth of MPNST Xenograft Without Inducing Apoptosis
The MEK inhibitors trametinib and TAK-733 and selumetinib were further evaluated in vivo. Following the successful establishment of S462TY xenografts, mice were gavaged with trametinib, TAK-733, or selumetinib for 3 weeks. As shown in Figures 5A, B, all the three MEK inhibitors reduced the tumor weight and tumor volume significantly. The inhibition in tumor volume persisted from the beginning to the end of administration with no obvious toxicity (Figure S7). Immunohistochemical analysis of the MPNST xenografts demonstrated that the three MEK inhibitors induced a robust decrease in p-ERK, Ki-67, and C-MYC at the protein level. However, no obvious alteration of cleaved Caspase 3 expression was observed (Figure 5C).
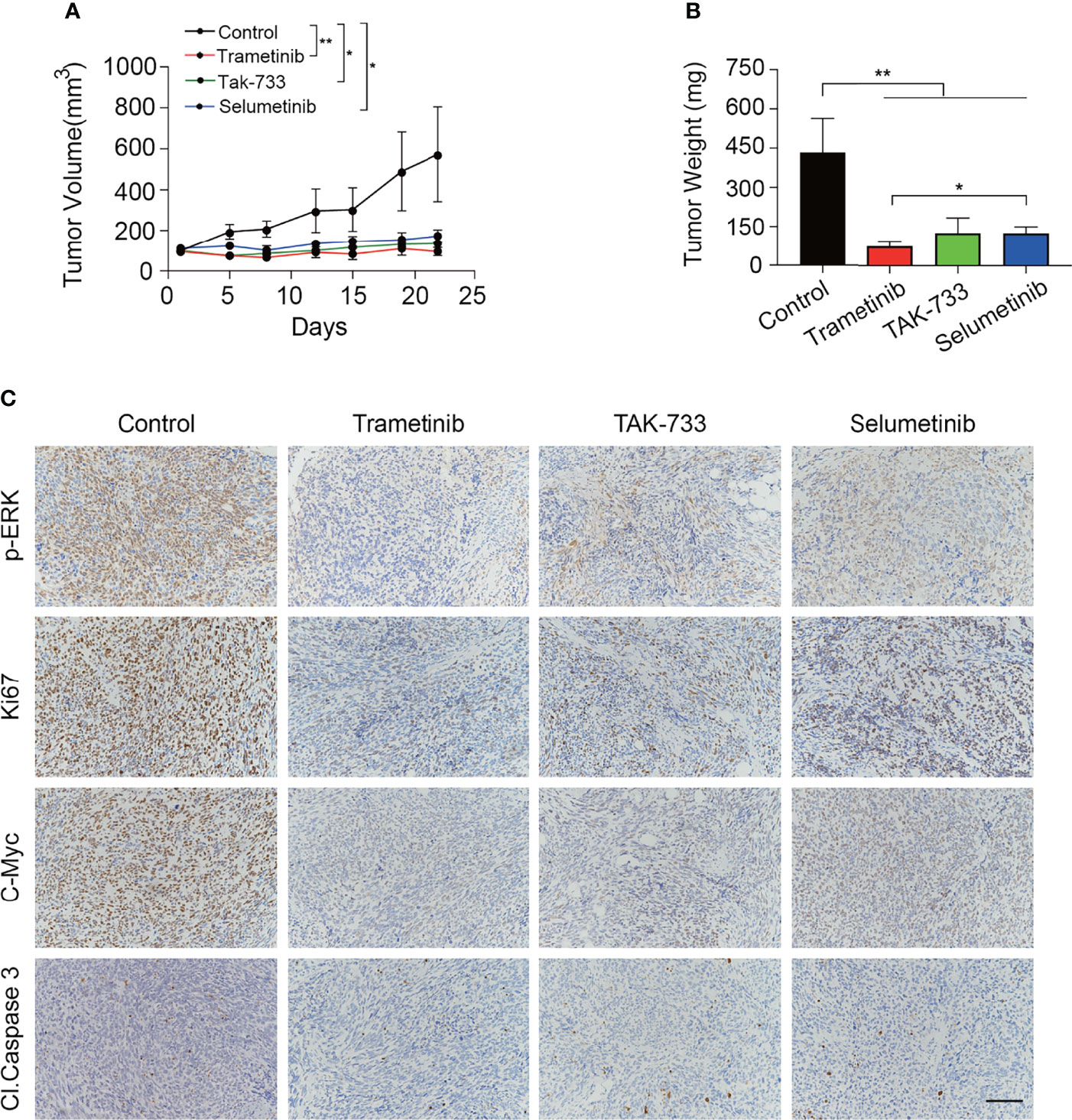
Figure 5 MEK inhibitors significantly inhibited the growth of MPNST xenograft without inducing apoptosis. (A, B) Tumor growth curves (A) and tumor weight (B) of mice treated with control, trametinib, TAK-733 or selumetinib for three weeks. Each point or column represents Mean ± SEM (n = 4). * Represents p < 0.05, and ** represents p < 0.01 compared with control group (Student’s t-test). (C) Representative images of p-ERK, Ki67, C-Myc, and Cl. Caspase3 immunohistochemical staining from the extracted tumor tissues in the four groups (scale bar = 100 μm).
Discussion
In this study, we investigated the efficacy of 8 different MEK inhibitors in treating MPNST using cell lines and xenograft models. We identified p-ERK as a biomarker for predicting the prognosis of MPNST patients as well as an effective therapeutic target. Moreover, we identified trametinib as the most potent MEK inhibitor for MPNST and demonstrated that the reduced reactivation of the MAPK pathway and activation of the parallel pathways contributed to better efficacy.
According to previous studies, genetic alterations including loss of NF1, EED, and SUZ12, have been demonstrated to activate the RAS/MAPK pathway in various human cancers including MPNSTs (7, 28, 29). Our findings also confirmed the overactivation of the MAPK pathway in MPNSTs as evidenced by the detection of p-ERK expression. In addition, our result first identified p-ERK as an indicator of a worse prognosis, indicating that the MAPK pathway could serve as a potential target in MPNSTs.
Inhibiting the RAS/MAPK pathway using MEK inhibitors has emerged as an effective strategy in a variety of cancers (30–34), as indicated in several clinical trials. However, the low incidence of MPNSTs added difficulties to the recruitment of clinical trials evaluating MEK inhibitors for MPNSTs. Therefore, we evaluated a panel of MEK inhibitors in various MPNST models and investigated the mechanism underlying the difference in efficacy. We included all the FDA-approved MEK inhibitors, and several potent MEK inhibitors proved to be efficient in clinical trials. Among the 8 inhibitors evaluated in vitro, trametinib was the most effective, likely from the significant reduction in p-ERK and downstream cyclin D1 levels. In addition, trametinib induced the lowest increase of p-MEK in S462 cells because it uniquely blocked Raf-dependent MEK phosphorylation, resulting in its potent efficacy in MAPK inhibition. Our in vivo experiments also confirmed the efficiency of MEK inhibitors. According to the results above, trametinib turned out to be the optimal choice for clinical application considering its excellent anti-tumor capacity in vitro and in vivo, as well as its mild reactivation of the MAPK pathway. As demonstrated by a recent case report, trametinib resulted in sustained complete response in an NF1 patient with recurrent and metastatic MPNST (35). In addition, trametinib has been approved by FDA for the treatment of malignant melanoma. Studies are being carried out to evaluate the effectiveness and to screen combinational drugs of MEK inhibitors in cancers including ovarian cancer and lung cancer (19, 32, 36–38).
Despite the potent efficacy of MAPK inhibition, trametinib caused significant activation of PI3K/Akt, a parallel pathway of MAPK signaling. It has been demonstrated in a variety of cancers that parallel pathways, especially the PI3K/Akt signaling pathway, could promote tumor growth and result in adaptive resistance after inhibition of the MAPK signaling (39, 40). Concurrent inhibition of MAPK and PI3K/Akt signaling using combinational therapies increased the efficacy of MEK inhibitors in multiple RAS-driven cancer models including MPNSTs (41, 42). Currently, a clinical trial is being carried out to test the effectiveness of selumetinib (AZD6244) and the mTOR inhibitor sirolimus in patients with unresectable or metastatic MPNST(NCT03433183).
Selumetinib is an FDA-approved MEK inhibitor that has shown excellent therapeutic efficacy in the treatment of pNF, the precursor lesion of MPNSTs. Compared to trametinib, selumetinib induced a more robust compensatory increase in p-MEK levels, indicating the occurrence of reactivation of the MAPK pathway, which might explain the less effective tumor-killing capacity. Wang et al. reported that in liver cancer, combinational therapy of sorafenib and selumetinib could increase the anti-tumor effectiveness through the synergistic inhibition of p-ERK (43). This combinational therapy might also improve the efficacy of selumetinib in MPNSTs.
It is also worth noticing that the MEK inhibitors inhibited the growth of MPNST xenografts without inducing tumor shrinkage or tumor cell apoptosis in vivo. It was inconsistent with the findings in Annexin V/PI assays showing that MEK inhibitors induced cell apoptosis significantly in vitro. It could be speculated that the tumor microenvironment including immune cells and extracellular matrix played an important role in limiting the efficacy of MEK inhibitors. Previous studies have suggested that microenvironment components are associated with drug resistance and are considered key obstacles that limit the efficacy of targeted therapy (44–49). For example, fibroblast-derived ECM abrogated anti-proliferative responses to BRAF/MEK inhibition in BRAFV600-mutated advanced melanoma and pharmacological targeting of key molecules could overcome drug resistance (50). Therefore, the role of the tumor microenvironment in limiting the efficacy of MEK inhibitors in treating MPNSTs and the underlying mechanisms deserve further investigation.
In conclusion, the variety of MEK inhibitors and the lack of evaluation of their efficacy in MPNSTs limited the research progress and clinical application of MEK inhibitors in MPNSTs. This study identified the most promising MEK inhibitor trametinib, for the treatment of MPNSTs and clarified the mechanism underlying the difference in efficacy. Our results provide a basis for the further investigation of MEK inhibitors for treating MPNSTs.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Ethics Committee of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine.
Author Contributions
YG, WW, YL, and HL generated and analyzed the data. ZG, CW, ML, MC, and RA assessed the TMA IHC staining and collected the clinical information. YG, WW, YL, and HL interpreted the results and wrote the manuscript. QL and ZW supervised the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from National Natural Science Foundation of China (82102344; 82172228); Shanghai Rising Star Program supported by Science and Technology Commission of Shanghai Municipality (20QA1405600); Science and Technology Commission of Shanghai Municipality (19JC1413); Natural Science Foundation of Shanghai (22ZR1422300); “Chenguang Program” supported by Shanghai Education Development Foundation (SHEDF) (19CG18); Shanghai Municipal Key Clinical Specialty (shslczdzk00901); Innovative research team of high-level local universities in Shanghai (SSMU-ZDCX20180700); the Project of Biobank (YBKA201901) from Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Prof. Vincent Keng and Prof. Jilong Yang for providing cell lines.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.903177/full#supplementary-material
References
1. Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A, et al. Malignant Peripheral Nerve Sheath Tumors. Oncol (2014) 19(2):193–201. doi: 10.1634/theoncologist.2013-0328
2. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis Type 1. Nat Rev Dis Primers (2017) 3(1):17004. doi: 10.1038/nrdp.2017.4
3. Kolberg M, Holand M, Agesen TH, Brekke HR, Liestol K, Hall KS, et al. Survival Meta-Analyses for >1800 Malignant Peripheral Nerve Sheath Tumor Patients With and Without Neurofibromatosis Type 1. Neuro Oncol (2013) 15(2):135–47. doi: 10.1093/neuonc/nos287
4. Vitolo V, Fiore MR, Barcellini A, Vischioni B, Iannalfi A, Facoetti A, et al. Carbon Ion Radiotherapy in the Management of the Tumors of the Peripheral Nervous System. Anticancer Res (2019) 39(2):909–13. doi: 10.21873/anticanres.13193
5. Widemann BC. Current Status of Sporadic and Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Curr Oncol Rep (2009) 11(4):322–8. doi: 10.1007/s11912-009-0045-z
6. Blakeley JO, Plotkin SR. Therapeutic Advances for the Tumors Associated With Neurofibromatosis Type 1, Type 2, and Schwannomatosis. Neuro Oncol (2016) 18(5):624–38. doi: 10.1093/neuonc/nov200
7. Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, et al. PRC2 Is Recurrently Inactivated Through EED or SUZ12 Loss in Malignant Peripheral Nerve Sheath Tumors. Nat Genet (2014) 46(11):1227–32. doi: 10.1038/ng.3095
8. Rahrmann EP, Watson AL, Keng VW, Choi K, Moriarity BS, Beckmann DA, et al. Forward Genetic Screen for Malignant Peripheral Nerve Sheath Tumor Formation Identifies New Genes and Pathways Driving Tumorigenesis. Nat Genet (2013) 45(7):756–66. doi: 10.1038/ng.2641
9. Mowery A, Clayburgh D. Malignant Peripheral Nerve Sheath Tumors: Analysis of the National Cancer Database. Oral Oncol (2019) 98:13–9. doi: 10.1016/j.oraloncology.2019.09.010
10. Schulte A, Ewald F, Spyra M, Smit DJ, Jiang W, Salamon J, et al. Combined Targeting of AKT and mTOR Inhibits Proliferation of Human NF1-Associated Malignant Peripheral Nerve Sheath Tumour Cells In Vitro But Not in a Xenograft Mouse Model In Vivo. Int J Mol Sci (2020) 21(4):1548. doi: 10.3390/ijms21041548
11. Bodempudi V, Yamoutpoor F, Pan W, Dudek AZ, Esfandyari T, Piedra M, et al. Ral Overactivation in Malignant Peripheral Nerve Sheath Tumors. Mol Cell Biol (2009) 29(14):3964–74. doi: 10.1128/MCB.01153-08
12. Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T, et al. Prognostic Significance of AKT/mTOR and MAPK Pathways and Antitumor Effect of mTOR Inhibitor in NF1-Related and Sporadic Malignant Peripheral Nerve Sheath Tumors. Clin Cancer Res (2013) 19(2):450–61. doi: 10.1158/1078-0432.CCR-12-1067
13. Barbosa R, Acevedo LA, Marmorstein R. The MEK/ERK Network as a Therapeutic Target in Human Cancer. Mol Cancer Res (2021) 19(3):361–74. doi: 10.1158/1541-7786.MCR-20-0687
14. Ney GM, McKay L, Koschmann C, Mody R, Li Q. The Emerging Role of Ras Pathway Signaling in Pediatric Cancer. Cancer Res (2020) 80(23):5155–63. doi: 10.1158/0008-5472.CAN-20-0916
15. Butler E, Schwettmann B, Geboers S, Hao G, Kim J, Nham K, et al. Functional Imaging of RAS Pathway Targeting in Malignant Peripheral Nerve Sheath Tumor Cells and Xenografts. Pediatr Blood Cancer (2020) 67(12):e28639. doi: 10.1002/pbc.28639
16. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK Inhibition in Melanoma With BRAF V600 Mutations. N Engl J Med (2012) 367(18):1694–703. doi: 10.1056/NEJMoa1210093
17. Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med (2016) 375(26):2550–60. doi: 10.1056/NEJMoa1605943
18. Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, et al. MEK Inhibition Exhibits Efficacy in Human and Mouse Neurofibromatosis Tumors. J Clin Invest (2013) 123(1):340–7. doi: 10.1172/JCI60578
19. Han J, Liu Y, Yang S, Wu X, Li H, Wang Q. MEK Inhibitors for the Treatment of non-Small Cell Lung Cancer. J Hematol Oncol (2021) 14(1):1. doi: 10.1186/s13045-020-01025-7
20. Kim C, Giaccone G. MEK Inhibitors Under Development for Treatment of Non-Small-Cell Lung Cancer. Expert Opin Investig Drugs (2018) 27(1):17–30. doi: 10.1080/13543784.2018.1415324
21. Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in Children With Inoperable Plexiform Neurofibromas. N Engl J Med (2020) 382(15):1430–42. doi: 10.1056/NEJMoa1912735
22. Ahsan S, Ge Y, Tainsky MA. Combinatorial Therapeutic Targeting of BMP2 and MEK-ERK Pathways in NF1-Associated Malignant Peripheral Nerve Sheath Tumors. Oncotarget (2016) 7(35):57171–85. doi: 10.18632/oncotarget.11036
23. Wang J, Pollard K, Allen AN, Tomar T, Pijnenburg D, Yao Z, et al. Combined Inhibition of SHP2 and MEK Is Effective in Models of NF1-Deficient Malignant Peripheral Nerve Sheath Tumors. Cancer Res (2020) 80(23):5367–79. doi: 10.1158/0008-5472.CAN-20-1365
24. Fischer-Huchzermeyer S, Dombrowski A, Wilke G, Stahn V, Streubel A, Mautner VF, et al. MEK Inhibitors Enhance Therapeutic Response Towards ATRA in NF1 Associated Malignant Peripheral Nerve Sheath Tumors (MPNST) in-Vitro. PloS One (2017) 12(11):e0187700. doi: 10.1371/journal.pone.0187700
25. Castellsague J, Gel B, Fernandez-Rodriguez J, Llatjos R, Blanco I, Benavente Y, et al. Comprehensive Establishment and Characterization of Orthoxenograft Mouse Models of Malignant Peripheral Nerve Sheath Tumors for Personalized Medicine. EMBO Mol Med (2015) 7(5):608–27. doi: 10.15252/emmm.201404430
26. Kun E, Tsang YTM, Ng CW, Gershenson DM, Wong KK. MEK Inhibitor Resistance Mechanisms and Recent Developments in Combination Trials. Cancer Treat Rev (2021) 92:102137. doi: 10.1016/j.ctrv.2020.102137
27. Wang J, Pollard K, Calizo A, Pratilas CA. Activation of Receptor Tyrosine Kinases Mediates Acquired Resistance to MEK Inhibition in Malignant Peripheral Nerve Sheath Tumors. Cancer Res (2021) 81(3):747–62. doi: 10.1158/0008-5472.CAN-20-1992
28. Rasmussen SA, Overman J, Thomson SA, Colman SD, Abernathy CR, Trimpert RE, et al. Chromosome 17 Loss-Of-Heterozygosity Studies in Benign and Malignant Tumors in Neurofibromatosis Type 1. Genes Chromosomes Cancer (2000) 28(4):425–31. doi: 10.1002/1098-2264(200008)28:4<425::AID-GCC8>3.0.CO;2-E
29. Sohier P, Luscan A, Lloyd A, Ashelford K, Laurendeau I, Briand-Suleau A, et al. Confirmation of Mutation Landscape of NF1-Associated Malignant Peripheral Nerve Sheath Tumors. Genes Chromosomes Cancer (2017) 56(5):421–6. doi: 10.1002/gcc.22446
30. Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 Inhibitors and Cancer Therapy: The Long and Winding Road. Nat Rev Cancer (2015) 15(10):577–92. doi: 10.1038/nrc4000
31. Fuse MA, Dinh CT, Vitte J, Kirkpatrick J, Mindos T, Plati SK, et al. Preclinical Assessment of MEK1/2 Inhibitors for Neurofibromatosis Type 2-Associated Schwannomas Reveals Differences in Efficacy and Drug Resistance Development. Neuro Oncol (2019) 21(4):486–97. doi: 10.1093/neuonc/noz002
32. Chesnokov MS, Khan I, Park Y, Ezell J, Mehta G, Yousif A, et al. The MEK1/2 Pathway as a Therapeutic Target in High-Grade Serous Ovarian Carcinoma. Cancers (Basel) (2021) 13(6):1369. doi: 10.3390/cancers13061369
33. Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A Phase I Trial of the MEK Inhibitor Selumetinib (AZD6244) in Pediatric Patients With Recurrent or Refractory Low-Grade Glioma: A Pediatric Brain Tumor Consortium (PBTC) Study. Neuro Oncol (2017) 19(8):1135–44. doi: 10.1093/neuonc/now282
34. Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, et al. Five-Year Outcomes With Dabrafenib Plus Trametinib in Metastatic Melanoma. N Engl J Med (2019) 381(7):626–36. doi: 10.1056/NEJMoa1904059
35. Nagabushan S, Lau LMS, Barahona P, Wong M, Sherstyuk A, Marshall GM, et al. Efficacy of MEK Inhibition in a Recurrent Malignant Peripheral Nerve Sheath Tumor. NPJ Precis Oncol (2021) 5(1):9. doi: 10.1038/s41698-021-00145-8
36. Liu S, Zou Q, Chen JP, Yao X, Guan P, Liang W, et al. Targeting Enhancer Reprogramming to Mitigate MEK Inhibitor Resistance in Preclinical Models of Advanced Ovarian Cancer. J Clin Invest (2021) 131(20):e145035. doi: 10.1172/JCI145035
37. Planchard D, Besse B, Groen HJM, Souquet PJ, Quoix E, Baik CS, et al. Dabrafenib Plus Trametinib in Patients With Previously Treated BRAF(V600E)-Mutant Metastatic non-Small Cell Lung Cancer: An Open-Label, Multicentre Phase 2 Trial. Lancet Oncol (2016) 17(7):984–93. doi: 10.1016/S1470-2045(16)30146-2
38. Kauko O, O'Connor CM, Kulesskiy E, Sangodkar J, Aakula A, Izadmehr S, et al. PP2A Inhibition Is a Druggable MEK Inhibitor Resistance Mechanism in KRAS-Mutant Lung Cancer Cells. Sci Transl Med (2018) 10(450):eaaq1093. doi: 10.1126/scitranslmed.aaq1093
39. Irvine M, Stewart A, Pedersen B, Boyd S, Kefford R, Rizos H. Oncogenic PI3K/AKT Promotes the Step-Wise Evolution of Combination BRAF/MEK Inhibitor Resistance in Melanoma. Oncogenesis (2018) 7(9):72. doi: 10.1038/s41389-018-0081-3
40. Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, et al. MEK Inhibition Leads to PI3K/AKT Activation by Relieving a Negative Feedback on ERBB Receptors. Cancer Res (2012) 72(13):3228–37. doi: 10.1158/0008-5472.CAN-11-3747
41. Watson AL, Anderson LK, Greeley AD, Keng VW, Rahrmann EP, Halfond AL, et al. Co-Targeting the MAPK and PI3K/AKT/mTOR Pathways in Two Genetically Engineered Mouse Models of Schwann Cell Tumors Reduces Tumor Grade and Multiplicity. Oncotarget (2014) 5(6):1502–14. doi: 10.18632/oncotarget.1609
42. Arozarena I, Wellbrock C. Overcoming Resistance to BRAF Inhibitors. Ann Transl Med (2017) 5(19):387. doi: 10.21037/atm.2017.06.09
43. Wang C, Jin H, Gao D, Lieftink C, Evers B, Jin G, et al. Phospho-ERK is a Biomarker of Response to a Synthetic Lethal Drug Combination of Sorafenib and MEK Inhibition in Liver Cancer. J Hepatol (2018) 69(5):1057–65. doi: 10.1016/j.jhep.2018.07.004
44. Yang XH, Flores LM, Li Q, Zhou P, Xu F, Krop IE, et al. Disruption of Laminin-Integrin-CD151-Focal Adhesion Kinase Axis Sensitizes Breast Cancer Cells to ErbB2 Antagonists. Cancer Res (2010) 70(6):2256–63. doi: 10.1158/0008-5472.CAN-09-4032
45. Yu Q, Xiao W, Sun S, Sohrabi A, Liang J, Seidlits SK. Extracellular Matrix Proteins Confer Cell Adhesion-Mediated Drug Resistance Through Integrin Alpha V in Glioblastoma Cells. Front Cell Dev Biol (2021) 9:616580. doi: 10.3389/fcell.2021.616580
46. Cho C, Horzempa C, Jones D, McKeown-Longo PJ. The Fibronectin III-1 Domain Activates a PI3-Kinase/Akt Signaling Pathway Leading to Alphavbeta5 Integrin Activation and TRAIL Resistance in Human Lung Cancer Cells. BMC Cancer (2016) 16:574. doi: 10.1186/s12885-016-2621-6
47. Ye Y, Zhang R, Feng H. Fibronectin Promotes Tumor Cells Growth and Drugs Resistance Through a CDC42-YAP-Dependent Signaling Pathway in Colorectal Cancer. Cell Biol Int (2020) 44(9):1840–9. doi: 10.1002/cbin.11390
48. Gao Y, Li X, Zeng C, Liu C, Hao Q, Li W, et al. CD63(+) Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells Through Exosomal miR-22. Adv Sci (Weinh) (2020) 7(21):2002518. doi: 10.1002/advs.202002518
49. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discovery (2020) 10(9):1330–51. doi: 10.1158/2159-8290.CD-19-1384
Keywords: neurofibromatosis type 1, malignant peripheral nerve sheath tumors (MPNST), mek inhibitor, trametinib, target therapy
Citation: Gu Y, Wang W, Li Y, Li H, Guo Z, Wei C, Long M, Chung M, Aimaier R, Li Q and Wang Z (2022) Preclinical Assessment of MEK Inhibitors for Malignant Peripheral Nerve Sheath Tumors Reveals Differences in Efficacy and Adaptive Response. Front. Oncol. 12:903177. doi: 10.3389/fonc.2022.903177
Received: 24 March 2022; Accepted: 07 June 2022;
Published: 07 July 2022.
Edited by:
Laura Gatti, IRCCS Carlo Besta Neurological Institute Foundation, ItalyReviewed by:
Elisabetta Vergani, National Cancer Institute Foundation (IRCCS), ItalyCristina Corno, National Cancer Institute Foundation (IRCCS), Italy
Copyright © 2022 Gu, Wang, Li, Li, Guo, Wei, Long, Chung, Aimaier, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhichao Wang, c2htdXd6Y0AxNjMuY29t; ZHJfd2FuZ3poaWNoYW9AMTYzLmNvbQ==; Qingfeng Li, bGlxaW5nZmVuZ0BzaHNtdS5lZHUuY24=; ZHJfbGlxaW5nZmVuZzlAMTYzLmNvbQ==
†These authors have contributed equally to this work