 Abdellatif El Khayari
Abdellatif El Khayari Najat Bouchmaa
Najat Bouchmaa Bouchra Taib
Bouchra Taib Zhiyun Wei
Zhiyun Wei Ailiang Zeng
Ailiang Zeng Rachid El Fatimy
Rachid El Fatimy
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 14 July 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.901951
This article is part of the Research TopicReviews in Cancer MetabolismView all 8 articles
Glioblastoma multiforme (GBM), a highly invasive and incurable tumor, is the humans’ foremost, commonest, and deadliest brain cancer. As in other cancers, distinct combinations of genetic alterations (GA) in GBM induce a diversity of metabolic phenotypes resulting in enhanced malignancy and altered sensitivity to current therapies. Furthermore, GA as a hallmark of cancer, dysregulated cell metabolism in GBM has been recently linked to the acquired GA. Indeed, Numerous point mutations and copy number variations have been shown to drive glioma cells’ metabolic state, affecting tumor growth and patient outcomes. Among the most common, IDH mutations, EGFR amplification, mutation, PTEN loss, and MGMT promoter mutation have emerged as key patterns associated with upregulated glycolysis and OXPHOS glutamine addiction and altered lipid metabolism in GBM. Therefore, current Advances in cancer genetic and metabolic profiling have yielded mechanistic insights into the metabolism rewiring of GBM and provided potential avenues for improved therapeutic modalities. Accordingly, actionable metabolic dependencies are currently used to design new treatments for patients with glioblastoma. Herein, we capture the current knowledge of genetic alterations in GBM, provide a detailed understanding of the alterations in metabolic pathways, and discuss their relevance in GBM therapy.
Over the past few years, significant advances in understanding glioma at the molecular level have greatly improved our understanding of the genetic alterations that characterize this heterogeneous brain tumor. Since 2016, with the revised World Health Organization (WHO) classification of tumors of the Central Nervous System (CNS), different glioma entities are defined not only by histological features but also by genetic and molecular markers (1). Besides, the classification based on genetic alterations has helped distinguish primary and secondary gliomas, unlike that based on histopathological features (1, 2). Moreover, recent studies have shown that the genetic characteristics adopted in conjunction with histopathological data have remarkably improved the diagnosis and prognosis of gliomas (3), with the benefit of providing new insights into targeted strategies in the treatment of malignant gliomas. Nevertheless, the fourth-grade glioma, also known as glioblastoma multiform (GBM), has disposed of a particular challenge within the clinical setting due to its unique and distinctive cellular heterogeneity. The frequent occurrence of genetic and epigenetic alterations has made GBM immune to all conventional cancer therapies ranging from resection to chemotherapy and radiation therapy (Cancer Genome Atlas Research Network, 2008) (4). Alongside, GBM tumors rapidly develop therapeutic resistance, making this cancer intractable to date. Most of the available pre-clinical and clinical treatment strategy stages target host immune response stimulation to decrease tumor proliferation and invasion through immune and/or gene therapies (3, 4).
To date, therapies that target the angiogenic nature of the tumor, such as the use of monoclonal antibodies for Vascular Endothelial Growth Factor (VEGF) (Bevacizumab), have displayed some promising results in the clinical trials (5). Besides using alkaline agents such as Temozolomide and other targeted therapies like the inhibitors of growth factors, transcriptional and translational pathways coupled with radiation therapy are finding their way into pre-clinical and clinical trials (6). Further, the Epidermal Growth Factor Receptor (EGFR), a critical factor in tumor malignancy in several cancer types, including glioblastoma, has emerged as one of the most explored drug targets in cancer therapeutics over the past 20 years (7). For example, several EGFR inhibitor-based approaches are currently used to treat patients with EGFR-positive tumors, notably non-small cell lung cancer. In GBMs, abnormal EGFR activation due to somatic mutations and EGFR gene amplification is a reliable feature incriminated in various pathological processes. Recently, metabolic rewiring of GBM cells was linked mainly to these alterations, and a considerable body of evidence considered them among the most potentially targetable alterations in GBM (8, 9). However, the outcome of all available therapeutic options against GBM remains non-universal due to the disease’s rapid variations at the molecular, genetic, and epigenetic levels, which are unique across individuals (10).
Therefore, therapeutic solutions are driven primarily toward developing a personalized treatment. This emerges the need to acquire an in-depth understanding of gliomas’ molecular programming and reprogramming, including several deletions, amplifications, and mutations followed by metabolic alterations (10).
Metabolism-related gene signatures, which may more effectively predict patient prognosis, are still lacking in gliomas, especially in GBM cancer. Understanding the malignant phenotype of gliomas requires knowledge of these metabolic alterations to decipher the mechanisms underlying the complex relationship between molecular aberrations, metabolism profile, and tumor behavior. Indeed, recent evidence increasingly highlights the high frequency of alterations in metabolic genes, also revealing a strong correlation between the grade of malignancy and the occurrence of these alterations (11, 12). The extensive use of glucose by cancer cells for energy generation through aerobic glycolysis, a phenomenon is known as the “Warburg effect,” as opposed to mitochondrial oxidative phosphorylation (OXPHOS), is responsible for the rapid generation of adenosine triphosphate (ATP) and adaption to the hypoxic tumor environment (13). In the case of GBM, it is now clear that malignant cells adapt their bioenergetic metabolism to overcome the fluctuations/constraints in tumor microenvironmental conditions and maintain their high proliferative characteristic. Thus, the rate of aerobic glycolysis and OXPHOS is closely associated with tumor stage, microenvironment, and specifically activated oncogenes (14). The balance between high glycolysis and OXPHOS drives the bioenergetics of the tumor, enhancing the malignant processes such as cell proliferation and invasion.
Moreover, in GBM, several genes associated with glycolysis have been suggested to correlate with tumor proliferation, invasion, angiogenesis, and chemoresistance. Likewise, fatty acids and other lipid species are thought to contribute to the malignancy of GBM (15). In addition to their bioenergetic role, several proofs have shown the effective implication of glycolysis and lipid metabolism in modulating the tumor microenvironment and facilitating adaptation to hypoxic conditions. Also, inhibitors of some metabolic enzymes involved in these pathways have shown promising outcomes in enhancing radio-sensitivity in patients with gliomas (16, 17). However, integrated analyses have evidenced that glycolysis and lipid alterations are significantly associated with worse overall survival of patients (11, 18), suggesting the involvement of these pathways in the aggressiveness of glioblastoma. Therefore, shedding light on these altered metabolic processes is urgently needed to harness the metabolic clues provided by the available knowledge and the ongoing efforts to develop new therapeutic approaches for glioma cancers.
The purpose of this review is to capture an overview of the current understanding of the hallmark genetic alterations in GBM, and explore compelling evidence for the common dysregulated metabolic pathways such as lipid synthesis and glycolysis, with a particular focus on the complex interplay between genomic aberrations and altered cellular metabolism in GBM. Furthermore, the significance of established metabolic dependencies as emerging targets in advancing therapeutic solutions against GBM is thoroughly discussed.
GBM has been known as a highly nonhomogeneous cancer (3, 4), and it is obscure how genomic alterations conduce to this phenotype. With the growing effectiveness of genetically targeted precision therapy for other tumor malignancies (19), several prior studies have been first interested in investigating and identifying numerous credibly targetable mutations as well as copy number changes and epigenetic variants that are usually common in GBMs. In this respect, the new classification system, merging classical histologic classification and molecular genetic data alterations, divided GBMs into primary GBMs (IDH (Isocitrate dehydrogenase) wild-type) and secondary GBMs (IDH mutant) (10–12), allowing as well a better characterization of gliomas (20, 21). Furthermore, specific genetic rearrangements are believed to have clinical relevance and are further related to GBMs class II (20). Besides, in recent years, molecular alterations in metabolic gene IDH 1/2 have received much attention. Admittedly, mutations of IDH1/2 are considered early events in tumorigenesis (22) and may reveal tumor vulnerabilities that can be exploited for potential therapeutics (23). Significantly, in adults, molecular alterations in this class of metabolic gene are now recognized as a defining molecular event commonly frequent in the secondary GBMs (50%), which are also known by O6-methylguanine-DNA methyltransferase promoter methylation (MGMT) (75%) (20), as well as in the majority of lower-grade astrocytoma (WHO grade II and III gliomas) (80%), which are also both reported by tumor protein (TP53) mutations and functional loss (mutation or deletion) of α-Thalassemia/Mental Retardation Syndrome X-linked (ATRX) (24, 25). In contrast, primary GBM, which are nearly all IDH Wild-type, are typically displaying epidermal growth factor receptor (EGFR) and mouse double minute 2 homolog (MDM2) amplification or mutation, cyclin-dependent kinase inhibitor 2 A/B (CDKN2A/B) deletion, phosphatase, and tensin homolog (PTEN) loss and neurofibromatosis type 1(NF1), and telomerase reverse transcriptase (TERT) promoter mutations, along with the gain of chromosome 7 and loss of heterozygosity at q10 (LOH 10q) (chromosome 10) (25). Interestingly, such genetic alterations permit an accurate distinction between primary and secondary GBMs, even though they are histologically indistinguishable. Additionally, primary GBMs, representing about 90% of all GBM subsets, show other less-common mutations and/or deletions in genes such as TP53, PDGFRA, EGFRvIII, PIK3CA, PIK3R1, RB1, H3F3A, MET, CDK4, CDK6, and MDM4 (24–26).
Likewise, a recent investigation through mutually exclusive gene set analysis (MEGSA) reported the alterations in Spectrin Alpha Erythrocytic 1 (SPTA1) and Capping Actin Protein of Muscle Z-Line Subunit Alpha 2 (CAPZA2) are linked to GBMs. This investigation suggests that mutated SPTA1 may be related to abnormal cell proliferation and apoptosis, while amplified CAPZA2 negatively regulates cell invasion (27). Moreover, H3K27M (A methionine substitution of lysine at residue 27 of histone H3) mutation, regular in pediatric diffuse midline glioma, has also been recognized in malignant adult glioblastoma, with mutual exclusion to IDH1/2 alterations (28).
Considerably, understanding the metabolic reprogramming (MR) of aggressive gliomas, a phenomenon that could potentially perform as a hallmark of cancer progression, has potential functions for therapy in tandem with biomarkers. To demonstrate how tumor heterogeneousness and therapeutic resistance promote this GBM phenotype, we investigated numerous recent studies exploring the genomic and epigenetic alterations in these tumors. This examination has also revealed that common pathways are altered in GBM, specifically metabolism pathways (Figure 1).
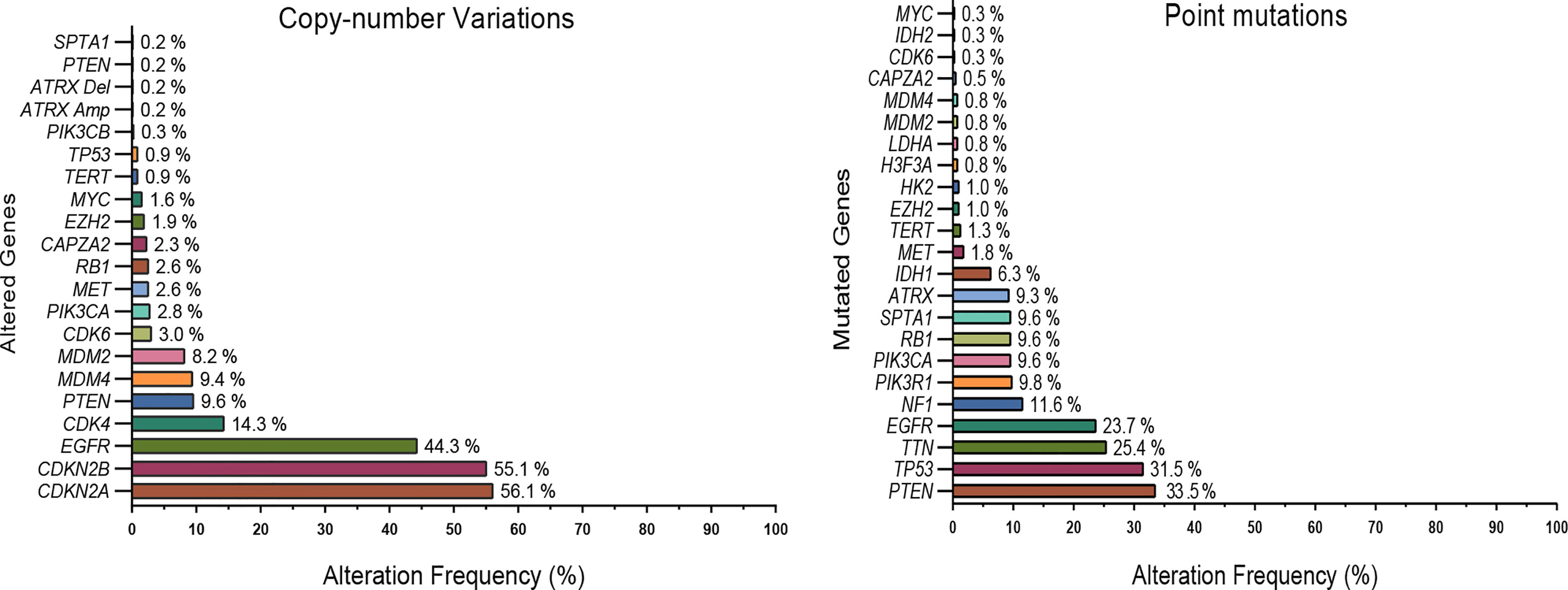
Figure 1 Commonly mutated genes and copy number variations in GBMs. Data (592 patients) from the TCGA dataset (Glioblastoma multiforme; TCGA- PanCancer Atlas) were explored using the cBioPortal platform. For clarity, only a subset of highly altered and metabolic genes are shown. EGFR, PTEN, TERT, and several metabolic genes such as IDH 1/2, HK2, MYC, LDHA, EZH2 were frequently altered in glioblastoma tumors.
Commonly, it is known that mutations that co-occur in driver genes trigger different cell signaling pathways that work together in carcinogenesis. Also, driver genes that display a mutually exclusive mutation pattern can have a redundant oncogenic function (29). For example, similar co-occurrence patterns that have been shown to collaborate in model organisms consist of alterations in KRAS and TP53 in pancreatic cancer, ERG translocations and PTEN deletions in prostate cancer, APC and KRAS in CRC, or MYC and TP53 in various cancer types (4, 30–34). Therefore, biological insights into the concurrent interactions of driver genes could also have crucial implications for improving novel therapeutic approaches. The common co-targeting of co-occurring and collaborating driver mutations or their related signaling pathways has long been a goal in preclinical and clinical research.
MR of tumors, an energetic process for maintaining the reduction-oxidation balance and macromolecular biosynthesis, is considered a hallmark of cancer progression required for cell growth, proliferation, and migration (18, 35). Therefore, malignancy cells must improve metabolic pathways by utilizing available nutrients to maintain energy demand, redox balance, and biosynthesis to prolong and achieve their proliferative potentiality. However, the altered mechanisms behind this metabolism shift remain complex and not completely understood. Nevertheless, a growing number of studies are underway to explore the potential of a metabolism-targeting strategy as a novel and promising approach against gliomas (36). However, accumulating evidence demonstrates that genetic alterations partially drive gliomas’ cell metabolism and contribute to the aggressive nature of some cancers like GBM.
The most studied example linking genetic alterations to gliomas metabolic changes is the acquired mutation in IDH1 and/or IDH2 genes. While IDH mutation has been detected in several cancer types and reported to have the same primary effects in those malignancies. However, gliomas remain the only cancer in which the IDH1/2 mutation constitutes a specific feature representing a propitious prognostic marker (37, 38). The natural enzymatic reaction catalyzed by the wild-type IDH1 and IDH2 is an oxidative decarboxylation of isocitrate into α-ketoglutarate (α-KG), and the production of the reduced form of the nicotinamide adenine dinucleotide phosphate (NADPH) (39). However, IDH mutation leads to the consumption of α-KG and NADPH to produce an oncometabolite, D-2-hydroxyglutarate (D2HG), directly involved in G-CIMP phenotype inducing and aberrant DNA methylation in gliomas (40, 41). In addition, D-2-hydroxyglutarate was reported as a competitive inhibitor for some α-ketoglutarate-dependent dioxygenases (42). Therefore, the pro-methylation effects of the IDH mutation may be linked to the extent to which a specific cell expresses the affected dioxygenases. For instance, in the study conducted by Unruh et al. (2019) aiming to determine the methylation and transcription patterns in IDH mutant gliomas, authors showed that of 365,092 analyzed CpG sites, about 19% were hypermethylated in gliomas with IDH mutation compared to wild-type gliomas (38). However, in acute myeloid leukemia, cholangiocarcinoma, and melanoma, only 3%, 4%, and 2% of CpG sites were hypermethylated in the IDH mutant cancers, respectively. Moreover, the hypermethylation degree was lower in mature astrocytes than in the undifferentiated neural progenitor cells, which may justify the importance of the IDH mutation as a helpful prognostic marker in gliomas (38).
D-2-hydroxyglutarate was also reported to inhibit the branched-chain amino acid aminotransferase (BCAT) transaminases and thus rendered gliomas cells addicted to glutamine and more sensitive to glutaminase inhibition (36, 43). Of note, primary GBM and other wild-type IDH gliomas display a similar addiction to glutamine, suggesting different players for the glutamine-dependent energy pathway observed in gliomas (36). Moreover, the metabolic dysregulations associated with IDH mutations could also be extended to glycolysis, TCA cycle metabolism, and phospholipid and Reactive oxygen species (ROS) production (44–46).
A growing body of proof suggests that oncogenic mutated epidermal growth factor receptor (EGFR) favors an MR to glycolysis, which is conceivably reversed via EGFR tyrosine-kinase inhibitors (TKIs), thereby inhibiting cancer cell growth.
Indeed, tyrosine phosphorylation is a metabolic process of multicellular organisms crucial for signal transduction. Tyrosine phosphorylation is, therefore, essential for many cellular mechanisms, such as differentiation, proliferation, migration, and survival (47).
Receptor tyrosine kinases (RTKs) are membrane-spanning proteins activated by the binding of a specific ligand to their extracellular domain. The change following this reaction led to the intracellular catalytic domain initiation, enabling the proteins occupation to trigger a signaling pathway in response to a specific cellular demand (48, 49). The epidermal growth factor receptor (EGFR) was highly altered in gliomas and the most amplified in GBM (50). As a result, the EGFR amplification and mutation led to several cellular processes modification, contributing to tumorigenesis and progression.
Despite the implication of proto-oncogene family MYC in several metabolic processes, including nucleotide synthesis, lipogenesis, glucose transport, and glycolysis, its role in some gliomas, such as adult GBM, for which the MYC is rarely amplified, was not fully understood until recently (8, 49). MYC regulates the expression of several genes involved in glucose transport and metabolism. This includes the glucose transporter GLUT1, hexokinase 2 (HK2), the muscle isoform of phosphofructokinase (PFKM), enolase 1 (ENO1), and lactate dehydrogenase A (LDHA) (51, 52), which leads to the Warburg effect characterized by an increase in the glucose uptake and its fermentation to lactate (53). Several works have revealed that EGFR mutation and amplification modulate GBM metabolism through MYC via three complementary mechanisms. This includes (1) EGFRvIII impacts an isoform arrangement by modulating the heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1). This protein leads to the splicing of the Delta max, which is an MYC-interacting partner that promotes glycolytic metabolism (54). (2) Simultaneously, EGFRvIII increases glycolysis in GBM via the mTORC2 signaling that regulates the cellular level of c-Myc through acetylation of forkhead box protein O1 (FoxO1) and FoxO3, leading to an upregulated expression of MYC protein (55). (3) The hyper-activated EGFRvIII promotes the transcription of SOX9 and FOXG1, which is implicated in regulating the MYC-dependent transcription (56). All the proofs, as mentioned above, show the pivotal player of MYC for the EGFRvIII-dependent tumorigenesis.
EGFR was also reported to influence glutamine metabolism in GBM cells. Indeed, after glutamine absorption by cancer cells, glutaminase converts it to glutamate, which enters the mitochondria or the cytoplasm to maintain the operational TCA cycle. Indeed, glutamate is an indispensable molecule in tumor cells due to its use as a forerunner to produce α-KG, which enters the TCA cycle to generate carbon and nitrogen for the cancer cells (57–59). The conversion of glutamate to the α-KG is catalyzed by glutamate dehydrogenase (GDH1). Furthermore, the study conducted by Yang et al. (2020) showed that phosphorylated ELK1 activated by the EGFR signaling pathway induced the transcription of GDH1, leading to the increase of glutamine metabolism (59).
Collectively, these studies show that EGFR is a critical factor in gliomas’ development and progression through its implication in the regulation of the proto-oncogene MYC family and the activation of the glutamine metabolism through the activation of ELK1.
Additionally, glioma cells involve further mutations to reprogram their metabolism and support high-energy demand. For example, PTEN loss has been seen to upregulate hexokinase 2 (HK2) and activate the autophosphorylated PGK1, promoting aerobic glycolysis in GBM (36, 60). Also, TERT promoter mutation is thought to regulate lipid metabolism through a histone H3K27 methyltransferase (EZH2), suggesting an essential role for TERT in MR of GBM cells (61).
In addition to genetic alterations, accumulated epigenetic events play a fundamental role in GBM pathobiology simply as associated with poor prognosis. Therefore, each new epigenetic event might be a valuable marker for glioblastoma prognosis. Furthermore, GBMs display intra- and inter-tumor heterogeneity, a phenomenon in which diverse cells inside the same tumor procure distinct epigenetic and genomic mutations (36, 62).
Numerous studies showed the implication of this phenomenon, one of the major factors of therapeutic resistance to anticancer therapies, to drive tumor genomics landscape misapprehension, which is a crucial fundamental confrontation towards personalized medicine and biomarker advancement.
Accordingly, large-scale genomic studies have described many epigenetic defects involved in GBM, including changes in DNA methylation profile, histone post-transcriptional modifications, and chromatin remodeling (63). However, chronicles of mutations in several epigenetic regulator genes have been reported in GBM. Notably, mutations in IDH 1/2, EGFR, H3F3A, MLL2-4, HDAC2, HDAC9, KDM4D, KDM5A/B/C, and KDM6A/B have been observed to promote DNA and histone methylation and modify chromatin state, thereby inducing aberrant modifications in the epigenetic pattern and consequently affecting the gene expression profile in GBM cells (10, 64). Moreover, earlier records have shown evidence for CpG island methylator phenotype (CIMP) in GBM. These studies further report the association of GBM with frequent hypermethylation at specific loci, especially at the promoters of several genes involved in GBM pathologies such as MGMT, CDKN2A-p14ARF and CDKN2A-p16INK4a, RB, and TIMP-3, which have direct implications in DNA repair, cell cycle regulation, tumor suppression, and inhibition of apoptosis, respectively (63, 65–67).
Given the critical role of non-coding RNAs (ncRNAs) as genetic regulators-controlling gene expression at the epigenetic level, microRNAs (miRNAs), a class of short ncRNAs, have recently emerged as key players in glioma pathogenesis. Recent studies revealed that approximately 351 miRNAs are dysregulated in GBM (68), and some of them have been associated with acquired malignant phenotypes such as tumor growth, proliferation, invasion, and angiogenesis. Among others, miRNA-21, miRNA-10b, miRNA-7, miRNA-100, miRNA-296, miRNA-210-3p, miRNA-128, and miRNA-221 are the prominent candidates that have been studied and investigated for miRNA-based therapeutic strategies in GBM (68–72). Alongside miRNAs, long ncRNAs (lncRNAs) have also been observed to control many cellular processes in glioma cells predominantly associated with GBM and its malignancy (68, 73, 74). Moreover, amassing proof approved the incrimination of miRNAs and LncRNAs as mediators in energy metabolism reprogramming of GBM tumors.
In addition to genetic and epigenetic variability among patients, three major molecular signaling pathways are altered in GBM: Growth Factor RTK pathway, Retinoblastoma (RB) pathway, and TP53 pathway (10, 63, 75). As a result, impaired phenotypes potentially contribute to different processes relevant to gliomagenesis, such as cell transformation, angiogenesis, invasion, and metabolic reprogramming (Figure 2).
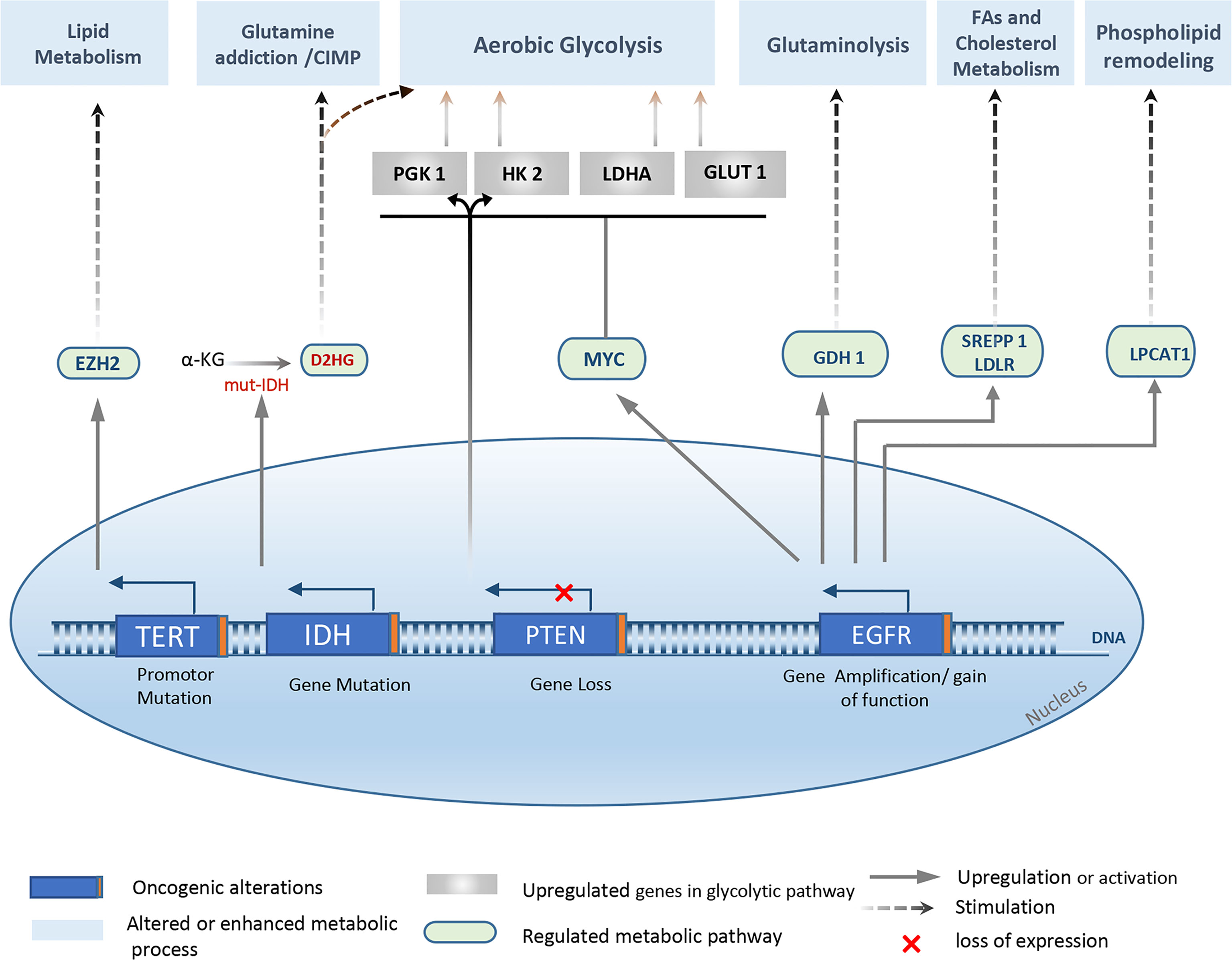
Figure 2 Overview of the major genetic alterations that drive metabolic reprogramming in Gliomas. Metabolic changes observed in gliomas and especially in glioblastoma are associated with several genetic abnormalities, mainly, IDH ½ mutation, EGFR amplification, mutation, or EGFRvIII activation. Also, PTEN loss, and TERT promoter mutation. Various metabolic pathways are affected, yielding a new metabolic profile that supports the high proliferative characteristic, cell adaption, and tumor progression. EZH2, Enhancer of zeste homolog 2; D2HG, d-2-hydroxyglutarate; SREPP 1, sterol regulatory element-binding protein 1; LPCAT1, lysophosphatidylcholine acyltransferase 1; LDLR, glutamate dehydrogenase 1; LDHA, Lactate dehydrogenase A; GDH 1, glutamate dehydrogenase 1; PGK 1, phosphoglycerate kinase 1.
Recently, a couple of Glutaminase isoenzymes have been proven to be linked to the expression of multiple miRNAs and could potentially promote tumor development or inhibition through various miRNA-mediated pathways (76). In GBM cells, miR-1297 repressed cell proliferation and glycolysis via targeting KPNA2 (77), a high oncogene in glioma tissues that was proven to be a high promoter of glycolytic metabolism in GBM cells as well (78). The downregulation of miR-1297 and upregulation of KPNA2 in glioma implied a regulatory association between them (78, 79).
Moreover, LncRNAs are considered an actor in cancer progression as miRNA decoys or targets. Furthermore, LncRNA HOX transcript antisense intergenic RNA (HOTAIR) was further observed upregulating the glutaminase expression level, which is crucial for glutamine metabolism and coming oncogenic approach. For instance, Liang Liu et al. (2018) reported the regulation function of HOTAIR on glutaminase (GLS) expression through sponging miR-126-5p in a competing endogenous RNA axis HOTAIR/miR-126-5p/GLS that is implicated in glioma progression (80). Similarly, It has been demonstrated that LINC00174 contributes to the carcinogenesis of glioma and further promotes glycolysis via miR-152-3p modulation, involving GLUT1 (SLC2A1) overexpression (81). Furthermore, He et al. also revealed that lncRNA UCA1/miR-182/PFKFB2 modulated glioblastoma-associated stromal cell-mediated glycolysis (82). In addition, referring to the existing literature, the oncogenic lncRNA PCAT19 and CARD8-AS1 were positively correlated with tryptophan metabolism and MDSC infiltration, suggesting their immunosuppressive function in the tumor microenvironment is potentially mediated by promoting immunosuppressive metabolism (83–85).
From the previous discussion, the Warburg effect represents a hallmark of different cancer types, including GBM. Indeed, this phenomenon is responsible for shifting glucose metabolism toward aerobic glycolysis to produce ATP and other metabolites required for cell proliferation, invasion, and adaptation to the hypoxic surrounding microenvironment (86). In line with this, it has been reported that GBM cells exhibit an increased glucose uptake compared to normal astrocytes and utilize cytosolic fermentation rather than mitochondrial oxidation, thereby producing large amounts of lactate, which is subsequently released into the extracellular space (87). Also, GBM-derived extracellular vesicles (GBM EVs) appear to play a pivotal role in reprogramming transformed cells toward a glycolytic state through horizontal mRNA transfer (extracellular RNA) and upregulation of several metabolic pathways (88).
Even though numerous reports have suggested that this unbalanced metabolic balance linking glycolysis with oxidative phosphorylation in gliomas cells is due solely to mitochondrial disorder (89), more current proof from in vitro and in vivo studies has also shown that either acquired genetic mutations or hypoxia increase glycolytic ratio and as a result of this re-modulate cellular energy metabolism of GBM cells.
Furthermore, immune cells, particularly macrophages, contribute to this metabolic adaptation by secreting interleukin-6 (IL-6) in the tumor microenvironment, phosphorylating thereby PGK1 and enhancing glycolysis in GBM cells (90). Moreover, GBMs are metabolically heterogeneous tumors and often show high metabolic plasticity depending on micro-environmental conditions (91). Hence, high rates of glycolysis occur in the tumor's central region whereas the peripheral region predominantly utilizes oxidative phosphorylation. Likewise, glioblastoma stem cells (GSCs) are less glycolytic than differentiated cells (92). Altogether, the last pieces of evidence support the crucial role of micro-environmental conditions, alongside the genetic alterations, in modulating metabolic pathways and regulating tumor growth (93).
However, recent reports have revealed a remarkable correlation between glycolysis and gliomas malignancy, whereby glycolytic profile was closely associated with poor prognosis and overall survival in patients with GBM (62, 94). Additionally, in vitro studies have demonstrated that inhibiting glycolysis sensitizes glioblastoma cells and significantly enhances the efficacy of radiotherapy and chemotherapy (temozolomide) against these highly malignant tumors (16, 17).
Rapid energy production has long been considered the Warburg effect’s main advantage in cancer cells. Currently, it is evidenced that cancer cells undergo aerobic glycolysis not only to meet their high energy demands but also as a critical adaptive process that enables cells to overcome the hypoxia conditions and effectively interact with other cells in the tumor microenvironment (95). Therefore, several metabolic pathways are ultimately activated to generate intermediate metabolites and macromolecules essential for cell proliferation or invasion (96, 97). In addition to ATP production, glycolysis fuels the pentose phosphate pathway by glucose-6-phosphate (G6P) and consequently accelerates the production of ribose-5-phosphate (R5P) and NADPH (96). These two molecules play an essential role in the adaptive metabolism of various cancers, including GBM (86). The ribose-5-phosphate (R5P) is an intermediate metabolite required for nucleotides synthesis, while NADPH acts as an antioxidant agent, combating high levels of intracellular ROS during cell proliferation. Additionally, NADPH is a crucial molecule for lipids biosynthesis, participating in 2-hydroxyglutarate (2‐HG) production in IDH-mutant glioblastoma (40). Further, it is thought to be involved inregulating apoptosis, invasion, and migration, thus promoting tumor cell proliferation and metastasis (98). Moreover, the increased lactate secretion resulting from aerobic glycolysis has stimulated angiogenesis and impaired tumor immunosurveillance in glioblastoma (97, 99) (Figure 3).
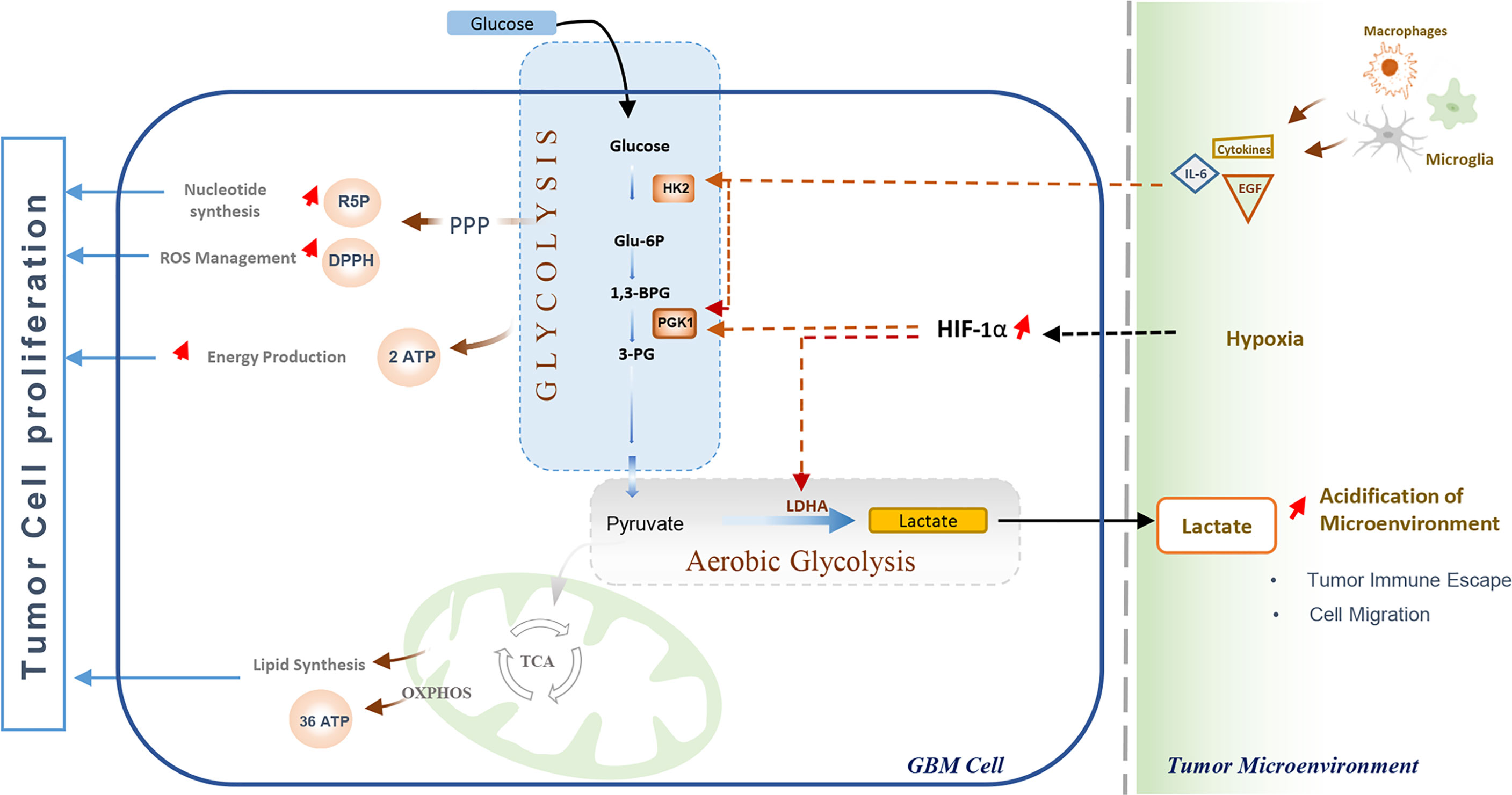
Figure 3 Glycolysis and its role in Glioblastoma cell proliferation and immune escape. The Warburg effect enables GBM cells to meet their energy demand through rapid ATP production, and promotes nucleotides synthesis and oxidative stress management, promoting cell proliferation. The elevated lactate production by tumor cells increases the acidification of the Tumor Microenvironment, which disturbs immune cells activation leading to impaired tumor immunosurveillance and cell migration. Immune cells (tumor-associated macrophages and Microglia) enhance glycolysis through Interleukin-6 (IL-6) and Epidermal Growth Factor (EGF), While hypoxic conditions in the Tumor microenvironment promote glycolysis and Warburg effect by activation and stabilization of hypoxia-inducible transcription factor (HIF-1α).
Recently, cancer lipid metabolism started to emerge as an opportunity to identify new therapeutic targets against various cancer types. Indeed, a growing body of studies started to explore how cancer cells support their tumorigenesis and cancer progression by reprogramming the metabolism of fatty acids, the most construction squares of several lipid species, counting phospholipids and triglycerides (100). Succeeding adipose tissues, the brain has the highest body lipid contents, which, beyond their vital role in building the brain structure, are also critical players as energy resources and as signaling molecules to maintain cell growth (101). However, the first study, performed by Brante et al. (1949), showed the presence of esterified cholesterols in brain tumors while absent in normal adult brain tissue (102). Since then, other studies have been conducted to analyze lipid composition in human intracranial tumors (103).
Several in vivo and in vitro studies have been conducted to better understand the metabolic profile of lipids in GBM. Using GBM cell lines, it has been shown that the lipid profile of GBM cells changes in response to the nutrient composition. Moreover, the lipidomic analysis showed an accumulation, of five main classes of lipids, including Ceramides, Phospholipids, Diacyl-glycerols, Triacyl-glycerols, and Sphingomyelins (104). In contrast, in ectopic and orthotopic mouse xenograft models, lipidomic examination distinguished 500 lipid species, with the most prominent forms dropping transcendently into four primary categories: glycosphingolipids, glycerophosphoethanolamines, triacylglycerols, and glycerophosphoserines. Interestingly, contrasts were observed in lipid profiles when the same tumor was engendered within the flank fronting the brain, suggesting the fundamental significance of the encompassing physiological environment on GBM cancer growth (105). Moreover, lipid composition analysis of human glioma appeared a principal sum of phosphatidylinositol, sphingomyelin, and lysophosphoglycerides, in contracts to typical cortex tissue, as well as a noteworthy increase in oleic, linoleic, and arachidonic acid (106), cholesterol esters (103), and phospholipids (107).
Lipid droplets (LDs) are intracellular organelles that regulate the storage and lipolysis of neutral lipids, such as triglycerides and sterol esters (108). Beyond their role of storage, deregulation of LDs metabolism has been associated with several types of tumor tissues from cancer patients (109). Recently, LDs have been raised as a potential biomarker of GBM. Most recently, using immunofluorescence, immunohistochemistry, and electronic microscopy, it has been shown that tumor tissues from GBM patients include massive amounts of LDs, which are not detectable in normal brain tissues and low-grade gliomas (110). Moreover, LDs prevalence was inversely correlated with GBM patient survival (110). In addition, nuclear magnetic resonance spectroscopy (NMR) revealed that triglycerides were present in biopsies from GBM brain patients but absent in healthy adult brain tissues (15). Moreover, a correlation has been established between lipid resonance spectra and the grade of malignancy (15). Alongside triglycerides, lipid droplets store cholesterol under their esterified form, cholesteryl ester, which is hydrolyzed by cholesteryl ester hydrolase, also known as hormone-sensitive lipase (111). Cholesterol Ester metabolism has also emerged as a promising pathway to target GBM treatment (9). Besides, in vitro studies have shown that densely plated astrocytes turn off the expression of genes associated with cholesterol synthesis and show a low cholesterol level, while glioma cells maintain a high cholesterol level (112).
In expanding de novo lipid synthesis, tumor cells can also use exogenous fatty acids to stimulate their growth. Recently, Acyl-CoA-binding protein, a regulator of LCFA intracellular metabolism in astrocytes very pronounced in GBM, reported controlling the accessibility of long-chain fatty acyl-CoAs to mitochondria, advancing fatty acid oxidation, tumor development, and poor survival through acyl-CoAs liaison, in several preclinical models (113, 114). Moreover, inhibiting fatty acid oxidation reduces proliferation in human glioma primary-cultured cells and prolongs survival in malignant glioma mouse models (115). On the other hand, as mentioned earlier, a mutation in IDH, the essential enzyme in the tricarboxylic acid cycle occurring in the mitochondria, is strongly associated with secondary GBM (116). Notably, a comprehensive metabolism investigation was performed on clinical IDH1 mutant glioma specimens. The results showed a reduction in the pool of fatty acyl chains, displayed as a reduction in triglyceride and sphingolipid levels, without any change in membrane phosphatidyl lipids level (116).
The focus of recent research has been on the interplay between lipid metabolism and cell death mechanisms. However, an emerging research area is understanding specific lipid-associated metabolic dependencies of malignant cells and harnessing their potential to disrupt the lipid homeostasis, which triggers lipotoxicity and induces cell death. For example, Ferroptosis, a newly described form of programmed cell death due to the increased iron-associated lipid peroxidation and ROS production, has been recently investigated as a new direction in gliomas treatment (117). Indeed, several ferroptosis-associated genes were recently established, and targeting them using diverse approaches has shown significant outcomes in low-grade glioma and GBM. For instance, inhibiting diacylglycerol-acyltransferase 1 (DGAT1), which is known to catalyze the storage of excess FFAs into TGs and lipid droplets (Figure 4), has been shown to suppress GBM growth in vitro and in vivo by triggering oxidative stress and ferroptosis (118, 119). Similarly, depletion of glutathione peroxidase 4 (GPX4), an enzyme with a pivotal role in preventing phospholipid oxidation, was powerfully efficient in inducing ferroptosis in GBM (120, 121). Moreover, past studies have demonstrated that overexpression of acyl-CoA synthetase long-chain family member 4 (ACSL4) results from ferroptosis induction, yielding a remarkable effect on inhibiting glioma cell proliferation.
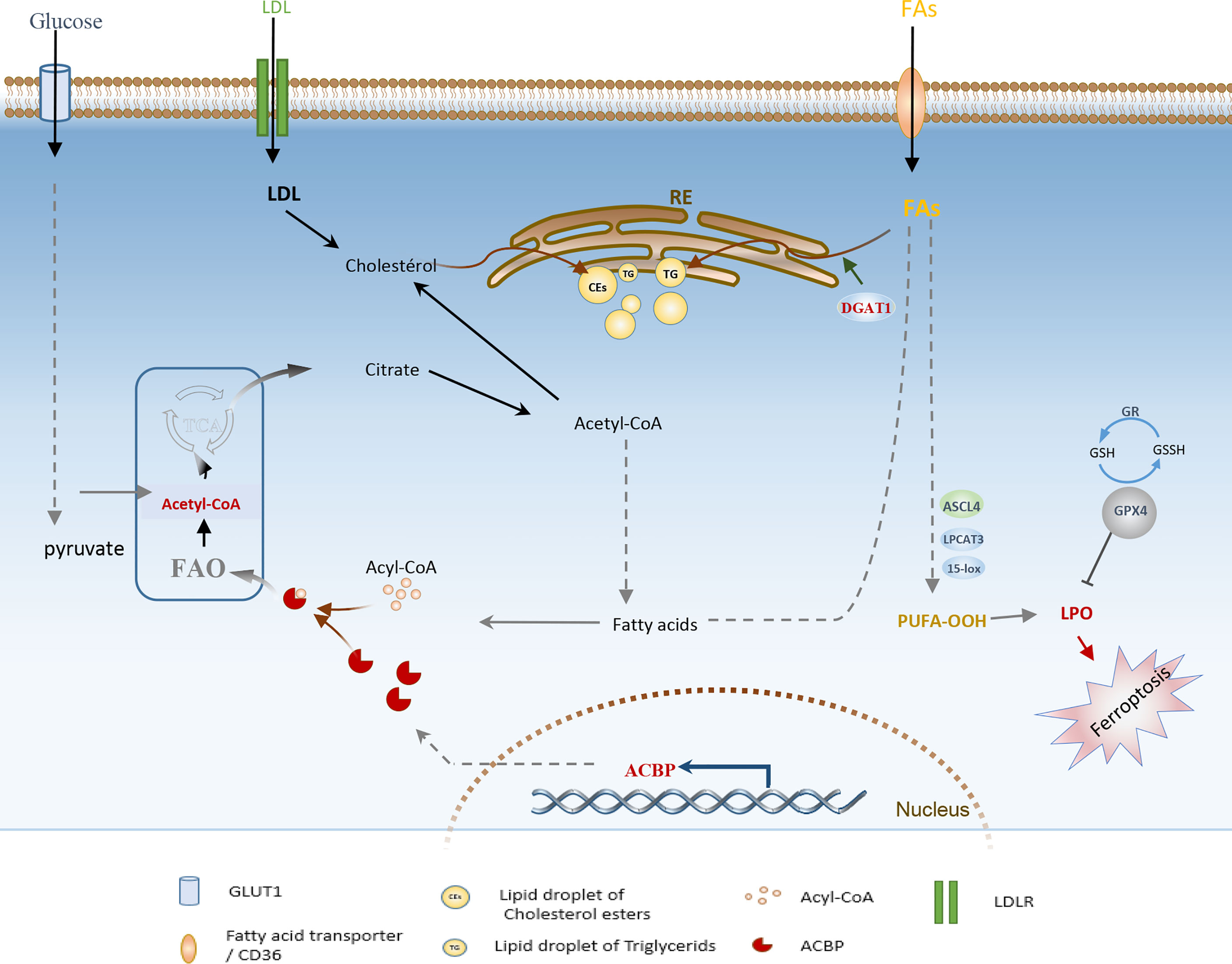
Figure 4 Schematic illustration of adaptive lipid metabolism in glioblastoma: role of the highly expressed ACBP, DGAT1, GPx4, and lipid droplets in lipid oxidation and ferroptosis induction. FAs, Fatty acids; FAO, Fatty acid oxidation; PUFA, Polyunsaturated fatty acids; DGAT1, diacylglycerol-acyltransferase 1; ACSL4, acyl-CoA synthetase long-chain family member 4; GPX4, glutathione peroxidase; GR, glutathione reductase; LD, lipid Droplet; LPO, lipid peroxides; ACBP, Acyl-CoA-binding protein; LDLR, low-density lipoprotein receptor; ER, Endoplasmic Reticulum.
Even though the evidence demonstrates the critical role of lipid metabolism on tumorigenic properties of GBM, the lipid metabolic pathways are not yet completely understood, and results still derive from in vitro studies, mouse genetic models, and xenograft models. Understanding how GBM cells adapt their underlying molecular mechanisms to reprogram lipid metabolism depending on their environment will be the key to further translating primary findings to clinical use.
Although the concept of targeting tumor metabolism is not entirely new, a better understanding of the metabolic landscape of malignant gliomas has opened the way for a growing number of investigations into the metabolic vulnerabilities of these tumors. Thus, considerable and continuous efforts are being directed toward exploiting specific dependencies to identify novel molecular targets and design new effective therapeutic strategies that help improve the patient’s clinical care with GBM (8, 35, 122). Indeed, the paradigm of focusing on cancer metabolism was freshly required beyond the dramatic flexibleness of tumor metabolic cascades reported in cancer cells challenged with multiplex inhibitors, including glycolysis and glutaminase inhibitors (123, 124).
However, recent research in gliomas and GBM clinical trials have focused mainly on the energetic aspect of cancer metabolism as a promising approach providing several potent targetable enzymes, aiming to overcome GBM heterogeneity and/or improve the outcomes of standard therapies. Currently, many drugs targeting glycolysis, OXPHOS, glutaminolysis, lipid, and nucleotide synthesis are being studied in clinical trials to treat gliomas (Table 1).
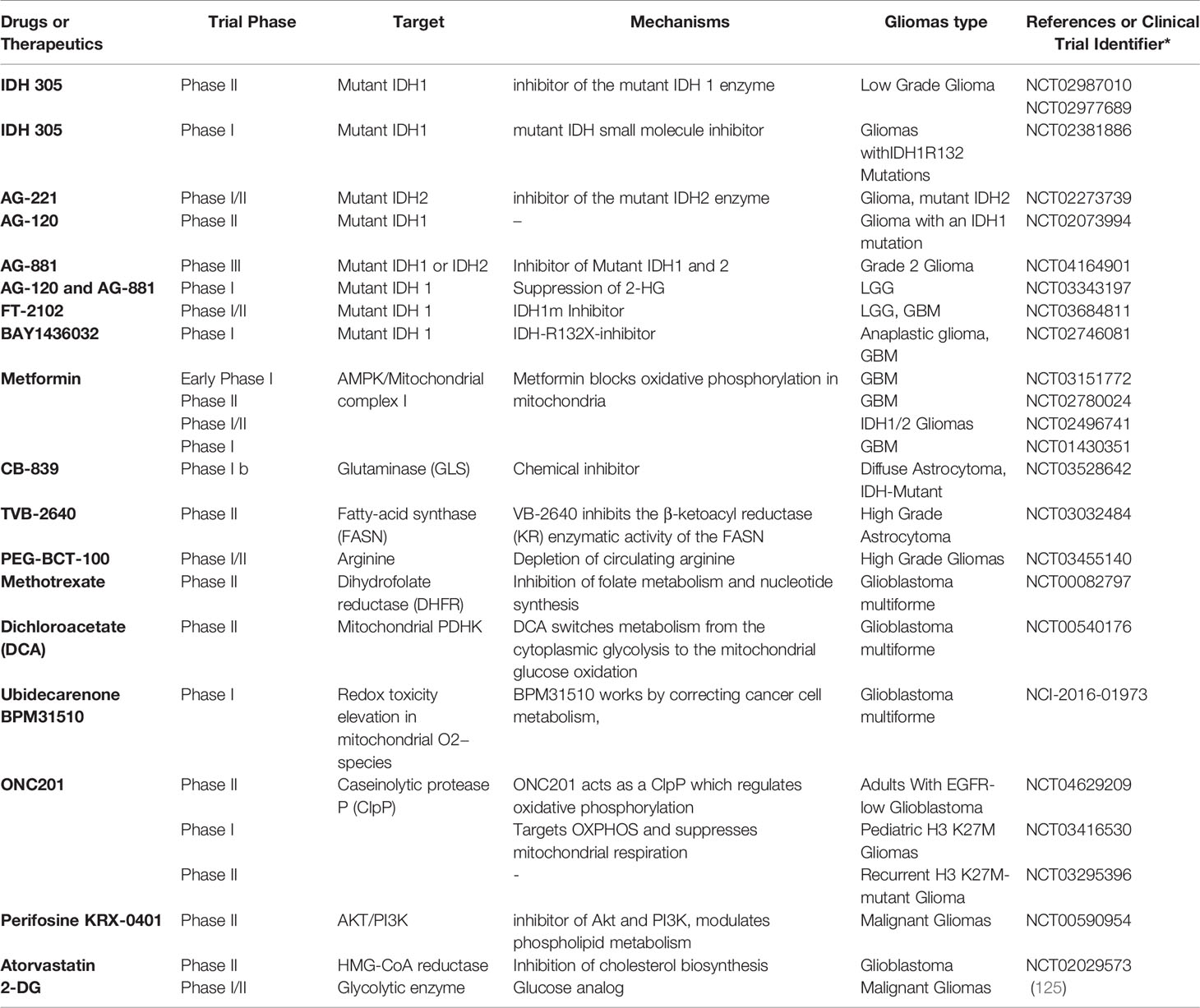
Table 1 Current Advances and main clinical trials in metabolism targeting for gliomas therapy.
Many current metabolism-based therapies against gliomas have attempted to harness the fact that IDH mutations are among the most common alterations that drive metabolism remodeling in low-grade gliomas and secondary GBM (44). Therefore, numerous researchers considered isocitrate dehydrogenase as an actionable target and have been performed over the past decade to take advantage of the inhibition of mutant IDH enzymes through pharmacological inhibitors or small molecules to disrupt tumor metabolism and alter thereby tumor growth. As a result, multiple IDH-selective inhibitors have been advanced to focalize tumors harboring IDH mutations. For instance, AG-881, a chemical inhibitor of IDH1, is the foremost enhanced IDH inhibitor for grade II gliomas that have launched a phase III clinical trial with a significant outcome in this grade of gliomas. In line with this, IDH-mutant glioma cells have been indirectly inhibited by (AGI-5198), an IDH1 inhibitor known to act specifically against R132H alterations (126, 127).
Currently, other clinical trials have discovered novel inhibitors targeting both IDH1 and IDH2. Thus, allowing the control of other malignancies harboring IDH mutations, such as gliomas invasiveness, has been shown, mechanistically, to decrease 2-HG levels (128). In this regard, IDH305 (Novartis) targets IDH1 mutations, and ongoing trials are presently being conducted in gliomas and other malignancies with IDH1 R132 mutations (NCT02381886). In addition, FT2102 (Forma Therapeutics) and BAY1436032 (Bayer) also target IDH1 R132 tumors (R132X for BAY1436032), and occurring trials are selecting patients with various tumor forms, including GBM (NCT03684811 and NCT02746081).
Other inhibitors are being developed to manage glioma tumors in the same context. Among these, AG221 (Enasidenib, CC90007; Agios) reported targeting IDH2 mutations and is currently beneath improvement to treat gliomas, in addition to AG881 (a pan-IDH1/-2 (Agios) inhibitor which is being developed for the administration of gliomas cancers. This molecule restrains the cytoplasmic IDH1 and IDH2 within the mitochondria; AG-881 can also penetrate the blood-brain barrier.
Undoubtedly, glutamine metabolism can indirectly influence IDH1/2 function since glutamine-derived glutamate is a precursor of ketoglutarate, the amounts of which determine the rate of IDH1/2 activity (123). In fact, beneath certain metabolic circumstances, such as hypoxia, cancer cells preferentially employ glutamine-derived chemicals in lipogenesis over the preferred IDH1 pathway-producing molecule (58, 129). Patients with IDH-mutated GBM may benefit from glutamine metabolism targeting since it will efficiently disrupt de novo lipids synthesis, crucial to sustaining cancer cell proliferation and tumor growth.
Besides clinical trials targeting directly mutant IDH enzymes, a wide range of emerging assays has been alternatively attempting to harness other metabolic aspects by acting on different effectors such as mTOR and EGFR, which are extensively targeted using various classes of molecules, including monoclonal antibodies (mAbs) and tyrosine kinase inhibitors (TKI) (130). Similarly, several metabolic enzymes have emerged and clinically tested, notably those involved in glucose oxidation, oxidative phosphorylation, fatty acids, and nucleotide synthesis. For instance, a phase II clinical trial has been led to evaluate the safety and efficiency of Dichloroacetate (DCA), which is an inhibitor of the mitochondrial pyruvate dehydrogenase kinase that is believed to shift metabolism from the cytoplasmic glycolysis to mitochondrial glucose oxidation, thereby altering the GBM metabolism and inhibiting the resistance to apoptosis (131).
Up to this point, the metabolic-driven treatment strategies had been challenging to implement clinically and had no longer proven effective thus far. Consequently, there may be a growing need to test innovative metabolism-driven approaches that target diverse elements of glioma metabolism, including glutamate-glutamine metabolism, for example, the utilization of a systemic glutaminases inhibitors therapy.
Of note, glutamine metabolism targets are also being developed as therapeutic approaches. For instance, targeting glutaminase (GLS), which catalyzes the primary stage of glutamine metabolism, leads to the development of a new drug, the CB-839 hydrochloride (CB-839), which is currently in phase 1b trial. This trial focused on combining CB-839 with radiotherapy and temozolomide to evaluate the efficacy and safety of treating IDH-mutated diffuse astrocytoma. The election of the most relevant players in glutamine metabolism will assist in the layout of novel treatments that might lethally disturb GBM cells and impair disease development.
In addition to all anterior researched targets clinically tested, recent evidence from preclinical models and in vitro studies reveals many new potential metabolic targets for glioblastoma, showing encouraging results with a remarkable inhibitory effect on tumor growth and malignancy (8, 9, 88, 119, 132). Notably, among the proposed actionable targets, several glycolytic enzymes such as hexokinase2 (HK2), phosphofructokinase-1 (PFK1), and phosphoinositide-dependent kinase-1 (PDK1) were considered for therapeutic use and experimentally evaluated (Table 2). Further, previous in vitro and in vivo studies have noted that targeting OXPHOS proteins, lipoxygenases, and many other metabolic effectors involved in cholesterol and amino acid metabolism, fatty acids, and nucleotide synthesis, have been able to disrupt the altered metabolism of GBM, yielding significant results that should be harnessed for better therapeutic solutions (8, 152).
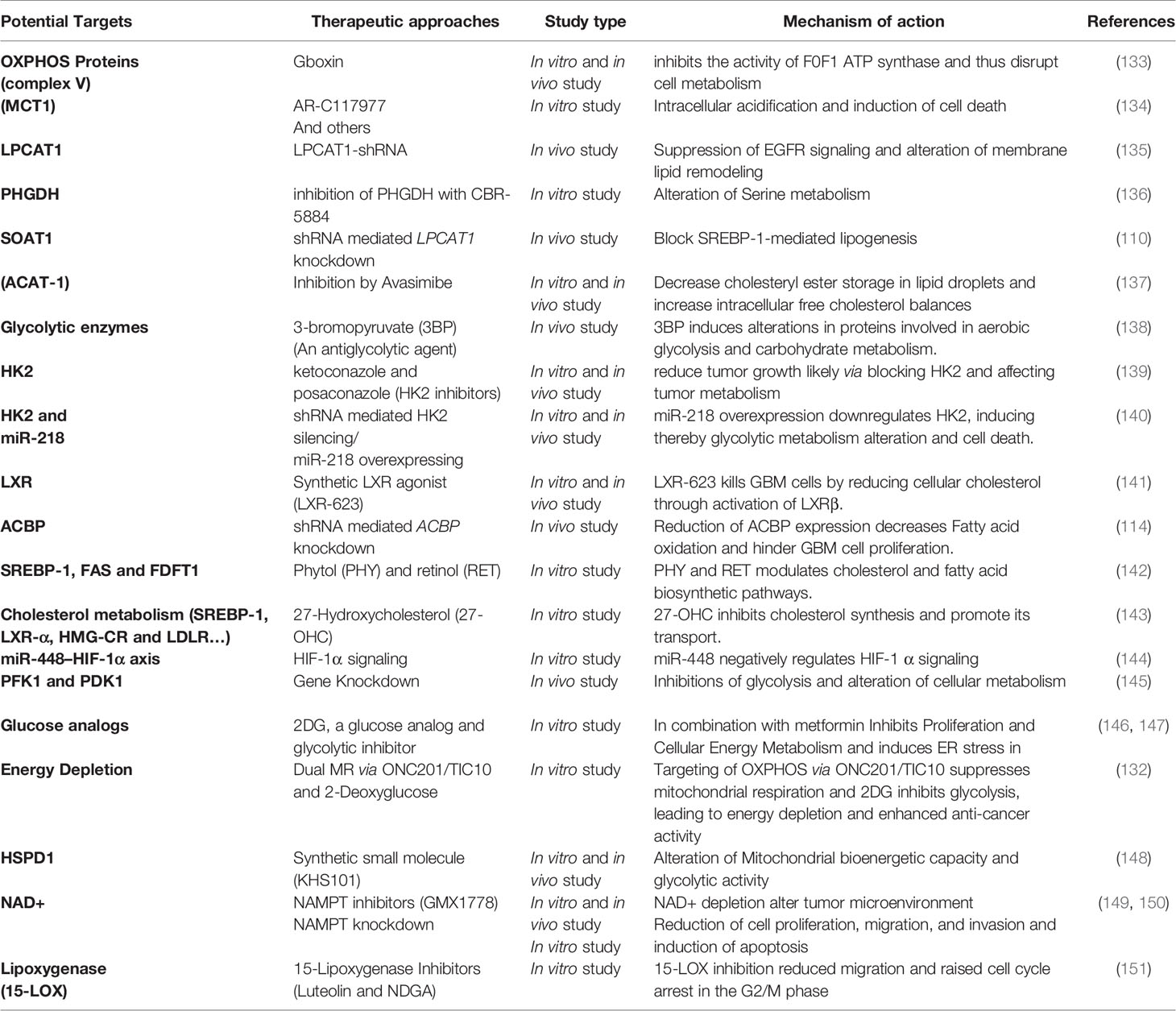
Table 2 Potential targets and promising agents against glioblastoma metabolism.
While mutated genes, especially those encoding metabolic enzymes and other vital metabolites such as the IDH1/2, were widely investigated in several malignancies, recent evidence highlights the relevance of unmutated forms of these enzymes in metabolic alterations as a hallmark of cancer additionally. Calvert et al., for instance, demonstrated via in silico and wet-bench investigations that non-mutated isocitrate dehydrogenase 1 (IDH1) is often overexpressed in primary GBM and that genetic and pharmacological inactivation leads to reduction of GBM tumor growth (153). Therefore, rather than focusing exclusively on mutated genes, new treatment approaches and future research should consider investigating and addressing the unmutated enzymes’ implications and potential.
Given that various cancers share a broad spectrum of genomic alterations, most metabolic abnormalities and actionable targets recently evidenced in glioblastoma are likely to be non-tumor specific (154). Moreover, the heterogeneous nature of GBM is another challenge in the way of establishing efficient metabolic-based therapeutic approaches. Thus, focusing on specific genetic mutations and their downstream metabolic pathways may offer great opportunities for more targeted therapy. For instance, in low-grade gliomas, the IDH1/2 is a significant target. A dozen clinical trials have been conducted to explore its potential in IDH-mutant gliomas (Table 1). In addition, EGFR amplification and EGFRvIII, a tumor-specific mutations frequently reported in glioblastoma, have been demonstrated to be involved in metabolism remodeling and recently regarded as promising clues for metabolism-based therapy in GBM (47). On the other hand, the emerging link between cancer genetic alterations and metabolic dependencies raises the critical need for coupling new metabolic therapeutics with specific biomarkers to deliver more targeted personalized treatments.
Overall, dysregulated metabolic pathways in glioblastoma offer considerable opportunities to improve treatment outcomes, whereas developing effective approaches is still challenging. However, small-molecule inhibitors and monoclonal antibodies remain the main therapeutic strategies being used so far, and many of them are undergoing clinical trials intending to inhibit key metabolic targets in gliomas. Genome editing and RNA therapy have recently emerged as powerful tools to screen targetable genes and understand the molecular mechanisms underlying the pathological phenotype and can be exploited for new therapeutic advances (155). Indeed, research in metabolism-targeting has benefited from the emerging technology of CRISPR/Cas9 and RNA therapeutics (ASO, siRNA, and shRNA). Meanwhile, recent studies on GBM cell lines and animal models have applied these targeted tools to establish the proof-of-concept and assess the efficiency of several metabolic candidates. Nevertheless, these versatile strategies remain lacking from ongoing clinical studies.
In conclusion, emerging evidence linking genetic and epigenetic alterations in cancer to altered metabolism has significantly enhanced our understanding of metabolic rewiring, a hallmark of gliomas tumors. Indeed, GBM cells shift their energetic metabolism towards a variety of metabolic states characterized by rapid and enhanced ATP generation as well as increased anabolic metabolism (8, 92). These changes help cells sustain the high proliferation rate and adapt to continuous fluctuations in the tumor microenvironment, particularly changes in nutrients and oxygen availability (95–97). Besides, numerous studies suggested a key role of these metabolic adaptations in immune cell dysfunction and cancer immune escape via acidification of the TME, secretion of immunosuppressive cytokines, and induction of epigenetic modifications in immune cells (156–158). Alongside this, similar metabolic reprogramming occurs on the surrounding immune cells, affecting their activation and function, altering the anti-cancer immunity, and thereby enabling tumor cells to evade the immunosurveillance system (159). Additionally, the altered metabolic landscape is tightly linked to the ability of tumor cells to maintain the cellular redox balance and escape apoptosis under tumor conditions (43, 58).
Importantly, GBM cells are metabolically heterogeneous within the tumor. They display distinct metabolic profiles and intrinsic plasticity to hypoxic and nutritionally deficient conditions through compensatory enhancement of alternative metabolic pathways (91, 160). In this sense, therapeutic approaches based on co-targeting several metabolic dependencies may represent a powerful strategy to tackle this dynamic metabolic nature of GBM cells.
From a therapeutic perspective, dysregulated metabolic pathways in GBM offer considerable opportunities to improve treatment outcomes, while the development of effective approaches is still lacking. However, several challenges, such as the rapid and continuous alterations at the genetic and epigenetic level leading to increased crosstalk between pathways, have made such therapies non-ubiquitous. This ending raises the need for exploring and exploiting new horizons in GBM metabolism. In this review, we focused on the most common and well-characterized genetic patterns affecting glioma’s cell metabolism. However, more research is being conducted to explore the relevance and the potential of other effectors, notably non-coding RNAs and epigenetic alterations, as new and promising aspects that may offer exploitable vulnerabilities to disrupt GBM metabolism with translational implications (161–164).
Conceptualization: RE and AE; Methodology: RE, AE, NB, BT, ZW, and AZ; Writing: All authors; Supervision: RE. Funding acquisition: RE. All authors contributed to the article and approved the submitted version.
This work received UM6P’s internal welcoming funds and the FFI from OCP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (Berl) (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
2. Tanboon J, Williams EA, Louis DN. The Diagnostic Use of Immunohistochemical Surrogates for Signature Molecular Genetic Alterations in Gliomas. J Neuropathol Exp Neurol (2016) 75:4–18. doi: 10.1093/jnen/nlv009
3. Delgado-Martín B, Medina MÁ. Advances in the Knowledge of the Molecular Biology of Glioblastoma and Its Impact in Patient Diagnosis, Stratification, and Treatment. Adv Sci (2020) 7(9):1902971. doi: 10.1002/advs.201902971
4. Cancer Genome Atlas Research Network. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature (2008) 455:1061–8. doi: 10.1038/nature07385
5. Liao K-L, Huang S, Wu Y-P. The Prognosis for Patients With Newly Diagnosed Glioblastoma Receiving Bevacizumab Combination Therapy: A Meta-Analysis. OncoTargets Ther (2018) 11:3513–20. doi: 10.2147/OTT.S156723
6. Liu H, Qiu W, Sun T, Wang L, Du C, Hu Y, et al. Therapeutic Strategies of Glioblastoma (GBM): The Current Advances in the Molecular Targets and Bioactive Small Molecule Compounds. Acta Pharm Sin B (2022) 12:1781–804. doi: 10.1016/j.apsb.2021.12.019
7. Huang L, Jiang S, Shi Y. Tyrosine Kinase Inhibitors for Solid Tumors in the Past 20 Years (2001–2020). J Hematol OncolJ Hematol Oncol (2020) 13:143. doi: 10.1186/s13045-020-00977-0
8. Bi J, Chowdhry S, Wu S, Zhang W, Masui K, Mischel PS. Altered Cellular Metabolism in Gliomas — an Emerging Landscape of Actionable Co-Dependency Targets. Nat Rev Cancer (2020) 20:57–70. doi: 10.1038/s41568-019-0226-5
9. Pirmoradi L, Seyfizadeh N, Ghavami S, Zeki AA, Shojaei S. Targeting Cholesterol Metabolism in Glioblastoma: A New Therapeutic Approach in Cancer Therapy. J Investig Med Off Publ Am Fed Clin Res (2019) 67:715–9. doi: 10.1136/jim-2018-000962
10. Filbin MG, Suvà ML. Gliomas Genomics and Epigenomics: Arriving at the Start and Knowing It for the First Time. Annu Rev Pathol Mech Dis (2016) 11:497–521. doi: 10.1146/annurev-pathol-012615-044208
11. Qi Y, Chen D, Lu Q, Yao Y, Ji C. Bioinformatic Profiling Identifies a Fatty Acid Metabolism-Related Gene Risk Signature for Malignancy, Prognosis, and Immune Phenotype of Glioma. Dis Markers (2019) 2019:e3917040. doi: 10.1155/2019/3917040
12. Shao W, Gu J, Huang C, Liu D, Huang H, Huang Z, et al. Malignancy-Associated Metabolic Profiling of Human Glioma Cell Lines Using 1H NMR Spectroscopy. Mol Cancer (2014) 13:197. doi: 10.1186/1476-4598-13-197
13. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
14. Jose C, Bellance N, Rossignol R. Choosing Between Glycolysis and Oxidative Phosphorylation: A Tumor’s Dilemma? Biochim Biophys Acta BBA - Bioenerg (2011) 1807:552–61. doi: 10.1016/j.bbabio.2010.10.012
15. Tugnoli V, Tosi MR, Tinti A, Trinchero A, Bottura G, Fini G. Characterization of Lipids From Human Brain Tissues by Multinuclear Magnetic Resonance Spectroscopy. Biopolymers (2001) 62:297–306. doi: 10.1002/bip.10005
16. Koukourakis M, Tsolou A, Pouliliou S, Lamprou I, Papadopoulou M, Ilemosoglou M, et al. Blocking LDHA Glycolytic Pathway Sensitizes Glioblastoma Cells to Radiation and Temozolomide. Biochem Biophys Res Commun (2017) 491(4):932–938. doi: 10.1016/j.bbrc.2017.07.138
17. Zhang L, Yang H, Zhang W, Liang Z, Huang Q, Xu G, et al. Clk1-Regulated Aerobic Glycolysis Is Involved in Glioma Chemoresistance. J Neurochem (2017) 142(4):574–88. doi: 10.1111/jnc.14096
18. Zhou Z, Huang R, Chai R, Zhou X, Hu Z, Wang W, et al. Identification of an Energy Metabolism-Related Signature Associated With Clinical Prognosis in Diffuse Glioma. Aging (2018) 10:3185–209. doi: 10.18632/aging.101625
19. Calabrese E, Villanueva-Meyer JE, Cha S. A Fully Automated Artificial Intelligence Method for non-Invasive, Imaging-Based Identification of Genetic Alterations in Glioblastomas. Sci Rep (2020) 10:11852. doi: 10.1038/s41598-020-68857-8
20. Montemurro N. Glioblastoma Multiforme and Genetic Mutations: The Issue Is Not Over Yet. An Overview of the Current Literature. J Neurol Surg Part Cent Eur Neurosurg (2020) 81:64–70. doi: 10.1055/s-0039-1688911
21. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics (2017) 14:284–97. doi: 10.1007/s13311-017-0519-x
22. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 Mutations are Early Events in the Development of Astrocytomas and Oligodendrogliomas. Am J Pathol (2009) 174:1149–53. doi: 10.2353/ajpath.2009.080958
23. Ruiz-Rodado V, Seki T, Dowdy T, Lita A, Zhang M, Han S, et al. Metabolic Landscape of a Genetically Engineered Mouse Model of IDH1 Mutant Glioma. Cancers (2020) 12(6):1633. doi: 10.3390/cancers12061633
24. Velázquez Vega JE, Brat DJ. Molecular-Genetic Classification of Gliomas and its Practical Application to Diagnostic Neuropathology. In: Diffuse Low-Grade Gliomas in Adults. Cham: Springer International Publishing (2017). p. 73–100. doi: 10.1007/978-3-319-55466-2_4
25. Masui K, Mischel PS, Reifenberger G. Molecular Classification of Gliomas. In: Handbook of Clinical Neurology. Elsevier B.V (2016). p. 97–120. doi: 10.1016/B978-0-12-802997-8.00006-2
26. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. An Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell (2010) 17:98. doi: 10.1016/j.ccr.2009.12.020
27. Gao Q, Cui Y, Shen Y, Li Y, Gao X, Xi Y, et al. Identifying Mutually Exclusive Gene Sets With Prognostic Value and Novel Potential Driver Genes in Patients With Glioblastoma. BioMed Res Int (2019) 2019:4860367–4860367. doi: 10.1155/2019/4860367
28. Lu VM, Akinduro OO, Daniels DJ. H3K27M Mutation in Adult Cerebellar Glioblastoma. J Clin Neurosci (2020) 71:316–7. doi: 10.1016/j.jocn.2019.11.016
29. El Tekle G, Bernasocchi T, Unni AM, Bertoni F, Rossi D, Rubin MA, et al. Co-Occurrence and Mutual Exclusivity: What Cross-Cancer Mutation Patterns can Tell Us. Trends Cancer (2021) 7:823–36. doi: 10.1016/j.trecan.2021.04.009
30. Ulz P, Heitzer E, Speicher MR. Co-Occurrence of MYC Amplification and TP53 Mutations in Human Cancer. Nat Genet (2016) 48:104–6. doi: 10.1038/ng.3468
31. Wang L, Hu H, Pan Y, Wang R, Li Y, Shen L, et al. PIK3CA Mutations Frequently Coexist With EGFR/KRAS Mutations in Non-Small Cell Lung Cancer and Suggest Poor Prognosis in EGFR/KRAS Wildtype Subgroup. PLoS One (2014) 9:e88291. doi: 10.1371/journal.pone.0088291
32. Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, et al. ETS Factors Reprogram the Androgen Receptor Cistrome and Prime Prostate Tumorigenesis in Response to PTEN Loss. Nat Med (2013) 19:1023–9. doi: 10.1038/nm.3216
33. Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, et al. Aberrant ERG Expression Cooperates With Loss of PTEN to Promote Cancer Progression in the Prostate. Nat Genet (2009) 41:619–24. doi: 10.1038/ng.370
34. Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, et al. Combined MYC and P53 Defects Emerge at Medulloblastoma Relapse and Define Rapidly Progressive, Therapeutically Targetable Disease. Cancer Cell (2015) 27:72–84. doi: 10.1016/j.ccell.2014.11.002
35. Fumarola C, Petronini PG, Alfieri R. Impairing Energy Metabolism in Solid Tumors Through Agents Targeting Oncogenic Signaling Pathways. Biochem Pharmacol (2018) 151:114–25. doi: 10.1016/j.bcp.2018.03.006
36. Quinones A, Le A. The Multifaceted Metabolism of Glioblastoma. In: Le A, editor. The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology. Cham: Springer International Publishing (2018). p. 59–72. doi: 10.1007/978-3-319-77736-8_4
37. Lugowska I, Teterycz P, Mikula M, Kulecka M, Kluska A, Balabas A, et al. IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma. J Cancer (2018) 9:998–1005. doi: 10.7150/jca.22915
38. Unruh D, Zewde M, Buss A, Drumm MR, Tran AN, Scholtens DM, et al. Methylation and Transcription Patterns are Distinct in IDH Mutant Gliomas Compared to Other IDH Mutant Cancers. Sci Rep (2019) 9(1):1–11. doi: 10.1038/s41598-019-45346-1
39. Bouchmaa N, Mrid RB, Boukharsa Y, Bouargalne Y, Nhiri M, Idir A, et al. Reactive Oxygen Species-Mediated Apoptosis and Cytotoxicity of Newly Synthesized Pyridazin-3-Ones In P815 (Murin Mastocytoma) Cell Line. Drug Res (2019) 69:528–36. doi: 10.1055/a-0762-3775
40. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature (2009) 462:739–44. doi: 10.1038/nature08617
41. Masui K, Onizuka H, Cavenee WK, Mischel PS, Shibata N. Metabolic Reprogramming in the Pathogenesis of Glioma: Update: Metabolic Reprogramming in Gliomas. Neuropathology (2019) 39:3–13. doi: 10.1111/neup.12535
42. Ye D, Guan K-L, Xiong Y. Metabolism, Activity, and Targeting of D-And L-2-Hydroxyglutarates. Trends Cancer (2018) 4:151–65. doi: 10.1016/j.trecan.2017.12.005
43. McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell (2018) 175:101–16.e25. doi: 10.1016/j.cell.2018.08.038
44. Fack F, Tardito S, Hochart G, Oudin A, Zheng L, Fritah S, et al. Altered Metabolic Landscape in IDH-Mutant Gliomas Affects Phospholipid, Energy, and Oxidative Stress Pathways. EMBO Mol Med (2017) 9:1681–95. doi: 10.15252/emmm.201707729
45. Biedermann J, Preussler M, Conde M, Peitzsch M, Richter S, Wiedemuth R, et al. Mutant IDH1 Differently Affects Redox State and Metabolism in Glial Cells of Normal and Tumor Origin. Cancers (2019) 11(12):2028. doi: 10.3390/cancers11122028
46. Dekker LJM, Wu S, Jurriëns C, Mustafa DAN, Grevers F, Burgers PC, et al. Metabolic Changes Related to the IDH1 Mutation in Gliomas Preserve TCA-Cycle Activity: An Investigation at the Protein Level. FASEB J (2020) 34:3646–57. doi: 10.1096/fj.201902352R
47. Eskilsson E, Røsland GV, Solecki G, Wang Q, Harter PN, Graziani G, et al. EGFR Heterogeneity and Implications for Therapeutic Intervention in Glioblastoma. Neuro-Oncol (2018) 20:743–52. doi: 10.1093/neuonc/nox191
48. Hunter T. Tyrosine Phosphorylation: Thirty Years and Counting. Curr Opin Cell Biol (2009) 21:140–6. doi: 10.1016/j.ceb.2009.01.028
49. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The Somatic Genomic Landscape of Glioblastoma. Cell (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
50. An Z, Aksoy O, Zheng T, Fan Q-W, Weiss WA. Epidermal Growth Factor Receptor and EGFRvIII in Glioblastoma: Signaling Pathways and Targeted Therapies. Oncogene (2018) 37:1561–75. doi: 10.1038/s41388-017-0045-7
51. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by C-Myc. J Biol Chem (2000) 275:21797–800. doi: 10.1074/jbc.C000023200
52. Kim J, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, et al. Evaluation of Myc E-Box Phylogenetic Footprints in Glycolytic Genes by Chromatin Immunoprecipitation Assays. Mol Cell Biol (2004) 24:5923–36. doi: 10.1128/MCB.24.13.5923-5936.2004
53. Kim J, Gao P, Liu Y-C, Semenza GL, Dang CV. Hypoxia-Inducible Factor 1 and Dysregulated C-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol Cell Biol (2007) 27:7381–93. doi: 10.1128/MCB.00440-07
54. Babic I, Anderson ES, Tanaka K, Guo D, Masui K, Li B, et al. EGFR Mutation-Induced Alternative Splicing of Max Contributes to Growth of Glycolytic Tumors in Brain Cancer. Cell Metab (2013) 17:1000–8. doi: 10.1016/j.cmet.2013.04.013
55. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, et al. mTOR Complex 2 Controls Glycolytic Metabolism in Glioblastoma Through FoxO Acetylation and Upregulation of C-Myc. Cell Metab (2013) 18:726–39. doi: 10.1016/j.cmet.2013.09.013
56. Liu F, Hon GC, Villa GR, Turner KM, Ikegami S, Yang H, et al. EGFR Mutation Promotes Glioblastoma Through Epigenome and Transcription Factor Network Remodeling. Mol Cell (2015) 60:307–18. doi: 10.1016/j.molcel.2015.09.002
57. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-Driven Oxidative Phosphorylation is a Major ATP Source in Transformed Mammalian Cells in Both Normoxia and Hypoxia. Mol Syst Biol (2013) 9:712. doi: 10.1038/msb.2013.65
58. Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, et al. Reductive Carboxylation Supports Redox Homeostasis During Anchorage-Independent Growth. Nature (2016) 532:255–8. doi: 10.1038/nature17393
59. Yang R, Li X, Wu Y, Zhang G, Liu X, Li Y, et al. EGFR Activates GDH1 Transcription to Promote Glutamine Metabolism Through MEK/ERK/ELK1 Pathway in Glioblastoma. Oncogene (2020) 39:2975–86. doi: 10.1038/s41388-020-1199-2
60. Qian X, Li X, Shi Z, Xia Y, Cai Q, Xu D, et al. PTEN Suppresses Glycolysis by Dephosphorylating and Inhibiting Autophosphorylated Pgk1. Mol Cell (2019) 76:516–27.e7. doi: 10.1016/j.molcel.2019.08.006
61. Ahmad F, Patrick S, Sheikh T, Sharma V, Pathak P, Malgulwar PB, et al. Telomerase Reverse Transcriptase (TERT) - Enhancer of Zeste Homolog 2 (EZH2) Network Regulates Lipid Metabolism and DNA Damage Responses in Glioblastoma. J Neurochem (2017) 143:671–83. doi: 10.1111/jnc.14152
62. Hoang-Minh LB, Siebzehnrubl FA, Yang C, Suzuki-Hatano S, Dajac K, Loche T, et al. Infiltrative and Drug-Resistant Slow-Cycling Cells Support Metabolic Heterogeneity in Glioblastoma. EMBO J (2018) 37(23):e98772. doi: 10.15252/embj.201798772
63. Crespo I, Vital AL, Gonzalez-Tablas M, Patino M del C, Otero A, Lopes MC, et al. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am J Pathol (2015) 185:1820–33. doi: 10.1016/j.ajpath.2015.02.023
64. Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, et al. Driver Mutations in Histone H3.3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature (2012) 482:226–31. doi: 10.1038/nature10833
65. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG Island Methylator Phenotype That Defines a Distinct Subgroup of Glioma. Cancer Cell (2010) 17:510–22. doi: 10.1016/j.ccr.2010.03.017
66. Butler M, Pongor L, Su Y-T, Xi L, Raffeld M, Quezado M, et al. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer (2020) 6:380–91. doi: 10.1016/j.trecan.2020.02.010
67. Gusyatiner O, Hegi ME. Glioma Epigenetics: From Subclassification to Novel Treatment Options. Semin Cancer Biol (2018) 51:50–8. doi: 10.1016/j.semcancer.2017.11.010
68. Paulmurugan R, Malhotra M, Massoud TF. The Protean World of non-Coding RNAs in Glioblastoma. J Mol Med (2019) 97:909–25. doi: 10.1007/s00109-019-01798-6
69. Masoudi MS, Mehrabian E, Mirzaei H. MiR-21: A Key Player in Glioblastoma Pathogenesis. J Cell Biochem (2018) 119:1285–90. doi: 10.1002/jcb.26300
70. Khani P, Nasri F, Khani Chamani F, Saeidi F, Sadri Nahand J, Tabibkhooei A, et al. Genetic and Epigenetic Contribution to Astrocytic Gliomas Pathogenesis. J Neurochem (2019) 148:188–203. doi: 10.1111/jnc.14616
71. El Fatimy R, Subramanian S, Uhlmann EJ, Krichevsky AM. Genome Editing Reveals Glioblastoma Addiction to MicroRNA-10b. Mol Ther J Am Soc Gene Ther (2017) 25:368–78. doi: 10.1016/j.ymthe.2016.11.004
72. El Fatimy R, Zhang Y, Deforzh E, Ramadas M, Saravanan H, Wei Z, et al. A Nuclear Function for an Oncogenic microRNA as a Modulator of snRNA and Splicing. Mol Cancer (2022) 21:17. doi: 10.1186/s12943-022-01494-z
73. Li Q, Jia H, Li H, Dong C, Wang Y, Zou Z. LncRNA and mRNA Expression Profiles of Glioblastoma Multiforme (GBM) Reveal the Potential Roles of lncRNAs in GBM Pathogenesis. Tumor Biol (2016) 37:14537–52. doi: 10.1007/s13277-016-5299-0
74. Qin J, Jiang C, Cai J, Meng X. Roles of Long Noncoding RNAs in Conferring Glioma Progression and Treatment. Front Oncol (2021) 11:688027. doi: 10.3389/fonc.2021.688027
75. Appin CL, Brat DJ. Biomarker-Driven Diagnosis of Diffuse Gliomas. Mol Aspects Med (2015) 45:87–96. doi: 10.1016/j.mam.2015.05.002
76. de los Santos-Jiménez J, Campos-Sandoval JA, Márquez-Torres C, Urbano-Polo N, Brøndegaard D, Martín-Rufián M, et al. Glutaminase Isoforms Expression Switches microRNA Levels and Oxidative Status in Glioblastoma Cells. J BioMed Sci (2021) 28:14. doi: 10.1186/s12929-021-00712-y
77. Li H, Yuan H. MiR-1297 Negatively Regulates Metabolic Reprogramming in Glioblastoma via Repressing KPNA2. Hum Cell (2020) 33:619–29. doi: 10.1007/s13577-019-00316-7
78. Li J, Liu Q, Liu Z, Xia Q, Zhang Z, Zhang R, et al. KPNA2 Promotes Metabolic Reprogramming in Glioblastomas by Regulation of C-Myc. J Exp Clin Cancer Res (2018) 37:194. doi: 10.1186/s13046-018-0861-9
79. Wang J, Xu X, Mo S, Tian Y, Wu J, Zhang J, et al. Involvement of microRNA-1297, a New Regulator of HMGA1, in the Regulation of Glioma Cell Growth In Vivo and In Vitro. Am J Transl Res (2016) 8:2149–58. Retrieved from http://https://e-century.us/web/journal.php?journal=ajtr
80. Liu L, Cui S, Wan T, Li X, Tian W, Zhang R, et al. Long non-Coding RNA HOTAIR Acts as a Competing Endogenous RNA to Promote Glioma Progression by Sponging miR-126-5p. J Cell Physiol (2018) 233:6822–31. doi: 10.1002/jcp.26432
81. Shi J, Zhang Y, Qin B, Wang Y, Zhu X. Long non-Coding RNA LINC00174 Promotes Glycolysis and Tumor Progression by Regulating miR-152-3p/SLC2A1 Axis in Glioma. J Exp Clin Cancer Res (2019) 38:395. doi: 10.1186/s13046-019-1390-x
82. He Z, You C, Zhao D. Long non-Coding RNA UCA1/miR-182/PFKFB2 Axis Modulates Glioblastoma-Associated Stromal Cells-Mediated Glycolysis and Invasion of Glioma Cells. Biochem Biophys Res Commun (2018) 500:569–76. doi: 10.1016/j.bbrc.2018.04.091
83. Zhang Y, Mao Q, Xia Q, Cheng J, Huang Z, Li Y, et al. Noncoding RNAs Link Metabolic Reprogramming to Immune Microenvironment in Cancers. J Hematol OncolJ Hematol Oncol (2021) 14:169. doi: 10.1186/s13045-021-01179-y
84. Xie Y, Hu J. Suppression of Long non-Coding RNA PCAT19 Inhibits Glioma Cell Proliferation and Invasion, and Increases Cell Apoptosis Through Regulation of MELK Targeted by miR-142-5p. Genes Genomics (2020) 42:1299–310. doi: 10.1007/s13258-020-01003-w
85. Lin X, Jiang T, Bai J, Li J, Wang T, Xiao J, et al. Characterization of Transcriptome Transition Associates Long Noncoding RNAs With Glioma Progression. Mol Ther - Nucleic Acids (2018) 13:620–32. doi: 10.1016/j.omtn.2018.10.009
86. Agnihotri S, Zadeh G. Metabolic Reprogramming in Glioblastoma: The Influence of Cancer Metabolism on Epigenetics and Unanswered Questions. Neuro-Oncol (2016) 18:160–72. doi: 10.1093/neuonc/nov125
87. Bhowmick R, Subramanian A, Sarkar RR. Exploring the Differences in Metabolic Behavior of Astrocyte and Glioblastoma: A Flux Balance Analysis Approach. Syst Synth Biol (2015) 9:159–77. doi: 10.1007/s11693-015-9183-9
88. Zeng A, Wei Z, Rabinovsky R, Jun HJ, El Fatimy R, Deforzh E, et al. Glioblastoma-Derived Extracellular Vesicles Facilitate Transformation of Astrocytes via Reprogramming Oncogenic Metabolism. iScience (2020) 23:101420. doi: 10.1016/j.isci.2020.101420
89. Guntuku L, Naidu GM, Yerra VG. Mitochondrial Dysfunction in Gliomas: Pharmacotherapeutic Potential of Natural Compounds. Curr Neuropharmacol (2016) 14:567–83. doi: 10.2174/1570159X14666160121115641
90. Zhang Y, Yu G, Chu H, Wang X, Xiong L, Cai G, et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol Cell (2018) 71:201–15.e7. doi: 10.1016/j.molcel.2018.06.023
91. Shibao S, Minami N, Koike N, Fukui N, Yoshida K, Saya H, et al. Metabolic Heterogeneity and Plasticity of Glioma Stem Cells in a Mouse Glioblastoma Model. Neuro-Oncol (2018) 20:343–54. doi: 10.1093/neuonc/nox170
92. Strickland M, Stoll EA. Metabolic Reprogramming in Glioma. Front Cell Dev Biol (2017) 5:43. doi: 10.3389/fcell.2017.00043
93. Duan K, Liu Z-J, Hu S-Q, Huo H-Y, Xu Z-R, Ruan J-F, et al. Lactic Acid Induces Lactate Transport and Glycolysis/OXPHOS Interconversion in Glioblastoma. Biochem Biophys Res Commun (2018) 503:888–94. doi: 10.1016/j.bbrc.2018.06.092
94. Toyonaga T, Yamaguchi S, Hirata K, Kobayashi K, Manabe O, Watanabe S, et al. Hypoxic Glucose Metabolism in Glioblastoma as a Potential Prognostic Factor. Eur J Nucl Med Mol Imaging (2017) 44:611–9. doi: 10.1007/s00259-016-3541-z
95. Caruso JP, Koch BJ, Benson PD, Varughese E, Monterey MD, Lee AE, et al. Ph, Lactate, and Hypoxia: Reciprocity in Regulating High-Affinity Monocarboxylate Transporter Expression in Glioblastoma. Neoplasia (2017) 19:121–34. doi: 10.1016/j.neo.2016.12.011
96. Kathagen-Buhmann A, Schulte A, Weller J, Holz M, Herold-Mende C, Glass R, et al. Glycolysis and the Pentose Phosphate Pathway are Differentially Associated With the Dichotomous Regulation of Glioblastoma Cell Migration Versus Proliferation. Neuro-Oncol (2016) 18:1219–29. doi: 10.1093/neuonc/now024
97. El Hassouni B, Granchi C, Vallés-Martí A, Supadmanaba IGP, Bononi G, Tuccinardi T, et al. The Dichotomous Role of the Glycolytic Metabolism Pathway in Cancer Metastasis: Interplay With the Complex Tumor Microenvironment and Novel Therapeutic Strategies. Semin Cancer Biol (2020) 60:238–48. doi: 10.1016/j.semcancer.2019.08.025
98. Hao S. NADPH Promotes the Rapid Growth of the Tumor. Infect Int (2018) 6:57–60. doi: 10.1515/ii-2017-0164
99. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
100. Koundouros N, Poulogiannis G. Reprogramming of Fatty Acid Metabolism in Cancer. Br J Cancer (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
101. Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CRH, Shimizu T, et al. Update of the LIPID MAPS Comprehensive Classification System for Lipids. J Lipid Res (2009) 50:S9–S14. doi: 10.1194/jlr.R800095-JLR200
102. Brante G. Studies on Lipids in the Nervous System With Special Reference to Quantitative Chemical Determination and Topical Distribution. J Am Med Assoc (1950) 144:507–8. doi: 10.1001/jama.1950.02920060069044
103. Gopal K, Grossi E, Paoletti P, Usardi M. LIPID COMPOSITION OF HUMAN INTRACRANIAL TUMORS: A BIOCHEMICAL STUDY. Acta Neurochir (Wien) (1963) 11:333–47. doi: 10.1007/BF01402012
104. Taïb B, Aboussalah AM, Moniruzzaman M, Chen S, Haughey NJ, Kim SF, et al. Lipid Accumulation and Oxidation in Glioblastoma Multiforme. Sci Rep (2019) 9:19593. doi: 10.1038/s41598-019-55985-z
105. Ha SJ, Showalter G, Cai S, Wang H, Liu WM, Cohen-Gadol AA, et al. Lipidomic Analysis of Glioblastoma Multiforme Using Mass Spectrometry. Anal Chem (2007) 79:8423–30. doi: 10.1021/ac071413m
106. Ledwozyw A, Lutnicki K. Phospholipids and Fatty Acids in Human Brain Tumors. Acta Physiol Hung (1992) 79:381–7.
107. Lou HOC, Clausen J, Bierring F. Phospholipids and Glycolipids of Tumours in the Central Nervous System. J Neurochem (1965) 12:619–27. doi: 10.1111/j.1471-4159.1965.tb04255.x
108. Walther TC, Farese RV. Lipid Droplets And Cellular Lipid Metabolism. Annu Rev Biochem (2012) 81:687–714. doi: 10.1146/annurev-biochem-061009-102430
109. Petan T, Jarc E, Jusović M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Mol J Synth Chem Nat Prod Chem (2018) 23(8):1941. doi: 10.3390/molecules23081941
110. Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22:5337–48. doi: 10.1158/1078-0432.CCR-15-2973
111. Cruz ALS, Barreto E de A, Fazolini NPB, Viola JPB, Bozza PT. Lipid Droplets: Platforms With Multiple Functions in Cancer Hallmarks. Cell Death Dis (2020) 11:1–16. doi: 10.1038/s41419-020-2297-3
112. Kambach DM, Halim AS, Cauer AG, Sun Q, Tristan CA, Celiku O, et al. Disabled Cell Density Sensing Leads to Dysregulated Cholesterol Synthesis in Glioblastoma. Oncotarget (2017) 8:14860–75. doi: 10.18632/oncotarget.14740
113. Bouyakdan K, Taïb B, Budry L, Zhao S, Rodaros D, Neess D, et al. A Novel Role for Central ACBP/DBI as a Regulator of Long-Chain Fatty Acid Metabolism in Astrocytes. J Neurochem (2015) 133:253–65. doi: 10.1111/jnc.13035
114. Duman C, Yaqubi K, Hoffmann A, Acikgöz AA, Korshunov A, Bendszus M, et al. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab (2019) 30:274–89.e5. doi: 10.1016/j.cmet.2019.04.004
115. Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, et al. Fatty Acid Oxidation is Required for the Respiration and Proliferation of Malignant Glioma Cells. Neuro-Oncol (2017) 19:43–54. doi: 10.1093/neuonc/now128
116. Zhou L, Wang Z, Hu C, Zhang C, Kovatcheva-Datchary P, Yu D, et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma With IDH1 Mutation. J Proteome Res (2019) 18:960–9. doi: 10.1021/acs.jproteome.8b00663
117. Huang R, Dong R, Wang N, He Y, Zhu P, Wang C, et al. Adaptive Changes Allow Targeting of Ferroptosis for Glioma Treatment. Cell Mol Neurobiol (2021) 2021:1–20. doi: 10.1007/s10571-021-01092-5
118. Cheng X, Geng F, Guo D. DGAT1 Protects Tumor From Lipotoxicity, Emerging as a Promising Metabolic Target for Cancer Therapy. Mol Cell Oncol (2020) 7:1805257. doi: 10.1080/23723556.2020.1805257
119. Cheng X, Geng F, Pan M, Wu X, Zhong Y, Wang C, et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab (2020) 32:229–242.e8. doi: 10.1016/j.cmet.2020.06.002
120. Zhao H, Ji B, Chen J, Huang Q, Lu X. Gpx 4 is Involved in the Proliferation, Migration and Apoptosis of Glioma Cells. Pathol - Res Pract (2017) 213:626–33. doi: 10.1016/j.prp.2017.04.025
121. Yi R, Wang H, Deng C, Wang X, Yao L, Niu W, et al. Dihydroartemisinin Initiates Ferroptosis in Glioblastoma Through GPX4 Inhibition. Biosci Rep (2020) 40(6). doi: 10.1042/BSR20193314
122. Bi J, Khan A, Tang J, Armando AM, Wu S, Zhang W, et al. Targeting Glioblastoma Signaling and Metabolism With a Re-Purposed Brain-Penetrant Drug. Cell Rep (2021) 37(5):109957. doi: 10.1016/j.celrep.2021.109957
123. Martins F, Gonçalves LG, Pojo M, Serpa J. Take Advantage of Glutamine Anaplerosis, the Kernel of the Metabolic Rewiring in Malignant Gliomas. Biomolecules (2020) 10:1370. doi: 10.3390/biom10101370
124. Shi R, Pan P, Lv R, Ma C, Wu E, Guo R, et al. High-Throughput Glycolytic Inhibitor Discovery Targeting Glioblastoma by Graphite Dots–Assisted LDI Mass Spectrometry. Sci Adv (2022) 8:eabl4923. doi: 10.1126/sciadv.abl4923
125. Dwarakanath BS, Singh D, Banerji AK, Sarin R, Venkataramana NK, Jalali R, et al. Clinical Studies for Improving Radiotherapy With 2-Deoxy-D-Glucose: Present Status and Future Prospects. J Cancer Res Ther (2009) Suppl 1:S21–6. doi: 10.4103/0973-1482.55136
126. Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, et al. Mutant IDH1 Regulates the Tumor-Associated Immune System in Gliomas. Genes Dev (2017) 31:774–86. doi: 10.1101/gad.294991.116
127. Urban DJ, Martinez NJ, Davis MI, Brimacombe KR, Cheff DM, Lee TD, et al. Assessing Inhibitors of Mutant Isocitrate Dehydrogenase Using a Suite of Pre-Clinical Discovery Assays. Sci Rep (2017) 7:12758. doi: 10.1038/s41598-017-12630-x
128. Dang L, Yen K, Attar EC. IDH Mutations in Cancer and Progress Toward Development of Targeted Therapeutics. Ann Oncol Off J Eur Soc Med Oncol (2016) 27:599–608. doi: 10.1093/annonc/mdw013
129. Kaminska B, Czapski B, Guzik R, Król SK, Gielniewski B. Consequences of IDH1/2 Mutations in Gliomas and an Assessment of Inhibitors Targeting Mutated IDH Proteins. Molecules (2019) 24:968. doi: 10.3390/molecules24050968
130. Cruz Da Silva E, Mercier M-C, Etienne-Selloum N, Dontenwill M, Choulier L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers (2021) 13:1795. doi: 10.3390/cancers13081795
131. Sanchez WY, McGee SL, Connor T, Mottram B, Wilkinson A, Whitehead JP, et al. Dichloroacetate Inhibits Aerobic Glycolysis in Multiple Myeloma Cells and Increases Sensitivity to Bortezomib. Br J Cancer (2013) 108:1624–33. doi: 10.1038/bjc.2013.120
132. Pruss M, Dwucet A, Tanriover M, Hlavac M, Kast RE, Debatin K-M, et al. Dual Metabolic Reprogramming by ONC201/TIC10 and 2-Deoxyglucose Induces Energy Depletion and Synergistic Anti-Cancer Activity in Glioblastoma. Br J Cancer (2020) 122:1146–57. doi: 10.1038/s41416-020-0759-0
133. Shi Y, Lim SK, Liang Q, Iyer SV, Wang H-Y, Wang Z, et al. Gboxin is an Oxidative Phosphorylation Inhibitor That Targets Glioblastoma. Nature (2019) 567:341–6. doi: 10.1038/s41586-019-0993-x
134. Takada T, Takata K, Ashihara E. Inhibition of Monocarboxylate Transporter 1 Suppresses the Proliferation of Glioblastoma Stem Cells. J Physiol Sci (2016) 66:387–96. doi: 10.1007/s12576-016-0435-6
135. Bi J, Ichu T-A, Zanca C, Yang H, Zhang W, Gu Y, et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers Dependent on Membrane Lipid Remodeling. Cell Metab (2019) 30:525–38.e8. doi: 10.1016/j.cmet.2019.06.014
136. Engel AL, Lorenz NI, Klann K, Münch C, Depner C, Steinbach JP, et al. Serine-Dependent Redox Homeostasis Regulates Glioblastoma Cell Survival. Br J Cancer (2020) 122:1391–8. doi: 10.1038/s41416-020-0794-x
137. Liu J, Fu W, Zheng X, Li W, Ren L, Wang J, et al. Avasimibe Exerts Anticancer Effects on Human Glioblastoma Cells via Inducing Cell Apoptosis and Cell Cycle Arrest. Acta Pharmacol Sin (2021) 42:97–107. doi: 10.1038/s41401-020-0404-8
138. Chiasserini D, Davidescu M, Orvietani PL, Susta F, Macchioni L, Petricciuolo M, et al. 3-Bromopyruvate Treatment Induces Alterations of Metabolic and Stress-Related Pathways in Glioblastoma Cells. J Proteomics (2017) 152:329–38. doi: 10.1016/j.jprot.2016.11.013
139. Agnihotri S, Mansouri S, Burrell K, Li M, Mamatjan Y, Liu J, et al. Ketoconazole and Posaconazole Selectively Target HK2-Expressing Glioblastoma Cells. Clin Cancer Res (2019) 25:844–55. doi: 10.1158/1078-0432.CCR-18-1854
140. Liu H, Liu N, Cheng Y, Jin W, Zhang P, Wang X, et al. Hexokinase 2 (HK2), the Tumor Promoter in Glioma, is Downregulated by miR-218/Bmi1 Pathway. PLoS One (2017) 12(12):e0189353. doi: 10.1371/journal.pone.0189353
141. Villa GR, Hulce JJ, Zanca C, Bi J, Ikegami S, Cahill GL, et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell (2016) 30(5):683–93. doi: 10.1016/j.ccell.2016.09.008
142. Facchini G, Ignarro RS, Rodrigues-Silva E, Vieira AS, Lopes-Cendes I, Castilho RF, et al. Toxic Effects of Phytol and Retinol on Human Glioblastoma Cells are Associated With Modulation of Cholesterol and Fatty Acid Biosynthetic Pathways. J Neurooncol (2018) 136:435–43. doi: 10.1007/s11060-017-2672-9
143. An Y, Zhang D-D, Yu H-L, Ma W-W, Lu Y-H, Liu Q-R, et al. 27-Hydroxycholesterol Regulates Cholesterol Synthesis and Transport in C6 Glioma Cells. NeuroToxicology (2017) 59:88–97. doi: 10.1016/j.neuro.2017.02.001
144. Lan F, Qin Q, Yu H, Yue X. Effect of Glycolysis Inhibition by miR-448 on Glioma Radiosensitivity. J Neurosurg (2019) 132:1456–64. doi: 10.3171/2018.12.JNS181798
145. Sanzey M, Abdul Rahim SA, Oudin A, Dirkse A, Kaoma T, Vallar L, et al. Comprehensive Analysis of Glycolytic Enzymes as Therapeutic Targets in the Treatment of Glioblastoma. PLoS One (2015) 10(5):e0123544. doi: 10.1371/journal.pone.0123544
146. Kim EH, Lee J-H, Oh Y, Koh I, Shim J-K, Park J, et al. Inhibition of Glioblastoma Tumorspheres by Combined Treatment With 2-Deoxyglucose and Metformin. Neuro-Oncol (2017) 19:197–207. doi: 10.1093/neuonc/now174
147. Shah SS, Rodriguez GA, Musick A, Walters WM, de Cordoba N, Barbarite E, et al. Targeting Glioblastoma Stem Cells With 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR). Cancers (2019) 11(2):159. doi: 10.3390/cancers11020159
148. Polson ES, Kuchler VB, Abbosh C, Ross EM, Mathew RK, Beard HA, et al. KHS101 Disrupts Energy Metabolism in Human Glioblastoma Cells and Reduces Tumor Growth in Mice. Sci Transl Med (2018) 10(454):eaar2718. doi: 10.1126/scitranslmed.aar2718
149. Li M, Kirtane AR, Kiyokawa J, Nagashima H, Lopes A, Tirmizi ZA, et al. Local Targeting of NAD+ Salvage Pathway Alters the Immune Tumor Microenvironment and Enhances Checkpoint Immunotherapy in Glioblastoma. Cancer Res (2020) 80:5024–34. doi: 10.1158/0008-5472.CAN-20-1094
150. Guo Q, Han N, Shi L, Yang L, Zhang X, Zhou Y, et al. NAMPT: A Potential Prognostic and Therapeutic Biomarker in Patients With Glioblastoma. Oncol Rep (2019) 42:963–72. doi: 10.3892/or.2019.7227
151. Souza F da C, Ferreira MT, Colquhoun A. Influence of Lipoxygenase Inhibition on Glioblastoma Cell Biology. Int J Mol Sci (2020) 21:8395. doi: 10.3390/ijms21218395
152. Garofano L, Migliozzi S, Oh YT, D’Angelo F, Najac RD, Ko A, et al. Pathway-Based Classification of Glioblastoma Uncovers a Mitochondrial Subtype With Therapeutic Vulnerabilities. Nat Cancer (2021) 2:141–56. doi: 10.1038/s43018-020-00159-4
153. Calvert AE, Chalastanis A, Wu Y, Hurley LA, Kouri FM, Bi Y, et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep (2017) 19:1858–73. doi: 10.1016/j.celrep.2017.05.014
154. Jang M, Kim SS, Lee J. Cancer Cell Metabolism: Implications for Therapeutic Targets. Exp Mol Med (2013) 45:e45–5. doi: 10.1038/emm.2013.85
155. Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of Genome Editing Technology in the Targeted Therapy of Human Diseases: Mechanisms, Advances and Prospects. Signal Transduct Target Ther (2020) 5:1–23. doi: 10.1038/s41392-019-0089-y
156. Dastmalchi F, Deleyrolle LP, Karachi A, Mitchell DA, Rahman M. Metabolomics Monitoring of Treatment Response to Brain Tumor Immunotherapy. Front Oncol (2021) 11:691246. doi: 10.3389/fonc.2021.691246
157. Qiu R, Zhong Y, Li Q, Li Y, Fan H. Metabolic Remodeling in Glioma Immune Microenvironment: Intercellular Interactions Distinct From Peripheral Tumors. Front Cell Dev Biol (2021) 9:693215. doi: 10.3389/fcell.2021.693215
158. Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the Immune System in Glioblastoma. Br J Cancer (2018) 119:1171–81. doi: 10.1038/s41416-018-0258-8
159. Guerra L, Bonetti L, Brenner D. Metabolic Modulation of Immunity: A New Concept in Cancer Immunotherapy. Cell Rep (2020) 32:107848. doi: 10.1016/j.celrep.2020.107848
160. Pelaz SG, Jaraíz-Rodríguez M, Álvarez-Vázquez A, Talaverón R, García-Vicente L, Flores-Hernández R, et al. Targeting Metabolic Plasticity in Glioma Stem Cells In Vitro and In Vivo Through Specific Inhibition of C-Src by TAT-Cx43266-283. eBioMedicine (2020) 62:103134. doi: 10.1016/j.ebiom.2020.103134
161. Xu C, Zhao J, Song J, Xiao M, Cui X, Xin L, et al. lncRNA PRADX is a Mesenchymal Glioblastoma Biomarker for Cellular Metabolism Targeted Therapy. Front Oncol (2022) 12:888922:888922. doi: 10.3389/fonc.2022.888922
162. Cheng Z, Luo C, Guo Z. LncRNA-XIST/microRNA-126 Sponge Mediates Cell Proliferation and Glucose Metabolism Through the IRS1/PI3K/Akt Pathway in Glioma. J Cell Biochem (2020) 121:2170–83. doi: 10.1002/jcb.29440
163. Dong Z, Cui H. Epigenetic Modulation of Metabolism in Glioblastoma. Semin Cancer Biol (2019) 57:45–51. doi: 10.1016/j.semcancer.2018.09.002
Keywords: GBM, genetic alteration, metabolic genes, glycolysis, glioma therapy
Citation: El Khayari A, Bouchmaa N, Taib B, Wei Z, Zeng A and El Fatimy R (2022) Metabolic Rewiring in Glioblastoma Cancer: EGFR, IDH and Beyond. Front. Oncol. 12:901951. doi: 10.3389/fonc.2022.901951
Received: 22 March 2022; Accepted: 21 June 2022;
Published: 14 July 2022.
Edited by:
Alison Colquhoun, University of São Paulo, BrazilReviewed by:
Shuhua Zheng, Northwestern University, United StatesCopyright © 2022 El Khayari, Bouchmaa, Taib, Wei, Zeng and El Fatimy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachid El Fatimy, cmFjaGlkLmVsZmF0aW15QHVtNnAubWE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.