 Letizia Mezzasoma
Letizia Mezzasoma Ilaria Bellezza
Ilaria Bellezza Rita Romani
Rita Romani Vincenzo Nicola Talesa
Vincenzo Nicola Talesa- Department of Medicine and Surgery, University of Perugia, Perugia, Italy
Extracellular vesicles (EVs) are membrane enclosed spherical particles devoted to intercellular communication. Cancer-derived EVs (Ca-EVs) are deeply involved in tumor microenvironment remodeling, modifying the inflammatory phenotype of cancerous and non-cancerous residing cells. Inflammation plays a pivotal role in initiation, development, and progression of many types of malignancies. The key feature of cancer-related inflammation is the production of cytokines that incessantly modify of the surrounding environment. Interleukin-1β (IL-1β) is one of the most powerful cytokines, influencing all the initiation-to-progression stages of many types of cancers and represents an emerging critical contributor to chemoresistance. IL-1β production strictly depends on the activation of inflammasome, a cytoplasmic molecular platform sensing exogenous and endogenous danger signals. It has been recently shown that Ca-EVs can activate the inflammasome cascade and IL-1β production in tumor microenvironment-residing cells. Since inflammasome dysregulation has been established as crucial regulator in inflammation-associated tumorigenesis and chemoresistance, it is conceivable that the use of inflammasome-inhibiting drugs may be employed as adjuvant chemotherapy to counteract chemoresistance. This review focuses on the role of cancer-derived EVs in tuning tumor microenvironment unveiling the intricate network between inflammasome and chemoresistance.
Introduction
In the complexity of cancer progression, the intricate interplay between inflammation and tumor microenvironment (TME), depicts an extraordinary multifaceted scenario in the development of acquired drug resistance and in the clinical outcome of malignant processes (1, 2). Extracellular vesicles (EVs), in particular cancer-derived (Ca-EVs), represent signal transducer or messengers in cell-cell communication (3–5), responsible for the continuous modification of TME (6, 7). TME includes cancerous and non-cancerous cellular components such as fibroblasts, stromal, immune, and endothelial cells. The cross-talk between TME components can induce a dysregulated inflammatory and immune response (1, 2). Inflammation, indeed, plays a pivotal role in tumor initiation, by dynamically and incessantly modifying TME via the release of cytokines and soluble mediators generating a “vicious cycle”. This, in turn, endorses oncogenic plasticity toward immune-suppression, more aggressive phenotype and reduction of therapeutic efficacy. One of the main mechanisms contributing to inflammation is mediated by cytoplasmatic complexes known as inflammasomes. Inflammasomes are activated by endogenous/exogenous danger signals and changes in cytoplasm homeostasis. Upon activation, inflammasomes act as “signal integrators” by the release of inflammasome-effectors cytokines. Inflammasomes are pivotal hubs of innate immunity and modulate immune/inflammatory responses by cross-talking with different cellular components. Inflammasome inappropriate activation, creating a pro-inflammatory TME and suppressing local immunity, appears as an emerging player in all the initiation to progression stages of cancer (8–11). Crucial novel modulators of inflammasome are EVs that, on the basis of the different nature of their cellular source, positively or negatively affect inflammasome cascade in diverse cancerous and non-cancerous recipient cells (12–14). In this scenario, EVs, with their Janus face behavior, strongly contribute to the immune/inflammation-associated modification of TME, and play a critical role in tumorigenesis and chemoresistance.
EVs: Another Brick in the Wall
EVs are a heterogeneous group of membrane enwrapped spherical particles, produced by nearly all types of cells. There are no unique markers able to classify EVs on the bases on their biogenesis (ectosomes, exosomes, apoptotic bodies), for this reason the MISEV2018 guidelines suggest classifying EVs based on physical parameters, such as size (small and medium/large EVs) density or biochemical composition (15–17). EVs, found in body fluids and in cell culture media, carry various biomolecules, including proteins, lipids, metabolites, RNA, and DNA (16, 18). Upon interaction with target cell, EVs deeply impact cellular recipient cells responses, highlighting the pivotal role of EVs as signal transducers or messengers in cell-cell communication at close or distant sites. Intercellular communication is a key feature of tumor progression and metastasis. Cancer cells can release EVs that enter the circulation and reach distant organs, where they can generate favorable environmental conditions, enabling the outgrowth of disseminated tumor cells. This process, known as pre-metastatic niche formation, requires a series of predefined steps involving induction of vascular leakiness, alteration of stromal components and immune-escape (19, 20).
Cancer-derived EVs (Ca-EVs) ability to suppress immune anti-tumor activity, is guaranteed by the exchange of EVs between cancerous and non-cancerous TME-residing cells, and by the secretion of immune-modulating molecules (14, 21). Furthermore, the “exosome-immune suppression” and the Ca-EVs-mediated transfer of oncogenes or oncometabolites from one cell to other is also involved in the unrestrained cell proliferation and, subsequently, in the metastatic spread (14). On the other hand, Ca-EVs, may also carry tumor-associated antigens, damage associated molecular pattern (DAMPs), and immune-stimulating molecules, that can induce an immune anti-tumor response (22, 23) via the recruitment and activation of immune cells in TME (24, 25). Although the pro-inflammatory and the immune-suppressive role of Ca-EVs seem to be contrasting, pro-inflammatory EVs may still contribute to TME maintenance (26, 27).
Interleukin-1β and Chemoresistance: An Old Cytokine With a Novel Role
Interleukin-1β (IL-1β) is one of the most abundant and influential cytokines of TME. IL-1β expression and secretion are induced by different stimuli such as toll-like receptors (TLRs) ligands, tumor necrosis factor-α or IL-1β itself. IL-1β production/secretion are fine-tune controlled by a two-steps transcriptional and post-translational regulation, requiring the activation of both nuclear factor kappa B (NF-κB) and nucleotide-binding oligomerization domain (NOD)-like receptor pyrin domain-containing 3 (NLRP3) inflammasome-caspase-1 platform. NF-κB activation by inflammatory stimuli induces biologically inactive pro-IL-1β production which must be proteolytically cleaved, by inflammasome-activated caspase-1 (28). Tumor cells can directly produce IL-1β or can “instruct” cells within TME, such as stromal ones, to secrete it (26, 29). An uncontrolled increase in IL-1β release exerts immune-suppressive effects and influences all the initiation-to-progression stages of many types of cancers and represents an emerging critical contributor to chemoresistance (30, 31). Depending on tumor cell types, several “in vitro” and “in vivo” models highlighted multiple mechanisms for IL-1β promoted chemoresistance. In pleural mesothelioma, the IL-8/IL-1β signaling controls chemoresistance by inducing the overexpression of ATP-binding cassette transporter (ABC) G2, that determines resistance to cisplatin and pemetrexed (32). Prostate carcinoma cells engage bone marrow adipocytes in a functional cyclooxygenase-2 (COX-2)-dependent cross-talk that promotes IL-1β expression, leading to docetaxel resistance (33). IL-1β can also induce a reinforcement of NF-κB signaling. In fact, IL-1β induces a sustained NF-κB that has been related to chemoresistance in ovarian carcinoma (34), in acute myeloid leukaemia (35) and in renal cell carcinoma (36). In pancreatic cancer, IL-1β confers chemoresistance not only by activating NF-κB (37), but also by up-regulating COX-2 (38), an enzyme linked to chemoresistance also in cervical carcinoma (39) and in colon cancer cell lines (40). In bladder cancer, cisplatin-resistance has been linked to IL-1β-induced increase in aldo-keto reductase 1C1 levels (41). In breast cancer, IL-1β-induced chemoresistance has been attributed to several mechanisms including: methylation of the estrogen receptor α, which increases tamoxifen resistance (42); activation of β-catenin signaling, which increases cisplatin resistance (43); and induction of epithelial to mesenchymal transition (EMT), which increases doxorubicin resistance (44). In melanoma cells, ABCB5 controls IL-1β/IL-8 signaling (45) which, in turn, influences chemoresistance by activating Smad/DNA binding protein 1 signaling (46).
Considering the implication of IL-1β in influencing all the initiation-to-progression stages of many tumors and chemoresistance, this cytokine is considered a promising therapeutic target for many types of cancers (30).
The Inflammasome: A Double-Edge Sword
As already mentioned, inflammasome activation is the mandatory event for IL-1β maturation and secretion. Inflammasomes are cytoplasmic molecular platforms devoted to detecting pathogen associated molecular patterns (PAMPs) and DAMPs, playing a key role in innate immunity (47). The inflammasome platform is composed by a danger sensor receptor, an adaptor protein (Apoptosis-associated speck-like protein containing a CARD, ASC), and an effector enzyme (caspase-1). The receptor family includes the nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) family, composed of at least 22 members, the most characterized of which is NLRP3 (47). Upon activation, NLRP3 oligomerizes and assembles into a multimeric platform including a core unit comprehending ASC and the effector pro-caspase-1. The oligomerization of inflammasome components culminates in the autocatalytic activation of caspase-1, responsible for IL-1β and IL-18 maturation (47–49). Inflammasome activation may also induce the processing of gasdermin-D (GSDMD), leading to pyroptosis, an inflammatory form of cell death (50). The physical interaction among inflammasome components is mediated by the adaptor protein ASC which holds a pyrin (PYD) and a CARD domain which, assembling into a speck, consents the connection between NLRP3 and caspase-1. NLRP3 possesses three domains: an N-terminal effector PYD, involved in ASC recruitment via PYD-PYD interaction, a central NACHT domain carrying an ATPase activity essential for NLRP3 activation and platform assembly, and a C-terminal leucine-rich repeats domain, possibly involved in auto-regulation, protein-protein interaction, and signal sensing (51). Inflammasome-platform assembly is also regulated by the phosphorylation of Ser-295 of NLRP3. This post-translational modification, accomplished by several protein kinases (PKs) including PKA, PKD and PKG (52, 53), impedes inflammasome platform assembly. Because component assembly is mandatory for inflammasome activation, it represents an attractive target for the development of selective NLRP3 inhibitors, as discussed later. NLRP3 involvement in cancer is currently a very debated topic. NLRP3 and NLRP3-associated pyroptosis have been defined “a double-edge sword” (54) on the basis of their capability to achieve both an anti-tumorigenic and a pro-tumorigenic activity in different types of malignancies (9–11, 54, 55). The contrasting roles of NLRP3 inflammasome can be due to multiple factors such as type, heterogeneity and stage of cancer cells, or TME characteristics (8–11). Metabolites, cytokines and EVs released by TME residing cells, represent the key drivers of NLRP3 hyper-activation. NLRP3 dysregulated activation can induce a chronic inflammatory environment that boosts tumor progression and extinguishes local immunity (9).
Recently the uncontrolled inflammasome activation has also been associated to chemoresistance. In oral squamous carcinoma NLRP3 activation promotes 5-Fluorouracil resistance “in vitro” and “in vivo” (56); and NLRP3 inflammasome has been detected in cisplatin-resistant lung cancer cell lines (57). Conversely, NLRP3-induced pyroptosis, sensitizes gastric and epatocellular carcinoma to cisplatin (58, 59).
NLRP3 inflammasome has been also linked to cardiotoxicity of anticancer agents. The inhibition of NLRP3, as well as of the oligomerization of the myeloid differentiation primary response gene 88 (MyD88), reduces the cardiotoxicity and increases the anticancer properties of sunitinib, in renal cancer-bearing mice (60). Myd88 is molecular platform which oligomerization and assembly induces NF-κB activation and the release of cytokine and factors involved in cancer cell survival and chemoresistance (60). Pharmacological reduction of NLRP3 activity has been suggested as a tool to alleviate doxorubicin-induced cardiotoxicity while preserving or even improving its anti-cancer activity (61). On the other hand, pyroptosis‐associated cytokines can induce either an evasion of immune surveillance, or an effective immune response (62).
EVs and Inflammasome: Two Pieces of the Same Puzzle
Increasing evidence highlights the pivotal role of Ca-EVs on NLRP3 activation in different types of cancers (summarized in Table 1). Prostate cancer derived-EVs, by inducing NLRP3 activation and IL-1β maturation, modify the inflammatory response of ME residing cells in a tumor-promoting fashion (12). Furthermore, prostate cancer tumor progression is characterized by increased inflammasome activation (62). Lung cancer-derived EVs induce NLRP3 activation in macrophages, thus providing a positive feedback loop to promote cancer progression via IL-1β secretion in mice (63, 64). EVs released by primary cultures of human glioblastoma, up-regulate microglial inflammasome signaling and influence both microglial cells polarization and glioma-microglia crosstalk (65). Furthermore, EVs derived from colon adenocarcinoma cells mediate radiation-induced antitumor immunity by inducing NLRP3 activation in mice (66).

Table 1 Roles of EVs and drugs in inflammasome modulation.
The role of EVs in immune-escape and immune-stimulation also relies on their ability to modulate inflammasome cascade positively or negatively and IL-1β production in recipient cells (12–14) (summarized in Table 1). This different effect is both related to the nature of the EVs-releasing and -receiving cells and to the different EVs mechanisms of action (Figure 1). In fact, on the one hand, Ca-EVs activate NLRP3 inflammasome platform in non-immune receiving cells via ERK1/2-mediated pathway (12), on the other hand non-cancerous cell-derived EVs negatively modulate NLRP3 inflammasome activation in immune cells (13). This latter effect is mediated by EVs intrinsic metabolic activity that, through adenosine production, induces the activation of the adenosine A2a receptor, a member of the purinergic P1 receptor family (13). This novel mechanism of action highlights the active role of EVs in microenvironment homeostasis, via the autonomous synthesis of metabolic products able to alter microenvironment composition and cell behavior (13). Furthermore, the involvement of A2a receptor in this EVs effect, offers a novel point of view on the roles of EVs/purinergic receptors on cancer immunology (12). In fact, up to date, only the connection between EVs/type P2 purinergic receptors (P2Rs) and tumor-inflammatory signaling has been demonstrated (27). Only few reports demonstrate that the activation of P2Rs on immune cells induces the release of: (i) EVs containing IL-1β and IL-18, exerting a pro-inflammatory action, favor tumor progression at the expense of an effective immune response; (ii) EVs presenting P2Rs on their surfaces which activation, by extracellular ATP, can lead to the release of IL-1β, IL-18 and ATP itself (27). As discussed below, inflammasome and IL-1β dysregulation are crucial players in inflammation-associated tumorigenesis and chemoresistance. In this scenario, EVs by exerting an interaction-dependent effect on the receiving cells or by releasing immune-metabolites, can be considered novel crucial players in determining tumorigenesis and chemoresistance. The functional link between NLRP3 activation and EVs is further demonstrated by the finding that embryonic stem cell-derived EVs ameliorate the cardio-toxicity induced by the antineoplastic agent doxurobcin, by inhibiting NLRP3 signaling in mice (67). Nonetheless, further research is needed to increase the knowledge in this emerging research area.
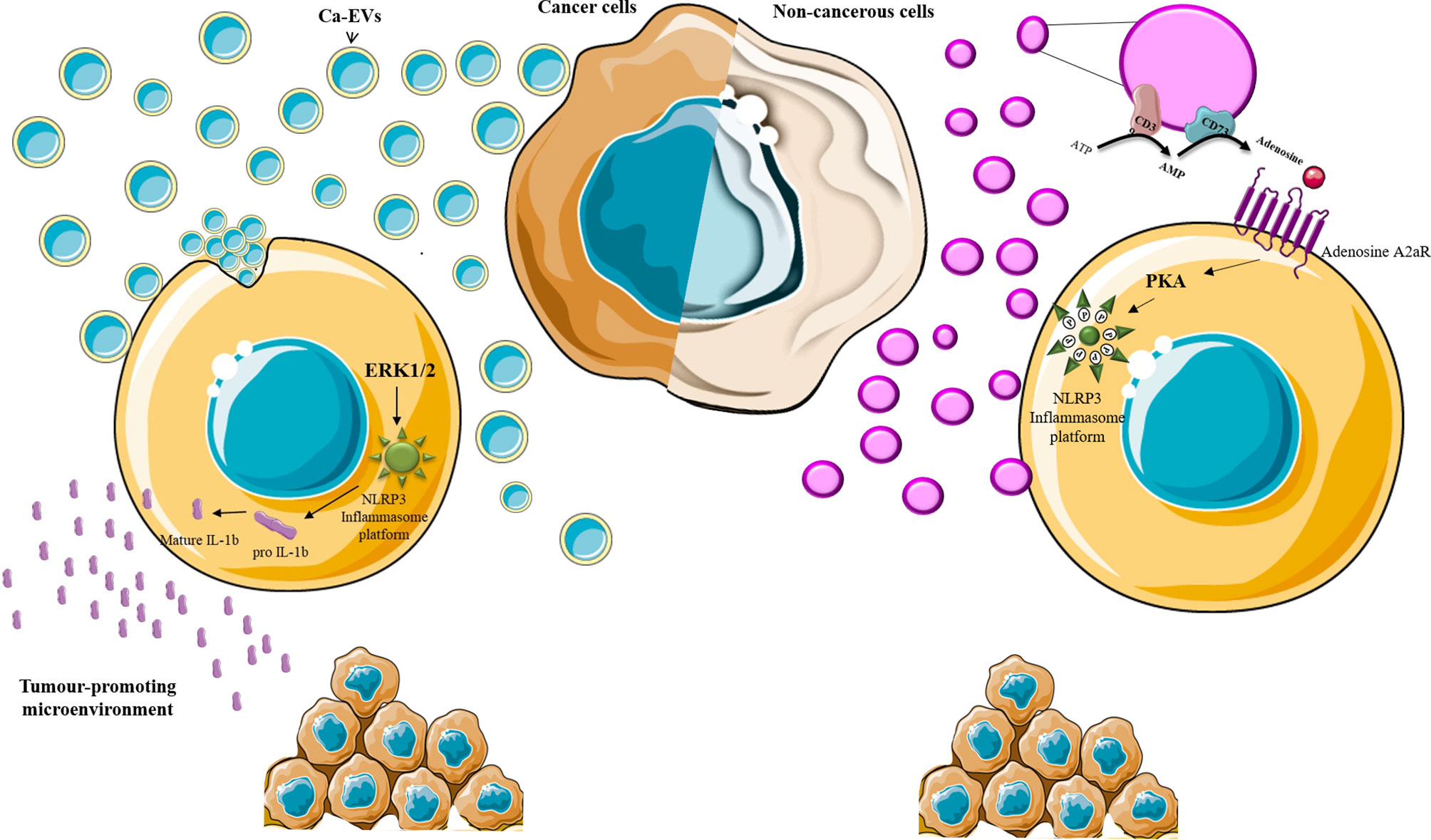
Figure 1 EVs and inflammasome. Schematic representation of EVs-induced effect on inflammasome activation. Ca-EVs (light blue) are up-taken by non-cancerous cells and, via the induction of intracellular signaling pathways, including ERK1/2 MAPK, induce inflammasome platform assembly and the maturation of IL-1β which release affects the microenvironment in a tumor-promoting fashion. EVs released by non-cancerous cells violet may produce soluble factor (Adenosine) that, via receptor (adenosine A2a receptor) engagement on the target cell, activates protein kinases (PKA) which impedes inflammasome platform assembly through NLRP3 phosphorylation (P).
Inflammasome Targeting Drugs: Potential Anti-Cancer Therapeutic
Inflammation sustained by inflammasome activation has been implicated in the insurgence or progression of several human pathologies, including cancer. For this reason, several efforts have been made to identify potential effective inhibitors of inflammasome to be used as new anti-cancer therapeutics (summarized in Table 1). Each step leading to inflammasome activation, may represent a good candidate for therapeutic targeting.
Several small molecules and natural compounds have been identified as inhibitors of the interaction between NLRP3 inflammasome monomers. As examples MCC950 and OLT1177, block NLRP3 oligomerization by inhibiting ATP hydrolysis via the NACHT domain, which is pivotal for receptor oligomerization and anti-cancer effects (68). MC950 inhibits LPS-induced inflammasome activation in pancreatic cancer cell lines (69), delays cell growth in a mouse model of head and neck squamous cell carcinoma (70) and inhibits pituitary prolactinoma growth and prolactin expression/secretion in rats (71). Similarly, inhibition of NLRP3 by OLT1177 enhances antitumor immunity, thus reducing melanoma growth (68). Oridonin, a natural terpenoids found in traditional Chinese herbal medicine, impedes inflammasome assembly by forming covalent bond with NLRP3 Cys279 (72). Oridonin administration effectively prevents the formation of colorectal cancer liver metastasis (73) and improves oxaliplatin efficacy (74). Oridonin derivative, with potent anticancer effects, has been very recently synthesized (75).
ASC polymerization can be another target for broad-spectrum therapeutics. MM01 (under patent procedure: application number, 20382237.4-1109) is a small-molecule interfering with ASC speck formation (76). Xantone, used in the early twentieth century as an ovicide and larvicide (85) can inhibit ASC speck formation without affecting inflammasome components expression (77). Although MM01 and xantone can be useful for the treatment of a broad range of diseases based on inflammasome dysregulation, they have not yet been tested on cancer models.
Caspase-1 activation can be targeted for impeding IL-1β maturation. Caspase-1 inhibition by the small-molecule VX-765 prevents pyroptosis in a multiple sclerosis model (78) and in monocytes and macrophages (79). Ritonavir, originally used as protease inhibitor for the treatment of HIV, effectively block caspase-1 (80), and induces apoptosis in pancreatic cancer (81). However, the quite unspecific actions of protease inhibitors should be taken into account to avoid deleterious side effects.
Specific monoclonal antibodies directed toward IL-1 receptor, including anakinra, rilonacept, canakinumab and gevokizumab have been developed to inhibit IL-1β signaling (82). Anakinra, a recombinant IL-1Ra, blocking the binding of IL-1 to IL-1 receptor, is under clinical investigation for the treatment of metastatic cancers (ClinicalTrials.gov Identifier: NCT00072111). An up-to date list of clinical trials involving IL-1 blockade has been recently published (86). Nevertheless, the blockade of IL-1 receptor, although displaying a favourable safety profile, caused a reduction in neutrophil counts with an overall increased risk for fatal infections.
Besides the possibility to inhibit inflammasome components, several strategies aimed to inhibit the pathways leading to inflammasome activation. Antioxidant compounds can inhibit ROS-mediated inflammasome platform assembly, P2X7 receptor antagonist can be used to impede K+ efflux known to be involved in NLRP3 activation (86). A strategy, explored by our group, is the induction of NLRP3 phosphorylation. We have indeed showed that, natriuretic peptides (NPs), by binding to NPs Receptor-1, can induce an increase in cGMP levels which culminates in the activation of PKG (53, 83, 84). Moreover, we showed that EVs, isolated from amniotic fluid-derived stem cells can activate PKA via A2a adenosine receptor in immune cells (13). Both PKA and PKG can phosphorylate NLRP3 at Ser295, thus leading to the inhibition of inflammasome assembly and IL-1β secretion (13, 53). Furthermore, NPs are able to counteract both the constitutive and EVs-induced NLRP3 activation in cancerous and non-cancerous prostate cells (62), supporting the critical role of these molecules in prostate cancer (87). Based on the fact that NPs analogues are already in clinical use for cardiovascular diseases (88, 89) and of the growing interest toward the use of EVs as therapeutics (90), further studies are needed to better define the potential anti-cancer efficacy of NPs and EVs.
Conclusion
Chemoresistance represents a major challenge in the clinic. Cancer cells response to therapy is deeply influenced by immune/inflammation-associated TME modifications. Therefore, the management of TME-mediated resistance may deeply affect the efficacy of cancer therapies. The key players that trigger TME modifications are multiple and strictly interconnected via a complex network of cell-cell communication. Given the pivotal role of inflammasome and related cytokines in TME re-modeling, they represent promising therapeutic targets for the development of novel anticancer approaches aimed to re-educate TME toward a favorable inflammatory/immune anti-tumorigenic phenotype. EVs have been recently discovered as novel active contributors of inflammasome/IL-1β modulation. Further studies are needed to better define the potential anti-cancer therapeutic efficacy of inflammasome-modulating drugs as adjuvant chemotherapy to counteract chemoresistance in a multidrug approach.
Author Contributions
LM, IB, and RR contributed to conception of the study and wrote the manuscript. VT supervised the study. All authors listed contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79:4557–66. doi: 10.1158/0008-5472.CAN-18-3962
3. Han L, Lam EW-F, Sun Y. Extracellular Vesicles in the Tumor Microenvironment: Old Stories, But New Tales. Mol Cancer (2019) 18:59. doi: 10.1186/s12943-019-0980-8
4. Lopatina T, Favaro E, Danilova L, Fertig EJ, Favorov AV, Kagohara LT, et al. Extracellular Vesicles Released by Tumor Endothelial Cells Spread Immunosuppressive and Transforming Signals Through Various Recipient Cells. Front Cell Dev Biol (2020) 8:698. doi: 10.3389/fcell.2020.00698
5. Lopatina T, Grange C, Cavallari C, Navarro-Tableros V, Lombardo G, Rosso A, et al. Targeting IL-3Rα on Tumor-Derived Endothelial Cells Blunts Metastatic Spread of Triple-Negative Breast Cancer via Extracellular Vesicle Reprogramming. Oncogenesis (2020) 10:90. doi: 10.1038/s41389-020-00274-y
6. Lombardo G, Gili M, Grange C, Cavallari C, Dentelli P, Togliatto G, et al. IL-3R-Alpha Blockade Inhibits Tumor Endothelial Cell-Derived Extracellular Vesicle (EV)-Mediated Vessel Formation by Targeting the β-Catenin Pathway. Oncogene (2018) 9:1175–91. doi: 10.1038/s41388-017-0034-x
7. Bebelman MP, Smit MJ, Pegtel DM, Baglio SR. Biogenesis and Function of Extracellular Vesicles in Cancer. Pharmacol Ther (2018) 188:1–11. doi: 10.1016/j.pharmthera.2018.02.013
8. Zhiyu W, Wang N, Wang Q, Peng C, Zhang J, Liu P, et al. The Inflammasome: An Emerging Therapeutic Oncotarget for Cancer Prevention. Oncotarget (2016) 7:50766–80. doi: 10.18632/oncotarget.9391
9. Lin T-Y, Tsai M-C, Tu W, Yeh H-C, Wang S-C, Huang S-P, et al. Role of the NLRP3 Inflammasome: Insights Into Cancer Hallmarks. Front Immunol (2020) 11:610492. doi: 10.3389/fimmu.2020.610492
10. Sharma BR, Kanneganti T-D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat Immunol (2021) 22:550–9. doi: 10.1038/s41590-021-00886-5
11. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 Inflammasome in Cancer. Mol Cancer (2018) 17:158. doi: 10.1186/s12943-018-0900-3
12. Mezzasoma L, Costanzi E, Scarpelli P, Talesa VN, Bellezza I. Extracellular Vesicles From Human Advanced-Stage Prostate Cancer Cells Modify the Inflammatory Response of Microenvironment-Residing Cells. Cancers (2019) 11:E1276. doi: 10.3390/cancers11091276
13. Mezzasoma L, Bellezza I, Orvietani P, Manni G, Gargaro M, Sagini K, et al. Amniotic Fluid Stem Cell-Derived Extracellular Vesicles Are Independent Metabolic Units Capable of Modulating Inflammasome Activation in THP-1 Cells. FASEB J (2022) 36:e22218. doi: 10.1096/fj.202101657R
14. Engin A. Dark-Side of Exosomes. Adv Exp Med Biol (2021) 1275:101–31. doi: 10.1007/978-3-030-49844-3_4
15. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J Extracell Vesicles (2018) 1:1535750. doi: 10.1080/20013078.2018.1535750
16. Mathieu M, Martin-Jaular L, Lavieu G, Théry C. Specificities of Secretion and Uptake of Exosomes and Other Extracellular Vesicles for Cell-to-Cell Communication. Nat Cell Biol (2019) 21:9–17. doi: 10.1038/s41556-018-0250-9
17. van Niel G, D’Angelo G, Raposo G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat Rev Mol Cell Biol (2018) 19:213–28. doi: 10.1038/nrm.2017.125
18. Costanzi E, Romani R, Scarpelli P, Bellezza I. Extracellular Vesicles-Mediated Transfer of Mirna Let-7b From PC3 Cells to Macrophages. Genes (2020) 12:1495. doi: 10.3390/genes11121495
19. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat Rev Cancer (2017) 17:302–17. doi: 10.1038/nrc.2017.6
20. Bellezza I, Aisa MC, Palazzo R, Costanzi E, Mearini E, Minelli A. Extracellular Matrix Degrading Enzymes at the Prostasome Surface. Prostate Cancer Prostatic Dis (2005) 4:344–8. doi: 10.1038/sj.pcan.4500828
21. Maacha S, Bhat AA, Jimenez L, Raza A, Haris M, Uddin S, et al. Extracellular Vesicles-Mediated Intercellular Communication: Roles in the Tumor Microenvironment and Anti-Cancer Drug Resistance. Mol Cancer (2019) 18:55. doi: 10.1186/s12943-019-0965-7
22. Lin W, Xu Y, Chen X, Liu J, Weng Y, Zhuang Q, et al. Radiation-Induced Small Extracellular Vesicles as ‘Carriages’ Promote Tumor Antigen Release and Trigger Antitumor Immunity. Theranostics (2020) 10:4871–84. doi: 10.7150/thno.43539
23. Squadrito ML, Cianciaruso C, Hansen SK, De Palma M. EVIR: Chimeric Receptors That Enhance Dendritic Cell Cross-Dressing With Tumor Antigens. Nat Methods (2018) 15:183–6. doi: 10.1038/nmeth.4579
24. Bobrie A, Colombo M, Raposo G, Théry C. Exosome Secretion: Molecular Mechanisms and Roles in Immune Responses. Traffic (2011) 12:1659–68. doi: 10.1111/j.1600-0854.2011.01225.x
25. Théry C, Ostrowski M, Segura E. Membrane Vesicles as Conveyors of Immune Responses. Nat Rev Immunol (2009) 9:581–93. doi: 10.1038/nri2567
26. Giusti I, Di Francesco M, Poppa G, Esposito L, D’Ascenzo S, Dolo V. Tumor-Derived Extracellular Vesicles Activate Normal Human Fibroblasts to a Cancer-Associated Fibroblast-Like Phenotype, Sustaining a Pro-Tumorigenic Microenvironment. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.839880
27. Graner MW. Extracellular Vesicles in Cancer Immune Responses: Roles of Purinergic Receptors. Semin Immunopathol (2018) 40:465–75. doi: 10.1007/s00281-018-0706-9
28. Dinarello CA. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
29. Apte RN, Voronov E. Immunotherapeutic Approaches of IL-1 Neutralization in the Tumor Microenvironment. J Leukoc Biol (2017) 102:293–306. doi: 10.1189/jlb.3MR1216-523R
30. Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int J Mol Sci (2018) 19(8):2155. doi: 10.3390/ijms19082155
31. Gelfo V, Romaniello D, Mazzeschi M, Sgarzi M, Grilli G, Morselli A, et al. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int J Mol Sci (2020) 21(17):E6009. doi: 10.3390/ijms21176009
32. Milosevic V, Kopecka J, Salaroglio IC, Libener R, Napoli F, Izzo S, et al. Wnt/IL-1β/IL-8 Autocrine Circuitries Control Chemoresistance in Mesothelioma Initiating Cells by Inducing ABCB5. Int J Cancer (2020) 146:192–207. doi: 10.1002/ijc.32419
33. Herroon MK, Diedrich JD, Rajagurubandara E, Martin C, Maddipati KR, Kim S, et al. Prostate Tumor Cell-Derived IL1β Induces an Inflammatory Phenotype in Bone Marrow Adipocytes and Reduces Sensitivity to Docetaxel via Lipolysis-Dependent Mechanisms. Mol Cancer Res (2019) 17:2508–21. doi: 10.1158/1541-7786.MCR-19-0540
34. Bondong S, Kiefel H, Hielscher T, Zeimet AG, Zeillinger R, Pils D, et al. Prognostic Significance of L1CAM in Ovarian Cancer and Its Role in Constitutive NF-κb Activation. Ann Oncol Off J Eur Soc Med Oncol (2012) 23:1795–802. doi: 10.1093/annonc/mdr568
35. Turzanski J, Grundy M, Russell NH, Pallis M. Interleukin-1beta Maintains an Apoptosis-Resistant Phenotype in the Blast Cells of Acute Myeloid Leukaemia via Multiple Pathways. Leukemia (2004) 18:1662–70. doi: 10.1038/sj.leu.2403457
36. Teixeira LFS, Peron JPS, Bellini MH. Silencing of Nuclear Factor Kappa B 1 Gene Expression Inhibits Colony Formation, Cell Migration and Invasion via the Downregulation of Interleukin 1 Beta and Matrix Metallopeptidase 9 in Renal Cell Carcinoma. Mol Biol Rep (2020) 47:1143–51. doi: 10.1007/s11033-019-05212-9
37. Arlt A, Vorndamm J, Müerköster S, Yu H, Schmidt WE, Fölsch UR, et al. Autocrine Production of Interleukin 1beta Confers Constitutive Nuclear Factor Kappab Activity and Chemoresistance in Pancreatic Carcinoma Cell Lines. Cancer Res (2002) 62:910–6.
38. Angst E, Reber HA, Hines OJ, Eibl G. Mononuclear Cell-Derived Interleukin-1 Beta Confers Chemoresistance in Pancreatic Cancer Cells by Upregulation of Cyclooxygenase-2. Surgery (2008) 144:57–65. doi: 10.1016/j.surg.2008.03.024
39. Nagai N, Tian X, Mukai K, Hirata E, Kusuda T, Shiroyama Y, et al. Overexpression of Cyclooxygenase-2 Protein and Its Relationship to Apoptosis in Cervical Carcinoma Treated With Neoadjuvant Chemotherapy. Int J Mol Med (2003) 12:709–14. doi: 10.3892/ijmm.12.5.709
40. Saikawa Y, Sugiura T, Toriumi F, Kubota T, Suganuma K, Isshiki S, et al. Cyclooxygenase-2 Gene Induction Causes CDDP Resistance in Colon Cancer Cell Line, HCT-15. Anticancer Res (2004) 24:2723–8.
41. Matsumoto R, Tsuda M, Yoshida K, Tanino M, Kimura T, Nishihara H, et al. Aldo-Keto Reductase 1C1 Induced by Interleukin-1β Mediates the Invasive Potential and Drug Resistance of Metastatic Bladder Cancer Cells. Sci Rep (2016) 6:34625. doi: 10.1038/srep34625
42. Jiménez-Garduño AM, Mendoza-Rodríguez MG, Urrutia-Cabrera D, Domínguez-Robles MC, Pérez-Yépez EA, Ayala-Sumuano JT, et al. Il-1β Induced Methylation of the Estrogen Receptor Erα Gene Correlates With EMT and Chemoresistance in Breast Cancer Cells. Biochem Biophys Res Commun (2017) 490:780–5. doi: 10.1016/j.bbrc.2017.06.117
43. Mendoza-Rodríguez MG, Ayala-Sumuano JT, García-Morales L, Zamudio-Meza H, Pérez-Yepez EA, Meza I. Il-1β Inflammatory Cytokine-Induced TP63 Isoform ΔNP63α Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells. Int J Mol Sci (2019) 20:E270. doi: 10.3390/ijms20020270
44. Mendoza-Rodríguez M, Arévalo Romero H, Fuentes-Pananá EM, Ayala-Sumuano J-T, Meza I. Il-1β Induces Up-Regulation of BIRC3, A Gene Involved in Chemoresistance to Doxorubicin in Breast Cancer Cells. Cancer Lett (2017) 390:39–44. doi: 10.1016/j.canlet.2017.01.005
45. Wilson BJ, Saab KR, Ma J, Schatton T, Pütz P, Zhan Q, et al. ABCB5 Maintains Melanoma-Initiating Cells Through a Proinflammatory Cytokine Signaling Circuit. Cancer Res (2014) 74:4196–207. doi: 10.1158/0008-5472.CAN-14-0582
46. Lu L, Wang P, Zou Y, Zha Z, Huang H, Guan M, et al. Il-1β Promotes Stemness of Tumor Cells by Activating Smad/ID1 Signaling Pathway. Int J Med Sci (2020) 17:1257–68. doi: 10.7150/ijms.44285
47. Broz P, Dixit VM. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
48. van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LAB. Inflammasome Activation and IL-1β and IL-18 Processing During Infection. Trends Immunol (2011) 32:110–6. doi: 10.1016/j.it.2011.01.003
49. Zheng D, Liwinski T, Elinav E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discovery (2020) 6:36. doi: 10.1038/s41421-020-0167-x
50. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in Pyroptosis. Acta Pharm Sin B (2021) 11:2768–82. doi: 10.1016/j.apsb.2021.02.006
51. Oroz J, Barrera-Vilarmau S, Alfonso C, Rivas G, de Alba E. ASC Pyrin Domain Self-Associates and Binds NLRP3 Protein Using Equivalent Binding Interfaces. J Biol Chem (2016) 291:19487–501. doi: 10.1074/jbc.M116.741082
52. Song N, Li T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front Immunol (2018) 9:2305. doi: 10.3389/fimmu.2018.02305
53. Mezzasoma L, Talesa VN, Romani R, Bellezza I. ANP and BNP Exert Anti-Inflammatory Action via NPR-1/Cgmp Axis by Interfering With Canonical, Non-Canonical, and Alternative Routes of Inflammasome Activation in Human THP1 Cells. Int J Mol Sci (2020) 22:E24. doi: 10.3390/ijms22010024
54. Hamarsheh S, Zeiser R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front Immunol (2020) 11:1444. doi: 10.3389/fimmu.2020.01444
55. Li L, Jiang M, Qi L, Wu Y, Song D, Gan J, et al. Pyroptosis, a New Bridge to Tumor Immunity. Cancer Sci (2021) 112:3979–94. doi: 10.1111/cas.15059
56. Feng X, Luo Q, Zhang H, Wang H, Chen W, Meng G, et al. The Role of NLRP3 Inflammasome in 5-Fluorouracil Resistance of Oral Squamous Cell Carcinoma. J Exp Clin Cancer Res (2017) 36:81. doi: 10.1186/s13046-017-0553-x
57. Kong H, Wang Y, Zeng X, Wang Z, Wang H, Xie W. Differential Expression of Inflammasomes in Lung Cancer Cell Lines and Tissues. Tumour Biol (2015) 36:7501–13. doi: 10.1007/s13277-015-3473-4
58. Ren N, Jiang T, Wang C, Xie S, Xing Y, Piao D, et al. Lncrna ADAMTS9-AS2 Inhibits Gastric Cancer (GC) Development and Sensitizes Chemoresistant GC Cells to Cisplatin by Regulating Mir-223-3p/NLRP3 Axis. Aging (2020) 12:11025–41. doi: 10.18632/aging.103314
59. Wang F, Xu C, Li G, Lv P, Gu J. Incomplete Radiofrequency Ablation Induced Chemoresistance by Up-Regulating Heat Shock Protein 70 in Hepatocellular Carcinoma. Exp Cell Res (2021) 409:112910. doi: 10.1016/j.yexcr.2021.112910
60. Quagliariello V, Berretta M, Buccolo S, Iovine M, Paccone A, Cavalcanti E, et al. Polydatin Reduces Cardiotoxicity and Enhances the Anticancer Effects of Sunitinib by Decreasing Pro-Oxidative Stress, Pro-Inflammatory Cytokines, and NLRP3 Inflammasome Expression. Front Oncol (2021) 11:680758. doi: 10.3389/fonc.2021.680758
61. Maayah ZH, Takahara S, Dyck JRB. The Beneficial Effects of Reducing NLRP3 Inflammasome Activation in the Cardiotoxicity and the Anti-Cancer Effects of Doxorubicin. Arch Toxicol (2021) 95:1–9. doi: 10.1007/s00204-020-02876-2
62. Mezzasoma L, Talesa VN, Costanzi E, Bellezza I. Natriuretic Peptides Regulate Prostate Cells Inflammatory Behavior: Potential Novel Anticancer Agents for Prostate Cancer. Biomolecules (2021) 11:794. doi: 10.3390/biom11060794
63. Chen J, Sun W, Zhang H, Ma J, Xu P, Yu Y, et al. Macrophages Reprogrammed by Lung Cancer Microparticles Promote Tumor Development via Release of IL-1β. Cell Mol Immunol (2020) 17:1233–44. doi: 10.1038/s41423-019-0313-2
64. Liang M, Chen X, Wang L, Qin L, Wang H, Sun Z, et al. Cancer-Derived Exosomal TRIM59 Regulates Macrophage NLRP3 Inflammasome Activation to Promote Lung Cancer Progression. J Exp Clin Cancer Res (2020) 39:176. doi: 10.1186/s13046-020-01688-7
65. Arienti C, Pignatta S, Zanoni M, Zamagni A, Cortesi M, Sarnelli A, et al. High-Pressure Oxygen Rewires Glucose Metabolism of Patient-Derived Glioblastoma Cells and Fuels Inflammasome Response. Cancer Lett (2021) 506:152–66. doi: 10.1016/j.canlet.2021.02.019
66. Han C, Godfrey V, Liu Z, Han Y, Liu L, Peng H, et al. The AIM2 and NLRP3 Inflammasomes Trigger IL-1-Mediated Antitumor Effects During Radiation. Sci Immunol (2021) 6:eabc6998. doi: 10.1126/sciimmunol.abc6998
67. Singla DK, Johnson TA, Tavakoli Dargani Z. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells (2019) 8:E1224. doi: 10.3390/cells8101224
68. Tengesdal IW, Menon DR, Osborne DG, Neff CP, Powers NE, Gamboni F, et al. Targeting Tumor-Derived NLRP3 Reduces Melanoma Progression by Limiting Mdscs Expansion. Proc Natl Acad Sci USA (2021) 118:e2000915118. doi: 10.1073/pnas.2000915118
69. Yaw ACK, Chan EWL, Yap JKY, Mai CW. The Effects of NLRP3 Inflammasome Inhibition by MCC950 on LPS-Induced Pancreatic Adenocarcinoma Inflammation. J Cancer Res Clin Oncol (2020) 146:2219–29. doi: 10.1007/s00432-020-03274-y
70. Chen L, Huang C-F, Li Y-C, Deng W-W, Mao L, Wu L, et al. Blockage of the NLRP3 Inflammasome by MCC950 Improves Anti-Tumor Immune Responses in Head and Neck Squamous Cell Carcinoma. Cell Mol Life Sci (2018) 75:2045–58. doi: 10.1007/s00018-017-2720-9
71. Wang X, Ma L, Ding Q-Y, Zhang W-Y, Chen Y-G, Wu J-H, et al. Microglial NLRP3 Inflammasome Activation-Mediated Inflammation Promotes Prolactinoma Development. Endocr Relat Cancer (2021) 28:433–48. doi: 10.1530/ERC-21-0137
72. Li X, Zhang C-T, Ma W, Xie X, Huang Q. Oridonin: A Review of Its Pharmacology, Pharmacokinetics and Toxicity. Front Pharmacol (2021) 12:645824. doi: 10.3389/fphar.2021.645824
73. Yang Y-S, Wen D, Zhao X-F. Preventive and Therapeutic Effect of Intraportal Oridonin on Balb/C Nude Mice Hemispleen Model of Colon Cancer Liver Metastasis. Transl Cancer Res (2021) 10:1324–35. doi: 10.21037/tcr-20-3042
74. Wen D, Yang YS, Gao DZ, Wang Z, Jiang QW, Zhao XF. Oridonin Enhances the Anti-Metastasis Effect of Oxaliplatinliplatin on Colorectal Cancer Liver Metastasis. Bull Exp Biol Med (2021) 172:26–32. doi: 10.1007/s10517-021-05324-y
75. Liu J, Xie S, Shao X, Xue S, Du P, Wu H, et al. Identification of New Potent Anticancer Derivatives Through Simplifying the Core Structure and Modification on Their 14- Hydroxyl Group From Oridonin. Eur J Med Chem (2022) 231:114155. doi: 10.1016/j.ejmech.2022.114155
76. Soriano-Teruel PM, García-Laínez G, Marco-Salvador M, Pardo J, Arias M, DeFord C, et al. Identification of an ASC Oligomerization Inhibitor for the Treatment of Inflammatory Diseases. Cell Death Dis (2021) 12:1155. doi: 10.1038/s41419-021-04420-1
77. Cui W, Chen S, Chi Z, Guo X, Zhang X, Zhong Y, et al. Screening-Based Identification of Xanthone as a Novel NLRP3 Inflammasome Inhibitor via Metabolic Reprogramming. Clin Transl Med (2021) 11:e496. doi: 10.1002/ctm2.496
78. McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, et al. Caspase-1 Inhibition Prevents Glial Inflammasome Activation and Pyroptosis in Models of Multiple Sclerosis. Proc Natl Acad Sci USA (2018) 115:E6065–74. doi: 10.1073/pnas.1722041115
79. Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JCR, et al. IL-Converting Enzyme/Caspase-1 Inhibitor VX-765 Blocks the Hypersensitive Response to an Inflammatory Stimulus in Monocytes From Familial Cold Autoinflammatory Syndrome Patients. J Immunol (2005) 175:2630–4. doi: 10.4049/jimmunol.175.4.2630
80. Kast RE. Ritonavir and Disulfiram may be Synergistic in Lowering Active Interleukin-18 Levels in Acute Pancreatitis, and Thereby Hasten Recovery. JOP (2008) 9:350–3.
81. Batchu RB, Gruzdyn OV, Bryant CS, Qazi AM, Kumar S, Chamala S, et al. Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways. Pharm Basel Switz (2014) 1:46–57. doi: 10.3390/ph7010046
82. Gottschlich A, Endres S, Kobold S. Therapeutic Strategies for Targeting IL-1 in Cancer. Cancers (2021) 13:477. doi: 10.3390/cancers13030477
83. Mezzasoma L, Antognelli C, Talesa VN. Atrial Natriuretic Peptide Down-Regulates LPS/ATP-Mediated IL-1β Release by Inhibiting NF-Kb, NLRP3 Inflammasome and Caspase-1 Activation in THP-1 Cells. Immunol Res (2016) 64:303–12. doi: 10.1007/s12026-015-8751-0
84. Mezzasoma L, Antognelli C. Talesa VN. A Novel Role for Brain Natriuretic Peptide: Inhibition of IL-1β Secretion via Downregulation of NF-Kb/Erk 1/2 and NALP3/ASC/Caspase-1 Activation in Human THP-1 Monocyte. Mediators Inflammation (2017) 2017:5858315. doi: 10.1155/2017/5858315
85. Steiner LF, Summerland SA. Xanthone as an Ovicide and Larvicide for the Codling Moth. J Econ Entomol (1943) 36:435–9. doi: 10.1093/jee/36.3.435
86. Lee C, Do HTT, Her J, Kim Y, Seo D, Rhee I. Inflammasome as a Promising Therapeutic Target for Cancer. Life Sci (2019) 231:116593. doi: 10.1016/j.lfs.2019.116593
87. Mezzasoma L, Peirce MJ, Minelli A, Bellezza I. Natriuretic Peptides: The Case of Prostate Cancer. Molecules (2017) 22:1680. doi: 10.3390/molecules22101680
88. Suwa M, Seino Y, Nomachi Y, Matsuki S, Funahashi K. Multicenter Prospective Investigation on Efficacy and Safety of Carperitide for Acute Heart Failure in the ‘Real World’ of Therapy. Circ J (2005) 69:283–90. doi: 10.1253/circj.69.283
89. Nomura F, Kurobe N, Mori Y, Hikita A, Kawai M, Suwa M, et al. Multicenter Prospective Investigation on Efficacy and Safety of Carperitide as a First-Line Drug for Acute Heart Failure Syndrome With Preserved Blood Pressure: COMPASS: Carperitide Effects Observed Through Monitoring Dyspnea in Acute Decompensated Heart Failure Study. Circ J (2008) 72:1777–86. doi: 10.1253/circj.CJ-07-0760
Keywords: extracellular vesicles, inflammasome, tumor microenvironment, IL-1β, chemoresistance
Citation: Mezzasoma L, Bellezza I, Romani R and Talesa VN (2022) Extracellular Vesicles and the Inflammasome: An Intricate Network Sustaining Chemoresistance. Front. Oncol. 12:888135. doi: 10.3389/fonc.2022.888135
Received: 02 March 2022; Accepted: 23 March 2022;
Published: 22 April 2022.
Edited by:
Stefano Falone, University of L’Aquila, ItalyReviewed by:
Maria Felice Brizzi, University of Turin, ItalyKaren Sfanos, Johns Hopkins Medicine, United States
Hee Sung Kim, Chung-Ang University, South Korea
Marzia Pucci, University of Palermo, Italy
Copyright © 2022 Mezzasoma, Bellezza, Romani and Talesa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Letizia Mezzasoma, bGV0aXppYS5tZXp6YXNvbWFAdW5pcGcuaXQ=
†These authors have contributed equally to this work and share first authorship