
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 02 June 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.903016
This article is part of the Research Topic Insights in Cancer Molecular Targets and Therapeutics: 2022 View all 8 articles
 Michela Chiappa1
Michela Chiappa1 Serena Petrella1
Serena Petrella1 Giovanna Damia1
Giovanna Damia1 Massimo Broggini2*
Massimo Broggini2* Federica Guffanti1†
Federica Guffanti1† Francesca Ricci1†
Francesca Ricci1†Polo-like kinase 1 (PLK1) is the principle member of the well conserved serine/threonine kinase family. PLK1 has a key role in the progression of mitosis and recent evidence suggest its important involvement in regulating the G2/M checkpoint, in DNA damage and replication stress response, and in cell death pathways. PLK1 expression is tightly spatially and temporally regulated to ensure its nuclear activation at the late S-phase, until the peak of expression at the G2/M-phase. Recently, new roles of PLK1 have been reported in literature on its implication in the regulation of inflammation and immunological responses. All these biological processes are altered in tumors and, considering that PLK1 is often found overexpressed in several tumor types, its targeting has emerged as a promising anti-cancer therapeutic strategy. In this review, we will summarize the evidence suggesting the role of PLK1 in response to DNA damage, including DNA repair, cell cycle progression, epithelial to mesenchymal transition, cell death pathways and cancer-related immunity. An update of PLK1 inhibitors currently investigated in preclinical and clinical studies, in monotherapy and in combination with existing chemotherapeutic drugs and targeted therapies will be discussed.
Human Polo-like kinases (PLKs) are a family of serine/threonine protein kinases comprising five members: PLK1, PLK2, PLK3, PLK4 and PLK5, with PLK1 is the most studied one. PLK1 has numerous different functions, the best known ones being its key role in mitotic entry, in centrosome regulation, in coordinating the spindle assembly, in segregation of the chromosomes and in cytokinesis (1). Other functions of the protein beyond the cell cycle have also been described based on emerging new substrates found phosphorylated by PLK1.
PLK1 dysregulation has been reported in different tumor types, contributing to tumor development and progression. PLK1 is reported to be overexpressed (at both mRNA and protein level) in many tumors compared to the normal tissue counterpart, and its overexpression has been associated with poor patient outcome (2). In addition, its overexpression has in some cases been associated with resistance to therapy and its inhibition to re-sensitization to chemo- and radio-therapy (3–5).
This review discusses the recently discovered functions of PLK1, mainly in relation to DNA damage response, EMT (epithelial to mesenchymal transition), cell death (apoptosis/autophagy) and immune system, and looks at the preclinical activity of PLK1 inhibitors, both as single agents and in combination, as well as their clinical development.
Very recent well written reviews have been published on PLK1 structure, regulation and its role in cancer development and therapy (6–9), and here we summarize some key features of the PLK1 protein that could help clarifying the rationale for the design of PLK1 inhibitors and their potential side effects.
PLK1 is a very highly conserved polo-like protein whose activity is a requisite for mitotic entry. PLK1 phosphorylates Cdc25C, WEE1 and MYT1 to promote activation of the CyclinB1/CDK1 complex in triggering prophase and, later on, in G2 and mitosis (10–13). PLK1 is involved in maturation of the centrosome, kinetochore formation, condensation of the chromosome, spindle assembly and cytokinesis (14). PLK1 maximum kinase activity is in the mitotic phase of the cell cycle and its expression is regulated by different phosphorylation events (15, 16).
The PLK1 protein (603 amino acids) comprises two polo-box domains (PBDs) at the C-terminal and a kinase domain (KD) at the N-terminal portion of the protein (Figure 1). The PBD determines the specific cellular localization where PLK1 can interact with phospho-epitopes on target substrates. Different PLK1 interacting proteins are implicated in multiple cellular processes (16). Mutations in its PBD interfere with PLK1 localization and function (17). PBD not only drives PLK1 substrate recognition and protein localization, but also abrogates the inhibitory interaction between KD and PBD, so the T-loop region of PLK1 (encompassing Thr210) becomes phosphorylated (Ser137 and Thr210) by upstream kinases (the most important being Aurora A and its co-factor Bora), achieving protein full activation (18). Interestingly, PLK1 can phosphorylate substrates already phosphorylated by itself (self-priming) or by other up-stream kinases (non self-priming); for example, the scaffold centrosome PBIP1 protein is first phosphorylated by PLK1 at Thr78, thus favouring in such a way the interaction of the PBD of PLK1 with PBIP1 and the correct localization of PLK1 to kinetochores (19). An example of non-self-priming is the WEE1 protein, which is first phosphorylated by a cyclin-dependent kinase (CDK) leading the binding motif for PBD of PLK1; in mitosis, when PLK1 levels rise, PLK1 binds to WEE1 at the PBD docking site and phosphorylates it, leading to its degradation (20). The initial phosphorylation of PLK1 by Aurora kinase A and its co-factor Bora in its kinase domain takes place in the cytoplasm and also leads to the exposure of a nuclear localization signal that allows PLK1 to be translocated into the nucleus (21). The phosphorylation status of PLK1 is not only important for its kinase activity and interaction with other proteins, but also for ubiquitination representing an important post-translation modification, affecting PLK1’s timely localization and degradation to allow correct cell cycle progression (22).

Figure 1 PLK1 protein domains. PLK1 structure includes two functional polo-box domains (PBDs) at C-terminal and the kinase domain at N-terminal.
Very recent data also suggest transient dimerization of PLK1 as a new mechanism underlying the activation of cytoplasmic PLK1 during G2 phase (23). These data suggest that in early G2 phase Bora facilitates PLK1 dimerization, acting as an allosteric modulator of the PLK1, that shifts from dimeric to monomeric active state; in late G2, PLK1 Thr210 phosphorylation by Aurora kinase A triggers dimer dissociation and the PLK1 monomers generated foster mitotic entry. These data are important as they may suggest the design of new allosteric compounds to mimic the Bora-PLK1 interaction, stabilizing and/or preventing the dimeric inactive conformation, hence inhibiting PLK1 activity.
PLK1 is a well conserved master regulator of cell division in eukaryotic cells, particularly involved in mitosis, where its functions are well understood (21, 24); less known is its function during the interphase and its role as modulator of the DNA damage response (DDR) and checkpoint resolution after DNA damage.
The DDR comprises a complex network of proteins that sense specific types of DNA lesions and respond to them by activating the necessary DNA repair mechanisms, and timely regulating cell cycle progression through checkpoint activation, with the final aim of repairing the damage, or if the damage is too extensive to induce cell death (25, 26). It is hardly surprising, that proteins like PLK1 involved in the surveillance mechanisms of cell cycle progression are also involved in the DDR, due to the close interactions among these mechanisms. Both DDR and cell cycle regulation have the ultimate aim of preventing genomic instability and the transmission of altered DNA to daughter cells. In the last few years mounting evidence has suggested PLK1 is involved in the DNA damage checkpoints and in DNA repair mechanisms activated during the interphase, when nuclear PLK1 levels start to rise (during S-phase), and mitosis, when PLK1 reaches its peak of expression (15). During S/G2 and M phases PLK1 interacts with and regulates by phosphorylation several key factors involved in these pathways (27).
DNA is replicated during S-phase of the cell cycle, and faithful duplication of the genome is critical for the maintenance of genomic stability. Pre-replication complexes, assembled during G1 phase at the replication origins along the genome are remodeled in active replication forks (RFs) and closely regulated by Cdc7 and various S-phase CDKs (28). Subsequently, mini-chromosome maintenance (MCM) helicase complexes are recruited at the active RFs to unwind DNA into two single filaments (ssDNA), where RPA proteins can bind and stabilize them. This leaves the RF ready to load replicative DNA polymerases and proliferating cell nuclear antigen (PCNA) proteins to initiate DNA replication (28). PLK1 expression does not increase during replication until DNA synthesis is completed, but it has been recently demonstrated that PLK1 function is also needed during S-phase, though at low level (29, 30). Once replication starts, CDK2 activity promotes DNA replication, CDK1 and PLK1 activities (31, 32). At the same time, DNA replication restricts CDK1 and PLK1 activities, causing a contrasting feed-forward loop to prevent mitosis starting until DNA replication is completed, and to promote PLK1 activation immediately after S-phase, thus favoring a smooth progression from DNA replication to mitosis (30). Moreover, in the G1 and S phases PLK1 phosphorylates and regulates different factors involved in the formation of the pre-replicative complexes at DNA replication origins. These targets include Orc2, a component of the origin recognition complex (ORC) (33), MCM2-7, parts of the MCM complex (34), DBF4, which couples with Cdc7 to selectively phosphorylate MCM2 subunit for its release from DNA once replication is completed (34).
The importance of PLK1 regulation during DNA replication is proved by the fact that PLK1 inhibition is associated with impaired replication and slowing of S-phase progression in vitro, and that PLK1 phosphorylates Orc2 under replication stress to maintain replication and promote genomic stability (33). Recently, PLK1 was shown in an in vitro system to be essential in regulating the spatio-temporal replication program by interacting with several origin firing factors (i.e. RIF1, TRESLIN; TopBP1), and the immuno-depletion of PLK1 reduced the number of replication forks and origins firing (35), giving additional proof of the PLK1’s role also in S-phase.
In normal conditions, mitotic entry depends on the activation and accumulation of cyclin B1/CDK1 complex, otherwise kept inactive through the inhibitory phosphorylation of CDK1 at Thr14 and Tyr15 by the kinase WEE1 and membrane-associated tyrosine-and threonine-specific Cdc2-inhibitory kinase (MYT1) kinase. Cyclin B1/CDK1 complex is activated when Cdc25C phosphatase overcomes the inhibitory effect of WEE1/MYT1. In fact, once activated, CDK1 triggers a positive feedback loop by phosphorylating both Cdc25C and WEE1/MYT1 (14, 36, 37). PLK1 takes part in this positive feedback loop, promoting CDK1 activation and mitotic entry by upregulating CDK1, promoting activation of Cdc25C and inhibition of both MYT1 and WEE1, and by degradation through E3 ubiquitin ligase SCF βTrCP (20, 38, 39). PLK1 activation is boosted by Aurora kinase A which in cooperation with Bora, mediates its phosphorylation on Thr210 (18, 40). In parallel, cyclin-B1/CDK1 complex targets Bora to promote its interaction with Aurora kinase A, fostering PLK1 activation (41).
Upon DNA damage, checkpoints are activated and cell cycle progression is halted. DNA double strand breaks (DSBs) or stalled replication forks (RFs) cause local alteration of the chromatin structure, recruitment of sensors like the Mre11-Rad50-Nbs1 (MRN) complex at the damaged site and activation of two main checkpoint pathways: ATR/CHK1 and ATM/CHK2 (42). ATM/ATR are two phosphoinositide 3-kinase (PI3K)-related protein kinases that activate CHK1/CHK2 which phosphorylate inactivating Cdc25C, preventing the de-phosphorylation and activation of nuclear CDK1 and blocking cells in the G2/M phase (43). Cell cycle arrest allows time for DNA repair and rescue of stalled RFs. Local activation of ATM at the DSB site recruits DSB repair factors like 53BP1, BRCA1 and activation of CHK2, and lack of entry in mitosis (44, 45). 53BP1 facilitates DNA repair by the error-prone non-homologous end joining (NHEJ) pathway, while BRCA1 is important for the error-free homologous recombination (HR) pathway during the S/G2-phases (46).
PLK1 is one of the numerous kinases downstream effectors of the ATM and ATR cascades (47, 48). In normal conditions, PLK1 is inhibited after DNA damage recognition through two mechanisms. The first involves Bora ubiquitination, induced by direct phosphorylation of ATM/ATR on Thr501, leading to p-Bora and degradation by E3 ubiquitin ligase SCF-β-TRCP (49); the second mechanism involves the ubiquitination of several mitotic factors, including PLK1, by the anaphase-promoting complex/cyclosome (APC/C), an E3 ubiquitin ligase (50).
Recently, a complex interplay has been described between PLK1 and the HR repair. Zou and colleagues described an interaction between BRCA1 and PLK1, showing that during the activation of the DDR, BRCA1 promptly down-regulates PLK1 kinase activity, affecting its dynamic interactions with Aurora kinase A and Bora; in BRCA1-depleted cells PLK1 activity was higher than in control-siRNA treated cells (51). However, it remains to be defined how BRCA1 binding to Aurora kinase A-Bora-PLK1 inhibits PLK1 activity. The involvement of phosphatases, such as myosin phosphatase targeting subunit 1 (MYPT1), and/or ubiquitin-mediated proteolysis, have been put forward (50, 52). Mono-methylation of PLK1 on Lys209 by methyltransferase G9 inactivates PLK1 by antagonizing Aurora kinase A-Bora’s activation through phosphorylation at Thr210 (53); an increasing amount of methylated PLK1 in cells exposed to genotoxic agents has indeed been reported. PLK1 methylation is necessary for DNA replication and for the timely removal of DSB repair DNA binding proteins, like RPA2 and RAD51, or BRCA2 before cell cycle progression (47, 54).
PLK1 can actively regulate several proteins essential for DNA DSB repair via HR. For instance, PLK1 phosphorylates RAD51 on Ser14 during S/G2-phase and in response to DNA damage (55). A transient increase of p-Ser14 was seen 20–40 min after DNA damage and allowed subsequent RAD51 phosphorylation at Thr13 by casein kinase 2. This double phosphorylation of RAD51 favors direct binding to the Nijmegen breakage syndrome (Nbs1) protein, part of the MRN complex involved in the early phase of the DDR (55). PLK1 inhibition before DNA damage has been associated with increased sensitivity to ionizing radiation and reduction of BRCA1 foci formation, suggesting PLK1 as a possible regulator of HR activation (56). This hypothesis has been recently further supported, since PARP1 and CHK1 seem to be responsible for PLK1 modification to promote HR (57). Peng et al. first observed in vitro a timely coordinated activity of PARP1 a few seconds after DSB induction, triggering PARylation of PLK1. As a consequence, PLK1 is protected from degradation. After 10 min, PARG had removed PAR chains from PLK1, allowing CHK1 to activate PLK1 by phosphorylating residues Ser137 and Thr210, and promoting PLK1–mediated phosphorylation on Ser14 of RAD51 (57). Interestingly, the primed p-Ser14 RAD51 allowed its subsequent phosphorylation at Thr309 directly by CHK1 (57). These phosphorylation events fully activate RAD51 and promote HR repair. Using an innovative technique of RF proteome analysis to clarify the composition of RF challenged by DSB, Nakamura et al. described a new signaling pathway that illustrates the mechanism underlying HR-dependent recovery of the stalled RF after TOP1 inhibitor-induced damage (58). In their model, there was an ATM-dependent recruitment of PLK1 at the broken fork and PLK1-dependent limitation of NHEJ in mitosis, induced by phosphorylation of 53BP1 (59) and XRCC4 (60), that prevent error-prone NHEJ, and support CtIP-mediated HR (58). After prolonged replication stress in S phase, PLK1 has been reported to interact with 53BP1 and BRCA1, which counteract each other to protect stalled RFs and promote replication restart through two distinct pathways. One is induced by 53BP1, that remodels the stalled RF without inducing DNA breakage, and the second is mediated by BRCA1, that triggers the fork-cleavage-coupled break-induced replication (BIR) pathway, mediated by the endonuclease MUS81 (61). In the early stage of replication stress, the balance favors the 53BP1-dependent pathway, but when replication stress is prolonged, PLK1 determines the switch from 53BP1-mediated pathway to the BRCA1-cleavage pathway (61).
These findings are consistent with PLK1 regulatory function not only in DDR through the HR pathway, but also in the replication stress response when replication origins stall in the presence of a DSB. In the metazoan, the ATM/ATR-dependent intra-S-phase checkpoint usually inhibits the firing of new replication origins, but in the presence of replication stress the dormant origins can be activated, following transient suppression of the intra-S-phase checkpoint, in order to preserve genomic stability (62). ATM/ATR phosphorylate MCM2, promoting PLK1 binding to the MCM complex (63, 64). PLK1 and the MCM complex, localized at the stalled RFs, promote the release of CHK1-mediated suppression of nearby dormant origins, even if this later interaction is not completely explained. PLK1-mediated phosphorylation of RAD51 is important for the protection of nascent ssDNA at stalled RFs following hydroxyurea (HU) treatment, in a BRCA2-dependent manner, from late-S-phase to mitosis, to improve genomic stability (65). All this evidence supports the critical role of PLK1 in DDR and consequently in maintaining genomic stability.
EMT is a process by which epithelial cells lose their characteristics (e.g. cell-cell and cell-extracellular matrix adhesion, cell polarity and morphology) and acquire a mesenchymal cell phenotype (fibroblast-like morphology, increased cell-matrix adhesions, and motility). Although EMT is involved in fundamental early processes including embryonic development, tissue formation and tissue fibrosis, it has also be implicated in tumor cell growth and proliferation, drug resistance and metastasis formation (66–68). In the last years, PLK1 emerged as one of the triggers of this process (69) (Figure 2).
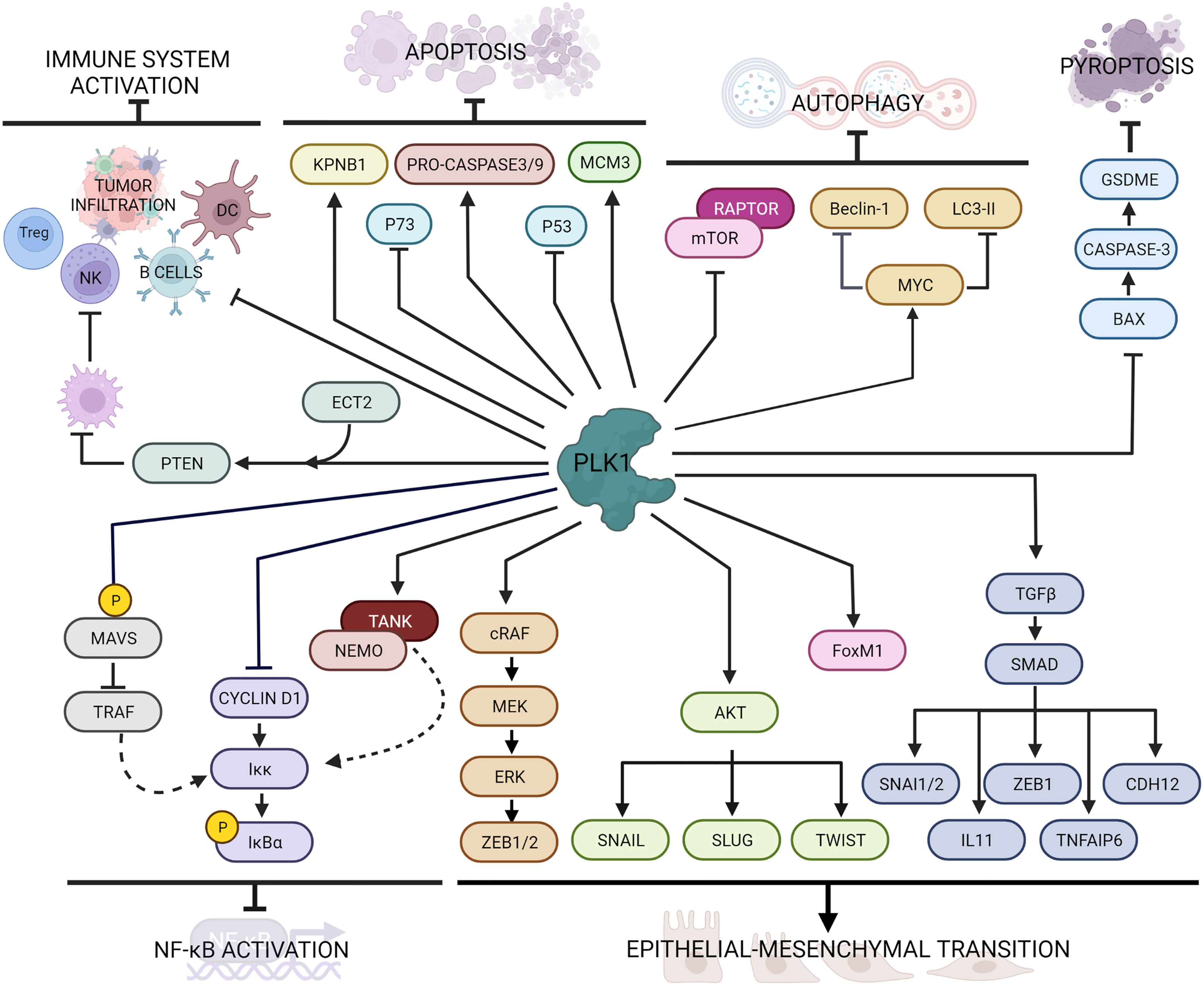
Figure 2 Main PLK1 functions in cellular processes beyond mitosis and DNA damage response. PLK1 interacts with several intracellular factors regulating cell death pathways (i.e. apoptosis, autophagy and pyroptosis), immune response, inflammation and epithelial to mesenchymal transition.
The molecular evidence of EMT is downregulation of epithelial markers, such as E-cadherin and some cytokeratin isoforms, and the overexpression of mesenchymal markers, such as N-cadherin and vimentin (70). PLK1 overexpression in prostate epithelial cells was associated with downregulation of E-cadherin and cytokeratin 19 and upregulation of N-cadherin, vimentin, fibronectin, and SM22, while its downregulation enhanced epithelial characteristics, reversed the EMT and inhibited cell motility (71). A comparison between cells expressing constitutively active wild-type or kinase-defective protein indicated that the induction of EMT requires PLK1-mediated phosphorylation events. In particular, PLK1 phosphorylates cRAF, which induces the MEK/ERK cascade eventually activating ZEB1 and ZEB2 transcription factors, leading to the expression of EMT genes (71).
The PLK1-driven EMT has also been reported in gastric carcinoma cells, where the overexpression of PLK1 induced down-regulation of E-cadherin and up-regulation of N-cadherin, Slug and Twist, through the induction of PLK1-mediated AKT phosphorylation (72). Instead, in non-small cell lung cancer (NSCLC) the role of PLK1 in promoting EMT and metastasis formation correlated with upregulation of the TGFβ/SMAD pathway (73, 74). Shin et al. demonstrated that the active PLK1 (p-Thr210 PLK1) is abundant in TGFβ-induced metastatic NSCLC and its presence promoted in vivo metastasis formation; active PLK1 led to an increase of the levels of TGFβ cascade effectors, including SNAI1, SNAI2, ZEB1, CDH2, IL11, and TNFAIP6, whose depletion resulted in a reversion of the EMT induced by active PLK1 (73). Moreover, PLK1 is responsible of the phosphorylation of FoxM1 (31), another promoter of EMT in different tumor types including lung (75, 76), prostate (77), gastrointestinal cancer (78), pancreas (79), glioblastoma (80) and glioma (81). Taken together, PLK1 activity promotes the epithelial-mesenchymal transition in different type of tumors.
Cell death through regulated molecular pathways (i.e. autophagy and apoptosis) occurs in physiological conditions in order to preserve the organism’s homeostasis. However, in cancer cells these pathways are impaired, especially after drug treatment (82, 83).
PLK1 has a role of in the inhibition of cell death pathways including autophagy and apoptosis (Figure 2). Autophagy is a conserved, adaptive process of self-degradation for the maintenance of cellular homeostasis, that can also occur in response to cellular stress (i.e. nutrient deprivation, growth factor depletion, infection, hypoxia) (84). Alterations in this pathway can lead to the initiation and development of tumors. Among the signaling pathways involved in this process, the mTOR (mammalian target of rapamycin) pathway is the most important and conserved. The Ser/Thr kinase mTOR and its regulatory protein RAPTOR forms the multiprotein complex mTORC1, which can inhibit autophagy under normal conditions (85). In HeLa cells PLK1 was identified as a physical interactor of mTORC1 by mTOR direct binding (86). In that study, Ruf et al. demonstrated that PLK1 and mTORC1 co-localize in the lysosomes and that the inhibition of PLK1 by shRNA or drug treatment promoted mTOR lysosomal localization and reduced autophagy, while PLK1 overexpression inhibited mTORC1 and contributed positively to autophagy.
In glioma cells, knock-out of PLK1 by shRNA had an inhibitory effect on autophagy through phosphorylation of the mTORC1 substrate RPS6KB (87). On the other hand, in esophageal squamous cell carcinoma (ESCC) (88) and acute myeloid leukemia (AML) cells (89) there was an opposite correlation between mTOR and PLK1 in autophagy. In ESCC, the suppression of PLK1 downregulated mTOR activity, suggesting that PLK1 activates the mTOR signaling pathway both in vitro and in vivo. Similarly, in AML cells, the inhibition of PLK1 led to autophagy induction through mTORC1 dephosphorylation.
In a recent study by Wang et al. PLK1 inhibition suppressed radiation-induced autophagy in breast cancer cells, while its overexpression increased it (90). In osteosarcoma cells, PLK1 overexpression led to downregulation of autophagy-related proteins such as Beclin-1 and LC3-II (91) by regulating MYC stabilization (92). Similarly, in ovarian clear cells carcinoma, PLK1 silencing resulted in lower LC3B-II induction with an impairment of autophagy (93). In that study, PLK1 knock-down correlated with an increase of apoptotic cells due to an increase of caspase 3 cleavage. These contrasting evidence may suggest that the PLK1-mediated regulation of autophagy may be cell type-specific, but further investigations are still needed.
Apoptosis is a programmed cell death pathway that leads to orderly and efficient removal of damaged cells, such as those following DNA damage or during development (94). The apoptosis machinery is complex and involves many signaling pathways, whose alterations result in deregulation of development, progression of cancer, and tumor resistance to therapy, and has been listed as an hallmark of cancer (95).
The tumor-suppressor p53 mediates apoptosis by activating mitochondrial and death receptor-induced apoptotic pathways with the activation of caspase signaling (96). Ando et al. not only demonstrated the physical interaction between PLK1 and p53, but also showed that the pro-apoptotic function of p53 was inhibited by the kinase activity of PLK1 (97). In castration-resistant prostate cancer cells the inhibition of PLK1 by the small molecule BI2536 resulted in an increased p53-induced cellular death (98). Recently, in TNBC too, the downregulation of PLK1 by the microRNA miR-183-5p resulted in an increase of apoptosis through the DNMT1-p53 axis (99).
As a member of the p53 family, p73 as well can induce apoptosis (100). PLK1 inhibits p73 pro-apoptotic activity by phosphorylating at Thr27 (101), while its silencing through small interference RNA led to increased p73 levels (102).
PLK1 seems also to interfere with caspase activity. During mitosis cyclinB1/CDK1 complex phosphorylates the pro-caspase-8, generating a phospho-epitope for the binding of PLK1, that interferes with caspase-8 auto-activation resulting in inhibition of apoptosis (103). In human airway smooth muscle cells the expression of PLK1 correlated with increased levels of pro-caspase-9 and -3, as indicated by the enhanced apoptosis resulting from PLK1 knockdown (104).
A regulatory role of PLK1 in cell death has also been described through action on other interactors. In renal carcinoma cells PLK1 suppressed apoptosis by phosphorylating the mini-chromosome maintenance 3 protein (MCM3) (105), while in lung adenocarcinoma it regulated the expression of karyopherin beta 1 (KPNB1) (106). However, in contrast with previous evidence, in some other studies PLK1 seemed to promote cell death. It was reported that PLK1 phosphorylates the Fas-associated death domain (FADD) after treatment with taxol, triggering caspase-mediated cell death (107, 108). In HeLa cells, phosphorylated FADD induces not only caspase-8, but also causes proteasomal degradation of PLK1 as a negative feedback loop (107). Similarly, Gupta et al. demonstrated that PLK1 modulates the phosphorylation of the receptor-interacting protein kinase 3 (RIPK3) involved in the induction of different cell death pathways (109).
Beside apoptosis, pyroptosis (or inflammatory cell necrosis) is another programmed cell death pathway activated by the inflammatory caspases (caspase-1 and caspase-11 in mice and caspase-1, caspase-4, and caspase-5 in humans) (110). Activation of this pathway causes cells to start swell, with a rapid destabilization of plasma membrane integrity, leading to the release of cell content including danger-associated molecular patterns (DAMPs) and cytokines that trigger a robust inflammatory response (111). Pyroptosis can also influence the proliferation, invasion and metastasis of different tumors, as recently reviewed (112). Inhibition of PLK1 by BI2536 treatment in esophageal squamous cell carcinoma (ESCC) induced pyroptosis both in vitro and in vivo through the BAX/caspase-3/GSDME pathway and boosted the sensitivity to cisplatin (113).
In recent years, the immune response has emerged as an important factor in tumor progression and treatment (114, 115). The possible role of PLK1 in modulating immunity has also been investigated.
From a screening in more than 30 different cancers, a correlation was seen between high levels of PLK1 and inhibition of immune cell infiltration and antitumor immunity (116). In particular, tumors with elevated expression of PLK1 displayed lower immune activity, such as lower expression of Human Leukocyte Antigens (HLA), fewer B cells, NK cells and tumor-infiltrating lymphocytes and reduction of T-regulatory cells. In addition, in vitro treatment with PLK1 inhibitors upregulated the expression of HLA molecules in different cancer cells. In lung cancer PLK1 expression not only negatively correlated with numerous immune cell lineages, but was also crucial in antigen processing and presentation (117). Its inhibition with BI2536 resulted in increased maturation of dendritic cells (DC) and enrichment of T cells infiltration, and promoted immune cell infiltration and activation in vivo, supporting the possibility that PLK1 blockade may act as an immune activator. In hepatocellular carcinoma the PLK1/PTEN axis activated by the cell transformation sequence 2 (ECT2) protein promoted M2 macrophage polarization, resulting in suppression of both NK and T cells functions (118).
The nuclear factor kappa light chain enhancer of activated B cells (NF-κB) is an ubiquitous transcription factor known for its role in the regulation of inflammation and innate immunity, since its stimulation triggers the expression of inflammatory mediators including cytokines, chemokines and cell adhesion molecules (119). The activity of NF-κB is tightly regulated through inhibitory IκB proteins and the kinase that phosphorylates IκBs, namely, the IκB kinase (IKK) complex. Stimulation through TLR4, TNF-α receptor (TNFR) and interleukin-1 receptor (IL-1R) activation leads to phosphorylation of IKK and release of NF-κB dimers (119).
In the last few years, PLK1 has been reported as a negative regulator of NF-κB transcriptional activation. Constitutively active expression of PLK1 in mammalian cells reduced tumor necrosis factor (TNF)-induced IKK activation, through inhibition of cyclin D1 expression, resulting in decreased phosphorylation of endogenous IκBα, and consequently reduced NF-κB activation (120). However, the suppression by PLK1 on the NF-κB signaling pathway was also promoted by the interaction of PLK1 with the TRAF-associated NF-κB activator (TANK) (121). Mechanistically, PLK1 binds the IKK adaptor protein NEMO, preventing its ubiquitination through the formation of a ternary complex with TANK, negatively regulating the TNF-induced IKK activation.
PLK1 also regulates the NF-κВ and the interferon regulatory factor 3 (IRF3) pathway by modulating mitochondrial antiviral-signaling (MAVS) protein activity (122). Briefly, the PBD domain of PLK1 associates with two different domains of MAVS in both dependent and independent phosphorylation events. The phospho-independent binding strongly disrupts the association of MAVS with its downstream partner TRAF3, which is essential for activation of an alternative IKK complex responsible for IRF3 phosphorylation. In Figure 2 are graphically summarized the interactions of PLK1 with factors involved in the immune response and NF-κB activator pathway.
Considering the pleiotropic roles of PLK1 in many cellular pathways whose involvement in cancer has been clearly demonstrated, PLK1 is a potential therapeutic target and in the last decade various PLK1 inhibitors have been developed by drug companies and academic research groups.
Two types of small molecule have been developed as PLK1 inhibitors: ATP-competitors target the kinase domain of the protein and non-ATP competitors target the PBD domain. Several drugs targeting the ATP-binding domain have been identified and some have progressed to clinical trials. However, there are important potential drawbacks with these molecules as they also inhibit the catalytic domain of the other PLKs; this could increase the toxic side effects and, in some cases-considering the different possible contrasting effects of the PLKs- might reduce the antitumor effects. In addition, like with other ATP competitors, point mutation (C67V) in the ATP-binding domain can confer resistance to other structurally unrelated ATP-inhibitors (123). Inhibitors targeting the PBD domain need to be more specific, as the PLK1-3 PBDs have unique substrate specificity allowing the design of target therapeutics (124), and could potentially suggest a novel biological basis on the molecular recognition of PLK1 and its substrates.
Different compounds have been synthesized (6, 125, 126). At the moment there are more than ten available PLK1 specific inhibitors, four of which (BI2536, BI6727-volasertib, GSK461364 and NMS-1286937-onvasertib- all ATP competitors) have reached the clinical trials (listed in Table 1).

Table 1 Clinical trials based on PLK1 inhibitors.
BI2536 is an adenosine triphosphate (ATP)–competitive kinase inhibitor derived from the novel chemical series of dihydropteridinone (140). It is a potent PLK1 inhibitor. BI2536 induces G2/M arrest and the formation of abnormal mitotic figures, such as monopolar spindles (141). Its effect has been observed in vitro, at nanomolar concentrations, and also in vivo (at a nanomolar concentration as well) with an acceptable safety profile (140). In Phase I studies, BI2536’s dose-limiting toxicity was reversible neutropenia, the most frequent adverse event at the maximum tolerated dose (grade 3 to 4; 56%); nausea, fatigue and anorexia were also frequent, but mostly mild to moderate (127). The toxicity profile was similar when combined with pemetrexed in NSCLC patients (142). These studies hinted at antitumor activities. However, in 21 patients with relapsed small-cell lung cancer enrolled in a Phase II study with BI2536, no responses were observed and disease progressed in all the patients (129).
No objective response or considerable tumor regression was observed in patients with advanced solid tumors (colorectal, melanoma, hepatoma and ovarian cancer) in a Phase I study (128). There was also no activity in chemo-naïve patients with unresectable exocrine adenocarcinoma of the pancreas (6). All these data were discouraging and have not fostered any further drug clinical investigation.
Volasertib (BI6727) is an ATP-competitive kinase inhibitor belonging to the same dihydropteridinone class as BI2536 (143), whose development was discontinued in favor of volasertib. Volasertib cytotoxic effects have been observed at nanomolar concentrations (144) in acute myeloid leukemia cells (144), and in carcinoma cancer cells (145) with both the induction of cell cycle arrest and cell death (146).
Volasertib had a better pharmacokinetic profile than BI2536, with a high volume of distribution, deep tissue penetration, and a long terminal half-life (147). In vivo preclinical data indicated that volasertib has antitumor activities in different tumors with quite a safe toxicological profile (148), fostering its clinical development. Phase I studies in monotherapy defined the maximum tolerated doses (400-450 mg every two or three weeks), with the most frequent side effects being haematological toxicities (anemia, neutropenia, thrombocytopenia), being reversible and manageable with standard care (130, 131). Phase II studies as single agent, however, showed only modest antitumor activity (132, 133).
NMS-1286937 (onvansertib), a pyrazoloquinazoline, is a third generation PLK1 ATP-competitor with an in vitro IC50 of 36 nmol/L, and had a strong cytotoxic effect in AML cells, for which it was originally registered by FDA as orphan drug (149). Onvansertib induced a mitotic cell-cycle arrest followed by apoptosis in cancer cells; it inhibited xenograft tumor growth at well tolerated oral doses (150). In addition, it potentiated cytarabine antitumor activity in a disseminated model of AML (149). On the basis of these promising results a Phase I trial with escalating drug doses was conducted in 21 patients with advanced tumors (134). This allowed the definition of the maximum tolerated dose, and dose limiting toxicities (mainly thrombocytopenia and neutropenia) with disease stabilization in several patients as the best treatment response (134).
GSK461364 is thiophene amide, an ATP-competitor PLK1 inhibitor, which promotes G2/M arrest in tumor tissues (151); in addition, the drug enhanced the radio sensitivity of breast cancer cells in vivo (90). A Phase I trial was conducted with two different schedules and GSK461364 doses in 40 patients with solid tumors; the dose-limiting toxicities were haematological (neutropenia and trombocytopenia) and venous thrombotic emboli; prolonged disease stabilization was the best activity reported in 15% of patients, including four with esophageal cancers (135).
Tak960 is a recently synthesized ATP-competitor PLK1 inhibitor, orally available and PLK1 selective (152). Tak960 arrests the cell cycle in G2/M phase, and accumulates cells with aberrant spindles (153). It has been shown in vitro cytototoxic activity in different cells and xenograft models, with favourable tolerability and PK/PD profiles (153). It is currently under Phase I investigation in advanced non-haematological malignancies (NCT01179399).
Rigosertib (ON01910) is a benzyl sulfone analog that acts as a Ras mimetic, and non- ATP- competitive molecule inhibiting both PLK1 and PI3K (154). Rigosertib induces mitotic arrest in different cancer cells by inducing spindle abnormalities, cell cycle arrest and apoptosis (155–157).
In Phase I/II trials was well tolerated and showed some activity in selected patients with advanced solid tumors (136, 137). Rigosertib had good tolerance in high-risk myelodysplastic syndrome and AML, with hints of antitumor activity (138). The most common side effect was urinary toxicity (139). In a Phase I trial, rigosertib’s most common side effects involved urothelial irritation and the dose-limiting toxicities were haematuria and dysuria (139).
Inhibition of the PBD domain of PLK1 is another strategy to target PLK1. As said, this domain is a docking site for phosphorylated substrates and is a druggable interface, as demonstrated in different studies (139, 154, 158). Poloxin, a synthetic thymoquinone derivative, was one of the first compounds shown to interfere with the interaction between the PBD of PLK1 and an optimal phosphor-peptide (126). It was later seen that also thymoquinone, with its similar chemical structure, had similar effects. Poloxin caused centrosome fragmentation, abnormal spindle, chromosome misalignment, mitotic arrest, and apoptosis in cancer cell lines and had a significantly effect in suppressing xenograft growth in vivo (159).
Different inhibitors of PLK1 have been generated through high-throughput screening approaches (9, 126, 155). However, many of these molecules could not be further developed because they had only modest activity in preclinical models and were shown to be non-specific protein alkylators (160). Recently, using REPLACE (Replacement with Partial Ligand Alternatives through Computational Enrichment), PLK1 inhibitors have been synthesized and showed to have promising PBD binding activity and cytotoxic activity in in vitro cell lines (6).
The use of PLK1 inhibitors has been widely used in preclinical studies, but has not been successfully translated to the clinic on account of its limited effect and resistance [reviewed in (161)]. To overcome this, researchers have started to investigate the possibility of combining PLK1 inhibitors with other agents. Combination therapy offers the opportunity to target different pathways, to eliminate different cancer cell populations, and possibly obtain additive/synergistic anticancer effects.
Investigations have also been examined possibility to boosting the response to therapy in different cancers by combining PLK1 inhibitors with chemotherapies (Table 2). The most widely studied PLK1 inhibitors were BI2536, volasertib and onvansertib. The ability of PLK1 inhibitors to induce G2/M cell cycle arrest was exploited in cisplatin-resistant gastric cancer cells, where the combination of BI2536 and cisplatin inhibited cell growth and invasion ability (162). Volasertib too potentiated the activity of cisplatin in cervical cancer (167).
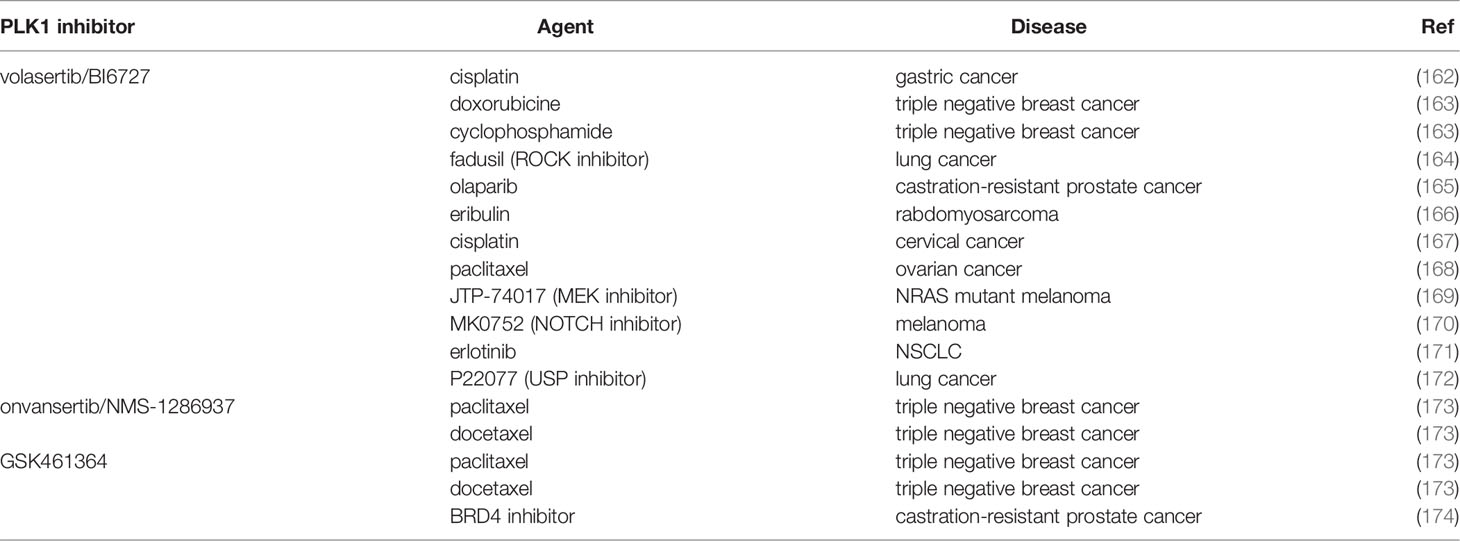
Table 2 Combination strategies with PLK1 inhibitors: in vitro data.
In ovarian cancer cells with CCNE1 amplification the combination of volasertib with paclitaxel synergistically triggered mitotic arrest, initiating mitochondrial apoptosis (168). Onvansertib too showed synergistic activity with paclitaxel, as reported by different groups, including ours (173, 175). Giordano et al. tested onvansertib and another PLK1 inhibitor (GSK461364) in combination with taxanes (paclitaxel and docetaxel) in a set of triple negative breast cancer cell lines in vitro and in vivo. Both the PLK1 inhibitors synergized with taxanes specifically inhibiting the G2/M transition, inducing aberrant mitotic exit and apoptosis, and also eliminating stem-like resistant clones (173). In another work, our group further showed that onvansertib and paclitaxel acted synergistically both in vitro and in vivo in xenografts, causing tumor regression and tumor growth inhibition in a model of mucinous ovarian cancer (175). The PLK1 inhibitor GSK461364 was also tested in combination with a BRD4 small inhibitor in castration-resistant prostate cancer both in vitro and in vivo, showing a strong synergistic effect (174).
Dual targeting of mitosis caused a synergistic induction of apoptosis for BI2536-eribulin co-treatment in rhabdomyosarcoma in vitro (166). In a triple negative breast cancer, the PLK1 inhibitor BI2536 impaired tumor growth also in vivo as single agent, but when combined with doxorubicin and cyclophosphamide this treatment gave a faster complete response and prevented relapses (163).
The possibility of combining PLK1 inhibitors to increase its efficacy and avoid the development of resistance was also tested in combination with targeted therapies. BI2536 was combined in tumors with defined pathways alterations (i.e. KRAS mutated cancers). Wang et al. showed that the inhibition of PLK1 and ROCK in KRAS-mutated lung cancer cells (but not in the wild type), through the upregulation of p21 protein, reduced viability (164). BI2536 was also tested with olaparib, a FDA-approved PARP inhibitor, mostly used in BRCA-deficient tumors and this combination synergistically inhibited the growth of xenograft tumors derived from BRCA-mutated castration-resistant prostate cancer (165).
In NRAS mutant melanoma the combination of volasertib and a MEK inhibitor (JTP-74017) had antitumor effects both in vitro and in vivo (169). Again, the synergistic effect was due to cell cycle arrest and greater induction of apoptosis. Su et al. found that the expression of PLK1 and NOTCH was associated with poor overall and disease-free survival in melanoma, and the combination of BI6727 with the NOTCH inhibitor MK0752 resulted in a synergistic antiproliferative response in BRAF mutated, BRAF and TP53 mutated, and NRAS mutated melanoma cells (170).
In different NSCLC cells mutated in the epidermal growth factor receptor (EGFR) volasertib reversed resistance to erlotinib, causing G2/M arrest and apoptosis, and reduced tumor growth in vivo (171). Volasertib had a strong synergistic effect also when combined with a USP7 inhibitor, counteracting resistance to taxane. In paclitaxel resistant lung cancer cells the combination counteracted the resistance to mitotic catastrophe through downregulation of MDR1/ABCB1 protein (172).
All these preclinical studies uphold the use of PLK1 inhibitor combinations in clinic (Table 3). In a Phase I trial BI6727 was combined with escalating doses of decitabine to investigate the maximum tolerated dose, safety and pharmacokinetics (176). The drug was also tested in a Phase II trial in AML in combination with low-dose cytarabine (177). Patients treated with the combination had a higher response rate (31%) than with low-dose cytarabine monotherapy (13%). This study launched a Phase III trial (NCT01721876), which aimed to investigate the efficacy, safety, and pharmacokinetics of volasertib with low-dose cytarabine in patients over 65 years of age with untreated AML (178). In 2013 the Food and Drug Administration granted volasertib breakthrough therapy status for combined treatment with cytarabine in AML.
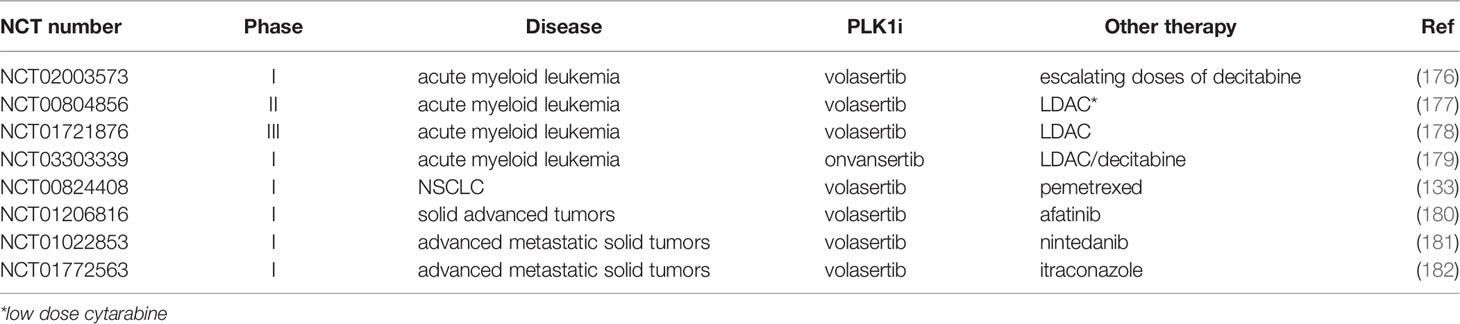
Table 3 Clinical trials based on PLK1 inhibition combination therapy.
Escalating doses of the third-generation PLK1 inhibitor onvansertib were tested in a Phase Ib study, alone and in combination with low-dose cytarabine (LDAC) or decitabine for AML. The combination was well tolerated and achieved a 24% complete remission rate (5 of the 21 evaluable patients) (179), supporting its further investigation in the ongoing phase II trial. Recently onvansertib was granted by a Fast Track Designation for the second-line treatment of patients with KRAS-mutant metastatic colorectal cancer in combination with 5-fluorouracil, leucovorin, irinotecan and bevacizumab (183).
The combination of PLK1 inhibitors with other drugs was also studied in solid tumors, but so far, they have not gone beyond Phase I. The combination of volasertib with pemetrexed for advanced/metastatic NSCLC did not have any greater toxicity, but did not improve the efficacy compared with pemetrexed single-agent (133). A combination of volasertib and afatinib (an oral ERBB family blocker) was tested in a Phase I trial in 57 patients with advanced solid tumors. However, only two patients achieved partial responses and eight experienced stable disease (180). Volasertib was recently combined with nintedanib a potent inhibitor of PDGF, VEGF and bFGF receptor, in patients with advanced solid tumors in a Phase I dose escalation study. It gave a well-tolerated safety profile with no unexpected or overlapping side effects and with significant tumor stabilization (181).
PLK1 is the most studied of the PLKs. Its main role is in the progression of mitosis, with an established regulatory function in mitotic entry, maturation of the centrosome, spindle assembly and cytokinesis. Recent works implicate PLK1 in many of the cellular pathways as we have discussed briefly. While PLK1 mutations are extremely rare in human cancers, it is often found overexpressed, especially in advanced cancers. This overexpression is often correlated with aggressiveness and poor patient prognosis. These evidence all have points to PLK1 as a promising therapeutic target in oncology. Small interfering RNA, CRISPR/Cas9 deleted PLK1 and chemical inhibitors of PLK1 have an impact on cell proliferation, cause mitotic arrest, cell death and in vivo tumor growth inhibition.
As summarized here, a number of inhibitors (ATP-competitors) have been synthesized and showed activity in both in vitro and in vivo preclinical models, fostering their clinical development. Their toxicological profiles are quite similar, the most frequent reported dose limiting toxicities in most cases being haematological toxicities (neutropenia and thrombocytopenia). As regards clinical efficacy, the results were not as expected when used in second and higher lines of therapy. However, there were hints of activity in specific subsets of patients and this has allowed volasertib and onvansertib to be granted by FDA as respectively breakthrough therapy and orphan drug status. The various new functions of PLK1 in many different cellular pathways suggest potential new combination approaches aimed at target tumor cell vulnerabilities/hallmarks (for example, with immunotherapy).
Lastly, the search for PLK1 synthetic lethal partners could be another strategy, still little developed. Recent data suggest this could be a complementary, efficacious approach. An unforeseen synthetic lethal interaction has in fact been reported between PLK1 and BRCA1 from screening a kinase inhibitors library (184). The authors found that BRCA1 downregulation and inhibition of PLK1 induced aberrant mitotic phenotypes, centrosomal duplication and altered cytokinesis, resulting in reduced clonogenicity of these cells. These data suggest the use of PLK1 inhibitors in subsets of patients (BRCA1-mutated, in triple negative breast and ovarian cancer). Emerging evidence also suggest that tumors with activated KRAS seem to be addicted to PLK1 activity (185) potentially opening the way to target KRAS mutated tumors with PLK1 inhibitors.
MC, SP, GD, FG, FR and MB contributed conception and design of the review; MC, SP, GD, FG and FR wrote the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
We acknowledge the support by the Italian Association for Cancer Research (AIRC, IG19797 project, PI GD; AIRC, IG24347, PI MB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Judith Baggott for English revision.
1. Goroshchuk O, Kolosenko I, Vidarsdottir L, Azimi A, Palm-Apergi C. Polo-Like Kinases and Acute Leukemia. Oncogene (2019) 38:1–16. doi: 10.1038/s41388-018-0443-5
2. Liu Z, Sun Q, Wang X. PLK1, A Potential Target for Cancer Therapy. Trans Oncol (2017) 10:22–32. doi: 10.1016/j.tranon.2016.10.003
3. Yu C, Luo D, Yu J, Zhang M, Zheng X, Xu G, et al. Genome-Wide CRISPR-Cas9 Knockout Screening Identifies GRB7 as a Driver for MEK Inhibitor Resistance in KRAS Mutant Colon Cancer. Oncogene (2022) 41:191–203. doi: 10.1038/s41388-021-02077-w
4. Rödel F, Keppner S, Capalbo G, Bashary R, Kaufmann M, Rödel C, et al. Polo-Like Kinase 1 as Predictive Marker and Therapeutic Target for Radiotherapy in Rectal Cancer. Am J Pathol (2010) 177:918–29. doi: 10.2353/ajpath.2010.100040
5. Hagege A, Ambrosetti D, Boyer J, Bozec A, Doyen J, Chamorey E, et al. The Polo-Like Kinase 1 Inhibitor Onvansertib Represents a Relevant Treatment for Head and Neck Squamous Cell Carcinoma Resistant to Cisplatin and Radiotherapy. Theranostics (2021) 11:9571–86. doi: 10.7150/thno.61711
6. Craig SN, Baxter M, Chapagai D, Stafford JM, Nurmemmedov E, Altomare D, et al. Structure-Activity and Mechanistic Studies of non-Peptidic Inhibitors of the PLK1 Polo Box Domain Identified Through REPLACE. Eur J Med Chem (2022) 227:113926. doi: 10.1016/j.ejmech.2021.113926
7. Iliaki S, Beyaert R, Afonina IS. Polo-Like Kinase 1 (PLK1) Signaling in Cancer and Beyond. Biochem Pharmacol (2021) 193:114747. doi: 10.1016/j.bcp.2021.114747
8. Raab CA, Raab M, Becker S, Strebhardt K. Non-Mitotic Functions of Polo-Like Kinases in Cancer Cells. Biochim Biophys Acta Rev Cancer (2021) 1875:188467. doi: 10.1016/j.bbcan.2020.188467
9. Su S, Chhabra G, Singh CK, Ndiaye MA, Ahmad N. PLK1 Inhibition-Based Combination Therapies for Cancer Management. Transl Oncol (2022) 16:101332. doi: 10.1016/j.tranon.2021.101332
10. Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, et al. M-Phase Kinases Induce Phospho-Dependent Ubiquitination of Somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U.S.A. (2004) 101:4419–24. doi: 10.1073/pnas.0307700101
11. Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E. Identification of a Consensus Motif for Plk (Polo-Like Kinase) Phosphorylation Reveals Myt1 as a Plk1 Substrate. J Biol Chem (2003) 278:25277–80. doi: 10.1074/jbc.C300126200
12. Lobjois V, Jullien D, Bouché J-P, Ducommun B. The Polo-Like Kinase 1 Regulates CDC25B-Dependent Mitosis Entry. Biochim Biophys Acta (2009) 1793:462–8. doi: 10.1016/j.bbamcr.2008.12.015
13. Toyoshima-Morimoto F, Taniguchi E, Nishida E. Plk1 Promotes Nuclear Translocation of Human Cdc25C During Prophase. EMBO Rep (2002) 3:341–8. doi: 10.1093/embo-reports/kvf069
14. Archambault V, Glover DM. Polo-Like Kinases: Conservation and Divergence in Their Functions and Regulation. Nat Rev Mol Cell Biol (2009) 10:265–75. doi: 10.1038/nrm2653
15. Schmucker S, Sumara I. Molecular Dynamics of PLK1 During Mitosis. Mol Cell Oncol (2014) 1:e954507. doi: 10.1080/23723548.2014.954507
16. Shakeel I, Basheer N, Hasan GM, Afzal M, Hassan MI. Polo-Like Kinase 1 as an Emerging Drug Target: Structure, Function and Therapeutic Implications. J Drug Target (2021) 29:168–84. doi: 10.1080/1061186X.2020.1818760
17. Lowery DM, Clauser KR, Hjerrild M, Lim D, Alexander J, Kishi K, et al. Proteomic Screen Defines the Polo-Box Domain Interactome and Identifies Rock2 as a Plk1 Substrate. EMBO J (2007) 26:2262–73. doi: 10.1038/sj.emboj.7601683
18. Seki A, Coppinger JA, Jang C-Y, Yates JR, Fang G. Bora and the Kinase Aurora a Cooperatively Activate the Kinase Plk1 and Control Mitotic Entry. Science (2008) 320:1655–8. doi: 10.1126/science.1157425
19. Kang YH, Park J-E, Yu L-R, Soung N-K, Yun S-M, Bang JK, et al. Self-Regulated Plk1 Recruitment to Kinetochores by the Plk1-PBIP1 Interaction is Critical for Proper Chromosome Segregation. Mol Cell (2006) 24:409–22. doi: 10.1016/j.molcel.2006.10.016
20. Watanabe N, Arai H, Iwasaki J-I, Shiina M, Ogata K, Hunter T, et al. Cyclin-Dependent Kinase (CDK) Phosphorylation Destabilizes Somatic Wee1 via Multiple Pathways. Proc Natl Acad Sci U.S.A. (2005) 102:11663–8. doi: 10.1073/pnas.0500410102
21. Kachaner D, Garrido D, Mehsen H, Normandin K, Lavoie H, Archambault V. Coupling of Polo Kinase Activation to Nuclear Localization by a Bifunctional NLS is Required During Mitotic Entry. Nat Commun (2017) 8:1701. doi: 10.1038/s41467-017-01876-8
22. Beck J, Maerki S, Posch M, Metzger T, Persaud A, Scheel H, et al. Ubiquitylation-Dependent Localization of PLK1 in Mitosis. Nat Cell Biol (2013) 15:430–9. doi: 10.1038/ncb2695
23. Raab M, Matthess Y, Raab CA, Gutfreund N, Dötsch V, Becker S, et al. A Dimerization-Dependent Mechanism Regulates Enzymatic Activation and Nuclear Entry of PLK1. Oncogene (2022) 41:372–86. doi: 10.1038/s41388-021-02094-9
24. Combes G, Alharbi I, Braga LG, Elowe S. Playing Polo During Mitosis: PLK1 Takes the Lead. Oncogene (2017) 36:4819–27. doi: 10.1038/onc.2017.113
25. Harper JW, Elledge SJ. The DNA Damage Response: Ten Years After. Mol Cell (2007) 28:739–45. doi: 10.1016/j.molcel.2007.11.015
26. Carrassa L, Colombo I, Damia G, Bertoni F. Targeting the DNA Damage Response for Patients With Lymphoma: Preclinical and Clinical Evidences. Cancer Treat Rev (2020) 90:102090. doi: 10.1016/j.ctrv.2020.102090
27. Li H, Wang Y, Liu X. Plk1-Dependent Phosphorylation Regulates Functions of DNA Topoisomerase IIalpha in Cell Cycle Progression. J Biol Chem (2008) 283:6209–21. doi: 10.1074/jbc.M709007200
28. Takeda DY, Dutta A. DNA Replication and Progression Through S Phase. Oncogene (2005) 24:2827–43. doi: 10.1038/sj.onc.1208616
29. Mandal R, Strebhardt K. Plk1: Unexpected Roles in DNA Replication. Cell Res (2013) 23:1251–3. doi: 10.1038/cr.2013.130
30. Lemmens B, Hegarat N, Akopyan K, Sala-Gaston J, Bartek J, Hochegger H, et al. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol Cell (2018) 71:117–128.e3. doi: 10.1016/j.molcel.2018.05.026
31. Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, et al. Plk1-Dependent Phosphorylation of FoxM1 Regulates a Transcriptional Programme Required for Mitotic Progression. Nat Cell Biol (2008) 10:1076–82. doi: 10.1038/ncb1767
32. Gheghiani L, Loew D, Lombard B, Mansfeld J, Gavet O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell Rep (2017) 19:2060–73. doi: 10.1016/j.celrep.2017.05.031
33. Song R, Hou G, Yang J, Yuan J, Wang C, Chai T, et al. Effects of PLK1 on Proliferation, Invasion and Metastasis of Gastric Cancer Cells Through Epithelial-Mesenchymal Transition. Oncol Lett (2018) 16:5739–44. doi: 10.3892/ol.2018.9406
34. Jares P, Donaldson A, Blow JJ. The Cdc7/Dbf4 Protein Kinase: Target of the S Phase Checkpoint? EMBO Rep (2000) 1:319–22. doi: 10.1093/embo-reports/kvd076
35. Ciardo D, Haccard O, Narassimprakash H, Cornu D, Guerrera IC, Goldar A, et al. Polo-Like Kinase 1 (Plk1) Regulates DNA Replication Origin Firing and Interacts With Rif1 in Xenopus. Nucleic Acids Res (2021) 49:9851–69. doi: 10.1093/nar/gkab756
36. Barr FA, Silljé HHW, Nigg EA. Polo-Like Kinases and the Orchestration of Cell Division. Nat Rev Mol Cell Biol (2004) 5:429–40. doi: 10.1038/nrm1401
37. Potapova TA, Sivakumar S, Flynn JN, Li R, Gorbsky GJ. Mitotic Progression Becomes Irreversible in Prometaphase and Collapses When Wee1 and Cdc25 are Inhibited. Mol Biol Cell (2011) 22:1191–206. doi: 10.1091/mbc.E10-07-0599
38. Roshak AK, Capper EA, Imburgia C, Fornwald J, Scott G, Marshall LA. The Human Polo-Like Kinase, PLK, Regulates Cdc2/Cyclin B Through Phosphorylation and Activation of the Cdc25c Phosphatase. Cell Signal (2000) 12:405–11. doi: 10.1016/s0898-6568(00)00080-2
39. Petronczki M, Lénárt P, Peters J-M. Polo on the Rise-From Mitotic Entry to Cytokinesis With Plk1. Dev Cell (2008) 14:646–59. doi: 10.1016/j.devcel.2008.04.014
40. Tavernier N, Thomas Y, Vigneron S, Maisonneuve P, Orlicky S, Mader P, et al. Bora Phosphorylation Substitutes in Trans for T-Loop Phosphorylation in Aurora A to Promote Mitotic Entry. Nat Commun (2021) 12:1899. doi: 10.1038/s41467-021-21922-w
41. Thomas Y, Cirillo L, Panbianco C, Martino L, Tavernier N, Schwager F, et al. Cdk1 Phosphorylates SPAT-1/Bora to Promote Plk1 Activation in C. Elegans and Human Cells. Cell Rep (2016) 15:510–8. doi: 10.1016/j.celrep.2016.03.049
42. Carrassa L, Damia G. DNA Damage Response Inhibitors: Mechanisms and Potential Applications in Cancer Therapy. Cancer Treat Rev (2017) 60:139–51. doi: 10.1016/j.ctrv.2017.08.013
43. Aressy B, Ducommun B. Cell Cycle Control by the CDC25 Phosphatases. Anticancer Agents Med Chem (2008) 8:818–24. doi: 10.2174/187152008786847756
44. Shibata A, Jeggo PA. Roles for 53BP1 in the Repair of Radiation-Induced DNA Double Strand Breaks. DNA Repair (Amst) (2020) 93:102915. doi: 10.1016/j.dnarep.2020.102915
45. Turan V, Oktay K. BRCA-Related ATM-Mediated DNA Double-Strand Break Repair and Ovarian Aging. Hum Reprod Update (2020) 26:43–57. doi: 10.1093/humupd/dmz043
46. Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JTF, Tkáč J, et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol Cell (2013) 49:872–83. doi: 10.1016/j.molcel.2013.01.001
47. Lee M, Daniels MJ, Venkitaraman AR. Phosphorylation of BRCA2 by the Polo-Like Kinase Plk1 is Regulated by DNA Damage and Mitotic Progression. Oncogene (2004) 23:865–72. doi: 10.1038/sj.onc.1207223
48. Jang Y-J, Ji J-H, Choi Y-C, Ryu CJ, Ko S-Y. Regulation of Polo-Like Kinase 1 by DNA Damage in Mitosis. Inhibition of Mitotic PLK-1 by Protein Phosphatase 2A. J Biol Chem (2007) 282:2473–82. doi: 10.1074/jbc.M605480200
49. Qin B, Gao B, Yu J, Yuan J, Lou Z. Ataxia Telangiectasia-Mutated- and Rad3-Related Protein Regulates the DNA Damage-Induced G2/M Checkpoint Through the Aurora A Cofactor Bora Protein. J Biol Chem (2013) 288:16139–44. doi: 10.1074/jbc.M113.456780
50. Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 Axis Controls the G2 DNA-Damage-Response Checkpoint. Cell (2008) 134:256–67. doi: 10.1016/j.cell.2008.05.043
51. Zou J, Rezvani K, Wang H, Lee KS, Zhang D. BRCA1 Downregulates the Kinase Activity of Polo-Like Kinase 1 in Response to Replication Stress. Cell Cycle (2013) 12:2255–65. doi: 10.4161/cc.25349
52. Yamashiro S, Yamakita Y, Totsukawa G, Goto H, Kaibuchi K, Ito M, et al. Myosin Phosphatase-Targeting Subunit 1 Regulates Mitosis by Antagonizing Polo-Like Kinase 1. Dev Cell (2008) 14:787–97. doi: 10.1016/j.devcel.2008.02.013
53. Li W, Wang H-Y, Zhao X, Duan H, Cheng B, Liu Y, et al. A Methylation-Phosphorylation Switch Determines Plk1 Kinase Activity and Function in DNA Damage Repair. Sci Adv (2019) 5:eaau7566. doi: 10.1126/sciadv.aau7566
54. Chaudhury I, Koepp DM. Recovery From the DNA Replication Checkpoint. Genes (Basel) (2016) 7:E94. doi: 10.3390/genes7110094
55. Yata K, Lloyd J, Maslen S, Bleuyard J-Y, Skehel M, Smerdon SJ, et al. Plk1 and CK2 Act in Concert to Regulate Rad51 During DNA Double Strand Break Repair. Mol Cell (2012) 45:371–83. doi: 10.1016/j.molcel.2011.12.028
56. Chabalier-Taste C, Brichese L, Racca C, Canitrot Y, Calsou P, Larminat F. Polo-Like Kinase 1 Mediates BRCA1 Phosphorylation and Recruitment at DNA Double-Strand Breaks. Oncotarget (2016) 7:2269–83. doi: 10.18632/oncotarget.6825
57. Peng B, Shi R, Bian J, Li Y, Wang P, Wang H, et al. PARP1 and CHK1 Coordinate PLK1 Enzymatic Activity During the DNA Damage Response to Promote Homologous Recombination-Mediated Repair. Nucleic Acids Res (2021) 49:7554–70. doi: 10.1093/nar/gkab584
58. Nakamura K, Kustatscher G, Alabert C, Hödl M, Forne I, Völker-Albert M, et al. Proteome Dynamics at Broken Replication Forks Reveal a Distinct ATM-Directed Repair Response Suppressing DNA Double-Strand Break Ubiquitination. Mol Cell (2021) 81:1084–1099.e6. doi: 10.1016/j.molcel.2020.12.025
59. Benada J, Burdová K, Lidak T, von Morgen P, Macurek L. Polo-Like Kinase 1 Inhibits DNA Damage Response During Mitosis. Cell Cycle (2015) 14:219–31. doi: 10.4161/15384101.2014.977067
60. Terasawa M, Shinohara A, Shinohara M. Canonical non-Homologous End Joining in Mitosis Induces Genome Instability and is Suppressed by M-Phase-Specific Phosphorylation of XRCC4. PloS Genet (2014) 10:e1004563. doi: 10.1371/journal.pgen.1004563
61. Xu Y, Ning S, Wei Z, Xu R, Xu X, Xing M, et al. 53BP1 and BRCA1 Control Pathway Choice for Stalled Replication Restart. Elife (2017) 6:e30523. doi: 10.7554/eLife.30523
62. Shechter D, Costanzo V, Gautier J. ATR and ATM Regulate the Timing of DNA Replication Origin Firing. Nat Cell Biol (2004) 6:648–55. doi: 10.1038/ncb1145
63. Yoo HY, Shevchenko A, Shevchenko A, Dunphy WG. Mcm2 is a Direct Substrate of ATM and ATR During DNA Damage and DNA Replication Checkpoint Responses. J Biol Chem (2004) 279:53353–64. doi: 10.1074/jbc.M408026200
64. Trenz K, Errico A, Costanzo V. Plx1 is Required for Chromosomal DNA Replication Under Stressful Conditions. EMBO J (2008) 27:876–85. doi: 10.1038/emboj.2008.29
65. Yata K, Bleuyard J-Y, Nakato R, Ralf C, Katou Y, Schwab RA, et al. BRCA2 Coordinates the Activities of Cell-Cycle Kinases to Promote Genome Stability. Cell Rep (2014) 7:1547–59. doi: 10.1016/j.celrep.2014.04.023
66. Babaei G, Aziz SG-G, Jaghi NZZ. EMT, Cancer Stem Cells and Autophagy; The Three Main Axes of Metastasis. Biomed Pharmacother (2021) 133:110909. doi: 10.1016/j.biopha.2020.110909
67. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell (2009) 139:871–90. doi: 10.1016/j.cell.2009.11.007
68. Bracken CP, Goodall GJ. The Many Regulators of Epithelial–Mesenchymal Transition. Nat Rev Mol Cell Biol (2022) 23:89–90. doi: 10.1038/s41580-021-00442-x
69. Fu Z, Wen D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers (Basel) (2017) 9:131. doi: 10.3390/cancers9100131
70. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, et al. Epithelial—mesenchymal and Mesenchymal—Epithelial Transitions in Carcinoma Progression. J Cell Physiol (2007) 213:374–83. doi: 10.1002/jcp.21223
71. Wu J, Ivanov AI, Fisher PB, Fu Z. Polo-Like Kinase 1 Induces Epithelial-to-Mesenchymal Transition and Promotes Epithelial Cell Motility by Activating CRAF/ERK Signaling. eLife (2016) 5:e10734. doi: 10.7554/eLife.10734
72. Cai XP, Chen LD, Song HB, Zhang CX, Yuan ZW, Xiang ZX. PLK1 Promotes Epithelial-Mesenchymal Transition and Metastasis of Gastric Carcinoma Cells. Am J Transl Res (2016) 8:4172–83.
73. Shin S-B, Jang H-R, Xu R, Won J-Y, Yim H. Active PLK1-Driven Metastasis is Amplified by TGF-β Signaling That Forms a Positive Feedback Loop in non-Small Cell Lung Cancer. Oncogene (2020) 39:767–85. doi: 10.1038/s41388-019-1023-z
74. Jang H-R, Shin S-B, Kim C-H, Won J-Y, Xu R, Kim D-E, et al. PLK1/vimentin Signaling Facilitates Immune Escape by Recruiting Smad2/3 to PD-L1 Promoter in Metastatic Lung Adenocarcinoma. Cell Death Differ (2021) 28:2745–64. doi: 10.1038/s41418-021-00781-4
75. Nilsson MB, Sun H, Robichaux J, Pfeifer M, McDermott U, Travers J, et al. A YAP/FOXM1 Axis Mediates EMT-Associated EGFR Inhibitor Resistance and Increased Expression of Spindle Assembly Checkpoint Components. Sci Transl Med (2020) 12:eaaz4589. doi: 10.1126/scitranslmed.aaz4589
76. Kong F-F, Zhu Y-L, Yuan H-H, Wang J-Y, Zhao M, Gong X-D, et al. FOXM1 Regulated by ERK Pathway MediatesTGF-β1-Induced EMT in NSCLC. Oncol Res (2014) 22:29–37. doi: 10.3727/096504014X14078436004987
77. Tang C, Liu T, Wang K, Wang X, Xu S, He D, et al. Transcriptional Regulation of FoxM1 by HIF-1α Mediates Hypoxia-Induced EMT in Prostate Cancer. Oncol Rep (2019) 42:1307–18. doi: 10.3892/or.2019.7248
78. Zhang J, Niu Y, Huang C. Role of FoxM1 in the Progression and Epithelial to Mesenchymal Transition of Gastrointestinal Cancer. Recent Patents Anti-Cancer Drug Discovery (2017) 12:247–59. doi: 10.2174/1574892812666170424144352
79. Huang C, Xie D, Cui J, Li Q, Gao Y, Xie K. FOXM1c Promotes Pancreatic Cancer Epithelial-To-Mesenchymal Transition and Metastasis via Upregulation of Expression of the Urokinase Plasminogen Activator System. Clin Cancer Res (2014) 20:1477–88. doi: 10.1158/1078-0432.CCR-13-2311
80. Wang Z, Zhang S, Siu TL, Huang S. Glioblastoma Multiforme Formation and EMT: Role of FoxM1 Transcription Factor. Curr Pharm Des (2015) 21:1268–71. doi: 10.2174/1381612821666141211115949
81. Zhang X LV Q-L, Huang Y-T, Zhang L-H, Zhou H-H. Akt/FoxM1 Signaling Pathway-Mediated Upregulation of MYBL2 Promotes Progression of Human Glioma. J Exp Clin Cancer Res (2017) 36:105. doi: 10.1186/s13046-017-0573-6
82. Kashyap D, Garg VK, Goel N. “Chapter Four - Intrinsic and Extrinsic Pathways of Apoptosis: Role in Cancer Development and Prognosis.,”. In: Donev R, editor. Advances in Protein Chemistry and Structural Biology. Apoptosis in Health and Disease - Part A. Academic Press (2021). p. 73–120. doi: 10.1016/bs.apcsb.2021.01.003
83. Li Y-J, Lei Y-H, Yao N, Wang C-R, Hu N, Ye W-C, et al. Autophagy and Multidrug Resistance in Cancer. Chin J Cancer (2017) 36:52. doi: 10.1186/s40880-017-0219-2
84. Dikic I, Elazar Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat Rev Mol Cell Biol (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
85. Wang Y, Zhang H. utophagy: Biology and Diseases: Basic Science. Advances in Experimental Medicine and Biology. A (2019) 1206:67–83. doi: 10.1007/978-981-15-0602-4_3
86. Ruf S, Heberle AM, Langelaar-Makkinje M, Gelino S, Wilkinson D, Gerbeth C, et al. PLK1 (Polo Like Kinase 1) Inhibits MTOR Complex 1 and Promotes Autophagy. Autophagy (2017) 13:486–505. doi: 10.1080/15548627.2016.1263781
87. Wu Z-Y, Wei N. Knockdown of PLK1 Inhibits Invasion and Promotes Apoptosis in Glioma Cells Through Regulating Autophagy. Eur Rev Med Pharmacol Sci (2018) 22:2723–33. doi: 10.26355/eurrev_201805_14969
88. Liu T-T, Yang K-X, Yu J, Cao Y-Y, Ren J-S, Hao J-J, et al. Co-Targeting PLK1 and mTOR Induces Synergistic Inhibitory Effects Against Esophageal Squamous Cell Carcinoma. J Mol Med (2018) 96:807–17. doi: 10.1007/s00109-018-1663-4
89. Tao Y-F, Li Z-H, Du W-W, Xu L-X, Ren J-L, Li X-L, et al. Inhibiting PLK1 Induces Autophagy of Acute Myeloid Leukemia Cells via Mammalian Target of Rapamycin Pathway Dephosphorylation. Oncol Rep (2017) 37:1419–29. doi: 10.3892/or.2017.5417
90. Wang B, Huang X, Liang H, Yang H, Guo Z, Ai M, et al. PLK1 Inhibition Sensitizes Breast Cancer Cells to Radiation via Suppressing Autophagy. Int J Radiat OncologyBiologyPhys (2021) 110:1234–47. doi: 10.1016/j.ijrobp.2021.02.025
91. Jin B, Jin D, Zhuo Z, Zhang B, Chen K. MiR-1224-5p Activates Autophagy, Cell Invasion and Inhibits Epithelial-To-Mesenchymal Transition in Osteosarcoma Cells by Directly Targeting PLK1 Through PI3K/AKT/mTOR Signaling Pathway. Onco Targets Ther (2020) 13:11807–18. doi: 10.2147/OTT.S274451
92. Mo H, He J, Yuan Z, Wu Z, Liu B, Lin X, et al. PLK1 Contributes to Autophagy by Regulating MYC Stabilization in Osteosarcoma Cells. Onco Targets Ther (2019) 12:7527–36. doi: 10.2147/OTT.S210575
93. Chan K-K, Wong OG-W, Wong ES-Y, Chan KK-L, Ip PP-C, Tse K-Y, et al. Impact of iASPP on Chemoresistance Through PLK1 and Autophagy in Ovarian Clear Cell Carcinoma. Int J Cancer (2018) 143:1456–69. doi: 10.1002/ijc.31535
94. Fuchs Y, Steller H. Programmed Cell Death in Animal Development and Disease. Cell (2011) 147:742–58. doi: 10.1016/j.cell.2011.10.033
95. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
96. Wang X, Simpson ER, Brown KA. P53: Protection Against Tumor Growth Beyond Effects on Cell Cycle and Apoptosis. Cancer Res (2015) 75:5001–7. doi: 10.1158/0008-5472.CAN-15-0563
97. Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, et al. Polo-Like Kinase 1 (Plk1) Inhibits P53 Function by Physical Interaction and Phosphorylation*. J Biol Chem (2004) 279:25549–61. doi: 10.1074/jbc.M314182200
98. Chen L, Ahmad N, Liu X. Combining P53 Stabilizers With Metformin Induces Synergistic Apoptosis Through Regulation of Energy Metabolism in Castration-Resistant Prostate Cancer. Cell Cycle (2016) 15:840–9. doi: 10.1080/15384101.2016.1151582
99. Kudo M, Zalles N, Distefano R, Nigita G, Veneziano D, Gasparini P, et al. Synergistic Apoptotic Effect of miR-183-5p and Polo-Like Kinase 1 Inhibitor NMS-P937 in Breast Cancer Cells. Cell Death Differ (2022) 29:407–19. doi: 10.1038/s41418-021-00864-2
100. Rozenberg JM, Zvereva S, Dalina A, Blatov I, Zubarev I, Luppov D, et al. Dual Role of P73 in Cancer Microenvironment and DNA Damage Response. Cells (2021) 10:3516. doi: 10.3390/cells10123516
101. Koida N, Ozaki T, Yamamoto H, Ono S, Koda T, Ando K, et al. Inhibitory Role of Plk1 in the Regulation of P73-Dependent Apoptosis Through Physical Interaction and Phosphorylation. J Biol Chem (2008) 283:8555–63. doi: 10.1074/jbc.M710608200
102. Tyagi S, Bhui K, Singh R, Singh M, Raisuddin S, Shukla Y. Polo-Like Kinase1 (Plk1) Knockdown Enhances Cisplatin Chemosensitivity via Up-Regulation of P73α in P53 Mutant Human Epidermoid Squamous Carcinoma Cells. Biochem Pharmacol (2010) 80:1326–34. doi: 10.1016/j.bcp.2010.07.025
103. Matthess Y, Raab M, Knecht R, Becker S, Strebhardt K. Sequential Cdk1 and Plk1 Phosphorylation of Caspase-8 Triggers Apoptotic Cell Death During Mitosis. Mol Oncol (2014) 8:596–608. doi: 10.1016/j.molonc.2013.12.013
104. Liao G, Wang R, Tang DD. Plk1 Regulates Caspase-9 Phosphorylation at Ser-196 and Apoptosis of Human Airway Smooth Muscle Cells. Am J Respir Cell Mol Biol (2022) 66:223–34. doi: 10.1165/rcmb.2021-0192OC
105. Gao Z, Man X, Li Z, Bi J, Liu X, Li Z, et al. PLK1 Promotes Proliferation and Suppresses Apoptosis of Renal Cell Carcinoma Cells by Phosphorylating MCM3. Cancer Gene Ther (2020) 27:412–23. doi: 10.1038/s41417-019-0094-x
106. Sekimoto N, Suzuki Y, Sugano S. Decreased KPNB1 Expression is Induced by PLK1 Inhibition and Leads to Apoptosis in Lung Adenocarcinoma. J Cancer (2017) 8:4125–40. doi: 10.7150/jca.21802
107. Jang M-S, Lee S-J, Kim C-J, Lee C-W, Kim E. Phosphorylation by Polo-Like Kinase 1 Induces the Tumor-Suppressing Activity of FADD. Oncogene (2011) 30:471–81. doi: 10.1038/onc.2010.423
108. Jang M-S, Lee S-J, Kang NS, Kim E. Cooperative Phosphorylation of FADD by Aur-A and Plk1 in Response to Taxol Triggers Both Apoptotic and Necrotic Cell Death. Cancer Res (2011) 71:7207–15. doi: 10.1158/0008-5472.CAN-11-0760
109. Gupta K, Liu B. PLK1-Mediated S369 Phosphorylation of RIPK3 During G2 and M Phases Enables its Ripoptosome Incorporation and Activity. iScience (2021) 24:102320. doi: 10.1016/j.isci.2021.102320
110. de Gassart A, Martinon F. Pyroptosis: Caspase-11 Unlocks the Gates of Death. Immunity (2015) 43:835–7. doi: 10.1016/j.immuni.2015.10.024
111. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
112. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A New Frontier in Cancer. Biomed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
113. Wu M, Wang Y, Yang D, Gong Y, Rao F, Liu R, et al. A PLK1 Kinase Inhibitor Enhances the Chemosensitivity of Cisplatin by Inducing Pyroptosis in Oesophageal Squamous Cell Carcinoma. EBioMedicine (2019) 41:244–55. doi: 10.1016/j.ebiom.2019.02.012
114. Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell (2016) 164:1233–47. doi: 10.1016/j.cell.2016.01.049
115. Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation With Chemotherapy in the Era of Immune Checkpoint Inhibitors. Nat Rev Clin Oncol (2020) 17:725–41. doi: 10.1038/s41571-020-0413-z
116. Li M, Liu Z, Wang X. Exploration of the Combination of PLK1 Inhibition With Immunotherapy in Cancer Treatment. J Oncol (2018) 2018:3979527. doi: 10.1155/2018/3979527
117. Zhou J, Yang Q, Lu L, Tuo Z, Shou Z, Cheng J. PLK1 Inhibition Induces Immunogenic Cell Death and Enhances Immunity Against NSCLC. Int J Med Sci (2021) 18:3516–25. doi: 10.7150/ijms.60135
118. Xu D, Wang Y, Wu J, Zhang Z, Chen J, Xie M, et al. ECT2 Overexpression Promotes the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma via the ECT2/PLK1/PTEN Pathway. Cell Death Dis (2021) 12:162. doi: 10.1038/s41419-021-03450-z
119. Oeckinghaus A, Ghosh S. The NF-κb Family of Transcription Factors and Its Regulation. Cold Spring Harb Perspect Biol (2009) 1:a000034. doi: 10.1101/cshperspect.a000034
120. Higashimoto T, Chan N, Lee Y-K, Zandi E. Regulation of Iκb Kinase Complex by Phosphorylation of γ-Binding Domain of Iκb Kinase β by Polo-Like Kinase 1 *. J Biol Chem (2008) 283:35354–67. doi: 10.1074/jbc.M806258200
121. Zhang W, Wang J, Zhang Y, Yuan Y, Guan W, Jin C, et al. The Scaffold Protein TANK/I-TRAF Inhibits NF-κb Activation by Recruiting Polo-Like Kinase 1. Mol Biol Cell (2010) 21:2500–13. doi: 10.1091/mbc.E09-08-0715
122. Vitour D, Dabo S, Ahmadi Pour M, Vilasco M, Vidalain P-O, Jacob Y, et al. Polo-Like Kinase 1 (PLK1) Regulates Interferon (IFN) Induction by MAVS*. J Biol Chem (2009) 284:21797–809. doi: 10.1074/jbc.M109.018275
123. Burkard ME, Santamaria A, Jallepalli PV. Enabling and Disabling Polo-Like Kinase 1 Inhibition Through Chemical Genetics. ACS Chem Biol (2012) 7:978–81. doi: 10.1021/cb200551p
124. Lee KS, Burke TR, Park J-E, Bang JK, Lee E. Recent Advances and New Strategies in Targeting Plk1 for Anticancer Therapy. Trends Pharmacol Sci (2015) 36:858–77. doi: 10.1016/j.tips.2015.08.013
125. Ryu S, Park J-E, Ham YJ, Lim DC, Kwiatkowski NP, Kim D-H, et al. Novel Macrocyclic Peptidomimetics Targeting the Polo-Box Domain of Polo-Like Kinase 1. J Med Chem (2022) 65:1915–32. doi: 10.1021/acs.jmedchem.1c01359
126. Reindl W, Yuan J, Krämer A, Strebhardt K, Berg T. Inhibition of Polo-Like Kinase 1 by Blocking Polo-Box Domain-Dependent Protein-Protein Interactions. Chem Biol (2008) 15:459–66. doi: 10.1016/j.chembiol.2008.03.013
128. Frost A, Mross K, Steinbild S, Hedbom S, Unger C, Kaiser R, et al. Phase I Study of the Plk1 Inhibitor BI 2536 Administered Intravenously on Three Consecutive Days in Advanced Solid Tumours. Curr Oncol (2012) 19:28–35. doi: 10.3747/co.19.866
129. Awad MM, Chu QS-C, Gandhi L, Stephenson JJ, Govindan R, Bradford DS, et al. An Open-Label, Phase II Study of the Polo-Like Kinase-1 (Plk-1) Inhibitor, BI 2536, in Patients With Relapsed Small Cell Lung Cancer (SCLC). Lung Cancer (2017) 104:126–30. doi: 10.1016/j.lungcan.2016.12.019
130. Kobayashi Y, Yamauchi T, Kiyoi H, Sakura T, Hata T, Ando K, et al. Phase I Trial of Volasertib, a Polo-Like Kinase Inhibitor, in Japanese Patients With Acute Myeloid Leukemia. Cancer Sci (2015) 106:1590–5. doi: 10.1111/cas.12814
131. Ottmann OG, Müller-Tidow C, Krämer A, Schlenk RF, Lübbert M, Bug G, et al. Phase I Dose-Escalation Trial Investigating Volasertib as Monotherapy or in Combination With Cytarabine in Patients With Relapsed/Refractory Acute Myeloid Leukaemia. Br J Haematol (2019) 184:1018–21. doi: 10.1111/bjh.15204
132. Stadler WM, Vaughn DJ, Sonpavde G, Vogelzang NJ, Tagawa ST, Petrylak DP, et al. An Open-Label, Single-Arm, Phase 2 Trial of the Polo-Like Kinase Inhibitor Volasertib (BI 6727) in Patients With Locally Advanced or Metastatic Urothelial Cancer. Cancer (2014) 120:976–82. doi: 10.1002/cncr.28519
133. Ellis PM, Leighl NB, Hirsh V, Reaume MN, Blais N, Wierzbicki R, et al. A Randomized, Open-Label Phase II Trial of Volasertib as Monotherapy and in Combination With Standard-Dose Pemetrexed Compared With Pemetrexed Monotherapy in Second-Line Treatment for Non-Small-Cell Lung Cancer. Clin Lung Cancer (2015) 16:457–65. doi: 10.1016/j.cllc.2015.05.010
134. Weiss GJ, Jameson G, Von Hoff DD, Valsasina B, Davite C, Di Giulio C, et al. Phase I Dose Escalation Study of NMS-1286937, an Orally Available Polo-Like Kinase 1 Inhibitor, in Patients With Advanced or Metastatic Solid Tumors. Invest New Drugs (2018) 36:85–95. doi: 10.1007/s10637-017-0491-7
135. Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, et al. Phase I Study of GSK461364, a Specific and Competitive Polo-Like Kinase 1 Inhibitor, in Patients With Advanced Solid Malignancies. Clin Cancer Res (2011) 17:3420–30. doi: 10.1158/1078-0432.CCR-10-2946
136. Ohnuma T, Lehrer D, Ren C, Cho SY, Maniar M, Silverman L, et al. Phase 1 Study of Intravenous Rigosertib (ON 01910.Na), a Novel Benzyl Styryl Sulfone Structure Producing G2/M Arrest and Apoptosis, in Adult Patients With Advanced Cancer. (2013) 16:323–338.
137. Jimeno A, Li J, Messersmith WA, Laheru D, Rudek MA, Maniar M, et al. Phase I Study of ON 01910.Na, a Novel Modulator of the Polo-Like Kinase 1 Pathway, in Adult Patients With Solid Tumors. JCO (2008) 26:5504–10. doi: 10.1200/JCO.2008.17.9788
138. Navada SC, Fruchtman SM, Odchimar-Reissig R, Demakos EP, Petrone ME, Zbyszewski PS, et al. A Phase 1/2 Study of Rigosertib in Patients With Myelodysplastic Syndromes (MDS) and MDS Progressed to Acute Myeloid Leukemia. Leuk Res (2018) 64:10–6. doi: 10.1016/j.leukres.2017.11.006
139. Bowles DW, Diamond JR, Lam ET, Weekes CD, Astling DP, Anderson RT, et al. Phase I Study of Oral Rigosertib (ON 01910.Na), a Dual Inhibitor of the PI3K and Plk1 Pathways, in Adult Patients With Advanced Solid Malignancies. Clin Cancer Res (2014) 20:1656–65. doi: 10.1158/1078-0432.CCR-13-2506
140. Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krššák M, et al. BI 2536, a Potent and Selective Inhibitor of Polo-Like Kinase 1, Inhibits Tumor Growth In Vivo. Curr Biol (2007) 17:316–22. doi: 10.1016/j.cub.2006.12.037
141. Lénárt P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, et al. The Small-Molecule Inhibitor BI 2536 Reveals Novel Insights Into Mitotic Roles of Polo-Like Kinase 1. Curr Biol (2007) 17:304–15. doi: 10.1016/j.cub.2006.12.046
142. Ellis PM, Chu QS, Leighl N, Laurie SA, Fritsch H, Gaschler-Markefski B, et al. A Phase I Open-Label Dose-Escalation Study of Intravenous BI 2536 Together With Pemetrexed in Previously Treated Patients With Non–Small-Cell Lung Cancer. Clin Lung Cancer (2013) 14:19–27. doi: 10.1016/j.cllc.2012.04.003
143. Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, et al. In Vitro Biological Activity of a Novel Small-Molecule Inhibitor of Polo-Like Kinase 1. Mol Cancer Ther (2007) 6:450–9. doi: 10.1158/1535-7163.MCT-06-0543
144. Rudolph D, Impagnatiello MA, Blaukopf C, Sommer C, Gerlich DW, Roth M, et al. Efficacy and Mechanism of Action of Volasertib, a Potent and Selective Inhibitor of Polo-Like Kinases, in Preclinical Models of Acute Myeloid Leukemia. J Pharmacol Exp Ther (2015) 352:579–89. doi: 10.1124/jpet.114.221150
146. Brassesco MS, Pezuk JA, Morales AG, Carvalho de Oliveira J, Roberto GM, Nicioli da Silva G, et al. In Vitro Targeting of Polo-Like Kinase 1 in Bladder Carcinoma: Comparative Effects of Four Potent Inhibitors. Cancer Biol Ther (2013) 14:648–57. doi: 10.4161/cbt.25087
147. Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, et al. BI 6727, A Polo-Like Kinase Inhibitor With Improved Pharmacokinetic Profile and Broad Antitumor Activity. Clin Cancer Res (2009) 15:3094–102. doi: 10.1158/1078-0432.CCR-08-2445
148. Van den Bossche J, Lardon F, Deschoolmeester V, De Pauw I, Vermorken JB, Specenier P, et al. Spotlight on Volasertib: Preclinical and Clinical Evaluation of a Promising Plk1 Inhibitor: SPOTLIGHT ON VOLASERTIB. Med Res Rev (2016) 36:749–86. doi: 10.1002/med.21392
149. Casolaro A, Golay J, Albanese C, Ceruti R, Patton V, Cribioli S, et al. The Polo-Like Kinase 1 (PLK1) Inhibitor NMS-P937 Is Effective in a New Model of Disseminated Primary CD56+ Acute Monoblastic Leukaemia. PloS One (2013) 8:e58424. doi: 10.1371/journal.pone.0058424
150. Valsasina B, Beria I, Alli C, Alzani R, Avanzi N, Ballinari D, et al. NMS-P937, an Orally Available, Specific Small-Molecule Polo-Like Kinase 1 Inhibitor With Antitumor Activity in Solid and Hematologic Malignancies. Mol Cancer Ther (2012) 11:1006–16. doi: 10.1158/1535-7163.MCT-11-0765
151. Gilmartin AG, Bleam MR, Richter MC, Erskine SG, Kruger RG, Madden L, et al. Distinct Concentration-Dependent Effects of the Polo-Like Kinase 1–Specific Inhibitor GSK461364A, Including Differential Effect on Apoptosis. Cancer Res (2009) 69:6969–77. doi: 10.1158/0008-5472.CAN-09-0945
152. Nie Z, Feher V, Natala S, McBride C, Kiryanov A, Jones B, et al. Discovery of TAK-960: An Orally Available Small Molecule Inhibitor of Polo-Like Kinase 1 (PLK1). Bioorg Med Chem Lett (2013) 23:3662–6. doi: 10.1016/j.bmcl.2013.02.083
153. Hikichi Y, Honda K, Hikami K, Miyashita H, Kaieda I, Murai S, et al. TAK-960, a Novel, Orally Available, Selective Inhibitor of Polo-Like Kinase 1, Shows Broad-Spectrum Preclinical Antitumor Activity in Multiple Dosing Regimens. Mol Cancer Ther (2012) 11:700–9. doi: 10.1158/1535-7163.MCT-11-0762
155. Lu T, Laughton CA, Wang S, Bradshaw TD. In Vitro Antitumor Mechanism of (E)-N-(2-Methoxy-5-(((2,4,6-Trimethoxystyryl)Sulfonyl)Methyl)Pyridin-3-Yl)Methanesulfonamide. Mol Pharmacol (2015) 87:18–30. doi: 10.1124/mol.114.093245
156. Hyoda T, Tsujioka T, Nakahara T, Suemori S, Okamoto S, Kataoka M, et al. Rigosertib Induces Cell Death of a Myelodysplastic Syndrome-Derived Cell Line by DNA Damage-Induced G2/M Arrest. Cancer Sci (2015) 106:287–93. doi: 10.1111/cas.12605
157. Kowalczyk JT, Wan X, Hernandez ER, Luo R, Lyons GC, Wilson KM, et al. Rigosertib Induces Mitotic Arrest and Apoptosis in RAS-Mutated Rhabdomyosarcoma and Neuroblastoma. Mol Cancer Ther (2021) 20:307–19. doi: 10.1158/1535-7163.MCT-20-0525
158. García-Alvarez B, de Cárcer G, Ibañez S, Bragado-Nilsson E, Montoya G. Molecular and Structural Basis of Polo-Like Kinase 1 Substrate Recognition: Implications in Centrosomal Localization. Proc Natl Acad Sci U.S.A. (2007) 104:3107–12. doi: 10.1073/pnas.0609131104
159. Yuan J, Sanhaji M, Krämer A, Reindl W, Hofmann M, Kreis N-N, et al. Polo-Box Domain Inhibitor Poloxin Activates the Spindle Assembly Checkpoint and Inhibits Tumor Growth in Vivo. Am J Pathol (2011) 179:2091–9. doi: 10.1016/j.ajpath.2011.06.031
160. . Several inhibitors of the Plk1 Polo Box Domain turn out to be non specific protein alkylators. pdf.
161. Gutteridge REA, Ndiaye MA, Liu X, Ahmad N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol Cancer Ther (2016) 15:1427–35. doi: 10.1158/1535-7163.MCT-15-0897
162. Lian G, Li L, Shi Y, Jing C, Liu J, Guo X, et al. BI2536, a Potent and Selective Inhibitor of Polo-Like Kinase 1, in Combination With Cisplatin Exerts Synergistic Effects on Gastric Cancer Cells. Int J Oncol (2018) 52:804–14. doi: 10.3892/ijo.2018.4255
163. Maire V, Némati F, Richardson M, Vincent-Salomon A, Tesson B, Rigaill G, et al. Polo-Like Kinase 1: A Potential Therapeutic Option in Combination With Conventional Chemotherapy for the Management of Patients With Triple-Negative Breast Cancer. Cancer Res (2013) 73:813–23. doi: 10.1158/0008-5472.CAN-12-2633
164. Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang W, et al. Suppression of KRas-Mutant Cancer Through the Combined Inhibition of KRAS With PLK1 and ROCK. Nat Commun (2016) 7:11363. doi: 10.1038/ncomms11363
165. Li J, Wang R, Kong Y, Broman MM, Carlock C, Chen L, et al. Targeting Plk1 to Enhance Efficacy of Olaparib in Castration-Resistant Prostate Cancer. Mol Cancer Ther (2017) 16:469–79. doi: 10.1158/1535-7163.MCT-16-0361
166. Stehle A, Hugle M, Fulda S. Eribulin Synergizes With Polo-Like Kinase 1 Inhibitors to Induce Apoptosis in Rhabdomyosarcoma. Cancer Lett (2015) 365:37–46. doi: 10.1016/j.canlet.2015.04.011
167. Xie F-F, Pan S-S, Ou R-Y, Zheng Z-Z, Huang X-X, Jian M-T, et al. Volasertib Suppresses Tumor Growth and Potentiates the Activity of Cisplatin in Cervical Cancer. Am J Cancer Res (2015) 5:3548–59.
168. Noack S, Raab M, Matthess Y, Sanhaji M, Krämer A, Győrffy B, et al. Synthetic Lethality in CCNE1-Amplified High Grade Serous Ovarian Cancer Through Combined Inhibition of Polo-Like Kinase 1 and Microtubule Dynamics. Oncotarget (2018) 9:25842–59. doi: 10.18632/oncotarget.25386
169. Posch C, Cholewa BD, Vujic I, Sanlorenzo M, Ma J, Kim ST, et al. Combined Inhibition of MEK and Plk1 Has Synergistic Antitumor Activity in NRAS Mutant Melanoma. J Invest Dermatol (2015) 135:2475–83. doi: 10.1038/jid.2015.198
170. Su S, Chhabra G, Ndiaye MA, Singh CK, Ye T, Huang W, et al. PLK1 and NOTCH Positively Correlate in Melanoma and Their Combined Inhibition Results in Synergistic Modulations of Key Melanoma Pathways. Mol Cancer Ther (2021) 20:161–72. doi: 10.1158/1535-7163.MCT-20-0654
171. Wang Y, Singh R, Wang L, Nilsson M, Goonatilake R, Tong P, et al. Polo-Like Kinase 1 Inhibition Diminishes Acquired Resistance to Epidermal Growth Factor Receptor Inhibition in non-Small Cell Lung Cancer With T790M Mutations. Oncotarget (2016) 7:47998–8010. doi: 10.18632/oncotarget.10332
172. Shin S-B, Kim C-H, Jang H-R, Yim H. Combination of Inhibitors of USP7 and PLK1 has a Strong Synergism Against Paclitaxel Resistance. Int J Mol Sci (2020) 21:E8629. doi: 10.3390/ijms21228629
173. Giordano A, Liu Y, Armeson K, Park Y, Ridinger M, Erlander M, et al. Polo-Like Kinase 1 (Plk1) Inhibition Synergizes With Taxanes in Triple Negative Breast Cancer. PloS One (2019) 14:e0224420. doi: 10.1371/journal.pone.0224420
174. Mao F, Li J, Luo Q, Wang R, Kong Y, Carlock C, et al. Plk1 Inhibition Enhances the Efficacy of BET Epigenetic Reader Blockade in Castration-Resistant Prostate Cancer. Mol Cancer Ther (2018) 17:1554–65. doi: 10.1158/1535-7163.MCT-17-0945
175. Affatato R, Carrassa L, Chilà R, Lupi M, Restelli V, Damia G. Identification of PLK1 as a New Therapeutic Target in Mucinous Ovarian Carcinoma. Cancers (Basel) (2020) 12:E672. doi: 10.3390/cancers12030672
176. Cortes J, Podoltsev N, Kantarjian H, Borthakur G, Zeidan AM, Stahl M, et al. Phase 1 Dose Escalation Trial of Volasertib in Combination With Decitabine in Patients With Acute Myeloid Leukemia. Int J Hematol (2021) 113:92–9. doi: 10.1007/s12185-020-02994-8
177. Döhner H, Lübbert M, Fiedler W, Fouillard L, Haaland A, Brandwein JM, et al. Randomized, Phase 2 Trial of Low-Dose Cytarabine With or Without Volasertib in AML Patients Not Suitable for Induction Therapy. Blood (2014) 124:1426–33. doi: 10.1182/blood-2014-03-560557
178. . Available at: https://clinicaltrials.gov/ct2/show/NCT01721876.
179. Zeidan AM, Ridinger M, Lin TL, Becker PS, Schiller GJ, Patel PA, et al. A Phase Ib Study of Onvansertib, a Novel Oral PLK1 Inhibitor, in Combination Therapy for Patients With Relapsed or Refractory Acute Myeloid Leukemia. Clin Cancer Res (2020) 26:6132–40. doi: 10.1158/1078-0432.CCR-20-2586
180. Machiels J-P, Peeters M, Herremans C, Surmont V, Specenier P, De Smet M, et al. A Phase I Study of Volasertib Combined With Afatinib, in Advanced Solid Tumors. Cancer Chemother Pharmacol (2015) 76:843–51. doi: 10.1007/s00280-015-2860-2
181. de Braud F, Cascinu S, Spitaleri G, Pilz K, Clementi L, Liu D, et al. A Phase I, Dose-Escalation Study of Volasertib Combined With Nintedanib in Advanced Solid Tumors. Ann Oncol (2015) 26:2341–6. doi: 10.1093/annonc/mdv354
182. Lang I, Liu D, Fritsch H, Taube T, Chizhikov E, Liptai B. Potential Drug-Drug Interactions With Combination Volasertib + Itraconazole: A Phase I, Fixed-Sequence Study in Patients With Solid Tumors. Clin Ther (2020) 42:2214–24. doi: 10.1016/j.clinthera.2020.09.015
183. . Available at: https://bit.ly/2XBAd57.
184. Carbajosa S, Pansa MF, Paviolo NS, Castellaro AM, Andino DL, Nigra AD, et al. Polo-Like Kinase 1 Inhibition as a Therapeutic Approach to Selectively Target BRCA1-Deficient Cancer Cells by Synthetic Lethality Induction. Clin Cancer Res (2019) 25:4049–62. doi: 10.1158/1078-0432.CCR-18-3516
Keywords: cell cycle, G2/M checkpoint, DNA damage response, EMT, PLK1 inhibitors, drug combination, immune response
Citation: Chiappa M, Petrella S, Damia G, Broggini M, Guffanti F and Ricci F (2022) Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 12:903016. doi: 10.3389/fonc.2022.903016
Received: 23 March 2022; Accepted: 09 May 2022;
Published: 02 June 2022.
Edited by:
Brian Gabrielli, The University of Queensland, AustraliaReviewed by:
Chun-Han Chen, Taipei Medical University, TaiwanCopyright © 2022 Chiappa, Petrella, Damia, Broggini, Guffanti and Ricci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimo Broggini, bWFzc2ltby5icm9nZ2luaUBtYXJpb25lZ3JpLml0
†These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.